Embed Size (px)
Citation preview

Brain, Behavior, and Immunity 33 (2013) 112–122
Contents lists available at SciVerse ScienceDirect
Brain, Behavior, and Immunity
journal homepage: www.elsevier .com/locate /ybrbi
Intrathecal injection of adenosine 2A receptor agonists reversedneuropathic allodynia through protein kinase (PK)A/PKC signaling
0889-1591/$ - see front matter � 2013 Elsevier Inc. All rights reserved.http://dx.doi.org/10.1016/j.bbi.2013.06.004
⇑ Corresponding author. Address: Department of Psychology and Neuroscience,UCB 345, University of Colorado at Boulder, Boulder, CO 80309, USA. Fax: +1 303492 2967.
E-mail address: [email protected] (L.C. Loram).
Lisa C. Loram a,⇑, Frederick R. Taylor a, Keith A. Strand a, Jacqueline A. Harrison a,Rachael RzasaLynn b, Paige Sholar a, Jayson Rieger c, Steven F. Maier a, Linda R. Watkins a
a Department of Psychology and Neuroscience, Center for Neuroscience, University of Colorado-Boulder, Boulder, CO, USAb Department of Anesthesiology, University of Colorado, Anschutz Medical Campus, Aurora, CO, USAc Dogwood Pharmaceuticals, Inc., a Subsidiary of Forest Labs., Inc., Charlottesville, VA, USA
a r t i c l e i n f o
Article history:Received 25 January 2013Received in revised form 3 June 2013Accepted 21 June 2013Available online 28 June 2013
Keywords:Mechanical allodyniaProtein kinaseInterleukin-10ATL313IntrathecalRatsMicrogliaAstrocytes
a b s t r a c t
A single intrathecal dose of adenosine 2A receptor (A2AR) agonist was previously reported to produce amulti-week reversal of allodynia in a chronic constriction injury (CCI) model of neuropathic pain. Weaimed to determine if this long-term reversal was induced by A2AR agonism versus more generalizedacross adenosine receptor subtypes, and begin to explore the intracellular signaling cascades involved.In addition, we sought to identify whether the enduring effect could be extended to other models of neu-ropathic pain. We tested an A1R and A2BR agonist in CCI and found the same long duration effect withA2BR but not A1R agonism. An A2AR agonist (ATL313) produced a significant long-duration reversal ofmechanical allodynia induced by long established CCI (administered 6 weeks after surgery), spinal nerveligation and sciatic inflammatory neuropathy. To determine if ATL313 had a direct effect on glia, ATL313was coadministered with lipopolysaccharide to neonatal microglia and astrocytes in vitro. ATL313 signif-icantly attenuated TNFa production in both microglia and astrocytes but had no effect on LPS induced IL-10. Protein kinase C significantly reversed the ATL313 effects on TNFa in vitro in microglia and astrocytes,while a protein kinase A inhibitor only effected microglia. Both intrathecal PKA and PKC inhibitors signif-icantly reversed the effect of the A2AR agonist on neuropathic allodynia. Therefore, A2AR agonists admin-istered IT remain an exciting novel target for the treatment of neuropathic pain.
� 2013 Elsevier Inc. All rights reserved.
1. Introduction
Neuropathic pain affects approximately 4% of the global popu-lation (Global Industry Analysts, 2011). Neuropathic pain remainsintractable to available therapeutics as most available treatmentstarget neurons (Global Industry Analysts, 2011). Glially-drivenspinal neuroinflammation has received increasing attention asimportantly contributing to such pain (Scholz and Woolf, 2007).Activated microglia and astrocytes release neuroexcitatory media-tors such as pro-inflammatory cytokines and chemokines (Watkinset al., 2007). This contribution of glia in developing and maintain-ing neuropathic pain has led to the search for clinically relevant ap-proaches to suppress glially-driven pain amplification.
There are four adenosine receptor subtypes, A1 and A3 receptorsdecrease cAMP, while A2A and A2B receptors increase cAMP (Fred-holm et al., 2007). Adenosine receptors are found on all cell typeswithin the CNS including neurons and glia (Dare et al., 2007).
A2AR agonists recently attracted attention as potential glial inhibi-tors, based on their anti-inflammatory effects in peripheral im-mune cells. Remarkably, when A2AR agonists were administeredintrathecally (IT), a fascinating phenomenon was revealed; namely,a single intrathecal (IT) injection of A2AR-selective agonistsreversed neuropathic pain (mechanical allodynia, thermal hyperal-gesia) for more than 4 weeks in a classic animal model of neuro-pathic pain, the sciatic chronic constriction injury (CCI) (Loramet al., 2009). While many drugs briefly suppress neuropathic pain,none other induces such enduring pain inhibition. Intriguingly, wedemonstrated that A2AR agonism accounts for reversal of neuro-pathic pain initially, but continued A2AR activation cannot accountfor the enduring reversal (Loram et al., 2009). How a brief exposureto an A2AR agonist creates a strikingly persistent inhibition ofneuropathic pain remains unknown.
Therefore, the aim of this study was to further explore theunderlying mechanism for the remarkable long-acting effect ofthe A2AR agonist. We investigated whether the effects were uniqueto CCI, or if the long duration effect could be generalized to multi-ple neuropathic pain models. We further explored whether the ef-fects were generalizable to other adenosine receptors. Ourhypothesis, based on our previous findings, was that long-term

L.C. Loram et al. / Brain, Behavior, and Immunity 33 (2013) 112–122 113
reversal of allodynia was mediated by an attenuation of glial acti-vation within the spinal cord, resulting in reduced pro-inflamma-tory cytokine production (Loram et al., 2009). We havedemonstrated previously that IL-10 mRNA was significantly ele-vated following intrathecal A2AR agonist in CSF cells but not withinthe spinal cord tissue. It is possible that evaluating the wholespinal tissue diluted the potent change in IL-10 produced by glialcells alone. In order to further explore whether intrathecal A2ARagonism may affect glial cells within the spinal cord to produceIL-10, we tested an A2AR agonist on pure microglial cells, astrocytesand mixed astrocyte and microglial cells in vitro. Lastly, if the po-tential mechanism of the long duration of effect is via an increasein cAMP, then the intracellular kinases would be activated. Proteinkinase (PK) A is the canonical kinase stimulated by cAMP, which isknown to occur in peripheral immune cells following A2AR activa-tion. In addition PKC is also activated by A2AR activation in periph-eral immune cells (Cho et al., 2002; Goethe et al., 2007; Huchoet al., 2005; Tortora and Ciardiello, 2002a). Therefore, we aimedto explore whether PKA and/or PKC inhibitors administered intra-thecally could reverse the effect of the A2AR agonist on neuropathicallodynia in vivo. And furthermore, whether PKA and/or PKC inhib-itors would reverse the A2AR effect in glial cultures.
2. Materials and methods
2.1. Subjects
Pathogen-free male Sprague Dawley rats (325–350 g, HarlanLaboratories) were housed two per cage with standard rat chowand water ad libitum. Housing was in a temperature-controlledenvironment (23 ± 2 �C) with a 12 h light/dark cycle (lights on at7:00 AM). All procedures occurred in the light phase. All rats wereallowed 1 week of acclimation to the colony rooms before experi-mentation. The Institutional Animal Care and Use Committee of theUniversity of Colorado at Boulder approved all procedures.
2.2. Drugs
The A2AR agonist 4-(3-(6-amino-9-(5-cyclopropylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl)prop-2-ynyl)-piperidine-1-carboxylic acid methyl ester (ATL313) was a gift fromDogwood Pharmaceuticals. The A2AR agonist 2-p-(2-carboxy-ethyl)phenethylamino-50-N-ethylcarboxamido adenosine HCl(CGS21680) and CCPA were purchased from Tocris Bioscience. H-89 (PKA inhibitor) was purchased from Assay design (Ann Arbor,MI, USA). Lipopolysaccharide and chelerythrine (PKC inhibitor)were purchased from Sigma (St. Louis, MO, USA). BAY606583(A2BR agonist) was gifted by Bayer Healthcare (Wuppertal,Germany). All of the compounds except LPS were dissolved inDMSO to create 10 mM stock concentrations and stored at�20 �C. LPS was diluted in sterile saline and stored at 1 mg/mlaliquots at �20 �C. Fresh aliquots were diluted to the appropriateconcentration in sterile endotoxin-free isotonic saline (AbbottLaboratories).
2.3. Von Frey test for mechanical allodynia
Rats were habituated to the testing apparatus for 4 consecutivedays before testing. The Von Frey test was performed on the plan-tar surface of each hindpaw within the region of sciatic nerveinnervation, as described previously (Milligan et al., 2000). A loga-rithmic series of 10 calibrated Semmes–Weinstein monofilaments(Stoelting) were sequentially applied (from low- to high-intensitythreshold) to the left and right hind-paws in random order, eachfor 8 s at constant pressure to determine the stimulus intensity
threshold stiffness required to elicit a paw withdrawal response.Log stiffness of the hairs is determined by log10(mg � 10). Therange of monofilaments used in these experiments (0.407–15.136 g) produces a logarithmically graded slope when interpo-lating a 50% response threshold of stimulus intensity [expressedas log10(mg � 10)] (Chaplan et al., 1994). The stimulus intensitythreshold to elicit a paw withdrawal response was used to calcu-late the 50% paw withdrawal threshold (absolute threshold) usingthe maximum-likelihood fit method to fit a Gaussian integral psy-chometric function (Harvey, 1986). This method normalizes thewithdrawal threshold for parametric analyses (Harvey, 1986).The behavioral testing was performed blind with respect to thedrug administration.
2.4. Surgery
2.4.1. Chronic constriction injuryChronic constriction injury (CCI) (Bennett and Xie, 1988) of the
left sciatic nerve was aseptically performed under isoflurane anes-thesia. Four ligatures of 4–0 chromic gut were tied loosely aroundthe left sciatic nerve at the level of the midthigh, as described pre-viously (Milligan et al., 2004b).
