Embed Size (px)
Citation preview

Intramolecular Electronic Communication in a
Dimethylaminoazobenzene–Fullerene C60 Dyad:
An Experimental and TD-DFT Study
K. SENTHIL KUMAR, ARCHITA PATNAIK
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600 036, India
Received 16 April 2009; Revised 24 July 2009; Accepted 2 August 2009DOI 10.1002/jcc.21404
Published online 13 October 2009 in Wiley InterScience (www.interscience.wiley.com).
Abstract: An electronically push–pull type dimethylaminoazobenzene–fullerene C60 hybrid was designed and syn-
thesized by tailoring N,N-dimethylaniline as an electron donating auxochrome that intensified charge density on the
b-azonitrogen, and on N-methylfulleropyrrolidine (NMFP) as an electron acceptor at the 4 and 40 positions of the
azobenzene moiety, respectively. The absorption and charge transfer behavior of the hybrid donor-bridge-acceptor
dyad were studied experimentally and by performing TD-DFT calculations. The TD-DFT predicted charge transfer
interactions of the dyad ranging from 747 to 601 nm were experimentally observed in the UV-vis spectra at 721 nm
in toluene and dichloromethane. A 149 mV anodic shift in the first reduction potential of the N¼¼N group of the
dyad in comparison with the model aminoazobenzene derivative further supported the phenomenon. Analysis of the
charge transfer band through the orbital picture revealed charge displacement from the n(N¼¼N) (nonbonding) and p
(N¼¼N) type orbitals centered on the donor part to the purely fullerene centered LUMOs and LUMO1n orbitals, delo-
calized over the entire molecule. The imposed electronic perturbations on the aminoazobenzene moiety upon cou-
pling it with C60 were analyzed by comparing the TD-DFT predicted and experimentally observed electronic transi-
tion energies of the dyad with the model compounds, NMFP and (E)-N,N-dimethyl-4-(p-tolyldiazenyl)aniline
(AZNME). The n(N¼¼N) ? p*(N¼¼N) and p(N¼¼N) ? p*(N¼¼N) transitions of the dyad were bathochromically shifted with a
significant charge transfer character. The shifting of p(N¼¼N) ? p*(N¼¼N) excitation energy closer to the n ? p*(N¼¼N) in
comparison with the model aminoazobenzene emphasized the predominant existence of charge separated quinonoid-
like ground state electronic structure. Increasing solvent polarity introduced hyperchromic effect in the p(N¼¼N) ?p*(N¼¼N) electronic transition at the expense of transitions involved with benzenic states, and the extent of intensity
borrowing was quantified adopting the Gaussian deconvolution method. On a comparative scale, the predicted exci-
tation energies were in reasonable agreement with the observed values, demonstrating the efficiency of TD-DFT in
predicting the localized and the charge transfer nature of transitions involved with large electronically asymmetric
molecules with HOMO and LUMO centered on different parts of the molecular framework.
q 2009 Wiley Periodicals, Inc. J Comput Chem 31: 1182–1194, 2010
Key words: donor–acceptor conjugates; dimethylaminoazobenzene–fullerene (C60); electronic structure; TD-DFT;
charge transfer
Introduction
Synthesizing functional organic molecules for applications rang-
ing from artificial photosynthesis to molecular electronic compo-
nents has been actively pursued in view of their potential alter-
nates to the existing silicon based technologies.1–4 Molecular
components based on fullerenes offer a unique possibility of
combining the advantageous three-dimensional framework with
electron accepting character, enabled by the triply degenerate
LUMO.5–10 On the other hand, azobenzenes with two different
conformers, addressable by light or electric field were proven as
building blocks for the construction of stimuli-responsive materi-
als with innumerable applications, ranging from conformation
switching of proteins to data storage.11–15 Electronic spectra of
various azobenzene dyes and their impact on the design of opti-
Additional Supporting Information may be found in the online version of
this article
Correspondence to: A. Patnaik; e-mail: [email protected]
Contract/grant sponsor: Department of Science and Technology, New
Delhi; contract/grant number: SR/S2/CMP-57/2006
Contract/grant sponsor: IIT Madras
q 2009 Wiley Periodicals, Inc.

cal data storage materials were reported.16 Imahori et al.
reported ground state electronic interactions in an ortho linked
Porphyrine-Fullerene conjugate.17 We recently reported the
ground state electronic interactions in a didodecyloxybenzene-
C60 dyad.18 A variety of fullerene-azobenzene hybrids were
synthesized and studied19–21 among which the molecular system
investigated by Guldi and coworkers22 is the only report
concerned with electron transfer mediating ability of the pure
azobenzene bridge.
Covalent tailoring of azobenzene and fullerene could provide
novel stimuli-tunable materials with accessible redox states and
lower excitation energies, desirable for the construction of
optical data storage materials. Understanding of the electronic
perturbation imposed on the electronic structure of azobenzene
upon functionalization with fullerene and its consequences on
the light absorption behavior is important from the view point of
reading and writing of data in memory devices.
Among the methods available for the prediction of excitation
energies and oscillator strengths of molecules, TD-DFT23–25 was
reported with reasonable accuracy and affordable computational
time in comparison with the conventional highly correlated
wave function methods, using large active spaces. However, the
reliability of TD-DFT in predicting the long-range CT interac-
tions was questioned and was reported to depend on the chosen
exchange correlation functional.26–33 Calculations of excitation
energies, especially the energy corresponding to the charge
transfer interactions were challenging in the case of large
molecules with frontier molecular orbitals located on the
different parts of the molecular framework.34–37 Therefore, it is
mandatory to evaluate the performance of TD-DFT by
comparing the predicted excitation energies with the available
experimental data.
The present investigation designed a novel push–pull,
structurally nonentrosymmetric and electronically asymmetric
azobenzene molecular skeleton with many desirable features by
tailoring it with electron rich N,N-dimethylaniline as the donor
and fullerene C60 as an electron acceptor at 4 and 40 positionsrespectively. The aim of this investigation was (i) to probe the
interaction between the donor and acceptor in the ground state
using DFT and TD-DFT and (ii) to assess the utility of TD-DFT
in predicting the excitation energies of fullerene–azobenzene
hybrid by comparing the calculated results with the experimental
data. Further, experimental investigations were also carried out
to get an insight on the effect of solvent polarity in controlling
the electronic communication between the donor and the
acceptor.
Experimental Details
Electronic absorption spectra were measured with a Schimadzu
double beam UV-vis spectrophoto- meter. Differential pulse vol-
tammetry (DPV) was carried out with a CH instrument’s 660B
electrochemical analyzer. Toluene: Acetonitrile in 4:1 volume
ratio was used as the solvent and 0.1 M tetrabutylammonium
hexafluorophosphate was used as the supporting electrolyte with
a potential window ranging from 13 V to 23 V. The Glassy
Carbon electrode embedded in a Teflon rod (CHI 1.5 mm in
diameter) was polished with 0.1 lm c-alumina powder to mirror
finish and was used as the working electrode. Ag/AgNO3
nonaqueous electrode was used as the reference, whose potential
was calibrated with a Fc1/Fc internal redox couple. A pre-
cleaned Pt wire was used as the counter electrode. The analyte
concentration was fixed at 5 3 1024 M, unless otherwise stated.
Dissolved oxygen was removed by purging with high pure
nitrogen gas prior to experiments.
Computational Methodology
Density Functional calculations were carried out using Gaussian
03 set of programmes.38 The ground-state geometry of each
molecule was fully optimized using the hybrid B3LYP func-
tional with 3-21g (d, p) basis set, until the RMS residual force
became smaller than 2.533 3 1026 a.u. In B3LYP39 the
exchange is a combination of 20% HF exchange, Slater func-
tional, and Becke’s GGA correction,40 whereas the correlation
part combines VWN41 and LYP42 functionals. TD-DFT method-
ology was then used to compute the excitation energies, oscilla-
tor strengths, and the composition of electronic transitions.
The bulk solvent effects were evaluated during the TD-DFT
calculations by means of the standard Polarizable Continuum
Model (IEF-PCM).43,44 In PCM, the problem was divided into a
solute part lying inside a cavity and a solvent part represented
as a structureless material, characterized by its macroscopic
properties (dielectric constant, radius, density, and molecular
volume).
Results and Discussion
The Ground State Electronic Structures of NMFP,
AZNME, and the Dyad
The chemical and optimized structures of NMFP (N-methylful-
leropyrrolidine), AZNME ((E)-N,N-dimethyl-4-(p-tolyldiazenyl)-
aniline) and the dyad ((E)-N,N-dimethyl-4-(p-pyrrolidinofullere-
nediazenyl)aniline) are displayed in Figures 1I(a–c) and 1II
(a–c) respectively, where the azo part is seen in trans conforma-
tion. The present system was divided into two parts: NMFP as
the acceptor and AZNME as the donor. To mimic the 4, 40 func-tionalization of the azo part in the dyad, methyl group was
attached to the 40 position of AZNME.
