Embed Size (px)
Citation preview

1
INTRODUCTION
CHAPTER-I
Much attention has been devoted by bioinorganic as well as medicinal
chemists to present the relationship between the metal ions and their complexes as
antitumour [1] and antibacterial [2] agents. Several reviews showed that the
coordination chemistry of many compounds greatly influence their biological
action highlighting the catalytic function of metal in many biological processes.
Metal complexes have shown many useful evidences in medicinal aspects. This
thesis contributes a piece of work with a fact of usefulness of coordination
complexes.
1.1. Coordination complex
In chemistry, a coordination complex or metal complex, is a structure
consisting of a central atom or ion, bonded to a surrounding array of molecules or
anions (ligands, complexing agents). The atom within a ligand that is directly
bonded to the central atom or ion is called the donor atom. Polydentate ligands can
form a chelate complex. Compounds that contain a coordination complex are
called coordination compounds. The central atom or ion, together with all ligands
forms the coordination sphere. Coordination refers to the coordinate covalent
bonds between the ligands and the central atom. Originally, a complex implied a
reversible association of molecules, atoms or ions through such weak chemical
bonds. As applied to coordination chemistry, this meaning has evolved. Some
metal complexes are formed virtually irreversibly and many are bound together by
bonds that are quite strong [3].

2
1.1.1. History of coordination complexes
Coordination complexes were known although not understood in any sense
since the beginning of chemistry, e.g. Prussian blue and copper vitriol. The key
breakthrough occurred when Alfred Werner proposed in 1893, that Co(III) bears
six ligands in an octahedral geometry. His theory allows one to understand the
difference between coordinated and ionic in a compound, for example chloride in
the cobalt ammine chlorides and to explain many of the previously inexplicable
isomers. In 1914, Werner resolved the first coordination complex, called hexol,
into optical isomers, overthrowing the theory that chirality was necessarily
associated with carbon compounds.
1.1.2. Applications of coordination complexes
The applications of coordination complexes are of great importance. These
complexes are widely present in the mineral, plant and animal worlds are known to
play many important functions in the area of analytical chemistry, metallurgy,
biological function system, industry and medicine. A compound containing single
or additional organic bonds, which is a connection among a couple of electrons in
which together electrons are donated by one of the atoms.
The interest of the coordination complexes of Schiff bases, stems from their
versatile catalytic reactions for organic synthesis [4] and its use in degradation of
organic substances [5] and in radiopharmaceuticals [6] and their ability to
reversibly bind oxygen [7] and photochromic properties [8], and the complexing
ability towards transition metals.

3
Importance and applications of coordination compounds are of great
importance in biological system. The pigment responsible for
photosynthesis ie., chlorophyll is a coordinated compound of magnesium.
Haemoglobin, the red pigment of blood which acts as oxygen carrier is a
coordination compound of iron. Coordination compounds are used a s
catalysts for many industrial processes.
Importance and applications of coordination compounds find use in many
qualitative and quantitative chemical analyses. Many familiar color
reactions are given by metal ions with number of ligands. Similarly
purification of metal can be achieved through formation and subsequence
decomposition of their coordination compounds.
There is a growing interest in the user of chelate therapy in medicinal
chemistry. An example is the treatment of problem caused by the presence
of metal in toxic proportion in plant and animal. Some coordination
compounds of platinum effectively inhibit the growth of tumours.
1.2. Ligand
In coordination chemistry, a ligand is an ion or molecule that binds to a
central metal atom to form a coordination complex. The bonding between metal
and ligand generally involves formal donation of one or more of the ligand's
electron pairs. The nature of metal-ligand bonding can range from covalent to
ionic. Furthermore, the metal-ligand bond order can range from one to three.
Ligands are viewed as Lewis bases, although rare cases are known involving Lewis
acidic ligands. Metal and metalloids are bound to ligands virtually in all

4
circumstances, although gaseous naked metal ions can be generated in high
vacuum. Ligands in a complex dictate the reactivity of the central atom, including
ligand substitution rates, the reactivity of the ligands themselves, and redox.
Ligand selection is a critical consideration in many practical areas, including
bioinorganic and medicinal chemistry, homogeneous catalysis and environmental
chemistry.
Ligands are classified in many ways: their charge, size (bulk), the identity
of the coordinating atom and the number of electrons donated to the metal
(denticity or hapticity). For example, a diagrammatic representation of cobalt
complex is shown below.
Figure.1.1. Cobalt complex [HCo(CO)4] with five ligands
1.2.1. Chelation
Chelation is the binding or complexation of a bi- or multidentate ligand.
These ligands, which are often organic compounds called chelants, chelators,
chelating agents or sequestering agents. Chelating agents form multiple bonds with
a single metal ion. The ligand forms a chelate complex with the substrate. The term
is reserved for complexes in which the metal ion is bound to two or more atoms of
the chelant.

5
1.3. Schiff bases
Coordination compounds containing polydentate ligands are called chelates
and their formation is termed chelation. Chelates are particularly stable and very
useful. Schiff base ligand is also called as chelate type ligand. Schiff base
(azomethine), named after Hugo Schiff, is a functional group that contains a
carbon nitrogen double bond with the nitrogen atom connected to an aryl group or
an alkyl group but not hydrogen.
Schiff bases are of the general formula R1R2C=N-R3, where R3 is an aryl
or alkyl group that makes the Schiff base a stable imine. Schiff bases can be
synthesized from an aromatic amine and a carbonyl compound by nucleophilic
addition followed by a dehydration reaction to generate an imine.
1.3.1. Applications of Schiff bases
Schiff bases have a wide variety of applications in many fields, e.g.,
biological, inorganic and analytical chemistry [9]. Application of many new
analytical devices requires the presence of organic reagents as essential compounds
of the measuring system. They are used, e.g., in optical and electrochemical
sensors, as well as in various chromatographic methods, to enable detection of
enhance selectivity and sensitivity [10-12]. Among the organic reagents actually
used, Schiff bases possess excellent characteristics, structural similarities with
natural biological substances, relatively simple preparation procedures and the
synthetic flexibility that enables design of suitable structural properties [13-18].

6
A large number of Schiff bases and their complexes have been investigated
for their interesting and important properties, such as their ability to reversibly bind
oxygen, catalytic activity in the hydrogenation of olefins, photochromic properties
and complexing ability towards some toxic metals. Furthermore, complexes of
Schiff bases showed promising biological activity and biological modeling
applications. The Schiff base ligands with sulphur and nitrogen donor atoms in
their structures act as good chelating agents for the transition metal ions.
Coordination of such compounds with metal ions, such as copper, nickel,
manganese and vanadium, often enhances their activities. There is a continuing
interest in metal complexes of Schiff bases. Because of the presence of both hard
nitrogen or oxygen and soft sulphur donor atoms in the backbones of these ligands,
they are readily coordinate with a wide range of transition metal ions yielding
stable and intensely coloured metal complexes. Some of which have been shown to
exhibit interesting physical and chemical properties and potentially useful
biological activities.
In recent years there has been considerable attention focused on the
chemistry of metal complexes of Schiff bases containing nitrogen and oxygen
[19, 20]. Schiff base ligands and their metal complexes have been found to have a
variety of applications in many fields including biology [21], materials synthesis,
photochemistry, magnetism [22], medical imaging [23], and industrial use as
catalysts [24] etc.,

7
1.4. Transition metal complexes
Transition metals have an esteemed place in medicinal chemistry. This
property of transition metals resulted in the foundation of coordination complexes.
Metal complex or coordination compound is a structure consisting of a central
metal atom, bonded to a surrounding array of molecules or anions. Sophus
Jorgensen in Denmark synthesized metal conjugates for the first time in the mid
1870‟s. In 1893 the major breakthrough in this field occurred when Alfred Werner
investigated a series of compounds, which contained cobalt, chlorine and
ammonia. He was awarded the Noble Prize in 1913 for his work.
Transition metal complexes having unique electronic and spectroscopic
signatures which offer a multitude of coordination geometries and mechanism of
cytotoxic action which is related to DNA binding affinity [25] and can also vary
accordingly as the biological activity is strongly dependent on structure–activity
relationship. Besides this, metal complexes also utilize or create open coordination
positions for DNA binding and hydrolysis generates reactive oxygen-containing
species or other radicals for DNA oxidation [26].
The interaction of transition metal complexes with DNA have been
extensively studied for their usage as probes for DNA structure and their potential
application in chemotherapy and are potent catalytic inhibitors of DNA gyrase
[27-29]. One of the important DNA related activity of the transition metal
complexes is that some of the complexes show the ability to cleave DNA.

8
1.4.1. Transition metal complexes as anticancer agents
Metal complexes have been widely applied in clinics for centuries,
although their molecular mechanism has not yet been entirely understood [30, 31].
The great successes achieved with platinum-based antitumor agents, mainly
including cisplatin, carboplatin and oxaliplatin, have promoted the development of
metal-based drugs. However, all these Pt-based drugs are associated with severe
side effects and evolution of drug resistance during therapy processes, which has
stimulated inorganic chemists to seek and develop more effective, less toxic and
target-specific metal-based anticancer drugs [32].
1.4.2. Platinum based anticancer drugs
Platinum(II) complexes have been used as anticancer drugs. Among them,
cisplatin has proved to be a highly effective chemotherapeutic agent for treating
various types of cancers [33]. Cisplatin moves into the cell through diffusion and
active transport. Inside the cell it causes platination of DNA, which involves
interstrand and intrastrand cross-linking as well as formation of adducts, usually
through guanine, as it is the most electron rich site and hence, easily oxidized.
Formation of cisplatin DNA adducts causes distortion and results in inhibition of
DNA replication [34]. Cisplatin DNA adducts also serve as binding site for cellular
proteins such as repair enzymes, histones, transcription factors and HMG-domain
proteins [35,36]. The binding of HMG protein to cisplatin DNA adduct has been
suggested to enhance anticancer effect of the drug [37].

9
1.4.3. Non-platinum anticancer agents
Platinum is not the only transition metal used in the treatment of cancer,
various other transition metals have been used in anticancer drugs [38]. Titanium
complexes such as Titanocene dichloride have been recognized as active
anticancer drug against breast and gastrointestinal carcinomas. Gold complexes
also show anticancer activity. These complexes act through a different mechanism
as compared to cisplatin [39].
The 2-[(dimethylamino) methyl] phenyl gold (III) complex has also been
proven to be anti-tumor agent against human cancers [40]. Gold nanoparticles
when used in combination with radio therapy or chemotherapy enhance DNA
damage and make the treatment target specific [41]. Copper as an essential element
for human with its bioessential activity and oxidative nature has attracted
numerous inorganic chemists to address Cu(II) complexes with the aim of medical
applications [42-45].
Ansari et al [46], studied some complexes of Mn(III) induce tumor
selective apoptosis of human cells. Many ruthenium complexes were studied
which showed anti-proliferative effects in human ovarian cancers. Ruthenium
complexes with oxidation state +2 or +3 show antitumor activity against
metastasis cancers. The relative binding of ruthenocene derivatives were very
high and even better than hydroxyl tamoxifen which is novel antagonist for
estrogen [47].

10
1.4.4. Transition metal complexes as anti-infective agents
Transition metals like silver have been used as anti-microbial agents. Silver
has low toxicity as compared to other transition metals. Silver nitrate is still given to
the infants to prevent the development of opthalmia neonatorum. One of the most
commonly used compounds of silver is silver (I) sulfazine; it is used to treat severe
burns to prevent them from bacterial infection. Chlorohexidine- Silver Sulfadiazine is
an anti-infective metal complex against catheter infections in vivo [48, 49].
1.4.5. Transition metal complexes as anti-inflammatory agents
Transition metals have also been used as anti-inflammatory and anti-
arthritic agents. The transition metals complexes of Cu and Fe are capable of
catalyzing dismutation of the superoxide anion. A manganese(II) complex with bis
(cyclohexylpyridine)- substituted macrocyclic ligand has designed as a functional
mimic of the superoxide dismutase (SOD) enzymes that normally remove these
radicals [50]. Manganese complexes have also been used to treat cell and tissue
oxidative injuries by acting as superoxide anion scavenger [51]. Magnesium is
used for the treatment of asthma in children. Some Cu complexes are also active
against inflammation but their use is limited [52]. Cu (II) complexes tend to
dissociate and bind to natural ligands such as albumins [53]. Zinc has been proved
to be involved in the inhibition of pro-inflammatory cytokines [54].
1.5. Multinuclear Schiff base metal complexes
The chemistry of multinuclear Schiff base metal complexes, especially of
coupled systems is of special interest in various fields of science. The main reason

11
probably is due to the phenomenon of interaction between metal centres lies at the
crossover point of two widely separated areas, namely the physics of the magnetic
materials and the role of polynuclear reaction sites in biological processes [55,56].
Trinuclear transition metal complexes bridged by polyatomic Schiff base ligands
have gained much attention in the recent years, towards synthesis and
characterization [57]. In particular, transition metal complexes have been the subject
for thorough investigation because of their extensive applications in wide ranging
areas from material science to biological sciences [58]. Metal complexes are well
known to accelerate drug action and the efficiency of a therapeutic agent can often
be enhanced upon coordination with a metal ion [59]. The pharmacological activity
has been found to be highly dependent on the nature of the metal ion and the donor
sequence of the ligands, with different ligands showing widely dissimilar biological
properties, although they may vary only slightly in their molecular structure.
1.6. Biological importance of Copper
The chemical nature of copper is very important in determining its
biological availability, both in the environment and in food. Some of the uses of
copper come from its ability to control the growth of organisms. This occurs when
copper is biologically available and at concentrations that are detrimental. As a
result, copper is used in a range of cidal agents. For example, copper has been
demonstrated to be an effective antibacterial, antiplaque agent in mouthwashes and
toothpastes. Copper also continues to be widely used for the control of unwanted
organisms in fish farming. Evidence in both fresh water and salt water indicates
that there were no hazardous effects on consumers of fish. Copper antifouling

12
agents used on fish net pens have been considered a source of metal to the
sediments but there is little evidence that they provide an important source of
dissolved copper when there is adequate water exchange for fish farming.
Few dietary components are more misunderstood than copper. Although
copper is the third most abundant essential trace mineral in the body, after iron and
zinc, most people consider it unimportant. Even worse, many people have actually
taken steps to exclude it from their diets and dietary supplements, believing it to be
nothing more than a cause of free radical reactions. This is surprising, because
copper has been recognized as an essential nutrient since the 1920's [60]. In the
past seventy years, much has been learned about the important biological roles of
copper and the copper-dependent enzymes. In fact, copper is emerging as one of
the most important minerals in our diet. While unbound, free copper does generate
free radicals in vitro, the relevance of this in the body has been called more
imaginary than real [61]. In fact, copper has an entirely different role in the body,
being a component of two of our most important antioxidant enzymes, copper-zinc
superoxide dismutase and ceruloplasmin [62].
Unbound, free copper is not found in large quantities in the human body.
Instead, almost all of the copper in our bodies is bound to transport proteins
(ceruloplasmin and copper-albumin), storage proteins (metallothioneins), or copper
containing enzymes [63]. A substantial number of copper metalloenzymes have
been found in the human body. Copper is essential for the proper functioning of
these copper-dependent enzymes, including cytochrome C oxidase (energy
production), superoxide dismutase (antioxidant protection), tyrosinase

13
(pigmentation), dopamine hydroxylase (catecholamine production), lysyl oxidase
(collagen and elastin formation), clotting factor V (blood clotting), and
ceruloplasmin (antioxidant protection, iron metabolism and copper transport) [64].
Most features of severe copper deficiency can be explained by a failure of one or
more of these copper-dependent enzymes. For instance, depigmentation can be
explained by a tyrosinase deficiency and the defects of collagen and elastin causing
abnormalities in the connective tissue and vascular system can be explained by a
lysyl oxidase deficiency.
Unfortunately, most research into copper deficiency has focused on acute,
severe deficiency. This is relatively rare in humans and animals on typical, varied
diets. Marginal, chronic deficiency, however, is much more common. The
determination of copper needs and marginal deficiency is complicated by the fact
that while copper deficiency doesn't necessarily lower the level of copper
dependent enzymes, it does significantly lower their activity [65]. As an example
in lysyl oxidase is one of the most important and best understood roles of copper in
the body. This is the main enzyme involved in the necessary cross-linking of
connective tissue. Optimal functioning of lysyl oxidase ensures the proper cross
linking of collagen and elastin, vital for the strength and flexibility of our
connective tissue. A reduction in lysyl oxidase activity affects the integrity of
numerous tissues, including our skin, bones, and blood vessels. In copper
deficiency the level of lysyl oxidase isn't altered, but the activity of the enzyme can
be reduced by more than fifty percent [66]. Not surprisingly, some of the hallmarks
of copper deficiency are connective tissue disorders, osteoporosis and blood vessel
damage.