2.4.2. Spinal nerve ligationSpinal nerve ligation of L5 and L6 left spinal nerve roots was
conducted as described by Kim and Chung (1992). 125–150 g maleSprague–Dawley rats were used for this experiment. Under isoflu-rane anesthesia, the left L5 and L6 nerve roots were isolated andtightly ligated with 6–0 silk suture. The rats were allowed 2 weeksrecovery before drug delivery.
2.4.3. Sciatic inflammatory neuropathySciatic inflammatory neuropathy was performed as described
previously (Milligan et al., 2004a). Chronic perisciatic catheterswere constructed and implanted at mid-thigh level of the left hind-leg as previously described (Chacur et al., 2001). This method al-lows multi-day recovery of the rats from anesthesia and surgerybefore the experiments.
2.4.4. Intrathecal drug deliveryRats were lightly anesthetized with isoflurane. The lumbar re-
gion was shaved and cleaned. An 18 Gauge guide needle, withthe hub removed, was inserted into the L5/6 intervertebral space.A PE-10 catheter was inserted into the guide needle, premarkedsuch that the proximal end of the PE-10 tubing rested over theL4–L6 lumbar spinal cord. All drugs were administered over 20 s(1 ll of drug followed by 2 ll of sterile saline flush) with a 30 s de-lay before removing the catheter and guide needle. Each rat wasanesthetized for a maximum of 5 min, and none incurred observa-ble neurological damage from the procedure.
2.5. Glial cultures
Neonatal microglia and astrocytes were cultured from P0/1 dayold male Sprague–Dawley pups as described previously (Hutchin-son et al., 2010). While differences in responses to pro-inflamma-tory stimuli have been identified between neonatal and adultcortical glia cultures (Loram et al., 2012; Schwarz et al., 2012),these experiments provide information regarding the effect ofadenosine receptor agonist and protein kinase inhibitors fromCNS derived immunocompetent cells subsequent to LPS stimula-tion. At confluence, (7–10 days), the microglia were separated fromthe astrocytes by shaking the cells for 90 min on an orbital shaker.The microglia were rinsed and then plated at 40,000–50,000 cells/well in a 96-well plate in 100 ll MEM media with 100 U/ml peni-cillin, 100 lg/ml streptomycin and 10% FBS. Once the microglia

114 L.C. Loram et al. / Brain, Behavior, and Immunity 33 (2013) 112–122
were removed, DMEM (10% FBS and 5 mM L-LME) was added tothe flasks containing the astrocytes. The cells were incubated for2 h to remove any residual microglia remaining in the flask. Thecells were washed with DPBS twice and removed with trypsin(0.05%) for 5 min. The cells were centrifuged at 200g for 10 minat RT. The cells were resuspended in DMEM/F12 (100 U/ml penicil-lin, 100 lg/ml streptomycin, 10% FBS). The cells were counted withtrypan exclusion and plated in 24-well tissue culture plates in 1 mlmedia at 100,000 cells/well. All cells were incubated for 48 h at37 �C and 5% CO2 until the experiment was conducted.
2.6. Enzyme linked immunosorbant assay (ELISA)
IL-10 protein in rat CSF was analyzed using a commerciallyavailable ELISA kit specific for rat IL-10 (R&D Systems, Minneapolis,MN, USA). TNFa and IL-10 protein were analyzed in the superna-tant of the glial cultures using a commercially available ELISA kitspecific for rat TNFa and rat IL-10 (R&D Systems, Minneapolis,MN, USA). The sensitivity of the TNFa assay is 5 pg/ml and for IL-10 is 10 pg/ml.
2.7. Statistical analysis
Behavioral measures were normalized as described above andanalyzed using repeated measures 2-way ANOVA with time andtreatment as main effects. ELISA data from the CSF were analyzedusing an unpaired t-test. Bonferroni post-hoc tests were usedwhere appropriate and P < 0.05 was considered statisticallysignificant.
2.8. Experimental procedures
2.8.1. Experiment 1: Effect of A1R and A2BR agonist on peripheralneuropathy-induced mechanical allodynia
Baseline behavioral measures were recorded after 4 days of40 min/day habituation to the testing environment. CCI or shamsurgery was then conducted and behavioral responses to mechan-ical stimuli or thermal stimuli were tested, in separate groups ofrats, at 4 and 10 days after surgery. At 10–14 days after surgery,an acute IT administration of CCPA (A1R agonist) at 1 or 10 pmol,or BAY606583 (A2BR agonist) at 1 or 10 pmol or equivolume vehi-cle was given (n = 4–6 rats per group) in groups tested for mechan-ical allodynia. The behavioral responses were measured 1, 3, 24,72 h and 1 week after CCPA administration and 1, 3 days andweekly for 5 weeks after BAY606583 administration.
2.8.2. Experiment 2: Effect of A2AR agonist on spinal nerve ligation,sciatic inflammatory neuropathy and established chronic constrictioninjury-induced mechanical allodynia
Baseline behavioral measures were recorded after 4 days of40 min/day habituation to the testing environment.
2.8.2.1. CCI. CCI or sham surgery was then conducted and behav-ioral responses to mechanical stimuli were tested, 1, 2, 4 and6 weeks after surgery. The rats were then injected IT withATL313 (0, 1 or 10 pmol in 1 ll) under brief isoflurane anesthesia.Behavioral testing was done 3 days and weekly for 6 weeks after ITadministration (n = 6–7 rats per group). We have demonstratedpreviously that ATL313 has no effect on sham-operated rats so onlysham + vehicle were included.
2.8.2.2. SNL. SNL surgery was then conducted and behavioral re-sponses to mechanical stimuli were tested 1 and 2 weeks after sur-gery. The rats were then injected IT with ATL313 (0, 1 or 10 pmol in1 ll) under brief isoflurane anesthesia. Behavioral testing was done
3 days and weekly for 6 weeks after IT administration (n = 6–7 ratsper group). Sham-operated rats received vehicle only.
2.8.2.3. SIN. Gel foam wraps were surgically implanted as de-scribed above. 4–5 days after surgery the rats were tested forbehavioral responses. Injections of zymosan (160 lg) over the leftsciatic nerve were performed in awake, unrestrained rats as previ-ously described (Chacur et al., 2001; Milligan et al., 2003). Zymosan(160 lg in 50 ll incomplete Freund’s adjuvant) was injected 4–5 days after surgery and administered every alternate day, fourtimes. ATL313 (10 pmol in 1 ll) or vehicle (IT) was administered24 h after the first zymosan injection, and immediately afterbehavioral testing, under isoflurane anesthesia.
2.8.3. Experiment 3: Effect of A2AR agonist on lipopolysaccharide (LPS)induced TNFa and IL-10 production in microglia and astrocyte cultures
Microglia and astrocytes were separately incubated with LPS (0,100 ng/ml) and ATL313 (0, 0.01 lM, 0.1 lM, 1 lM) for 24, 48 or72 h. The cells were centrifuged at 1000g for 10 min at 4 �C. Thesupernatant was processed for TNFa and IL-10 protein. In a sepa-rate experiment, microglia and astrocytes were either separatelycultured or co-cultured as a mixed glial population in 24 well plateor plated in transwell 24 well plates with the microglia plated onthe insert and the astrocytes in the well. The cultures were incu-bated with 0 or 100 ng/ml LPS and 0 or 1 lM ATL313 for 24 h.The supernatant was collected and processed for TNFa and IL-10protein. For the transwell incubation, the supernatant was col-lected from the media within the well rather than above the insert.
2.8.4. Experiment 4: Effect of A2AR agonist on IL-10 release into the CSFof neuropathic rats
Rats underwent CCI or SNL surgery as described above. For CCIrats 10–14 days after surgery they received ATL313 (10 pmol in1 ll) or equivolume vehicle IT. For the SNL rats, 2 weeks after sur-gery they received ATL313 (10 pmol in 1 ll) or equivolume vehicleIT. 1 week after IT administration, lumbar CSF was collected using asimilar protocol to that described for intrathecal injections. Underisoflurane anesthesia, an 18-g needle was inserted between L5 andL6 vertebrae for use as a guide cannula. PE-10 tubing was insertedinto this guide and threaded rostrally to the lumbosacral enlarge-ment as described for IT injections. A 3 ml syringe with attached30-G needle was connected to the free end of the PE-10 tubingand CSF gently withdrawn into the catheter and syringe. Approxi-mately 100 ll of CSF was obtained from each rat and placed into aneppendorf tube and frozen in liquid nitrogen for IL-10 proteinanalysis.
2.8.5. Experiment 5: Effect of inhibiting PKA and PKC in glial culturesUsing contained single cell cultures allows for the initial explo-
ration of whether protein kinase A or C are involved in the reduc-tion in TNFa following A2AR administration. H-89 is a PKAinhibitor, as it is a competitive inhibitor to the ATP binding siteon the catalytic subunit of the PKA molecule. Chelerythrine, aPKC inhibitor, is a competitive inhibitor on the substrate but anon-competitive inhibitor with respect to ATP (Herbert et al.,1990). To explore their effects on primary glia, microglia culturesand astrocyte cultures were each incubated with LPS (0, 100 ng/ml) and ATL313 (0, 1 lM) for 24 h as described in experiment 3.In addition, PKA inhibitor (H-89, 0, 0.1 and 1 lM) or PKC inhibitor(chelerythrine, 0, 0.1 and 1 lM) were added to the cultures at thesame time as LPS and ATL313. The cells were centrifuged at 1000gfor 10 min at 4 �C. The supernatants were analyzed for TNFa andIL-10 protein.