In Figure 1II(a), the optimized structure of NMFP shows it to
be deviated largely from planarity with a C��C��N��C dihedral
angle of 898 in comparison to a very large 1718 in the dyad
with minimum puckering, vide Figure 1I(c). Interestingly, the
extent of puckering in the dyad is very similar to that calculated
for N-methylpyrrolidine (NMP) as shown in Figure 1II(d) and in
Supporting Information Figure S2. This fact validates a signifi-
cant steric role of the donor substituent on the NMFP framework
in regaining back the almost planar configuration of the dyad. A
closer observation of the bent structure in Figure 1IIa reveals
that the methyl group could rotate around the N��C axis and
that an equatorial position of the N��CH3 group has been pre-
ferred, with one C��H bond of the methyl group in trans posi-
tion to the nitrogen lone pair. The almost similar CNC bond
1183Intramolecular Electronic Communication in a Dimethylaminoazobenzene – Fullerene C60 Dyad
Journal of Computational Chemistry DOI 10.1002/jcc
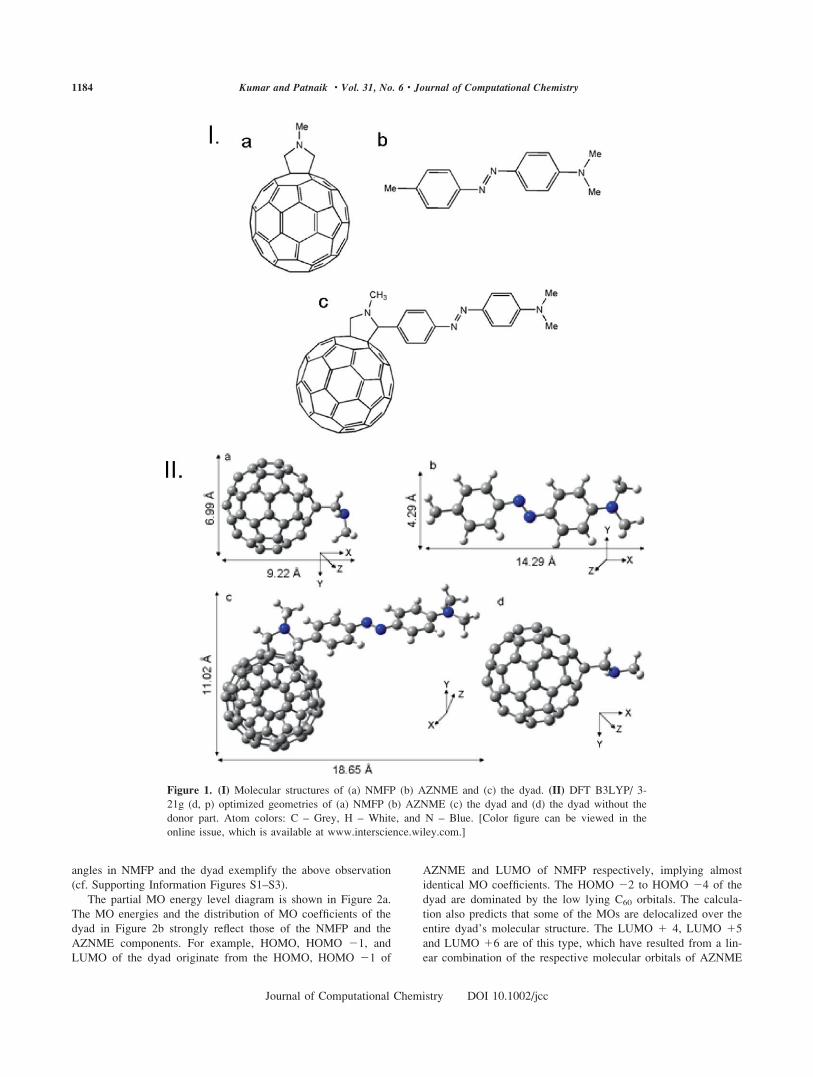
angles in NMFP and the dyad exemplify the above observation
(cf. Supporting Information Figures S1–S3).
The partial MO energy level diagram is shown in Figure 2a.
The MO energies and the distribution of MO coefficients of the
dyad in Figure 2b strongly reflect those of the NMFP and the
AZNME components. For example, HOMO, HOMO 21, and
LUMO of the dyad originate from the HOMO, HOMO 21 of
AZNME and LUMO of NMFP respectively, implying almost
identical MO coefficients. The HOMO 22 to HOMO 24 of the
dyad are dominated by the low lying C60 orbitals. The calcula-
tion also predicts that some of the MOs are delocalized over the
entire dyad’s molecular structure. The LUMO 1 4, LUMO 15
and LUMO 16 are of this type, which have resulted from a lin-
ear combination of the respective molecular orbitals of AZNME
Figure 1. (I) Molecular structures of (a) NMFP (b) AZNME and (c) the dyad. (II) DFT B3LYP/ 3-
21g (d, p) optimized geometries of (a) NMFP (b) AZNME (c) the dyad and (d) the dyad without the
donor part. Atom colors: C – Grey, H – White, and N – Blue. [Color figure can be viewed in the
online issue, which is available at www.interscience.wiley.com.]
1184 Kumar and Patnaik • Vol. 31, No. 6 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
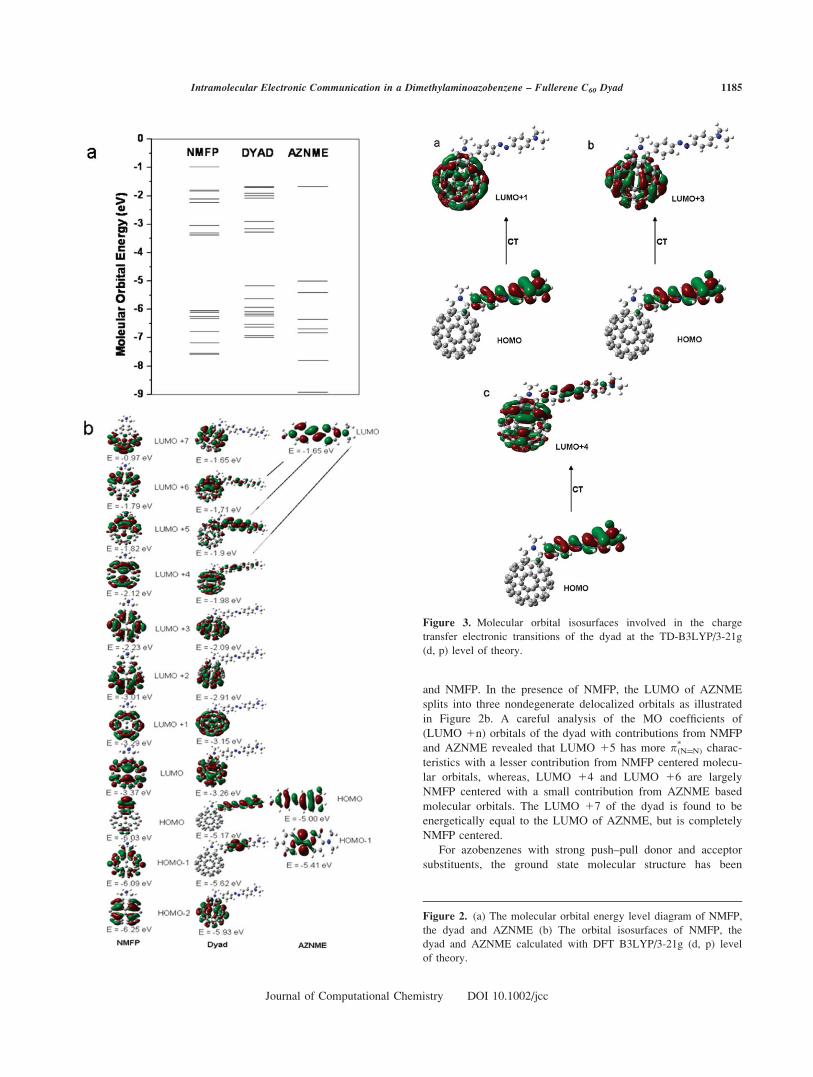
and NMFP. In the presence of NMFP, the LUMO of AZNME
splits into three nondegenerate delocalized orbitals as illustrated
in Figure 2b. A careful analysis of the MO coefficients of
(LUMO 1n) orbitals of the dyad with contributions from NMFP
and AZNME revealed that LUMO 15 has more p*(N¼¼N) charac-
teristics with a lesser contribution from NMFP centered molecu-
lar orbitals, whereas, LUMO 14 and LUMO 16 are largely
NMFP centered with a small contribution from AZNME based
molecular orbitals. The LUMO 17 of the dyad is found to be
energetically equal to the LUMO of AZNME, but is completely
NMFP centered.
For azobenzenes with strong push–pull donor and acceptor
substituents, the ground state molecular structure has been
Figure 2. (a) The molecular orbital energy level diagram of NMFP,
the dyad and AZNME (b) The orbital isosurfaces of NMFP, the
dyad and AZNME calculated with DFT B3LYP/3-21g (d, p) level
of theory.
