Embed Size (px)
Citation preview

The Pennsylvania State University
The Graduate School
College of Medicine
INVESTIGATING THE FUNCTION AND REGULATION OF PROSTATE
APOPTOSIS RESPONSE-4 IN CANCER
A Dissertation in
Molecular Medicine
by
Jeffrey Nguyen
© 2016 Jeffrey Nguyen
Submitted in Partial Fulfillment
of the Requirements
for the Degree of
Doctor of Philosophy
December 2016

ii
This dissertation was reviewed and approved* by the following:
Rosalyn Irby
Associate Professor of Medicine
Department of Medicine
Dissertation Advisor
Chair of Committee
Jennifer Baccon
Associate Professor of Pathology
Arun Sharma
Associate Professor of Pharmacology
Robert Levenson
Distinguished Professor of Pharmacology
Charles Lang
Distinguished Professor of Cellular and Molecular Physiology and Surgery
Chair of Molecular Medicine PhD Program
*Signatures are on file in the Graduate School

iii
Abstract
Cancer is a disease where normal cells proliferate uncontrollably, which can
ultimately lead to significant morbidity and death. The aggressiveness and mortality of
cancers vary by type: certain cancers respond well to treatment, such as pediatric
leukemias, whereas pancreatic cancer and glioblastoma have a high mortality and low
five-year survival. In an effort to improve current cancer therapies, I focus on elucidating
the function and regulation of prostate apoptosis response-4 (Par-4) in various cancers.
Par-4 is a tumor-suppressor that has been shown to induce cancer cell selective
apoptosis and to sensitize cancer cells to apoptotic stimuli, such as chemotherapeutics
and radiation, and therefore has therapeutic potential. In the first part of this work, I
focused on studying the effect of Par-4 on cell migration, invasion, and the epithelial-
mesenchymal transition in colon cancer cells. I found that ectopic expression of Par-4
inhibited both cell migration and cell invasion, while knocking down Par-4 promoted cell
migration in SW480 and SW620 colon cancer cells. In addition, I found that Par-4
overexpression appeared to induce a mesenchymal-epithelial transition in SW620 cells.
In the second part of this work, I sought to identify novel regulators of Par-4 and
elucidate the mechanism of regulation. I identified Trim21 as a novel binding partner in
colon cancer cells, and show that Trim21 overexpression in the presence of cisplatin
downregulates Par-4 in colon and pancreatic cancer cell lines, and show that
modulating levels of Trim21 and Par-4 affects the sensitivity of cancer cells to cisplatin.
Finally, I demonstrate that Trim21 mRNA levels correlate with survival in pancreatic
cancer patients, with lower Trim21 levels correlating with increased overall survival and
disease-free survival and high Trim21 levels correlating with reduced disease-free

iv
survival. In the third part of this work, I sought to determine whether Par-4 could
enhance the effectiveness of chemotherapeutics and small molecule drugs. I chose to
focus on glioma due to the lack of effective therapeutics. I show that ectopic Par-4
expression alone is sufficient to reduce cell viability and to induce apoptosis in glioma
cell lines, A172 and SNB19. Furthermore, I demonstrate that Par-4 transfected glioma
cells are sensitized to 5-fluorouracil and ISC-4. Taken together, the results presented in
this dissertation suggest novel roles and regulatory mechanisms of Par-4 in cancer, and
provide rationale for its use in cancer treatment; as well as suggesting a novel
prognostic marker for pancreatic cancer.

v
Table of Contents List of Figures ............................................................................................................................................. viii
Abbreviations ............................................................................................................................................... ix
Acknowledgements ...................................................................................................................................... xi
1. Chapter 1 ............................................................................................................................................... 1
1.1. Prostate apoptosis response-4 ..................................................................................................... 1
1.1.1. Domains ...................................................................................................................................... 2
1.1.2. Intracellular functions of Par-4 ................................................................................................... 3
1.1.3. Extracellular functions of Par-4 ................................................................................................... 4
1.1.4. Mechanisms of regulation .......................................................................................................... 5
1.1.5. Non-apoptosis related functions ................................................................................................ 7
1.2. Colon cancer ................................................................................................................................. 9
1.2.1. Par-4 and Colon Cancer ............................................................................................................. 13
1.3. Pancreatic cancer ........................................................................................................................ 13
1.3.1. Par-4 and Pancreatic Cancer ..................................................................................................... 16
1.4. Glioblastoma ............................................................................................................................... 17
1.4.1. Classification ............................................................................................................................. 18
1.4.2. Prognosis and Treatment .......................................................................................................... 19
1.4.3. Molecular Genetics ................................................................................................................... 20
1.5. Par-4 and glioma ......................................................................................................................... 21
1.6. Apoptosis .................................................................................................................................... 22
1.6.1. Mechanism of apoptosis ........................................................................................................... 22
1.7. Conclusion ................................................................................................................................... 24
2. Chapter 2 ............................................................................................................................................. 26
2.1. Introduction ................................................................................................................................ 26
2.2. Materials and Methods ............................................................................................................... 27
2.2.1. Cell culture and transfection ..................................................................................................... 27
2.2.2. Western blot analyses ............................................................................................................... 27
2.2.3. MTT assay .................................................................................................................................. 28
2.2.4. Scratch assay ............................................................................................................................. 29
2.2.5. Boyden Chamber assays ........................................................................................................... 29
2.2.6. RT-PCR analyses ........................................................................................................................ 30

vi
2.2.7. Cell proliferation assay .............................................................................................................. 30
2.2.8. Statistical analyses .................................................................................................................... 30
2.3. Results ......................................................................................................................................... 30
2.3.1. Par-4 increases susceptibility of metastatic SW620 cells to 5-FU ............................................ 30
2.3.2. Par-4 inhibits cell migration and invasion in SW480 and SW620 cells ..................................... 34
2.3.3. Par-4 induces a mesenchymal-epithelial transition in SW620 cells ......................................... 40
2.3.4. Par-4 regulates tight-junction protein expression in SW620 cells ............................................ 45
2.4. Discussion .................................................................................................................................... 48
2.5. Acknowledgements ..................................................................................................................... 51
2.6. Conflict of Interest ...................................................................................................................... 51
2.7. Publication Note ......................................................................................................................... 52
3. Chapter 3 ............................................................................................................................................. 53
3.1. Introduction ................................................................................................................................ 53
3.2. Results ......................................................................................................................................... 55
3.2.1. Trim21 is a novel interacting partner of Par-4 .......................................................................... 55
3.2.2. Trim21 interacts with Par-4 through its PRYSPRY domain ....................................................... 59
3.2.3. Trim21 is not sufficient to downregulate Par-4 levels .............................................................. 62
3.2.4. Ectopic expression of Trim21 downregulates Par-4 in the presence of cisplatin ..................... 65
3.2.5. Cisplatin downregulates Par-4 in a dose- and proteaseome-dependent manner ................... 68
3.2.6. Cisplatin downregulates Par-4 in both the cytoplasmic and nuclear compartments .............. 71
3.2.7. Cisplatin downregulates Par-4 in pancreatic cancer cells ......................................................... 73
3.2.8. Trim21 is a potential therapeutic target in colon and pancreatic cancer................................. 76
3.3. Discussion .................................................................................................................................... 80
3.4. Materials/Methods ..................................................................................................................... 82
3.4.1. Cell culture, transfection, plasmids, reagents, and antibodies ................................................. 82
3.4.2. Western blot analyses ............................................................................................................... 83
3.4.3. Co-IP/Mass-Spec ....................................................................................................................... 84
3.4.4. MTT assay .................................................................................................................................. 85
3.4.5. Immunofluoresence .................................................................................................................. 85
3.4.6. Nuclear-Cytoplasmic Fractionation ........................................................................................... 86
3.4.7. Statistical analyses .................................................................................................................... 86
3.5. Acknowledgements ..................................................................................................................... 87

vii
4. Chapter 4 ............................................................................................................................................. 88
4.1. Introduction ................................................................................................................................ 88
4.2. Materials and Methods ............................................................................................................... 89
4.2.1. Cell culture and transfection ..................................................................................................... 89
4.2.2. Western blot analyses ............................................................................................................... 89
4.2.3. MTT viability assay .................................................................................................................... 90
4.2.4. PE Annexin V apoptosis assay ................................................................................................... 91
4.2.5. Statistical analysis ..................................................................................................................... 91
4.3. Results ......................................................................................................................................... 91
4.3.1. Akt inhibitors reduce cell viability in glioblastoma cells ........................................................... 91
4.3.2. Par-4 is sufficient to reduce cell viability in GBM cells ............................................................. 95
4.3.3. Par-4 sensitizes GBM cells to ISC-4 and 5-FU ............................................................................ 98
4.3.4. Par-4, ISC-4, and their combinations induce apoptosis in GBM cells ..................................... 101
4.4. Discussion .................................................................................................................................. 104
5. Chapter 5 ........................................................................................................................................... 106
References ................................................................................................................................................ 112

viii
List of Figures
Figure 2-1. Overexpression of Par-4 increases susceptibility of metastatic SW620 cells to 5-FU. ............. 33
Figure 2-2. Par-4 overexpression inhibits metastatic processes in SW620 cells. ....................................... 37
Figure 2-3. Par-4 inhibits migration in SW480 cells. ................................................................................... 39
Figure 2-4. Par-4 induces a mesenchymal-epithelial transition in SW620 cells. ........................................ 43
Figure 2-5. Par-4 induces E-cadherin transcription and tight-junction protein upregulation. ................... 47
Figure 3-1. Trim21 is a novel interacting partner of Par-4. ......................................................................... 58
Figure 3-2. Trim21 interacts with Par-4 via its PRY-SPRY domain. ............................................................. 60
Figure 3-3. Trim21 is not sufficient to downregulate Par-4 protein levels. ................................................ 63
Figure 3-4. Ectopic expression of Trim21 downregulates Par-4 in the presence of cisplatin in colon cancer
cells. ............................................................................................................................................................ 66
Figure 3-5. Cisplatin downregulates Par-4 in a dose- and proteasome-dependent manner. .................... 69
Figure 3-6. Cisplatin downregulates Par-4 in both the cytoplasmic and nuclear compartments. ............. 72
Figure 3-7. Cisplatin downregulates Par-4 in pancreatic cancer cells. ....................................................... 75
Figure 3-8. Trim21 is a potential therapeutic target in colon and pancreatic cancer. ............................... 79
Figure 4-1. Akt inhibitors decrease cell viability in glioblastoma cells. ....................................................... 94
Figure 4-2. Par-4 is sufficient to reduce cell viability in GBM cells. ............................................................ 96
Figure 4-3. Par-4 sensitizes GBM cells to 5-FU and ISC-4. ........................................................................ 100
Figure 4-4. Par-4, ISC-4, and their combinations induce apoptosis in GBM cells. .................................... 102

ix
Abbreviations
Par-4 – Prostate apoptosis response-4; Trim21-Tripartite motif-containing protein 21;
EMT-Epithelial-mesenchymal transition; MET-Mesenchymal-epithelial transition; CDDP-
cis-diamminedichloridoplatinum (II); MTT-3-(4,5-dimethylthiazol-2-yl)-2,5-
diphenyltetrazolium bromide; 5-FU-5-fluorouracil; ISC-4-Isoselenocyanate; DNA-
Deoxyribonucleic acid; cDNA-complementary DNA; RNA-Ribonucleic acid; shRNA-
short hairpin RNA; NF-κB-Nuclear factor kappa-light-chain-enhancer of activated B
cells; PARP-Poly (ADP-ribose) polymerase; VEGF-Vascular endothelial growth factor;
PDGF-Platelet-derived growth factor; EGFR-Epidermal growth factor receptor; Bcl-2-B-
cell lymphoma 2; DR5-Death receptor 5; IRF-Interferon regulatory factor; c-FLIP-FLICE-
like inhibitory protein; PI3K-Phosphatidylinositol-4,5-bisphosphate 3-kinase; NLS-
Nuclear localization signal; SAC-Selective for apoptosis in cancer; FADD-Fas-
associated protein with death domain; TRADD-Tumor necrosis factor receptor type 1-
associated death domain; PKC-Protein kinase C; WT1-Wilm’s tumor 1; TOP1-DNA
topoisomerase 1; ER-Endoplasmic reticulum; GRP78-78 kDa glucose-regulated protein;
PKA-Protein kinase A; Akt1-RAC-alpha serine/threonine-protein kinase; FBXO45-F-box
only protein 45; IDH1-Isocitrate dehydrogenase 1;Foxo3a-Forkhead box O3a; miRNA-
microRNA; UTR-Untranslated region; TGF-β-Transforming growth factor-β; ROS-
Reactive oxygen species; APC-Adenomatous polyposis coli; k-RAS-V-Ki-ras2 Kirsten
rat sarcoma viral oncogene homolog; TP53-Tumor protein p53; MSH-MutS protein
homolog; MLH-MutL homolog; PDAC-Pancreatic ductal adenocarcinoma; BRCA-Breast
cancer susceptibility gene; PanIN-Pancreatic intraepithelial neoplasia; RB-
Retinoblastoma gene; Apaf-1-Apoptotic protease activating factor 1; ATP-Adenosine

x
triphosphate; TNF-Tumor necrosis factor; RING-Really interesting new gene finger
domain; RT-PCR-Reverse transcription polymerase chain reaction; TRAMP-Transgenic
adenocarcinoma of the mouse prostate; RPMI-Roswell park memorial institute medium;
FBS-Fetal bovine serum; PBS-Phosphate buffered saline; BCA-bicinchoninic acid;
SDS-Sodium dodecyl sulfate; TBS-Tris buffered saline; HRP-Horseradish peroxidase;
ECl-Enhanced Chemiluminescence; MT1-MMP-Membrane-type 1 matrix
metalloproteinase; bHLH-Basic helix-loop-helix; ZO-1-Zonula occludens 1; DDX41-
DEAD-Box helicase 41; dsDNA-double stranded DNA; TCGA-The cancer genome
atlas; DMSO-Dimethyl sulfoxide; FITC-Fluorescein isothiocyanate; Ro-Rhodamine;
DAPI-4',6-diamidino-2-phenylindole; GBM-Glioblastoma; TMZ-Temozolomide; PrPc-
Cellular prion protein; IL-2-Interleukin 2; CD-28-Cluster of differentiation 28

xi
Acknowledgements
I would like to thank Dr. Rosalyn Irby for her guidance in helping me develop as a
scientist. After suddenly finding myself in the position of being forced to switch labs after
a year into my PhD, she graciously accepted me as a student. She has always been
very patient with me, and has allowed me to pursue my own ideas. I am very grateful for
that. I also would like to thank my committee for their helpful guidance throughout the
process. In addition, I would like to thank my family, especially my parents, for their
support and encouragement throughout my life. They instilled in me the values which
have shaped me into the person that I am and have led me to this point. Finally, I would
like to thank my wife, Felicia, for her patience, encouragement, and love throughout this
process. It is because of her that I have been able to find the resolve to get through the
more difficult moments during my PhD.

1
1. Chapter 1
Introduction
1.1. Prostate apoptosis response-4
Prostate apoptosis response-4 (Par-4) is a gene that was originally discovered in
rat prostate cancer cells that were induced to undergo apoptosis.1 Par-4 was one of a
group of early-upregulated genes in these prostate cancer cells, and was the only one
that had not yet been described. The initial characterization of Par-4 demonstrated that it
is highly expressed in a wide variety of normal tissues.2 Initial studies showed that Par-4
expression was induced in response to physiologic stimuli, such as androgen withdrawal.2
Studies in cancer cells revealed that Par-4 could sensitize cells toward apoptosis-inducing
agents, such as chemotherapeutics and radiation.3-6 In certain types of cancer cells, such
as androgen-independent cancer cell lines, ectopic Par-4 expression is sufficient to
induce apoptosis.7 Interestingly, Par-4 protein has also been shown to be secreted, and
this extracellular Par-4 protein is sufficient to induce apoptosis.8 An attractive property of
the apoptosis-inducing ability of Par-4 is that it is selective for cancer cells. In other words,
it leaves normal cells unaffected (for mechanisms, see sections 1.1.3 and 1.1.4). This
selectivity for cancer cells is ideal therapeutically, since several of the toxic side-effects
of commonly used chemotherapies is due to their non-selective mechanism.9 More recent
studies on Par-4 have begun to examine its role in regulating cellular processes beyond
apoptosis, including migration and autophagy.10-13 Taken together, the vast majority of

2
studies on Par-4 function suggest that it functions as a tumor suppressor. This is
supported by expression data from clinical specimens. Par-4 is shown to be
downregulated in renal cancer,14 leukemia, lung cancer,15 and endometrial cancer.16 In
addition, oncogenes have been shown to downregulate Par-4.17, 18 The tumor-
suppressive function of Par-4 in also suggested from work in animal models: Par-4
knockout mice (KO) are prone to tumor development in a variety of tissues, especially
hormone-dependent tissues, such as the endometrium and prostate; Par-4 KO mice are
also more sensitive to developing carcinogen-induced tumors.19
1.1.1. Domains
Par-4 is a 343 amino acid protein with several conserved domains. At the C-
terminus, there is a leucine-zipper domain, which, like other leucine zippers, is an alpha-
helix that contains a series of heptad repeats, where every fourth position is a leucine.20
These regularly spaced leucines give the alpha-helix a hydrophobic face that allows for
dimerization – with itself and other proteins. This leucine zipper domain of Par-4 is
responsible for mediating almost all of the protein-protein interactions of Par-4.21 Also at
the C-terminus is a nuclear export sequence, though the function of this sequence has
not been studied. Near the N-terminus, there are two localization sequences, NLS1 and
NLS2: NLS1 is not required for the nuclear localization of Par-4, whereas NLS2 is
required.21 There is also a domain in the middle of Par-4 termed the selective-for-
apoptosis-in-cancer domain (SAC). This domain was discovered by serially deleting
fragments from either termini in order to discover the minimal sequence that was sufficient
for Par-4 apoptotic activity.21 This 59 amino acid SAC domain has demonstrated apoptotic
activity against cancer cells when overexpressed intracellularly and when cells are treated

3
with recombinant SAC protein, which is comparable to the apoptotic activity of full-length
Par-4.8 Additionally, ectopic expression of the SAC domain has been shown to induce
apoptosis in cells that were resistant to full-length Par-4.22 Par-4 also contains many other
putative domains and sites for post-translational modification, such as phophorylation
sites and glycosylation sites; however, their function has yet to experimentally validated.23
1.1.2. Intracellular functions of Par-4
The vast majority of studies on Par-4 function have focused on the intracellular
role of Par-4 in apoptosis. Par-4 affects apoptosis by regulating both the intrinsic and
extrinsic pathways of apoptosis.
Par-4 modulates the extrinsic pathway by facilitating the trafficking of both Fas and
FasL to the cell membrane.24 Fas and FasL, in turn, recruit FADD and activate the
extrinsic pathway of apoptosis. Another mechanism by which Par-4 regulates apoptosis
is by inhibiting NF-κB.25 NF-κB is a transcription factor that upregulates pro-survival and
pro-inflammatory genes in response to cytokines.26 Given its pro-survival function, NF-κB
is also, not surprisingly, found to be upregulated in a wide variety of cancers. Par-4 inhibits
NF-κB function in multiple ways. For example, Par-4 can inhibit NF-κB transcriptional
activity in the nucleus,27 though the exact mechanism for this is currently unknown. It
could act via direct DNA binding and inhibition of NF-κB activity, or it could act by
recruiting co-repressors, which in turn inhibit NF-κB activity. In addition, Par-4 has been
shown to inhibit NF-κB activation in the cytoplasm.25 Furthermore, Par-4 can influence
NF-κB activity indirectly by binding to and inhibiting protein kinase C zeta (PKCζ).25, 28

4
PKCζ is an atypical isoform of the PKC family of kinases that activates NF-κB by
phophorylating IκB.
Though its ability to affect Fas/FasL translocation and its ability to inhibit NF-κB
are its most well-known functions, Par-4 also has been described to act on other
intracellular proteins. For example, Par-4 enhances the repressor activity of Wilm’s Tumor
1 (WT1).29, 30 WT1 is a transcription factor that acts as a tumor suppressor by repressing
the transcription of genes and thereby inhibiting proliferation. Par-4 has also been shown
to interact with topoisomerase 1 (TOP1).31 TOP1 functions to relax supercoiled DNA,
which is essential for DNA replication to proceed. By interacting with TOP1, Par-4 inhibits
its ability to relax DNA supercoils.
1.1.3. Extracellular functions of Par-4
The function of extracellular Par-4 is a relatively recent finding. Studies have
shown that Par-4 is secreted by both normal cells and cancer cells.8 Like other secreted
proteins, Par-4 is synthesized and secreted from the cell via the classical ER-Golgi
pathway. It was also found that ER stress inducers increase Par-4 secretion. Importantly,
it was found that secreted Par-4 can induce cancer-cell selective apoptosis by binding to
glucose-regulated protein 78 (GRP78), which in turn activates the extrinsic apoptosis
pathway.8, 32 GRP78 is normally found in the endoplasmic reticulum and normally acts as
a sensor for ER stress and activator of the unfolded protein response, but for unknown
reasons, a significant fraction is found at the cell surface in cancer cells.33, 34 It is this
cancer-cell specific membrane distribution of GRP78 that is thought to confer the ability
of extracellular Par-4 to induce cancer-cell selective apoptosis.9

5
1.1.4. Mechanisms of regulation
Though most studies of Par-4 have focused on elucidating its function, there has
also been some work done examining how Par-4 expression and activity is regulated.
Gene regulation can be categorized as transcriptional, post-transcriptional, or post-
translational, depending on at what point in the process of gene expression that a
regulatory mechanism acts.
Most studies of Par-4 regulation have focused on the post-translational
mechanisms that control Par-4 activity. One of the most common post-translational
mechanisms to control protein activity is phosphorylation. The phosphorylation of Par-4
by protein kinase A (PKA) was one of the first regulatory mechanisms discovered.35 PKA
phosphorylates Par-4 at a threonine residue located at position 155. This position is
located within the SAC domain, and was found to be essential for the nuclear
translocation of Par-4, essential for the inhibition of NF-κB, essential for translocation of
Fas/FasL to the cell membrane, and essential for its pro-apoptotic activity.35 This
modification is significant, because ability of ectopically expressed Par-4 to induce
apoptosis selectively in cancer cells is attributed to the differential expression of PKA
between cancer cells and normal cells: cancer cells, in general, have higher PKA levels
than normal cells, and it is thought that this activates the apoptotic functionality of Par-4.
In contrast, phosphorylation of Par-4 at a serine residue at position 249 by another kinase,
AKT1,36 is responsible for inhibiting Par-4 activity. In this case, once Par-4 is
phosphorylated by AKT1, it is sequestered by 14-3-3δ protein, and it is this sequestration
in the cytosol that prevents Par-4 from carrying out its apoptotic activity. Other
mechanisms of post-translational regulation of Par-4 include ubiquitination37 and