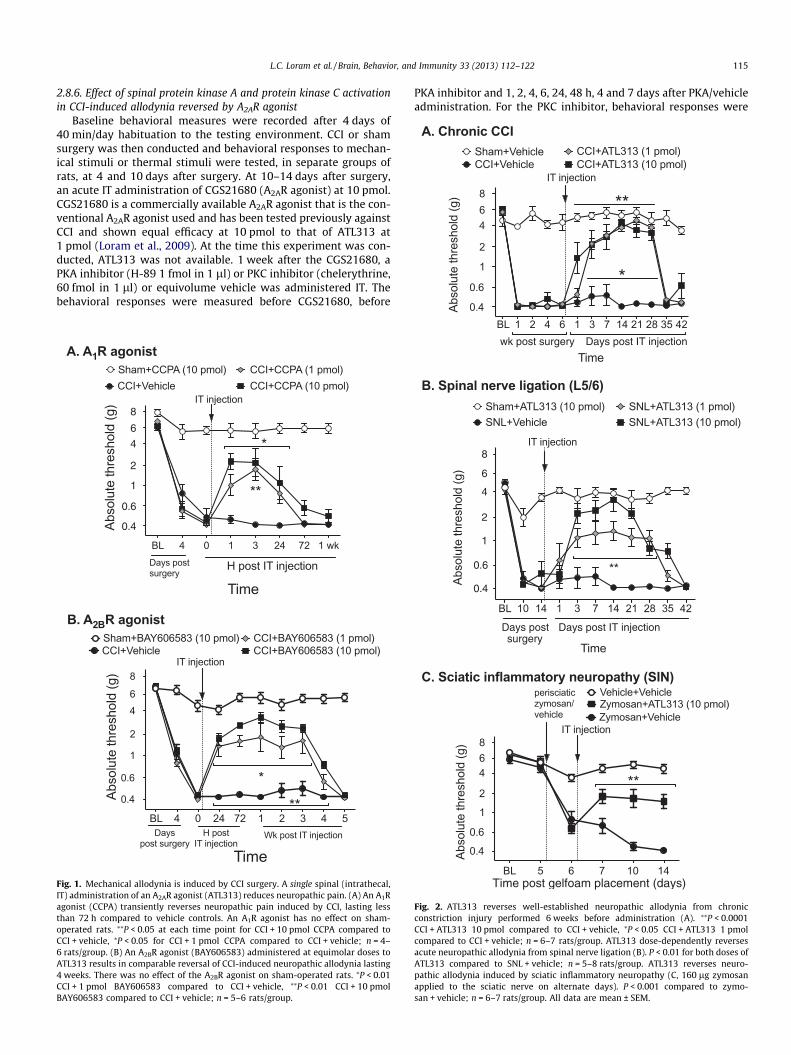
0.4
0.6
1
2
468
CCI+ATL313 (10 pmol)CCI+VehicleSham+Vehicle CCI+ATL313 (1 pmol)
IT injection
*
**
bsol
ute
thre
shol
d (g
)
A. Chronic CCI
L.C. Loram et al. / Brain, Behavior, and Immunity 33 (2013) 112–122 115
2.8.6. Effect of spinal protein kinase A and protein kinase C activationin CCI-induced allodynia reversed by A2AR agonist
Baseline behavioral measures were recorded after 4 days of40 min/day habituation to the testing environment. CCI or shamsurgery was then conducted and behavioral responses to mechan-ical stimuli or thermal stimuli were tested, in separate groups ofrats, at 4 and 10 days after surgery. At 10–14 days after surgery,an acute IT administration of CGS21680 (A2AR agonist) at 10 pmol.CGS21680 is a commercially available A2AR agonist that is the con-ventional A2AR agonist used and has been tested previously againstCCI and shown equal efficacy at 10 pmol to that of ATL313 at1 pmol (Loram et al., 2009). At the time this experiment was con-ducted, ATL313 was not available. 1 week after the CGS21680, aPKA inhibitor (H-89 1 fmol in 1 ll) or PKC inhibitor (chelerythrine,60 fmol in 1 ll) or equivolume vehicle was administered IT. Thebehavioral responses were measured before CGS21680, before
BL 4 0 1 3 24 72 1 wk
0.4
0.6
1
2
4
6
8
CCI+CCPA (10 pmol) CCI+CCPA (1 pmol)
CCI+Vehicle
Days postsurgery
IT injection
H post IT injection
Sham+CCPA (10 pmol)
*
**
Time
Abso
lute
thre
shol
d (g
)
BL 4 0 24 72 1 2 3 4 5
0.4
0.6
1
2
46
8
CCI+VehicleCCI+BAY606583 (1 pmol)CCI+BAY606583 (10 pmol)
H post IT injection
Days post surgery
IT injection
Sham+BAY606583 (10 pmol)
*
**
Wk post IT injection
Time
Abso
lute
thre
shol
d (g
)
B. A2BR agonist
A. A1R agonist
Fig. 1. Mechanical allodynia is induced by CCI surgery. A single spinal (intrathecal,IT) administration of an A2AR agonist (ATL313) reduces neuropathic pain. (A) An A1Ragonist (CCPA) transiently reverses neuropathic pain induced by CCI, lasting lessthan 72 h compared to vehicle controls. An A1R agonist has no effect on sham-operated rats. ⁄⁄P < 0.05 at each time point for CCI + 10 pmol CCPA compared toCCI + vehicle, ⁄P < 0.05 for CCI + 1 pmol CCPA compared to CCI + vehicle; n = 4–6 rats/group. (B) An A2BR agonist (BAY606583) administered at equimolar doses toATL313 results in comparable reversal of CCI-induced neuropathic allodynia lasting4 weeks. There was no effect of the A2BR agonist on sham-operated rats. ⁄P < 0.01CCI + 1 pmol BAY606583 compared to CCI + vehicle, ⁄⁄P < 0.01 CCI + 10 pmolBAY606583 compared to CCI + vehicle; n = 5–6 rats/group.
PKA inhibitor and 1, 2, 4, 6, 24, 48 h, 4 and 7 days after PKA/vehicleadministration. For the PKC inhibitor, behavioral responses were
BL 1 2 4 6 1 3 7 14 21 28 35 42wk post surgery Days post IT injection
Time
A
BL 10 14 1 3 7 14 21 28 35 42
0.4
0.6
1
2
4
6
8
SNL+VehicleSNL+ATL313 (1 pmol)
IT injection
SNL+ATL313 (10 pmol)Sham+ATL313 (10 pmol)
Days postsurgery
Days post IT injection
**
Time
Abso
lute
thre
shol
d (g
)
BL 5 6 7 10 14
0.4
0.6
1
2
468
Zymosan+VehicleZymosan+ATL313 (10 pmol)Vehicle+Vehicleperisciatic
zymosan/vehicle
IT injection
**
Time post gelfoam placement (days)
Abso
lute
thre
shol
d (g
)
C. Sciatic inflammatory neuropathy (SIN)
B. Spinal nerve ligation (L5/6)
Fig. 2. ATL313 reverses well-established neuropathic allodynia from chronicconstriction injury performed 6 weeks before administration (A). ⁄⁄P < 0.0001CCI + ATL313 10 pmol compared to CCI + vehicle, ⁄P < 0.05 CCI + ATL313 1 pmolcompared to CCI + vehicle; n = 6–7 rats/group. ATL313 dose-dependently reversesacute neuropathic allodynia from spinal nerve ligation (B). P < 0.01 for both doses ofATL313 compared to SNL + vehicle; n = 5–8 rats/group. ATL313 reverses neuro-pathic allodynia induced by sciatic inflammatory neuropathy (C, 160 lg zymosanapplied to the sciatic nerve on alternate days). P < 0.001 compared to zymo-san + vehicle; n = 6–7 rats/group. All data are mean ± SEM.

116 L.C. Loram et al. / Brain, Behavior, and Immunity 33 (2013) 112–122
measured before CGS21680, before PKA inhibitor and 20 and40 min, 1, 2, 3, 4, 6 and 24 h after PKC/vehicle administration. BothPKA and PKC inhibitors were tested in sham-operated rats withvehicle to ensure the drug alone induced no allodynia.
3. Results
3.1. Long duration of reversal of neuropathic allodynia extends to A2BRbut not A1R agonism
A first important question is whether this enduring effect orreversal of neuropathic allodynia only occurs with A2AR or does itextend to the other adenosine receptors more generally. BothA2AR and A2BR agonists increase cAMP while A1R and A3R agonistsreduce cAMP. In order to determine if this long duration of effecton reversal of mechanical allodynia only occurs with A2AR or if itoccurs via an increased cAMP such that the same effect would oc-cur with an A2BR agonist, we tested A1R (CCPA) and A2BR (BAY606583) agonists against CCI-induced allodynia, each at two dosesequimolar or higher than ATL313, an A2AR agonist that producesmore than 4 weeks of pain reversal. The A1R agonist, which pre-dominantly affects neurons (Cunha, 2008), transiently reversedallodynia for less than 3 days (Interaction: F21,119 = 7.19,P < 0.001, n = 4–6/gp, Fig. 1A). In contrast, the A2BR agonist pro-duced a prolonged reversal of the allodynia for more than 3 weeks(Interaction: F27,171 = 17.31, P < 0.0001, n = 5/6/gp, Fig. 1B) com-pared to CCI + vehicle. The results of the A2BR agonist are compara-ble to that seen by ATL313 (Loram et al., 2009). Thus, thephenomenon of the long-duration effect of A2AR agonism can begeneralized to adenosine receptors elevating cAMP, thus beyondjust A2AR.