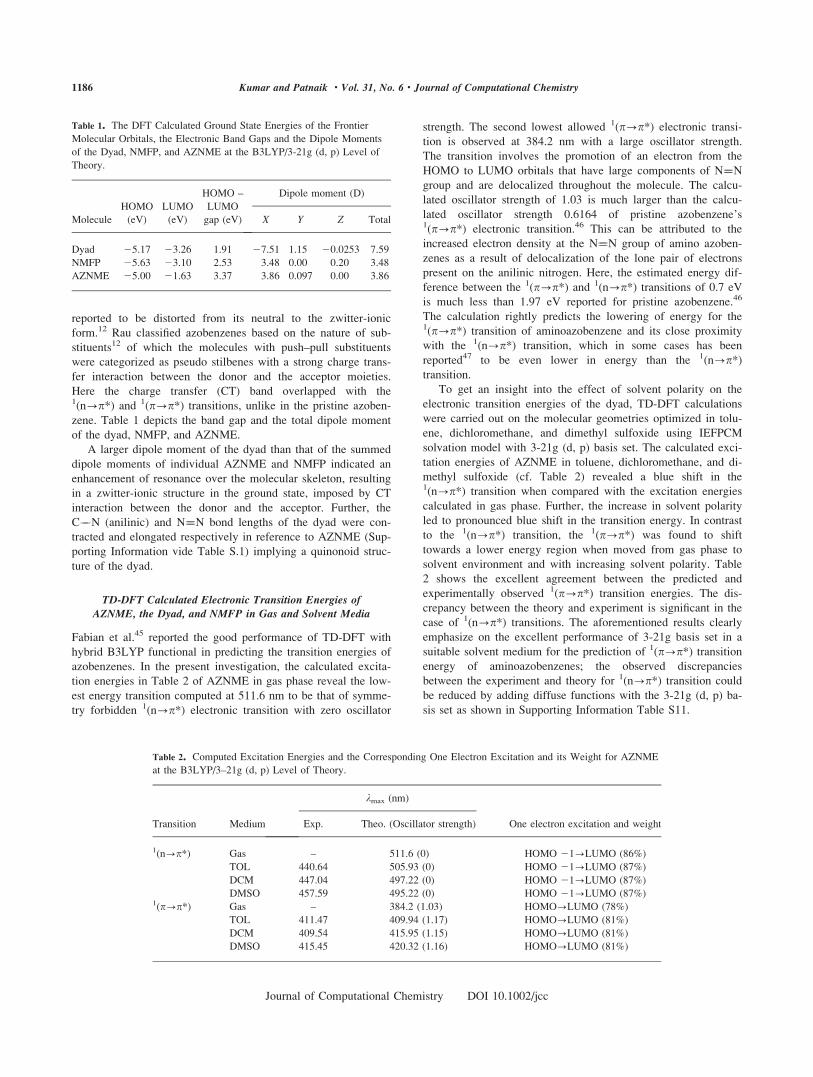
Figure 3. Molecular orbital isosurfaces involved in the charge
transfer electronic transitions of the dyad at the TD-B3LYP/3-21g
(d, p) level of theory.
1185Intramolecular Electronic Communication in a Dimethylaminoazobenzene – Fullerene C60 Dyad
Journal of Computational Chemistry DOI 10.1002/jcc
reported to be distorted from its neutral to the zwitter-ionic
form.12 Rau classified azobenzenes based on the nature of sub-
stituents12 of which the molecules with push–pull substituents
were categorized as pseudo stilbenes with a strong charge trans-
fer interaction between the donor and the acceptor moieties.
Here the charge transfer (CT) band overlapped with the1(n?p*) and 1(p?p*) transitions, unlike in the pristine azoben-
zene. Table 1 depicts the band gap and the total dipole moment
of the dyad, NMFP, and AZNME.
A larger dipole moment of the dyad than that of the summed
dipole moments of individual AZNME and NMFP indicated an
enhancement of resonance over the molecular skeleton, resulting
in a zwitter-ionic structure in the ground state, imposed by CT
interaction between the donor and the acceptor. Further, the
C��N (anilinic) and N¼¼N bond lengths of the dyad were con-
tracted and elongated respectively in reference to AZNME (Sup-
porting Information vide Table S.1) implying a quinonoid struc-
ture of the dyad.
TD-DFT Calculated Electronic Transition Energies of
AZNME, the Dyad, and NMFP in Gas and Solvent Media
Fabian et al.45 reported the good performance of TD-DFT with
hybrid B3LYP functional in predicting the transition energies of
azobenzenes. In the present investigation, the calculated excita-
tion energies in Table 2 of AZNME in gas phase reveal the low-
est energy transition computed at 511.6 nm to be that of symme-
try forbidden 1(n?p*) electronic transition with zero oscillator
strength. The second lowest allowed 1(p?p*) electronic transi-
tion is observed at 384.2 nm with a large oscillator strength.
The transition involves the promotion of an electron from the
HOMO to LUMO orbitals that have large components of N¼¼N
group and are delocalized throughout the molecule. The calcu-
lated oscillator strength of 1.03 is much larger than the calcu-
lated oscillator strength 0.6164 of pristine azobenzene’s1(p?p*) electronic transition.46 This can be attributed to the
increased electron density at the N¼¼N group of amino azoben-
zenes as a result of delocalization of the lone pair of electrons
present on the anilinic nitrogen. Here, the estimated energy dif-
ference between the 1(p?p*) and 1(n?p*) transitions of 0.7 eV
is much less than 1.97 eV reported for pristine azobenzene.46
The calculation rightly predicts the lowering of energy for the1(p?p*) transition of aminoazobenzene and its close proximity
with the 1(n?p*) transition, which in some cases has been
reported47 to be even lower in energy than the 1(n?p*)transition.
To get an insight into the effect of solvent polarity on the
electronic transition energies of the dyad, TD-DFT calculations
were carried out on the molecular geometries optimized in tolu-
ene, dichloromethane, and dimethyl sulfoxide using IEFPCM
solvation model with 3-21g (d, p) basis set. The calculated exci-
tation energies of AZNME in toluene, dichloromethane, and di-
methyl sulfoxide (cf. Table 2) revealed a blue shift in the1(n?p*) transition when compared with the excitation energies
calculated in gas phase. Further, the increase in solvent polarity
led to pronounced blue shift in the transition energy. In contrast
to the 1(n?p*) transition, the 1(p?p*) was found to shift
towards a lower energy region when moved from gas phase to
solvent environment and with increasing solvent polarity. Table
2 shows the excellent agreement between the predicted and
experimentally observed 1(p?p*) transition energies. The dis-
crepancy between the theory and experiment is significant in the
case of 1(n?p*) transitions. The aforementioned results clearly
emphasize on the excellent performance of 3-21g basis set in a
suitable solvent medium for the prediction of 1(p?p*) transitionenergy of aminoazobenzenes; the observed discrepancies
between the experiment and theory for 1(n?p*) transition could
be reduced by adding diffuse functions with the 3-21g (d, p) ba-
sis set as shown in Supporting Information Table S11.
Table 2. Computed Excitation Energies and the Corresponding One Electron Excitation and its Weight for AZNME
at the B3LYP/3–21g (d, p) Level of Theory.
Transition Medium
kmax (nm)
One electron excitation and weightExp. Theo. (Oscillator strength)
1(n?p*) Gas – 511.6 (0) HOMO 21?LUMO (86%)
TOL 440.64 505.93 (0) HOMO 21?LUMO (87%)
DCM 447.04 497.22 (0) HOMO 21?LUMO (87%)
DMSO 457.59 495.22 (0) HOMO 21?LUMO (87%)1(p?p*) Gas – 384.2 (1.03) HOMO?LUMO (78%)
TOL 411.47 409.94 (1.17) HOMO?LUMO (81%)
DCM 409.54 415.95 (1.15) HOMO?LUMO (81%)
DMSO 415.45 420.32 (1.16) HOMO?LUMO (81%)
Table 1. The DFT Calculated Ground State Energies of the Frontier
Molecular Orbitals, the Electronic Band Gaps and the Dipole Moments
of the Dyad, NMFP, and AZNME at the B3LYP/3-21g (d, p) Level of
Theory.
Molecule
HOMO
(eV)
LUMO
(eV)
HOMO –
LUMO
gap (eV)
Dipole moment (D)
X Y Z Total
Dyad 25.17 23.26 1.91 27.51 1.15 20.0253 7.59
NMFP 25.63 23.10 2.53 3.48 0.00 0.20 3.48
AZNME 25.00 21.63 3.37 3.86 0.097 0.00 3.86
1186 Kumar and Patnaik • Vol. 31, No. 6 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
In Table 3, the excitation energies and one electron excitations
of the 1(p?p*) and 1(n?p*) transitions of the dyad are presented.