14
Although most research utilizing copper complexes has been to determine
anti-inflammatory activity, copper complexes have shown potential as a
physiological approach to the treatment of numerous chronic diseases. This
potential has been expanded to include, in addition to inflammatory diseases,
gastrointestinal ulcers, cancers, carcinogenesis and diabetes. In these conditions
much of the research interest has centered on finding that many copper complexes
demonstrate superoxide dismutase (SOD) activity. Because of this, many of these
compounds have been designated as SOD-mimetics. One of the better recent
reviews on this topic of copper complexes is a good example of the breadth of
research that has been published on this topic. Unfortunately, despite the
tremendous promise that copper complexes have in many varied diseases and
conditions, clinical interest in these compounds has been almost nonexistent.
While copper is slowly becoming less misunderstood, one can only hope that it
will eventually be properly utilized in its potential for maintaining health and
treating disease.
The polynuclear copper(II) complexes have aroused extensive interest due
to their importance in biological processes and in inorganic material science [67].
The metal ion dependent oxidative DNA cleavage by Cu(II) complexes is of
topical interest. In the presence of suitable chelating ligands, the d9 electronic state
of Cu(II) may easily accept an electron to produce stable d10 Cu(I) species. Such
redox-active systems may achieve the decomposition of H2O2 to reactive oxygen
species (ROS) which can oxidize DNA by multiple attacks at the sugars and
nucleobases [68]. The coordination geometry around the Cu(II) ion plays a major
role as well in the redox-mediated formation of reactive oxygen species. The

15
activation of molecular oxygen by a mononuclear Cu(II) complex in the presence
of DNA is expected to lead to the abstraction of a proton/hydrogen from the sugar
backbone or from the bulk solvent.
1.7. Biological importance of Nickel
Although not recognized until the 1970s, nickel plays important roles in the
biology of microorganisms and plants [69]. Nickel compounds are present in the
active sites of urease and are used extensively in the design and construction of
new magnetic materials. The study of nickel compounds is of great importance in
various aspects of chemistry. The NiFe-hydrogenases contain nickel in addition to
iron-sulfur clusters. Such [NiFe]-hydrogenases characteristically oxidise H2.
A nickel-tetrapyrrole coenzyme, F430, is present in the methyl coenzyme
reductase which powers methanogenic archaea. One of the carbon monoxide
dehydrogenase enzymes consists of an Fe-Ni-S cluster [70]. Other nickel
containing enzymes include a class of superoxide dismutase [71] and a glyoxalase
[72].
In plants and microorganisms, the importance of nickel has been well
documented. In many cases, nickel is needed for the proper functioning of various
plant enzymes such as urease and hydrogenase. In the decreased presence of
urease, due to the lack of adequate nickel, urea accumulation leads to necrosis of
the plant [73]. In soyabeans, where hydrogenase activity was depressed due to
nickel-depletion, only low levels of nitrogen-fixation occurred, which resulted in
slow plant growth and decreased crop yields. Nickel depletion has also been linked
to necrosis of the leaves and stems of a variety of plants, lack of grain viability,

16
and depressed vigor of seedlings [74]. Because of the broad distribution of plants
that exhibit a nickel requirement, the authors of the aforementioned studies suggest
that this element is an essential micronutrient for all higher plants.
Nickel complexes draw much attention due to the environmental toxicity
and carcinogenic nature of certain nickel compounds and the chemotherapentic
properties of other group VIII metal complexes [75]. Some researchers have shown
that bound proteins or synthetic ligands may increase the toxic effect of nickel
ions. However, up to now, the exact mechanism to involve carcinogenesis has not
been fully elucidated [76]. The elucidation of the mechanism is essential not only
for the risk assessment [77], but also for developing novel nickel complexes that
have potential applications in medicine and research, such as inhibitors of cancer
proliferation and useful DNA or RNA probes [78]. Therefore, further studies by
employing various ligands with different structures to evaluate and understand
those factors that can determine the DNA binding modes and cleavage mechanism
are necessary.
Nickel complexes in the presence of oxidants have been extensively used
for DNA cleavage reactions [79]. Nickel is an essential trace element for many
species. Chicks and rats raised on nickel deficient diets are found to have liver
problems. Enzymes known as hydrogenases in bacteria contain nickel. Nickel is
also an important additive agent in plant ureases. Nickel complexes in the presence
of oxidants have been extensively used for DNA cleavage reactions.

17
1.8. Biological importance of Manganese
Manganese is an essential trace nutrient in all forms of life [80]. The
classes of enzymes that have manganese cofactors are very broad and include
oxidoreductases, transferases, hydrolases, lyases, isomerases, ligases, lectins and
integrins. The reverse transcriptases of many retroviruses (though not lentiviruses
such as HIV) contain manganese. The best known manganese containing
polypeptides may be arginase, the diphtheria toxin, and Mn-containing superoxide
dismutase (Mn-SOD).
Mn-SOD is the type of SOD present in eukaryotic mitochondria and also in
most bacteria (this fact is in keeping with the bacterial-origin theory of
mitochondria). The Mn-SOD enzyme is probably one of the most ancient, for
nearly all organisms living in the presence of oxygen use it to deal with the toxic
effects of superoxide, formed from the 1-electron reduction of dioxygen.
Exceptions include a few kinds of bacteria such as Lactobacillus plantarum and
related lactobacilli, which use a different non-enzymatic mechanism, involving
manganese (Mn2+) ions complexed with polyphosphate directly for this task,
indicating how this function possibly evolved in aerobic life. The human body
contains about 10 mg of manganese, which is stored mainly in the liver and
kidneys. In the human brain the manganese is bound to manganese metalloproteins
most notably glutamine synthetase in astrocytes [81].
It is also important in photosynthetic oxygen evolution in chloroplasts in
plants. The oxygen evolving complex (OEC) is a part of Photosystem II contained
in the thylakoid membranes of chloroplasts; it is responsible for the terminal

18
photooxidation of water during the light reactions of photosynthesis and has a
metalloenzyme core containing four atoms of manganese [82]. For this reason,
most broad-spectrum plant fertilizers contain manganese.
The chemistry of manganese complexes is of interest as models for
biological redox systems involving manganese ions,such as the oxygen-evolving
center (OEC) of Photosystem II in green plants, superoxide dismutases (SOD) and
Mn-catalases (CAT). 4H2O + hν → 4H+ + O2 is considered the most simple written
and important reaction on the planet at this moment. This is because we owe our
life to the oxygen released from this reaction through photosynthesis. However, 2.7
billion years ago this reaction was the most polluting factor for the environment of
that day. The polluting factor was the oxygen. It was being discarded from the
prokaryotes biological systems in their attempt to consume as much H2O as
possible, to use as an energy source (food). This pollutant (O2) destroyed the
atmospheric environment consisting of CO2 and H2. As a result of this change, the
next step of evolution started, with the creation of higher and more complicated
biological systems, first plants, then animals and human beings. It has been written
that human beings are the highest and most perfect biological system on the planet.
I know that plants can survive without our presence, but we cannot survive without
the presence of plants, having a crucial defect, that of not having chloroplasts.
Coming to the chemical factors implicated in the above given reaction we can find
a metal, i.e. manganese, playing a key role. It catalyzes the cleavage of water in
oxygen evolving complexes (OECs). Besides that, manganese is an essential
element in many other biological processes. Concerning manganese, two
functional values can be distinguished; Mn(II) as a Lewis acid and in higher

19
oxidation states [Mn(III), Mn(IV)] as an oxidative catalyst. In most Mn redox
enzymes [83], Mn exists in the oxidation states +2, +3 and +4. Mn(II), in the
manganese-containing ribonucleotide reductase [84-87]; a trimanganese center in
inorganic pyrophosphatase; a binuclear Mn(II) site in the Mn thiosulfate oxidase
[88]; a single Mn(III) center in the manganese SOD [89] catalyzing the
dismutasion of superoxide radicals to oxygen and hydrogen peroxide; Mn(III)
heme center in the manganese peroxidase (MnP) [90,91] capable for the oxidative
degradation of lignin; two Mn(III) single centers in the non heme manganese
catalase [92–95] and four Mn(II)–Mn(IV) in the OEC [96].
The coordination chemistry of manganese in the +2, +3 and +4 oxidation
states is receiving considerable attention due to the biological importance of these
ions [97]. The synthesis and characterization of manganese coordination
complexes to model the structure, reactivity and spectroscopy of manganese in its
various oxidation states, with various ligand types and nuclearities, has contributed
substantially to our understanding of the role and mechanism of manganese
enzyme.
The polynuclear manganese complexes are of great current interest due to
rich biochemistry [98] and remarkable magnetic properties [99]. In order to
understand the fundamental coordination chemistry in manganese systems that is
relevant to catalytic and biochemical functions, efforts have been centered on the
controlled preparation and characterization of manganese carboxylate complexes
with nitrogen donor ligands. A great deal of recent research in polynuclear mixed
valent manganese complexes has focussed on two main lines of enquiry. Firstly, in

20
the bioinorganic area, many model complexes have been made to replicate the
structure and function of manganese containing proteins and enzymes including,
significantly, the water oxidation complex (WOC) in the Photosystem II (PSII) of
green plants and cyanobacteria, which is responsible for the catalysis of the light
driven oxidation of H2O to O2 [100, 101].
1.9. Biological importance of Vanadium
Vanadium has been recognized as a metal of biological importance. Its
importance has been recognized at the beginning of 20th Century. Vanadium plays
an important role in the biological system. Considerable effort has been made in
studying the function of vanadium in biological systems as well as its role in
catalytic and pharmaceutical applications [102, 103]. Salen type ligands, one of
the oldest classes of ligands in coordination chemistry, have been used extensively
to complex transition and main group metals [104]. Schiff base complexes
containing different metal ions such as Ni and Cu have been studied in great details
for their various crystallographic feature, structure/redox relationships and
enzymatic reactions, mesogenic characteristics and catalysis properties [105, 106].
Vanadium has a rich and varied chemistry which includes compounds in all
oxidation states from -1 to +5.
Among the transition elements, vanadium has been widely used in a
number of industrial system. Because of its extreme resistance to corrosive agents,
vanadium is extensively used in steel alloys [107]. Vanadium is an important
catalyst in oxidation reactions such as oxidation of naphthalene to phthalic acid
and conversion of toluene to benzaldehyde. V2O5 is an important catalyst in the

21
manufacture of SO3 by the contact process. Recently vanadium has been widely
used in bioinorganic chemistry. Though the requirement of vanadium in mammals
is at nano to pico molar level, it is involved in promontory and inhibitory
biochemical processes [108, 109]. Vanadium compounds are also of major concern
because of their adverse effect on the hydro processing catalysis used in the
refining of crude oil [110]. There has been considerable interest in the coordination
chemistry of vanadium involving nitrogen and oxygen donor ligands due to the
increasing recognition of the role of this metal in biological systems. Compounds
in which vanadium is found in different oxidation states from -1 to +5, the higher
oxidation state is that corresponding to the total number of 3d and 4s electrons, that
is vanadium(V), which exists only in oxide species, such as V2O5 and is strong
oxidizing. Among these the most stable and generally important state is +4. which
exists only in oxo species such as (VO)2+
ie., oxovanadium (IV) ion. The
oxovanadium(IV) ion, VO2+, is considered to be the most stable oxocation of the
first row transition metal ion. It forms stable anionic, cationic and neutral
complexes with various types of ligands.
A square pyramidal geometry has been established or more frequently
assumed, for the five co-ordinate oxovanadium(IV) chelate complexes. The
electrolytic behaviour and polymerization tendency of the metal ion is well
documented and many polynuclear complexes of the metal ion have been isolated
and characterized. Because of the stability of the +4 state, compounds of vanadium
in the +3 and +5 states have a strong tendency to get oxidized/reduced to the +4
state.

22
Biochemical role of vanadium has now become a widely chosen topic of
bioinorganic chemistry. Though, the requirement of vanadium in mammals is at
nano to picomolar level, its involvement in promontory and inhibitory biochemical
processes, viz., enzymes-nitrogens and halperoxidases, phosphates and ATPase
inhibitor, lowering of hyperglycemia and hyperlipidemida, etc. have enough
driving force for intensive research aimed at establishing its role as a micronutrient
in human beings.
The element has also been identified in the sun, stars and in meteorites. It
is found as a by-product in petrol refining process. Significant amounts are also
found in milk, cereals, seafood‟s and in vegetables. It has a natural affinity for fats
and oils. Food oils have high concentrations of vanadium. When excess amount
of vanadium are taken in the diet, the concentration of vanadium in the red cells
tends to increase. Parental administration increases levels in the lever and kidneys.
However, such increased amounts may only be transient. The lung tissues may
contain some vanadium, depending upon the exposure and route, but normally the
other organs contain its negligible amount. In a variety of plant and animal life, it
appears to play an important role.
1.10. DNA Interactions
The interest of the bioinorganic community in the field of metal / nucleic
acid interactions has burgeoned in the last decade. This interest and the resulting
progress have come about primarily because of the tremendous advances that have
occurred in nucleic acid technology. Bioinorganic chemistry has itself been
evolving from a field focused on delineating metal centers in biology to one that

23
includes also the application of inorganic chemistry to probe biological structures
and function. In the past decades it has become clear that nucleic acids,
structurally, functionally and even remarkably in terms of catalysis, play active and
diverse roles in nature. Transition metal chemistry, both in the cell and in the
chemist‟s test tube, provides a valuable tool both to accomplish and to explore
these processes.
There are also many practical motivations behind the study of how metal
ions and complexes interact with nucleic acids. Heavy metal toxicity in our
environment arises in part from the covalent interactions of heavy metal ions with
nucleic acids. In addition, these heavy metals interface with metalloregulatory
proteins and in so doing disrupt gene expression. We need to understand the
functioning of the natural metalloregulators of gene expression and we need to
design new metal specific ligands, which like the proteins themselves, capture
heavy metals before their damage is done. Heavy metal interactions with nucleic
acids indeed have provided the basis also for the successful application of cisplatin
and its derivates as anticancer chemotherapeutic agents. The design of new
pharmaceuticals like cisplatin requires a detailed understanding of how platinum
and other metal ions interact with nucleic acids and nucleic acid processing.
Furthermore, we are finding that metal complexes can be uniquely useful in
developing spectroscopic and reactive probes of nucleic acids, and hence may
become valuable in developing new diagnostic agents. Finally, Nature itself takes
advantage of metal / nucleic acid chemistry, from the biosynthesis of natural
products such as bleomycin, which chelates redox active metal ions to target and
damage foreign DNA, to the development of basic structural motifs for eukaryotic

24
regulatory proteins, the zinc finger proteins, which bind to DNA and regulate
transcription. In all these endeavors, we need first to develop an understanding of
how transition metal ions and complexes interact with nucleic acids and how this
chemistry may best be exploited.
Metal ions and complexes associate with DNA in a variety of ways. Both
strong covalent interactions and week non covalent complexes are observed. Each
may yield a significant perturbation in the nucleic acid and / or may be exploited to
obtain a site specific response. Clearly there are some general guidelines, based on
principles of coordination chemistry that may be helpful in sorting out these
interactions.
Most prevalent among covalent complexes with DNA are those involving
coordination between soft metal ions and nucleophilic positions on the bases. The
structure of cis – (NH3) 2Pt-dGpG is an example for its platinum center coordinates
to the N7 position of the guanine basis. In terms of interactions with the full
polynucleotide, it is likely that the cis – diammineplatinum center, with two
coordination sites available, would yield an intrastrand crosslink between
neighbouring guanine residues on a strand. Other nucleophilic sites targeted by
soft metal ions on the bases include the N7 position of adenine, the N3 position on
cytosine, and the deprotonated N3 position on thymine and uracil. Some additional
covalent binding to the N1 positions of the purines has also been observed.
Indeed, coordination by the metal to one site on the heterocyclic base lowers the
pKa and increases the metal binding affinity to secondary sites. It is noteworthy,
however, that in base paired double helical DNA only N7 positions on the purines

25
are easily accessible in the major groove of the helix. Base binding at the purine
N7 positions is, of course, not limited to soft metal ions such as Pt (II), and Ru(II).
Coordination at these sites has been evident also with first row transition metal
ions such as Cu (II) and Zn (II). For these, as is consistent with basic coordination
chemistry, the liability of complexes formed is higher.
1.10.1. DNA Cleavage Studies
Identifying molecules that intercalate into DNA helices has attracted
considerable interest over the last few decades. Intercalation was first proposed by
Lerman and is defined as insertion between base pairs [111]. The cleavage of
nucleic acids may be considered as an enzymatic reaction which comprises of
various biological processes as well as the biotechnological manipulation of
genetic material. The application of artificial DNA cleaving agents is manifold:
biotechnology, structural studies of nucleic acids, or development of new drug
[112]. Compounds showing the property of effective binding as well as cleaving
double stranded DNA under physiological conditions are of importance since these
could be used as diagnostic agents in medicinal and genomic research [113].
Noting the very sensitive nature of DNA towards oxidative cleavage, the majority
of the studies on artificial DNAs have been centered around molecules capable of
cleaving DNA with an oxidative mechanism. Several efficient cleaving agents
have been developed in course of time. These involve reactive oxygen species
(ROS) or free radicals that are able to induce an oxidative pathway. The antitumor
antibiotic leinamycin and its analogues have been shown to play the role of

26
chemical nuclease exhibiting reduction of molecular oxygen to form reactive
hydroxyl species [114].
Biochemistry also provides very sensitive techniques that have been
invaluable in characterizing interactions of metal complexes with nucleic acids.
First are simply gel electrophoresis experiments, which permits and assessment of
changes in the nucleic acid conformation, through its changes in gel mobility, as a
function of metal binding. A classic illustration is that of the unwinding of super
helical DNA as a function of intercalation. Closed circular DNA has much the
same topological constraints on it as does a rope or a telephone card; the DNA
helices can wind up in coils. We define the duplex turning in a double helix as
secondary helical turns and turns of the helices about one another as the super coils
or tertiary turns. As long as a DNA double helix is closed in a circle (Form I), the
total winding ie., the total number of secondary and tertiary turns, is fixed.
Molecules with differing extents of winding have different super helical densities.
In a circular molecule with one strand scission, what we call form II (nickel) DNA,
the topological constraints are relaxed and no super coils are apparent. The same,
by analogy, can be said telephone card off the phone receiver, which can turn
about itself to relax its many supercoils. Now let us consider a DNA unwinding
experiment, monitored by gel electrophoresis. Supercoil Form I DNA can be
distinguished from nicked DNA (Form II) in a agarose gel because of their
differing mobilities; the wound- up supercoiled molecule moves easily through the
gelatinous matrix to the positive pole, whereas the nicked species is more floppy
and thus is inhibited in its travels down the gel.