3.2. A2AR agonism produced long duration reversal of allodynia inmultiple models of neuropathic pain
The results presented above demonstrate that the long durationreversal of mechanical allodynia occurs following agonism of aden-osine receptors (A2AR and A2BR) that increase cAMP. The remainderof the experiments were designed to further explore the effects
0 50 700 800 900
Vehicle+Vehicle
Vehicle+ATL313 (1 µM)
LPS+ATL313 (1 µM)
LPS+ATL313 (0.1 µM)
LPS+ATL313 (0.01 µM)
LPS+Vehicle
**
******
TNF pg/ml
0 50 100 150
Vehicle+Vehicle
Vehicle+ATL313 (1 µM)
LPS+ATL313 (1 µM)
LPS+ATL313 (0.1 µM)
LPS+ATL313 (0.01 µM)
LPS+Vehicle
**
IL-10 (pg/ml)
V
L
V
L
A. TNF from microglia
C. IL-10 from microglia
Fig. 3. An A2AR agonist downregulates TNFa in central immune cells. (A) TNFa release (pgis attenuated by co-administration of ATL313. n = 3/4 wells/group and the experimeLPS + vehicle. IL-10 release (pg/ml) from neonatal cortical microglia (C) and astrocytesupregulated by co-administration of ATL313. ⁄P < 0.05, ⁄⁄P < 0.01. ⁄⁄⁄P < 0.001 compared t
following A2AR agonism given its long duration of effect. If A2ARagonists have potential clinical relevance, their effects must gener-alize to other models with distinct underlying mechanisms (Kimet al., 1997), as well as to long-established neuropathic pain. Todate, only an acute CCI model has been tested (Loram et al.,2009). Here, a single IT ATL313 injection was tested against long-established CCI (6 weeks post-injury), spinal nerve ligation (SNL),and sciatic inflammatory neuropathy (Fig. 2). The A2AR agonist,ATL313, at both doses tested, produced a prolonged reversal ofallodynia for at least 4 weeks in the chronic CCI rats (Interaction:F36,252 = 16.06, P < 0.0001, n = 6/gp, Fig. 2A) compared to CCI + vehi-cle. The same duration of effect of both doses of ATl313 was iden-tified in the SNL rats (Interaction: F30,240 = 7.86, P < 0.001, n = 5–8/gp, Fig. 2B). In the SIN model, ATL313 significantly reversed themechanical allodynia for 1 week after IT administration, whichwas the full duration of the experiment (Interaction:F10,95 = 14.14, P < 0.0001, n = 6–7/gp, Fig. 2C).
3.3. A2AR agonism decreases TNFa release from glial cells in vitro
While diverse CNS cell types express A2AR (Fredholm et al.,2007; Palmer and Trevethick, 2008), we have shown previouslythat IT A2AR agonism reduced glial activation in the spinal cordof rats with CCI (Loram et al., 2009). A2AR agonists can attenuatepro-inflammatory cytokine production induced in microglial cellsbut it is not known if there is an upregulation of IL-10 (Chen andPedata, 2008). This may be important as glial pro-inflammatorycytokines serve a critical role in the development and maintenanceof neuropathic pain (Watkins et al., 2007) and intrathecal IL-10 iseffective in attenuating neuropathic pain (Milligan et al., 2006).Therefore, we tested the effects of an A2AR agonist on proinflam-matory cytokine release in microglia and astrocyte culturesin vitro. When microglia and astrocytes were incubated separatelyfor 24 h, 1 lM ATL313 significantly attenuated LPS-stimulatedTNFa release on the microglia (F5,11 = 379.2, P < 0.0001, Fig. 3A)and all doses of ATL313 significantly attenuated TNFa release fromthe astrocytes (F5,18 = 573.2, P < 0.0001, Fig. 3B). LPS significantlyincreased IL-10 release (F5,12 = 4.03, P < 0.05, Fig. 3C). There wasno significant effect of ATL313 on IL-10 release in the presence or
0 25 500 600 700 800
Vehicle+Vehicle
ehicle+ATL313 (1 µM)
LPS+ATL313 (1 µM)
LPS+ATL313 (0.1 µM)
PS+ATL313 (0.01 µM)
LPS+Vehicle
***
******
******
TNF (pg/ml)
40 50 60 70 80
Vehicle+Vehicle
ehicle+ATL313 (1 µM)
LPS+ATL313 (1 µM)
LPS+ATL313 (0.1 µM)
PS+ATL313 (0.01 µM)
LPS+Vehicle
******
********
IL-10 (pg/ml)
B. TNF from astrocytes
D. IL-10 from astrocytes
/ml) from neonatal cortical microglia and (B) astrocytes incubated for 24 h with LPSnt was replicated at least twice. ⁄P < 0.05, ⁄⁄P < 0.01. ⁄⁄⁄P < 0.001 compared to
(D) incubated for 24 h with LPS is upregulated and maintained by ATL313 but noto LPS + vehicle. n = 3/4 wells/group and the experiment was replicated at least twice.
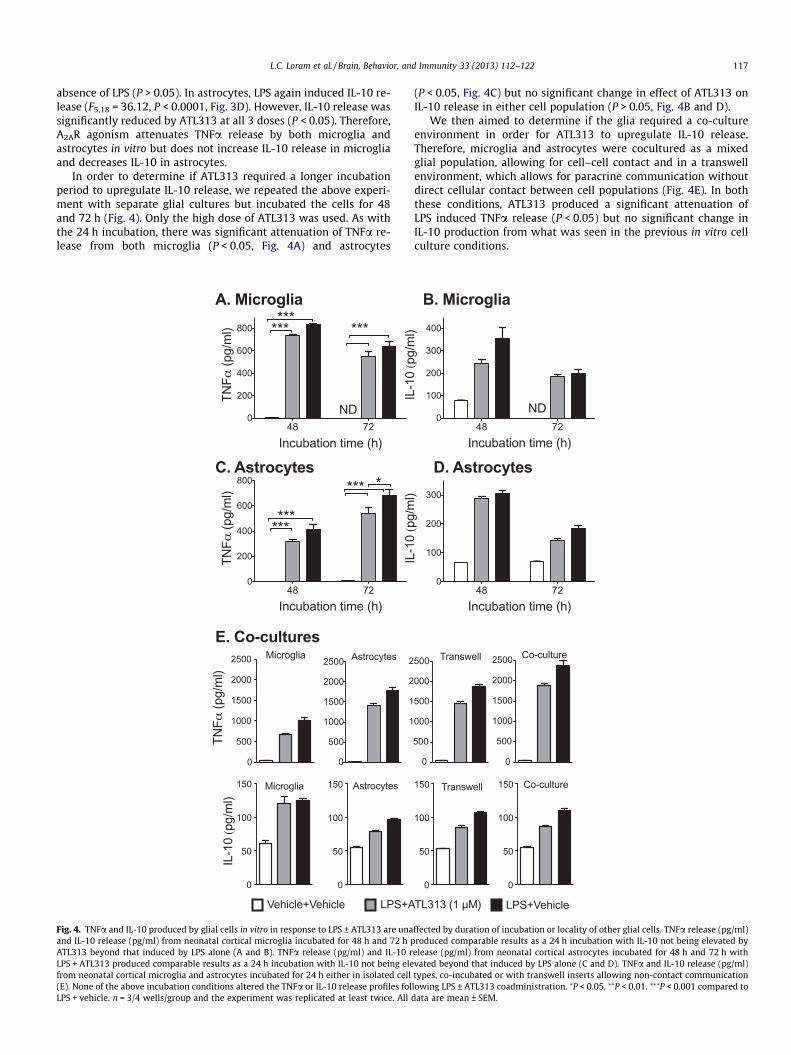
L.C. Loram et al. / Brain, Behavior, and Immunity 33 (2013) 112–122 117
absence of LPS (P > 0.05). In astrocytes, LPS again induced IL-10 re-lease (F5,18 = 36.12, P < 0.0001, Fig. 3D). However, IL-10 release wassignificantly reduced by ATL313 at all 3 doses (P < 0.05). Therefore,A2AR agonism attenuates TNFa release by both microglia andastrocytes in vitro but does not increase IL-10 release in microgliaand decreases IL-10 in astrocytes.
In order to determine if ATL313 required a longer incubationperiod to upregulate IL-10 release, we repeated the above experi-ment with separate glial cultures but incubated the cells for 48and 72 h (Fig. 4). Only the high dose of ATL313 was used. As withthe 24 h incubation, there was significant attenuation of TNFa re-lease from both microglia (P < 0.05, Fig. 4A) and astrocytes
48 720
200
400
600
800
ND
*********
Incubation time (h)
TNF
(pg/
ml)
48 720
200
400
600
800 *
***
***
***
Incubation time (h)
TNF
(pg/
ml)
A. Microglia
0
500
1000
1500
2000
2500 Microglia
TNF
(pg/
ml)
0
50
100
150 Microglia
IL-1
0 (p
g/m
l)
0
500
1000
1500
2000
2500 Astrocytes
1
1
2
2
0
50
100
150 Astrocytes
LPS+AVehicle+Vehicle
C. Astrocytes
E. Co-cultures
Fig. 4. TNFa and IL-10 produced by glial cells in vitro in response to LPS ± ATL313 are unaand IL-10 release (pg/ml) from neonatal cortical microglia incubated for 48 h and 72 h pATL313 beyond that induced by LPS alone (A and B). TNFa release (pg/ml) and IL-10 rLPS + ATL313 produced comparable results as a 24 h incubation with IL-10 not being elefrom neonatal cortical microglia and astrocytes incubated for 24 h either in isolated cell(E). None of the above incubation conditions altered the TNFa or IL-10 release profiles folLPS + vehicle. n = 3/4 wells/group and the experiment was replicated at least twice. All
(P < 0.05, Fig. 4C) but no significant change in effect of ATL313 onIL-10 release in either cell population (P > 0.05, Fig. 4B and D).
We then aimed to determine if the glia required a co-cultureenvironment in order for ATL313 to upregulate IL-10 release.Therefore, microglia and astrocytes were cocultured as a mixedglial population, allowing for cell–cell contact and in a transwellenvironment, which allows for paracrine communication withoutdirect cellular contact between cell populations (Fig. 4E). In boththese conditions, ATL313 produced a significant attenuation ofLPS induced TNFa release (P < 0.05) but no significant change inIL-10 production from what was seen in the previous in vitro cellculture conditions.
48 720
100
200
300
400
ND
Incubation time (h)
IL-1
0 (p
g/m
l)
48 720
100
200
300
Incubation time (h)
IL-1
0 (p
g/m
l)
0
500
000
500
000
500 Transwell
0
500
1000
1500
2000
2500 Co-culture
0
50
100
150 Transwell
0
50
100
150 Co-culture
TL313 (1 µM) LPS+Vehicle
B. Microglia
D. Astrocytes
ffected by duration of incubation or locality of other glial cells. TNFa release (pg/ml)roduced comparable results as a 24 h incubation with IL-10 not being elevated by
elease (pg/ml) from neonatal cortical astrocytes incubated for 48 h and 72 h withvated beyond that induced by LPS alone (C and D). TNFa and IL-10 release (pg/ml)types, co-incubated or with transwell inserts allowing non-contact communicationlowing LPS ± ATL313 coadministration. ⁄P < 0.05, ⁄⁄P < 0.01. ⁄⁄⁄P < 0.001 compared todata are mean ± SEM.