6
proteolytic cleavage.38, 39 Ubiquitination of target proteins by E3 ligases leads to their
proteolytic cleavage by the proteasome.40 Par-4 was shown to a substrate of the E3
ligase, FBXO45, and the ubiquitination and subsequent downregulation of Par-4 by
FBXO45 was shown to regulate cancer cell apoptosis, survival, and colony formation.37
Multiple reports have shown the ability of Par-4 to be cleaved by caspases in response
to various apoptotic stimuli.38, 39 Interestingly, cleaved Par-4 appears to have some
interesting characteristics: it contains the essential SAC domain, it localizes to the
nucleus, and appears to have greater apoptotic activity than full-length Par-4.41, 42
While most reports on Par-4 regulation have focused on its post-translational
regulation, a few reports have highlighted transcriptional mechanisms of Par-4 regulation.
As an example of the transcriptional regulation of Par-4, in endometrial epithelial cells
during a normal menstrual cycle, Par-4 is very highly expressed and this was correlated
with NF-κB activity.43 Furthermore, Par-4 levels in endometrial carcinoma cells were also
correlated with NF-κB activity despite overall downregulation of Par-4 levels relative to
normal endometrium. Further, in vitro studies showed that Par-4 is a direct target of NF-
κB. As another example, it was found that gliomas with mutant isocitrate dehydrogenase
1 (IDH1) had significantly lower levels of Par-4 relative to gliomas without mutant IDH1.44
In vitro studies showed that the product of mutant IDH1 protein, D-2-hydroxyglutarate,
suppresses Par-4 transcription through inhibition of promoter activity. In addition, it
enhances Par-4 mRNA degradation. In another study, Par-4 was shown to be a
transcriptional target of FOXO3a.45 In this study, treatment with Withaferin A, a small
herbal molecule that inhibits AKT1, promotes FOXO3a translocation to the nucleus, which

7
in turn induces Par-4 transcription. Finally, one group reported on the activation of Par-4
transcription by targeting the Par-4 promoter with small-activating RNAs.46
No studies have reported on the post-transcriptional regulation of Par-4, such as
regulation by microRNAs (miRNAs). miRNAs are endogenous short 20-22 nucleotide
sequences of RNA that negatively regulate target genes by binding to the 3’- UTRs of
target mRNAs and negatively regulate gene expression by leading to mRNA cleavage or
by inhibiting mRNA translation.47 Like protein-coding genes, the spectrum of activity of
individual miRNAs can be classified as tumor-suppressive or oncogenic, and miRNAs
have been shown to play a role in cancer progression.48 Undoubtedly, Par-4 is the target
of some miRNAs. It would be interesting to see what role miRNAs play in regulating Par-
4 expression during cancer development.
1.1.5. Non-apoptosis related functions
Recent work on Par-4 has attempted to look beyond its role in regulating apoptosis
and has begun to examine its role in other cellular process, such as migration, and
autophagy.10-13, 49
Cell migration is an important phenotype, especially in the context of cancer. As
cancer cells accumulate mutations, they also gain the ability to escape from their primary
site and to metastasize and colonize distant sites.50 The epithelial-mesenchymal
transition (EMT) is thought to play in important role in the process of cancer cells
developing a metastatic phenotype.51 EMT, which was originally discovered as a
developmental process, is a process by which epithelial cells lose their characteristic
epithelial features, such as cell-cell contacts, and acquire mesenchymal features, such

8
as spindle-shaped cells with minimal cell-cell contacts. Par-4 was first shown to regulate
EMT in a study that was examining the mechanisms of cisplatin resistance in pancreatic
cancer.10 In that study, a cisplatin resistant pancreatic cell line was created, and the
resistant cells acquired a mesenchymal phenotype, which correlated with Par-4
downregulation. Further in vitro studies showed that ectopic Par-4 expression in these
resistant cells reversed EMT and cisplatin resistance, and they demonstrated that Par-4
regulated EMT in a PI3K/Akt-dependent manner. Finally, Par-4 knock-down in cisplatin
sensitive cells induced EMT and cisplatin resistance. In endometrial and breast cancer
cell lines, the opposite effect of Par-4 on EMT was found. In that study, Par-4 mediated
TGF-β induced EMT.11 In this study, Par-4 transcription was induced by SMAD, and Par-
4 in turn upregulated Vimentin and other mesenchymal markers. In chapter 2 of this
dissertation, I examine the role of Par-4 in regulating migration and EMT in colon cancer.
Autophagy is the process by which the cell recycles damaged or senescent
components.52 During this regulated process, old components are surrounded by a
double membrane, resulting in an autophagosome. Later, the autophagosome fuses with
the lysosome, and the contents are broken down. In the context of disease, autophagy
can be pro-survival or anti-survival. In some instances, autophagy is a cellular response
to stress, such as in times of starvation; however, in some cases it can promote cell-
death.52 The first study to link Par-4 to autophagy was a study that examined the
mechanisms by which Par-4 enhanced response to chemoradiotherapy in
hypopharyngeal carcinoma cells.13 The results from that study showed that Par-4 induced
both apoptosis and autophagy and sensitized cells toward both chemotherapeutics and
x-ray irradiation. Another study showed that Par-4 induced autophagic cell death in glioma

9
cells in response to curcumin.12 In this study, Par-4 expression was induced in a reactive
oxygen species-dependent (ROS) manner in response to curcumin, which led to
autophagic cell death. In this case, Par-4 overexpression sensitized glioma cells toward
curcumin, whereas antioxidants blunted the ROS-dependent Par-4 induction and
autophagic cell death.12
1.2. Colon cancer
Colon cancer is a type of cancer that originates from the epithelial cells lining the
colon. The most distal portion of the gastrointestinal tract, called the rectum, is also a
site of malignancy. Collectively, colon and rectal cancers are referred to as colorectal
cancer. Combined, colorectal cancer is one of the leading causes of mortality among all
types of cancer, second only to lung cancer.53 The median age at diagnosis is 68 years.
The incidence of colorectal cancer is about 41 per 100,000 per year. In the year 2014,
136,830 people are expected to be diagnosed with colon cancer, while 50,310 people
are estimated to die from the disease. Overall, the five-year survival rate is at 64.7%
with the survival rate inversely proportional to disease stage at diagnosis – localized
disease, where cancer cells are confined to the primary site of origin, has a survival rate
of 90.1%, while metastatic disease, where cancer cells have spread to distant organs,
has a survival rate of 13.5%.54
Colon cancer arises from a series of mutations in key genes.55, 56 Over time, this
leads to a series of morphological changes that convert normal colonic epithelium into a
carcinoma, which is a general term that refers to a cancer that arises from epithelial
tissue. Although the exact genetic changes and the order in which they occur may vary
from individual to individual, the transition from epithelium to carcinoma is thought to

10
occur through one of two distinct pathways: the adenoma-carcinoma sequence or the
mismatch-repair pathway.55 The pathways differ by the genes that are mutated and the
mutational mechanisms that give rise to them. The adenoma-carcinoma sequence is
characterized by chromosomal instability, which gives rise to disease-causing mutations
in genes, such as APC, K-RAS, TP53, and CTNNB157. These accumulated genetic
changes, in turn, result in a series of morphological changes at the cellular and tissue
level. Initially, there is focal epithelial proliferation. With time this epithelial proliferation
creates a physical protrusion, called a polyp, consisting of dysplastic cells. As mutations
continue to accumulate, the protrusion grows and breaks through adjacent tissue layers,
thereby becoming an invasive cancer. In contrast, the mismatch-repair pathway of colon
carcinogenesis is characterized by mutations in DNA mismatch repair genes. Mutations
in mismatch repair genes, such as MSH2, MSH6, or MLH1, are most likely the initiating
genetic lesions that give rise to this pathway of colon carcinogenesis.56 Macroscopically,
the progression from normal tissue to cancerous tissue does not occur in a stepwise
progression, unlike the adenoma-carcinoma pathway. Carcinomas arising through the
mismatch-repair pathway can arise with no detectable precursor lesion in some
instances; in other instances, cancer can arise from a precursor lesion, called a sessile
serrated adenoma, a term that describes a polyp with a flat morphology without a
detectable stalk.
A large component of the risk of developing colon cancer is related to
environmental factors, such as diet.57 Specifically, consuming excess amounts of
calories, red meat, and fat puts one at higher risk of developing colon cancer. Though
many hypotheses have been proposed to explain the link, the mechanistic basis for how

11
these risk factors enhance the development of colon cancer is still unclear. Other risk
factors include heritable cancer predisposition syndromes, such as Lynch syndrome,
where germline mutations in DNA mismatch repair genes lead to a higher risk of
developing colorectal cancer and cancers in other sites.58 In addition, people with a
history of inflammatory bowel disease are at high risk of developing colorectal cancer,59
highlighting the role that chronic inflammation of the intestines plays in colon cancer
development.60
Patients with colon cancer can present with a variety of symptoms based on the
location of the tumor. For example, patients with tumors in the ascending colon can
become sizable since stool is relatively liquid and little water has been absorbed by the
intestines at that point. Likewise, patients with such tumors will be unlikely to have
experienced any changes in bowel movements. Instead, due to the ability of a tumor at
that location to grow large, these tumors tend to ulcerate, leading to chronic blood loss.
Thus, patients can experience fatigue, palpitations, and anemia secondary to this blood
loss. In contrast to tumors arising in the ascending colon, tumors arising in the
transverse or descending colon tend to cause a different set of symptoms. For example,
since stool has less water and is more solid at these portions of the colon, patients with
tumors arising at these locations tend to experience abdominal pain, cramping, and
obstructive symptoms, such as changes in bowel habits and changes in the size and
caliber of stool.
Once the diagnosis of colorectal cancer is made, the prognosis and ultimate
treatment depend on the stage of the tumor. Staging, in turn, is determined by the depth
of penetration of the tumor into normal tissue. Normal colonic tissue consists of multiple

12
layers. The layer that is in contact with the lumen of the colon is the epithelium. Just
underneath the epithelial layer is a thin layer of muscle, called the muscularis mucosa.
Underneath the muscularis mucosa layer lie the submucosa, muscularis propria, and
serosa, respectively. Finally, the serosa is in contact with a connective tissue layer
consisting of fat, blood vessels, and lymph nodes.61 Stage I colon cancer can be divided
into two subgroups, T1 or T2: T1 disease is defined as a tumor that has not penetrated
completely through the submucosa; T2 disease is defined as a tumor that has not
penetrated completely through the muscularis propria. Stage II colon cancer is defined
as a tumor that has completed breached the muscularis propria, but has not yet spread
to the lymph nodes. Like stage I, stage III colon cancer can also be divided into two
subgroups, N1 or N2: N1 disease is defined as a tumor that has spread to 1-3 lymph
nodes; N2 disease is defined as a tumor that has spread to greater than 4 lymph nodes.
Finally, a patient has stage IV colon cancer if there is evidence of metastatic spread to
distant sites, such as the liver.61
Like prognosis, treatment also depends on the stage of the tumor at diagnosis. In all
cases, surgical resection of the tumor is indicated. Depending on the stage of disease,
further treatment may be warranted. With stage I disease, where there is no evidence of
any local or distant spread, no chemotherapy is indicated.61 In patients with stage II
disease, additional chemotherapy is controversial. In patients with stage III and IV
disease, chemotherapy is an essential part of treatment - 5-FU, which is the backbone
of treatment, in combination with other drugs, such as oxaliplatin or irinotecan, can
reduce the rate of the recurrence and prolong survival in such settings.61 Chemotherapy
drugs used in the treatment of colon cancer act through a variety of mechanisms. For

13
example, 5-fluorouracil inhibits thymidylate synthase, whereas oxaliplatin and irinotecan
are a DNA cross-linking agent and topoisomerase inhibitor, respectively.61
1.2.1. Par-4 and Colon Cancer
Much work has been done on the role of Par-4 in colon cancer. The first study to
report on the role of Par-4 in colon cancer demonstrated that treatment of HCA-7 colon
cancer cells with cyclooxygenase inhibitors upregulated Par-4 expression.62 Future
studies examined the ability of Par-4 to sensitize colon cancer cells to
chemotherapeutics. Specifically, Par-4 plasmid delivered with nanoliposomes sensitized
HT-29 colon tumor cells to 5-FU in a nude mouse model.63 A microarray study showed
that Par-4 sensitizes to 5-FU by inhibiting NF-κB and regulating a network of miRNAs.64
Later studies showed that both ISC-465 and a combination of Src inhibitor and 5-FU66
can activate Par-4 resulting in reduced tumor growth and cell death, respectively.
1.3. Pancreatic cancer
Pancreatic cancer is an umbrella term for a family of different cancers arising from
cells of the pancreas. The various types are categorized by histology and cell-of-origin.
Infiltrating ductal adenocarincoma (PDAC) is the most common and constitutes greater
than ninety-percent of all pancreatic cancer cases.67 In this dissertation, PDAC will be a
focus.
PDAC is a type of cancer that originates from the ductal epithelial cells of the
exocrine pancreas.67 The median age at diagnosis is 70.54 Pancreatic cancer is one of
the most aggressive forms of cancer and the fourth leading cause of cancer death with
an incidence 12.4 per 100,000 per year. This leads to an estimated 53,070 new cases

14
in 2016. In 2016, about 41,780 people are expected to die from pancreatic cancer.54
Thus, virtually all patients will die from their disease. The five-year overall survival for
pancreatic cancer is 7.7%. Stratified by stage, the five-year survival is: 29.3% for
localized disease, 11.1% for regional disease, and 2.6% for metastatic disease.54
Like colon cancer, pancreatic cancer also arises as a consequence of the
accumulation of genetic mutations.68, 69 As mutations accumulate, the pancreatic ductal
cell acquires the ability to proliferate uncontrollably and to infiltrate adjacent structures.
The most common mutations involved in pancreatic cancer are KRAS, P16, P53,
SMAD4, and BRCA2.70, 71 Also, like colon cancer, as the mutations accumulate, the
pancreatic ductal cell progresses through a series of histologically defined stages on its
way to becoming an invasive cancer. These stages are called pancreatic intraepithelial
neoplasisas (PanINs), and together the sequence of pancreatic carcinogenesis is
termed the PanIN-carcinoma sequence.67 The stages represent a series of increasingly
dysplastic pancreatic epithelium, PanIN 1 through 3, finally terminating in invasive
pancreatic adenocarcinoma. In this progression, activating mutations in KRAS are
thought to occur relatively early, whereas the inactivating mutations of tumor-suppressor
genes, such as P16, P53, SMAD4, and BRCA2 occur relatively late in the sequence.72-
75
Risk factors for pancreatic cancer include smoking, diabetes, obesity, chronic
pancreatitis, and a diet high in fats.76 Smoking is the biggest risk factor and accounts for
roughly 25% of pancreatic cancer patients, and is thought to double the risk of
developing pancreatic cancer. Though diabetes and chronic pancreatitis increase one’s

15
risk, it is unclear whether the disease processes have a causal role in pancreatic
carcinogenesis, since they both can also occur secondary to pancreatic cancer.77 Age,
socioeconomic status, and race also play a role in increasing one’s risk for pancreatic
cancer. Roughly 80% of cases occur in patients between the ages of 60 and 80.54
Pancreatic cancer is more common in blacks compared to whites, and also is more
common in Ashkenazi jews. Finally, various cancer predisposition syndromes can
increase one’s risk for developing pancreatic cancer - examples of such syndromes
include: familial pancreatic cancer syndrome, Peutz-Jager’s syndrome, familial
pancreatitis, Li-Fraumeni syndrome, and Lynch syndrome.78, 79
Depending on the location of the tumor, patients can experience a variety of
symptoms. When the tumor originates in the head of the pancreas, for example, the bile
duct can be obstructed, which can lead to abdominal pain, itchiness, lethargy, and
weight loss. Obstruction of the pancreatic duct from a pancreatic tumor can lead to
symptoms, such as epigastric pain, new-onset diabetes mellitus, and acute pancreatitis.
Finally, constriction of the junction between the distal stomach and duodenum can result
in nausea and vomiting.
Imaging is used to diagnose pancreatic cancer, usually a spiral CT scan. Most
patients are diagnosed with pancreatic cancer at a late stage: only 9% have localized,
resectable disease, whereas the remainder have either metastatic or locally advanced
disease.54
As with colon cancer, the prognosis depends on the stage at diagnosis. Staging of
pancreatic cancer utilizes the TNM staging system: stage I disease is defined as a

16
tumor without lymph node involvement, and such tumors are usually around 2 cm; once
the tumor extends beyond the pancreas and/or involves the lymph nodes, it is stage II,
by definition;61 if the celiac axis or superior mesenteric artery is involved, then the tumor
is stage III; if there is evidence of distant metastatic lesions, then the tumor is stage IV.61
The five-year survival rate by stage is inversely proportional to the stage of disease at
diagnosis: if a patient has localized, resectable, stage I disease, the five-year survival
rate is 16%; if a patient has stage II or III disease, the five-year survival rate is 8%; if the
patient has stage IV disease, the five-year survival rate is 2%.61
Like prognosis, treatment of pancreatic cancer also depends on stage. Stage I
disease is, by definition, resectable. Therefore, surgical resection is warranted, followed
by adjuvant chemotherapy, since adjuvant chemotherapy in this setting has been shown
to improve survival. Stage II disease warrants neoadjuvant chemotherapy, followed by
surgical resection and adjuvant chemotherapy. Finally, stage III and IV disease call
purely for chemotherapy, since surgical resection is not curative in this setting. In all
cases, chemotherapy used in the treatment of pancreatic cancer consists of a
combination of gemcitabine, 5-FU, and radiation.61
1.3.1. Par-4 and Pancreatic Cancer
A significant body of work has been done on the role of Par-4 in pancreatic cancer.
For example, when oncogenic KRAS was expressed in pancreatic cancer cell lines,
Par-4 expression was downregulated.18 In addition, in an analysis of Par-4 and KRAS
expression in clinical specimens, KRAS mutational status was shown to correlate with
Par-4 expression.18 Additionally, Par-4 expression had prognostic significance: Par-4

17
expression was correlated with prolonged survival.18 Finally, small molecule inducers of
Par-4 expression sensitized pancreatic cancer cells to chemotherapeutics.80
1.4. Glioblastoma
The brain is a complex tissue consisting of many different cell types, which can be
classified into two types: neurons and glial cells. Neurons are cells that communicate
with one another and are responsible for our cognition, and through nerves, coordinate
and control all of our bodily functions. Glial cells are supportive cells that serve a variety
of functions, such as structural support for neuronal cells. In addition, they help to
regulate blood flow by constricting local vasculature and function as insulation for
neuronal cells. Finally, certain glial cells act as a defense against foreign pathogens.
Some examples of glial cells are astrocytes, Schwann cells, oligodendrocytes,
ependymal cells, and satellite cells.
Brain tumors can arise from any cell type, neurons or glia. The annual incidence
of primary brain tumors is about 10-17 per 100,000 per year.81 About half of patients
that are diagnosed with brain cancer have a primary tumor, while half have metastatic
disease from another primary site.82 Primary brain tumors have an uneven distribution
among age groups: while they comprise only 1-2% of all cancers, they make up about
20% of all childhood cancers.82
Brain cancers have several features that distinguish them from most other
cancers. Unlike colon cancer, and similar to pancreatic cancer, the distinction between
benign and malignant tumors is obscure. All grades of brain cancer have a relatively
poor prognosis.82 In addition, due to its sensitive location and the infiltrating nature of

18
brain cancers, the ability to resect a given tumor is limited. Finally, primary brain tumors
rarely metastasize outside of the central nervous system, even grade 4 brain cancers.82
Patients that develop primary brain tumors can present with a variety of
symptoms based on the location of the tumor. Usually patients present with a focal
neurologic deficit, such as paralysis.61, 82 These focal neurologic deficits are the result of
the compression of adjacent neurons and nerves. Inflammation and edema around the
tumor can also contribute to this compression. Patients can also present with seizures.
Seizures result from a growth of a tumor that disrupts the neuronal connections leading
to an imbalance between excitatory and inhibitory circuits, shifting the balance in favor
of over-excitation.61 Likewise, patients can present with non-focal neurologic symptoms
such as headache, altered mental status, or a change in personality. These non-focal
symptoms are the result of increased intracranial pressure secondary to the tumor. The
tumor can lead to increased intracranial pressure by a variety of mechanisms: tumor
growth compressing adjacent brain tissue, inflammation and edema, tumor hemorrhage,
or obstruction of cerebrospinal fluid pathways.61, 82
1.4.1. Classification
Brain tumors are classified according to the histology and cell type.83 Of the glial
tumors, the most common types arise from astrocytes, oligodendrocytes, and
ependymal cells.82 Astrocytic, specifically glioblastoma (discussed later), tumors will be
a focus of this dissertation. Tumors arising from other types of glial cells or tumors
arising from neuronal cells are outside the scope of this dissertation and will not be
discussed further.

