Embed Size (px)
Citation preview

SURFACE SCIENCE 28 (I9711 395-408 0 North-Holland Publishing Co.
INVESTIGATION OF LONGTER~ PROCESSES ON A REAL
GERMANIUM SURFACE
S. N. KOZLOV, V. F. KISELEV and YU. F. NOVOTOTSKII-VLASOV
Physics Department, Moscow State University, Moscow, U.S.S.R.
Received 14 April 1971; revised manuscript received 30 June 1971
The surface charging of germanium and the kinetics of slow relaxation in various gaseous media have been investigated. The experimental data are compared with different slow relaxation models. It is shown that the electron capture model on the heterogenous surface of a semiconductor is in a good agreement with the experimental data. The possible mechanisms of semiconductor surface charging by adsorption are discussed,
1. Introduction
It is common knowledge that absorption of certain molecules leads to a
change in the surface charge of germaniuml). The additional surface charge
AQ, usually arising in adsorption is almost completely determined by the
charge in the slow surface states (AQss). Therefore investigation of the
surface charging mechanism in adsorption is intimately connected with the
study of the nature of slow states. In this sphere, a great deal of information
is gained by investigating the kinetics of slow relaxation in adsorption and in
the field effect. Kinetic measurements supply data on the mechanism of
charge exchange between the semiconductor volume and surface states.
Despite the great number of investigations of slow surface states [see
reviewsI-s)] our knowledge of their nature and parameters is very scanty.
These investigations were, for the greatest part, carried out in humid media,
often with uncontrolled compositions and pressures. As it is well knowns),
water interacts in a complex manner with a real germanium surface, and
this greatly complicates the interpretation of the data obtained. A number of
mechanisms of slow relaxation have been suggested, but none of them can
explain the whole set of laws observed.
With a view to constructing a better substantiated model of germanium
surface charging in adsorption, we found it expedient to extend substantially
the variety of adsorbate molecules to be investigated and also to ascertain
the effect of the hydrate cover of the oxide film on the slow relaxation. To
obtain more extensive information on the nature of slow states, we used
395

396 S. i-4. KOZLOV, V. F. KISELEV AND YU. F. NOVOTOTSKII-VLA~V
adsorption and rad~osp~~troscopic methods of investigation together with
the electrophysical methods.
2. Experimental
In studying the electrophysical parameters of the surface, we used equip-
ment which enabled us to follow simultaneously the changes in the system
of fast and slow states under active external effects. The conductance of
single-crystal germanium specimens was recorded by the bridge method.
The surface potential and capture by fast and, sometimes, slow states were
investigated by the method of the large-signal field effect on a sinusoidal
signal. In our set-up, the passing over from the bridge measurement method
to the ac field effect method took only few seconds, and thus we were able to
check the absence of the accumulation effect4). Mica and a vacuum (gas) gap
were used as dielectrics in the field capacitor. Check-up experiments showed
that under our experimental conditions the contact of the specimen surface
with the mica did not substantially affect the electrophysical parameters of
the surface (including the kinetics of slow relaxation). Adsorption was
measured by the volumetric method. Heats of absorption were determined
by a differential-type Kalve calorimeter. EPR spectra were taken on a
standard radiospectrometer RE 1301.
The adsorbates used included thorougly purified water, ammonia, carbon
monoxide, carbon dioxide, nitrogen oxide, parabensoquinone (p-BQ) di-
phenylamine (DPA), oxygen, and iodine. In electrophysical investigations
use was made of single-crystal plates (20 x 5 x 0.5 mm3 in size) of high-resis-
tivity n-germanium cut out parallel to the (Ill) plane and treated in a
peroxide etchant. Adsorption measurements were made with coarsely
dispersed, non-aggregated 5, germanium powders (of average particle size ex-
ceeding the Debye screening length), treated, as the single crystals, in H,O,.
After etching, both the single crystals and the powders of germanium were
outgassed in vacuum (N lop6 Torr) at three different temperatures viz.
27°C (Ge-27), 200°C (Ge-200), and 400°C (Ge-400). According to refs. 5
and 6 the water content in these specimens was anZo= 1.5 x 101’, 7 x lOi
and 3 x 1014 molecules cm-’ 3 respectively. Similar values of xHzo for GeO,
were obtained in ref. 7.