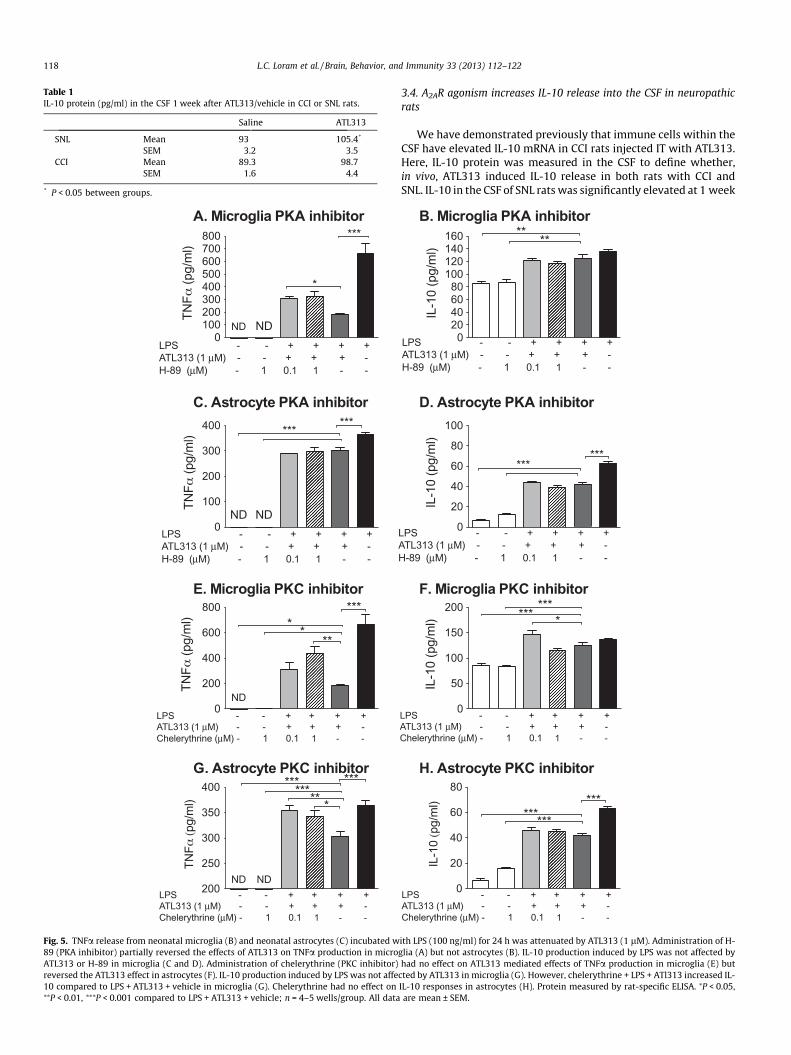
Table 1IL-10 protein (pg/ml) in the CSF 1 week after ATL313/vehicle in CCI or SNL rats.
Saline ATL313
SNL Mean 93 105.4*
SEM 3.2 3.5CCI Mean 89.3 98.7
SEM 1.6 4.4
* P < 0.05 between groups.
0100200300400500600700800
ND ND
*
***
++++--SPLATL313 (1 µM) - - + + + -H-89 (µM) - 1 0.1 1 - -
TNF
(pg/
ml)
0
100
200
300
400 ******
ND ND++++--SPL
ATL313 (1 µM) - - + + + -H-89 (µM) - 1 0.1 1 - -
TNF
(pg/
ml)
0
200
400
600
800
++++--SPLATL313 (1 µM) - - + + + -Chelerythrine (µM) - 1 0.1 1 - -
ND
TNF
(pg/
ml) *
***
***
200
250
300
350
400
TNF
(pg/
ml)
++++--SPLATL313 (1 µM) - - + + + -Chelerythrine (µM) - 1 0.1 1 - -
NDND
*
***********
A. Microglia PKA inhibitor
C. Astrocyte PKA inhibitor
E. Microglia PKC inhibitor
G. Astrocyte PKC inhibitor
Fig. 5. TNFa release from neonatal microglia (B) and neonatal astrocytes (C) incubated w89 (PKA inhibitor) partially reversed the effects of ATL313 on TNFa production in microATL313 or H-89 in microglia (C and D). Administration of chelerythrine (PKC inhibitor)reversed the ATL313 effect in astrocytes (F). IL-10 production induced by LPS was not affe10 compared to LPS + ATL313 + vehicle in microglia (G). Chelerythrine had no effect on⁄⁄P < 0.01, ⁄⁄⁄P < 0.001 compared to LPS + ATL313 + vehicle; n = 4–5 wells/group. All data
118 L.C. Loram et al. / Brain, Behavior, and Immunity 33 (2013) 112–122
3.4. A2AR agonism increases IL-10 release into the CSF in neuropathicrats
We have demonstrated previously that immune cells within theCSF have elevated IL-10 mRNA in CCI rats injected IT with ATL313.Here, IL-10 protein was measured in the CSF to define whether,in vivo, ATL313 induced IL-10 release in both rats with CCI andSNL. IL-10 in the CSF of SNL rats was significantly elevated at 1 week
020406080
100120140160 **
**
++++--SPLATL313 (1 µM) - - + + + -H-89 (µM) - 1 0.1 1 - -
IL-1
0 (p
g/m
l)
0
20
40
60
80
100
******
++++--SPLATL313 (1 µM) - - + + + -H-89 (µM) - 1 0.1 1 - -
IL-1
0 (p
g/m
l)
0
20
40
60
80
++++--SPLATL313 (1 µM) - - + + + -Chelerythrine (µM) - 1 0.1 1 - -
IL-1
0 (p
g/m
l)
*********
0
50
100
150
200
++++--SPLATL313 (1 µM) - - + + + -Chelerythrine (µM) - 1 0.1 1 - -
IL-1
0 (p
g/m
l) ****
***
B. Microglia PKA inhibitor
D. Astrocyte PKA inhibitor
H. Astrocyte PKC inhibitor
F. Microglia PKC inhibitor
ith LPS (100 ng/ml) for 24 h was attenuated by ATL313 (1 lM). Administration of H-glia (A) but not astrocytes (B). IL-10 production induced by LPS was not affected byhad no effect on ATL313 mediated effects of TNFa production in microglia (E) but
cted by ATL313 in microglia (G). However, chelerythrine + LPS + ATl313 increased IL-IL-10 responses in astrocytes (H). Protein measured by rat-specific ELISA. ⁄P < 0.05,are mean ± SEM.
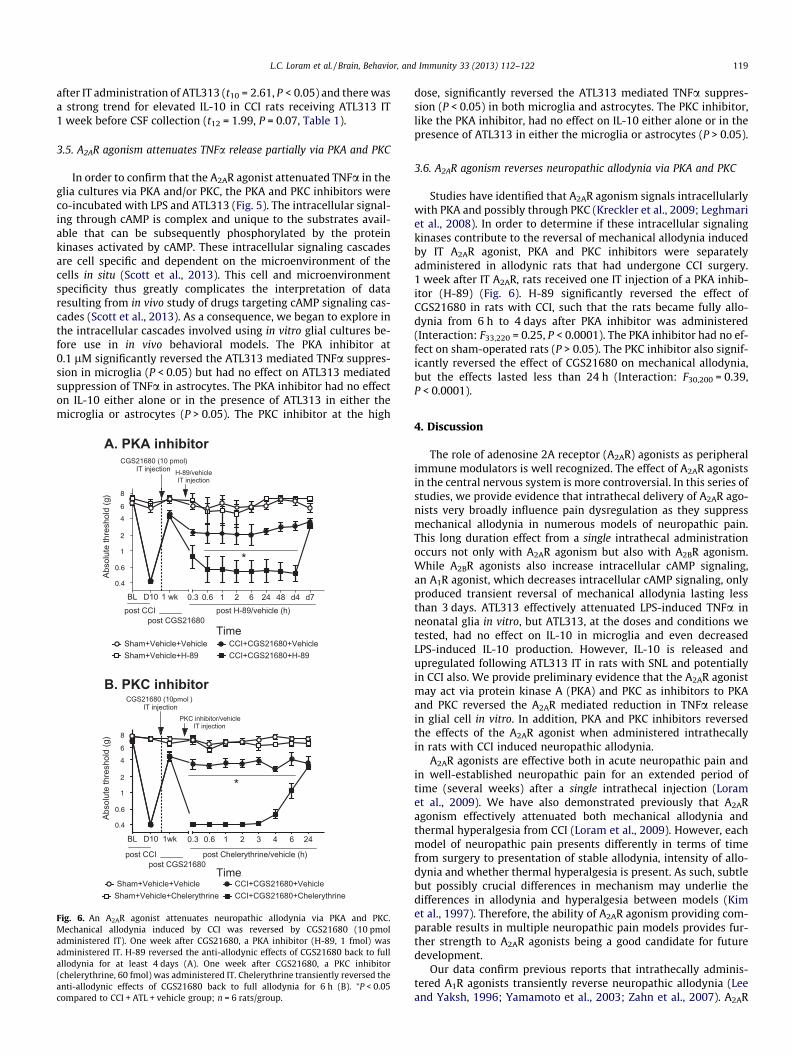
L.C. Loram et al. / Brain, Behavior, and Immunity 33 (2013) 112–122 119
after IT administration of ATL313 (t10 = 2.61, P < 0.05) and there wasa strong trend for elevated IL-10 in CCI rats receiving ATL313 IT1 week before CSF collection (t12 = 1.99, P = 0.07, Table 1).
