Embed Size (px)
Citation preview

Investigating haemoglobinopathies

Carrier frequencies of thalassaemia alleles (%)
Region β-Thalassaemia α0-Thalassaemia α+-Thalassaemia
Americas 0–3 0–5 0–40
Eastern Mediterranean
2–18 0–2 1–60
Europe 0–19 1–2 0–12
Southeast Asia 0–11 1–30 3–40
Sub-Saharan Africa
0–12 0 10–50
Western Pacific 0–13 0 2–60
Weatherall D, et al. Inherited Disorders of Hemoglobin. In: Disease Control Priorities in Developing Countries.2nd ed. New York: Oxford University Press; 2006: 663-80. Available from: www.dcp2.org/pubs/DCP.
7% of the world’s population are carriers of haemoglobin disorders

Types of haemoglobinopathies
Thalassaemias – result from an imbalance in and globin gene production, most commonly due to
– Point mutations in the gene
– Deletion of one or more genes
Haemoglobin variants - result from point mutation in the or genes leading to amino acid substitution, producing a different haemoglobin, sometimes with different properties
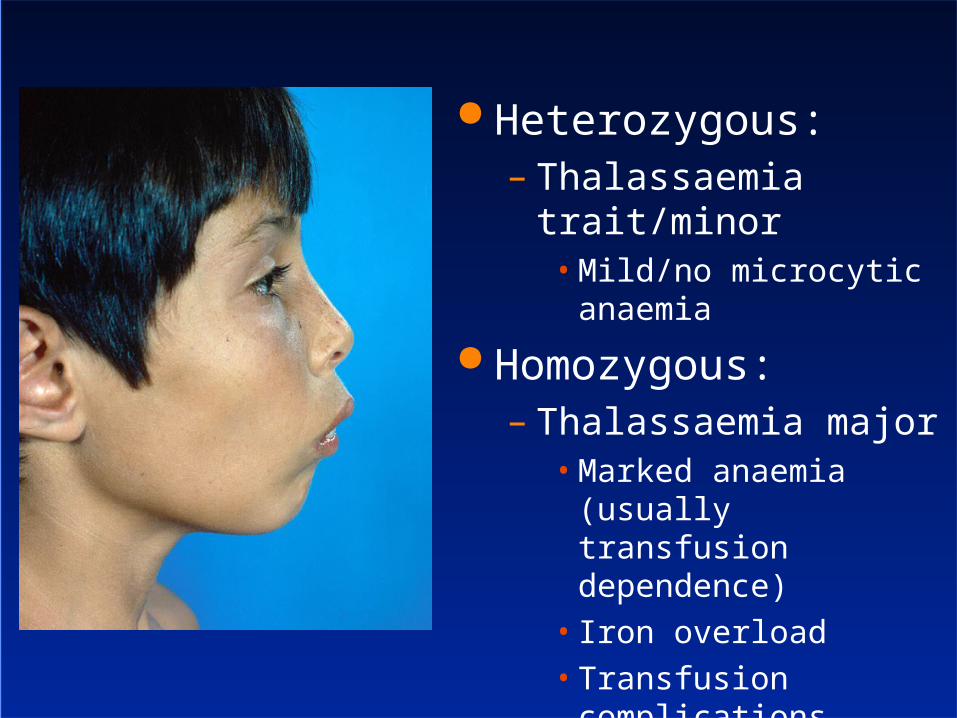
Heterozygous:– Thalassaemia trait/minor
• Mild/no microcytic anaemia
Homozygous:– Thalassaemia major
• Marked anaemia (usually transfusion dependence)
• Iron overload• Transfusion complications