3. Results and discussion
3.1. INVESTIO.~TION INTO KINETICS 0~ 5x0~ RELAXATION
The investigations showed that in all gaseous media employed in the
present work and with all surface treatments used, the kinetics of slow

LONG-TERM PROCESSESON Ge SURFACE 397
relaxation after switching off the transverse field (surface potential variations
AY< 1.5) is well approximated by the empirical functions suggested by
Koc8)
AQss = AQ& ev[- W4‘T (1) with parameter a=0.3. Under our experimental conditions the value of
z eR in eq. (1) was independent of the amplitude and polarity of the external
field (see fig. 1 b).
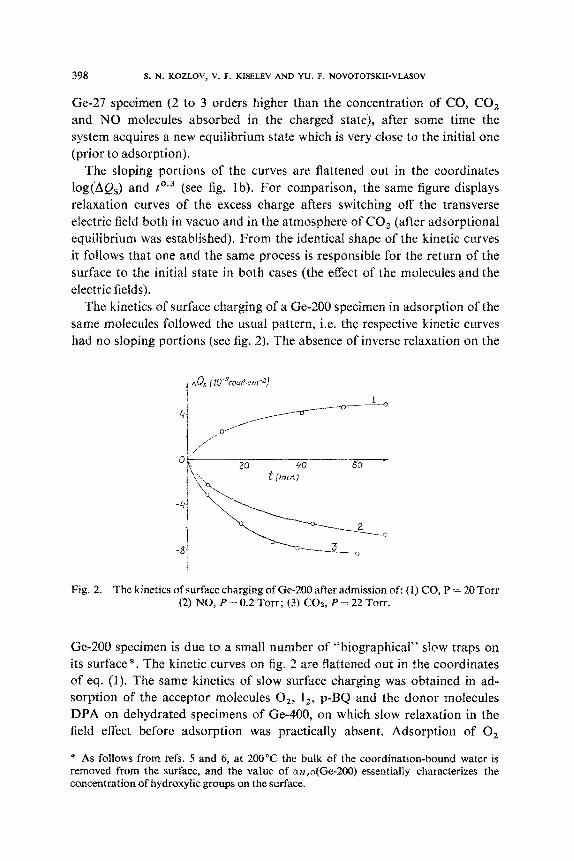
The kinetics of surface charging of a Ge-27 specimen on admission of
CO2 at three different pressures is shown in fig. 1. Similar curves were ob-
2.4 t (mrnjo3
Fig. 1. The kinetics of surface charging of Ge-27 on admission of COZ, P = OS-50 Torr (a and b, curves l-3), and after switching off the transverse electric fields of different am-
plitudes and polarities (b, curves 4-7).
tained in adsorption of CO and NO. As can be seen from fig. la, admission
of the gas is immediately followed by a relatively fast (2 to 4 min) negative
charging of the surface with a subsequent gradual drop in excess charge
which had arisen in adsorption. The sloping relaxation branches (fig. la) may
be attributed to the fact that in adsorption of CO, (and also of CO and NO)
the system of “biographical” slow states is thrown out of equilibrium.
According to refs. 5 and 6, as a result of pumping-out at 27°C only water
molecules weakly bound by the hydrogen bond with the surface hydroxyls
desorb from the germanium surface. The great majority of the active centres
are blocked by water molecules coordination-bound with the surface atoms
of Ge. These molecules may be responsible for the system of “biographical”
states. Owing to the high concentration of the latter on the surface of the
398 S. N. KOZLOV, V. F. KISELEV AND YU. F. NOVOTOTSKII-VLASOV
Ge-27 specimen (2 to 3 orders higher than the concentration of CO, CO, and NO molecules absorbed in the charged state), after some time the system acquires a new equilibrium state which is very close to the initial one (prior to adsorption).
The sloping portions of the curves are flattened out in the coordinates log(AQs) and toS3 (see fig. lb). For comparison, the same figure displays relaxation curves of the excess charge afters switching off the transverse electric field both in vacua and in the atmosphere of CO, (after adsorptional equilibrium was established). From the identical shape of the kinetic curves it follows that one and the same process is responsible for the return of the surface to the initial state in both cases (the effect of the molecules and the electric fields).
The kinetics of surface charging of a Ge-200 specimen in adsorption of the same molecules followed the usual pattern, i.e. the respective kinetic curves had no sloping portions (see fig. 2). The absence of inverse relaxation on the
Fig. 2. The kinetics of surface charging of Ge-200 after admission of: (1) CO, P = 20 Torr (2) NO, P = 0.2 Torr; (3) CO%, P = 22 Torr.