3.5. A2AR agonism attenuates TNFa release partially via PKA and PKC
In order to confirm that the A2AR agonist attenuated TNFa in theglia cultures via PKA and/or PKC, the PKA and PKC inhibitors wereco-incubated with LPS and ATL313 (Fig. 5). The intracellular signal-ing through cAMP is complex and unique to the substrates avail-able that can be subsequently phosphorylated by the proteinkinases activated by cAMP. These intracellular signaling cascadesare cell specific and dependent on the microenvironment of thecells in situ (Scott et al., 2013). This cell and microenvironmentspecificity thus greatly complicates the interpretation of dataresulting from in vivo study of drugs targeting cAMP signaling cas-cades (Scott et al., 2013). As a consequence, we began to explore inthe intracellular cascades involved using in vitro glial cultures be-fore use in in vivo behavioral models. The PKA inhibitor at0.1 lM significantly reversed the ATL313 mediated TNFa suppres-sion in microglia (P < 0.05) but had no effect on ATL313 mediatedsuppression of TNFa in astrocytes. The PKA inhibitor had no effecton IL-10 either alone or in the presence of ATL313 in either themicroglia or astrocytes (P > 0.05). The PKC inhibitor at the high
BL D10 1 wk 0.3 0.6 1 2 6 24 48 d4 d7
0.4
0.6
1
2
4
6
8
CGS21680 (10 pmol)IT injection H-89/vehicle
IT injection
post CCIpost CGS21680
post H-89/vehicle (h)
CCI+CGS21680+VehicleCCI+CGS21680+H-89Sham+Vehicle+H-89
Sham+Vehicle+Vehicle
*
Time
Abso
lute
thre
shol
d (g
)
BL D10 1wk 0.3 0.6 1 2 3 4 6 24
0.4
0.6
1
2
4
6
8
CGS21680 (10 pmol )IT injection
PKC inhibitor/vehicle IT injection
CCI+CGS21680+VehicleCCI+CGS21680+ChelerythrineSham+Vehicle+Chelerythrine
Sham+Vehicle+Vehicle
post CCIpost CGS21680
post Chelerythrine/vehicle (h)
*
Time
Abso
lute
thre
shol
d (g
)
A. PKA inhibitor
B. PKC inhibitor
Fig. 6. An A2AR agonist attenuates neuropathic allodynia via PKA and PKC.Mechanical allodynia induced by CCI was reversed by CGS21680 (10 pmoladministered IT). One week after CGS21680, a PKA inhibitor (H-89, 1 fmol) wasadministered IT. H-89 reversed the anti-allodynic effects of CGS21680 back to fullallodynia for at least 4 days (A). One week after CGS21680, a PKC inhibitor(chelerythrine, 60 fmol) was administered IT. Chelerythrine transiently reversed theanti-allodynic effects of CGS21680 back to full allodynia for 6 h (B). ⁄P < 0.05compared to CCI + ATL + vehicle group; n = 6 rats/group.
dose, significantly reversed the ATL313 mediated TNFa suppres-sion (P < 0.05) in both microglia and astrocytes. The PKC inhibitor,like the PKA inhibitor, had no effect on IL-10 either alone or in thepresence of ATL313 in either the microglia or astrocytes (P > 0.05).
3.6. A2AR agonism reverses neuropathic allodynia via PKA and PKC
Studies have identified that A2AR agonism signals intracellularlywith PKA and possibly through PKC (Kreckler et al., 2009; Leghmariet al., 2008). In order to determine if these intracellular signalingkinases contribute to the reversal of mechanical allodynia inducedby IT A2AR agonist, PKA and PKC inhibitors were separatelyadministered in allodynic rats that had undergone CCI surgery.1 week after IT A2AR, rats received one IT injection of a PKA inhib-itor (H-89) (Fig. 6). H-89 significantly reversed the effect ofCGS21680 in rats with CCI, such that the rats became fully allo-dynia from 6 h to 4 days after PKA inhibitor was administered(Interaction: F33,220 = 0.25, P < 0.0001). The PKA inhibitor had no ef-fect on sham-operated rats (P > 0.05). The PKC inhibitor also signif-icantly reversed the effect of CGS21680 on mechanical allodynia,but the effects lasted less than 24 h (Interaction: F30,200 = 0.39,P < 0.0001).
4. Discussion
The role of adenosine 2A receptor (A2AR) agonists as peripheralimmune modulators is well recognized. The effect of A2AR agonistsin the central nervous system is more controversial. In this series ofstudies, we provide evidence that intrathecal delivery of A2AR ago-nists very broadly influence pain dysregulation as they suppressmechanical allodynia in numerous models of neuropathic pain.This long duration effect from a single intrathecal administrationoccurs not only with A2AR agonism but also with A2BR agonism.While A2BR agonists also increase intracellular cAMP signaling,an A1R agonist, which decreases intracellular cAMP signaling, onlyproduced transient reversal of mechanical allodynia lasting lessthan 3 days. ATL313 effectively attenuated LPS-induced TNFa inneonatal glia in vitro, but ATL313, at the doses and conditions wetested, had no effect on IL-10 in microglia and even decreasedLPS-induced IL-10 production. However, IL-10 is released andupregulated following ATL313 IT in rats with SNL and potentiallyin CCI also. We provide preliminary evidence that the A2AR agonistmay act via protein kinase A (PKA) and PKC as inhibitors to PKAand PKC reversed the A2AR mediated reduction in TNFa releasein glial cell in vitro. In addition, PKA and PKC inhibitors reversedthe effects of the A2AR agonist when administered intrathecallyin rats with CCI induced neuropathic allodynia.
A2AR agonists are effective both in acute neuropathic pain andin well-established neuropathic pain for an extended period oftime (several weeks) after a single intrathecal injection (Loramet al., 2009). We have also demonstrated previously that A2ARagonism effectively attenuated both mechanical allodynia andthermal hyperalgesia from CCI (Loram et al., 2009). However, eachmodel of neuropathic pain presents differently in terms of timefrom surgery to presentation of stable allodynia, intensity of allo-dynia and whether thermal hyperalgesia is present. As such, subtlebut possibly crucial differences in mechanism may underlie thedifferences in allodynia and hyperalgesia between models (Kimet al., 1997). Therefore, the ability of A2AR agonism providing com-parable results in multiple neuropathic pain models provides fur-ther strength to A2AR agonists being a good candidate for futuredevelopment.
Our data confirm previous reports that intrathecally adminis-tered A1R agonists transiently reverse neuropathic allodynia (Leeand Yaksh, 1996; Yamamoto et al., 2003; Zahn et al., 2007). A2AR

120 L.C. Loram et al. / Brain, Behavior, and Immunity 33 (2013) 112–122
and A2BR, but not A1R, activation enduringly reverses neuropathicpain. As A2BR and A2AR, but not A1R, activation increases cAMP(Hasko and Cronstein, 2004), this suggests that multi-week reversalof pain may potentially be linked to elevated cAMP. Adenosinereceptors are found on all cells within the CNS including on neu-rons. A2AR on neurons are pleiotropic with complex interactionswith other neuropeptide receptors both at the presynaptic andpostsynaptic sites (Sebastiao and Ribeiro, 2009a,b).
Adenosine receptors have been identified in the spinal cordwith the dominant receptor being A1 receptor in the dorsal hornof the spinal cord (Sebastiao et al., 2011). In addition, A2AR havebeen demonstrated to be upregulated in the spinal cord followingspinal cord injury (Paterniti et al., 2011). In the spinal cord injurymodel, the A2AR was colocalized to some neurons, but not all (Pat-erniti et al., 2011). These studies extend to the underlying mecha-nism by which these A2AR agonists work within the spinal cord asdogma dictates that elevated cAMP, as seen following A2AR andA2BR activation, results in increased neuronal excitability (Mominand McNaughton, 2009). The studies examining adenosine recep-tors on neurons have been done from cells or tissue from the brain.Therefore, it is not known if the same results occur from neuronalcells in the spinal cord. Activation of A2AR on neurons enhances therelease of presynaptic glutamate and the subsequent increase incalcium influx. Both can enhance neuronal cell death (Dai andZhou, 2011). There is also evidence of an increase in noradrenalinerelease following A2AR activation and thus an impact on sympa-thetic nervous activity in the spinal cord (Brooke et al., 2004).However, when looking at in vivo models of brain injury or ische-mia, there is bidirectional effect with some studies showing pro-tection and some showing enhancement of injury (Dai and Zhou,2011). If A2AR activation had an effect on neurons in the spinalcord, the effect would most likely increase excitatory neurotrans-mitter release. However, the present evidence suggests that theelevated cAMP within glial cells produces the opposite effect, withan attenuation of pro-inflammatory cytokine production and sub-sequent reduction in pain.
The studies here demonstrate that A2AR agonism in single glialpopulations or co-cultured glia, where functional interactions mayoccur (Schubert et al., 2000), significantly attenuated TNFa. Bothstimulation of peripheral immune cells with cAMP in vitro andinduction of intracellular cAMP by a non-selective adenosine ago-nist (NECA), attenuated LPS induced pro-inflammatory mediators(Kreckler et al., 2009, 2006). In addition, studies with peripheralimmune cell in vitro indicate that elevated cAMP can reduce pro-inflammatory cytokines and upregulate IL-10 through PKA (Krec-kler et al., 2009; Palmer and Trevethick, 2008) and PKC (Dinizet al., 2008) both of which activate the cAMP response elementbinding protein (CREB) among other intracellular mediators (Avniet al., 2010; Wen et al., 2010). The protein kinase inhibitors usedin this studies have predominantly protein kinase A or C inhibition,but do have some non selectivity for other intracellular kinases(Davies et al., 2000). Therefore, the signaling cascade initiated bycAMP may be important to attenuate pro-inflammatory cytokinesin immune cells, but further studies would further elucidate theassociated intracellular signaling cascades.