19
Astrocytic tumors are the most common of the primary glial tumors and are
divided into four grades based on histology. A grade I astrocytoma is called a pilocytic
astrocytoma and is well circumscribed on histology and imaging. A pilocytic astrocytoma
is considered benign. A grade II astrocytoma is called a diffuse astrocytoma. Instead of
being well circumscribed, the borders of a diffuse astrocytoma are infiltrative on
histology and imaging. A grade III astrocytoma is called an anaplastic astrocytoma. On
histology an anaplastic astrocytoma has infiltrative borders and the presence of
numerous mitotic figures. A grade IV astrocytoma is called a glioblastoma. On histology,
a glioblastoma has an infiltrative border, numerous mitotic figures, and the presence of
vascular proliferation and/or necrosis. Grades III & IV astrocytomas are considered
malignant.82
1.4.2. Prognosis and Treatment
As with colon cancer and pancreatic cancer, the prognosis and treatment of
primary astrocytic tumors depend on how advanced the cancer is.
Pilocytic astrocytomas are the most common type of glial neoplasms in children.
They tend to affect the cerebellum and optic nerves. They are often well circumscribed
and are slow-growing with an excellent prognosis with total excision.61
Diffuse astrocytomas make up about 10-15% of all glial neoplasms and have a
peak incidence in the fourth decade. The edges of the tumor are not well defined due to
the infiltrative nature of the cancer cells. The mean survival of patients with diffuse
astrocytomas is 6-8 years. Most patients ultimately progress to anaplastic astrocytoma
or glioblastoma as mutations in their tumor accumulate.61

20
Anaplastic astrocytomas have a peak incidence in the fifth and sixth decades. As
with diffuse astrocytomas, the edges of the tumor are also ill-defined and infiltrative. In
addition, the presence of mitotic figures without evidence of vascular proliferation or
necrosis by histology characterizes anaplastic astrocytomas. The mean survival of
patients with anaplastic astrocytoma is 3 years.82
Glioblastomas have a peak incidence in the sixth and seventh decades, and
make up the majority of gliomas (50-60%). As with diffuse and anaplastic astrocytomas,
the borders of glioblastomas are irregular and on histology; however, there is also
evidence of dedifferentiation, mitotic figures, and either vascular proliferation, necrosis,
or both. The mean survival of patients with glioblastoma is 8-10 months.82
Treatment for pilocytic astrocyomas is surgical resection, which though not
necessarily curative, allows for a very favorable prognosis, with a five-year survival in
some cohorts over 90%.61 Treatment for diffuse astrocytomas, anaplastic astrocytomas,
and glioblastomas, consists of combinations of surgical resection and
chemoradiotherapy.84 Unfortunately, due to the infiltrative nature of the more advanced
grades of astrocytomas, treatment is rarely curative and the tumors almost always
recur.61
1.4.3. Molecular Genetics
The progression from low grade to high grade astrocytomas correlate with a series
of mutations in the tumor.85 Low-grade astrocytomas are associated with inactivating
mutations in P53 and activating mutations in PDGF and its receptor. In contrast, high-

21
grade astrocytomas are additionally associated with inactivating mutations in RB and
P16.86
There are two clinically distinct subsets of gliobastomas: new-onset glioblastomas,
also called primary glioblastomas; and secondary glioblastomas, which are
glioblastomas that present in patients with a prior history of a lower grade astrocytoma.
Primary glioblastomas tend to occur in older patients, whereas secondary glioblastomas
tend to occur in younger patients and have a better prognosis.61 Though primary and
secondary glioblastomas have different clinical courses and prognoses, they share
some common genetic abnormalities, such as mutations in P53. In addition, they have
unique genetic abnormalities. For example, whereas activating mutations in platelet
derived growth factor receptor more often are seen in secondary glioblastomas,
activating mutations in epidermal growth factor receptor are found more often in primary
glioblastoma.82
1.5. Par-4 and glioma
Very little work has been done on the role of Par-4 in glioma. Temozolomide, the
drug used in the treatment of glioblastoma, has been shown to induce PrPc expression
in glioma cell lines, which in turn inhibits Par-4 activation.87 In this study, knocking down
Par-4 enhances cell death in response to Temozolomide. Par-4 has also been shown to
mediate glioma cell death in response to curcumin12 and tamoxifen.88 Interestingly,
IDH1 mutation gliomas, which have a favorable prognosis, have downregulated Par-4
expression.44

22
1.6. Apoptosis
Apoptosis is an evolutionarily conserved cell-death process that was originally
discovered in 1842 that is characterized by a series of morphological changes.89 Such
classic morphological features of apoptosis include: membrane blebbing, cell shrinkage,
nuclear fragmentation, chromatin condensation, chromosomal DNA fragmentation, and
global mRNA decay. These morphological changes are due to a fundamental series of
biochemical processes that occur in the cell.89, 90
Apoptosis is a process that plays an important role in human development and in
health and disease. The purpose of apoptosis is to eliminate aged, damaged, harmful, or
unwanted cells. For example, during development, apoptosis plays a role in
organogenesis, developmental involution, and implantation.91 In the adult organism,
apoptosis is responsible for maintaining homeostasis of cell number in tissues that consist
of proliferating cells, for involution of hormone-dependent tissues during hormone
withdrawal, and for eliminating self-reactive lymphocytes. In disease settings, apoptosis
serves to eliminate cells that are damaged beyond repair - for example, in response to
DNA damage by chemotherapeutics, in response to the immune response against a
virally-infected cell, or in response to an accumulation of unfolded proteins.82
1.6.1. Mechanism of apoptosis
Apoptosis can be broadly divided into two pathways: the extrinsic pathway and intrinsic
pathway.92 This classification is based on the nature of the apoptosis-inducing stimulus
and the sensor/effector components involved. A family of cysteine proteases, called
caspases, are the main effectors of apoptosis regardless of the pathway.90, 93

23
In the intrinsic pathway, a cell stimulus leads to an increase in mitochondrial
permeability, which in turn leads to leakage of initiators,94, 95 such as cytochrome C, from
the mitochondria into the cytosol. Upon release, cytochrome C binds to apoptotic
protease activating factor-1 (APAF1) and ATP, which then binds to procaspase 9, itself
an initiator capsase, forming the apoptosome.96 Formation of the apoptosome leads to
the cleavage of pro-caspase 9 into its active form. Active caspase 9 then cleaves pro-
caspase 3 into active caspase 3. Caspase 3 is an effector caspase, which leads to
cleavage of multiple intracellular components, including DNA, resulting in cell death.
Thus, regulating mitochondrial permeability is important for regulating the intrinsic
pathway, since it is the initial step in the intrinsic pathway. The BCL family of proteins is
responsible for regulating mitochondrial permeability, which consists of groups that are
pro-apoptotic and anti-apoptotic.97 Certain conditions, such as presence of growth
factors, induce the production of anti-apoptotic molecules over pro-apoptotic molecules;
however, under certain stressors - such as heat, radiation, or nutrient deprivation – the
balance of BCL family proteins shifts toward pro-apoptotic effectors.
The extrinsic pathway is mainly initiated by receptor-ligand interactions, where
death receptors at the cell surface bind to their respective ligands and initiate apoptosis.98-
100 The cell-surface death receptors are members of the TNF receptor family. There are
two main ligands that have been demonstrated to initiate apoptosis: Fas ligand and TNF.
When a ligand binds its death receptor, the receptor trimerizes, allowing the recruitment
of adaptor proteins, such as FADD or TRADD, to the cytoplasmic domain of the receptors.
These adaptor proteins then bind to multiple inactive forms of caspase 8, an initiator
caspase.101 The aggregation of caspase 8 molecules leads to their activation, by

24
proteolytic cleavage, to yield active caspase 8. Active caspase 8 then activates various
effector caspases and cell death results from caspase cleavage of intracellular
components. The end result of apoptosis is the formation of apoptotic bodies, which are
phagocytosed by immune cells, leaving no inflammation or damage to surrounding cells
and tissues.102
1.7. Conclusion
From the body of work presented above, it is clear that advanced colon cancer,
pancreatic cancer, and glioblastoma have very poor prognoses, and there is a need for
novel and effective therapies. In addition, Par-4 is a positive regulator of apoptosis with
cancer cell selective properties, and Par-4 plays a role in colon cancer, pancreatic
cancer, and glioblastoma. Given its tumor-suppressive properties and therapeutic
potential, further study of Par-4 in the context of those cancers is warranted. To study
Par-4, three different approaches will be taken:
1. Elucidating novel Par-4 function.
2. Identifying and characterizing new regulators of Par-4.
3. Demonstrating the usefulness of Par-4 in increasing the sensitivity to
chemotherapeutics.
My central hypothesis is that Par-4 acts as a general tumor-suppressor and inhibits
cancer progression and can be used to increase the effectiveness of
chemotherapeutics. Toward this end, I chose to pursue three specific aims:

25
1. Determine the impact of Par-4 on colon cancer cell migration, invasion, and
EMT.
2. Determine novel regulators of Par-4 in colon and pancreatic cancer.
3. Determine whether Par-4 can enhance the efficacy of therapeutics and
radiation toward glioma cell death.
Specific aims 1, 2, and 3 will be covered in chapters 2, 3, and 4 of this dissertation,
respectively. Chapter 1 has been previously published.82

26
2. Chapter 2
Overexpression of the pro-apoptotic protein Prostate Apoptosis
Response-4 (Par-4) in colon cancer cells can inhibit metastasis by
upregulating E-cadherin expression
2.1. Introduction
The poor prognosis of advanced colorectal cancer previously discussed
underscores the need for novel strategies to inhibit colorectal cancer metastases.
As also previously mentioned, Par-4 plays a role in apoptosis in a cell-type-
specific manner. Par-4 overexpression is sufficient to induce apoptosis in vitro and in
vivo in a myriad of cancer cell types: breast cancer22, 35, androgen-independent and
androgen-dependent prostate cancer cell line, TRAMP, lung cancer, cervical cancer,
nasopharyngeal cancer, and melanoma7. In other cell types - Jurkat T lymphocytes4,
androgen-dependent prostate cancer cell line, LNCaP24, 36, melanoma cells103, and
renal carcinoma - Par-4 increases the susceptibility of cancer cells to pro-apoptotic
stimuli, including UV irradiation, serum-withdrawal, ionizing radiation, doxorubicin, and
camptothecin. In colon cancer cells, Par-4 overexpression increases apoptosis in
response to the chemotherapeutic agent 5-fluorouracil63.
Par-4 not only induces cell death in cancer cells, but it may also inhibit their
metastasis. This was suggested in a previous study, where mRNA and microRNA
microarray analyses on Par-4 overexpressing HT-29 colorectal cancer cells showed that
Par-4 altered the expression of genes involved in cell movement, including cell

27
migration and invasion64. In addition, Par-4 induced the upregulation of 13 and
downregulation of 9 microRNA’s. Among the predicted target mRNAs of these
dysregulated microRNAs, a significant number are involved in the WNT/β-catenin
pathway, a pathway that has been strongly implicated in colon cancer metastasis. In
vivo, recombinant Par-4 protein inhibits the formation of lung nodules by mouse Lewis
lung carcinoma cells in a tail vein metastasis model32. The goal of this study is to
uncover the mechanisms by which Par-4 inhibits metastasis.
2.2. Materials and Methods
2.2.1. Cell culture and transfection
SW480 and SW620 colorectal cancer cells were maintained in RPMI + 10% fetal
bovine serum (FBS) + 1% penicillin-streptomycin. The cells were transiently transfected
with empty vector (mock), a plasmid vector encoding for human Par-4 (OriGene
Technologies, Rockville, MD), or a plasmid vector encoding for anti-Par-4 shRNA
(Thermo Scientific, Waltham, MA) using either Lipofectamine 2000 Transfection
Reagent (Life Technologies, Grand Island, NY) or PolyJet DNA Transfection Reagent
(SignaGen Laboratories, Rockville, MD), according to the manufacturer’s instructions.
Stable transfectants were isolated using geneticin selection 24 hrs post-transfection.
2.2.2. Western blot analyses
Cells were washed twice with PBS and were lysed into lysis buffer (50mm
HEPES, 100 mm NaCl, 10 mm EDTA, 0.5 % NP40, 10% glycerol, supplemented with
0.0001% Tween 20, 0.1 mM PMSF, 0.1 mM NaVO4, 0.5 mM NaF, 5 μg/ml leupeptin,
0.1 mm DTT). The proteins were quantified according to the BCA Assay (Thermo

28
Scientific Inc., Rockford, IL) and loaded equally onto 10% SDS-polyacrylamide gels.
Proteins were electrophoresed at 150 V and transferred to nitrocellulose membranes
using a semi-dry blotter (BioRad, Hercules, CA). Membranes were blocked with 5%
non-fat dry milk or 5% BSA for 1 hr and incubated with primary antibody overnight. The
blots were washed 3X in TBS with 0.1% Tween20 and incubated for 1 hr in appropriate
HRP-conjugated secondary antibodies (Amersham, Piscataway, NJ). Blots were
washed 3X and chemiluminescent detection was performed using Amersham ECL
Prime Western Blotting Detection Reagent (Thermo Scientific Inc., Rockford, IL). The
blots were either exposed to autoradiography film (GE Healthcare Life Sciences,
Pittsburgh, PA) and scanned or imaged using the Molecular Imager Gel Doc XR System
(Bio-Rad). Densitometric analyses were performed with NIH Image J software104.
Primary antibodies used were: E-Cadherin, Claudin-1, Occludin, ZO-1, Vimentin (Cell
Signaling, Danvers, MA), Par-4 (Santa Cruz Biotechnology, Santa Cruz, CA), and Actin
(Sigma-Aldrich, Saint Louis, MO).
2.2.3. MTT assay
SW620 cells were seeded at a density of 6 x 103 cells/well in a 96-well culture
plate. After 24 hrs, the medium was replaced with medium containing different
concentrations of 5-fluorouracil (5-FU). Forty-eight hours later, MTT reagent was added
(Calbiochem) and the cells were incubated for 3.5 hrs at 37◦ C. After incubation, the
media was aspirated off, crystals were dissolved in MTT solvent (4 mM HCl, 0.1%
Nonidet P-40, in isopropanol), and viability was assessed by measuring the absorbance
at 570 nm with 630 nm absorbance as the reference.

29
2.2.4. Scratch assay
Mock, Par-4, and anti-Par-4 shRNA transfected cells were plated in either 6-well
or 12-well plates and grown to confluence. A scratch was made with sterile pipette tip.
The wells were washed with PBS and photomicrographs were collected under 200x
magnification. The cells were incubated for 24 hrs and the scratch areas were again
photographed.
2.2.5. Boyden Chamber assays
Matrigel-coated inserts (BD Biosciences) were prepared according to the
manufacturer’s instructions. The inserts were aseptically-transferred into 12 well culture
plates containing medium with 10% FBS. Mock and Par-4 transfected SW620 cells
were suspended in culture medium with 0.1% FBS and seeded into each insert. The
cells were allowed to invade for 72 hrs. After removing the culture medium and
scrubbing the cells off of the inside of the insert, the cells on the underside of the insert
were stained with 1% crystal violet in 50% methanol and counted under a microscope.
Cell culture inserts without matrigel were aseptically transferred into 24 well
culture plates containing 10% FBS-RPMI. Mock, Par-4 and shRNA transfected SW480
cells (1x105) were suspended in 1% FBS-RPMI and seeded into the inserts. The cells
were allowed to migrate for 24 hrs. After removing the culture medium, cells were fixed
with 4% paraformaldehyde, permeabilized with 100% methanol, and stained with 0.1%
crystal violet in 50% methanol. After scrubbing off non-migrated cells from the inside of
the insert with a cotton swab, the number of migrated cells on the outside of the insert
was quantified at 100X magnification.

30
2.2.6. RT-PCR analyses
RNA was isolated from mock and Par-4-transfected SW620 cells using RNeasy
kit (Qiagen). cDNA was synthesized using a High Capacity cRNA reverse transcription
kit (Applied Biosystems). Real-time quantitative PCR was performed using ABI Gene
Expression Assay primers on the ABI7900 HT Sequence Detection System. Using the
2ΔΔCt analysis method with the ABI SDS2.2.2 software, relative amounts of target
mRNA were quantitated using actin as an internal control.
2.2.7. Cell proliferation assay
SW480 and SW620 cells were seeded into a 12-well culture dish (1 x 105
cells/well). At the appropriate timepoints, cells were trypsinized, and the number of
viable cells was quantitated using a hemacytometer and trypan blue.
2.2.8. Statistical analyses
The statistical analyses were carried out using GraphPad Prism software, version
6.04 (GraphPad Software, Inc., San Diego, CA, USA). Unpaired two-tailed t-tests are
carried out in order to determine statistically significant differences between control and
transfectants, unless otherwise noted. The threshold for significance is a P < 0.05.
*P<0.05; **P<0.01; ***P<0.001.
2.3. Results
2.3.1. Par-4 increases susceptibility of metastatic SW620 cells to 5-FU
Stable Par-4 transfectants and control transfectants were created by transfecting
SW620 colon cancer cells with Par-4 plasmid or control plasmid, respectively, followed
by geneticin selection. First, Par-4 expression in the stable clones was validated by

31
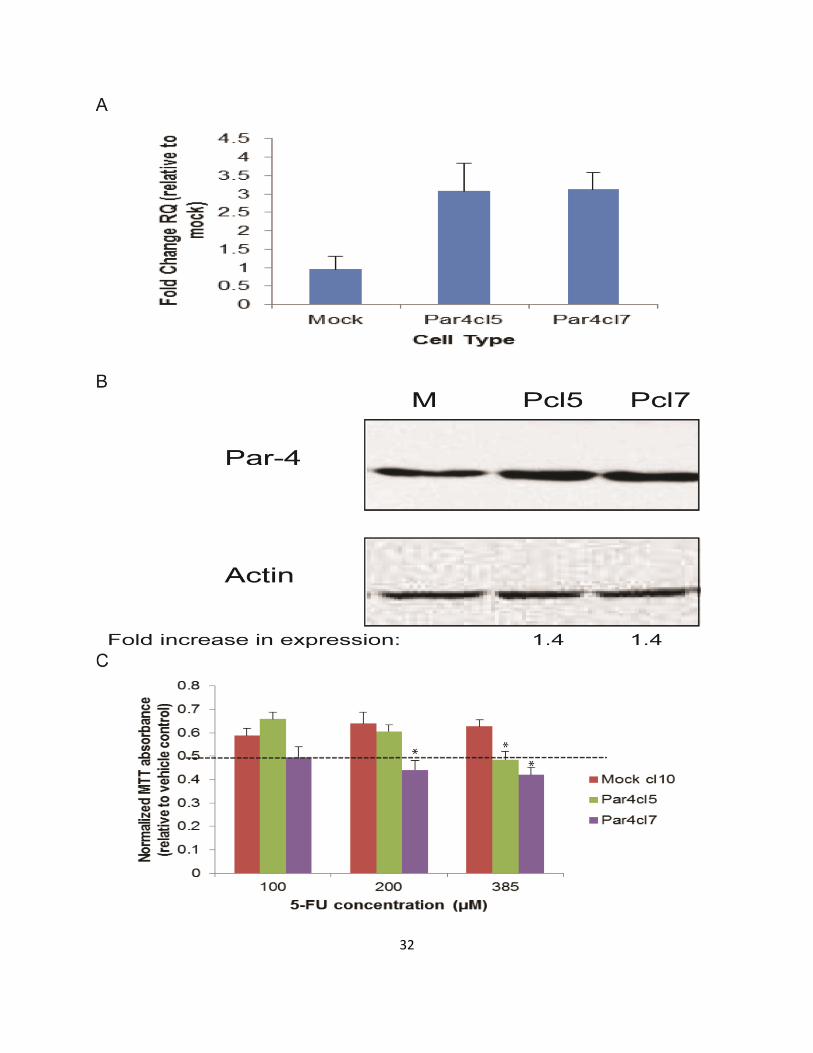
measuring mRNA and protein levels by qRT-PCR and Western blotting, respectively.
The Par-4 clones (Pcl5 and Pcl7) had increased Par-4 mRNA levels (Fig. 2-1a) and
protein levels (Fig. 2-1b) relative to the control transfectants (Mock). The susceptibility
of mock and Par-4 transfected SW620 cells to the chemotherapeutic agent 5-
fluorouracil (5-FU) was assessed by performing MTT viability assays on treated cells.
As can be seen in Fig. 2-1c, both Par-4-overexpressing clones tested had lower viability
in comparison to a mock-transfected clone when treated with 385 μM 5-FU for 48 hours.
32

33
Figure 2-1. Overexpression of Par-4 increases susceptibility of metastatic SW620 cells to 5-FU.
SW620 colorectal cancer cells were transfected either with a plasmid vector containing a
human Par-4 construct or an empty vector (mock). A) Par-4 mRNA expression was assessed in
mock-(M) and Par-4-transfected (Pcl5 and Pcl7) cells by RT-PCR analysis. The bars in the graph
reflect the fold upregulation of Par-4 mRNA expression in Par-4-transfected cells over the
expression in mock-transfected cells. Data shown are means + SE of three biological replicates.
B) Western blot analyses were performed to assess Par-4 protein expression in mock- and Par-
4-transfected cells. The intensities of the Par-4 bands were normalized against the respective
intensities of the bands for the loading control, actin, to calculate the -fold increase in
expression. C) The susceptibility of mock- and Par-4 transfected SW620 cells to the
chemotherapeutic agent 5-FU was assayed by MTT. Cells were treated with either the vehicle
control (DMSO) or with the indicated concentrations of 5-FU for 48 hours. The bars in the graph
reflect the absorbance readings of the 5-FU-treated cells normalized against those of the
vehicle-treated cells. Data shown are means + SE of at least three biological replicates. The data
presented in this figure is the work of Christina Leah Kline.