Ge-200 specimen is due to a small number of “‘biographical” slow traps on its surface*. The kinetic curves on fig. 2 are flattened out in the coordinates of eq. (I). The same kinetics of slow surface charging was obtained in ad- sorption of the acceptor molecules 0 2, I,, p-BQ and the donor molecules DPA on dehydrated specimens of Ge-400, on which slow relaxation in the field effect before adsorption was practically absent. Adsorption of 0,
* As follows from refs. 5 and 6, at 200°C the bulk of the coordination-bound water is removed from the surface, and the value of aB%o(Ge-200) essentially characterizes the concentration of hydroxylic groups on the surface.
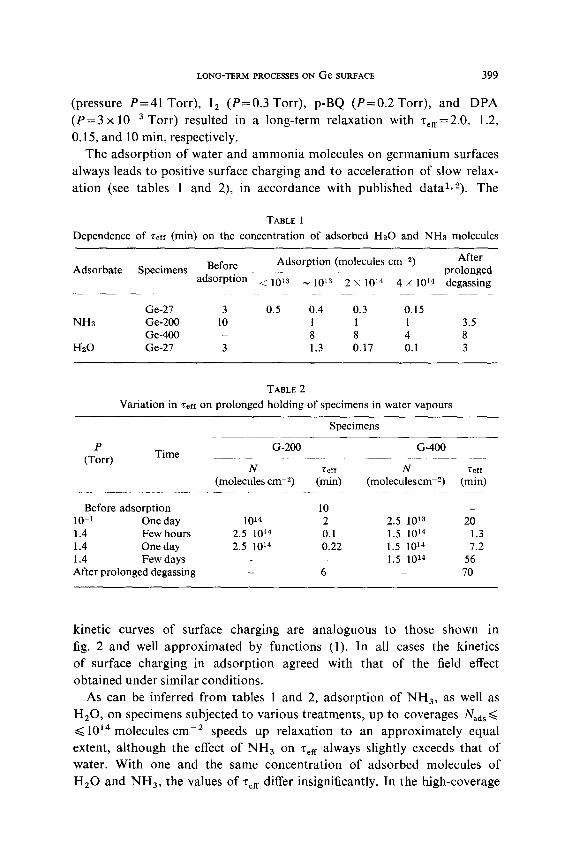
LONG-TERM PROCESSESON Ge SURFACE 399
(pressure P=41 Torr), I, (P=O.3 Torr), p-BQ (P=O.2 Torr), and DPA
(P= 3 x 10m3 Torr) resulted in a long-term relaxation with z,~= 2.0, 1.2,
0.15, and 10 min, respectively.
The adsorption of water and ammonia molecules on germanium surfaces
always leads to positive surface charging and to acceleration of slow relax-
ation (see tables 1 and 2), in accordance with published dataly2). The
TABLE 1
Dependence of reef (min) on the concentration of adsorbed Ha0 and NH3 molecules
Adsorbate Specimens Before Adsorption (molecules cm-2) After ~_____
adsorption < 1013 prolonged
N 1Ol3 2 x lOI* 4 x 1014 degassing
Ge-27 3 0.5 0.4 0.3 0.15 NH3 Ge-200 10 1 1 1 3.5
Ge-400 - 8 8 4 8 Hz0 Ge-27 3 1.3 0.17 0.1 3
TABLE 2
Variation in T~II on prolonged holding of specimens in water vapours
Specimens
P (Torr) Time
G-200
N (molecules cm-2)
Before adsorption 10-l One day 1.4 Few hours 1.4 One day 1.4 Few days After prolonged degassing
1014
2.5 1014
2.5 10’4
G-400
ren N teec
(min) (moleculescm-z) (min)
10 -
2 2.5 1Ol3 20 0.1 1.5 10’4 1.3 0.22 1.5 1014 7.2 - 1.5 1014 56 6 _ 70
kinetic curves of surface charging are analoguous to those shown in
fig. 2 and well approximated by functions (1). In all cases the kinetics
of surface charging in adsorption agreed with that of the field effect
obtained under similar conditions.
As can be inferred from tables 1 and 2, adsorption of NH,, as well as
H,O, on specimens subjected to various treatments, up to coverages Nads d
< 1014 molecules cmm2 speeds up relaxation to an approximately equal
extent, although the effect of NH, on z,~ always slightly exceeds that of
water. With one and the same concentration of adsorbed molecules of
H,O and NH,, the values of tetf differ insignificantly. In the high-coverage

400 S. N. KOZLOV, V. F. KISELEV AND YU. F. NOVOTOTSKII-VLASOV
region (Nads> 1014 molecules cm-‘) adsorption of H,O leads to a sharper
reduction in z,~ than adsorption of NH,. On prolonged holding of dehy-
drated specimens of Ge-200, and especially Ge-400, in water vapours
(table 2) a marked increase in retf in observed (see also ref. 9). This effect is not
observed in adsorption of NH,. In accordance with refs. 5 and 6, during
adsorption of water on specimens dehydrated at temperatures above
300°C slow rehydration processes take place which result in partial recovery
of the hydrate cover of the oxide.