The transition predicted at 520.1 nm agrees with the calculated1(n?p*) energy of AZNME located at 511.6 nm (cf. Table 2),
implying the lowering of 1(n?p*) excitation energy in the dyad
as a result of C60 substitution. The1(n?p*) transition of the dyad
has gained some oscillator strength unlike ‘‘0’’ in AZNME. The
nonlocalized nature of this transition from HOMO 21 to LUMO
15 with a weightage of 61% was of p* (N¼¼N) character and was
found to have contributions also from the fullerene centered orbi-
tals. The other two orbitals LUMO 14 and LUMO 16 contrib-
uted 16% and 7%, respectively, and have resulted from linear
combination of orbitals centered largely on fullerene and
AZNME. This showed that the calculated 1(n?p*) transition of
the dyad has finite charge transfer character and the delocalization
of LUMO 14, LUMO 15, and LUMO 16 orbitals over the entire
complex relaxed the symmetry constraints enabling it to gain
some intensity/oscillator strength.
The transition at 405.4 nm is from HOMO to LUMO15 and
LUMO 16 with a significant contribution of (62%) from
LUMO 15 (cf. Table 3 and Supporting Information Table S.3).
As mentioned above, the orbitals LUMO 15 and LUMO 16 of
the dyad are a resultant of linear combination of the respective
LUMO 15 and LUMO 16 orbitals of NMFP and LUMO of
AZNME. An inspection of the MO coefficients of LUMO 15
revealed the predominant p* (N¼¼N) character of this orbital,
leading us to assign the transition to be of 1(p?p*) in nature. In
comparison with the predicted 1(p?p*) transition of AZNME,
this transition is found to be 21.3 nm bathochromically shifted.
Further, the transition is of charge transfer in nature involving
electron transition from an orbital completely localized on the
donor (HOMO) to orbitals delocalized on the donor and the
acceptor. Interestingly, the calculated oscillator strength of 0.575
is almost half of the calculated oscillator strength of 1(p?p*)transition of AZNME. Furthermore, a higher energy transition at
387.2 nm with an oscillator strength of 0.5857, almost equal to
the aforementioned predominantly 1(p?p*) cum charge transfer
transition of the dyad was noted (Supporting Information Table
S.3). The 54% weight of HOMO to LUMO16 along with a
small contribution from HOMO to LUMO15 excitation revealed
the dominant charge transfer character of this band with a slight1(p?p*) character. The above facts bring about the following
points: (1) the 1(p?p*) transition, similar to the 1(n?p*) transi-tion is also bathochromically shifted with a significant charge
transfer character in comparison with the 1(p?p*) and 1(n?p*)transitions of AZNME. (2) the calculated oscillator strength for
the 1(p?p*) electronic transition of the dyad is almost half in
magnitude in comparison with AZNME. This was attributed to
the splitting of the LUMO of AZNME into three LUMO 1n
orbitals in the dyad and also to the coupling of CT interactions
with the 1(p?p*) transition of the dyad.
Apart from 1(n?p*) and 1(p?p*) transitions, two other
types of excited states were identified: (i) the charge transfer
complex state and (ii) the locally excited fullerene state.
Selected transitions with remarkable oscillator strengths associ-
ated with these states are discussed as follows. The transition
located around 701.2 nm is of HOMO to LUMO 11 type (cf.
Table 4) and correspond to the excitation of p electrons of
N¼¼N to fullerene centered unoccupied orbital as shown in
Figure 3a. The TD-DFT predicted values are in excellent agree-
ment with our experimentally observed values discussed in
Absorption Characteristics of the Dyad: Effect of Fullerene C60
Substitution on the Eletronic Transitions of AZNME and Intra-
molecular Charge Transfer Section and with that for a covalently
Table 4. Computed Excitation Energies (nm) and the Corresponding One
Electron Excitation and its Weight for the Charge Transfer Transitions
of the Dyad at the B3LYP/3-21g (d, p) Level of Theory.
Medium kmax
Oscillator
strength
One electron excitation
and weight
Gas 701.2 0.0080 HOMO?LUMO 11 (99%)
440.8 0.0128 HOMO?LUMO 13 (99%)
425 0.0621 HOMO?LUMO 14 (90%)
TOL 729.36 0.0043 HOMO?LUMO 11 (100%)
451.36 0.0054 HOMO?LUMO 13 (98%)
433.67 0.0405 HOMO?LUMO 14 (98%)
DCM 729.61 0.0040 HOMO?LUMO 11 (100%)
451.78 0.0061 HOMO?LUMO 13 (98%)
433.44 0.0562 HOMO?LUMO 14 (97%)
DMSO 720.76 0.0092 HOMO?LUMO 11 (96%)
449.59 0.0540 HOMO?LUMO 13 (98%)
434.8 0.6957 HOMO?LUMO 14 (77%)
Table 3. Computed Excitation Energies (nm) and the Corresponding One Electron Excitation and its Weight for the
Dyad at the B3LYP/3–21g (d, p) Level of Theory.
Transition Medium
kmax (nm)
One electron excitation and weightExp. Theo. (Oscillator strength)
1(n?p*) Gas – 520.1 (ca. 0) HOMO 21?LUMO 15 (61%)
TOL 447.07 519.85 (ca. 0) HOMO 21?LUMO 15 (68%)
DCM 449.41 510.95 (ca. 0) HOMO 21?LUMO 15 (55%)
DMSO 464.98 501.4 (ca. 0) HOMO 21?LUMO 15 (62%)1(p?p*) Gas - 405.4 ( 0.58) HOMO?LUMO 15 (62%)
TOL 414.42 417.06 (0.93) HOMO?LUMO 15 (82%)
DCM 416.41 420.07 (1.09) HOMO?LUMO 15 (79%)
DMSO 430.19 425.79 (0.56) HOMO?LUMO 15 (83%
1187Intramolecular Electronic Communication in a Dimethylaminoazobenzene – Fullerene C60 Dyad
Journal of Computational Chemistry DOI 10.1002/jcc
linked porphyrine-fullerene dyad with a charge transfer transi-
tion energy �1.66 eV.17 The transitions listed at 440.8 and 425
nm in Table 4 are concluded to be charge transfer in nature after
analyzing the one electron excitations involved with these transi-
tions, wherein p electrons of N¼¼N are transferred to the fuller-
ene centered unoccupied orbitals as shown in Figures 3b and 3c.
Further, the transition predicted at 598.1 nm is of HOMO –2
to LUMO type with as oscillator strength of 0.0025. Excellent
agreement of the above transition with the predicted first elec-
tronic transition of NMFP at 598.1 nm and the nature of molec-
ular orbitals involved there-in, guided us to attribute this transi-
tion to be of localized fullerene centered of type (ii). The calcu-
lated electronic transition energies of the dyad in toluene,
dichloromethane and dimethyl sulfoxide are listed in Tables 3
and 4. Even though the character of the transition remains same,
the charge transfer transition which involved the p orbital
(HOMO) of AZNME moiety of the dyad was found to be red
shifted in solvents compared to vacuum. The charge transfer
transition predicted in gas phase at 701 nm was found around
729 nm in toluene and dichloromethane and at 720.76 nm in di-
methyl sulfoxide. Other charge transfer transitions located at
440.8 and 420 nm in gas phase were also bathochromically
shifted when moved to solvent media and with increasing sol-
vent polarity.
The dominant transitions of the dyad with large oscillator
strengths are 1(p?p*) in nature, coupled with charge transfer
character as discussed above. The 1(p?p*) transitions predicted
in the solvent phase are bathochromically shifted in comparison
to the gas phase. The transitions which involved the n orbitals
of the AZNME moiety of the dyad are found to be hypso-
chromically shifted when moving from vacuum to solvent phase
and with increasing solvent polarity. Calculations in solvents
also allowed us to get an insight into the impact of fullerene
moiety on the electronic transitions of AZNME. The blue shifts
observed in the electronic transitions involving the ‘‘n’’ orbitals
of the azo group in the dyad are seen to be less predominant
than the shifts observed in AZNME. Further the 1(n?p*) transi-tion was shifted to the lower energy part of the spectrum in
comparison with AZNME due to the localized nature of the
transition in the latter as against the dyad with a delocalized
charge transfer character. The 1(p?p*) transitions of the dyad
are seen to be red shifted in comparison with AZNME with
increasing solvent polarity which could also be explained by the
charge transfer character associated with the transition.