34
2.3.2. Par-4 inhibits cell migration and invasion in SW480 and SW620 cells
Next, the effects of Par-4 overexpression on two key steps of metastasis,
migration and invasion, were examined. To examine migration, scratch assays on
SW620 cells were performed and the number of cells that migrated into the area of the
scratch after 24 hours were quantified. Fewer Par-4 overexpressing cells than mock-
transfected cells migrated into the scratch area (Fig. 2-2a and b). The scratch assay
showed that Par-4 overexpression inhibited the migratory ability of SW620 cells. Next,
the invasive ability of mock- and Par-4-transfected SW620 cells was assessed in a
Matrigel assay. As can be seen in Fig. 2-2c, fewer Par-4 overexpressing cells were able
to invade through the Matrigel than mock transfected cells. It is possible that more of the
mock-transfected cells appeared to invade through the Matrigel, because increased
Par-4 expression reduced cell proliferation. To assess this, the cell growth of mock- and
Par-4 transfected SW620 cells (Fig. 2-2d) was monitored. No significant differences in
cell proliferation were observed as a result of increased Par-4 expression.
To extend the previous observations beyond a single cell line, SW480 cells were
transiently transfected with either control or anti-Par-4 shRNA, and scratch assays were
performed to assess migratory ability. Consistent with the inhibitory effect of Par-4 on
migration in SW620 cells, Par-4 knock-down in SW480 cells enhanced migration as
evidenced by complete closure of the scratch relative to the control transfected cells
(Fig. 2-3a). To validate this observation, SW480 cells were transiently transfected with
control plasmid, Par-4 expression plasmid, or anti-Par-4 shRNA, then seeded into a
Boyden chamber, and the number of cells that migrated across the Boyden chamber
after 24 hrs was quantified (Fig. 2-3b). As can be seen in Fig. 2-3b, Par-4

35
overexpression inhibited migration, whereas knock-down of Par-4 enhanced migration
across the Boyden chamber. Western blots confirming overexpression and knock-down
of Par-4 are also shown in Fig. 2-3b. Cell proliferation assays were performed to
compare the growth of Par-4 and control transfected SW480 cells. Although there is a
statistically significant difference in cell number by day 3 post-transfection, the 24-hr
time point of the Boyden chamber assay corresponds to day 1 post-transfection;
therefore, differences in proliferation cannot account for the inhibition of migration seen
in Par-4 transfected SW480 cells in Fig. 2-3b.
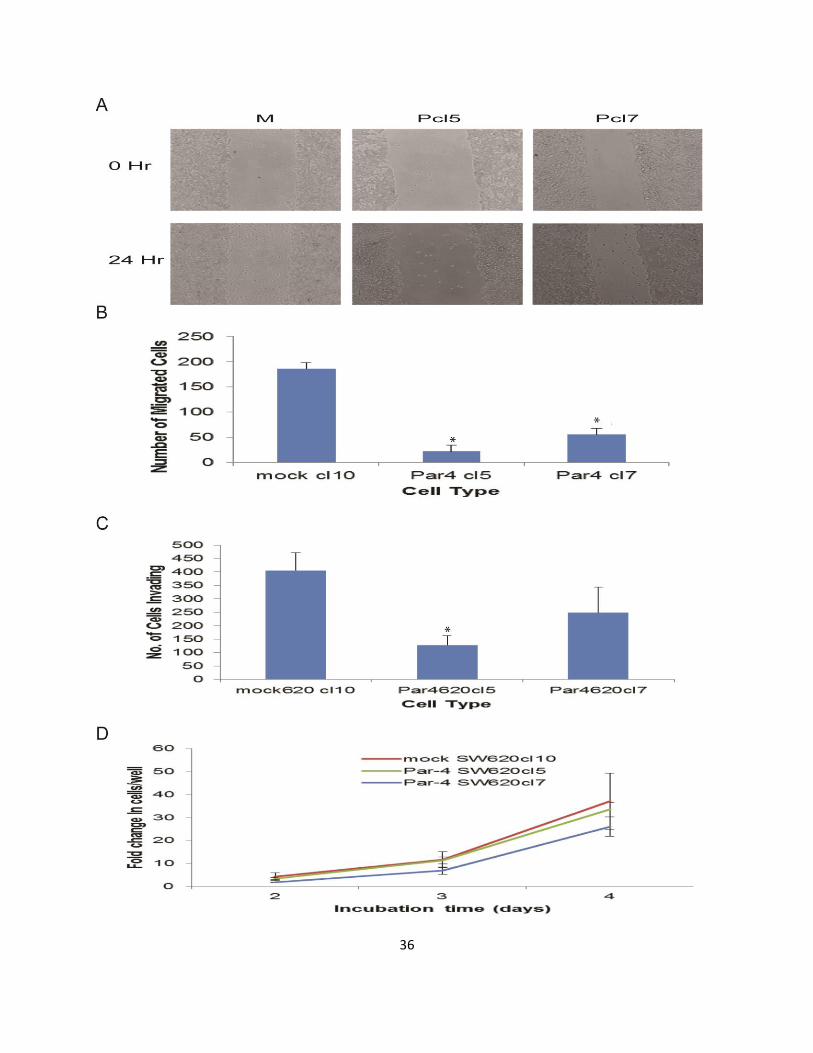
36

37
Figure 2-2. Par-4 overexpression inhibits metastatic processes in SW620 cells.
Par-4- and mock-transfected SW620 cells were seeded into six-well or twenty-four-well plates,
and migration and invasion assays were performed to measure the effect of Par-4 on these
metastatic processes. A) Scratch assays were performed to compare the ability of mock- and
Par-4 transfected cells to migrate in 24 hours. The lines are drawn to indicate the boundaries of
the scratch made at the 0 hour timepoint. B) The number of cells that migrated into the scratch
area after 24 hours were graphed. Data shown are means + SE of at least two biological
replicates and a total of six scratch areas were imaged. C) Mock- and Par-4 transfected cells
were cultured in low-serum (0.1%) media in Matrigel-coated inserts to assess invasive
capability. The bars in the graphs reflect the number of cells that invaded through the insert
after 72 hours. Data shown are means + SE of at least 5 biological replicates. D) Cell
proliferation was assessed by counting viable cells in trypan blue assays. Data shown are means
± SE of at least two replicates. The data presented in this figure is the work of Christina Leah
Kline.
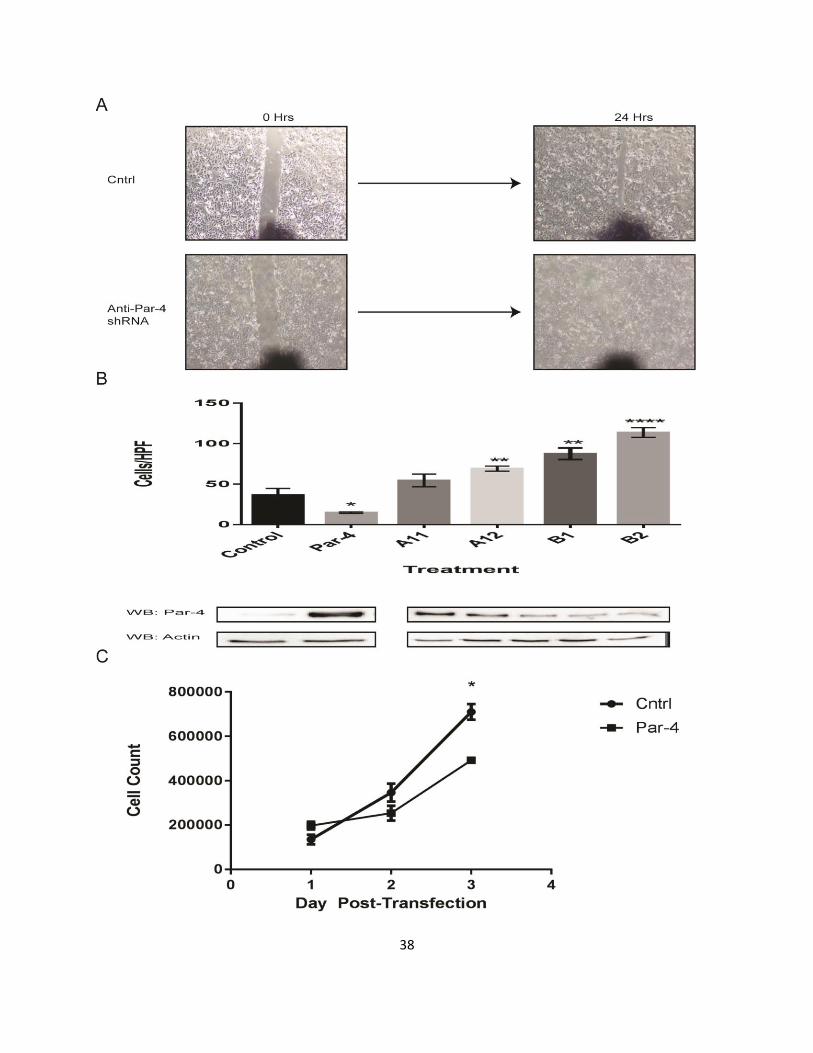
38

39
Figure 2-3. Par-4 inhibits migration in SW480 cells.
A) Scratch assays were performed to compare the ability of mock- and anti-Par-4 shRNA-
transfected cells to migrate in 24 hours. A representative image of the scratch at the 0 and 24
hour timepoint is shown. B) SW480 cells that were transiently transfected with either Par-4
expression plasmid, anti-Par-4 shRNA (A11, A12, B1, and B2), or empty vector were cultured in
media with 1% FBS in the inner chamber of a Boyden chamber, while media with 10% FBS in the
lower chamber acted as a chemoattractant. Cells are allowed to migrate for 24 hrs, then the
number of migrated cells were quantitated (# cells/high-powered field). Data shown are the
means ± SE of five random fields. Western blots confirming the overexpression and knock-down
of Par-4 levels are also shown. C) Cell proliferation was assessed by counting viable cells in trypan
blue assays. Data shown are means ± SE of at least two replicates.

40
2.3.3. Par-4 induces a mesenchymal-epithelial transition in SW620 cells
Given the novel observations that Par-4 inhibits migration and invasion of colon
cancer cells, the mechanisms behind these effects were investigated. Par-4
overexpression altered the morphology of SW620 cells (Fig. 2-4a). This is significant,
since the SW620 cell line is one that was derived from a metastatic site, a lymph node,
from a patient with colorectal cancer. Both mock-transfected clones exhibited a mixture
of round cells and elongated cells with minimal cell-to-cell contacts. However, with Par-4
overexpression, the cells demonstrate more cell-to-cell contacts. It is proposed that a
cancer cell that has gained the ability to metastasize undergoes a phenotypic change in
a process referred to as epithelial-mesenchymal transition (EMT). One characteristic of
this change is a morphologic change where the epithelial cell loses cell-to-cell contacts
and becomes more elongated. Thus, the morphologic changes observed in the SW620
cell line with Par-4 overexpressed are suggestive of an EMT reversal, namely a
mesenchymal-epithelial transition (MET).
Another hallmark of EMT is the loss of the epithelial protein, E-cadherin,
concomitant with the gain of the mesenchymal marker, vimentin. Thus, the levels of E-
cadherin and vimentin were examined. Western blot analyses showed that E-cadherin
protein expression was increased in the Par-4 overexpressing cells, while vimentin
levels were reduced (Fig. 2-4b). Par-4 overexpression in SW480 cells also increased
the expression of E-cadherin (Fig. 2-4c). Immunofluorescence studies on the
localization of E-cadherin in the mock and Par-4 transfected SW620 cells shows that
more E-cadherin can be detected in the membranes of Par-4 overexpressing cells than
in mock-transfected cells (Fig. 2-4d). E-cadherin associates with β-catenin at the cell

41
membrane. This association inhibits the activation of the WNT/β-catenin pathway, a
pathway that is involved in colon cancer progression. Immunofluorescence results show
that there is increased localization of β-catenin at the membrane of Par-4
overexpressing cells (Fig. 2-4e). Taken together, these data further support the
observation of an MET phenotypic change.
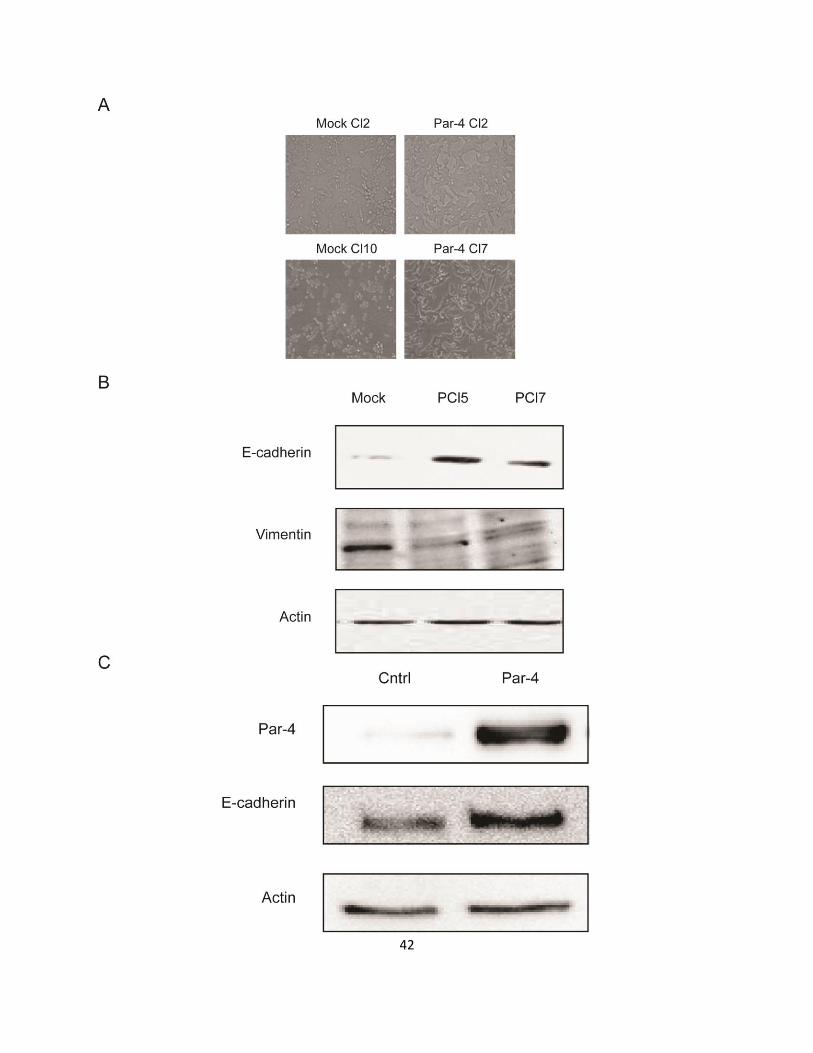
42

43
Figure 2-4. Par-4 induces a mesenchymal-epithelial transition in SW620 cells.
A) Phase-contrast microscope images of mock and Par-4-transfected SW620 cells were
collected, and B) lysates were processed for Western blot analyses of E-cadherin, the
mesenchymal marker, vimentin, and actin. C) SW480 cells were transfected with Par-4 and
Western blot analyses were performed for Par-4, E-cadherin, and actin. D) Par-4 overexpression
increases E-cadherin localization in the membrane. Immunofluorescence for the epithelial

44
marker E-cadherin (green) was performed with mock and Par-4-transfected cells. Nuclei were
stained with DAPI. E) Par-4 overexpression increases β-catenin localization in the membrane.
Immunofluorescence for the E-cadherin-interacting protein β-catenin was performed. The
percentage of cells expressing β-catenin in their surface was quantitated. The data shown
indicate mean + SE of at least 3 sections.

45
2.3.4. Par-4 regulates tight-junction protein expression in SW620 cells
E-cadherin protein expression can be altered by different mechanisms, including
transcriptional, post-transcriptional, and epigenetic. To determine whether Par-4
increases E-cadherin mRNA expression, qRT-PCR analyses were performed on mock
and Par-4 transfected SW620 cells. As can be seen in Fig. 2-5a, E-cadherin mRNA
levels were increased in the Par-4 overexpressing SW620 clones. Western blot
analyses were performed to identify the transcriptional repressor(s) that may be altered
in response to Par-4 overexpression. Snail is a transcription factor that promotes the
repression of E-cadherin, thus regulating EMT during embryonic development. Snail
upregulation and nuclear localization have been shown to downregulate E-cadherin and
upregulate vimentin. Western blot analyses indicated that Snail expression may be
slightly decreased (Fig. 2-5b). Like Snail, Slug is also a transcriptional repressor of E-
cadherin during embryonic development. Western blot analyses of Par-4
overexpressing cells show that Slug expression was not changed (Fig. 2-5b). Even if
Snail expression was not markedly altered, I assessed the transcriptional activity of
Snail by examining the expression of its other targets -- claudin, occludin, and ZO-1.
Par-4 overexpression caused the upregulation of these proteins, especially occludin and
ZO-1 (Fig. 2-5c).

46

47
Figure 2-5. Par-4 induces E-cadherin transcription and tight-junction protein upregulation.
A) RT-PCR analyses for E-cadherin mRNA expression in mock and Par-4-transfected cells were
performed. The bars in the graph reflect the fold upregulation of E-cadherin mRNA expression
in Par-4-transfected cells over the expression in mock-transfected cells. Data shown are means
+ SE of three biological replicates. B) Par-4 overexpression does not alter the expression of a
subset of E-cadherin transcriptional repressors. Western blot analyses for E-cadherin
transcriptional repressors Snail, Slug, and Zeb1 were performed. C) Western blot analyses for
tight junction proteins, claudin 1, occludin, ZO-1, and actin were performed.

48
2.4. Discussion
The potential of Par-4 in cancer therapy has been increasingly appreciated given
its ability to induce cell death by itself or in combination with chemotherapeutics and
radiation 105. In this study, we have shown that SW620 cells are sensitized to 5-FU upon
ectopic Par-4 expression. This sensitization corroborates our prior findings in HT-29
cells in response to 5-FU and ISC-4 treatment, respectively63-66. Although it has been
shown that recombinant Par-4 can inhibit metastasis32, the mechanisms behind this
effect have been scarcely elucidated. Furthermore, ectopic Par-4 expression in HT-29
cells was shown to deregulate mRNA’s and microRNA’s involved in cell migration and
motility64. We have shown in this study that Par-4 can inhibit the ability of colon cancer
cell lines, SW480 and SW620, to migrate and invade.
The ability of SW620 cells to migrate and invade was reduced with increased
expression of Par-4. Moreover, the ability to SW480 cells to migrate was also inhibited
by Par-4 overexpression, while downregulation of Par-4 resulted in increased migration.
One of the mechanisms behind this reduction in migration and invasion may be the Par-
4-induced upregulation of E-cadherin. Increased expression of E-cadherin in mammary
and prostate epithelial carcinoma cells has been shown to inhibit migration and invasion
106, 107. Small molecules that can restore E-cadherin expression in SW620 cells have
been shown to reduce invasion 108. The ability of E-cadherin to inhibit cell migration and
invasion has been shown to be independent of its role in mediating cell-cell adhesions,
and instead is due to its role in downregulating the β-catenin/TCF pathway 107. E-
cadherin sequesters β- catenin, keeping β- catenin from interacting with actinin-4. On
the other hand, in the absence of E-cadherin or when E-cadherin is downregulated, β-

49
catenin has been found to colocalize with actinin-4 in bleb-like membrane protrusions in
colorectal cancer cells 109. Overexpression of actinin-4 has been shown to increase
motility of colorectal cancer cells 110. The mechanism underlying the β-catenin/actinin-4
complex-induced migration remains to be elucidated. Another mechanism by which E-
cadherin can inhibit migration and invasion is due to downregulation of hNanos1.
hNanos1 is a zinc finger protein that acts a translational repressor by binding to the 3’-
UTR of mRNA targets. Overexpression of hNanos1 is sufficient to induce invasion in
collagen type I gels and increase migration 111. This may be due in part to the hNanos1-
induced expression of the matrix metalloproteinase MT1-MMP (membrane type1-matrix
metalloproteinase) 112.
The loss or heterogenous expression of E-cadherin in colorectal cancer tissue
samples has been correlated to an advanced clinical stage and liver metastasis 113. This
makes the upregulation of E-cadherin induced by Par-4 overexpression in SW480 and
SW620 cells a significant finding. E-cadherin expression is regulated via different
mechanisms 114. Although the CpG island on the E-cadherin promoter is methylated in a
number of cancers 115, including colorectal cancer 116, 117, this may not be the dominant
mechanism of E-cadherin downregulation in colorectal cancer 118. E-cadherin
expression is also inhibited by a number of transcriptional repressors, including
members of the ZEB and basic helix-loop-helix (bHLH) families 119. In this study, we
reported the transcriptional upregulation of E-cadherin as a result of Par-4
overexpression. In addition, the expression levels of other tight junction proteins –
claudin-1, occludin, and ZO-1 – were upregulated. The mechanisms by which Par-4
regulates the expression of these genes is an area for further study.

50
Increasing Par-4 levels in SW620 cells was sufficient to alter the cell morphology
from a rounded phenotype with minimal cell-to-cell contacts to one with many cell-to-cell
contacts, which is characteristic of epithelial cells. Par-4-overexpressing cells had a
decreased expression of mesenchymal markers, such as vimentin, and upregulation of
the epithelial cell protein, E-cadherin. This is significant, because it suggests an MET
transition, the opposite of the epithelial-mesenchymal transition (EMT) that primary
tumor cells must undergo in order to metastasize. In colorectal cancer, EMT has been
observed in cells at the invasive front of primary tumors and lymph node metastases 120.
Characteristics of cells undergoing EMT include the decreased expression of epithelial
markers, in particular E-cadherin, and upregulation of mesenchymal markers, like
vimentin. Another characteristic of EMT is the increased nuclear localization of β-
catenin. In this study, we report that by increasing the levels of Par-4 in the metastatic
cell line, SW620, MET was induced, which is significant in that it helps to resolve a
discrepancy in the literature regarding the regulation of EMT by Par-4. In a study by Tan
et. al., Par-4 downregulation was shown to induce EMT in the cell line, BxPc-3,
concomitant with the acquisition of cisplatin resistance10; however, in a more recent
study, Par-4 was shown to mediate TGF-β-induced EMT11. In that study, TGF-β induced
EMT resulted in a Smad-dependent Par-4 upregulation, which in turn downregulated
epithelial markers and upregulated mesenchymal markers with a concomitant increase
in cell motility in several different cell lines. Subsequent overexpression and knockdown
of Par-4 showed that these phenotypic changes were mediated by Par-411. Thus, our
results corroborate the data by Tan et. al. and contradict the data by Chaudhry et. al. A
potential explanation for this discrepancy is that Par-4 regulates EMT in a cell-type

51
dependent manner. In the study by Tan et. al., the studies were carried out in a
pancreatic cancer cell line10, whereas in the study by Chaudhry et. al., endometrial,
cervical, ovarian, and breast cancer cell lines were used11. Our study was carried out in
colon cancer cell lines. The exact, cell-specific mechanisms by which Par-4 regulates
EMT remain to be elucidated. Taken together, in this study, the increased cell-to-cell
contacts due to the increased expression of tight junction proteins along with the EMT
reversal help to explain the observed inhibitory effect of Par-4 on the migration and
invasion of colon cancer cells.
In summary, our data show that Par-4 inhibits the migration and invasion of
SW480 and SW620 colon cancer cells, and the upregulation of epithelial markers and
downregulation of mesenchymal markers concomitant with a mesenchymal-epithelial
transition can partially explain the observed phenotype. These findings provide an
additional impetus to explore the use of Par-4 in colon cancer therapy. A potential
limitation of this study is that the migration and invasion experiments were entirely in
vitro. Future experiments in in vivo models, such as a tail-vein injection model, will help
to address this limitation. Future experiments should also examine the effect of Par-4 on
migration and invasion in other types of cancer and further elucidate the cell-specific
mechanisms by which Par-4 regulates migration.
2.5. Acknowledgements
This work was supported by Penn State Hershey start-up funds of Rosalyn B. Irby.
2.6. Conflict of Interest
The authors declare no conflicts of interest.

52
2.7. Publication Note
The work contained within this chapter was published in the Journal of Colon and Rectal
Cancer.121

53
3. Chapter 3
Trim21 is a novel regulator of Par-4 in colon and pancreatic cancer
cells
3.1. Introduction
As discussed above, metastatic colon cancer and pancreatic cancer both have a
very grim prognosis. The poor prognoses are due to a variety of factors. In colon
cancer, inherent chemoresistance and acquired resistance to treatment both play a
role.122 The inherent difficulty in treating pancreatic cancer is related to late diagnosis
and the resistance of pancreatic cancer cells to chemotherapy.123 This underscores an
important need for more effective therapeutics and for identifying novel therapeutic
targets.
Cancer care is shifting from the use of traditional chemotherapy with a broad-
based mechanism of action to targeted therapy, which is designed to interfere
selectively with cancer cells thereby limiting side-effects. Some examples of targeted
therapies used in the treatment of colorectal cancer and pancreatic cancer include
Avastin and Erlotinib. Avastin is a monoclonal antibody directed against vascular
endothelial growth factor (VEGF), thereby inhibiting angiogenesis, and is used in the
treatment of colorectal cancer.122 Erlotinib is a small-molecule inhibitor of epidermal
growth factor receptor (EGFR), thereby inhibiting cell survival and proliferation, and is
used in the treatment of pancreatic cancer.123 While the combination of standard

54
chemotherapy with targeted therapy improves survival compared with standard therapy
alone, resistance often develops.
Par-4 is a tumor suppressor with cancer-cell selective properties. In colon cancer
cells, Par-4 overexpression sensitizes cells to apoptosis in response to the
chemotherapeutic agent, 5-fluorouracil63, and AKT inhibitor, ISC-465. In pancreatic
cancer cells, treatment with inhibitors of NF-κB and BCL-2 induce Par-4 expression,
which in turn sensitizes cells to chemotherapeutic-induced apoptosis.80, 124
While most Par-4 studies have focused on characterizing the function of Par-4,
its regulation is a relatively understudied area. Par-4 is upregulated in response to
treatment with various natural products and small-molecules.12, 62, 125 Recently, Par-4
has also been shown to be transcriptionally upregulated by FOXO3a in response to
treatment with Withaferin A.45 Ubiquitination of target proteins by E3 ligases followed by
proteasomal degradation is a common mechanism for downregulating protein levels
and activity. The ubiquitination of Par-4 by FBXO45 and the subsequent proteasomal
degradation of Par-4 regulates cancer cell survival.37
Trim21, as an E3 ligase, also functions by ubiquitination of target substrates. For
example, Trim21 regulates innate immune signaling through ubiquitination of DDX41,
an intracellular DNA sensor, thereby inhibiting the innate immune response to
intracellular dsDNA.126 In addition, multiple members of the IRF family of proteins, which
is a family of transcription factors that are activated and act downstream of toll-like
receptors, are substrates of Trim21.127, 128 Thus, Trim21 negatively regulates the innate
immune response to foreign pathogens.