Numerous studies have identified that in vitro cAMP inducedPKA (Cho et al., 2002; Tortora and Ciardiello, 2002a,b) and to someextent PKC (Goethe et al., 2007; Gold et al., 1998; Hucho et al.,2005) and that PKA induction reduces pro-inflammatory mediatorproduction in a model of traumatic brain injury (Atkins et al.,2007). However, we are the first to demonstrate that the reversalof allodynia by A2AR agonist is in part via PKA activation in vivo.While the interpretation of the in vivo kinase inhibitor data needto be made within the context of the complex interaction of theprotein kinases with numerous substrates (Scott et al., 2013),which is further complicated by the microenvironment in which
it is activated (Scott et al., 2013). The results remain intriguing,as even though the PKA inhibitor did indeed reverse the effectsof the A2AR agonist on neuropathic pain, even after 4 days of rever-sal, the allodynic reversal effects of the A2AR agonist returned.cAMP is also known to increase PKC phosphorylation and thePKC inhibitor was effective in reversing the A2AR mediated reversalof allodynia in vivo, though with shorter duration than that of thePKA inhibitor. It is possible that inhibiting both kinases may resultin a persistent resetting of the immune cells as has been identifiedto occur in peripheral macrophages (Csoka et al., 2012). It is inter-esting to note that at 1 week after the A2AR agonist was adminis-tered, the A2AR antagonist had no effect on the mechanicalallodynia (Loram et al., 2009) but the PKA and PKC inhibitor effec-tively abolished the effect of the A2AR agonist. These results do sug-gest that subsequent to A2AR binding, some enduring intracellularchanges occur downstream of A2AR, but independent of A2AR acti-vation, that induce the long term reversal of the allodynia. Futurestudies to explore whether these intracellular kinases, activatedby numerous cAMP mediated receptors and not just A2AR, arekey in inducing long-term reversal of allodynia (Antoni, 2012).
The PKA and PKC inhibitors significantly suppressed ATL313-in-duced TNFa attenuation in both microglia and astrocytes in vitro,but had no effect on IL-10. Together, these studies suggest thatATL313 may, via PKA and PKC in glial cells in the spinal cord dorsalhorn of neuropathic rats, enduringly suppress pro-inflammatorycytokines. The glial in vitro results presented here contrast withdata from peripheral immune cells where IL-10 protein was upreg-ulated by an A2AR agonist (Csoka et al., 2007; Link et al., 2000) andthat it was mediated by PKA (Kreckler et al., 2009; Palmer andTrevethick, 2008).
Previous studies have demonstrated that microglia stimulatedin vitro with higher doses of adenosine or with an A2BR agonistboth upregulated IL-10 production when co-administered withLPS (Koscso et al., 2012). We have shown that A2AR agonism upreg-ulates IL-10 mRNA in cerebrospinal fluid (CSF) cells (Loram et al.,2009) and here that IL-10 protein was moderately elevated in theCSF 1 week after ATL313 IT in SNL rats. Therefore, elevated CSFIL-10 following ATL313 may not be produced by glia, as we identi-fied in spinal cord mRNA previously, but rather from immunocom-petent cells, largely macrophages, of the intrathecal space (Loramet al., 2009). The in vitro experiments were done in neonatal corti-cal glia. The literature has identified differences in both morphol-ogy and responses to inflammatory stimuli between neonataland adult cortical glia (Loram et al., 2012; Schwarz et al., 2012),with neonatal cortical glia demonstrating a more pro-inflamma-tory phenotype. In rodent models, neonates are less susceptibleto neuropathic pain induction compared to adult rodents, with lessspinal glial activation in the neonatal spinal cord (Moss et al.,2007). However, this does imply that neonatal spinal glia are notexcitable were they to have received the proper activation signals(Costigan et al., 2009). Nonetheless, the neonatal cortical glia, usedin culture conditions were used to mimic a pro-inflammatory envi-ronment to evaluate the effect of A2AR agonist and the subsequentprotein kinases on TNF and IL-10 levels. Therefore, we cannot ex-clude the possibility of adult spinal glia producing IL-10 in vivo.However, the data do demonstrate the effect of A2AR agonismattenuating LPS-induced TNFa production, and that the PKA andPKC inhibitors had the ability to reverse the effects of A2AR agon-ism. The potential of exploring the effect of A2AR agonists on spinalglia ex vivo is worth future investigation.
Together, these data demonstrate that single IT A2AR agonistdoses enduringly attenuate neuropathic pain across multiple mod-els. The presence of an A2AR agonist does not alter normal cellularresponses when the cells are in a homeostatic surveying state.Thus, the potent effect seen by A2AR agonism requires priorneuroinflammation. The proposed underlying mechanism involves

L.C. Loram et al. / Brain, Behavior, and Immunity 33 (2013) 112–122 121
a significant downregulation of TNFa, by glia, as demonstratedhere, and CSF immune cells (Loram et al., 2009). A modest eleva-tion in IL-10 in the CSF may be produced by immune cells withinthe CSF but may not be produced by glial cells. This unique effecthas implications in understanding the role of A2AR in modulatingneuropathic pain.
Acknowledgments
Financial support for these studies was provided by DogwoodPharmaceuticals, LLC, Bayer Pharmaceuticals and by NIH grantsDA02422 and RC1-NS067807 and Department of Defense grantDM102108.
References
Antoni, F.A., 2012. New paradigms in cAMP signalling. Mol. Cell. Endocrinol. 353, 3–9.
Atkins, C.M., Oliva Jr., A.A., Alonso, O.F., Pearse, D.D., Bramlett, H.M., Dietrich, W.D.,2007. Modulation of the cAMP signaling pathway after traumatic brain injury.Exp. Neurol. 208, 145–158.
Avni, D., Ernst, O., Philosoph, A., Zor, T., 2010. Role of CREB in modulation ofTNFalpha and IL-10 expression in LPS-stimulated RAW264.7 macrophages. Mol.Immunol. 47, 1396–1403.
Bennett, G.J., Xie, Y.K., 1988. A peripheral mononeuropathy in rat that producesdisorders of pain sensation like those seen in man. Pain 33, 87–107.
Brooke, R.E., Deuchars, J., Deuchars, S.A., 2004. Input-specific modulation ofneurotransmitter release in the lateral horn of the spinal cord via adenosinereceptors. J. Neurosci. 24, 127–137.
Chacur, M., Milligan, E.D., Gazda, L.S., Armstrong, C., Wang, H., Tracey, K.J., Maier,S.F., Watkins, L.R., 2001. A new model of sciatic inflammatory neuritis (SIN):induction of unilateral and bilateral mechanical allodynia following acuteunilateral peri-sciatic immune activation in rats. Pain 94, 231–244.
Chaplan, S.R., Pogrel, J.W., Yaksh, T.L., 1994. Role of voltage-dependent calciumchannel subtypes in experimental tactile allodynia. J. Pharmacol. Exp. Ther. 269,1117–1123.
Chen, J.F., Pedata, F., 2008. Modulation of ischemic brain injury andneuroinflammation by adenosine A2A receptors. Curr. Pharm. Des. 14, 1490–1499.
Cho, Y.S., Kim, M.K., Tan, L., Srivastava, R., Agrawal, S., Cho-Chung, Y.S., 2002. Proteinkinase A RIalpha antisense inhibition of PC3M prostate cancer cell growth: Bcl-2hyperphosphorylation, Bax up-regulation, and Bad-hypophosphorylation. Clin.Cancer Res. 8, 607–614.
Costigan, M., Moss, A., Latremoliere, A., Johnston, C., Verma-Gandhu, M., Herbert,T.A., Barrett, L., Brenner, G.J., Vardeh, D., Woolf, C.J., Fitzgerald, M., 2009. T-cellinfiltration and signaling in the adult dorsal spinal cord is a major contributor toneuropathic pain-like hypersensitivity. J. Neurosci. 29, 14415–14422.
Csoka, B., Nemeth, Z.H., Virag, L., Gergely, P., Leibovich, S.J., Pacher, P., Sun, C.X.,Blackburn, M.R., Vizi, E.S., Deitch, E.A., Hasko, G., 2007. A2A adenosine receptorsand C/EBPbeta are crucially required for IL-10 production by macrophagesexposed to Escherichia coli. Blood 110, 2685–2695.
Csoka, B., Selmeczy, Z., Koscso, B., Nemeth, Z.H., Pacher, P., Murray, P.J., Kepka-Lenhart, D., Morris Jr., S.M., Gause, W.C., Leibovich, S.J., Hasko, G., 2012.Adenosine promotes alternative macrophage activation via A2A and A2Breceptors. FASEB J. 26, 376–386.
Cunha, R.A., 2008. Different cellular sources and different roles of adenosine: A1receptor-mediated inhibition through astrocytic-driven volume transmissionand synapse-restricted A2A receptor-mediated facilitation of plasticity.Neurochem. Int. 52, 65–72.
Dai, S.S., Zhou, Y.G., 2011. Adenosine 2A receptor: a crucial neuromodulator withbidirectional effect in neuroinflammation and brain injury. Rev. Neurosci. 22,231–239.
Dare, E., Schulte, G., Karovic, O., Hammarberg, C., Fredholm, B.B., 2007. Modulationof glial cell functions by adenosine receptors. Physiol. Behav. 92, 15–20.
Davies, S.P., Reddy, H., Caivano, M., Cohen, P., 2000. Specificity and mechanism ofaction of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105.
Diniz, C., Borges, F., Santana, L., Uriarte, E., Oliveira, J.M., Goncalves, J., Fresco, P.,2008. Ligands and therapeutic perspectives of adenosine A(2A) receptors. Curr.Pharm. Des. 14, 1698–1722.
Fredholm, B.B., Chern, Y., Franco, R., Sitkovsky, M., 2007. Aspects of the generalbiology of adenosine A2A signaling. Prog. Neurobiol. 83, 263–276.
Global Industry Analysts, I., 2011. Global Pain Management Market to Reach US$60Billion by 2015. Global Industry Analysts, I.
Goethe, R., Basler, T., Phi-van, L., 2007. Regulation of C/EBPbeta mRNA expressionand C/EBPbeta promoter activity by protein kinases A and C in amyelomonocytic cell line (HD11). Inflamm. Res. 56, 274–281.
Gold, M.S., Levine, J.D., Correa, A.M., 1998. Modulation of TTX-R INa by PKC and PKAand their role in PGE2-induced sensitization of rat sensory neurons in vitro. J.Neurosci. 18, 10345–10355.