As shown in ref. 8, the time constant reff varies with temperature according
to the law:
zeff = zeff ’ exp (AE,/kT) . (2)
The value of AE, characterized the activation energy of the effective relax-
ation time, whose numerical values for different experimental conditions are
given in table 3. From this table it follows that under identical conditions the
temperature dependence of zctf in the atmosphere of H,O and NH, for
Ge-27 and Ge-200 specimens is characterized by similar values of AEz. For
Ge-400 specimens, reff is altogether independent of temperature (within the
range from - 25 “C to + 50 “C). Long-term processes in the atmosphere of
various gases on identically treated surfaces are characterized by widely
differing values of AE,. We will note here one sufficiently general regularity:
prolonged pumping-out usually led to a reduction in the temperature
dependence of slow relaxation, along with an increase in z,e (see table 3).
TABLE 3
Activation energy AEr of teff in the environment of different gases and vapours
Adsorbate P
(Torr) Ge-27
Specimens
Ge-200 Ge-400
Hz0 10-1-l 0.6 0.65 (0.3)* 0 (O)*
NH3 10-1-l 0.6 0.6 (0.25)* 0 (O)* 02 4-l 0.9
PBQ 10-l 0.4 (0.15)*
DPA 10-S 0.2
12 10-l 0.4
* In the parentheses, AE, values are given after prolonged degassing.
3.2. SLOW RELAXATION MODELS
Various slow relaxation models are proposed in the literaturelo-IT), but
none of them can explain the whole set of laws observed by us. Some in-
vestigators1sp17) believe that the entire responsibility for slow processes on
the real surface of semiconductors lies on water molecules. This point of

LONG-TERMPROCESSESON Ge SURFACE 401
view does not agree with our experimental data, which indicate that slow relaxation is observed on the strongly dehydrated, at 300-4OO”C, surfaces in the atmosphere of various gases and vapors, the kinetics of relaxation being the same in all cases. The authors of ref. 16 associate the slow relaxation with the disturbance of electron-ion equilibrium in the oxide layer, which is regarded as an electrolyte. From this standpoint it is impossible to explain the equal effect on slow relaxation of ammonia and water molecules (at IV&< lO’4 molecules cm-‘), which differ drastically in dissociation con- stants and in the ability of chemical interaction with the oxide. As has been indicated above, differences in slow relaxation in the atmosphere of water and ammonia vapours appear only in the region of higher coverages (NaldS > > lOI molecules cm-‘) where the ion processes in the case of adsorption of HZ0 may play a certain role.
Naturally, all the foregoing remarks can be applied as well to the “field” mechanisms of slow relaxation 9917 ). These models are actually particular cases of the electron-ion equilibrium model, when the process limiting slow relaxation is the dissociation of adsorbed molecules17) or the drift of ions in the oxide layera) under the effect of the electric field. According to ref. 17 the electric field is absent in the bulk of the oxide in adsorption of molecules. The “field” mechanisms of relaxation cannot account for the equal kinetics of surface charging (see fig. 1) in the field effect (when the field is present in the oxide) and in adsorption of different molecules [when the field is absent in the oxide7)]. These experimental facts and, besides, a decrease in the temperature dependence of z,~ after pumping-out of the specimens (see table 3) are also in contradiction with the model proposed in refs. 13 and 14.
All our experimental data are in a good agreement with the electron cap- ture modello-la>l*). This model implies that the limiting stage of a slow process is the capture of carriers by surface states separated from the semi- conductor volume by potential barriers and therefore possessing small capture cross section. It is the traditionally accepted view that slow states are localized on the external surface of the oxide film, and the barrier is deter- mined by the forbidden band of the oxide lO~rl>. From this point of view it is difficult to explain the drastically differing temperature dependences of slow relaxation in adsorption of different molecules on one and the same surface (see table 3). Also, this conception does not agree with a set of other datalg-24). Apparently, at least, part of the slow states are localized near the germanium-oxide interface. So far it is hard to say anything definite about the nature of the barriers separating these states from the semiconductor volume. The investigations carried out by us into the temperature dependence of relaxation testify to an important role of the tunnelling mechanism of penetration of carriers through the barrier. The weak dependence of z,~ on

402 S. N. KOZLOV, V. F. KISELEV AND YU. F. NOVOTOTSKII-VLASOV
the temperature observed in some cases (table 3) indicates this. The poten-
tial barriers separating the slow states from the semiconductor are not uni-
form, as it follows from the data of refs. 18 and 25. Taking into account the
inhomogeneity of a real surface we have recentlyls) showed that the theo-
retical relaxation curves obtained from the solution of the general kinetics
equation are well approximated by Koc’s empirical functions (1). This im-
portant conclusion is well confirmed by experimental data considered in the
present paper (fig. 1) and reported in the literatures,s~rs,r4).