55
While most studies have examined the role of Trim21 in regulating innate
immune signaling, some studies have implicated Trim21 in the regulation of other
cellular processes. For example, Trim21 has been shown to positively regulate
apoptosis via ubiquitination of apoptosis inhibitors, such as BCL-2 and c-FLIP, leading
to their degradation.129, 130 Furthermore, Trim21 is a negative regulator of B-cell
proliferation.131 The above findings suggest a possible tumor-suppressive role of
Trim21. In line with this, two recent studies have demonstrated that reduced Trim21
expression is correlated with poor prognosis in hepatocellular carcinoma and diffuse
large B-cell lymphoma.132, 133
Identifying novel regulators of Par-4 represents a potential avenue for identifying
new drug targets for colon and pancreatic cancer. In this study, I identify Trim21 as a
novel interaction partner of Par-4 in colon cancer cells. Furthermore, I show that Trim21
can regulate Par-4 levels in response to cisplatin in both pancreatic cancer and colon
cancer cell lines. Finally, I show that Trim21 may represent a potentially novel
therapeutic target and biomarker.
3.2. Results
3.2.1. Trim21 is a novel interacting partner of Par-4
In order to discover new regulators of Par-4, I sought to identify novel binding
partners of Par-4. To identify novel binding partners of Par-4, I performed an
immunoprecipitation of Par-4 from HCT-116, HT-29, and KM12C colon cancer whole
cell lysates that had been transiently transfected with Par-4 plasmid. The purpose of the
transfection was to increase the signal. Proteins that co-precipitated with Par-4 were
analyzed by mass spectrometry for identification. Trim21 was identified as an interacting

56
protein in all three cell lines tested. A representative list of the top identified proteins in
the pull-down and the negative control pull-down is shown in Fig. 3-1A. To validate the
mass-spectrometry result and to confirm that the interaction between Par-4 and Trim21
was not an artifact of the ectopic Par-4 expression, reciprocal co-immunoprecipitations
were performed with endogenous Par-4 in HCT-116, HT-29, and KM12C whole cell
lysates. As can be seen in Fig. 3-1B through Fig. 3-1D, Par-4 and Trim21 interact
endogenously in colorectal cancer cells. To corroborate the interaction data, an
immunofluoresence analysis was performed in order to examine the co-localization of
Trim21 and Par-4 proteins intracellularly. The co-localization of Par-4 and Trim21 further
suggests that Trim21 and Par-4 proteins interact endogenously (Fig. 3-1E).
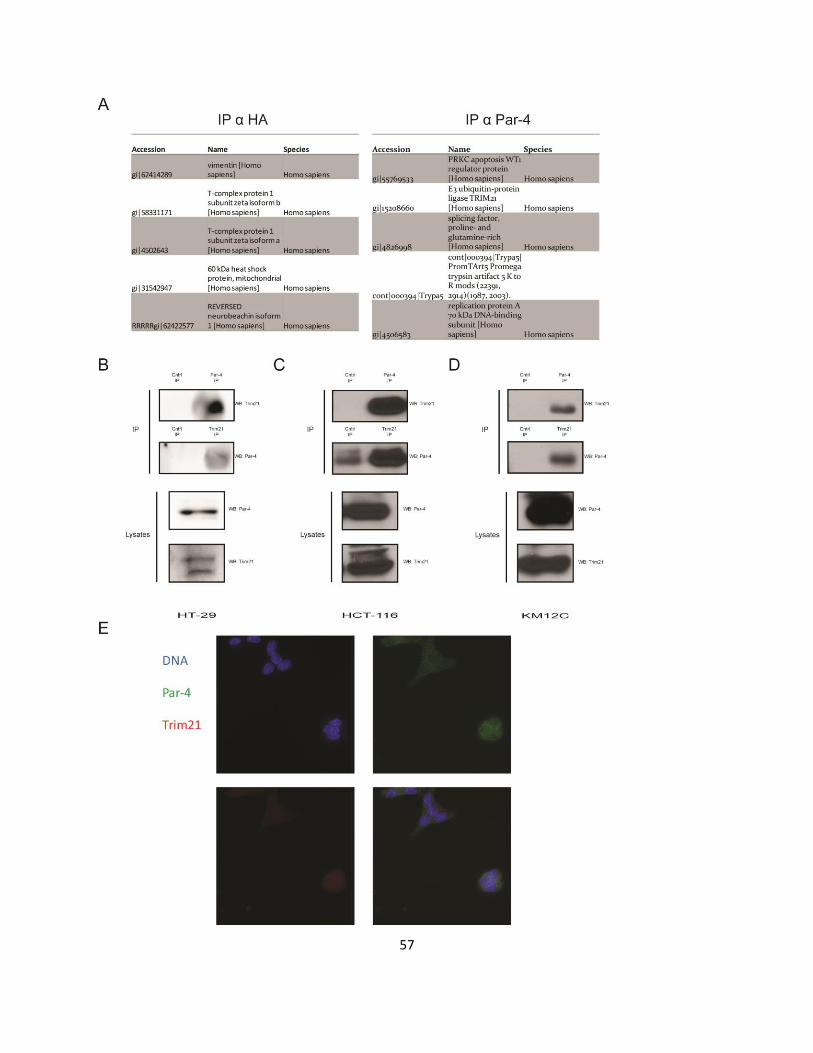
57

58
Figure 3-1. Trim21 is a novel interacting partner of Par-4.
HCT-116, HT-29, and KM12C cells are grown and either transfected with Par-4 expression
plasmid or control plasmid for 48 hrs. Par-4 was immunoprecipitated from whole cell lysates
using anti-Par-4 antibody and protein G magnetic beads. An in-solution trypsin digestion was
performed on the eluted proteins, and the digests were sent for mass spectrometry analysis. A)
A representative list of the most abundant proteins identified in the digest reveals Trim21 as a
potential novel interacting partner of Par-4. Co-immunoprecipitations are performed in HT-29,
HCT-116, and KM12C cells (B, C, D, respectively) in order to validate the Trim21/Par-4
interaction. KM12C cells were grown on coverslips, fixed, permeabilized, and probed for Par-4
and Trim21. E) A representative image showing co-localization of Par-4 and Trim21 in KM12C
cells.

59
3.2.2. Trim21 interacts with Par-4 through its PRYSPRY domain
Next, I sought to characterize the Par-4/Trim21 interaction. Specifically, I wanted
to identify which domain of Trim21 is responsible for mediating its interaction with Par-4.
Trim21 is an E3 ligase that is part of the Tripartite motif (TRIM) family. TRIM family
members contain a RING finger domain, a B-box domain, and a coiled-coil domain.
Trim21 is unique in that it also contains a C-terminal PRYSPRY domain, which is a
protein-protein interaction domain.40 A schematic of Trim21 can be seen in Fig. 3-2A.
To determine which domain of Trim21 mediates its interaction with Par-4, I
obtained three Trim21 constructs: full-length, exon 1 deletion (Trim21ΔEx1), and exon 6
(Trim21ΔEx6) deletion. The exon 1 deletion does not contain the RING domain nor the
B-box domain. These domains are responsible for the E3 ligase activity of Trim21. The
exon 6 deletion does not contain the PRYSPRY domain. A schematic of these
constructs can also be seen in Fig. 3-2A. To test which domain of Trim21 is responsible
for its interaction with Par-4, I co-transfected HCT-116 cells with combinations of the
Par-4 plasmid and each of the three Trim21 constructs. Then, I performed a co-
immunoprecipitation against Par-4, and probed for the presence of Trim21 by Western
blotting. The co-immunoprecipitation performed in cells with the Trim21ΔEx6 mutant
shows decreased binding of Trim21 to Par-4. This suggests that the PRYSPRY domain
of Trim21 is required for Trim21/Par-4 binding (Fig. 3-2B).

60
Figure 3-2. Trim21 interacts with Par-4 via its PRY-SPRY domain.
HCT-116 cells are co-transfected with Par-4 plasmid and different Trim21 constructs for 48 hrs.
Par-4 is immunoprecipitated from whole cell lysates using anti-Par-4 antibody and protein G
magnetic beads. The proteins were eluted from the beads and a Western blot analysis is
performed using anti-Trim21 antibody. A) A diagram representing the various Trim21
constructs that are used B) Western blots demonstrating that when co-transfecting with the
Trim21 construct that does not contain the PRY-SPRY domain, Trim21 does not interact with

61
Par-4; in contrast, when co-transfecting with the Trim21 constructs that contain the PRY-SPRY
domain, Trim21 does interact with Par-4.

62
3.2.3. Trim21 is not sufficient to downregulate Par-4 levels
Given that Trim21 is an E3 ligase and interacts with Par-4, I hypothesized that
Trim21 may downregulate Par-4 protein levels via ubiquitination and subsequent
proteasomal degradation. The endogenous expression patterns of Par-4 and Trim21
across a panel of colorectal cancer cell lines appears to show an inverse relationship
(Fig. 3-3A). This inverse relationship suggests that Trim21 could downregulate Par-4
levels. To test this hypothesis, I ectopically expressed the three Trim21 constructs in
SW480 cells by transient transfection, and probed for the expression of Par-4 by
Western blotting. The Par-4 protein expression was not significantly changed in the
Trim21 transfectants compared to the control, which demonstrate that Trim21
overexpression is not sufficient to downregulate Par-4 levels (Fig. 3-3B). These results
were corroborated by repeating the experiment using HCT-116 cells (data not shown).
This conclusion is further supported by the observation that ectopic Trim21 expression
and Trim21 knock-down do not affect the half-life of Par-4.37
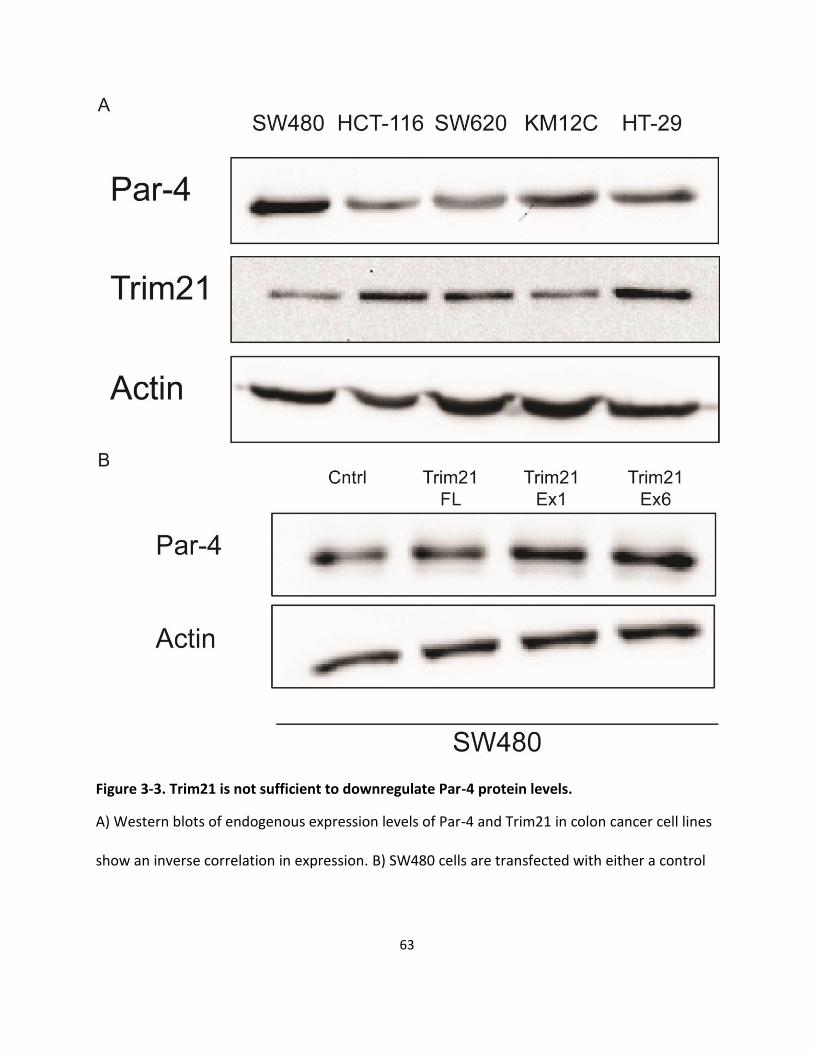
63
Figure 3-3. Trim21 is not sufficient to downregulate Par-4 protein levels.
A) Western blots of endogenous expression levels of Par-4 and Trim21 in colon cancer cell lines
show an inverse correlation in expression. B) SW480 cells are transfected with either a control

64
plasmid or various Trim21 constructs for 48 hours. Whole cell lysates are collected and Par-4
protein levels are analyzed by Western blot with actin as a loading control.

65
3.2.4. Ectopic expression of Trim21 downregulates Par-4 in the presence of
cisplatin
Given that Trim21 was not sufficient to downregulate Par-4 levels, I hypothesized
that Trim21 may regulate Par-4 levels in response to a stimulus. Cisplatin (CDDP) is
one such stimulus that has been shown to affect Par-4 activity via promoting its
cleavage by caspase 3.41 Furthermore, Par-4 downregulation induces cisplatin
resistance in pancreatic cancer cells via a PI3K/AKT-dependent EMT pathway.10
To test whether Trim21 modulates Par-4 levels in response to cisplatin treatment,
I transfected HCT-116 and HT-29 cells with Trim21 full-length plasmid or control
plasmid, and treated the transfected cells with increasing concentrations of cisplatin for
24 hrs. I probed for changes in Par-4 levels by Western blotting. As can be seen in Fig.
3-4A and Fig. 3-4B, in the presence of Trim21 overexpression, cisplatin downregulates
Par-4 levels relative to control transfected cells. In HT-29 cells, this downregulation is
evident even at the lowest cisplatin dose tested, and this downregulation occurs at all
doses up to 15 μg/ml (Fig. 3-4A). In HCT-116 cells, this downregulation occurs at all
doses at or above 3.75 μg/ml with the exception of the 7.5 μg/ml dose (Fig. 3-4B).

66
Figure 3-4. Ectopic expression of Trim21 downregulates Par-4 in the presence of cisplatin in colon cancer cells.
Colon cancer cell lines, transfected with or without Trim21 expression plasmid for 48 hrs, were
treated with increasing doses of cisplatin for 24 hrs. The cisplatin concentrations are in units of

67
μg/ml. Western blots of Par-4 and Trim21 levels are shown with actin shown as a loading
control. A) HT-29 B) HCT-116.

68
3.2.5. Cisplatin downregulates Par-4 in a dose- and proteaseome-
dependent manner
Given the previous observations that cisplatin downregulates Par-4 at multiple
doses, I asked whether cisplatin downregulates Par-4 in a dose-dependent manner. To
determine this, I treated Trim21-transfected HCT-116 cells with increasing doses of
cisplatin and performed a time-course. Par-4 levels from whole cell lysates were then
examined via Western blotting. The data show that cisplatin-induced Par-4
downregulation occurs in a dose-dependent manner: Par-4 levels are reduced more
quickly at higher doses (Fig. 3-5A).
Given that Trim21 is an E3 ligase, I asked whether the downregulation of Par-4
occurs through proteasomal degradation. To test this hypothesis, I transfected HCT-116
cells with control plasmid or Trim21 plasmid, treated cells with two different doses of
cisplatin, and treated cells either with or without 10 μM MG132, a proteasome inhibitor.
The effects on Par-4 levels were determined by Western blotting. The data show that
the Par-4 downregulation in reponse to Trim21/cisplatin is abrogated by co-treatment
with MG132 (Fig. 3-5B). This suggests that the downregulation of Par-4 in response to
cisplatin occurs at least partially through the proteasome pathway.

69
Figure 3-5. Cisplatin downregulates Par-4 in a dose- and proteasome-dependent manner.
A) Western blots showing Par-4 expression levels in Trim21-transfected HCT-116 cells over time
at different doses of cisplatin. Cisplatin doses are in units of μg/ml. Time is in units of hours.
Actin is shown as a loading control. B) HCT-116 cells were transfected with or without Trim21
expression plasmid for 48 hrs, then treated with the indicated doses of cisplatin in μg/ml for 24

70
hrs, and with or without 10 μM MG132. Blot shows Par-4 expression levels, and actin is shown
as a loading control. A vertical line demarcates the boundary between separate blots.

71
3.2.6. Cisplatin downregulates Par-4 in both the cytoplasmic and nuclear
compartments
To determine an intracellular location of the Par-4 downregulation in response to
cisplatin, I transfected HCT-116 cells with or without Trim21 and treated with 3.75 μg/ml
of cisplatin or carrier, then performed a nuclear-cytoplasmic fractionation to determine
whether the Par-4 downregulation was localized to a specific subcellular compartment.
Par-4 protein levels were examined by Western blotting. As can be seen in Fig. 3-6, the
Par-4 downregulation occurs in both the cytoplasmic and nuclear compartments, with
the downregulation being slightly more pronounced in the nuclear compartment. Lamin
A/B and β-tubulin were probed to confirm the integrity of the subcellular fractionation
and to ensure that there was no cross-contamination between compartments during the
fractionation.

72
Figure 3-6. Cisplatin downregulates Par-4 in both the cytoplasmic and nuclear compartments.
HCT-116 cells were either transfected with plasmid encoding Trim21 or control plasmid for 48
hrs and treated with or without 3.75 μg/ml of cisplatin. Then, nuclear-cytoplasmic fractionation
was performed and Par-4 and Trim21 levels were examined by Western blotting, with Lamin
A/B and β-tubulin serving as markers validating the integrity of the fractionation.

73
3.2.7. Cisplatin downregulates Par-4 in pancreatic cancer cells
Given that Par-4 was downregulated in response to cisplatin in the presence of
overexpressed Trim21 in colon cancer cells, I wanted to examine whether I could
extend this observation to a different type of cancer. To test that hypothesis, I
transfected AsPc-1, BxPc-3, and MiaPaca-2 cells, which are all pancreatic cancer cell
lines, with or without Trim21 for 48 hrs, and treated the cells with increasing doses of
cisplatin for 24 hrs, and probed whole cell lystates for Par-4 expression via Western
blotting. I observed an enhanced dose-dependent downregulation of Par-4 in response
to cisplatin treatment in Trim21-transfected cells compared to control cells (Fig. 3-7A-C).

74

75
Figure 3-7. Cisplatin downregulates Par-4 in pancreatic cancer cells.
Pancreatic cancer cell lines were either transfected with Trim21 plasmid or control plasmid for
48 hrs, and then treated with increasing concentrations of cisplatin for 24 hrs. Cisplatin doses
are in units of μg/ml. Western blots showing Par-4 expression are shown with actin as a loading
control. Western blots validating Trim21 overexpression are also included. A) BxPc-3 B)
MiaPaca-2 C) AsPc-1.

76
3.2.8. Trim21 is a potential therapeutic target in colon and pancreatic
cancer
Given the effect of Trim21 on Par-4 in response to cisplatin, I hypothesized that
Trim21 could contribute to cisplatin resistance. To address this question, I performed an
MTT viability assay on HCT-116 cells that had either been transfected with Trim21
plasmid or control plasmid, and then treated with increasing concentrations of cisplatin.
The data show that Trim21 decreases the sensitivity of HCT-116 cells to cisplatin (Fig.
3-8A). Specifically, HCT-116 cells had statistically significant increases in viability at
cisplatin doses between 0.5-10 μg/ml, with a right shift in IC50 from approximately 5
μg/ml to 10 μg/ml between the control and Trim21 transfected cells, respectively.
Likewise, I performed an analogous experiment in Panc-1 cells, a pancreatic cancer cell
line. To my surprise, ectopic Trim21 expression in Panc-1 cells had no effect on Panc-1
cell viability in response to cisplatin (data not shown). Despite that finding, I show in Fig.
3-8B that ectopic Par-4 expression sensitizes Panc-1 cells to cisplatin-induced
apoptosis as indicated by PARP cleavage, specifically at the 15 and 30 μg/ml doses.
PARP cleavage was examined by Western blotting and the percentage of the cleavage
product was determined using densitometry. Densitometric analysis revealed that Par-4
overexpression increased the levels of apoptosis in Panc-1 cells by two- to three-fold
relative to the control transfected cells at the 15 and 30 μg/ml doses of cisplatin (Fig. 3-
8B).
Given the effects of Trim21 on Par-4, and in light of the recent literature
suggesting that Trim21 is a favorable prognostic marker in cohorts of hepatocellular
carcinoma and diffuse large B-cell lymphoma patients,132, 133 I asked whether Trim21

77
has prognostic significance in either colorectal cancer or pancreatic cancer. To answer
this question, I downloaded data from the TCGA database, stratified by Trim21
expression: Trim21 high (Z-score > 2), Trim21 low (Z-score < -2), and Trim21
intermediate (-2 < Z-score < 2). Then, I plotted Kaplain Meier survival curves of these
three Trim21 expression groups in the colorectal and pancreatic cancer cohorts. There
was no statistically significant difference in overall survival or disease-free survival in the
colorectal cancer cohort between the three Trim21 expression groups (data not shown).
In contrast, in pancreatic cancer there was a dramatic increase in overall survival in the
Trim21 low cohort relative to the intermediate and high cohorts (p = 0.0043), as can be
seen in Fig. 3-8C. Though the difference in overall survival was not significantly different
between the Trim21 high and intermediate groups (p = 0.2121), the Trim21 high group
seems to be trending toward reduced overall survival relative to the intermediate group
(Fig. 3-8C). Likewise, the trends in disease-free survival follow the same patterns as the
trends in overall survival in the pancreatic cancer cohort. Specifically, low Trim21
expression correlates with increased disease-free survival, whereas high Trim21
expression correlates with reduced disease-free survival relative to the Trim21
intermediate cohort (Fig. 3-8D). In the disease-free survival curve, the Trim21 high,
intermediate, and low expression groups were all significantly different from each other
(p < 0.01).