Harvey, L.O., 1986. Efficient estimation of sensory thresholds. Behav. Res. Meth.Instrum. Comput. 18, 623–632.
Hasko, G., Cronstein, B.N., 2004. Adenosine: an endogenous regulator of innateimmunity. Trends Immunol. 25, 33–39.
Herbert, J.M., Augereau, J.M., Gleye, J., Maffrand, J.P., 1990. Chelerythrine is a potentand specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 172,993–999.
Hucho, T.B., Dina, O.A., Levine, J.D., 2005. Epac mediates a cAMP-to-PKC signaling ininflammatory pain: an isolectin B4(+) neuron-specific mechanism. J. Neurosci.25, 6119–6126.
Hutchinson, M.R., Loram, L.C., Zhang, Y., Shridhar, M., Rezvani, N., Berkelhammer, D.,Phipps, S., Foster, P.S., Landgraf, K., Falke, J.J., Rice, K.C., Maier, S.F., Yin, H.,Watkins, L.R., 2010. Evidence that tricyclic small molecules may possess toll-like receptor and myeloid differentiation protein 2 activity. Neuroscience 168,551–563.
Kim, K.J., Yoon, Y.W., Chung, J.M., 1997. Comparison of three rodent neuropathicpain models. Exp. Brain Res. 113, 200–206.
Kim, S.H., Chung, J.M., 1992. An experimental model for peripheral neuropathyproduced by segmental spinal nerve ligation in the rat. Pain 50, 355–363.
Koscso, B., Csoka, B., Selmeczy, Z., Himer, L., Pacher, P., Virag, L., Hasko, G., 2012.Adenosine augments IL-10 production by microglial cells through an A2Badenosine receptor-mediated process. J. Immunol. 188, 445–453.
Kreckler, L.M., Gizewski, E., Wan, T.C., Auchampach, J.A., 2009. Adenosinesuppresses lipopolysaccharide-induced tumor necrosis factor-alphaproduction by murine macrophages through a protein kinase A- andexchange protein activated by cAMP-independent signaling pathway. J.Pharmacol. Exp. Ther. 331, 1051–1061.
Kreckler, L.M., Wan, T.C., Ge, Z.D., Auchampach, J.A., 2006. Adenosine inhibits tumornecrosis factor-alpha release from mouse peritoneal macrophages via A2A andA2B but not the A3 adenosine receptor. J. Pharmacol. Exp. Ther. 317, 172–180.
Lee, Y.W., Yaksh, T.L., 1996. Pharmacology of the spinal adenosine receptor whichmediates the antiallodynic action of intrathecal adenosine agonists. J.Pharmacol. Exp. Ther. 277, 1642–1648.
Leghmari, K., Bennasser, Y., Tkaczuk, J., Bahraoui, E., 2008. HIV-1 Tat protein inducesIL-10 production by an alternative TNF-alpha-independent pathway inmonocytes: Role of PKC-delta and p38 MAP kinase. Cell. Immunol. 253 (1–2),45–53.
Link, A.A., Kino, T., Worth, J.A., McGuire, J.L., Crane, M.L., Chrousos, G.P., Wilder, R.L.,Elenkov, I.J., 2000. Ligand-activation of the adenosine A2a receptors inhibits IL-12 production by human monocytes. J. Immunol. 164, 436–442.
Loram, L.C., Harrison, J.A., Sloane, E.M., Hutchinson, M.R., Sholar, P., Taylor, F.R.,Berkelhammer, D., Coats, B.D., Poole, S., Milligan, E.D., Maier, S.F., Rieger, J.,Watkins, L.R., 2009. Enduring reversal of neuropathic pain by a single intrathecalinjection of adenosine 2A receptor agonists: a novel therapy for neuropathicpain. J. Neurosci. 29, 14015–14025.
Loram, L.C., Sholar, P.W., Taylor, F.R., Wiesler, J.L., Babb, J.A., Strand, K.A.,Berkelhammer, D., Day, H.E., Maier, S.F., Watkins, L.R., 2012. Sex and estradiolinfluence glial pro-inflammatory responses to lipopolysaccharide in rats.Psychoneuroendocrinology 37, 1688–1699.
Milligan, E.D., Maier, S.F., Watkins, L.R., 2004a. Sciatic inflammatory neuropathy inthe rat: surgical procedures, induction of inflammation, and behavioral testing.Methods Mol. Med. 99, 67–89.
Milligan, E.D., Mehmert, K.K., Hinde, J.L., Harvey, L.O., Martin, D., Tracey, K.J., Maier,S.F., Watkins, L.R., 2000. Thermal hyperalgesia and mechanical allodyniaproduced by intrathecal administration of the human immunodeficiencyvirus-1 (HIV-1) envelope glycoprotein, gp120. Brain Res. 861, 105–116.
Milligan, E.D., Sloane, E.M., Langer, S.J., Hughes, T.S., Jekich, B.M., Frank, M.G.,Mahoney, J.H., Levkoff, L.H., Maier, S.F., Cruz, P.E., Flotte, T.R., Johnson, K.W.,Mahoney, M.M., Chavez, R.A., Leinwand, L.A., Watkins, L.R., 2006. Repeatedintrathecal injections of plasmid DNA encoding interleukin-10 produceprolonged reversal of neuropathic pain. Pain 126, 294–308.
Milligan, E.D., Twining, C., Chacur, M., Biedenkapp, J., O’Connor, K., Poole, S., Tracey,K., Martin, D., Maier, S.F., Watkins, L.R., 2003. Spinal glia and proinflammatorycytokines mediate mirror-image neuropathic pain in rats. J. Neurosci. 23, 1026–1040.
Milligan, E.D., Zapata, V., Chacur, M., Schoeniger, D., Biedenkapp, J., O’Connor, K.A.,Verge, G.M., Chapman, G., Green, P., Foster, A.C., Naeve, G.S., Maier, S.F., Watkins,L.R., 2004b. Evidence that exogenous and endogenous fractalkine can inducespinal nociceptive facilitation in rats. Eur. J. Neurosci. 20, 2294–2302.
Momin, A., McNaughton, P.A., 2009. Regulation of firing frequency in nociceptiveneurons by pro-inflammatory mediators. Exp. Brain Res. 196, 45–52.
Moss, A., Beggs, S., Vega-Avelaira, D., Costigan, M., Hathway, G.J., Salter, M.W.,Fitzgerald, M., 2007. Spinal microglia and neuropathic pain in young rats. Pain128, 215–224.
Palmer, T.M., Trevethick, M.A., 2008. Suppression of inflammatory and immuneresponses by the A(2A) adenosine receptor: an introduction. Br. J. Pharmacol.153 (Suppl. 1), S27–S34.
Paterniti, I., Melani, A., Cipriani, S., Corti, F., Mello, T., Mazzon, E., Esposito, E.,Bramanti, P., Cuzzocrea, S., Pedata, F., 2011. Selective adenosine A2A receptoragonists and antagonists protect against spinal cord injury through peripheraland central effects. J. Neuroinflamm. 8, 31.
Scholz, J., Woolf, C.J., 2007. The neuropathic pain triad: neurons, immune cells andglia. Nature Neurosci. 10, 1361–1368.
Schubert, P., Morino, T., Miyazaki, H., Ogata, T., Nakamura, Y., Marchini, C., Ferroni,S., 2000. Cascading glia reactions: a common pathomechanism and itsdifferentiated control by cyclic nucleotide signaling. Ann. NY Acad. Sci. 903,24–33.

122 L.C. Loram et al. / Brain, Behavior, and Immunity 33 (2013) 112–122
Schwarz, J.M., Sholar, P.W., Bilbo, S.D., 2012. Sex differences in microglialcolonization of the developing rat brain. J Neurochem. 120 (6), 948–963.
Scott, J.D., Dessauer, C.W., Tasken, K., 2013. Creating order from chaos:cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 53,187–210.
Sebastiao, A.M., Assaife-Lopes, N., Diogenes, M.J., Vaz, S.H., Ribeiro, J.A., 2011.Modulation of brain-derived neurotrophic factor (BDNF) actions in the nervoussystem by adenosine A(2A) receptors and the role of lipid rafts. Biochim.Biophys. Acta 1808, 1340–1349.
Sebastiao, A.M., Ribeiro, J.A., 2009a. Adenosine receptors and the central nervoussystem. In: Handbook of Experimental Pharmacology, pp. 471–534.
Sebastiao, A.M., Ribeiro, J.A., 2009b. Tuning and fine-tuning of synapses withadenosine. Curr. Neuropharmacol. 7, 180–194.
Tortora, G., Ciardiello, F., 2002a. Protein kinase A as target for novel integratedstrategies of cancer therapy. Ann. N. Y. Acad. Sci. 968, 139–147.
Tortora, G., Ciardiello, F., 2002b. Protein kinase A type I: a target for cancer therapy.Clin. Cancer Res. 8, 303–304.
Watkins, L.R., Hutchinson, M.R., Milligan, E.D., Maier, S.F., 2007. ‘‘Listening’’ and‘‘talking’’ to neurons: implications of immune activation for pain control andincreasing the efficacy of opioids. Brain Res. Rev. 56, 148–169.
Wen, A.Y., Sakamoto, K.M., Miller, L.S., 2010. The role of the transcription factorCREB in immune function. J. Immunol. 185, 6413–6419.
Yamamoto, S., Nakanishi, O., Matsui, T., Shinohara, N., Kinoshita, H., Lambert, C.,Ishikawa, T., 2003. Intrathecal adenosine A1 receptor agonist attenuateshyperalgesia without inhibiting spinal glutamate release in the rat. Cell. Mol.Neurobiol. 23, 175–185.
Zahn, P.K., Straub, H., Wenk, M., Pogatzki-Zahn, E.M., 2007. Adenosine A1 but notA2a receptor agonist reduces hyperalgesia caused by a surgical incision in rats:a pertussis toxin-sensitive G protein-dependent process. Anesthesiology 107,797–806.





