27
Figure.1.2. Relaxation of supercoiled circular form (Form I) of DNA into nicked
circular form (Form II) and linear form (Form III).
DNA strand scission can also be sensitively monitored and even more
importantly the specific nucleotide position cleaved can be pinpointed by
biochemical methods. This methodology has been applied successfully in
monitoring both the efficiency strand scission by metal complexes and specific site
cleaved and hence where the complexes are specifically bound on the helical
strand.
Relative extents of cleavage of DNA by different metal complexes can be
easily assayed in an experiment that is an extension of the unwinding experiment
described above. One simply measures the conversion of supercoiled (form I)
DNA to nicked (form II) species. One strand cleavage on the DNA circle releases
the topological constraints on the circular molecule and relaxes the supercoils. Two
cleavage events within twelve base points on opposite strands will convert the
DNA to a linear (form III) which also has a distinguishable gel mobility.

28
Figure.1.3. Metal complexes exhibit high ability of DNA cleavage through the
effective cooperation of metal with the nucleic acid.
1.10.2. Binding Interactions with DNA
The interaction of metal complexes containing N4 Schiff base ligands has
been thoroughly considered [115]. DNA binding is the critical step for DNA
activity. To design effective chemotherapeutic agents and better anticancer drugs,
it is essential to explore the interactions of metal complexes with DNA [116, 117].
Recently, there has been tremendous interest in studies related to the interaction of
transition metal ions with nucleic acid because of their relevance in the
development of new reagents for biotechnology and medicine [118]. These studies
are also important to understand the toxicity of drugs containing metal ions.
The transition metal complexes are octahedral, substitutionally insert
complexes and as a result of this coordinative saturation the complexes bind to
double helical DNA through a mixture of noncovalent interactions. Tris
(phenanthroline) metal complexes bind to the double helix both by intercalation in
the major groove and through hydrophobic association in the minor groove.

29
Intercalation and minor groove binding are, in fact, the two most common modes
of noncovalent association of small molecules with nucleic acids. In addition, as
with other small molecules, a non specific electrostatic interaction between the
cationic complexes and the DNA polyanion serves to stabilize association.
The extent of intercalative versus groove binding is seen to depend upon
environmental conditions, such as temperature and ionic strength, the charge of the
metal center and the DNA base sequence; groove binding is favored at AT – rich
sequences [119]. Second generation mixed ligand derivatives of the tris
(phenanthroline) series has been prepared and their interactions with DNA have
provided useful insight into the factors important for promoting either intercalation
or groove binding [120]. Aromatic heterocyclic ligands with increased surface
areas that are planar bind DNA with increasing avidity through intercalation,
irrespective of the charge on the metal center. Intercalative binding constants
greater than 107 M-1 can be easily achieved with planar heterocyclic ligands that
just out from the metal center. Not surprisingly, complexes containing ligands of
increasing hydrophobicity that are not planar, favor minor groove binding.
Figure.1.4. DNA binding ability of Metal complexes.

30
Transition metal complexes with aromatic ligand also generally associate
by minor groove binding or through the mix of intercalative and groove-bound
ineractions. Cu(Phen)2, a tetrahedral complex appears to favor minor groove
binding over intercalation. Perhaps the tetrahedral coordination does not permit
appreciable overlap of the phenanthroline ring with the bases in an intercalative
mode. Metalloporphyrins, despite their large expense and the presence commonly
of nonplanar substituents appear to bind to double- helical DNA both by
intercalation and by minor groove binding at AT-Rich sequences. Occupation of
the porphyrins by transition metal ions such as Cu(II), which bind axial ligands,
leads to the favoring of groove binding over intercalation.
1.11. Antimicrobial activity
An anti-microbial is a substance that kills or inhibits the growth of
microorganisms such as bacteria, fungi or protozoans. Antimicrobial drugs either
kill microbes or prevent the growth of microbes. Generally transition metal
complexes have higher antimicrobial activity. Such increased activity of the metal
chelates can be explained on the basis of chelation theory [121]. The transition
metal complexes also disturb the respiration process of the cell and thus block the
synthesis of proteins, which restricts further growth of the organism.
Organic compounds and their metal complexes are well known for their
antimicrobial activity. The metal complexes of ligands containing electron with
drawing group exhibit improved biological activity [122]. Schiff bases and their
metal complexes have been found to possess important biological [123] and
catalytic activity.

31
Azomethines bind to the metal ions through nitrogen, oxygen or sulphur
atoms so form an important class of biologically active ligands and provide models
for metal ligand binding sites in several enzymes [124]. These ligands and their
metal complexes are known to function as antimicrobial [125, 126], antimalarial
[127], antitumor [128] and antileukemic agents [129].
Metal complexes are toxic to most microorganisms at specific
concentrations and often cause serious upsets in biological processes. Some of the
metal complexes are essential for the growth of microorganisms at very low
concentrations and certain metal ions inhibit the growth of many microorganisms
at higher concentrations.
The toxicity of the metal complexes depends mainly upon the nature of
metal ion, ligands and their concentration. Other factor such as pH and
temperature are also reported to affect the toxicity of metals [130]. Though to a
lesser degree, the metal complexes of ligands containing electron-withdrawing
group exhibit improved biological activity.
Some biologically active compounds act via chelation, but for most of them
little is known about how metal coordination influences their activity. The
trinuclear metal complexes have respective controls produce different inhibition
zones against the tested bacterial strains.

32
References
[1] V. L. Narayanan, M. Nasr and K. D. Paull, "Tin-Based Antumor Drugs",
M. Gielen (Ed), NATO ASI Series, Springer Verlag, Berlin. H 37 (1990).
[2] V. K. Patel, A. M. Vasanwola and C. R. Jejurkas, Indian. J. Chem., 28A
(1989) 719.
[3] Cotton, Frank Albert, Geoffrey Wilkinson and A. Carlos. Murillo,
Advanced Inorganic Chemistry. (1999) 1355.
[4] A. Nashinaga, H. Ohara, H. Tomita and T. Matsuura, Tetrahedron Lett., 24
(1983) 213.
[5] Y. K. Choi, W. S. Kim, K. I. Chung, M. W. Chung and H. P. Nam,
Microchem. J., 65 (2000) 3.
[6] M. A. Green, H. Luo and P. E. Fanwick, Inorg. Chem., 37 (1998) 1127.
[7] R. D. Jones, R. D Summerville and F. Basolo, Chem. Rev., 79 (1979) 139.
[8] J. D. Margerum and L. J. Miller, Photochromism, Interscience Wiley, New
York, 1971, 569.
[9] Z. Cimerman, S. Miljanic and N. Galic, Croat. Chem. Acta., 73 (2000) 81.
[10] L. A. Bigelow and H. Eatough, Org. Synthesis., 1 (1967) 80.
[11] M. N. Ibrahim, K. J. Hamad and S. H. Al-Joroshi, Asian. J. Chem., 18
(2006) 2404.
[12] C. E. White and R. J. Aragauer, "Fluorescence Analysis, a practical
Approach", Marcel Dekker, Inc., New York (1970).
[13] E. Bishop, "Indicators", Pergamon Press Ltd., Headington Hill Hall, Oxford
(1972).

33
[14] C. M. Metzler, A. Cahill and D. E. Metzler, J. Am. Chem. Soc., 102 (1980)
6075.
[15] G. O. Dudek and E. P. Dudek, Chem. Commun., (1965) 464.
[16] G. O. Dudek and E. P. Dudek, J. Am. Chem. Soc., 88 (1966) 2407.
[17] Z. Cimerman and Z. Stefanac, Polyhedron., 4 (1985) 1755.
[18] N. Galic, Z. Cimerman and V. Tomisic, Anal. Chem. Acta., 343 (1997)
135.
[19] S. Karuppasamy, S. Eringathodi and P. Mallayan, Inorg. Chim. Acta., 362
(2009) 199.
[20] J. Welby, L. N. Rusere, J. M. Tanski and L. A. Tyler, Inorg. Chim. Acta.,
362 (2009) 1405.
[21] M. S. Refat, S. A. El-Korashy, D. N. Kumar and A. S. Ahmed,
Spectrochim. Acta A., 70 (2008) 906.
[22] S. Mandal, G. Rosair, J. Ribas and D. Bandyopadhyay, Inorg. Chim. Acta.,
362 (2009) 2200.
[23] P. Mukherjee, C. Biswas, M. G. B. Drew and A. Ghosh, Polyhedron., 26
(2007) 3121.
[24] S. Chattopadhyay, G. Boclli, A. Musatti and A. Ghosh, Inorg. Chemi.
Commun., 9 (2006) 1053.
[25] J. Tan, B. Wang and L. Zhu, Bioorg. Med. Chem., 17 (2009) 614.
[26] L. J. K. Boerner and J. M. Zaleski, Curr. Opin. Chem. Biol., 9 (2005) 135.
[27] Y. Lia, Y. Wua, J. Zhaoa and Y. Pin, J. Inorg. Biochem., 101 (2007) 283.
[28] X. L. Wang, H. Chao, L. Hong, X. L. Hong, J. L. Nian and L. X. Yuan,
J. Inorg. Biochem., 98 (2004) 423.

34
[29] J. A. Cowan, Curr. Opin. Chem. Biol., 5 (6) (2001) 634.
[30] V. Milacic, D. Chen, L. Ronconi, K.R. Landis-Piwowar, D. Fregona and
Q.P. Dou, Cancer. Res., 66 (2006) 10478.
[31] I. Kostova, Anticancer Agents Med. Chem., 6 (2006) 19.
[32] B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature., 222
(1969) 385.
[33] E. R. Jamieson and S. J. Lippard, Chem. Rev., 99 (1999) 2467.
[34] K. B. Lee, D. Wang, S. J. Lippard and P. A. Sharp, Proc. Natl. Acad. Sci.,
99 (2002) 4239.
[35] A. Y. Louie and T. J. Meade, Chem. Rev., 99 (1999) 2711.
[36] W. A. Volkter and T. J. Hoffman, Chem. Rev., 99 (1999) 2269.
[37] B. Wong, J. E. Masse, Y. M. Yen, P. Giannikoupolous, J. Feigon and
R. C. Johnson, Biochem., 41(2002) 5404.
[38] D. Chen, V. Milacic, M. Frezza and Q. P. Dou, Curr. Pharm. Des., 15
(2009) 777.
[39] L. Au, D. Zheng, F. Zhou, Z. Y. Li, X. Li and Y. Xia, ACS Nano., 2 (2008)
1645.
[40] L. Messori, F. Abbate, G. Marcon, P. Orioli, M. Fontani, E. Mini and
S. Carroti, J. Med. Chem., 43 (2000) 3541.
[41] Y. Zheng and L. Sanche, Radiat. Res., 172 (2009) 114.
[42] D. R. Green and J. C. Reed, Science., 281 (1998) 1309.
[43] Y. Ma, L. Cao, T. Kavabata, T. Yoshino, B.B. Yang and S. Okada, Free.
Radic. Biol. Med., 25 (1998) 568.

35
[44] F. Liang, C. Wu, H. Lin, T. Li, D. Gao, Z. Li, J. Wei, C. Zheng and
M. Sun, Bioorg. Med. Chem. Lett., 13 (2003) 2469.
[45] J. Easmon, G. Purstinger, G. Heinisch, T. Roth, H.H. Fiebig, W. Holzer,
W. Jager, M. Jenny and J. Hofmann, J. Med. Chem., 44 (2001) 2164.
[46] K. I. Ansari, J. D. Grant, S. Kasiri, G. Woldemariam, B. Shrestha
and S. S. Mandal, J. Inorg. Biochem., 103 (2009) 818.
[47] M. J. Clarke, Coord. Chem., 236 (2003) 299.
[48] S. Bassetti, J. Hu, Jr. R. B. Agostino and R. J. Sherertz, Antimicrob. Agent.
Chemother., 45 (2001) 1535.
[49] P. M. Lorisean, D. G. Carciunescu, J. C. D. Villarejo, G.C. Fombona
and P. Gayyal, Trop. Med. Parasito., 43 (1992) 110.
[50] H. Li and Z. M. Qian, Med. Res. Rev., 22 (2002) 225.
[51] P. Failli, D. Bani, A. Bencini, M. Cantore, C. Di, L. Mannelli,
C. Ghelardini, C. Giorgi, F. Rugi, A. Spepi, R. Udisti and B. Valtancoli,
J. Med. Chem., 52 (2009) 7273.
[52] M. V. Angelusiu, G. L. Almajan, T. Rosu, M. Negoiu, E. R. Almajan and
J. Roy, Eur. J. Med. Chem., 44 (2009) 3323.
[53] B. Halova-Lajoie, V. Brumas, M. M. Fiallo and G. Berthon, J. Inorg.
Biochem., 100 (2006) 362.
[54] J. J. Haddad, Mol. Immunol., 47 (2009) 205.
[55] H. P. Zhang, H. Zhou, Z. Q. Pan, X. G. Meng and Y. Song, Transition.
Met. Chem., 33 (2008) 55.
[56] A. K. Panda, H. Mohanty and D. C. Dash, Indian. J. Chem., 34A (1995)
911.

36
[57] M. Liu, M. Wang, T. Zhang and H. Sun, Coord. Chem. Rev., 248
(2004)147.
[58] P. Tamilselvi and M. Palaniandavar, Inorg. Chim. Acta., 337 (2002) 420.
[59] B. Dede, F. Karipcin and M. Cengiz, J. Chem. Sci., 121 (2009) 163.
[60] E. B. Hart, H. Steenback and J. Waddell, J. Biol. Chem., 77 (1928) 797.
[61] J. R. J. Sorenson, Med Bio. 63 (1985) 40.
[62] E. D. Harris, "Trace Elements in Health." (ed. J. Rose), Butterworths,
London. (1983).
[63] C. J. Gubler, M. E. Lahey and M. E. Cartwright, J. Clin. Invest., 32 (1953)
405.
[64] N. W. Solomons, J. Am. Coll. Nutr., 4 (1985) 83.
[65] N. W. Solomons and L. H. Allen, Nutr Rev., 41 (1983) 33.
[66] S. N. Gacheru, P. C. Trackman and M. A. Shah, J. Biol. Chem., 265 (1990)
19022.
[67] E. L. Hegg, S. H. Mortimore, C. L. Cheung, J. E. Huyett, D. R. Powell and
J. N. Burstyn, Inorg. Chem., 38 (1999) 2961.
[68] K. A. Deal, A. C. Hengge and J. N. Burstyn, J. Am. Chem. Soc., 118
(1996) 1713.
[69] A. Sigel, H. Sigel and R. K. O. Sigel, Nickel and its Surprising Impact in
Nature, Metal Ions in Life Sciences, Wiley (2008).
[70] G. Jaouen, Bioorganometallics: Biomolecules, Labeling, Medicine. Wiley-
VCH: Weinheim, (2006).
[71] R. K. Szilagyi, P. A. Bryngelson, M. J. Maroney, B. Hedman,
K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 126 (2004) 3018.

37
[72] P. J. Thornalley, Biochem. Soc. Transactions., 31 (2003) 1343.
[73] D. L. Eskew, R. M. Welch and E. E. Cary, Science., 222 (1983) 621.
[74] P. H. Brown, R. M. Welch and E. E. Cary, Plant. Physiol., 85 (1987) 801.
[75] J. G. Muller, X. Chen, A.C. Dadiz, S. E. Rokita and C. J. Burrows,
J. Am. Chem. Soc., 114 (1992) 6407.
[76] S. S. Matkar, L. A. Wrischnik, P. R. Jones and U. H. Blumberg, Biochem.
Biophys. Res. Commun., 343 (2006) 754.
[77] H. Lu, X. Shi, M. Costa and C. Huang, Mol. Cell. Biochem., 279 (2005)
45.
[78] M. C. Rodrguez-Arguelles, M. B. Ferrari, F. Biscegli, C. Pellizi, G. Pelosi,
S. Pinelli and M. Sassi, J. Inorg. Biochem., 98 (2004) 313.
[79] S. Karabocek and N. Karabocek, Polyhedron., 17 (1998) 319.
[80] Emsley and John, "Manganese". Nature's Building Blocks: An A-Z Guide
to the Elements. Oxford, UK: Oxford University Press. (2001) 249.
[81] A. Takeda, Brain Research Reviews., 41 (2003) 79.
[82] G. Dismukes and T. Rogier, Encyclop of Inorg. Chem., (2006).
[83] V.L. Pecoraro (Ed.), Manganese Redox Enzymes, VCH, New York, 1992.
[84] J. Stube, J. Biol. Chem., 265 (1990) 5329.
[85] J. B. Lynch, C. J. Garcia, E. Munck and L. Que, J. Biol. Chem., 264 (1989)
8091.
[86] P. Nordlund, B. M. S. Berg and H. Eklund, Nature., 345 (1990) 593.
[87] M. Atta, P. Nordlund, A. Aberg, H. Eklund and M. Fontecave, J. Biol.
Chem., 267 (1992) 682.