Clinical impact of thalassaemia major
Transfusion dependence Iron overload
– Cardiac complications– Endocrine (diabetes, hypothyroidism,
hypogonadism, hypoparathyroidism)
Transfusion complications (eg hepatitis C) Osteoporosis – bone disease
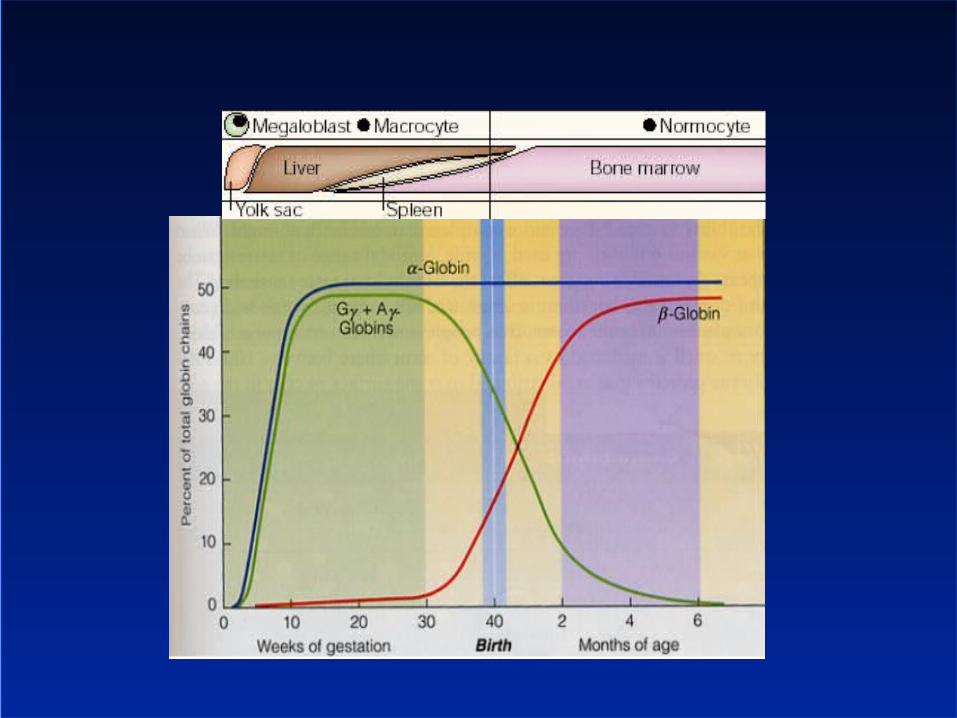
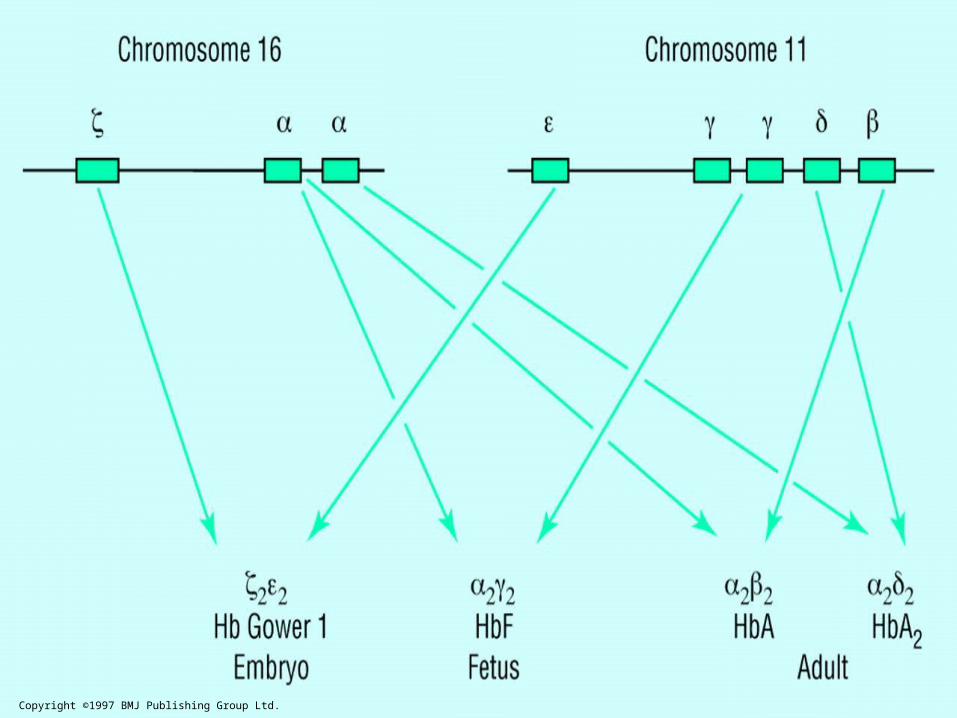
Copyright ©1997 BMJ Publishing Group Ltd.