3.3. THE EFFECTIVE PARAMETERS OF SLOW SURFACE STATES
At present there are no reliable methods for the evaluation of slow state
parameters. Proceeding from the theoretical consideration of the kinetics of
conductance ralaxation following switching on and off the transverse electric
field and also from experimental data, a method for calculating the effective
parameters of slow states (concentration Ns and effective energy level EJ can be
proposed (see Appendix). Data on the values of Ns and E, for the case of
ammonia adsorption are given in table 4. In the case of water adsorption the
following positions of the effective slow state energy level in the energy
spectrum of the surface have been obtained : for specimens of Ge-200, E, 2: - 1;
for Ge-400, E, ~5.
As it is seen from table 4, in the case of adsorption of NH,, there is good
agreement between the concentration of charged slow states N:, and the
total change in surface charge in adsorption AQ, determined from the field
effect on a sinusoidal signal. In the case of water adsorption this agreement
exists only at low surface coverages (NadSd 1014 molecules cm-“). With
Nads> 1014 molecules cm -’ in this case Nz considerably exceeds AQ,, which
fact, as noted above, is most probably associated with the increasing role of
ion processes. The effective levels in the cases of adsorption of H,O and
NH, under identical conditions were found to be similar, this agreeing with
the identical nature of relaxation in the atmosphere of these vapours. These
facts indicate that slow relaxation can be described satisfactorily provided
the nonhomogeneity of the surface is accounted for by a simple model of the
energy spectrum with a single effective level*. Incidentally, the non-uni-
formity of the surface in the sense of its kinetic behaviour does not imply
non-uniform distribution of E, over the surface, since the relaxation kinetics
is mainly determined by the parameters of the potential barrier separating
the slow state from the semiconductor volume. As mentioned above, the
model in question assumes that the potential barriers are not uniform in
different areas of the surface.
* Naturally, such a phenomenological model does not exclude the possibility of existence of two sets of slow states (acceptor and donor).
LONG-TERM PROCESSESON Ge SURFACE 403
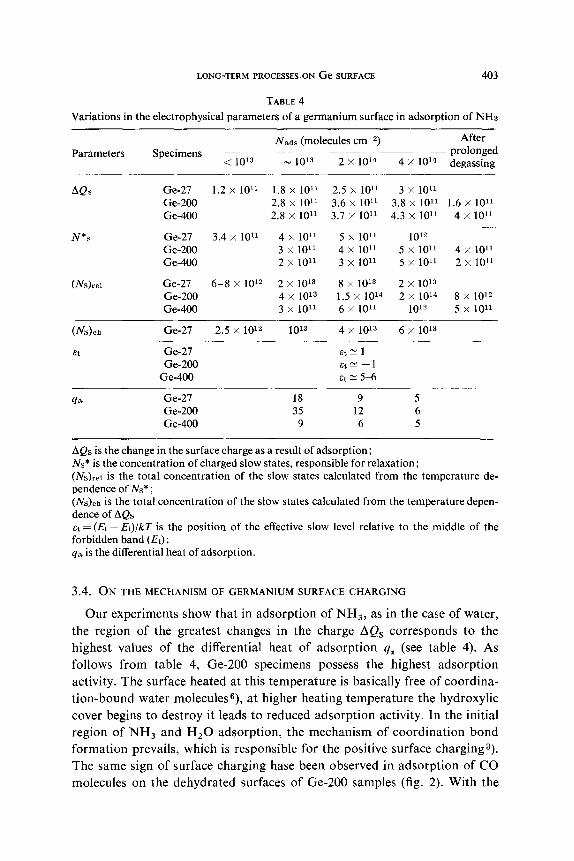
TABLE 4
Variations in the electrophysical parameters of a germanium surface in adsorption of NH3
Parameters Nads (molecules cm+?) After
Specimens -~~ prolonged < 1013 - 10’3 2 x 10’4 4 X 1014 degassing
AQs Ge-27 1.2 x 1Or’ 1.8 x 1O’r 2.5 x 10” 3 x 10” Ge-200 2.8 x 1011 3.6 x 10” 3.8 x 101’ 1.6 x 10” Ge-400 2.8 x 101’ 3.7 x 1O’l 4.3 x 10” 4 x 10’1
N*S Ge-27 3.4 x 1011 4 x 1011 5 x 1011 1012 Ge-200 3 x 10’1 4 x 1011 5 x 10” 4 x 10” Ge-400 2 x 101’ 3 x 10” 5 x 10” 2 x 10”
(N&e] Ge-27 6-8 x 1012 2 x 1013 8 x IOr 2 x 1013 Ge-200 4 x 1013 1.5 x 10’4 2 x 10’4 8 x lo= Ge-400 3 x 101’ 6 x 1O’r 1012 5 x 10”
(N&h
at
4a
Ge-27 2.5 x 1012 1013 4 x 10’3 6 x lo13 ~__~~~__
Ge-27 Et= 1 Ge-200 &tc -1
Ge-400 at ~5-6
Ge-27 18 9 5 Ge-200 35 12 6 Ge-400 9 6 5
AQs is the change in the surface charge as a result of adsorption; Ns* is the concentration of charged slow states, responsible for relaxation; (N&I is the total concentration of the slow states calculated from the temperature de- pendence of Ns* ; (N&h is the total concentration of the slow states calculated from the temperature depen- dence of AQs at = (Et ~ Ei)/kT is the position of the effective slow level relative to the middle of the forbidden band (Ei); qa is the differential heat of adsorption.