38
[88] R. Cammack, A. Chapman, L. Wei-Ping, A. Katagouni and D.P. Kelly,
FEBS Lett., 253 (1989) 239.
[89] M. L. Ludwig, A. L. Metzger, K. A. Pattridge and W. C. Stallings, J. Mol.
Biol., 219 (1991) 335.
[90] H. Wariishi and M. H. Gold, J. Biol. Chem. 265 (1990) 2070.
[91] H. Wariishi, H. B. Dunford, I. D. MacDonald and M. H. Gold, J. Biol.
Chem., 264 (1989) 3335.
[92] Y. Kono and I. Fridovich, J. Biol. Chem., 258 (1983) 13646.
[93] G. S. Waldo, R. M. Fronko and J. E. Penner-Hahn, Biochemistry., 30
(1991) 10486.
[94] R. M. Fronko, J. E. Penner-Hahn and C. J. Bender, J. Am. Chem. Soc., 110
(1988) 7554.
[95] D. R. Gamelin, M. L. Kirk, T. L. Stemmler, S. Pal, W. H. Armstrong,
J. E. Penner-Hahn and E. I.Solomon, J. Am. Chem. Soc., 116 (1994) 2392.
[96] J. Messinger, J. H. A. Nugent and M. C. W. Evans, Biochemistry., 36
(1997) 11055.
[97] V. L. Pecoraro, M. J. Baldwin and A. Gelaso, Chem. Rev. 94 (1994) 807.
[98] L. Que and A. E. True, Prog. Inorg. Chem., 38 (1990) 97.
[99] W. Wernsdorfer, N. E. Chakov and G. Christou, Phys. Rev., B 70 (2004)
1324.
[100] T. G. Carrell, A. M. Tyryshkin and G. C. Dismukes, J. Biol. Inorg. Chem.,
7 (2002) 2.
[101] M. Yagi and M. Kaneko, Chem. Rev., 101 (2001) 21.

39
[102] M. Melchier, K. H. Thompson, J. M. Jong, S. J. Rettig, E. Shuter, V.G. Yuen,
Y. Zhou, J. H. McNeill and C. Orvig, Inorg. Chem., 38 (1999) 2288.
[103] M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 99 (1999) 2511.
[104] E. J. Cambel and S. T. Nguyen, Tetrahedron Lett., 42 (2001) 1221.
[105] A. J. Stemmler and C. T. Burrows, J. Am. Chem. Soc., 121 (1999) 6956.
[106] I. C. Santos, M. Vilas-Boas, M. F. M. Piedade, C. Freire, M.T. Durate and
B. Castro, Polyhedron., 19 (2000) 655.
[107] J. Chakravarthy and S. Bhattacharya, Polyhedron., 13 (1994) 2671.
[108] A. M. Bond, R. Colton and D. R. Mann, Inrog. Chem., 29 (1990) 4665.
[109] A. Basu, T. G. Kasan and N. Y. Sapre, Inorg. Chem., 27 (1998) 4639.
[110] T. J. Meyer and V. Hung, Inrong. Chem., 42 (2003) 8140.
[111] L. S. Lerman, J. Mol. Biol., 3 (1961) 18.
[112] E. L. Hegg and J. N. Burstyn, Coord. Chem. Rev., 173 (1998) 133.
[113] A. M. Pyle, J. K. Barton, Prog Inorg Chem., 38 (1990) 413.
[114] N. Raman, R. Jayamurugan, A. Sakthivel and L. Mitu, Spectrochim. Acta. A.,
75 (2010) 97.
[115] A. Sigel, H. Sigel, Metal ions in biological systems, vol. 32, Marcel
Dekker, New York, 1996.
[116] N. Saglam, A. Colak, K. Serbest, S. Dulger, S. Guner, S. Karabocek and
A. O. Belduz, Biometals., 15 (2002) 357.
[117] B. C. Baguley and M. LeBert, Biochemistry., 23 (1984) 937.
[118] S. Deepalatha, P. Sambasiva Rao and R. Venkatesan, Spectrochim. Acta. A.,
64 (2006) 187.
[119] J. K. Barton, J. Am. Chem. Soc., 108 (1986) 2081.

40
[120] A. M. Pyel, J. Am. Chem. Soc., 111 (1989) 3051.
[121] N. Raman, J. D. Raja and R. Sakthivel, J. Chem. Sci., 119 (2007) 303.
[122] Wilson and Gisvold, Text Book of Organic Medicinal and Pharmaceutical
Chemistry, 9th Edn., J. B. Lippin Colt Co, 1 (1991).
[123] A. Imai and E. F. Cloyna, Wat. Res., 24 (1990) 1143.
[124] L. Mishra, A. Jha and A. K. Yadav, Trans. Met. Chem., 22 (1997) 406.
[125] G. Manaussakis, C. Bolos, L. Ecateriniatatou and C. J. Sarris, Med. Chem.,
22 (1987) 421.
[126] R. K. Parashar, R. C. Sharma, A. Kumar and G. Mohan, Inorg. Chim. Acta.,
151 (1988) 201.
[127] M. G. Bhowon, H. L. K. Wah and R. Narain, Polyhedron., 18 (1998) 341.
[128] S. G. Clarkson and F. Basolo, Inorg. Chem., 12 (1975) 1528.
[129] V. P. Sing, R. V. Sing and J. P. Tandon, Main Group. Met. Chem., 13 (1990)
135.
[130] M. Mohan, P. Sharma, M. Kumar and N. K. Jha, Inorg. Chim. Acta., 9
(1986) 125.

41
1.12. LITERATURE SURVEY
Kilic et al1
have reported the properties of the new Mn(II)–Co(II)–Mn(II)-type
hetero-trinuclear oxime metal complexes with N4 and N4O2 ligands. The metal
complexes were identified by combination of FT-IR spectra, UV–Vis spectra,
magnetic susceptibility measurements, mass spectra, X-ray powder diffraction
measurements, elemental analysis, molar conductivity measurements and their
morphology studied by SEM measurements. The aim of his study was to prepare
and characterize new different trinuclear metal–oxime complexes and then to
understand the structural properties of the coordination of different groups.
Zhang et al2 reported the synthesis, crystal structure and magnetic
characterization of a novel linear trinuclear copper(II) complex bridged by
phenoxy and benzyloxy oxygen atoms. The crystal structure of the complex
contains a linear trinuclear array of copper(II) ions in which the central copper(II)
ion is in an octahedron coordination sphere and lies on an inversion center of the
molecule, the terminal ones are in an identical square pyramid structure. Variable-
temperature magnetic data indicate that the complex displays a strong
antiferromagnetic coupling with J = −270(8) cm-1 between the metal ions.

42
Dede et al3
worked on the synthesis, characterization and extraction
studies of N,N‟‟-bis[1-biphenyl-2-hydroxyimino-2-(4-acetylanilino)-1-ethylidene]-
diamines and their homo-and heteronuclear copper(II) complexes. The homodi-,
homotrinuclear and heterodinuclear copper(II) perchlorate complexes of
tetradentate Schiff bases which possess N4 donor sets derived from the
condensation of 4-(arylaminoisonitrosoacetyl)biphenyl and diamine derivatives
were synthesized and characterized. Elemental analysis, FT–IR, ESR, molar
conductivity, magnetic moment measurements and thermal analyses studies were
utilized for the investigation of the complexes. Elemental analyses, stoichiometric
and spectroscopic data of the metal complexes indicated that the metal:ligand ratio
of dinuclear copper(II) complexes were found to be 2 : 1 while this ratio was 3 : 2
in trinuclear copper(II) complexes and the metal complexes indicated that the
metal ions are coordinated to the oxime and imine nitrogen atoms.
Saglam et al4 explored the oxidative cleavage of DNA by homo- and
heteronuclear Cu(II)-Mn(II) complexes of an oxime-type ligand. The novel
homodinuclear, heterodinuclear and homotrinuclear complexes with a novel
oxime-type ligand were prepared and their nucleolytic activities on pCYTEXP

43
were established by neutral agarose gel electrophoresis. The analyses of the
cleavage products obtained electrophoretically indicate that although the examined
complexes induces very similar conformational changes on supercoiled DNA by
converting supercoiled form to nicked form than linear form in a sequential
manner as the complex concentration or reaction period is increased.
Karabocek et al5 have reported the synthesis and structural studies of
(2E,3E)-3-[(6-{[(1E,2E)-2-(hydroxyimino)-1-methylpropylidene]amino}pyridin-
2yl)imino] butan-2-one oxime, ligand and its mono-, di- and trinuclear copper(II)
complexes. The dioxime ligand (H2Pymdo) and its copper(II) complexes were
characterized by 1H NMR, 13C NMR, elemental analyses, magnetic moments, IR
and mass spectral studies. The mononuclear copper(II) complex of H2Pymdo was
found to have a 1:1 metal:ligand ratio. Elemental analyses, stoichiometric and
spectroscopic data of the metal complexes indicated that the metal ions are
coordinated to the oxime and imine nitrogen atoms (C=N). In the dinuclear
complexes, in which the first Cu(II) ion was complexed with nitrogen atoms of the
oxime and imine groups, the second Cu(II) ion is ligated with dianionic oxygen

44
atoms of the oxime groups and are linked to the 1,10-phenanthroline nitrogen
atoms. The trinuclear copper(II) complex was formed by coordination of the third
Cu(II) ion with dianionic oxygen atoms of each of two molecules of the
mononuclear copper(II) complexes.
Yu et al6 identified the crystal structure of trinuclear nickel(II) complex
with Schiff base Ligand of N,N‟-Bis(Salicylidene)-1,3-Diiminopropane. All the
complexes were synthesized and characterized by elemental analysis,
thermogravimetric analysis, IR spectroscopy and X-ray diffraction techniques. All
the Ni(II) ions are coordinated in distorted octahedral geometry.
Deepalatha et al7 have reported the synthesis, physico-chemical and DNA
interaction studies of homo- and hetero-trinuclear complexes. Synthesis and

45
characterization of three new trinuclear metal complexes of type Cu3, Cu2Zn and
Cu2Ni was achieved by assembling simple mononuclear complexes, namely
2,2-bipyridyl 3,4-dihydroxo benzaldehyde copper(II) complex and
diethylenetriamine complexes of copper(II), nickel(II) and zinc(II) ions, through
the reaction of coordinated ligands. The FAB mass spectra for the complexes
showed fragmentation pattern in accordance with the molecular formula. The
frozen electron paramagnetic resonance spectrum of trinuclear copper complex
shows two sets of parallel lines with approximately 2:1 ratio. The simulation has
been carried out by considering dipolar interaction between the two types of
copper ions present in the complex. The trimetallic complexes, Cu3, Cu2Ni and
Cu2Zn show strong intercalation type of interaction with calf thymus DNA in
0.02 mol L-1
of phosphate buffer containing 60 mmol sodium chloride at pH 7.0 at
room temperature. The binding constant is found to be in the order Cu3
<Cu2Zn<Cu2Ni. The enhanced binding capability of Cu2Ni complex is attributed to
the increased symmetry in overall structure of the complex and the pronounced
binding character of positively charged Ni(II) ions with the purines.

46
3
Ray et al8 found three new mono-di-trinuclear cobalt complexes of
selectively and non-selectively condensed Schiff bases with N2O and N2O2 donor
sets: Syntheses, structural variations, EPR and DNA binding studies. In all the
three complexes psuedohalides (NCO− and N −) have been incorporated to
generate structural variation. Of the three Schiff bases, HL1 and HL2 were
obtained by selective condensation of two different 1,3-diamines with
2-hydroxyacetophenone and H2L3
resulted from non-selective condensation of
1,3-diamine with 2-hydroxyacetophenone. Only one –NH2 functionality of
1,3-diaminopropane and 1,3-diaminopentane was selectively condensed with
2-hydroxyacetophenone to generate Schiff bases HL1 and HL2, respectively. H2L3
was obtained by condensing both the amine functionality of 1,3-diaminopropane
with 2-hydroxyacetophenone. Therefore HL1
and HL2
behave as N2O donors,
whereas H2L3 provides a N2O2 donor coordination environment for the cobalt ions
in the respective complexes. The complexes have been characterised using IR, UV–
Vis spectroscopy and cyclic voltammetry. Structural aspects of the complexes have
been described by performing single crystal X-ray analysis. EPR analyses and DNA
binding abilities of all three cobalt complexes were studied in detail.
Jiang et al9 identified one trinuclear copper(II) complex derived from a new
Schiff base ligand based on the dianion of 4-chloro-6-(hydroxymethyl)-2-((3-
aminopropylimino)methyl)-phenol: Synthesis, structure, spectroscopic and
magnetic properties. A linear trinuclear copper(II) complex, prepared from a new
Schiff base ligand, namely the dianion of 4-chloro-6-(hydroxymethyl)-2-((3-
aminopropylimino)methyl)-phenol, was synthesized and characterized. The X-ray
structural study reveals that the geometry of the central Cu(II) ion is elongated

47
octahedral and that of the two side Cu(II) ions is distorted square pyramidal. The
magnetic susceptibility measurements from 2 to 300 K revealed a medium
antiferromagnetic interactions between the Cu(II) ions.
Bian et al10
have reported the synthesis, X-ray structure and magnetic
properties of trinuclear copper(II) tridentate Schiff base complexes containing a
partial cubane Cu3O4 core. Two copper(II) complexes with tridentate Schiff bases
AE and SE, which are condensed from N,N-dimethylethylenediamine and
acetylacetone or salicylaldehyde, respectively, was synthesized and characterized
by X-ray structural analysis, magnetic measurement, IR and UV spectra. Both of
the complexes contain a partial cubane Cu3O4 core consisting of [Cu(AE)] or
[Cu(SE)], nonbonded ClO4−
anions and water molecules.
Asada et al11 have reported the preparations and structures of trinuclear
manganese(II) complexes with N-2-pyridiylmethylidene-2-hydroxy-5-substituted
phenylamine. In the molecular structures of these complexes, two terminal
manganese ions are coordinated with one oxygen and two nitrogen atoms of
ligand, two oxygen atoms of OAc− and a solvent molecule, to form a distorted

48
octahedral structure where the central manganese ion resides on a center of
symmetry and is surrounded by an O6 donor set of four oxygen atoms from four
bridging OAc− and two phenolic oxygen atoms of two ligands.
Liu et al12 studied the synthesis, structural characterization and magnetic
properties of new tripeptide Schiff base heterotrinuclear Cu(II)–M(II)–Cu(II)
(M = Ni and Mn) complexes. Two novel heterotrinuclear complexes,
[Ni(H2O)4(CuL)2]7H2O (1), [Mn(H2O)4(CuL)2] 8H2O (2), have been obtained by
the self-assembly of the mononuclear tripeptide Schiff base complexes [CuL] with
bivalent metal ion M2+, where H3L=N-salicylideneglycylglycylglycine. Single
crystal X-ray diffraction experiments indicate that the structures both 1 and 2, are
made up of centrosymmetric trinuclear units with the M atom lying on an inversion
center, formed the 1D supramolecular chain structures via hydrogen bondings and
M----O weak interactions. The magnetic susceptibility data (2–300 K) revealed
antiferromagnetic interactions between copper(II) ions and the central metal.
Neelakantan et al13 worked on the spectral characterization, cyclic
voltammetry, morphology, biological activities and DNA cleaving studies of
amino acid Schiff base metal(II) complexes. Metal complexes are synthesized with
Schiff bases derived from o-phthalaldehyde (opa) and amino acids viz., glycine,
l-alanine, l-phenylalanine. Metal ions coordinate in a tetradentate or hexadentate
manner with these N2O2 donor ligands, which are characterized by elemental
analysis, molar conductance, magnetic moments, IR, electronic, 1H NMR and EPR
spectral studies. The biological activity of the complexes has been tested on eight
bacteria and three fungi. Cu(II) and Ni(II) complexes showed an increased activity

49
in comparison to the controls. The metal complexes of opapal Schiff base were
evaluated for their DNA cleaving activities with calf-thymus DNA under aerobic
conditions. Cu(II) and VO(II) complexes show more pronounced activity in
presence of an oxidant.
Leelavathy et al14 have reported the synthesis and characterization of a
new series of an unsymmetrical macrocyclic binuclearvanadyl complexes:
Electrochemical, antimicrobial, DNA binding and cleavage studies. A new series
of an unsymmetrical macrocyclic binuclear bis-phenoxo bridged
oxidovanadium(IV) complexes have been synthesized and characterized by
elemental and spectral techniques. These complexes significantly promote the
oxidative cleavage of supercoiled plasmid DNA under physiological conditions in
the presence of H2O2. All the complexes show noticeable growth inhibition of
some plant pathogenic fungal species and human pathogenic bacterial species.
Karipcin et al15 synthesized, characterized and studied the properties of
heteronuclear ruthenium(III) complexes of N,N‟-o-bis[1-p-diphenylmethane-2-
(arylamine)-1-ethiliden]-phenylenediamine quadridentate ligands. In this study,
they synthesized the amine compounds (1-p-diphenylmethane-2-hydroxyimino-2-
(1-arylamino)-1-ethanones) [HL1 and HL2] as the starting material, and then
quadridentate tetraaza Schiff base ligands [H2L3 and H2L4] were prepared from the
reactions of the amine compounds with 1,2-phenylenediamine. A new series of
mononuclear Ru(III), heterodinuclear Ru(III)–Mn(II), Ru(III)–Ni(II), Ru(III)–
Cu(II) and heterotrinuclear Ru(III)–Cu(II)–Ru(III) complexes were synthesized.