Mutations in thalassaemia
thalassaemia– 200 point mutations in the globin gene– Deletions are rare
thalassaemia– Deletion of one globin gene on an allele
• Common, many ethnic groups
– Deletion of both globin genes on an allele• Less common, some ethnic groups
– Occasional point mutations
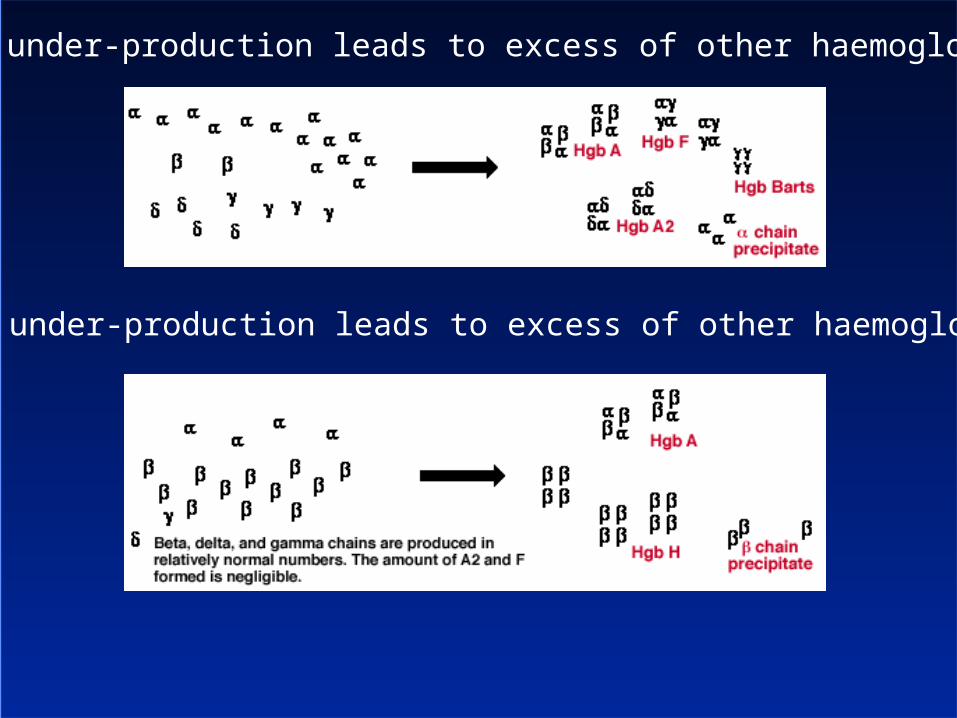
globin under-production leads to excess of other haemoglobins
globin under-production leads to excess of other haemoglobins