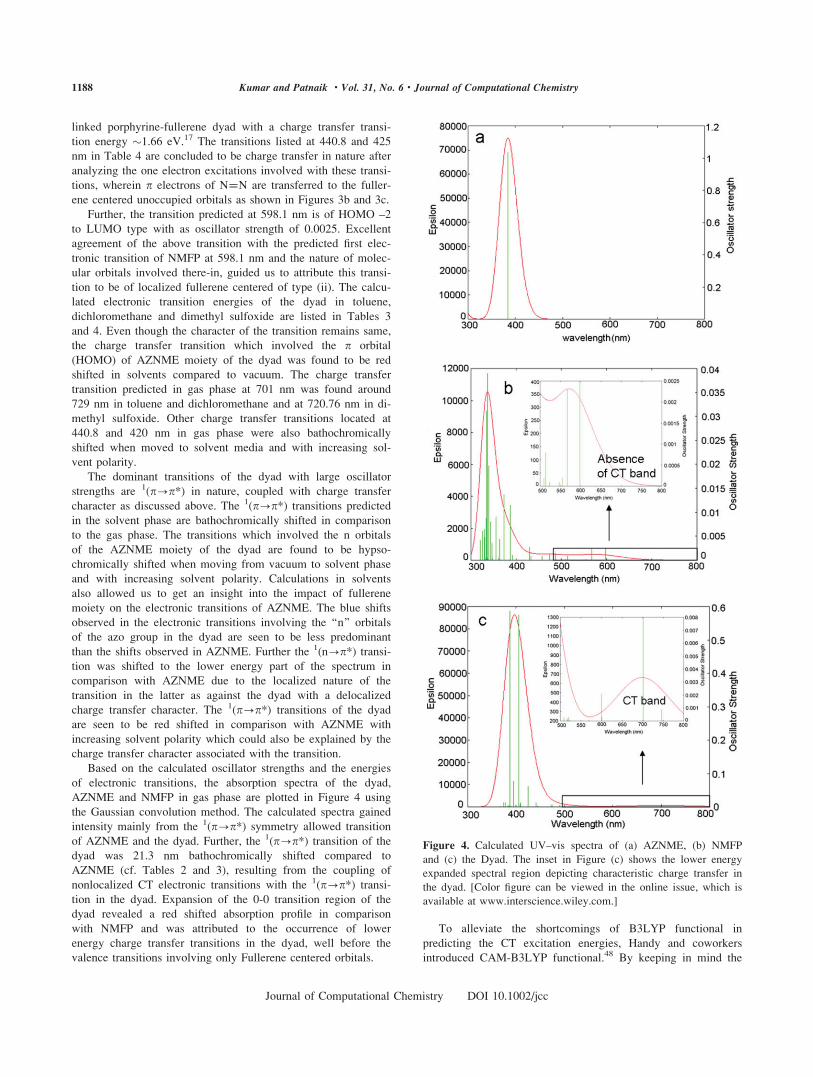
Based on the calculated oscillator strengths and the energies
of electronic transitions, the absorption spectra of the dyad,
AZNME and NMFP in gas phase are plotted in Figure 4 using
the Gaussian convolution method. The calculated spectra gained
intensity mainly from the 1(p?p*) symmetry allowed transition
of AZNME and the dyad. Further, the 1(p?p*) transition of the
dyad was 21.3 nm bathochromically shifted compared to
AZNME (cf. Tables 2 and 3), resulting from the coupling of
nonlocalized CT electronic transitions with the 1(p?p*) transi-
tion in the dyad. Expansion of the 0-0 transition region of the
dyad revealed a red shifted absorption profile in comparison
with NMFP and was attributed to the occurrence of lower
energy charge transfer transitions in the dyad, well before the
valence transitions involving only Fullerene centered orbitals.
To alleviate the shortcomings of B3LYP functional in
predicting the CT excitation energies, Handy and coworkers
introduced CAM-B3LYP functional.48 By keeping in mind the
Figure 4. Calculated UV–vis spectra of (a) AZNME, (b) NMFP
and (c) the Dyad. The inset in Figure (c) shows the lower energy
expanded spectral region depicting characteristic charge transfer in
the dyad. [Color figure can be viewed in the online issue, which is
available at www.interscience.wiley.com.]
1188 Kumar and Patnaik • Vol. 31, No. 6 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
problems associated, we justify the use of B3LYP functional in
the present study as follows: (1) the experimentally observed
sharp response of the transitions to the change in the solvent po-
larity were well reproduced by TD-B3LYP calculations. (2) The
agreement between the predicted and observed charge transfer
energies was found to be excellent. Further, the performance of
CAM-B3LYP in predicting 1(p?p*) electronic transition ener-
gies of donor–acceptor substituted azobenzenes as discussed by
Jacquemin et al.49 were found to underestimate the 1(p?p*)transition energy of the substituted azobenzenes with significant
donor–acceptor character. In line with the above, the results
obtained from the present study employing B3LYP functional
yielded much better results, i.e., the discrepancy between the
experiment and theory was negligible (�6 nm as shown in
Tables 2 and 3). Another possible approach will be the use of
post HF methods for the calculation of excitation energies; the
large molecular size of the present dyad system impeded the
above calculations.
Electronic Absorption Spectroscopy and Validation of the
TD-DFT Predicted Charge Transfer Interactions
Azobenzene has been known to possess the lowest energy for-
bidden 1(n?p*) transition around 440 nm and an allowed1(p?p*) transition at 314 nm.46 Substitution of electron donat-
ing groups with lone pair of electrons on the azobenzene skele-
ton has resulted in the lowering of the 1(p?p*) transition
energy. In the present investigation, toluene and dichlorome-
thane solvents with dielectric constants 2.5 and 7.5 respectively
with their solvent cut offs around 300 and 243 nm were the cho-
sen solvents along with dimethyl sulfoxide as the third solvent
of an intermediate dielectric constant 48 and a solvent cut off at
268 nm.
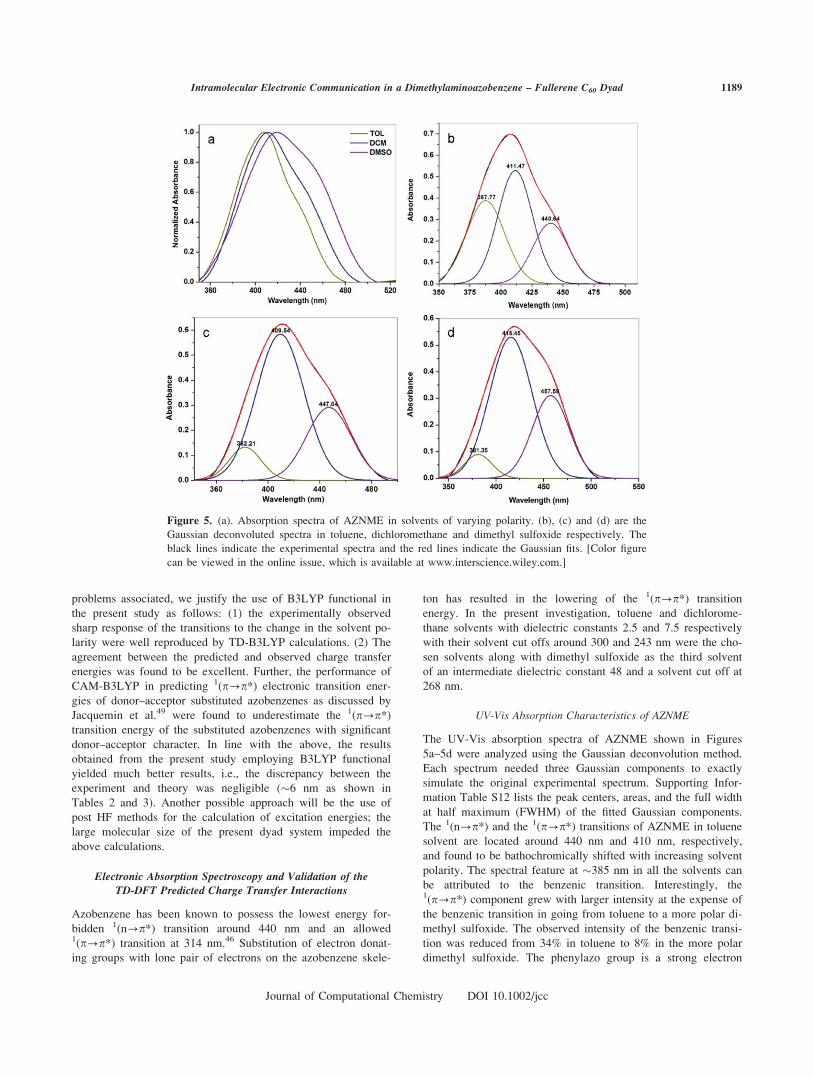
UV-Vis Absorption Characteristics of AZNME
The UV-Vis absorption spectra of AZNME shown in Figures
5a–5d were analyzed using the Gaussian deconvolution method.
Each spectrum needed three Gaussian components to exactly
simulate the original experimental spectrum. Supporting Infor-
mation Table S12 lists the peak centers, areas, and the full width
at half maximum (FWHM) of the fitted Gaussian components.