50
The complexes were characterized by elemental analysis, molar conductivity, IR,
ESR, ICP-OES, magnetic moment measurements and thermal analyses studies.
Mathur et al16 reported new homodinuclear complexes of the type
[LM2IICl2], heterotrinuclear complexes of the type [LM2
II SnIV Cl6] where M= CuII,
MnII, CoII, NiII and CuII and NiII , respectively which were synthesized and
characterized by elemental analysis and various spectroscopic techniques. The
homodinuclear complexes possess two different environments (N2 and N2O2 donor
sets) for holding the metal ions. The metal ion in N2 set exhibits square planar
geometry with two chloride ions in the inner sphere but rhombic structure is found
in tetradentate N2O2 Schiff base cavity while in heterotrinuclear complexes SnIV
atom is in the octahedral environment. The interaction of complexes with calf
thymus DNA was carried out by absorption spectroscopy and cyclic voltammetry.
Fluorescence studies along with viscosity measurements were also checked to
authenticate the binding of metal complexes with DNA.
Sreedaran et al17 have reported the N-functionalized cyclam based
trinuclear copper(II) complexes: electrochemical, magnetic, catalytic and
antimicrobial studies. A series of trinuclear Cu(II) complexes have been prepared
by Schiff base condensation of 1,8-[bis-(3-formyl-2-hydroxy-5-methyl)benzyl]-

51
l,4,8,11- tetraazacyclotetradecane and 1,8-[bis(3-formyl-2-hydroxy-5-bromo)-
benzyl]-l,4,8,11-tetraazacyclotetradecane with aromatic and aliphatic diamines,
Cu(II) perchlorate and triethylamine. The complexes were characterized by
elemental and spectroscopic analysis. All the complexes were screened for
antifungal and antibacterial activity.
Durmus et al18 explored the thermal decomposition of some linear
trinuclear schiff base complexes with acetate bridges. Ni(II)–M(II)–Ni(II) nuclear
structured complexes were prepared from N,N‟-bis(salicylidene)-1,3-
propanediamine (LH2) and its derivatives N,N‟-bis(salicylidene)-2,2‟-dimethyl-
1,3-propanediamine (LDMH2) and N,N‟-bis(salicylidene)-2-hydroxy-1,3-
propanediamine (LOH3), where M represents one of the following metal ions;
Mn(II), Co(II), Ni(II), Cu(II), Zn(II), Cd(II). Two different µ-bridges are found
between the metal nucleuses of the complexes. The phenolic oxygens and acetate
ions tend to form µ-bridges between the terminal Ni(II) ions and central metal(II)
ion. The coordinatively bonded DMF molecules, in the complexes, were observed
to abandon the structure between 160–180°C. Further heating resulted primarily in
the thermal decomposition of the complexes above 310°C, whereas metal oxide
residue mixtures were observed above 650°C.

52
Dulger et al19 studied the DNA cleavage by homo- and heterotetranuclear
Cu(II) and Mn(II) complexes with tetrathioether-tetrathiol moiety. Novel
homotetranuclear Cu(II) and heteronuclear Cu(II)-Mn(II) complexes with
tetrathioether-tetrathiol moiety has been prepared and their DNA relaxation
activities with plasmid pCYTEXP were electrophoretically established. The
cleavage products analyzed by neutral agarose gel electrophoresis indicated that
the interaction of the metal complexes with supercoiled plasmid DNA yielded
linear, nicked or degraded DNA. The relaxation activities of both homo- and
heterotetranuclear complexes are time and concentration dependent. His findings
suggest that a homo and heterotetranuclear complexes with potent nucleolytic
activity is a good nuclease substitute in the presence of cooxidant.
Mathur et al20 have reported a template synthesis of novel carboxamide
dinuclear copper(II) complex: spectral characterization and reactivity towards calf
thymus DNA. Dinuclear complexes Bis [aqua 1,8-(1,2-dicarboxamido benzene)

53
3,6-diazaoctane copper (II)/nickel (II)] tetrachloride (1 and 2) were synthesized by
a two component one-pot metal template condensation between phthalic anhydride
and 1,8-diamino 3,6-diazaoctane. Elemental analysis, molar conductance
measurements, electronic absorption, infra-red, electron paramagnetic resonance,
nuclear magnetic resonance, atomic absorption and electron spray mass spectral
studies have been performed to probe the nature and structure of the complexes.
The interaction of copper (II) complex with calf thymus has been studied by using
absorption, emission and circular dichoric spectral methods, viscometry and cyclic
voltammetry.
Prabahkara et al21 have studied the binding and photocleavage of DNA by
mixed ligand Co(III) and Ni(II) complexes of thiophene[2, 3-b] quinoline and
phenanthrolie/bipyridine. In order to systematically perform an experimental and
theoretical study on DNA binding and photocleavage properties of transition metal
complexes of the type [M(L)2(L1)](PF6)n xH2O (where M = Co(III) or Ni(II),
L = 1,10-phenanthroline or 2.2‟ bipryidine, L1 = Thiophene [2,3-b] quinoline (qt),
n = 3 or 2 and x = 5 or 2) have been synthesized and characterized by elemental
analysis, IR, 1H NMR, UV and magnetic susceptibility data. The DNA-binding
properties of these complexes have been investigated with UV-Vis, viscosity
measurements, thermal denaturation and cyclic voltametric studies. The
photocleavage studies with pUC19 DNA shows that all these complexes promoted
the conversion of SC form to NC form in absence of „inhibitors‟.
Liu et al22
have reported synthesis, DNA-binding and cleavage studies of
macrocyclic copper(II) complexes. Two macrocyclic copper(II) complexes have

54
been prepared and characterized by elemental analysis, UV–Vis., IR and mass
spectra. Absorption, fluorescence, circular dichroic spectra and viscosity
experiments have been carried out on the interaction of the two complexes with
calf thymus DNA. The results suggest that both complexes can bind to CT DNA
by intercalation via the aromatic moiety ring in the macrocycle into the base pairs
of DNA.
Mukherjee et al23
have reported three di-Schiff-base ligands, N,N‟-
bis(salicylidene)-1,3-propanediamine (H2Salpn), N,N‟-bis(salicylidene)-1,3-
pentanediamine (H2Salpen) and N,N‟-bis(salicylidine)-ethylenediamine (H2Salen)
react with Ni(SCN)2 4H2O in 2:3 molar ratios to form the complexes; mononuclear
[Ni(HSalpn)(NCS)(H2O)]H2O, trinuclear [{Ni(Salpen)}2Ni(NCS)2] and trinuclear
[{Ni(Salen)}2Ni(NCS)2] respectively. All the complexes have been characterized
by elemental analyses, IR and UV–VIS spectra, and room temperature magnetic
susceptibility measurements.

55
Maity et al24 have reported a novel trinuclear nickel(II) complex of an
unsymmetrical tetradentate ligand involving bridging oxime and acetylacetone
functions. The trinuclear nickel(II) complex, [Ni3(L)2(H2O)2](ClO4)2, where L is a
bridging unsymmetrical tetradentate ligand, involving o-phenylenediamine,
diacetyl monoxime and acetylacetone (H2L = 4-[2-(3-hydroxy-1-methyl-but-2-
enylideneamino)-phenylimino]-pentan-2-one oxime) has been synthesized and
characterized structurally. In the complex, an octahedral Ni(II) centre is held in the
middle by two square planar units with the aid of oxime and ketonic bridges.
Refat et al25 have reported Synthesis, infrared spectra and thermal
studies of Zn(II), Cd(II) and Hg(II) complexes with 2-aminobenzaldehyde
phenylhydrazone “nitrin” ligand. The nitrin, 2-aminobenzaldehyde
phenylhydrazone was synthesized by refluxing 2-nitrobenzaldehyde with
phenylhydrazine in ethanolic solvent. Three transition metal (II) complexes have
been prepared. Elemental analysis, molar conductivity, IR, UV, 1H NMR and mass
spectra, as well as TG/DTG have been used to characterize these complexes. The
ligand and its complexes have been studied for their possible biological activity
including antibacterial and antifungal activity.

56
Kwiatkowski et al26 have reported Nickel(II) and palladium(II) complexes
with singly condensed diprimary triamines and 2-aminobenzaldehyde. Six new
nickel(II) and one palladium(II) complexes are obtained by complexation of
unsymmetrical Schiff bases from 1:1 condensation of 2-aminobenzaldehyde with
1,5-diamino-3-azapentane, 1,7-diamino-4-azaheptane and 1,7-diamino-4-methyl-4-
azaheptane.
Dragancea et al27
have reported a Bis(o-aminobenzaldehyde)
thiocarbohydrazone (HL) forms with copper(II) nitrate a tetranuclear complex
[Cu2(L)(NO3)3]2 2H2O, in which two dinuclear units are joined by nitrate bridges.
The dihydrazone ligand behaves ditopically, providing NNS and NNN binding
sites, with the four coppers essentially in a squarepyramidal geometry. The
tetranuclear molecule displays intramolecular magnetic interactions, with the
antiferromagnetic exchange between the copper(II) ions within each dinuclear
moiety dominant over weak interdimer ferromagnetic coupling.
Abdallah et al28 have reported spectroscopic study of molecular structures
of novel Schiff base derived from o-phthaldehyde and 2-aminophenol and its
coordination compounds together with their biological activity. New Schiff base
(H2L) ligand is prepared via condensation of o-phthaldehyde and 2-aminophenol.
The metal complexes of Cr(III), Mn(II), Fe(II), Fe(III), Co(II), Ni(II), Cu(II) and
Zn(II) with the ligand are prepared in good yield from the reaction of the ligand
with the corresponding metal salts. They are characterized based on elemental
analyses, IR, solid reflectance, magnetic moment, electron spin resonance (ESR),
molar conductance, 1H NMR and thermal analysis (TGA).

57
Singh et al29 have reported synthesis and spectroscopic studies of
biologically active compounds derived from oxalyldihydrazide and benzil, and
their Cr(III), Fe(III) and Mn(III) complexes. A new series of complexes have been
synthesized by template condensation of oxalyldihydrazide and benzil in
methanolic medium in the presence of trivalent chromium, manganese and iron
salts forming complexes of the type [M(C32H24N8O4)X]X2 where M = Cr(III),
Mn(III), Fe(III) and X = Cl−1
, NO3 −1, CH3COO−1. The complexes have been
characterized with the help of elemental analyses, conductance measurements,
magnetic susceptibility measurements, electronic, NMR, infrared and far infrared
spectral studies. On the basis of these studies, a five coordinate square pyramidal
geometry has been proposed for all these complexes. The biological activities of
the metal complexes have been tested in vitro against a number of pathogenic
bacteria to assess their inhibiting potential. Some of these complexes have been
found to exhibit remarkable antibacterial activities.
Shebl et al30 have reported synthesis, spectroscopic characterization and
antimicrobial activity of mono-, bi- and tri-nuclear metal complexes of a new
Schiff base ligand. Condensation of o-acetoacetylphenol and 1,2-diaminopropane
in 1:1 molar ratio under condition of high dilution yielded the mono-condensed
dibasic Schiff base ligand with a N2O2 donors. The mono-condensed ligand has
been used for further condensation with 2-hydroxy-5-nitrobenzaldehyde to obtain
the new asymmetrical dicompartmental Schiff base ligand, H3L, with N2O3 donors.
The structure of the ligand was elucidated by analytical and spectroscopic tools
(IR, 1H and 13C NMR spectra) which indicated that the coordinating sites are
oxygen atoms of the phenolic OH groups, nitrogen atoms of the azomethine groups
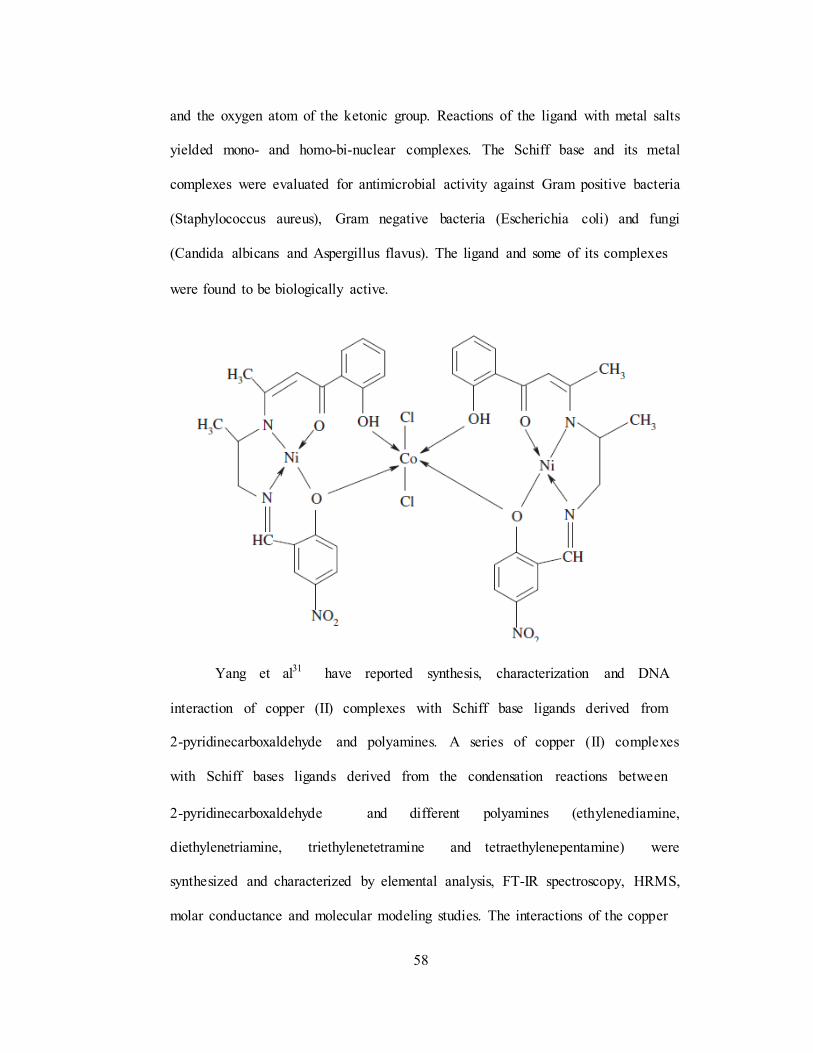
58
and the oxygen atom of the ketonic group. Reactions of the ligand with metal salts
yielded mono- and homo-bi-nuclear complexes. The Schiff base and its metal
complexes were evaluated for antimicrobial activity against Gram positive bacteria
(Staphylococcus aureus), Gram negative bacteria (Escherichia coli) and fungi
(Candida albicans and Aspergillus flavus). The ligand and some of its complexes
were found to be biologically active.
Yang et al31 have reported synthesis, characterization and DNA
interaction of copper (II) complexes with Schiff base ligands derived from
2-pyridinecarboxaldehyde and polyamines. A series of copper (II) complexes
with Schiff bases ligands derived from the condensation reactions between
2-pyridinecarboxaldehyde and different polyamines (ethylenediamine,
diethylenetriamine, triethylenetetramine and tetraethylenepentamine) were
synthesized and characterized by elemental analysis, FT-IR spectroscopy, HRMS,
molar conductance and molecular modeling studies. The interactions of the copper

59
complexes with DNA were investigated by the UV spectra, viscosity
measurements and gel electrophoresis under physiological conditions. The
experimental results indicated that the complexes could bind to DNA via an
intercalative mode and showed a different DNA cleavage activity.
Chaviara et al32 have exhibited DNA interaction studies and evaluation of
biological activity of homo- and hetero-trihalide mononuclear Cu(II) Schiff
base complexes. Quantitative structure – activity relationships. A new series of
mixed-ligand mono- or hetero-trihalide Cu(II) complexes of the type
[Cu(dienXX)Y(YZ2)], where dienXX = Schiff dibase of diethylenetriamine with
2-thiophene-carboxaldehyde (dienSS), 2-furaldehyde (dienOO) or 2-pyrrole-2-
carboxaldehyde (dienNN), Y = Cl, Br and Z = Br, I was synthesized by the
reaction of the precursors of the type [Cu(dienXX)Y]Y with iodine or bromine in
1:1 molar ratio. The distorted square pyramidal configuration of the new homo-
and hetero-trihalide Cu(II) mononuclear complexes was identified by C, H, N, Cu
analysis, spectroscopic methods (IR, UV–visible), molar conductivity and
magnetic measurements. The effect of the new compounds on the single stranded
(ss), double stranded (ds) and pDNA led either to the formation of a DNA-complex
cationic adduct, or to its degradation, evidenced by DNA electrophoretic mobility
and DNA interaction spectroscopic titration studies. Moreover, the antimicrobial
activity of Cu(II) complexes against Gram positive and Gram negative bacteria can
be attributed to the synergistic action of the dissociation species, namely the
cationic [Cu(dienXX)Y]+ and anionic [YZ2]− ones.

60
References
[1] A. Kilic, F. Durap, A. Baysal and M. Durgun, J. Incl. Phenom. Macrocycl.
Chem., 67 (2010) 423.
[2] H. P. Zhang, H. Zhou, Z. Q. Pan, X. G. Meng and Y. Song, Transition.
Met. Chem., 33 (2008) 55.
[3] B. Dede, F. Karipcin and M. Cengiz, J. Chem. Sci., 121 (2009) 163.
[4] N. Saglam, A. Colak, K. Serbest, S. Dulger, S. Guner, S. Karabocek and
A. O. Belduz, Biometals., 15 (2002) 357.
[5] N. Karabocek, A. Armutcu and S. Karabocek, Transition. Met. Chem., 31
(2006) 938.
[6] Z. Yu, M. He, P. Sun, W. Zhang and L. Chang, J. Chem. Crystallogr., 39
(2009) 885.
[7] S. Deepalatha, P. Sambasiva Rao and R. Venkatesan, Spectrochim. Acta.
A., 64 (2006) 187.
[8] A. Ray, G. M. Rosair, R. Kadam and S. Mitra, Polyhedron., 28 (2009) 796.
[9] J. Jiang, Z. Chu and W. Huang, Inorg. Chim. Acta., 362 (2009) 2933.
[10] H. D. Bian, J. Y. Xu, W. Gua, S. P. Yan, P. Cheng, D. Z. Liao and
Z. H. Jiang, Polyhedron., 22 (2003) 2927.
[11] H. Asada, K. Hayashi, S. Negoro, M. Fujiwara and T. Matsushita, Inorg.
Chem. Commun., 6 (2003) 193.
[12] W. L. Liu, Y. Zou, C. L. Ni, Y. Z. Li and Q. J. Meng, J. Mol. Struct., 751
(2005) 1.
[13] M.A. Neelakantan, F. Rusalraj, J. Dharmaraja, S. Johnsonraja,
T. Jeyakumar and M. S. Pillai, Spectrochim. Acta. Part A., 71 (2008) 1599.