Microcytosis
Thalassaemia trait– Anisocytosis, poikilocytosis– Target cells
Iron deficiency– Hypochromic red cells– Pencil cells
Anaemia of chronic disease (eg rheumatoid arthritis) – upregulation of hepcidin, functional iron deficiency (poor release of iron from enterocytes, hepatocytes)
Very rare: sideroblastic anaemia
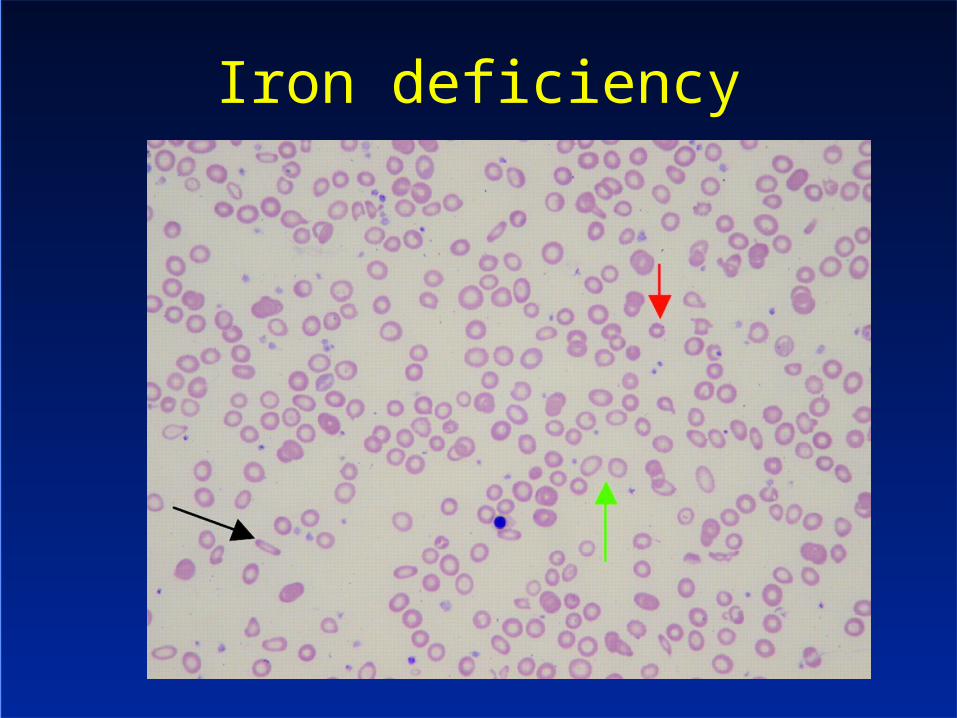
Iron deficiency
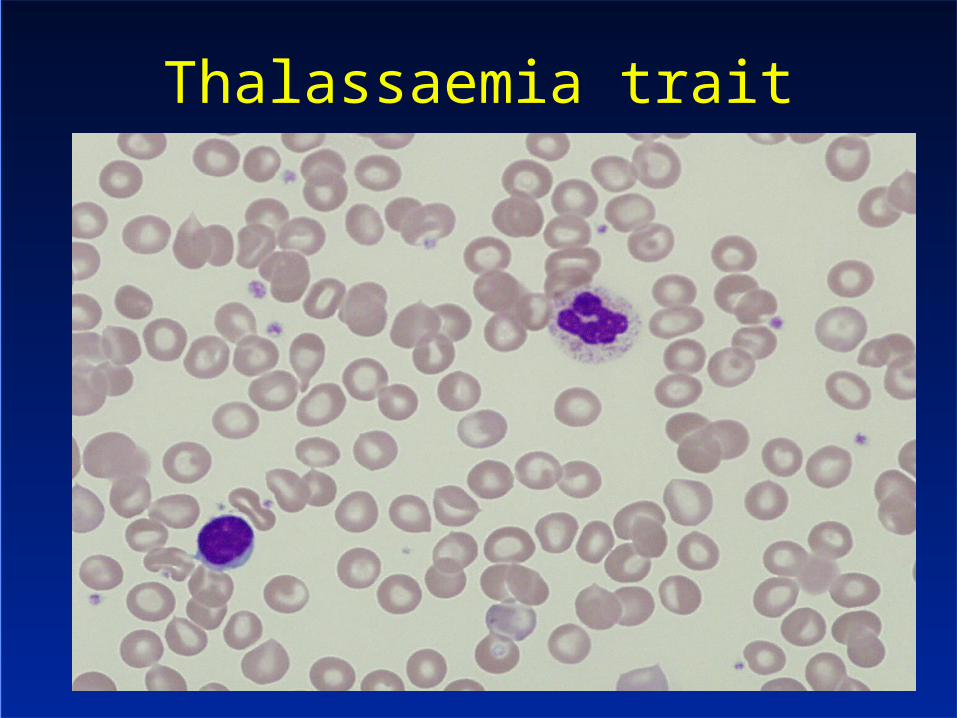
Thalassaemia trait
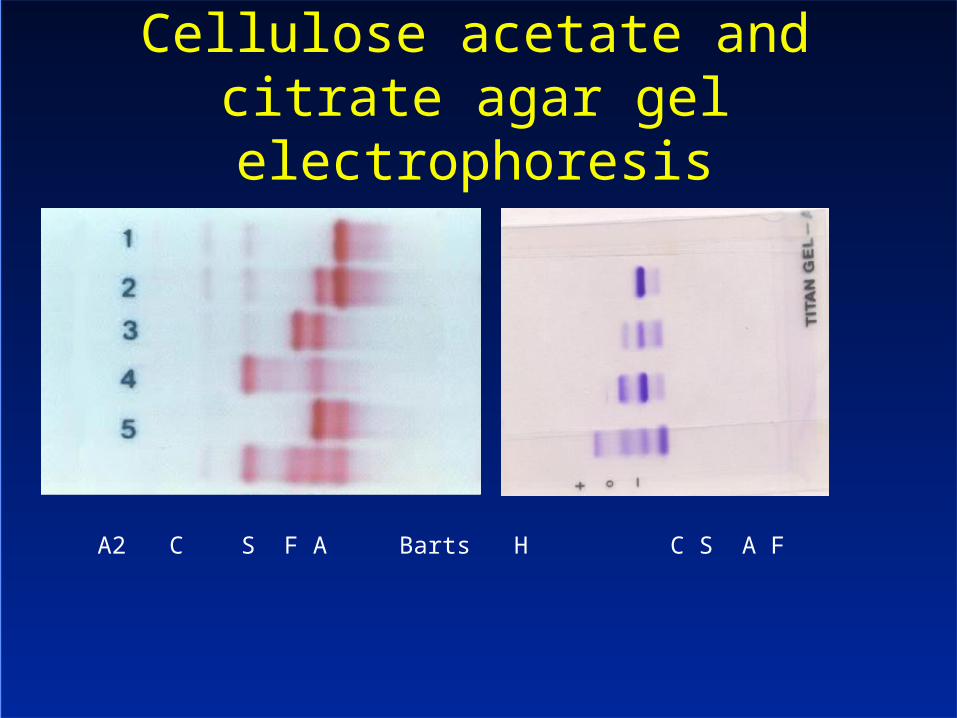
Cellulose acetate and citrate agar gel electrophoresis
A2 C S F A Barts H C S A F
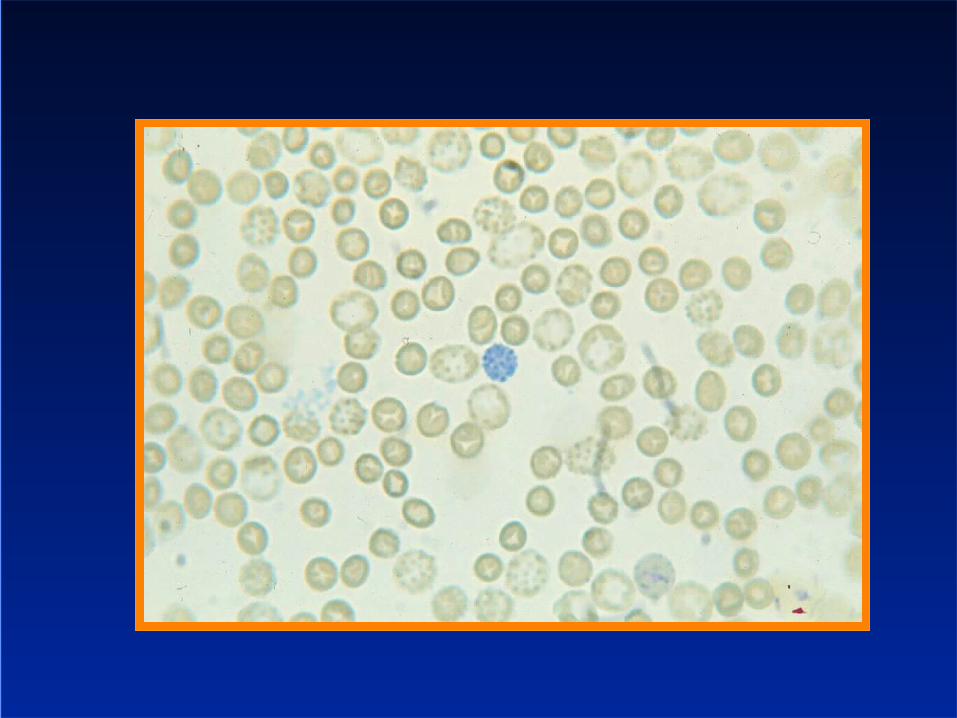