3.4. ON THE MECHANISM OF GERMANIUM SURFACE CHARGING
Our experiments show that in adsorption of NH,, as in the case of water,
the region of the greatest changes in the charge AQs corresponds to the
highest values of the differential heat of adsorption qa (see table 4). As
follows from table 4, Ge-200 specimens possess the highest adsorption
activity. The surface heated at this temperature is basically free of coordina-
tion-bound water moleculess), at higher heating temperature the hydroxylic
cover begins to destroy it leads to reduced adsorption activity. In the initial
region of NH, and H,O adsorption, the mechanism of coordination bond
formation prevails, which is responsible for the positive surface charginga).
The same sign of surface charging hase been observed in adsorption of CO
molecules on the dehydrated surfaces of Ge-200 samples (fig. 2). With the

404 S. N. KOZLOV, V. F. KISELEV AND YU. F, NOVOTOTSKII-VLASOV
coordination type of bond unshared electron pairs of H,O, NH,, and CO
molecules are drawn into vacant d-orbitafs of Ge atoms located near the
Ge-GeO, interface. Such Ge atoms acquire a certain effective charge
6 <q, where q is the electron charge, and can turn into focalization centres of
free holes. Effective dipoles ( +6, -6) may change the existing spectrum of
biographical surface states. At higher coverages of the surface by adsorbed
H,O and NH, molecules (Nads>, 10”” molecules cm-‘) the hydrogen bond
mechanism prevails3,26), 4, shifts toward the heat of condensation and the
effect of adsorption on AQ, decreases steeply {see table 3).
In the case of adsorption of H,O and NH, molecules, surface charging is
associated with the partial transfer of the charge from the semiconductor to
the orbitafs of the adsorbed particle (6 ~4)~). In adsorption of p-BQ and
DPA molecules we observed the formation of anion- and cation-radicals,
that is a complete transfer of the electron (6-+q) occurs and a charge-transfer
complex (CTC) arises. Adsorption of DPA at 300°C (P-10 Torr) on a
Ge-400 specimen resulted in positive charging of the surface (AQs- 10”
electrons cme2). At the same time, a structureless EPR signal with a fine
width of AB= 3.5 G and g =2.004 appeared. The spin concentration (A$,) is
in good agreement with the concentration of the charge captured on the
surface AQ,). In the case of adsorption of p-BQ (P- 10-l Torr, N&N IOr
molecules cm-‘) on a Ge-400 specimen at 27”C, we observed fast negative
surface charging (AQ, - 10” electrons cm-‘) had also the appearance of an
EPR signal with a smeared out hfs, AB=7 to 8 G and g =2.004. The spin
concentration was similar to the number of charges localized on the surface.
The unpaired electron is sufficiently delocalized inside the p-BQ molecules
and interacts weakly with the adsorption centre. Adsorption on a pure
germanium oxide (GeO,) fed to the appearance of an EPR signal corre-
sponding to a spin concentration lower by one order (IV,,- 10” spin cm-‘).
It follows herefrom that the semiconductor is responsible for the formation
of most of the spins. The good agreement between the number of ion-
radicals formed and the number of free charge carriers localized on the
surface, indicates unambiguously that in the formation of CTC free electrons
and holes of the semiconductor crystal are used regardless of the details of
capture mechanism.