78

79
Figure 3-8. Trim21 is a potential therapeutic target in colon and pancreatic cancer.
A) HCT-116 cells were either transfected with a plasmid encoding Trim21 or control plasmid for
48 hrs, and then treated with increasing doses of cisplatin for 24 hrs, and viability was assessed
by MTT assay. Viability is plotted as a percentage of control samples. Asterisks indicate
statistically significant differences in viability between Trim21 and control transfected cells. B)
Panc-1 cells were either transfected with a plasmid encoding Par-4 or control plasmid for 48
hrs, and then treated with increasing doses of cisplatin. Cisplatin doses are shown in units of
μg/ml. Levels of PARP and Par-4 levels were examined by Western blotting. Percentage of
cleaved PARP, as determined by densitometric analysis, is shown below the blots. Kaplan-Meier
survival curves showing overall survival, Fig. 3-8C and progression-free survival, Fig. 3-8D, of a
cohort of pancreatic cancer patients stratified by Trim21 mRNA expression levels. Data was
obtained from TCGA database.

80
3.3. Discussion
In this study, I identified Trim21 as a novel interaction partner of Par-4, and
demonstrate that they interact endogenously. I show that the binding occurs through the
PRYSPRY domain of Trim21. Though Trim21 is not sufficient to downregulate Par-4
levels, I show that in response to cisplatin Trim21 downregulates Par-4 in a dose- and
proteasome-dependent manner in both the nucleus and cytoplasm. Furthermore, I show
that Trim21 can increase the resistance of cancer cells to cisplatin and that by
overexpressing Par-4, cancer cells can be sensitized to cisplatin-induced apoptosis.
Finally, I demonstrate that Trim21 expression can predict overall- and disease-free
survival in a cohort of pancreatic cancer patients.
Par-4 is an important tumor-suppressor whose expression is downregulated in
several cancers.14-16 In addition, its ability to selectively induce and sensitize cancer
cells toward apoptosis underscores the tremendous therapeutic potential of Par-4.134-136
Therefore, understanding the mechanisms of Par-4 regulation is an important, yet
understudied, area of research, and this work represents an advance in that field. Chen
et. al. showed that Par-4 levels could be post-translationally regulated by the
proteasome through targeting by FBXO45.37 Brasseur et. al. showed that cleaved Par-4
is also regulated by the proteasome.42 I now report a stimulus-dependent regulation of
Par-4 by the proteasome. That stimulus is cisplatin. This observation is significant, since
cisplatin and its analogues are used in the treatment of colon and pancreatic cancer.
Additionally, the subcellular localization of Par-4 is important, because its nuclear
localization is correlated with its apoptotic function.21 Thus, the finding that its
downregulation is present in the nucleus, along with in the cytoplasm, is also significant.

81
By identifying a novel mechanism of regulation of Par-4, I in turn discovered a
potentially new therapeutic target: Trim21. This is especially important for pancreatic
cancer, where most patients ultimately succumb to their disease,54 and are therefore in
need of novel, effective therapies. Here, I show that low Trim21 levels correlate with
prolonged overall and disease-free survival in pancreatic cancer patients. Conversely, I
showed that high Trim21 levels correlate with lower disease-free survival. This intriguing
finding is the opposite of the protective effects of Trim21 in hepatcellular carcinoma and
diffuse large B-cell lymphoma.132, 133 This illustrates the complexity of disease biology,
and the prognostic differences of Trim21 may reflect differences in downstream Trim21
targets, such as Par-4, in different types of cancer. Further study of the role of Trim21 in
pancreatic cancer is needed.
Other work has been done on the relationship between cisplatin and Par-4. For
example, cisplatin has been shown to activate Par-4 via cleavage by caspase 3.41 In
this instance, the cleaved Par-4 had more apoptotic activity and a greater nuclear
localization than full-length Par-4. In another study, Par-4 downregulation was shown to
confer resistance to cisplatin through the PI3K/AKT pathway.10 In this example, Par-4
overexpression reversed the resistance phenotype. This work adds to the complex
relationship between cisplatin and Par-4, and gives rise to interesting questions. For
example, understanding the mechanisms immediately downstream of cisplatin-induced
DNA damage that determine whether Par-4 is cleaved to a more active form versus
downregulated would be an important area of study. Finding ways to switch from
cisplatin-induced Par4 downregulation to cisplatin-induced Par-4 cleavage could be
beneficial. In addition, it would be interesting to examine whether Par-4 downregulation

82
occurs solely in response to the DNA-crosslinking that occurs during cisplatin
treatment,137 or whether this response extends to other types of DNA damage, such as
double-strand breaks.
In conclusion, our data show that Trim21 is a novel interaction partner of Par-4
and a novel regulator of Par-4 in response to cisplatin treatment. Our data suggests that
Trim21 can influence sensitivity of cancer cells to cisplatin. Targeting Trim21 may
potentially be a way to enhance the effectiveness of cisplatin treatment, and may
represent an important therapeutic target, especially in pancreatic cancer.
3.4. Materials/Methods
3.4.1. Cell culture, transfection, plasmids, reagents, and antibodies
HCT-116, SW480, and HT-29 colorectal cancer cells were obtained from ATCC
and maintained in RPMI (Cellgro, Manassas, VA) + 10% fetal bovine serum (FBS).
Panc-1 cells were a kind gift from Dr. Arun Sharma and were maintained in DMEM
(Cellgro, Manassas, VA) + 10% FBS. FBS was obtained from Atlanta Biologicals
(Norcross, GA). Cell lines were grown at 37◦C and 5% CO2. For experiments, cells were
seeded in six-well plates at a seeding density of 300,000 cells/well. Transient
transfections were performed 24 hrs post-seeding. Drug treatments were performed 48
hrs post-seeding. The cells were transiently transfected using PolyJet DNA Transfection
Reagent (SignaGen Laboratories, Rockville, MD), according to the manufacturer’s
instructions. The Par-4 plasmid and anti-Trim21 shRNA plasmids were obtained from
Origene Technologies (Rockville, MD). The plasmids encoding for full-length Trim21
and deletion mutant Trim21 were kind gifts from Dr. Caroline Jefferies at the Royal
College of Surgeons in Ireland. The anti-Trim21, anti-Lamin A/C, and anti-Par-4

83
antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-
actin and anti-β-tubulin antibodies were obtained from Sigma-Aldrich (Saint Louis, MO).
The anti-PARP antibody was obtained from Cell-Signaling (Danvers, MA). Cisplatin was
obtained from Acros Organics (Thermo Fisher, Waltham, MA). MG132 was obtained
from Fisher (Thermo Fisher, Waltham, MA).
3.4.2. Western blot analyses
To examine changes in protein expression, cells were washed twice with PBS
and were lysed into lysis buffer (50mm HEPES, 100 mm NaCl, 10 mm EDTA, 0.5 %
NP40, 10% glycerol, supplemented with 0.0001% Tween20, 0.1 mM PMSF, 0.1 mM
NaVO4, 0.5 mM NaF, 5 μg/ml leupeptin, 0.1 mm DTT). The proteins were quantified
according to the BCA Assay (Thermo Scientific Inc., Rockford, IL) and loaded equally
onto 10% SDS-polyacrylamide gels. Proteins were electrophoresed at 150 V and
transferred to nitrocellulose membranes using a Trans-Blot SD Semi-Dry Transfer Cell
(BioRad, Hercules, CA). Membranes were blocked with 5% non-fat dry milk for 1 hr and
incubated with primary antibody overnight. The blots were washed 3X in TBS with 0.1%
Tween20 and incubated for 1 hr in appropriate HRP-conjugated secondary antibodies
(Amersham, Piscataway, NJ). Blots were washed 3X and chemiluminescent detection
was performed using Amersham ECL Prime Western Blotting Detection Reagent
(Thermo Scientific Inc., Rockford, IL). The blots were either exposed to autoradiography
film (GE Healthcare Life Sciences, Pittsburgh, PA) and scanned or imaged using the
Molecular Imager Gel Doc XR System (Bio-Rad, Hercules, CA).

84
3.4.3. Co-IP/Mass-Spec
To identify novel binding partners of Par-4, HCT-116, HT-29, and SW480 cells
were cultured in two 10-cm dishes and transfected with 5 μg of Par-4 plasmid per dish
using SignaGen Polyjet transfection reagent, according to the manufacturer’s
instructions. After 48 hours of transfection, cells were lysed in lysis buffer (50mm
HEPES, 100 mm NaCl, 10 mm EDTA, 0.5 % NP40, 10% glycerol, supplemented with
0.0001% Tween20, 0.1 mM PMSF, 0.1 mM NaVO4, 0.5 mM NaF, 5 μg/ml leupeptin, 0.1
mm DTT), and the total protein concentration was determined as above.
To perform the co-immunoprecipitation, approximately 3,000 μg of total protein
from the lysate was mixed with 2 μg of anti-Par-4 antibody. As a negative control, the
same amount of total protein from the lysate was mixed with anti-HA antibody. The
antibody-lysate mixtures were allowed to mix overnight at 4◦ C. The next day the
antibody-antigen complexes were pulled down using Dynabeads Protein G (Thermo
Fisher Scientific, Leesport, PA) and washed with lysis buffer, according to the
manufacturer’s instructions. Proteins were eluted from the beads by boiling at 100◦C for
10 mins in digestion buffer (6 M urea, 50 mM Tris-HCl, pH = 8.0). Samples were then
cooled on ice and eluate removed from beads.
To perform the in-solution digestion, 5 μl of 200 mM DTT/50 mM Tris-HCl/pH =
8.0 was added to solution, mixed, and allowed to incubate for 1 hr at room temperature.
After 1 hr, 20 μl of 200 mM Iodoacetamide/50 mM Tris-HCl/pH = 8.0 was added, mixed,
and allowed to incubate at room temperature in the dark for 1 hr. After 1 hr, 775 μl of 50
mM Tris-HCl/1 mM CaCl2/pH = 7.6. 2 microliters of trypsin (0.2 μg/ml) was added,
mixed, and allowed to incubate at 37◦C overnight. Formic acid was added to bring pH

85
down to 3-4. Solutions were snap frozen in liquid nitrogen and spun to dryness under
vacuum. Residue was resuspended in milliQ water (EMD Millipore, Billerica, MA), snap
frozen in liquid nitrogen, and dried using a speed vac. This process was repeated two
more times. The final residue was used for protein identification via MALDI-TOF mass
spectrometry. Proteins were identified from mass spectra using the Paragon Algorithm.
3.4.4. MTT assay
To examine changes in cell viability, cells were seeded at a density of 6 x 103
cells/well in a 96-well culture plate. After 24 hours, the cells were transfected with the
appropriate plasmid. Twenty- four hours post-transfection, the medium was replaced
with medium containing different concentrations of cisplatin. Twenty-four hours post-
cisplatin treatment, MTT reagent was added (Calbiochem) and the cells were incubated
for 3.5 hrs at 37◦ C. After incubation, the media was aspirated off, crystals were
dissolved in MTT solvent (4 mM HCl, 0.1% Nonidet P-40, in isopropanol), and viability
was assessed by measuring the absorbance at 570 nm with 630 nm absorbance as the
reference. Percent viability is expressed as the absorbance normalized to the DMSO
control.
3.4.5. Immunofluoresence
Cells were grown to approximately 50% confluence on 22 x 22 mm cover slips
(#1.5). Media was removed and cells were washed with PBS, followed by fixation with
4% paraformaldehyde in PBS for 15 mins at room temperature. Cells were washed in
PBS, then permeabilized 3 x 5 mins with 0.1% Triton X-100 in PBS. Cells were washed
with PBS, then blocked with 10% BSA in PBS for 1 hr at room temperature. After one

86
hour, the cells were washed with PBS, and incubated with a mixture of anti-Par-4
antibody (1:50) and anti-Trim21 antibody (1:50) in 4% BSA/PBS for two hours at room
temperature. After the incubation in primary antibody, the cells were washed 4X with
PBS for 10 mins each at room temperature with gentle shaking. After washing, the cells
were then incubated in a mixture of secondary antibody in 4% BSA/PBS: anti-rabbit
FITC (1:100) and anti-mouse Ro (1:100). The secondary antibody incubation was
carried out in the dark for two hours at room temperature. After incubation in secondary
antibody, the cells were washed 4X with PBS for 10 mins each at room temperature
with gentle shaking. After washing, the cells were incubated with DAPI (0.5 μg/ml in
PBS) for 5 mins at room temperature in the dark. After incubation with DAPI, the cells
were washed 4X with PBS for 10 mins each at room temperature with gentle shaking.
Finally, the coverslips were mounted on glass slides using ProLong Diamond Antifade
mountant (Thermo Fisher, Leesport, PA). Slides were imaged using DeltaVision Elite
Inverted Microscope. Six random fields are imaged to ensure representativeness.
3.4.6. Nuclear-Cytoplasmic Fractionation
To perform the nuclear-cytoplasmic fractionation, the NE-PER Nuclear and
Cytoplasmic Extraction Kit (Thermo Fisher, Leesport, PA) was used, and the
manufacturer’s instructions were followed. The resultant extracts were quantified and
loaded equally onto a 10% acrylamide gel, and proteins probed by Western blotting.
3.4.7. Statistical analyses
The statistical analyses were carried out using GraphPad Prism software, version
6.04 (GraphPad Software, Inc., San Diego, CA, USA). All plots were created using

87
GraphPad Prism. All experiments were repeated three times with representative
experiments shown in the results. For the MTT assay, two-way ANOVA was used to
determine differences between transfectants and controls, using the Sidak correction to
correct for multiple comparisons. For the Kaplan-Meier survival curves, the log-rank test
was used to determine differences between groups. The pancreatic cancer TCGA data
was accessed and downloaded from www.cbioportal.org. Trim21 high expression was
considered to be those that had a Z-score greater than 2. Trim21 low expression was
considered to be those than had a Z-score lower than -2. Patients with intermediate
Trim21 expression had Z-scores between +/- 2. Densitometric analysis was performed
using ImageJ.104 The threshold for statistical significance is P < 0.05.
3.5. Acknowledgements
The Penn State Hershey Imaging Core is acknowledged for their help in obtaining
images. This work was funded by start-up funds granted to Rosalyn B. Irby.

88
4. Chapter 4
Prostate apoptosis response-4 sensitizes glioma cells to
chemotherapeutics
4.1. Introduction
Despite the current standard of care - which consists of surgical resection,
chemoradiotherapy, and adjuvant temozolomide (TMZ) - the median survival for GBM
14.6 months.81 This highlights the need for novel monotherapies and combination
therapies.84
Prostate apoptosis response-4 is a tumor suppressor that can induce apoptosis
in cancer cells.23 While the role of Par-4 has been studied in a variety of cancers, its role
in glioma is relatively understudied. One of the earliest studies of Par-4 in glioma
showed that ectopic expression of Par-4 induced apoptosis in glioma and other types of
brain tumors.138 Another study showed that upon TMZ treatment, cellular prion protein
was upregulated, which bound to and inhibited Par-4 activation; knock-down of cellular
prion protein dramatically enhanced TMZ induced apoptosis, which was mediated by
Par-4.87 Other studies have demonstrated a role of Par-4 in drug-induced apoptosis of
tamoxifen and drug-induced autophagic cell death of curcumin.12, 88 These studies
underscore the importance of Par-4 in mediating cell death in glioma cells alone and in
response to treatment.
AKT/PKB is a serine/threonine protein kinase that acts downstream of various
growth factor receptors to mediate signaling that leads to cell growth, proliferation,

89
migration, and apoptosis-inhibition.139 Multiple studies have demonstrated that AKT is
activated in the majority of gliomas through mutations in upstream signaling proteins.85
Despite this, there are few AKT inhibitors with demonstrated efficacy in vitro and in
vivo.140
In this work, I demonstrate that the novel AKT inhibitor, ISC-4, and its analogs
show activity in reducing glioma cell viability, and Par-4 sensitizes glioma cells to ISC-4.
Furthermore, I show that Par-4 is sufficient to reduce cell viability, induce apoptosis, and
sensitize glioblastoma cells toward 5-fluorouracil.
4.2. Materials and Methods
4.2.1. Cell culture and transfection
A172 and SNB19 glioblastoma cells were obtained from ATCC and maintained in
RPMI/10% FBS in a 37°C incubator with 5% CO2 and a humidified atmosphere. The
cells were transfected with either empty vector or plasmid vector encoding for human
Par-4 (OriGene Technologies, Rockville, MD) using Signagen Polyjet DNA transfection
reagent (SignaGen Laboratories, Rockville, MD) according to the manufacturer’s
instructions.
4.2.2. Western blot analyses
To measure changes in protein expression, cells were washed twice with PBS and were
lysed into lysis buffer (50mm HEPES, 100 mm NaCl, 10 mm EDTA, 0.5 % NP40, 10%
glycerol, supplemented with 0.0001% Tween 20, 0.1 mM PMSF, 0.1 mM NaVO4, 0.5
mM NaF, 5 μg/ml leupeptin, 0.1 mm DTT). The proteins were quantified according to
the BCA Assay (Thermo Scientific Inc., Rockford, IL) and loaded equally onto 10%

90
SDS-polyacrylamide gels. Proteins were electrophoresed at 150 V and transferred to
nitrocellulose membranes using a semi-dry blotter (BioRad, Hercules, CA). Membranes
were blocked with 5% non-fat dry milk or 5% BSA for 1 hr and incubated with primary
antibody overnight. The blots were washed 3X in TBS with 0.1% Tween20 and
incubated for 1 hr in appropriate HRP-conjugated secondary antibodies (Amersham,
Piscataway, NJ). Blots were washed 3X and chemiluminescent detection was
performed using Amersham ECL Prime Western Blotting Detection Reagent (Thermo
Scientific Inc., Rockford, IL). The blots were either exposed to autoradiography film (GE
Healthcare Life Sciences, Pittsburgh, PA) and scanned or imaged using the Molecular
Imager Gel Doc XR System (Bio-Rad). Densitometric analyses were performed with
NIH Image J software104. Primary antibodies used were: phospho-Akt, phospho-Par-4,
and PARP (Cell Signaling, Danvers, MA), GRP78 (Santa Cruz Biotechnology, Santa
Cruz, CA), and Actin (Sigma-Aldrich, Saint Louis, MO).
4.2.3. MTT viability assay
To measure in vitro cytotoxic efficacy, we used 3-(4,5-Dimethylthiazol-2-yl)-2,5-
diphenyltetrazolium bromide to measure cell viability. A172 and SNB19 cells were
seeded at a density of 7 x 103 cells/well in a 96-well culture plate. After 24 hours, the
cells were transfected with Par-4 plasmid or control plasmid. Twenty-four hours later,
cells were treated with the indicated concentrations of drugs. Forty-eight hours later,
MTT reagent was added (Calbiochem) and the cells were incubated for 3.5 hrs at 37◦ C.
After incubation, the media was aspirated off, crystals were dissolved in MTT solvent (4
mM HCl, 0.1% Nonidet P-40, in isopropanol), and viability was assessed by measuring
the absorbance at 570 nm with 630 nm absorbance as the reference.

91
4.2.4. PE Annexin V apoptosis assay
To measure apoptosis, the PE Annexin V flow cytometry assay was used
according to the manufacturer’s protocol. Briefly, after cells seeded in six-well plates
were transfected and treated with drugs for the appropriate time period, cells were
trypsinized, washed twice with cold PBS, and resuspended in binding buffer at a
concentration of 1 x 106 cells/ml. One-hundred microliters of the cell suspension were
transferred to a 5 ml culture tube, and 5 microlitres of PE Annexin V and 5 microliters of
7-AAD were added. The mixture was vortexed gently and allowed to incubate for 15
mins at room temperature in the dark. Four-hundred microliters of binding buffer were
added, mixed, and analyzed by flow cytometry.
4.2.5. Statistical analysis
All data analysis was conducted using a student’s T-test.
4.3. Results
4.3.1. Akt inhibitors reduce cell viability in glioblastoma cells
A172 cells were seeded into 96-well plates and treated with varying
concentrations of the AKT inhibitors, NISC-6, ISC-4, NNITC-2, NNISC-2 for 48 hours.
Viability was determined using the MTT viability assay. As can be seen in Fig. 4-1A, the
AKT inhibitors all decreased cell viability with IC50 values ranging from 2 to 8 μM. I then
wanted to examine the effectiveness of AKT inhibition on a wider panel of GBM cell
lines, and ISC-4 was chosen given that it has been well-characterized.65 As can be seen
in Fig. 4-1B and Fig. 4-1C, ISC-4 is effective at decreasing cell viability across a wide

92
panel of GBM cell lines at 24 and 72 hours, though some cell lines, such as CRL1690,
seem to be resistant to ISC-4 even at 72 hours.

93

94
Figure 4-1. Akt inhibitors decrease cell viability in glioblastoma cells.
A) A172 cells were seeded into 96-well plates, then treated with various concentrations of
NISC-6, ISC-4, NNITC-2, and NNISC-2 for 48 hrs. MTT assay was used to determine cell viability.
Various GBM cell lines were seeded into 96-well plates and treated with different
concentrations of ISC-4 for 24 (B) and 72 (C) hrs. MTT assay was used to determine cell viability.

95
4.3.2. Par-4 is sufficient to reduce cell viability in GBM cells
A172 and SNB19 cells were seeded into 96-well plates and transfected with
various concentrations of Par-4 for 24, 48, and 72 hours. Viability was determined by
the MTT viability assay. As can be seen from Fig. 4-2A and Fig. 4-2B, Par-4 is sufficient
to reduce cell viability in a dose-dependent manner. A172 cells are particularly sensitive
to ectopic Par-4.

96
Figure 4-2. Par-4 is sufficient to reduce cell viability in GBM cells.

97
A172 (A) and SNB19 (B) cells were seeded into 96-well plates then transfected with various
concentrations of Par-4 plasmid for 24, 48, and 72 hours and viability was assessed by MTT
assay.

98
4.3.3. Par-4 sensitizes GBM cells to ISC-4 and 5-FU
After determining that ISC-4 and Par-4 were both sufficient at reducing cell
viability in GBM cell lines, I sought to determine whether Par-4 could sensitize GBM
cells to ISC-4 treatment. In addition, I sought to determine whether Par-4 could sensitize
GBM cells toward the commonly used therapies: TMZ and 5-FU. As can be seen in Fig.
4-3A and Fig. 4-3B, Par-4 sensitizes A172 cells toward 5-FU and SNB19 cells toward
ISC-4 treatment, respectively. Par-4 did not sensitize GBM cells toward TMZ treatment
(data not shown).