The 1(n?p*) and the 1(p?p*) transitions of AZNME in toluene
solvent are located around 440 nm and 410 nm, respectively,
and found to be bathochromically shifted with increasing solvent
polarity. The spectral feature at �385 nm in all the solvents can
be attributed to the benzenic transition. Interestingly, the1(p?p*) component grew with larger intensity at the expense of
the benzenic transition in going from toluene to a more polar di-
methyl sulfoxide. The observed intensity of the benzenic transi-
tion was reduced from 34% in toluene to 8% in the more polar
dimethyl sulfoxide. The phenylazo group is a strong electron
Figure 5. (a). Absorption spectra of AZNME in solvents of varying polarity. (b), (c) and (d) are the
Gaussian deconvoluted spectra in toluene, dichloromethane and dimethyl sulfoxide respectively. The
black lines indicate the experimental spectra and the red lines indicate the Gaussian fits. [Color figure
can be viewed in the online issue, which is available at www.interscience.wiley.com.]
1189Intramolecular Electronic Communication in a Dimethylaminoazobenzene – Fullerene C60 Dyad
Journal of Computational Chemistry DOI 10.1002/jcc

acceptor which upon coupling with N,N-dimethylaniline as an
electron donor results in a quinonoid structure (cf. Figure 6)
with a greater probability of interaction in polar solvents,
observed with dichloromethane and dimethyl sulfoxide.
Absorption Characteristics of the Dyad: Effect of Fullerene C60
Substitution on the Electronic Transitions of AZNME andIntramolecular Charge Transfer
The electronic absorption spectra of the dyad in solvents of
varying polarity shown in Figure 7 depict the 1(n?p*) transitionlocated at �447 nm in toluene, dichloromethane and a batho-
chromically shifted feature at 465 nm in dimethyl sulfoxide.
When compared to AZNME, the 1(n?p*) transition of the dyad
is bathochromically shifted in toluene and dimethyl sulfoxide
and remained almost unaffected in dichloromethane. The corre-
sponding 1(p?p*) features in the dyad were observed at 414
nm in toluene with a 15 nm red shift in dimethyl sulfoxide.
The electronic absorption features of the dyad showed a new
transition located at �434 nm in all solvents as indicated by
shaded areas in Figures 7b–7d. This was attributed to a fullerene
C60 centered transition via the absorption spectra of NMFP.50
The Gaussian deconvoluted 434 nm band in Figure 7 (Sup-
porting Information vide Table S13) is seen to be embedded in
between the 1(n?p*) and 1(p?p*) features and is largely of
fullerene centered transition with a minimum weightage. This
clearly demonstrates that in the visible region, both the chromo-
phores, the AZNME, and NMFP were simultaneously excited
and the energetically close proximity between the electronic
transitions involved there-in might have favored the charge
transfer interactions in the dyad from the donor to the acceptor.
The two new transitions observed around 479 and 490 nm in
dichloromethane and dimethyl sulfoxide, respectively, confirmed
the above prediction. The TD-DFT predicted charge transfer
transitions located at 450 and 433 nm were hypsochromically
shifted when compared to the experiment. However, only one
charge transfer band was identified experimentally as against
two predicted transitions. The reason could be the coupling of
the charge transfer transitions with the large 1(p?p*) and1(n?p*) transitions of the dyad.
Further proof of the CT behavior of the dyad was derived
from the deconvolution of the 700 nm region of the absorption
spectra, the region of absorption of mono-functionalized C60
derivatives. Imahori et al.17 reported ground state electronic
interactions in a Fullerene–Porphyrine conjugate where the
charge transfer band was observed at 721 and 724 nm in ben-
zene and benzonitrile, respectively. Figures 8a and 8b illustrate
a concrete charge transfer contribution from the dyad in toluene
and dichloromethane in comparison with NMFP, whereas, in di-
methyl sulfoxide (Figure 8c) the absorption maximum of the 0-0
transition of the dyad was found to be bathochromically shifted
in comparison with the 0-0 absorption feature of NMFP, indicat-
ing the close proximity between the pure fullerene centered and
charge transfer transitions in more polar solvents. The fitted
bands (Supporting Information vide Table S14) of the dyad in
Figures 9b–9d clearly show the presence of a new feature �720
nm (shaded areas) in the absorption spectra of the dyad in tolu-
ene and dichloromethane. However, in dimethyl sulfoxide, it is
coalesced with the 0-0 absorption band and is bathochromically
shifted to 708 nm, abiding by the TD-DFT predicted results. In
line with Imahori et al.,17 we attribute the 720 nm band to be
the charge transfer band with a notable hypsochromic shift of
the charge transfer band in more polar dimethyl sulfoxide. It is
to be noted here that the minor features at 732 nm and 726 nm
in Figures 9c and 9d are a repercussion of the confidence level
of the Gaussian fit applied to the data.
Solution Phase Redox Behavior and Intramolecular
Electron Transfer
In an attempt to identify the redox sites and to get an insight
into the mutual donor–acceptor interactions, the electrochemical
behaviour of the dyad was compared with NMFP and AZNME.
For Differential Pulse Voltammetry (DPV) measurements, the
Figure 6. Schematic representation of the benzenoid and quinonoid
forms of the donor substituted azobenzene.
Figure 7. (a). Absorption spectra of the dyad in solvents of varying polarity. (b), (c), and (d) are the
Gaussian deconvoluted spectra in toluene, dichloromethane and dimethyl sulfoxide, respectively. The
black lines indicate the experimental spectra and the red lines indicate the Gaussian fits. [Color figure
can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Figure 8. Absorption spectra of the dyad and NMFP in (a) toluene, (b) dichloromethane and (c)
dimethylsulfoxide between 660–740 nm. [Color figure can be viewed in the online issue, which is
available at www.interscience.wiley.com.]
1190 Kumar and Patnaik • Vol. 31, No. 6 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
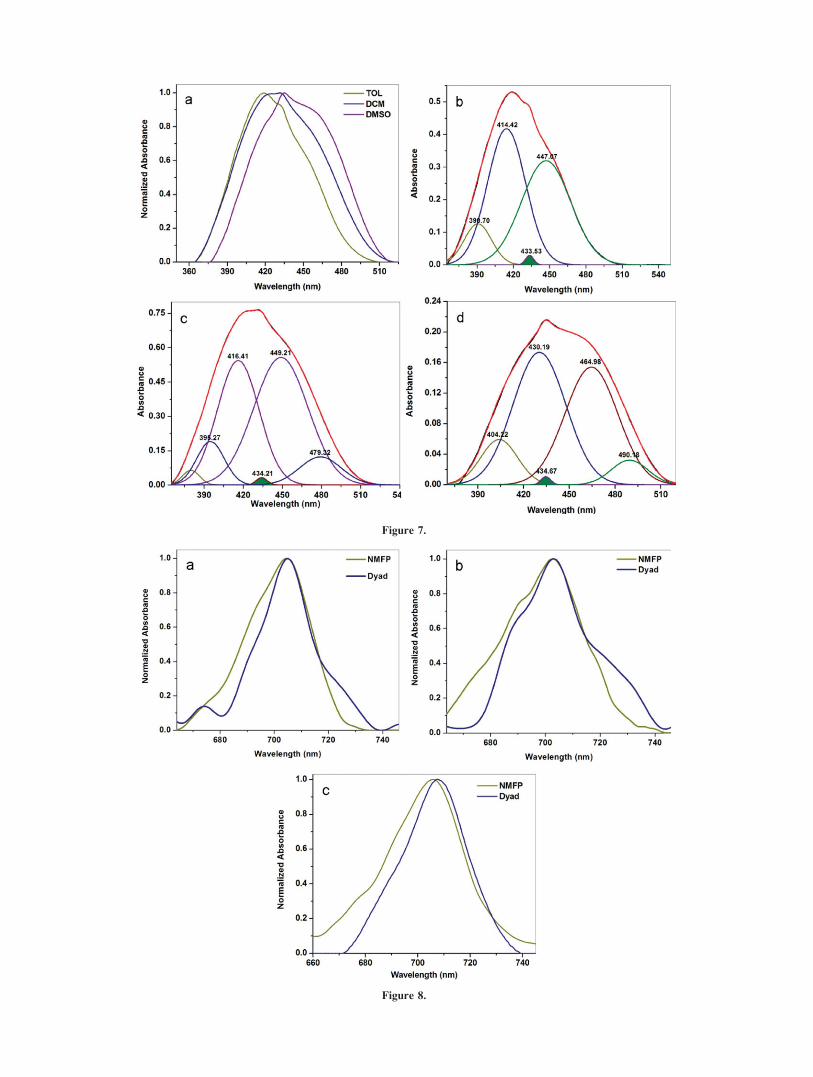
Figure 7.
Figure 8.