Analyzing the body of data available we may imagine the following
hypothetical picture of germanium surface charging in adsorption (fig. 3).
The adsorbed molecules freely penetrate through the sufficiently porous
GeO, film and settle down near the germanium-oxide interface. The similar
nature of charging in adsorption of molecules differs in size points to the
presence of comparatively wide pores. As we have already noted, in the
case of adsorption of such dissimilar molecules as H,O and NH, we observed

LONG-TERMPROCESSES ON Ge SURFACE 405
a striking agreement between the kinetic (z and AE,) and equilibrium
(AQ,, NC and E,) parameters of the appearing surface states. This most
probably indicates that complexes of molecules with adsorption centres,
rather than the adsorbed molecules themselves, are surface states. These
adsorption centres should be sufficiently inactive in oxidation reaction,
otherwise it would be difficult to explain the absence of overgrowth of pores
during the storage of the samples. Consequently, they cannot be atoms of a
regular germanium lattice or atoms with ruptured bonds. They are most
likely the extreme atoms of germanium hydrated in the course of genesis of
the oxide film (fig. 3). The presence of hydroxylic groups prevents the
oxidation of these surface elements. Such hydrated Ge atoms are coordi-
I I
Fig. 3. The scheme of the possible mechanism of interaction of the adsorbed molecules on the germanium surface.
nation-unsaturated and, according to refs. 3 and 6, may be adsorption
centres. Redistribution of the electron density in the complexes formed
(fig. 3) may result in the appearance of a slow state. In the case of water and
ammonia molecules, the values of the effective charges 6 <q, in the case of the
formation of CTC (P-BQ and DPA), 6 rq. The share of molecules taking
part in surface charging is small: AQJqAJ,,,- lo-’ to 10e3, which agrees
with previous estimates69 27).
Appendix
The method makes use of the fact that, at not very high perturbations
(AY< l), the relaxation kinetics on switching on the transverse electric field
relative to a new equilibrium state (to which the conductivity rz~r corresponds)
is identical to the relaxation kinetics to the initial state (a,J after switching
off the field. This enables us, by selecting the value of c1 (see fig. 4a) in such
a way that the relaxation kinetics on switching the transverse field on and off
should yield parallel lines in the coordinates 1ogAo and t0.3, to find the

406 S. N, KOZLOV, V. F. KISELEV AND YU. F. NOVOTOTSKII-VLASOV
equilibrium value of cl, corresponding to the field switched on (see fig. 4b) *. The new values of the surface potential (Y,), charge in the fast and slow states (Qrs and Qss) and in the space-charge layer (QsJ correspond to the new equilibrium value of 6r.
So far as the value A(QFs $ Qsc) is known from the data of field-effect on the sinusoidal signal, the change in charge of the slow states can be easily
Fig. 4. The slow relaxation kinetics after switching on and switching off the transverse electric field. (a) Dependence of conductivity on time: cro is the initial equilibria value of the conductivity, and ~1 is the equilibrium value of the conductivity after switching on the transverse electric field. Cb) The slow relaxation kinetics in the coordinates log Ao: versus t0.3: (I) the relaxation to the initial equilibrium state ((TO) after switching on the transverse field; (2) the relaxation to the initial state (CO) after switching off the trans- verse field; (3) the relaxation under switching on the field to a new equilibrium state (a~).
calculated :
ALIQSS = Qind -A(QFs + Qsc>~ (AlI where Qind is the charge induced in semiconductor.
We assume that slow relaxation kinetics is determined completely by the donor** surface states with effective energy level E, and density Ns. The difference in density of the charged slow states under switching on a trans- verse electric field (N,*,) and without any field (Nz) is:
AN; = N; - N,* = N,*:[exp(- AY) - l]
t. + exp Is* - Y, + In ( ni/no)] ’ WI
* AS a rule, direct determination of the value of 01 is jmpossible because of the low speed of the slow reIaxation processes. ** The type of the slow states under investigation can be determed if the sign of surface charging in adsorption is known.

LONG-TERM PROCESSES ON G‘Z SURFACE 401
where AY= Y, - Y,, E,= (Et-- E,)/kT, and the other symbols have their
usual meaningl).
The qualitative information about the disposition of the slow energy
level relative to the Fermi level can be received from the temperature
dependence of surface charging investigation. For example, the positive
surface charge usually increases with increasing temperature, if H,O or NH,
molecules are adsorbed on the germanium surfaces. Hence, the effective
energy levels of corresponding slow states are situated below the Fermi
level. In this case equation (A?) can be rewritten in the form*:
AN: AQss N”*=&p(-AY)- I q[exp(-AY)_’ (A3)
Thus N,* value can easily be calculated from the experimental data. From the
temperature dependence of this magnitude it is possible to find the total
concentration of the slow states responsible for relaxation (Ns) and the
position of the corresponding effective level E, in the energy spectrum of the
surface.