61
[14] L. Leelavathy, S. Anbu, M. Kandaswamy, N. Karthikeyan and N. Mohan,
Polyhedron., 28 (2009) 903.
[15] F. Karipcin and G. B. Akdogan, J. Incl. Phenom. Macrocycl. Chem., 64
(2009) 183.
[16] S. Mathur and S. Tabassum, Cent. Eur. J. Chem., 4 (2006) 502.
[17] S. Sreedaran, K. S. Bharathi, A. K. Rahiman, R. Prabu, R. Jagadesh,
N. Raman and V. Narayanan, Transition. Met. Chem., 34 (2009) 33.
[18] S. Durmus, U. Ergun, J. C. Jaud, K. C. Emregul, H. Fuess and O. Atakol,
J. Therm. Anal. Calorim., 86 (2006) 337.
[19] S. Dulger, N. Saglam, A. O. Belduz, S. Guner and S. Karabocek,
Biometals., 13 (2000) 261.
[20] S. Mathur and S. Tabassum, Biometals., 21 (2008) 299.
[21] M. C. Prabahkara and H. S. B. Naik, Biometals., 21 (2008) 675.
[22] J. Liu, T. B. Lu, H. Deng and L. N. Ji, Transition. Met. Chem., 28 (2003)
116.
[23] P. Mukherjee, C. Biswas, M. G. B. Drew and A. Ghosh, Polyhedron., 26
(2007) 3121.
[24] D. Maity, P. Mukherjee, A. Ghosh, M. G. B. Drew and G. Mukhopadhyay ,
Inorg. Chim. Acta., 361 (2008) 1515.
[25] M. S. Refat and A. A. Ibrahim, Spectrochim. Acta. A., 70 (2008) 234.
[26] E. Kwiatkowski, G. Romanowski and K. Suwinska, Polyhedron., 21 (2002)
2071.

62
[27] D. Dragancea, A. W. Addison, M. Zeller, M. E. Foster, M. J. Prushan,
L. K. Thompson, M. D. Revenco and A. D. Hunter, Inorg. Chim. Acta.,
363 (2010) 2065.
[28] S. M. Abdallaha, G. G. Mohamed, M.A. Zayedb and M. S. Abou El-Ela,
Spectrochim. Acta. Part. A., 73 (2009) 833.
[29] D. P. Singh, R. Kumar and J. Singh, Eur. J. Med. Chem., 44 (2009) 1731.
[30] M. Shebl, S. M. E. Khalil, S. A. Ahmed and H. A. A. Medien, J. Mol.
Struct., 980 (2010) 39.
[31] X. B. Yang, Y. Huang, J. S. Zhang, S. K. Yuan and R. Q. Zeng, Inorg.
Chem. Commun., 13 (2010) 1421.
[32] A. T. Chaviara, E. E. Kioseoglou, A. A. Pantazaki, A. C. Tsipis,
P. A. Karipidis, D. A. Kyriakidis and C. A. Bolos, J. Inorg. Biochem., 102
(2008) 1749.

63
1.13. SCOPE AND OBJECTIVES OF THE WORK
The chemistry of multinuclear Schiff base metal complexes, especially of
coupled systems is of special interest in various fields of science. The main reason
probably is due to the phenomenon of interaction between metal centres lies at the
crossover point of two widely separated areas, namely the physics of the magnetic
materials and the role of polynuclear reaction sites in biological processes.
Trinuclear transition metal complexes bridged by polyatomic Schiff base ligands
have gained much attention in the recent years, towards synthesis and
characterization.
The multinuclear copper(II) complexes have aroused extensive interest due
to their importance in biological processes and in inorganic material science. The
metal ion dependent oxidative DNA cleavage by Cu(II) complexes is of topical
interest. Nickel complexes in the presence of oxidants have been extensively used
for DNA cleavage reactions. Manganese plays an essential and versatile role in the
biochemistry of many organisms. The chemistry of manganese in various
oxidation states is currently receiving much attention owing to its importance in
many enzymatic systems such as superoxide dismutase and catalase. The vanadium
ions can play a role in biology as counter ions for protein, DNA, RNA and in
various biological organelles.
Hence the present work described in the thesis concerns with the reactions
of some of the Cu(II), Ni(II), VO(II) and Mn(II) complexes with p-phenylenediamine,
o-phthalaldehyde/2,3-pentanedione/benzil and 2-aminobenzaldehyde. The new homo

64
and heterotrinuclear Schiff base complexes formed with N4 donor sets. The structures
of these complexes have been investigated by using various physico chemical
methods.
The synthesised complexes have been effectively used as DNA cleavage
studies. The cleavage of nucleic acids may be considered as an enzymatic reaction
which comprises of various biological processes as well as the biotechnological
manipulation of genetic material. The application of artificial DNA cleaving agents
is manifold: biotechnology, structural studies of nucleic acids or development of
new drug.
Some of the complexes are used for DNA binding studies. These complexes
have effectively bind to CT DNA. DNA binding is the critical step for DNA
activity. To design effective chemotherapeutic agents and better anticancer drugs,
it is essential to explore the interactions of metal complexes with DNA. Recently,
it has been tremendous interest in studies related to the interaction of transition
metal ions with nucleic acid because of their relevance in the development of new
reagents for biotechnology and medicine. The antibacterial properties of the
complexes have also been screened against some pathogenic bacteria.
Objectives of the study
1. Template Synthesis of mononuclear Cu(II), Ni(II) and Mn(II) Schiff base
complexes derived from p-phenylenediamine and o-phthalaldehyde (or)
2,3-pentanedione (or) benzil.
2. Synthesis of homo and heterotrinuclear Schiff base metal complexes by
conventional methodology as given in the literature. The mononuclear

65
complex has been used as a ligand for further condensation with
2-aminobenzaldehyde and also react with metal salts to obtain the homo
and heterotrinuclear Schiff base metal complexes.
3. To characterize the homo and heterotrinuclear Schiff base metal complexes
using various analytical and spectral techniques such as elemental analysis,
molar conductance, FT-IR, UV-Visible spectroscopy, electrochemical,
thermal, magnetic and EPR studies. Several attempts to isolate crystals
suitable for X-ray diffraction were unsuccessful.
4. DNA binding Studies
The interaction of the homo and heterotrinuclear Schiff base metal
complexes with calf thymus DNA (CT-DNA) were studied using
absorption spectra, cyclic voltammetry and viscosity measurements.
5. DNA Cleavage studies
The cleavage study was monitored by gel electrophoresis method.
6. Antimicrobial activity
The in vitro biological screening effects of the synthesised compounds
were tested against the some Gram positive and Gram negative bacteria by
the well diffusion method.

66
1.14. EXPERIM ENTAL METHODS AND ANALYTICAL TECHNIQUES
This present chapter deals with the chemicals used, purification of the
solvents, synthesis of the compounds and their analytical and instrumental
techniques used in the determination of the stoichiometry of the complexes
prepared.
1.14.1. Analytical Reagents
The main reagents used are summarized in Table 1.1.
Table.1.1. Reagents used in this thesis
S.No
Product
Provider
1 CuCl2. 2H2O (A.R Loba)
2 NiCl2 6H2O (A.R Loba)
3 Cu(ClO4)2.6H2O (A.R Aldrich)
4 Ni(ClO4)2.6H2O (A.R Loba)
5 VOSO4. H2O (A.R Aldrich)
6 MnCl2. 4H2O (A.R Loba)
7 o-phthalaldehyde (A.R Aldrich)
8 2,3-pentanedione (A.R Aldrich)
9 Benzil (A.R Aldrich)
10 p-phenylenediamine (A.R Loba)
11 2-nitrobenzaldehyde (A.R Loba)
12 Ammonia (A.R Qualigens)
13 Ferrous sulphate heptahydrate (A.R Merck)
14 Sodium hydroxide (A.R Loba)
15 Triethylamine (L.R Qualigens)

67
16 Sodium hydroxide (A.R Loba)
17 Oxalic acid (A.R Loba)
18 Nitric acid (A.R Loba)
19 EDTA (A.R Merck)
20 Eriochrome Black-T (A.R Merck)
21 Ammonium Chloride (L.R SDS)
22 Ammonium Acetate (L.R SDS)
23 Tetrabutylammonium perchlorate (A.R Aldrich)
24 Hydrochloric acid (L.R Loba)
25 Potassium sulphate (L.R Qualigens)
26 Calcium sulphate (L.R Qualigens)
27 Diethyl dithiocarbamate (L.R Qualigens)
28 Bromine water (L.R Loba)
29 Nickelammonium sulphate (L.R Qualigens)
30 Hydroxylamine hydrochloride (A.R Loba)
1.14.2. Solvents
Solvents used in this thesis are gathered in Table 1.2
Table.1.2. Solvents used in this thesis
S.No Solvents Provider
1 Ethanol (A.R Loba)
2 Methanol (A.R Loba)
3 Dimethyl formamide (A.R SDS)
4
Dimethyl sulfoxide
(A.R Merck)
5
Acetonitrile
(A.R Merck)
6
Acetone
(A.R Merck)

68
1.14.2.1. Purification of solvents
Common solvents such as methanol, ethanol and DMF, all of commercial
grade were purified as usual [1]. Ethanol and methanol were purified by the
following procedures [2].
1.14.2.2. Ethanol
Commercial ethanol was distilled, collecting the middle fraction (80%). It
was then refluxed over ignited quicklime for 4 hours and again distilled at
atmospheric pressure, when 98% ethanol was obtained.
1.14.2.3. Methanol
The commercial grade methanol was refluxed for 4 hours with quicklime
and distilled. The middle fraction was collected and used.
1.14.3. Synthesis of 2-aminobenzaldehyde
Figure.1.5. Synthesis route of 2-aminobenzaldehyde
1.14.3.1. Procedure
A three necked flask is employed as a reaction vessel from which the
product can be steam-distilled immediately after completion of the reaction. It is
convenient to arrange the apparatus for the reaction and that for the steam
distillation on the same steam bath, with provision for the rapid connection of the

69
flask to the distillation assembly at the desired time. For use as a reaction vessel,
the flask is mounted on a steam bath and fitted with a mechanical stirrer and a
reflux condenser, third neck is closed by a cork.
In a steam-distillation assembly of the small necks of the flask is fitted with
a steam-inlet tube, connected through a water trap to a steam line, the other small
neck is closed by a cork. The central neck is fitted to Kjeldahl trap leading to a 50
cm. Allihn condenser set downward and connected in series to a 50 cm Liebig
condenser. The second condenser leads to a 500 ml three necked flask used as the
receiver. The received flask is immersed in an ice bath and fitted with an Allihn
reflux condenser.
When all the apparatus has been set up and tested the flask is connected to
the reaction assembly and 175 ml of water, 105 g of ferrous sulphate heptahydrate,
0.5 ml of concentrated hydrochloric acid and 6 g of o-nitrobenzaldehyde are
introduced in the order given. The stirrer is then started and the flask is heated by
means of the steam bath. When the temperature of the mixture reaches 90oC, 25 ml
of aq.NH3 is added to one portion and at 2-minute intervals, three 10 ml portions of
ammonium hydroxide are added. Stirring and heating are continued throughout.
The total reaction time is 8 minutes.
Immediately after the addition of last portion of ammonium hydroxide, the
reflux condenser and stirrer are removed and the flask is connected to the steam
distillation assembly. The mixture is steam-distilled as rapidly as possible and two
250 ml fractions of distillate are collected during a period of 12 minutes. The first
fraction is saturated with sodium chloride and the solution is stirred until

70
precipitation appears to be complete. The solid is collected on a Buchner funnel
and derived in the air. The product weights 2.5 g. (60 %) and metals at 38oC. The
second fraction of the distillate is saturated with sodium chloride and combined
with the filtrate remaining from the first fraction. The combined solution is
extracted with two 45 ml portions of ether. The combined ether extract is filtered,
dried over anhydrous sodium sulphate and concentrated by distillation, finally
under reduced pressure. The residue solidifies on cooling and weights 1.0 g, it can
be purified by steam distillation from 40 ml of saturated sodium chloride solution
until 100 ml of distillate is collected, saturated the distillate with sodium chloride,
cooled and filtered. The pure product so obtained weights 0.87 g. The total yield is
3.0 g (70 %). The submitters have obtained yields of about 70% in runs eight times
as large as that described. This amino aldehyde undergoes self-condensation on
standing, especially in desiccators and so the product should be used immediately.
1.14.4. Synthesis of trinuclear Schiff base complexes derived from
o-phthalaldehyde
Synthesis of homo and heterotrinuclear Schiff base metal complexes from
p-phenylenediamine, o-phthalaldehyde and 2-aminobenzaldehyde were carried
out in two stage processes. In the first stage mononuclear Cu(II) and Ni(II)
complexes were prepared. In second stage, the mononuclear complex has been
used as a ligand for further condensation with 2-aminobenzaldehyde and also react
with metal salts to obtain the homo and heterotrinuclear Schiff base metal
complexes.

71
Stage I
Template synthesis of mononuclear Schiff base complexes derived from
p-phenylenediamine and o-phthalaldehyde. The mononuclear complexes were
synthesized by the following general procedure [3]. The p-phenylenediamine (2 mmol,
0.216 g) and o-phthalaldehyde (1 mmol, 0.134 g) in ethanol were refluxed for
2 hours. Metal salt (0.5 mmol) in ethanol was added to the resulting solution and again
refluxed for one hour. The resulting solution was evaporated and dried in air. The solid
product was collected. Synthesis pathway is shown in Figure 1.6.
Figure.1.6. Synthesis approaches for mononuclear Schiff base metal complexes
derived from o-phthalaldehyde.

72
Stage II
The bis-(p-phenylenediaminephthalaldehyde)metal(II) complexes (1 mmol)
and 2-aminobenzaldehyde (4 mmol) in hot methanol were refluxed for 2 hours and
metal salts in methanol were then added drop-wise to the resulting solution. The
contents were again refluxed for one hour. It was cooled to room temperature. The
product was obtained. It was filtered and dried over in decicator. Synthesis
pathway is shown in Figure 1.7.
Figure.1.7. Synthesis route of trinuclear Schiff base metal complexes derived from
mononuclear complexes and 2-aminobenzaldehyde.

73
Table.1.3. Physical data of trinuclear Schiff base complexes derived from o-
phthalaldehyde
Compounds Molecular Formula Color
Yield
(%)
Melting
Point
(°C)
[Cu3(L1)Cl2]4ClO4 [(C68H56N12)Cu3] 4ClO4 Brown 75 >250
[Ni3(L1)Cl2]4ClO4 [(C68H56N12)Ni3] 4ClO4 Pale 70 >250
Brown
[NiCu2(L1)Cl2]4ClO4 [C68H56N12)CuNi2] 4ClO4 Black 65 >250
[CuNi2(L1)Cl2]4ClO4 [C68H56N12)NiCu2] 4ClO4 Black 70 >250
[Cu(VO)2(L1)Cl2]2SO4 [C68H56N12)Cu(VO)2] 2SO4 Brown 73 >250
1.14.5. Synthesis of trinuclear Schiff base complexes derived from
2,3-pentanedione
Synthesis of homo and heterotrinuclear Schiff base metal complexes
derived from p-phenylenediamine, 2,3-pentanedione and 2-aminobenzaldehyde
were carried out in two stage processes. In the first stage mononuclear Cu(II),
Ni(II) and Mn(II) complexes were prepared. In second stage, the mononuclear
complex has been used as a ligand for further condensation with
2-aminobenzaldehyde and also react with metal salts to obtain the homo and
heterotrinuclear Schiff base metal complexes.
Stage I
The ethanolic solution (20 ml) of p-phenylenediamine (2 mmol) and
ethanolic solution (20 ml) of 2,3- pentanedione (1 mmol) were mixed slowly with
constant stirring. This mixture was refluxed at 90°C for 2 hours. Metal chloride

74
(0.5 mmol) in ethanol (20 ml) were then added to the resulting solution and again
refluxed for one hour as shown in Figure 1.8. When it was cooled to room
temperature, the solid product was obtained.
Figure.1.8. Synthesis approaches for mononuclear Schiff base metal complexes
derived from 2,3-pentanedione.
Stage II
An ethanolic solution (20 ml) of bis-(p-phenylenediamine,
2,3-pentanedione) metal(II) complexes in hot condition (1 mmol) and
2-aminobenzaldehyde (4 mmol) were refluxed for 2 hours and metal chloride
(2 mmol) in ethanol(20 ml) were then added drop wise to the resulting solution.

75
The contents were again refluxed for one hour as shown in Figure 1.9. It was
cooled to room temperature. The precipitate was obtained. It was filtered and dried
over fused CaCl2.
Figure.1.9. Synthesis route of trinuclear Schiff base metal complexes derived from
mononuclear complexes and 2-aminobenzaldehyde.