Thalassaemia trait - phenotype
thalassaemia trait– Variable microcytosis, mild anaemia
thalassaemia– Single gene deletion: none/microcytosis– Two gene deletion: mild anaemia/variable
microcytosis– Three gene deletion (Hb H disease)
• Variable
– Four gene deletion• Hb Barts hydrops fetalis
Compound heterozygotes– Sickling disorders (HbSS, HbSC, HbS--thal

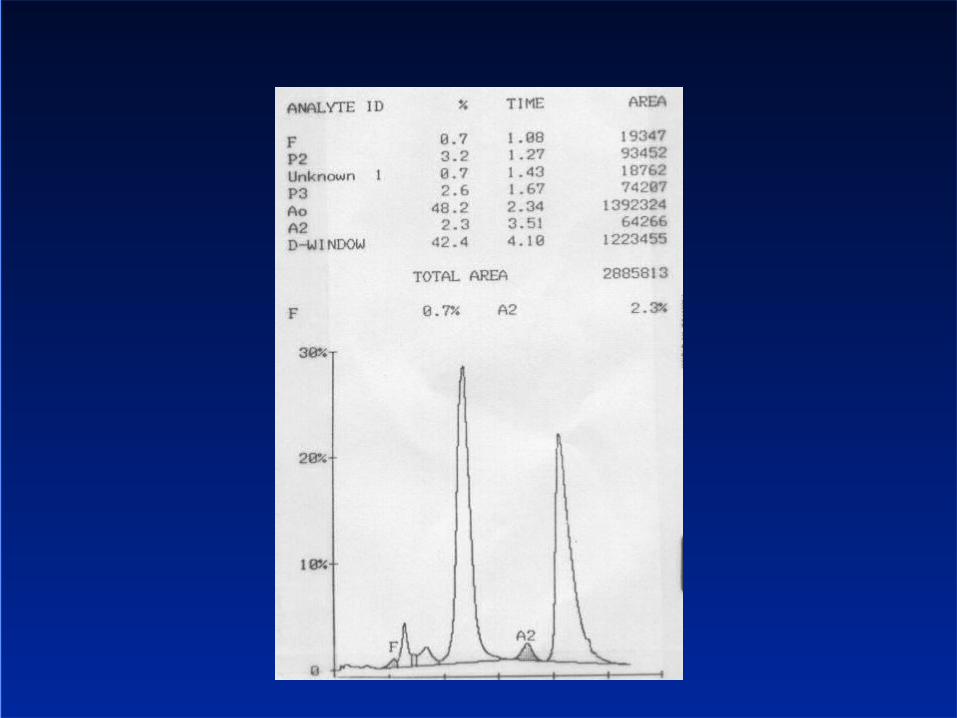
Interpreting the haemoglobin EPG An elevated HbA2 is diagnostic of thalassaemia trait Hb H inclusions are diagnostic of thalassaemia trait Elevated Hb F
– May be seen in thalassaemia trait– Other disorders of erythropoiesis– Pregnancy– Hereditary persistence of fetal haemoglobin
Abnormal bands – Haemoglobin E (with or without thalassaemia), Lepore
• Matters because of compound heterozygosity with thalassaemia trait
– Hb S, C• Matter because can contribute to sickling
– Other D, O, etc, etc• Often don’t matter• Some produce unstable haemoglobins• Can’t be easily characterised on standard HbEPG

Lab Testing FBC Characterisation of abnormal haemoglobins
– Haemoglobin electrophoresis– HPLC– Supravital staining for H inclusions
Iron studies– Ferritin– Transferrin/TIBC– Transferrin saturation– Serum iron
(inflammatory markers)

Common problems in Hb EPG interpretation
H inclusions are rare– thalassaemia cannot be excluded
The findings of thalassaemia may be masked by iron deficiency (reduction in Hb A2)
Rare problem of normal Hb A2 thalassaemia trait
Unexplained elevated Hb F

Solutions to difficult Hb EPG results
Family studies Repeat testing when iron replete DNA testing
– globin gene PCR testing– globin gene sequencing

When does it matter?
Pre-pregnancy Early pregnancy
– DNA testing is rapid if the mutations are known
– DNA testing is slow and may not yield a result if the mutation is not known
– CVS possible at 11 weeks– Second trimester amniocentesis– Genetic counselling takes time and causes
anxiety The role for screening?

Important patterns Microcytosis in early pregnancy
– Partner should have FBC, iron studies and Hb EPG (to exclude both thalassaemia trait and a sickling disorder).
– Don’t wait for the woman’s results Microcytosis (in a male or female)
– Test the partner– Test other family members – it may help someone else
Microcytosis in both partners– Is there a risk of Hb H disease or Hb Barts hydrops fetalis– “masked” thalassaemia major
Hb S or Hb E trait– Think of compound heterozygosity– Test the partner and other family members

















![Management of Thalassaemia[1]](https://img.pdfslide.us/doc/110x75/546a51dfb4af9ffa1d8b461b/management-of-thalassaemia1.jpg)