99

100
Figure 4-3. Par-4 sensitizes GBM cells to 5-FU and ISC-4.
A) A172 cells were either untreated, treated with 5-FU (700 μM), transfected with Par-4 (0.08
μg/ml), or treated with the combination for 48 hrs B) SNB19 cells were transfected with either
empty vector or Par-4 plasmid and treated with increasing amounts of ISC-4 for 48 hours. MTT
assays were used to determine cell viability

101
4.3.4. Par-4, ISC-4, and their combinations induce apoptosis in GBM cells
To gain insight into the mechanisms of the decrease in cell viability seen with
Par-4 transfection, drug treatments, or their combination, I sought to determine whether
cells were dying by apoptosis. A172 and SNB19 cells were seeded into six-well plates,
transfected with Par-4 plasmid or control, and then treated with DMSO control, ISC-4 (5
μM) or TMZ (200 μM) for 24 hrs. Then, the PE Annexin V flow cytometry assay was
used to determine the percentage of apoptotic cells under these conditions. As can be
seen in Fig. 4-4, Par-4, ISC-4, and their combinations led to increased levels of
apoptosis in A172 and SNB19 GBM cells. TMZ treatment alone did not lead to
significant levels of apoptosis in A172 and SNB19 cells.
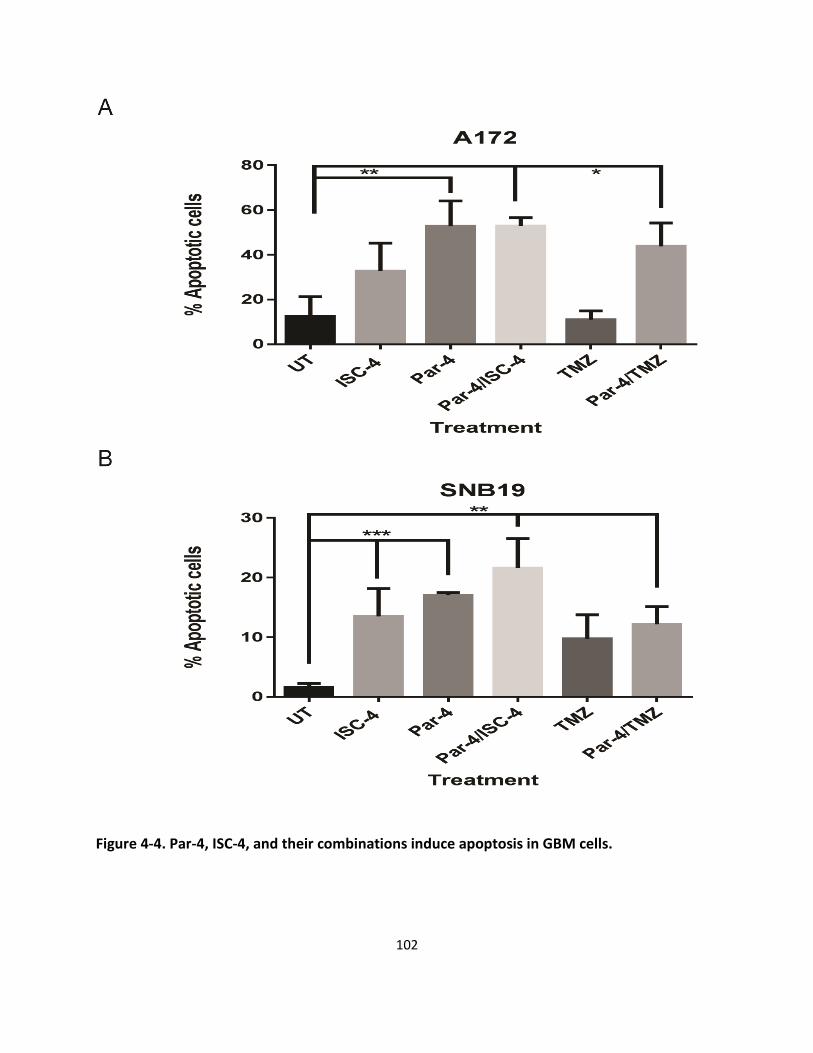
102
Figure 4-4. Par-4, ISC-4, and their combinations induce apoptosis in GBM cells.

103
A172 cells (A) and SNB19 cells (B) were either transfected with Par-4 plasmid or control plasmid
and then treated with ISC-4 (5 μM) or TMZ (200 μM). The percentage of apoptotic cells was
determined by PE Annexin V flow cytometry assay.

104
4.4. Discussion
In this study, I show that treating glioblastoma cells with selenocyanate AKT
inhibitors potently reduced cell viability. In addition, I showed that these types of
inhibitors are active across multiple GBM cell lines. I also found that ectopic Par-4
expression is sufficient to reduce cell viability in GBM and can sensitize cells toward
ISC-4 and 5-FU treatment. Finally, I show that the reduction in viability induced by Par-4
or its treatment combinations at least partially occurs through apoptosis.
These findings are significant, because little is known about the role and potential
therapeutic significance of Par-4 in gliomas, and much of that work is focused on glioma
stem cells. The first study on Par-4 in gliomas demonstrated that ectopic Par-4
expression induced apoptosis in a variety of different brain cancers, including
neuroblastoma, medulloblastoma, and gliomas.138 These findings corroborate the ability
of Par-4 to induce apoptosis in gliomas. Experiments are ongoing in our lab that are
attempting to elucidate the mechanisms by which Par-4 induces apoptosis. The study
by Vetterkind et. al. suggests that the mechanism is independent of BCL-2
downregulation and PKCζ inhibition, while being partially dependent on the Fas death
receptor pathway.138 This suggests that a novel mechanism of Par-4 induced apoptosis
is at play. Given the role of NF-κB activity in glioma141 and the essential role of NF-κB
inhibition in Par-4 activity,105 one possibility is enhanced inhibition of NF-κB
transcriptional activity in the nucleus.
Given that the PI3K/AKT pathway is activated in a majority of gliomas and given
that AKT inhibition has not been explored extensively, the finding that ISC-4 and its

105
derivatives reduce cell viability and is efficacious across a variety of glioma cell lines is
important. Furthermore, the ability of Par-4 to sensitize glioma cells to ISC-4 is
significant, because it underscores the therapeutic potential of Par-4 in glioma
treatment. Further experiments could expand on this by studying whether extracellular
Par-4 can sensitize to ISC-4 treatment. In addition, studies examining the efficacy of
ISC-4 alone and in combination with Par-4 in an in vivo model of glioma would be a
fruitful area of research.
In summary, our data show that Par-4 is sufficient to reduce cell viability alone
and in combination with ISC-4 and 5-FU. Given the lack of effective therapies for
glioblastoma patients, this work demonstrates the potential of using a combination of
AKT inhibitor, ISC-4, along with Par-4 in the treatment of glioma.

106
5. Chapter 5
Summary and Future Directions
Par-4 has been shown to have therapeutic potential in a number of cancers due
to its ability to induce cancer-cell selective apoptosis and its ability to sensitize cancer
cells to apoptosis-inducing agents, including chemotherapy and radiation.23 The long-
term goal of our lab is to utilize Par-4 to enhance cancer therapy, and at the start of this
dissertation the focus of the lab has been on studying Par-4 in colon cancer. At the time,
our lab had shown: 1) Delivery of Par-4 plasmid via nanoliposomes can sensitize colon
tumor cells to 5-fluorouracil.63 2) Par-4 overexpression leads to gene expression
changes and to changes in microRNA levels, which seemed to be mediated by NF-κB
inhibition, that could account for its sensitization to 5-fluorouracil.64 3) Endogenous Par-
4 can be activated by treatment with a combination of 5-fluorouracil and c-Src inhibitor
or by ISC-4, which can lead to cell death in vitro and in vivo.65, 66 The goal of this
dissertation was to expand on this foundation, and to elucidate novel mechanisms of
Par-4 activity, regulation, and potential applicability to cancer treatment. In the first part
of this work, I examined the effect of Par-4 on cell migration in colon cancer. Through
this work, I demonstrated that Par-4 inhibited migration, invasion, and induced a
mesenchymal-epithelial transition in colon cancer cells. In the second part of this work, I
sought to identify novel regulators of Par-4. In the work described here, I showed that
Trim21 is a novel interactor with Par-4, and that in response to cisplatin, Trim21
overexpression results in enhanced downregulation of Par-4. In addition, Trim21
expression can lead to cisplatin resistance. I also found that Trim21 levels correlated

107
with survival in a cohort of pancreatic cancer patients, with low Trim21 levels correlating
with prolonged overall and disease-free survival and high Trim21 levels correlating with
reduced disease-free survival. The third part of this work examined whether Par-4 could
enhance the effects of chemotherapy against glioblastoma cells. I found that Par-4 is
sufficient to reduce cell viability in GBM cells and that it also sensitizes GBM cells to the
AKT inhibitor, ISC-4, and chemotherapeutic, 5-FU. Taken together, these results
highlight novel tumor-suppressive mechanisms of Par-4, outside of its ability to regulate
apoptosis, which provides further rationale for its development as a cancer therapy.
Additionally, these results reveal a novel mechanism of Par-4 regulation, and in the
process uncovered a potentially novel biomarker for pancreatic cancer. Finally, the
results in glioma cells demonstrate the potential of Par-4 as gene therapy alone and in
combination with chemotherapeutics in the treatment of glioblastoma.
There is currently an unmet clinical need for novel, effective therapies for
advanced colon cancer, along with pancreatic cancer and glioblastoma.54, 81 With this
dissertation work, I illustrate the potential of Par-4 in these different contexts.
Understanding the role of Par-4 in regulating cell migration is a relatively new area of
research on Par-4 function. The vast majority of studies on Par-4 function have focused
on its ability to regulate apoptosis, and only recently have studies began to look at the
role that Par-4 plays in other processes such as migration and autophagy. Only two
other studies have reported on the role of Par-4 on migration. In one, a cisplatin
resistant pancreatic cancer cell line, BxPc-3, was created, and it was found that
phenotypic changes consistent with EMT resulted.10 In addition, Par-4 was
downregulated in the resistant cells relative to sensitive cells, and Par-4 overexpression

108
in resistant cells reversed these changes, while Par-4 knock-down in sensitive cells
conferred a similar cisplatin-resistant phenotype. In contrast, in the second study, TGF-
β mediated EMT in endometrial, breast, ovarian, and cervical cancer cell lines was
found to be dependent on Par-4.11 Overexpression of Par-4 induced the expression of
EMT markers and led to an increase in motility, whereas Par-4 knock-down decreased
the expression of EMT markers and prevented TGF-β induced EMT. These contrasting
results suggest that the ability of Par-4 to regulate EMT and motility depends on the
intrinsic cell-specific mutations and genetic background of different groups of cancers.
Future work is warranted to elucidate these cell-specific mechanisms and targets of
Par-4 that can account for these differences in Par-4 activity.
In my work on Trim21 as a regulator of Par-4, the finding that Trim21 correlates
with survival in pancreatic cancer patients is perhaps the most significant finding. This is
especially true given the current standard-of-care for pancreatic cancer. Despite the
current chemotherapy cocktail used for pancreatic cancer treatment, almost all
pancreatic cancer patients will die from their disease.54 Though this high mortality is due
partially to late diagnosis, it is also largely due to ineffective treatment.142 Knowing that
patients with tumors that have low Trim21 expression will survive significantly longer,
informing patients of their Trim21 status can be source of hope for patients and allow
them to cope with the psychological burden of a cancer diagnosis. Indeed, the survival
is prolonged such that the survival curve is essentially horizontal for the overall and
disease-free survival. Interesting basic science questions also arise from the data. For
example, most of the studies on Trim21 regulation of processes that are critical to
cancer development, such as apoptosis and proliferation, suggest that, overall, Trim21

109
acts as a tumor suppressor. This has been corroborated by studies that show that high
Trim21 expression predicts longer survival in cohorts of diffuse large B-cell lymphoma133
and hepatocellular carcinoma;132 however, my data show the opposite trend in
pancreatic cancer. One explanation for this difference in prognostic value of Trim21
across different cohorts could lie in the differences in expression of Trim21 targets
between pancreatic cancer compared to hepatocellular carcinoma and diffuse large B-
cell lymphoma. For example, while Trim21 overexpression inhibited proliferation in a B-
cell line,131, 133 other studies have shown that Trim21 can positively regulate cell
proliferation and cell cycle progression. In one study, Trim21 knock-down inhibited
progression through the cell-cycle via p27 accumulation.143 In another study, Trim21
overexpression enhanced IL-2 expression in response to CD28 treatment.144 Thus,
future studies could examine the effect of overexpression or knock-down of Trim21 on
pancreatic cell migration, proliferation, and resistance to cell death. Based on the data
in this dissertation, I hypothesize that Trim21 will be a positive regulator of cell migration
and proliferation and increase resistance to cell death. If, indeed, Trim21 appears to be
pro-oncogenic in the context of pancreatic cancer, developing small molecule inhibitors
of Trim21 would be warranted.
In the work of Par-4 and glioma, I demonstrated the utility of Par-4 in the
treatment of glioma. This work can be extended by moving to in vivo models of glioma.
In addition, elucidating the mechanism of Par-4-induced apoptosis and sensitization to
chemotherapeutics in glioma is warranted. Future studies could examine the role of NF-
ĸB inhibition and AKT inhibition, given the known roles of those two pathways in glioma
development and Par-4 activity.

110
Beyond the scope of the work in this dissertation, there are outstanding
questions in the Par-4 field that deserve further study. For example, given its recent
discovery, relatively little work has been done on the extracellular role of Par-4.8,22,32
Extracellular Par-4 inhibited lung carcinoma metastasis in vivo32, yet the mechanism of
inhibition was not elucidated. It could act via its reported receptor, GRP78, or through
some unknown receptor. Pursuing this avenue of research could be promising, since
extracellular Par-4 seems to have similar activity to intracellular Par-4: it both induces
apoptosis and inhibits metastasis of cancer cells. In addition, it has certain advantages
over delivering Par-4 plasmid as gene therapy, especially in the context of glioma –
given that Par-4 mediates neuronal cell death in models of neurodegeneration23, there
is a possibility of off-target neuronal cell death when using Par-4 as gene therapy in the
context of glioma; with extracellular Par-4 this side effect would not be a concern.
Another interesting research question would be clarifying the mechanisms of the
cancer-cell selectivity of Par-4. The prevailing paradigm is that the selectivity of
intracellular Par-4-induced apoptosis is due to the greater PKA activity in cancer cells
relative to normal cells.35 The explanation for the cancer-cell selectivity of extracellular
Par-4 is due to the selective membrane localization of GRP78 in cancer cells.8,32
However, this explanation is incomplete. Intracellular Par-4 does not induce apoptosis in
all cancer cells – only a subset. Furthermore, Par-4 nuclear localization correlates with
the sensitivity of cancer cells to Par-4-induced apoptosis. This suggests that there is an
intranuclear activity of Par-4 that is essential for its activity, yet little is known about its
roles in the nucleus besides interacting with TOP131, NF-ĸB25, and WT129,30. A co-
immunoprecipitation screen with Par-4 protein as bait and using nuclear lysates as

111
potential prey could begin to address this question with subsequent reporter assays to
explore the functional significance of the discovered interactions.
In conclusion, while more work remains before the goal of using Par-4 in cancer
treatment is realized, the work in this dissertation provides further rationale for
continuing to study the role of Par-4 in cancer and to develop it as a cancer treatment.

112
References
1. Sells SF, Wood DP, Jr., Joshi-Barve SS, Muthukumar S, Jacob RJ, Crist SA, Humphreys S, Rangnekar VM. Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research 1994; 5:457-66. 2. Boghaert ER, Sells SF, Walid AJ, Malone P, Williams NM, Weinstein MH, Strange R, Rangnekar VM. Immunohistochemical analysis of the proapoptotic protein Par-4 in normal rat tissues. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research 1997; 8:881-90. 3. Chendil D, Das A, Dey S, Mohiuddin M, Ahmed MM. Par-4, a pro-apoptotic gene, inhibits radiation-induced NF kappa B activity and Bcl-2 expression leading to induction of radiosensitivity in human prostate cancer cells PC-3. Cancer biology & therapy 2002; 1:152-60. 4. Boehrer S, Chow KU, Beske F, Kukoc-Zivojnov N, Puccetti E, Ruthardt M, Baum C, Rangnekar VM, Hoelzer D, Mitrou PS, et al. In lymphatic cells par-4 sensitizes to apoptosis by down-regulating bcl-2 and promoting disruption of mitochondrial membrane potential and caspase activation. Cancer research 2002; 62:1768-75. 5. Boehrer S, Brieger A, Schaaf S, Kukoc-Zivojnov N, Nowak D, Ruthardt M, Hoelzer D, Mitrou PS, Weidmann E, Chow KU. In the erythroleukemic cell line HEL Prostate-apoptosis-response-gene-4 (par-4) fails to down-regulate Bcl-2 and to promote apoptosis. Leukemia & lymphoma 2004; 45:1445-51. 6. Brieger A, Boehrer S, Schaaf S, Nowak D, Ruthardt M, Kim SZ, Atadja P, Hoelzer D, Mitrou PS, Weidmann E, et al. In bcr-abl-positive myeloid cells resistant to conventional chemotherapeutic agents, expression of Par-4 increases sensitivity to imatinib (STI571) and histone deacetylase-inhibitors. Biochemical pharmacology 2004; 68:85-93. 7. Lucas T, Pratscher B, Krishnan S, Fink D, Gunsberg P, Wolschek M, Wacheck V, Muster T, Romirer I, Wolff K, et al. Differential expression levels of Par-4 in melanoma. Melanoma research 2001; 11:379-83. 8. Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell 2009; 138:377-88. 9. Shrestha-Bhattarai T, Rangnekar VM. Cancer-selective apoptotic effects of extracellular and intracellular Par-4. Oncogene 2010; 29:3873-80. 10. Tan J, You Y, Xu T, Yu P, Wu D, Deng H, Zhang Y, Bie P. Par-4 downregulation confers cisplatin resistance in pancreatic cancer cells via PI3K/Akt pathway-dependent EMT. Toxicology letters 2013. 11. Chaudhry P, Fabi F, Singh M, Parent S, Leblanc V, Asselin E. Prostate apoptosis response-4 mediates TGF-beta-induced epithelial-to-mesenchymal transition. Cell death & disease 2014; 5:e1044. 12. Thayyullathil F, Rahman A, Pallichankandy S, Patel M, Galadari S. ROS-dependent prostate apoptosis response-4 (Par-4) up-regulation and ceramide generation are the prime signaling events associated with curcumin-induced autophagic cell death in human malignant glioma. FEBS open bio 2014; 4:763-76. 13. Wang LJ, Chen PR, Hsu LP, Hsu WL, Liu DW, Chang CH, Hsu YC, Lee JW. Concomitant induction of apoptosis and autophagy by prostate apoptosis response-4 in hypopharyngeal carcinoma cells. The American journal of pathology 2014; 184:418-30. 14. Cook J, Krishnan S, Ananth S, Sells SF, Shi Y, Walther MM, Linehan WM, Sukhatme VP, Weinstein MH, Rangnekar VM. Decreased expression of the pro-apoptotic protein Par-4 in renal cell carcinoma. Oncogene 1999; 18:1205-8.

113
15. Joshi J, Fernandez-Marcos PJ, Galvez A, Amanchy R, Linares JF, Duran A, Pathrose P, Leitges M, Canamero M, Collado M, et al. Par-4 inhibits Akt and suppresses Ras-induced lung tumorigenesis. The EMBO journal 2008; 27:2181-93. 16. Moreno-Bueno G, Fernandez-Marcos PJ, Collado M, Tendero MJ, Rodriguez-Pinilla SM, Garcia-Cao I, Hardisson D, Diaz-Meco MT, Moscat J, Serrano M, et al. Inactivation of the candidate tumor suppressor par-4 in endometrial cancer. Cancer research 2007; 67:1927-34. 17. Barradas M, Monjas A, Diaz-Meco MT, Serrano M, Moscat J. The downregulation of the pro-apoptotic protein Par-4 is critical for Ras-induced survival and tumor progression. The EMBO journal 1999; 18:6362-9. 18. Ahmed MM, Sheldon D, Fruitwala MA, Venkatasubbarao K, Lee EY, Gupta S, Wood C, Mohiuddin M, Strodel WE. Downregulation of PAR-4, a pro-apoptotic gene, in pancreatic tumors harboring K-ras mutation. International journal of cancer Journal international du cancer 2008; 122:63-70. 19. Garcia-Cao I, Duran A, Collado M, Carrascosa MJ, Martin-Caballero J, Flores JM, Diaz-Meco MT, Moscat J, Serrano M. Tumour-suppression activity of the proapoptotic regulator Par4. EMBO reports 2005; 6:577-83. 20. Rangnekar VM. Apoptosis mediated by a novel leucine zipper protein Par-4. Apoptosis : an international journal on programmed cell death 1998; 3:61-6. 21. El-Guendy N, Zhao Y, Gurumurthy S, Burikhanov R, Rangnekar VM. Identification of a unique core domain of par-4 sufficient for selective apoptosis induction in cancer cells. Molecular and cellular biology 2003; 23:5516-25. 22. Zhao Y, Burikhanov R, Qiu S, Lele SM, Jennings CD, Bondada S, Spear B, Rangnekar VM. Cancer resistance in transgenic mice expressing the SAC module of Par-4. Cancer research 2007; 67:9276-85. 23. Hebbar N, Wang C, Rangnekar VM. Mechanisms of apoptosis by the tumor suppressor Par-4. Journal of cellular physiology 2012. 24. Chakraborty M, Qiu SG, Vasudevan KM, Rangnekar VM. Par-4 drives trafficking and activation of Fas and Fasl to induce prostate cancer cell apoptosis and tumor regression. Cancer research 2001; 61:7255-63. 25. Diaz-Meco MT, Lallena MJ, Monjas A, Frutos S, Moscat J. Inactivation of the inhibitory kappaB protein kinase/nuclear factor kappaB pathway by Par-4 expression potentiates tumor necrosis factor alpha-induced apoptosis. The Journal of biological chemistry 1999; 274:19606-12. 26. Zhou J, Ching YQ, Chng WJ. Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: from molecular pathogenesis to therapeutic target. Oncotarget 2015; 6:5490-500. 27. Camandola S, Mattson MP. Pro-apoptotic action of PAR-4 involves inhibition of NF-kappaB activity and suppression of BCL-2 expression. Journal of neuroscience research 2000; 61:134-9. 28. Diaz-Meco MT, Municio MM, Frutos S, Sanchez P, Lozano J, Sanz L, Moscat J. The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell 1996; 86:777-86. 29. Johnstone RW, See RH, Sells SF, Wang J, Muthukkumar S, Englert C, Haber DA, Licht JD, Sugrue SP, Roberts T, et al. A novel repressor, par-4, modulates transcription and growth suppression functions of the Wilms' tumor suppressor WT1. Molecular and cellular biology 1996; 16:6945-56. 30. Cheema SK, Mishra SK, Rangnekar VM, Tari AM, Kumar R, Lopez-Berestein G. Par-4 transcriptionally regulates Bcl-2 through a WT1-binding site on the bcl-2 promoter. The Journal of biological chemistry 2003; 278:19995-20005. 31. Goswami A, Qiu S, Dexheimer TS, Ranganathan P, Burikhanov R, Pommier Y, Rangnekar VM. Par-4 binds to topoisomerase 1 and attenuates its DNA relaxation activity. Cancer research 2008; 68:6190-8.