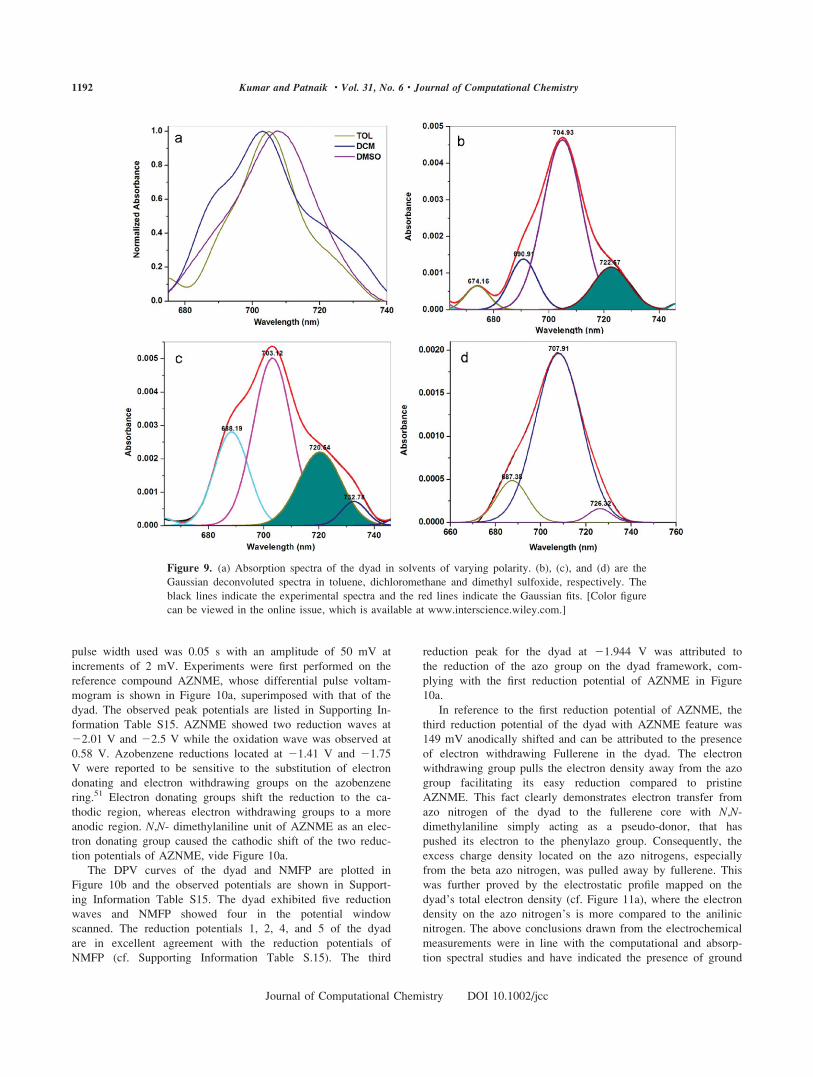
pulse width used was 0.05 s with an amplitude of 50 mV at
increments of 2 mV. Experiments were first performed on the
reference compound AZNME, whose differential pulse voltam-
mogram is shown in Figure 10a, superimposed with that of the
dyad. The observed peak potentials are listed in Supporting In-
formation Table S15. AZNME showed two reduction waves at
22.01 V and 22.5 V while the oxidation wave was observed at
0.58 V. Azobenzene reductions located at 21.41 V and 21.75
V were reported to be sensitive to the substitution of electron
donating and electron withdrawing groups on the azobenzene
ring.51 Electron donating groups shift the reduction to the ca-
thodic region, whereas electron withdrawing groups to a more
anodic region. N,N- dimethylaniline unit of AZNME as an elec-
tron donating group caused the cathodic shift of the two reduc-
tion potentials of AZNME, vide Figure 10a.
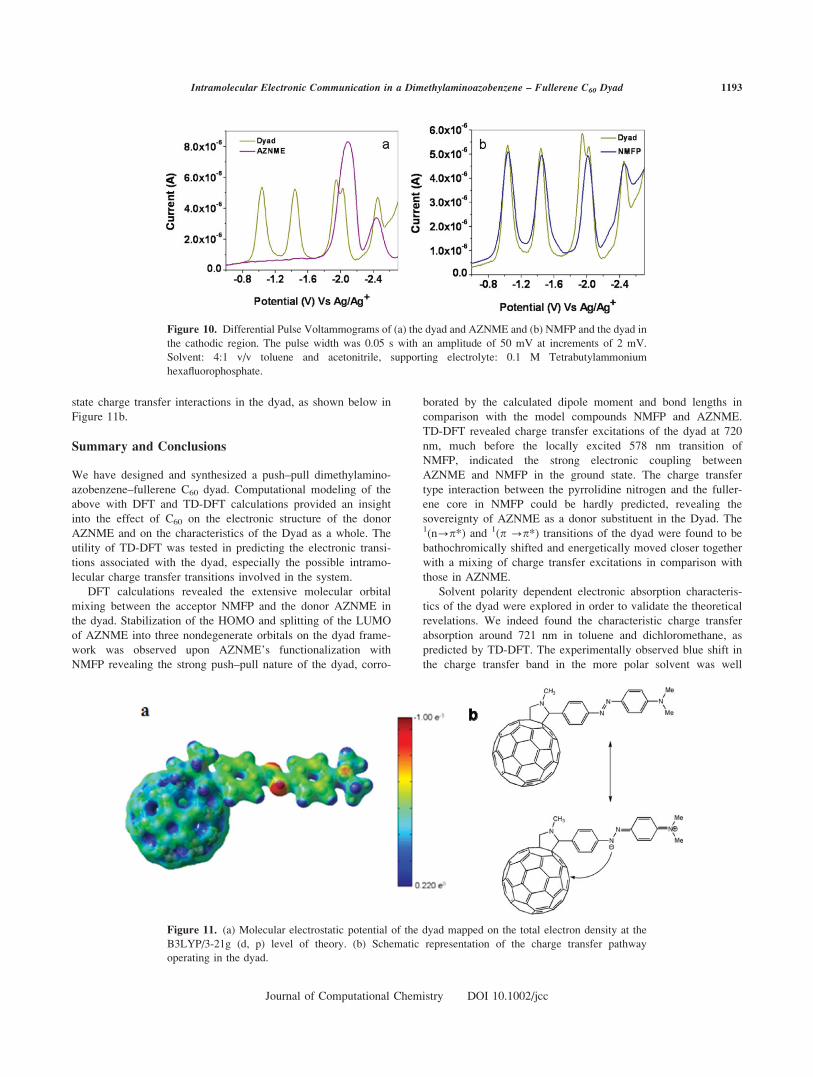
The DPV curves of the dyad and NMFP are plotted in
Figure 10b and the observed potentials are shown in Support-
ing Information Table S15. The dyad exhibited five reduction
waves and NMFP showed four in the potential window
scanned. The reduction potentials 1, 2, 4, and 5 of the dyad
are in excellent agreement with the reduction potentials of
NMFP (cf. Supporting Information Table S.15). The third
reduction peak for the dyad at 21.944 V was attributed to
the reduction of the azo group on the dyad framework, com-
plying with the first reduction potential of AZNME in Figure
10a.
In reference to the first reduction potential of AZNME, the
third reduction potential of the dyad with AZNME feature was
149 mV anodically shifted and can be attributed to the presence
of electron withdrawing Fullerene in the dyad. The electron
withdrawing group pulls the electron density away from the azo
group facilitating its easy reduction compared to pristine
AZNME. This fact clearly demonstrates electron transfer from
azo nitrogen of the dyad to the fullerene core with N,N-dimethylaniline simply acting as a pseudo-donor, that has
pushed its electron to the phenylazo group. Consequently, the
excess charge density located on the azo nitrogens, especially
from the beta azo nitrogen, was pulled away by fullerene. This
was further proved by the electrostatic profile mapped on the
dyad’s total electron density (cf. Figure 11a), where the electron
density on the azo nitrogen’s is more compared to the anilinic
nitrogen. The above conclusions drawn from the electrochemical
measurements were in line with the computational and absorp-
tion spectral studies and have indicated the presence of ground
Figure 9. (a) Absorption spectra of the dyad in solvents of varying polarity. (b), (c), and (d) are the
Gaussian deconvoluted spectra in toluene, dichloromethane and dimethyl sulfoxide, respectively. The
black lines indicate the experimental spectra and the red lines indicate the Gaussian fits. [Color figure
can be viewed in the online issue, which is available at www.interscience.wiley.com.]
1192 Kumar and Patnaik • Vol. 31, No. 6 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
state charge transfer interactions in the dyad, as shown below in
Figure 11b.
Summary and Conclusions
We have designed and synthesized a push–pull dimethylamino-
azobenzene–fullerene C60 dyad. Computational modeling of the
above with DFT and TD-DFT calculations provided an insight
into the effect of C60 on the electronic structure of the donor
AZNME and on the characteristics of the Dyad as a whole. The
utility of TD-DFT was tested in predicting the electronic transi-
tions associated with the dyad, especially the possible intramo-
lecular charge transfer transitions involved in the system.
DFT calculations revealed the extensive molecular orbital
mixing between the acceptor NMFP and the donor AZNME in
the dyad. Stabilization of the HOMO and splitting of the LUMO
of AZNME into three nondegenerate orbitals on the dyad frame-
work was observed upon AZNME’s functionalization with
NMFP revealing the strong push–pull nature of the dyad, corro-
borated by the calculated dipole moment and bond lengths in
comparison with the model compounds NMFP and AZNME.