76
Table.1.4. Physical data of trinuclear Schiff base complexes derived from
2,3-pentanedione
Compounds Molecular Formula Color
Yield
(%)
Melting
Point
(°C)
[Cu3(L2)Cl6] [(C62H60N12Cl6)Cu3] Black 81 >250
[Ni3(L2)Cl6] [C62H60N12Cl6)Ni3] Dark
brown
78 >250
[Mn3(L2)Cl6] [C62H60N12Cl6)Mn3] Brown 75 >250
[CuNi2(L2)Cl6] [C62H60N12Cl6)CuNi2] Black 74 >250
[NiCu2(L2)Cl6] [C62H60N12Cl6)NiCu2] Black 72 >250
1.14.6. Synthesis of trinuclear Schiff base complexes derived from benzil
Synthesis of homo and heterotrinuclear Schiff base metal complexes
derived from p-phenylenediamine, benzil and 2-aminobenzaldehyde were carried
out in two stage processes. In the first stage mononuclear Cu(II), Ni(II) and Mn(II)
complexes were prepared. In second stage, the mononuclear complex has been
used as a ligand for further condensation with 2-aminobenzaldehyde and also react
with metal salts to obtain the homo and heterotrinuclear Schiff base metal
complexes.
Stage I
An equimolar amount of the ethanolic (20 ml) solution of benzil and
p-phenylenediamine was refluxed at 90°C for 2 hours. CuCl2 (1 mmol) in ethanol
(20 ml) were then added to the resulting solution and again refluxed for one hour
as shown in Figure 1.10. The reaction mixture was cooled to room temperature,
filtered and filtrate on slow evaporation yielded greenish brown coloured solid was

obtained. It was filtered and dried over fused CaCl2.
77
obtained. The same procedure was adopted to the synthesis of mononuclear Ni(II)
and Mn(II) complexes.
Figure.1.10. Synthesis approaches for mononuclear Schiff base metal complexes
derived from benzil.
Stage II
A hot ethanolic solution (20 ml) of bis-(p-phenylenediamine, benzil)
metal(II) complexes (1 mmol) and 2-aminobenzaldehyde (4 mmol) in 1:4 molar
ratio were refluxed for 2 hours and metal chloride (2 mmol) in ethanol (20 ml)
were then added drop wise to the resulting solution. The contents were again
refluxed for one hour. It was cooled to room temperature. The precipitate was

from mononuclear complexes and 2-aminobenzaldehyde.
78
Figure.1.11. Synthesis routes of trinuclear Schiff base metal complexes derived

79
Table.1.5. Physical data of trinuclear Schiff base complexes derived from benzil
Compounds Molecular Formula Color Yield (%)
Melting Point
(°C)
[Cu3(L3)Cl6] [(C80H64N12Cl6)Cu3] Black 78 >250
[Ni3(L3)Cl6] [C80H64N12Cl6)Ni3] Brown 75 >250
[Mn3(L3)Cl6] [C80H64N12Cl6)Mn3] Brown 75 >250
[CuNi2(L3)Cl6]
[C80H64N12Cl6)CuNi2]
Black
72
0250
[NiCu2(L3)Cl6]
[C80H64N12Cl6)NiCu2]
Black
74
>250
1.14.7. Apparatus and Experimental conditions
1.14.7.1. Solubility
Solubility of the complexes were checked in hot and cold water,
dimethylsulfoxide (DMSO), dimethylforamide (DMF) and other organic solvents
by shaking a small amount of the complexes in a test tube.
1.14.7.2. Melting points
The melting points were determined by placing a fine powdered sample in
glass capillary and heated by using Technico Melting point apparatus.
1.14.7.3. Eleme ntal analysis
The elemental analyses (Carbon, Hydrogen and Nitrogen) were performed
using a Carlo Eraba 1106 instrument.
1.14.7.4. Molar conductivity
Molar conductances of the complexes in DMF solution were measured with
ELICO CM 185 conductivity Bridge. Conductance or conductivity can also be
explained as the power of electrolytes to conduct electrical current. Conductance is
the reciprocal of resistance. Its units are Mho or Ohm-1.

80
Where,
Λ = 1/R
Λ = conductance
R = Resistance
Conductance is affected by different factors including
(i) Concentration
(ii) Solvent
(ii) Temperature
Generally, conductance is decreased by increasing concentration and vice
versa. Concentration increases by dilution because on dilution, the ionic mobility
increases and interionic attractions decreased. The conductance of a solution of an
electrolyte generally increases with rise in temperature (2-3% of for one-degree
rise in temperature). The increase in conductance with rise in temperature is due to
the speed of ions with which they move towards the electrodes. With rise in
temperature velocity of the solvent decreases which makes the ions to move freely
towards the electrodes.
The molar conductivity of an electrolyte in a particular solvent depends on
various factors like viscosity, polarity and donor property of the solvent. Geary [4]
has given the ranges of molar conductivity of various types of electrolytes in
different solvents, which is given in Table.1.6.

81
Table.1.6. Molar conductance (S cm2 Mol-1) of various electrolytes in different
solvents
Electrolyte type
Solvents 1:1 1:2 1:3 1:4
Nitrobenzene
Acetone
20-30
100 – 140
50-60
160 – 200
70-82
260
90-100
270
Acetonitrile 120 – 160 220 – 300 340 – 420
500
DMF 65 – 90 130 – 170 200 – 240
300
Methanol
80 – 115
160 – 220
290 – 350
450
Ethanol 35 – 45 70 – 90
120
160
1.14.7.5. Infrared spectroscopy
Fourier Transform Infrared Spectroscopy (FT-IR) is a spectroscopic
technique that deals with the middle infrared region of the electromagnetic
spectrum (400 − 4000 cm−1
). Each covalent bond of organic functions and
inorganic species exhibits a characteristic frequency of vibration in FT-IR
spectroscopy which can be used to identify its component. The vibration forms of
molecules are primary two types: 1) stretching vibration (ν), 2) bending vibration
(δ). Generally, stretching vibration modes are observed at higher frequency than
that of bending vibrations for a same covalent bond.
The infrared spectra were recorded on the Perkin Elmer FT-IR-8300 model
spectrometer using KBr disc in the region 4000–400 cm−1. Infrared spectroscopy is
an extremely powerful analytical technique for both qualitative and quantitative

82
analysis. The infrared spectrum of any given substance is interpreted by the use of
known group frequencies. It is one of the most widely used tools for the detection
of functional group in pure compounds and mixtures, for compound comparison
and for the identification of the substances.
Infrared spectroscopy involves twisting, bending, rotational and vibrational
motion of the atomic groups in a molecule. Infrared radiation does not possess
sufficient energy to cause the excitation of electrons however, it causes atoms and
groups of atoms to vibrate faster about covalent bond or bond, which connect them.
The compounds absorb infrared energy in the particular region of spectrum. A highly
complex absorption spectrum is obtained which is characteristic of the functional
group comprising the molecule and overall configuration of the atom as well.
The infrared region constitutes three parts:
i. Near Infrared region: In the near infrared region, which meets the visible
region at about 12,500 cm−1
and extends to about 4000 cm−1
are found in
many absorption bands resulting from harmonic overtones of fundamentals
bands and combination bands often associated with hydrogen atoms.
Among these, the first overtones of the O-H and N-H stretching vibrations
are near 7140 cm−1 and 6667 cm−1 respectively, combination bands
resulting from C-H stretching and deformation vibration of alkyl groups at
4548 cm−1 and 3850 cm−1.
ii. Mid Infrared region: Middle infrared region is divided into the “group
frequency” region, 4000−1300 cm−1 and the finger print region, 300-650
cm−1.

83
iii. Far infrared region: Far infrared region between 667 cm−1 and
10 cm−1
contains the bending vibrations of carbon, nitrogen, oxygen and
fluorine with atoms heavier than mass 19 and additional motion in cyclic or
unsaturated systems.
The infrared spectrum can give a perfect picture of the structural formula
without a chemical investigation.
1.14.7.6. Electronic spectra
Electronic absorption spectra in the ultraviolet (UV) – Visible range were
recorded on Perkin Elmer Lambda-25 between 200 and 800 nm by using DMF as
the solvent. The spectra of transition metal complexes depend on the transition of
unpaired electrons from the ground state to an excited state. Most of the transition
metal complexes are colored; the color is observed due to a d-d transition in the
visible region. Transitions between a given set of p or d orbitals, where d-d
transitions are forbidden (e.g. in octahedral complexes), render such complexes
colorless.
The electronic structure of coordination compounds is mainly described by
the molecular orbital theory. The atomic overlap in metal-ligand bonds allows d-
electrons to penetrate from the central atom to the ligand. The transitions are
affected by the effect of ligands on the energies of the d orbital of the metal ions.
Since octahedral, square-planar and tetrahedral fields cause splitting of d orbitals in
different ways, the geometry will have a pronounced effect on the d-d transitions in
a metal ion complex. Thus, spectral data of these transitions provide useful
information about the structure of complexes.

84
Cu(II) complexes are usually blue or green, exception to this, is generally
caused by strong ultraviolet and charge transfer bands tailing off into the blue and
visible spectrum thus causing the substances to appear red or brown. The blue or
green colors are due to the presence of Cu d-d absorption bands in 500-800 nm
regions of the spectrum. The envelopes of these bands are generally unsymmetrical
seeming to encompass several overlapping transitions, but definite resolution into
the proper number of sub-bands with correct location is difficult.
1.14.7.7. Electron paramagnetic resonance (EPR) Spectroscopy
ESR spectra were recorded on a Varian JEOL-JES-TE100 ESR
spectrophotometer at X-band microwave frequencies for powdered samples at
room temperature. EPR is a valuable technique, both because of its high sensitivity
and its general capability to identify unambiguously paramagnetic species which
contain unpaired electrons. It can reveal the oxidation state, electronic
configuration and coordination number of a paramagnetic ion. This technique has
been used in catalytic chemistry, biological systems food production. Here, we will
focus on this technique and its characterization on homo-and heterotrinuclear
Schiff base metal complexes.
Principle
EPR is an absorption spectroscopy in which radiation having frequency in
microwave region is absorbed by paramagnetic species to induce transition
between magnetic energy level of electrons with unpaired spin. Each electron
processes a magnetic moment and a spin quantum number S = 1/2, with magnetic
components ms = +1/2 and ms = −1/2. In the presence of an external magnetic field

85
with strength B0, the electron‟s magnetic moment aligns itself either parallel
(ms = +1/2) or antiparallel (ms = −1/2) to the field. The separation between
ms = +1/2 and ms = -1/2 state is ∆E =geµBB0, where ge is the electron‟s so-called g-
factor, B0 is the magnetic field, µB is the Bohr magneton. An unpaired electron can
move between energy levels by either absorbing or emitting electromagnetic
radiation of energy ε = hν such that the resonance condition, ε = ∆E, is obeyed.
Therefore, the fundamental equation of EPR spectroscopy is obtained:
hν = geµBB0, In practice, the frequency (ν) of radiant energy is usually fixed to
approximately 9.5 GHz in X-band EPR and the external magnetic field (B0) is
varied until that the gap between the ms = +1/2 and ms = −1/2 energy states
matches the energy of microwaves.
Figure.1.12. Energy-level diagram for S = ½ and no zero field splitting.
By increasing an external magnetic field, the gap between the ms = +1/2
and ms = −1/2 energy states is widened until it matches the energy of the
microwaves, as represented by the double arrow in the diagram above. At this
point the unpaired electrons can move between their two spin states. Since there
are typically more electrons in the lower state, due to the Maxwell Boltzmann

86
distribution, there is a net absorption of energy and it is this absorption which is
monitored and converted into a spectrum.
Figure.1.13. An example for EPR spectra
As an example of how hν = geµBB0 can be used, consider the case of a free
electron, which has ge = 2.0023, and the simulated spectrum shown at the right in
two different forms. For the microwave frequency of 9388.2 MHz, the predicted
resonance position is a magnetic field of about B0 = hν/geµB = 0.3350 tesla = 3350
gauss, as shown. Note that while two forms of the same spectrum are presented
in the figure, most EPR spectra are recorded and published only as first
derivatives. Because of electron-nuclear mass differences, the magnetic moment of
an electron is substantially larger than the corresponding quantity for any nucleus,
so that a much higher electromagnetic frequency is needed to bring about a spin
resonance with an electron than with a nucleus, at identical magnetic field
strengths.

87
1.14.7.8. Magnetic susceptibility measurements
Magnetic susceptibility data were collected on powdered sample of the
compounds at room temperature with PAR 155 vibrating sample magnetometer.
Experimental methods to determine susceptibility
Volume magnetic susceptibility is measured by the force change felt upon
the application of a magnetic field gradient [5]. Early measurements were made
using the Gouy balance where a sample is hung between the poles of an
electromagnet. The change in weight when the electromagnet is turned on is
proportional to the susceptibility.
The behavior of a compound in a magnetic field can be classified as being
diamagnetic, paramagnetic, ferromagnetic or antiferromagnetic. We will be
concerned with the first two types of behavior. A diamagnetic substance is weakly
repelled by a magnet, whereas a paramagnetic substance is attracted by a magnet.
If the substance is diamagnetic the mass of the magnet as measured by the
balance will appear to increase, because the magnet is repelled by the sample. On
the other hand, if the substance is paramagnetic, the mass of the magnet will
appear to decrease, because the magnet is attracted to the sample. The force of
attraction between the sample and the magnet is given by
( H 2 − H 2 ) g∆m = µ o χ m nm
1 2
2l
(1)
where g is the acceleration due to gravity, ∆m is the apparent mass change
of the magnet, µo is the permeability of a vacuum, χm is the molar magnetic

88
susceptibility (its units are cm3mol-1), H1 and H2 are the magnetic field strengths at
the opposite ends of the sample, nm is the moles of sample and l is the height of the
sample in the sample tube.
The magnetic behavior of a substance is related to the electronic structure
of its atoms. A moving charged particle generates a magnetic field. Thus, the
electrons within atoms generate magnetic fields due to their orbital motion around
the nucleus and to their “spin”. Atoms with unpaired electrons are paramagnetic
since the unpaired electrons behave as tiny magnets and in the presence of an
external magnetic field these magnets become aligned with the external field and
cause the sample to be attracted to the external magnet. In this experiment it will
be concerned with the magnetic properties of compounds containing transition
metal ions. Many of these compounds are paramagnetic due to the presence of
unpaired electrons in the transition metal ion. In such compounds the electrons‟
orbital motion does not contribute to the paramagnetism and the permanent
magnetic moment of the compound, µ, is given by the “spin only” formula:
µ S = µ B n( n + 2 ) (2)
where, µS, is the spin contribution to the magnetic moment of an ion having n
unpaired electrons. µB, is the Bohr magneton, the fundamental unit of magnetic
moment.
Diamagnetism arise from the fact that in an external magnetic field, the
moving electrons in an atom experience an additional force which, in turn, results
in an induced magnetic field that opposes the external field. This causes the

89
2 1 m m
sample to be repelled by the external field. All atoms will exhibit diamagnetism
regardless of whether or not they also possess permanent magnetic moments.
A general expression for χm is
χ m = χ diamag + Nµ 2
3kT
(3)
where µ is the permanent magnetic moment per atom, N is Avogadro‟s
number, T is the absolute temperature, k is the Boltzmann constant and χdiamag is
the diamagnetic contribution to the molar susceptibility. In paramagnetic
substances, the first term is equation (3) is negligible compared to the second term,
and we have:
µ = 3kTχ m
N
= 2.84 µ B
Tχ m
(4)
As we have seen, for transition metal compounds, the paramagnetism is
due to the presence of unpaired electrons in the transition metal ion and we would
expect µ to have the “spin only” value given by equation (2). Thus, equations (1),
(2) and (4) provide us with an experimental method for determining the number of
unpaired electrons in a transition metal ion.
In practice, equation (1) is written as
χ m = 2 lg ∆m
= β ∆m
µo ( H 2 − H 2 ) n n (5)
Where β is a calibration constant that depends upon g, µo, the magnetic
field strength, the magnetic field gradient and the dimensions of the sample. Note

90
m
that in equations (1) and (2), ∆m and χm can be either positive or negative,
depending on whether the substance is diamagnetic or paramagnetic, respectively.
The value of β is determined experimentally, by measuring ∆m and nm for a
reference substance, then
χ mref
β = (∆m n
)ref
(6)
where the subscript “ref” refers to the reference substance. The reference
substance to be used is Mohr‟s salt, iron(II) ammonium sulfate hexahydrate,
FeSO4(NH4)2SO4.H2O. It‟s molar magnetic susceptibility is given by
χ m = 3.7278
T + 1 (7)
1.14.7.9. Thermal Studies
Thermal studies were carried out in the 100−900oC range using an
SETSYS Evolution -1750 model thermal analyzer. Thermal methods of analysis
may be defined as those techniques in which changes in physical and / or chemical
properties of a substance are measured as a function of temperature. Methods that
involve changes in weight, dimensions or changes in energy come within this
definition.
Thermogravimetry (TG) a technique in which a change in the weight of a
substance is recorded as a function of temperature or time.
Differential thermal analysis (DTA) a technique in which the temperature
difference between a substance and a reference material is measured as a function

91
of temperature whilst the substance and reference are subjected to a controlled
temperature programme.
The basic instrumental requirement for thermogravimetry is a prescision
balance with a furnace programmed for a linear rise of temperature with time. The
results may be presented as a thermogravimetric (TG) curve, in which the weight
change is recorded as a function of temperature or time, or as a derivative
thermogravimetric (DTG) curve, where the first derivative of the TG curve is
plotted with respect to either temperature or time.
1.14.7.10. DNA binding experiments
A solution of CT DNA in 0.5 mM NaCl/5 mM Tris–HCl (pH 7.0) gave a
ratio of UV absorbance at 260 and 280 nm of 1.8:1.9, indicating that the DNA was
sufficiently free of proteins [6]. A concentrated stock solution of DNA was
prepared in 5 mM Tris–HCl/50 mM
NaCl in water at pH 7.0, and the concentration of CT DNA was determined per
nucleotide by taking the absorption coefficient (6600 mol−1 cm−1) at 260 nm [7].
Absorption titration experiments were performed by varying the concentration of
the DNA with the complexes.
1.14.7.12. Viscosity experiments
Viscosity measurements were carried out from observed flow time of CT
DNA containing solution (t > 100 seconds) corrected for the flow time of buffer
alone (t0), using Ostwald‟s viscometer at 30 ± 0.1°C. Flow time was measured with
a digital stopwatch. Each complex was measured three times and an average flow

92
time was calculated. Data were presented as (η/η0) versus binding ratio
([complex]/[DNA]), where η is a viscosity of DNA in the presence of complex and
η0 is the viscosity of DNA alone. Viscosity values were calculated from the
η = t – t0 [8].
1.14.7.12. Gel electrophoresis
The cleavage of pUC18 DNA was determined by agarose gel
electrophoresis [9]. The gel electrophoresis experiments were performed by
incubation of the samples containing 40 µM pUC18 DNA, 50 µM metal complexes
and 50 µM H2O2 in Tris-HCl buffer (pH 7.2) at 37°C for 2 hours. After incubation,
the samples were electrophoresed for 2 hours at 50 V on 1% agarose gel using
Tris–acetic acid–EDTA buffer (pH 7.2). The gel was then stained using 1 µg/cm3
ethidium bromide and photographed under ultraviolet light at 360 nm. All the
experiments were performed at room temperature.
1.14.7.13. Cyclic voltammetry
All voltammetric experiments were performed with a CHI 760
electrochemical analyzer, in single compartmental cells using tetrabutylammonium
perchlorate as a supporting electrolyte. The redox behavior of the complexes has
been examined in absence and in presence of CT DNA at a scan rate of 0.1 Vs−1 in
the potential range +2.0 to –2.0 V. A three-electrode configuration was used,
comprising a glassy carbon electrode as the working electrode, a Pt-wire as the
auxiliary electrode and an Ag/AgCl electrode as the reference electrode. The
electrochemical data such as cathodic peak potential (Epc) and anodic peak
potential (Epa) were measured.