114
32. Zhao Y, Burikhanov R, Brandon J, Qiu S, Shelton BJ, Spear B, Bondada S, Bryson S, Rangnekar VM. Systemic Par-4 inhibits non-autochthonous tumor growth. Cancer biology & therapy 2011; 12:152-7. 33. Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer research 2007; 67:3496-9. 34. Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. The Journal of biological chemistry 2010; 285:15065-75. 35. Gurumurthy S, Goswami A, Vasudevan KM, Rangnekar VM. Phosphorylation of Par-4 by protein kinase A is critical for apoptosis. Molecular and cellular biology 2005; 25:1146-61. 36. Goswami A, Burikhanov R, de Thonel A, Fujita N, Goswami M, Zhao Y, Eriksson JE, Tsuruo T, Rangnekar VM. Binding and phosphorylation of par-4 by akt is essential for cancer cell survival. Molecular cell 2005; 20:33-44. 37. Chen X, Sahasrabuddhe AA, Szankasi P, Chung F, Basrur V, Rangnekar VM, Pagano M, Lim MS, Elenitoba-Johnson KS. Fbxo45-mediated degradation of the tumor-suppressor Par-4 regulates cancer cell survival. Cell death and differentiation 2014. 38. Thayyullathil F, Pallichankandy S, Rahman A, Kizhakkayil J, Chathoth S, Patel M, Galadari S. Caspase-3 mediated release of SAC domain containing fragment from Par-4 is necessary for the sphingosine-induced apoptosis in Jurkat cells. Journal of molecular signaling 2013; 8:2. 39. Treude F, Kappes F, Fahrenkamp D, Muller-Newen G, Dajas-Bailador F, Kramer OH, Luscher B, Hartkamp J. Caspase-8-mediated PAR-4 cleavage is required for TNFalpha-induced apoptosis. Oncotarget 2014; 5:2988-98. 40. Oke V, Wahren-Herlenius M. The immunobiology of Ro52 (TRIM21) in autoimmunity: a critical review. Journal of autoimmunity 2012; 39:77-82. 41. Chaudhry P, Singh M, Parent S, Asselin E. Prostate apoptosis response 4 (Par-4), a novel substrate of caspase-3 during apoptosis activation. Molecular and cellular biology 2012; 32:826-39. 42. Brasseur K, Fabi F, Adam P, Parent S, Lessard L, Asselin E. Post-translational regulation of the cleaved fragment of Par-4 in ovarian and endometrial cancer cells. Oncotarget 2016. 43. Saegusa M, Hashimura M, Kuwata T, Okayasu I. Transcriptional regulation of pro-apoptotic Par-4 by NF-kappaB/p65 and its function in controlling cell kinetics during early events in endometrial tumourigenesis. The Journal of pathology 2010; 221:26-36. 44. Liu Y, Gilbert MR, Kyprianou N, Rangnekar VM, Horbinski C. The tumor suppressor prostate apoptosis response-4 (Par-4) is regulated by mutant IDH1 and kills glioma stem cells. Acta neuropathologica 2014; 128:723-32. 45. Das TP, Suman S, Alatassi H, Ankem MK, Damodaran C. Inhibition of AKT promotes FOXO3a-dependent apoptosis in prostate cancer. Cell death & disease 2016; 7:e2111. 46. Yang K, Shen J, Chen SW, Qin J, Zheng XY, Xie LP. Upregulation of PAWR by small activating RNAs induces cell apoptosis in human prostate cancer cells. Oncology reports 2016; 35:2487-93. 47. Hollis M, Nair K, Vyas A, Chaturvedi LS, Gambhir S, Vyas D. MicroRNAs potential utility in colon cancer: Early detection, prognosis, and chemosensitivity. World journal of gastroenterology : WJG 2015; 21:8284-92. 48. Zhang L, Fan XM. The pathological role of microRNAs and inflammation in colon carcinogenesis. Clinics and research in hepatology and gastroenterology 2015; 39:174-9. 49. Sanz-Navares E, Fernandez N, Kazanietz MG, Rotenberg SA. Atypical protein kinase Czeta suppresses migration of mouse melanoma cells. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research 2001; 12:517-24. 50. Khanna C, Hunter K. Modeling metastasis in vivo. Carcinogenesis 2005; 26:513-23.

115
51. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nature reviews Cancer 2013; 13:97-110. 52. Lin L, Baehrecke EH. Autophagy, cell death, and cancer. Molecular & cellular oncology 2015; 2:e985913. 53. Surveillance, Epidemiology, and End Results (SEER) Program. 54. Surveillance, Epidemiology, and End Results (SEER) Program. National Cancer Institute. 55. Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proceedings of the National Academy of Sciences of the United States of America 2007; 104:18654-9. 56. Lassmann S, Weis R, Makowiec F, Roth J, Danciu M, Hopt U, Werner M. Array CGH identifies distinct DNA copy number profiles of oncogenes and tumor suppressor genes in chromosomal- and microsatellite-unstable sporadic colorectal carcinomas. J Mol Med (Berl) 2007; 85:293-304. 57. Willett WC. Diet and cancer: an evolving picture. Jama 2005; 293:233-4. 58. Vindigni SM, Kaz AM. Universal Screening of Colorectal Cancers for Lynch Syndrome: Challenges and Opportunities. Digestive diseases and sciences 2016; 61:969-76. 59. Sengupta N, Yee E, Feuerstein JD. Colorectal Cancer Screening in Inflammatory Bowel Disease. Digestive diseases and sciences 2016; 61:980-9. 60. Van Der Kraak L, Gros P, Beauchemin N. Colitis-associated colon cancer: Is it in your genes? World journal of gastroenterology : WJG 2015; 21:11688-99. 61. DL Kasper AF, SL Hauser, DL Longo, JL Jameson, J Loscalzo. Harrison's Principles of Internal Medicine. McGraw-Hill Education. 62. Zhang Z, DuBois RN. Par-4, a proapoptotic gene, is regulated by NSAIDs in human colon carcinoma cells. Gastroenterology 2000; 118:1012-7. 63. Kline CL, Shanmugavelandy SS, Kester M, Irby RB. Delivery of PAR-4 plasmid in vivo via nanoliposomes sensitizes colon tumor cells subcutaneously implanted into nude mice to 5-FU. Cancer biology & therapy 2009; 8:1831-7. 64. Wang BD, Kline CL, Pastor DM, Olson TL, Frank B, Luu T, Sharma AK, Robertson G, Weirauch MT, Patierno SR, et al. Prostate apoptosis response protein 4 sensitizes human colon cancer cells to chemotherapeutic 5-FU through mediation of an NF kappaB and microRNA network. Molecular cancer 2010; 9:98. 65. Sharma AK, Kline CL, Berg A, Amin S, Irby RB. The Akt inhibitor ISC-4 activates prostate apoptosis response protein-4 and reduces colon tumor growth in a nude mouse model. Clinical cancer research : an official journal of the American Association for Cancer Research 2011; 17:4474-83. 66. Kline CL, Irby RB. The pro-apoptotic protein Prostate Apoptosis Response Protein-4 (Par-4) can be activated in colon cancer cells by treatment with Src inhibitor and 5-FU. Apoptosis : an international journal on programmed cell death 2011; 16:1285-94. 67. Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Kloppel G, Longnecker DS, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. The American journal of surgical pathology 2001; 25:579-86. 68. Fu B, Luo M, Lakkur S, Lucito R, Iacobuzio-Donahue CA. Frequent genomic copy number gain and overexpression of GATA-6 in pancreatic carcinoma. Cancer biology & therapy 2008; 7:1593-601. 69. Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003; 425:846-51.

116
70. van Heek NT, Meeker AK, Kern SE, Yeo CJ, Lillemoe KD, Cameron JL, Offerhaus GJ, Hicks JL, Wilentz RE, Goggins MG, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. The American journal of pathology 2002; 161:1541-7. 71. Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nature reviews Cancer 2002; 2:897-909. 72. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008; 321:1801-6. 73. Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nature genetics 1994; 8:27-32. 74. Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, Kern SE. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer research 1994; 54:3025-33. 75. Iacobuzio-Donahue CA, Ashfaq R, Maitra A, Adsay NV, Shen-Ong GL, Berg K, Hollingsworth MA, Cameron JL, Yeo CJ, Kern SE, et al. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer research 2003; 63:8614-22. 76. Gold EB. Epidemiology of and risk factors for pancreatic cancer. The Surgical clinics of North America 1995; 75:819-43. 77. Chari ST, Leibson CL, Rabe KG, Ransom J, de Andrade M, Petersen GM. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 2005; 129:504-11. 78. Foulkes WD. Inherited susceptibility to common cancers. The New England journal of medicine 2008; 359:2143-53. 79. Rebours V, Boutron-Ruault MC, Schnee M, Ferec C, Maire F, Hammel P, Ruszniewski P, Levy P. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. The American journal of gastroenterology 2008; 103:111-9. 80. Azmi AS, Ahmad A, Banerjee S, Rangnekar VM, Mohammad RM, Sarkar FH. Chemoprevention of pancreatic cancer: characterization of Par-4 and its modulation by 3,3' diindolylmethane (DIM). Pharmaceutical research 2008; 25:2117-24. 81. Surveillance, Epidemiology, and End Results (SEER) Program. National Cancer Institute. 82. Kumar V, Abbas, A., Aster, J., Fausto, N. Robbins & Cotran Pathologic Basis of Disease. Saunders. 83. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica 2007; 114:97-109. 84. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine 2005; 352:987-96. 85. Louis DN. Molecular pathology of malignant gliomas. Annual review of pathology 2006; 1:97-117. 86. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455:1061-8. 87. Zhuang D, Liu Y, Mao Y, Gao L, Zhang H, Luan S, Huang F, Li Q. TMZ-induced PrPc/par-4 interaction promotes the survival of human glioma cells. International journal of cancer Journal international du cancer 2012; 130:309-18.

117
88. Jagtap JC, Dawood P, Shah RD, Chandrika G, Natesh K, Shiras A, Hegde AS, Ranade D, Shastry P. Expression and regulation of prostate apoptosis response-4 (Par-4) in human glioma stem cells in drug-induced apoptosis. PloS one 2014; 9:e88505. 89. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British journal of cancer 1972; 26:239-57. 90. McCarthy NJ, Evan GI. Methods for detecting and quantifying apoptosis. Current topics in developmental biology 1998; 36:259-78. 91. Wyllie AH. Apoptosis: an overview. British medical bulletin 1997; 53:451-65. 92. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004; 116:205-19. 93. Lavrik IN, Golks A, Krammer PH. Caspases: pharmacological manipulation of cell death. The Journal of clinical investigation 2005; 115:2665-72. 94. Shiozaki EN, Shi Y. Caspases, IAPs and Smac/DIABLO: mechanisms from structural biology. Trends in biochemical sciences 2004; 29:486-94. 95. Vaux DL, Silke J. Mammalian mitochondrial IAP binding proteins. Biochemical and biophysical research communications 2003; 304:499-504. 96. Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nature reviews Molecular cell biology 2007; 8:405-13. 97. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nature reviews Cancer 2002; 2:647-56. 98. Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annual review of immunology 1999; 17:331-67. 99. Nagata S. Fas ligand-induced apoptosis. Annual review of genetics 1999; 33:29-55. 100. Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell death and differentiation 2003; 10:26-35. 101. Elmore S. Apoptosis: a review of programmed cell death. Toxicologic pathology 2007; 35:495-516. 102. Ravichandran KS. "Recruitment signals" from apoptotic cells: invitation to a quiet meal. Cell 2003; 113:817-20. 103. Sells SF, Han SS, Muthukkumar S, Maddiwar N, Johnstone R, Boghaert E, Gillis D, Liu G, Nair P, Monnig S, et al. Expression and function of the leucine zipper protein Par-4 in apoptosis. Molecular and cellular biology 1997; 17:3823-32. 104. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods 2012; 9:671-5. 105. Irby RB, Kline CL. Par-4 as a potential target for cancer therapy. Expert opinion on therapeutic targets 2013; 17:77-87. 106. Mao Q, Zheng X, Yang K, Qin J, Bai Y, Jia X, Li Y, Xie L. Suppression of migration and invasion of PC3 prostate cancer cell line via activating E-cadherin expression by small activating RNA. Cancer Invest 2010; 28:1013-8. 107. Wong AS, Gumbiner BM. Adhesion-independent mechanism for suppression of tumor cell invasion by E-cadherin. J Cell Biol 2003; 161:1191-203. 108. Stoops SL, Pearson AS, Weaver C, Waterson AG, Days E, Farmer C, Brady S, Weaver CD, Beauchamp RD, Lindsley CW. Identification and optimization of small molecules that restore E-cadherin expression and reduce invasion in colorectal carcinoma cells. ACS Chem Biol 2011; 6:452-65. 109. Hayashida Y, Honda K, Idogawa M, Ino Y, Ono M, Tsuchida A, Aoki T, Hirohashi S, Yamada T. E-cadherin regulates the association between beta-catenin and actinin-4. Cancer Res 2005; 65:8836-45.

118
110. Honda K, Yamada T, Hayashida Y, Idogawa M, Sato S, Hasegawa F, Ino Y, Ono M, Hirohashi S. Actinin-4 increases cell motility and promotes lymph node metastasis of colorectal cancer. Gastroenterology 2005; 128:51-62. 111. Strumane K, Bonnomet A, Stove C, Vandenbroucke R, Nawrocki-Raby B, Bruyneel E, Mareel M, Birembaut P, Berx G, van Roy F. E-cadherin regulates human Nanos1, which interacts with p120ctn and induces tumor cell migration and invasion. Cancer Res 2006; 66:10007-15. 112. Bonnomet A, Polette M, Strumane K, Gilles C, Dalstein V, Kileztky C, Berx G, van Roy F, Birembaut P, Nawrocki-Raby B. The E-cadherin-repressed hNanos1 gene induces tumor cell invasion by upregulating MT1-MMP expression. Oncogene 2008; 27:3692-9. 113. Mohri Y. Prognostic significance of E-cadherin expression in human colorectal cancer tissue. Surg Today 1997; 27:606-12. 114. Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol 2009; 1:a003129. 115. Corn PG, Heath EI, Heitmiller R, Fogt F, Forastiere AA, Herman JG, Wu TT. Frequent hypermethylation of the 5' CpG island of E-cadherin in esophageal adenocarcinoma. Clin Cancer Res 2001; 7:2765-9. 116. Darwanto A, Kitazawa R, Maeda S, Kitazawa S. MeCP2 and promoter methylation cooperatively regulate E-cadherin gene expression in colorectal carcinoma. Cancer Sci 2003; 94:442-7. 117. Wheeler JM, Kim HC, Efstathiou JA, Ilyas M, Mortensen NJ, Bodmer WF. Hypermethylation of the promoter region of the E-cadherin gene (CDH1) in sporadic and ulcerative colitis associated colorectal cancer. Gut 2001; 48:367-71. 118. Liu Y, Zhao Y, Wu C, Ho KS, Koh PK, Chong SF, Eu KW. Modest promoter methylation of E-cadherin gene in sporadic colorectal cancers: a quantitative analysis. Cancer Biomark 2008; 4:111-20. 119. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007; 7:415-28. 120. Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proceedings of the National Academy of Sciences of the United States of America 2001; 98:10356-61. 121. Nguyen J, Kline, CL, Caballero, N, Irby, R. Overexpression Of Prostate Apoptosis Response Protein-4 In Colon Cancer Cells Can Inhibit Metastasis By Upregulating E-Cadherin Expression. Journal of Colon and Rectal Cancer 2015; 1:20-34. 122. Pai SG, Fuloria J. Novel therapeutic agents in the treatment of metastatic colorectal cancer. World journal of gastrointestinal oncology 2016; 8:99-104. 123. Kourie HR, Gharios J, Elkarak F, Antoun J, Ghosn M. Is metastatic pancreatic cancer an untargetable malignancy? World journal of gastrointestinal oncology 2016; 8:297-304. 124. Azmi AS, Wang Z, Burikhanov R, Rangnekar VM, Wang G, Chen J, Wang S, Sarkar FH, Mohammad RM. Critical role of prostate apoptosis response-4 in determining the sensitivity of pancreatic cancer cells to small-molecule inhibitor-induced apoptosis. Molecular cancer therapeutics 2008; 7:2884-93. 125. Srinivasan S, Ranga RS, Burikhanov R, Han SS, Chendil D. Par-4-dependent apoptosis by the dietary compound withaferin A in prostate cancer cells. Cancer research 2007; 67:246-53. 126. Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nature immunology 2013; 14:172-8.

119
127. Lazzari E, Korczeniewska J, Ni Gabhann J, Smith S, Barnes BJ, Jefferies CA. TRIpartite motif 21 (TRIM21) differentially regulates the stability of interferon regulatory factor 5 (IRF5) isoforms. PloS one 2014; 9:e103609. 128. Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol 2008; 181:1780-6. 129. Jauharoh SN, Saegusa J, Sugimoto T, Ardianto B, Kasagi S, Sugiyama D, Kurimoto C, Tokuno O, Nakamachi Y, Kumagai S, et al. SS-A/Ro52 promotes apoptosis by regulating Bcl-2 production. Biochemical and biophysical research communications 2012; 417:582-7. 130. Shibata N, Ohoka N, Sugaki Y, Onodera C, Inoue M, Sakuraba Y, Takakura D, Hashii N, Kawasaki N, Gondo Y, et al. Degradation of Stop Codon Read-through Mutant Proteins via the Ubiquitin-Proteasome System Causes Hereditary Disorders. The Journal of biological chemistry 2015; 290:28428-37. 131. Espinosa A, Zhou W, Ek M, Hedlund M, Brauner S, Popovic K, Horvath L, Wallerskog T, Oukka M, Nyberg F, et al. The Sjogren's syndrome-associated autoantigen Ro52 is an E3 ligase that regulates proliferation and cell death. J Immunol 2006; 176:6277-85. 132. Ding Q, He D, He K, Zhang Q, Tang M, Dai J, Lv H, Wang X, Xiang G, Yu H. Downregulation of TRIM21 contributes to hepatocellular carcinoma carcinogenesis and indicates poor prognosis of cancers. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine 2015. 133. Brauner S, Zhou W, Backlin C, Green TM, Folkersen L, Ivanchenko M, Lofstrom B, Xu-Monette ZY, Young KH, Moller Pedersen L, et al. Reduced expression of TRIM21/Ro52 predicts poor prognosis in diffuse large B cell lymphoma patients with and without rheumatic disease. Journal of internal medicine 2015. 134. Lee TJ, Jang JH, Noh HJ, Park EJ, Choi KS, Kwon TK. Overexpression of Par-4 sensitizes TRAIL-induced apoptosis via inactivation of NF-kappaB and Akt signaling pathways in renal cancer cells. Journal of cellular biochemistry 2010; 109:885-95. 135. Lee TJ, Lee JT, Kim SH, Choi YH, Song KS, Park JW, Kwon TK. Overexpression of Par-4 enhances thapsigargin-induced apoptosis via down-regulation of XIAP and inactivation of Akt in human renal cancer cells. Journal of cellular biochemistry 2008; 103:358-68. 136. Bergmann M, Kukoc-Zivojnov N, Chow KU, Trepohl B, Hoelzer D, Weidmann E, Mitrou PS, Boehrer S. Prostate apoptosis response gene-4 sensitizes neoplastic lymphocytes to CD95-induced apoptosis. Annals of hematology 2004; 83:646-53. 137. Dilruba S, Kalayda GV. Platinum-based drugs: past, present and future. Cancer chemotherapy and pharmacology 2016. 138. Vetterkind S, Boosen M, Scheidtmann KH, Preuss U. Ectopic expression of Par-4 leads to induction of apoptosis in CNS tumor cell lines. International journal of oncology 2005; 26:159-67. 139. Li X, Wu C, Chen N, Gu H, Yen A, Cao L, Wang E, Wang L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016. 140. McDowell KA, Riggins GJ, Gallia GL. Targeting the AKT pathway in glioblastoma. Current pharmaceutical design 2011; 17:2411-20. 141. Zanotto-Filho A, Braganhol E, Schroder R, de Souza LH, Dalmolin RJ, Pasquali MA, Gelain DP, Battastini AM, Moreira JC. NFkappaB inhibitors induce cell death in glioblastomas. Biochemical pharmacology 2011; 81:412-24. 142. Michl P, Gress TM. Current concepts and novel targets in advanced pancreatic cancer. Gut 2013; 62:317-26.

120
143. Sabile A, Meyer AM, Wirbelauer C, Hess D, Kogel U, Scheffner M, Krek W. Regulation of p27 degradation and S-phase progression by Ro52 RING finger protein. Molecular and cellular biology 2006; 26:5994-6004. 144. Ishii T, Ohnuma K, Murakami A, Takasawa N, Yamochi T, Iwata S, Uchiyama M, Dang NH, Tanaka H, Morimoto C. SS-A/Ro52, an autoantigen involved in CD28-mediated IL-2 production. J Immunol 2003; 170:3653-61.

CURRICULUM VITAE
JEFFREY NGUYEN
19 Rosedale Apartments, Hershey, PA 17033 | (309)706-0376 | [email protected]
EDUCATION
Pennsylvania State University College of Medicine, Hershey, PA
MD/PhD candidate 2009 - Present
Saint Louis University, Saint Louis, MO
B.S. Biochemistry 2007
TEACHING EXPERIENCE
Penn State College of Medicine, Hershey, PA
Undergraduate Summer Student Mentor 2012 - Present
ABSTRACTS AND PRESENTATIONS
Nguyen J, Irby R. Trim21: a novel regulator of Par-4 and oncogenic signaling. AACR Annual Meeting, Philadelphia, PA: April 18-22, 2015.
Nguyen J. Elucidating the role of prostate apoptosis response protein-4 in colon cancer. Penn State College of Medicine, MD/PhD seminar,
Hershey, PA: August 13, 2015.
Nguyen J. The regulation of EMT and colon cancer metastasis b y Par-4. Penn State College of Medicine, MD/PhD seminar, Hershey, PA:
March 13, 2014.
Nguyen J, Kline L, Irby R. The pro-apoptotic protein, Par-4, induces mesenchymal-epithelial transition and inhibits migration/invasion.
Medicine/Biochemistry Research Day, Penn State College of Medicine, Hershey, PA: May 2013.
PUBLICATIONS AND PAPERS
Nguyen J, Irby R. Trim21 is a novel regulator of Par-4 in colon and pancreatic cancer cells. Cancer Biology and Therapy. 2016. (Accepted/In
Press).
Nguyen, J, Kline, CL, Caballero, N, Irby R. Overexpression of prostate apoptosis response protein-4 in colon cancer cells can inhibit
metastasis by upregulating E-cadherin expression. Journal of Colon and Rectal Cancer. 2015; 1(1): 20-34.
EXTRACURRICULARS
APAMSA treasurer, Penn State College of Medicine, Hershey, PA: 2010-2011
PULSE, Penn State College of Medicine, Hershey, PA: 2010-2011
MEMBERSHIPS
American Association for Cancer Research





