A. Many, Y. Goldstein and N. B. Grover, Semiconductor Surfaces (North-Holland, Amsterdam, 1965). D. R. Frank], Electrical Properties of Semiconductor Surfaces (Pergamon, Oxford, 1967).
5)
6)
V. F. Kiselev, Surface Phenomena in Semiconductors and Dielectrics (Edit. Nauka, in Russian, 1970). A. V. Rzhanov, Yu. F. Novototskii-Vlasov and I. G. Neizvestnyi, J. Tech. Phys. (USSR) 27 (1957) 2440; S. Koc, Czech. J. Phys. B ll(l961) 289; G. Dorda, Phys. Status Solidi 5 (1964) 107; G. Dorda and S. Koc, Surface Sci. 2 (1964) 120. R. V. Prudnikov, V. F. Kiselev and M. M. Egorov, Dokl. Akad. Nauk SSSR 166 (1966) 395. V. F. Kiselev, S. N. Koslov, Yu. F. Novototskii-Vlasov and R. V. Prudnikov, Surface Sci. 11(1968) 111.
7) M. J. D. Low, N. Madison and R. Ramamurthy, Surface Sci. 13 (1969) 238. 8) S. Koc, Czech. J. Phys. B ll(l961) 193,287. 9) M. H. Pilkuhn, J. Appl. Phys. 34 (1963) 3302.
10) S. R. Morrison, in: Semiconductor Surface Physics (Philadelphia, Pa., 1957) p. 169.
11) R. H. Kingston and A. L. McWhorter, Phys. Rev. 98 (1955) 1191; 103 (1956) 534. 12) I. I. Abkevitch, Dokl. Akad. Nauk SSSR 127 (1959) 1199. 13) S. Koc, Phys. Status Solidi (1962) 1304. 14) G. Dorda, Czech. J. Phys. B 15 (1965) 581. 15) 0. JPntsch, J. Phys. Chem. Solids 26 (1965) 1233. 16) Yu. V. Fedorovitch and V. A. Fogel, Phys. Techn. Semicond. (USSR) 3 (1969) 840. 17) 0. S. Frolov, 0. V. Snitko and G. F. Romanova, Ukr. Fiz. Zh. 14 (1969) 585.
References
* Using eq. (A3), when et - Yl +ln(n&m) = - 1, leads to the miscalculation of N 37 ‘A; when it - YI +ln(nJno) = - 2 the miscalculation is equal to - 14 ‘A only.

408 S. N. KOZLOV, V. F. KISELEV AND YU. F. NOVOTOTSKII-VLASOV
18) S. N. Kozlov and Yu. F. Novototzkii-Vlasov, Phys. Status Solidi (a) 6 (1971) 345. 19) S. N. Kozlov and V. F. Kiselev, Phys. Techn. Semicond. (USSR) l(l967) 568. 20) O.V. Snitko, Ukr. Fiz. Zh. 2 (1957) 68. 21) A. Deubner and F. Schulz, Ann. Physik 5 (1960) 113;
K. Ztickler, Z. Physik 136 (1953) 40. 22) H. J. Krusemeyer, Phys. Rev. 114 (1959) 655; J. Phys. Chem. Solids 23 (1962) 767;
H. J. Van Hove, Surface Sci. 22 (1970) 76; H. J. Van Hove, D. Bohrmann and A. Luycks, Surface Sci. 7 (1967) 474.
23) G. W. Pratt and H. H. Kolm, in: Semiconductor Surface Physics (Philadelphia, Pa., 1957) p. 297.
24) J. J. Sparnaay, A. H. Boonstra and J. van Ruler, Surface Sci. 2 (1964) 56; D. R. Palmer, S. R. Morrison and Dauenbaugh, J. Phys. Chem. Solids 14 (1960) 27; Y. Margoninsky, Phys. Rev. 132 (1963) 1910.
25) V. I. Stricha and S. S. Kilchizkaja, Izv. Vuzov, Fiz. 11(1968) 122. 26) M. J. D. Low and Kun-ichi Matsushita, J. Phys. Chem. 75 (1969) 908. 27) J. Sochanski and H. C. Gatos, Surface Sci. 13 (1969) 393.





