93
1.14.7.14. Antimicrobial activity
The in vitro antibacterial activity of the ligand and the complexes were
tested against the bacteria Klebsiella pneumoniae, Escherichia coli and
Staphylococcus aureus by well diffusion method using nutrient agar as the medium
[10]. Streptomycin was used as standard component. The stock solution (10−1 mol
L−1
) was prepared by dissolving the compound in DMF, and the solution was
serially diluted to find minimum inhibitory concentration (MIC) values. In a
typical procedure, a well was made on the agar medium with microorganisms in a
petri plate. The well was filled with the test solution and the plate was incubated
for 24 h for bacteria at 35°C. During the period, the test solution diffused and the
growth of the inoculated micro-organisms was affected. The inhibition zone was
developed, at which the concentration was noted.
Glasswares
Glasswares used in the present investigation were thoroughly washed, dried
and were sterilized at 121oC for fifteen minutes in autoclave.
Grams staining
In the gram, staining test was performed and the morphology cell shape and
arrangement were observed.
Gram stain reagents
Crystal violet - 1.0 gm
5% Na2CO3 - 1.0 ml
Distilled water - 99 ml

94
Crystal violet was added to sodium bicarbonate into a mortar. Using a
pestle grind to a good paste. Distilled water was added and mixed well. It was
then filtered and stored in brown bottles.
Ethyl Alcohol
Ethyl alcohol (100%) 95 ml
Distilled water 5 ml
Saffranin
Saffranin 1 gm
Distilled water 100 ml
Grams iodine
Iodine crystals 2 g
NaOH 10 ml
Distilled water 90 ml
Sodium hydroxide was added to the iodine crystals kept in a mortar and
grind to a good paste. Distilled water was added and mixed well. It was then
filtered and stored in brown bottles.
1. A thin smear of sample is made on glass slide.
2. Sear is allowed to dry and heat fix it.
3. The slides are holded using slide rack.
4. Each smear is covered with crystal violet for 30 sec.
5. Each slide is then washed with distilled water.
6. Slides are covered with iodine solution.

95
7. Iodine solution is washed off by 95% ethyl alcohol.
8. Slides are washed by distilled water and drained.
9. Saffranin was flooded on the smear for 30 min.
10. Washed with distilled water and blot dried with absorbent paper.
11. The slides are allowed to dry.
12. Slides are observed under microscopically using oil immersion
objectives.
Motility test
Motility test was performed using hanging drop method. The motile and
non-motile organisms were differentiated.
1. Take two cavities slides and washed carefully.
2. Then cover slips are washed.
3. Dropped a small amount of culture on these cover slips each culture on
one cover slip.
4. Then applied Vaseline on the four corners of both the cover slips.
5. Then attached the cover slip by inverting it on the cavity slide to
scaffold the drop of culture inside the cavity.
6. Then the slide was examined under the microscope.
Catalase test
Some organism posses catalases and splits hydrogen peroxide into oxygen
and water.
1. Nutrient agar was prepared melted and pored into the test tube.
2. The test tubes were sterilized and then allowed in a stand position to
prepared slants.

96
3. After solidification, the organisms were streaked and stand position to
incubate at 370C for about 12 to 24 hours.
4. After incubation 3%, hydrogen peroxide is added to slant.
Oxidase test
The enzyme oxidase that forms the part of the electron transport system is
possessed by some bacteria. The enzyme oxidizes the reagent N-N tetramethylene
paraphenylene diamine dihydrochloride to a coloured product into phenol.
Sugar fermentation test
Some organisms produce small amount of acid either by fermentation or by
oxidation. These small amounts of acids are neutralized by large amount of
alkaline products released during protein metabolism and hence acid production is
not noticed. Thus, carbohydrate fermentation reaction can be tested in minimum
protein.
Indole test
Tryptophan present in peptones of the culture media is act upon by the enzyme
tryptophanase and converted into indole, skatol and induce acetic acid. Indole reacts
with aldehydes to produce a red coloured product. Therefore, organisms are grown in
tryptophan rich medium and tested for the presence of indole.
Tryptone broth
Tryptone 1.0 g
NaCl 0.5 g
Distilled water 100 ml

97
Kovacs reagent
Paradimethylamino benzaldehyde 5 g
Amyl alcohol 75 g
pH 7.4
Conc.HCl 25.5 ml
Dissolved the aldehyde in alcohol and added slowly. Stored in brown bottles.
Methyl Red Voges-proskauer Test
Enterobacteriaceae ferment glucose via pyruvate and produce mixed acids
and other products. Among them, one group produces mixed acids such as acetic,
lactic, succinic, formic acid, ethanol, CO2 and H2.
Because of the abundant acid production, the final pH of the broth drops to
less than 4.5, which can be detected by pH indicators.
Other group of organisms can produce the butylenes glycol and acetone,
which are more neutral in nature and not much drop in pH noticed. The end
products are detected by reagent.
MR-VB broth preparation
Peptone 7.0 g
Glucose 5.0 g
K2PO4 5.0 g
Distilled water 1000 ml
Final pH 6.9

98
MR-Reagents
Methyl red 0.1 g
Ethyl alcohol 300 ml
Distilled water 200 ml
Dissolved methyl red in alcohol and then add distilled water. Stored in brown
bottles at 40C.
VP reagent preparation
VP-A
Alpha napthol 5.0 g
Ethyl alcohol 100 ml
Dissolve alpha-naphthol in small amount of alcohol first and then add the
remaining alcohol to 100ml. Store in brown bottles at 40C.
VP-B
KOH 40.0 g
Distilled water 100 ml
Cooled the cylinder in cold-water bath with 80 ml of water and add KOH crystals,
dissolved and made upto 100 ml. Stored in polythene bottles at 40C.
Citrate utilization test
Certain organisms can utilize as sole carbon source and grow. During the
growth acetate and other alkaline carbonated are produced. This reaction is shown
by the change in colour of the indicator.

99
Simmon‟s citrate agar media
NaCl 5.0 g
MgSO4 0.2 g
NH4H2PO4 1.0 g
K2PO4 1.0 g
Sodium citrate 2.0 g
Distilled water 1000 ml
pH 6.9
TSI Test
Some bacteria liberate sulfur from sulfur containing amino acid or other
sulfur containing compounds. The sulfur is used as final hydrogen acceptor
leading to the formation of H2S. When the following conditions are present, H2S
can be detected by the test system.
Yellow - A (acid)
Yellow gas - AG+
Pink - AK (Alkali)
Black precipitation - H2S
TSI Medium
Beef extract 0.3
Yeast extract
0.3
Peptone
2.0
NaCl
0.5
Glucose
0.1

100
Lactose 1.0
Sucrose 1.0
Fe2(NH4)SO4 0.2
NaHSO4 2.5
Casein digests animal tissue 10.0 g
Phenol red 2.5
Distilled water 100 ml
Agar 20 g
Final pH 7.4
Urease test
Urea is diamide of carbonic acid. Urease the enzyme possessed by the
bacteria hydrolyses urea and release ammonia and carbon dioxide. NH4 reacts in
solution to form ammonium carbonate, which is alkaline leading to the increase in
the pH of phenol red which is incorporated in the medium changes its colour from
yellow to red in alkaline pH, thus indicates the presence of urease activity.
Christensons Urea agar
Peptone 0.1 g
Glucose 0.1 g
NaCl 0.5 g
K2PO4 0.2 g
Phenol red 1.0 g
Agar 2.0 g
Distilled water 1000 ml
pH 6.8

101
Primary culture media
Nutrient agar and Mac Conkey was used as the primary culture media.
Enriched media
Blood agar and citramide agar are used for isolation and identification of
the pathogenic microorganisms during the study.
Nutrient agar
Peptone 0.5 g
NaCl 0.5 g
Beef extract 0.3 g
Yeast extract 0.3 g
Agar 2 g
Distilled Water 100 ml
pH 7.2
Blood agar
Peptone 0.5 g
NaCl 0.5 g
Beef extract 0.5 g
Agar 2 g
Distilled water 100 ml
Sheep blood 5 ml
pH 7.4

102
Mac Conkey agar
Peptone 20 g
Lactose 10 g
Bile Salt 1.5 g
Neutral red 0.03 g
Crystal violet 0.001 g
Agar 15 g
Distilled water 1000 ml
pH 7.4
1.14.8. Metal Estimations
1.14.8.1. Estimation of copper [11]
The amount of copper in the complexes was estimated
spectrophotometrically by the following method. The addition of an aqueous
solution diethyldithiocarbamate to a slightly acidic or an ammoniacal solution of
the copper salt produces a brown precipitate of a sparingly soluble copper
derivative. With minute amounts of copper, a golden brown coloration is obtained.
The copper derivative is soluble in organic solvents such as CCl4 and the optical
density of the yellow-brown colored solution is compared with that of similar
standard solution.
Procedure
The complex was decomposed by adding 10 ml of concentrated sulphuric
acid and few drops of perchloric acid and the mixture was digested in a kjeldahl‟s
flask until the solution become colorless. The solution was cooled and made up to

103
50 ml. Dilution was effected by taking 5 ml of the solution in a 50 ml volumetric
flask and making up to the mark by addition of water. An aliquot of 2 ml of the
solution was transferred into a beaker and rendered alkaline using a dilute solution
of ammonia. The excess ammonia was boiled off. The pH of the solution was
adjusted to 8.5 using very dilute solutions of ammonia and sulphuric acid. 15 ml of
4% solution of EDTA (disodium salt) was then added and the solution was allowed
to cool to room temperature. The solution was transferred to a separatory funnel
and treated with 10 ml of 0.2% aqueous solution of diethyl dithiocarbamate. The
solution was thoroughly mixed by shaking for 45 seconds during which a yellow-
brown color developed in the solution, was immediately extracted with 5 ml of
CCl4. The lower organic layer was collected in a dry vessel. Extraction was
repeated until the entire complex was removed.
The extracted organic layer was washed by shaking with 20 ml of 5%
solution of sulphuric acid for 15 seconds in a reparatory funnel. After allowing
enough time for the two layers to separate, the organic layer was separated and
made up to a total volume of 25 ml. The copper content was then determined by
measuring the optical density of the made up solution at 435 nm and compared
with the standard solutions of copper treated similarly. The standard solution was
prepared by dissolving 0.0623 g of AR grade copper (II) sulphate pentahydrate in
100 ml of water in a standard flask. A calibration curve was obtained by measuring
the optical density of solutions prepared as above using different volumes of the
standard solution.

104
1.14.8.2. Estimation of nickel [12]
The nickel content in the complex was estimated colorimetrically using
dimethylglyoxime after the complex had been decomposed completely by
dissolving in 10 ml of warm 1:1 nitric acid, boiling the solution and was then
cooled and made upto 250 ml with distilled water in a volumetric flask.
An aliquot of 5 ml of the solution was taken in a 50 ml volumetric flask. To
that was added, with mixing after each addition, 5 mlof the 10% citric acid
solution, 2 ml of saturated bromine water, 2 ml of 1:1 ammonium hydroxide
solution and finally 1 ml of the 1% solution of dimethylglyoxime in rectified spirit.
Nickel content was then determined by measuring the optical density of the made
up solution at 530 nm and compared with the O.D of standard solution of nickel
similarly treated. The standard solution was prepared by dissolving 0.0673 g of
nickel ammonium sulphate in 100 ml of water. A calibration curve was obtained
by measuring the O.D of the solution prepared as above, using different volumes of
the standard solutions.
1.14.8.3. Estimation of manganese [13]
About 0.075 g of the complex was taken and dissolved in dilute nitric acid
(1:3) in a 250 ml conical flask. The solution was boiled for 5 minutes and cooled.
Then 0.5 g of A.R sodium bismuthate was added again and again boiled for
5minutes. A pink color was produced due to the formation of permanganic acid.
Then a concentrated solution of freshly prepared sodium sulphite was added drop
wise until the solution became clear. The solution was boiled to expell all oxides of
nitrogen and cooled to about 15ºC.

105
To the cooled solution, A.R Sodium bismuthate was added until no further
appreciable alteration in color was noticed. Then, excess of 0.5 g of sodium
bismuthate was added and the mixture was stirred for 2-3 minutes. To this, 50 ml
of the prepared 3% nitric acid was added and filtered through a sintered crucible
into a 350 ml conical flask. The residue was washed well with about 100 ml of the
3% nitric acid until it is free from permanganic acid.
Then the pink colored solution was added slowly with 0.003N FAS in
slight excess as indicated by the disappearance of pink color. This solution was
titrated immediately with a standard solution of (0.03) KMnO4 to the first faint
pink color.
The FAS solution was standardized by running a blank. In a conical flask,
50 ml of 1:3 nitric acid was taken, then a little of sodium bismuthate was added
and diluted with 10 ml of 3% nitric acid. To this an equal volume of the FAS was
added and titrated against the standard KMnO4. The difference between the
titrations gives the amount of permanganate equivalent to that of permanganic acid
formed in the determination.
1ml of KMnO4 = 0.0109 of Mn
1.14.8.4. Estimation of vanadium [14]
The amount of vanadium in the complexes was estimated volumetrically by
the following method. About 0.055g of the complex was weighed out and
dissolved in 2N H2SO4 acid. Approximately 0.1 N KMnO4 solutions was prepared
and standardized by titrating against standard solution of oxalic acid. The complex

106
solution was titrated against KMnO4 solution. The amount of vanadium was
calculated by the following formula.
Percentage of Vanadium = Titre value × N/100 ×51 × 100/W
Where
N – Strength of KMnO4
W – Weight of the vanadium complex

107
References
[1] A. Weissberger, E. S. Proskaver, J. A. Riddick and E. E. Toops, “Organic
solvents in techniques of Organic Chemistry”, 3rd Edn., Interscience., 7 (1970).
[2] A. I. Vogel “A Text book of practical organic analysis”, Longmans (1971).
[3] A. K. Panda, D. C. Dash, P. Mishra and H. Mohanty, Ind. J. Chem., 35
(1996) 848.
[4] W. J. Geary, Coord. Chem. Rev., 7 (1971) 81.
[5] L. N. Mulay, A. Weissberger and B. W. Rossiter, Techniques of Chemistry.
4. Wiley-Interscience: New York. p. 431, (1972).
[6] J. Marmur, J. Mol. Biol., 3 (1961) 208.
[7] M. E. Reicmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc.,
76 (1954) 3047.
[8] S. Mathur and S. Tabassum, Biometal., 21 (2008) 299.
[9] N. Raman, J. Raja and A. Sakthivel, J. Chem. Sci., 119 (2007) 303.
[10] G. G. Mohamed, M. M. Omar and A. A. Ibrahim, Spectrochim. Acta. A., 75
(2010) 678.
[11] A. I.Vogel, A Text Book of Quantitative Inorganic analysis, ELBS 3rd
Edn,
794 (1975).
[12] A. I.Vogel, A Text book of Quantitative Inorganic analysis, ELBS 3rd Edn,
796 (1975).
[13] A. I.Vogel, A Text Book of Quantitative Inorganic analysis, ELBS 4th Edn,
160 (1979).
[14] A. I.Vogel, A Text book of Quantitative Inorganic analysis, ELBS 3rd Edn,
(1975).





