TD-DFT revealed charge transfer excitations of the dyad at 720
nm, much before the locally excited 578 nm transition of
NMFP, indicated the strong electronic coupling between
AZNME and NMFP in the ground state. The charge transfer
type interaction between the pyrrolidine nitrogen and the fuller-
ene core in NMFP could be hardly predicted, revealing the
sovereignty of AZNME as a donor substituent in the Dyad. The1(n?p*) and 1(p ?p*) transitions of the dyad were found to be
bathochromically shifted and energetically moved closer together
with a mixing of charge transfer excitations in comparison with
those in AZNME.
Solvent polarity dependent electronic absorption characteris-
tics of the dyad were explored in order to validate the theoretical
revelations. We indeed found the characteristic charge transfer
absorption around 721 nm in toluene and dichloromethane, as
predicted by TD-DFT. The experimentally observed blue shift in
the charge transfer band in the more polar solvent was well
Figure 10. Differential Pulse Voltammograms of (a) the dyad and AZNME and (b) NMFP and the dyad in
the cathodic region. The pulse width was 0.05 s with an amplitude of 50 mV at increments of 2 mV.
Solvent: 4:1 v/v toluene and acetonitrile, supporting electrolyte: 0.1 M Tetrabutylammonium
hexafluorophosphate.
Figure 11. (a) Molecular electrostatic potential of the dyad mapped on the total electron density at the
B3LYP/3-21g (d, p) level of theory. (b) Schematic representation of the charge transfer pathway
operating in the dyad.
1193Intramolecular Electronic Communication in a Dimethylaminoazobenzene – Fullerene C60 Dyad
Journal of Computational Chemistry DOI 10.1002/jcc

reproduced by TD-DFT. Considering the large, electronically
asymmetric nature of the dyad with HOMO and LUMO centered
on different parts of the molecular framework, the result is very
significant and this is the first report demonstrating the utility of
TD-DFT in predicting the excitation energies of the Fullerene-
azobenzene hybrid.
To sum up, the charge transfer pathway on the dyad framework
was predicted as follows: an initial ET from electron rich dimethy-
laniline to the electron withdrawing phenylazo group in AZNME
with a resonance driven large electronic population on the b-azo-nitrogen. The peak observed around 477 nm upon the Gaussian
deconvolution of the absorption spectra of AZNME in solvents of
varying polarity exemplified the above process. Substitution of
AZNME with electron deficient fullerene C60 of NMFP, para to
the electron rich azo group, enabled the charge transfer from
AZNME centered HOMOs to the NMFP based LUMOs. Further
support came from the 149 mV anodic shift in the first reduction
potential of the azo group in the dyad in comparison with
AZNME, substantiated by DFT and TD-DFT calculations.
References
1. Wasielewski, M. R. Chem Rev 1992, 92, 435.
2. Kinbara, K.; Aida, T. Chem Rev 2005, 105, 1377.
3. Anthony, J. E. Chem Rev 2006, 106, 5028.
4. Bendikov, M.; Wudl, F.; Perepichka, D. F.; Aida, T. Chem Rev
2004, 104, 4891.
5. Echegoyen, L.; Echegoyen, L. E. In Organic Electrochemistry;
Lund, H.; Hammerich, O., Eds.; Marcel Dekker: New York, 2001;
pp. 323–340.
6. Guldi, D. M. Chem Commun 2000, 321.
7. Prato, M. J Mater Chem 1997, 7, 1097.
8. Kadish, K. M.; Ruoff, R. M., Eds. Fullerenes: Chemistry, Physics
and Technology; Wiley-Interscience: New York, 2000.
9. Guldi, D. M. Chem Soc Rev 2002, 31, 22; and references therein.
10. Figueira-Duarte, T. M.; Gegout, A.; Nierengarten, J. F. Chem Com-
mun 2007, 109.
11. Flint, D. G.; Kumita, J. R.; Smart, O. S.; Woolley, G. A. Chem Biol
2002, 9, 39.
12. Rau, H. In Photocromism, Molecules and Systems; Durr, H.; Bou-
nas-Laurent, H., Eds.; Elsevier: Amsterdam, 1990; Chapter 4, pp.
165–192.
13. Ikeda, T.; Tsutsumi, O. Science 1995, 268, 1873.
14. Liu, Z. F.; Hashimoto, K.; Fujishima, K. Nature 1990, 347, 658.
15. Kawata, S.; Kawata, Y. Chem Rev 2000, 100, 1777.
16. Astrand, P.-O.; Ramanujam, P. S.; Hvilsted, S.; Bak, K. L.; Sauer,
P. A. S. J Am Chem Soc 2000, 122, 3482.
17. Imahori, H.; Tkachenko, N. V.; Vehmanen, V.; Tamaki, K.; Lemme-
tyinen, H.; Sakata, Y.; Fukuzumi, S. J Phy Chem A 2001, 105,
1750.
18. Gayathri, S.; Patnaik, A. Chem Phy Lett 2005, 414, 198.
19. Kay, K.-Y.; Han, K.-J.; Yu, Y.-J.; Park, Y.-D. Tetrahedron Lett
2002, 43, 5053.
20. Oh-Ishi, K.; Okamura, J.; Ishi-I, T.; Sano, M.; Shinkai, S. Langmuir
1999, 15, 2224.
21. Shirai, Y.; Sasaki, T.; Guerrero, J. M.; Yu, B.-C.; Hodge, P.; Tour,
J. M. ACS Nano 2008, 2, 97.
22. Schuster, D. I.; Li, K.; Guldi, D. M.; Palkar, A.; Echegoyen, L.;
Stanisky, C.; Cross, R. J.; Niemi, M.; Tkachenko, N. V.; Lemmetyi-
nen, H. J Am Chem Soc 2007, 129, 15973.
23. Runge, E.; Gross, E. K. U. Phys Rev Lett 1984, 52, 997.
24. Casida, M. In Recent Advances in Density Functional Methods;
Chong, D. P., Ed.; World Scientific: Singapore, 1995, 155.
25. Casida, M. In Recent Developments and Applications of Modern
Density Functional Theory; Seminario, J. M., Ed.; Elsevier: Amster-
dam, 1996, 391.
26. Stratmann, R. R.; Scuseria, G. E.; Frisch, M. J. J Chem Phys 1998,
109, 8218.
27. Bauernschmitt, R.; Ahlrichs, R.; Hennrich, F. H.; Kappes, M. M. J
Am Chem Soc 1998, 120, 5052.
28. Autschbach, J. ChemPhysChem 2009, 10, 1757.
29. Cohen, A. J.; Mori-Sanch, P.; Yang, W. Science 2008, 321, 792.
30. Elliott, P.; Burke, K.; Furche, F. In Reviews of Computational
Chemistry; Lipko-Witz, K. B.; Cundari, T. R., Eds.; Wiley: Hobo-
ken, 2009, 26, 91.
31. Dreuw, A.; Weisman, J. L.; Head-Gordon, M. J Chem Phys 2003,
119, 2943.
32. Tozer, D. J. J Chem Phys 2003, 119, 12697.
33. Gritsenko, O.; Baerends, E. J. J Chem Phys 2004, 121, 655.
34. Dreuw, A.; Head-Gordon, M. J Am Chem Soc 2004, 126, 4007.
35. Tozer, D. J.; Amos, R. D.; Handy, N. C.; Roos, B. J.; Serrano-
Andres, L. Mol Phys 1999, 97, 859.
36. Sobolewski, A. L.; Domcke, W. Chem Phys 2003, 294, 73.
37. Zandler, M. E.; D’souza, F. C. R. Chimie 2006, 9, 960.
38. Gaussian 03, Revision C.02, Frisch, M. J.; Trucks, G. W.; Schlegel,
H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgom-
ery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J.
M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.;
Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.;
Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Naka-
jima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox,
J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gom-
perts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.;
Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G.
A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.;
Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A.
D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul,
A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Lia-
shenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.;
Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challa-
combe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M. W.;
Gonzalez, C.; Pople, J. A. Gaussian, Inc., Wallingford CT, 2004.
39. Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J.
J Phys Chem 1994, 98, 11623.
40. Becke, A. D. Phy Rev A 1988, 38, 3098.
41. Vosko, S. H.; Wilk, L.; Nusair, M. Can J Phys 1980, 58, 1200.
42. Lee, C.; Yang, W.; Parr, R. G. Phys Rev B 1988, 37, 785.
43. Amovilli, C.; Barone, V.; Cammi, R.; Cances, E.; Cossi, M.; Mennucci,
B.; Pomelli, C. S.; Tomasi, J. Adv Quantum Chem 1998, 32, 227.
44. Cossi, M.; Barone, V. J Chem Phys 2001, 115, 4708.
45. Fabian, J.; Diaza, L. A.; Seifert, G.; Niehaus, T. J. Mol Struct
(Theochem) 2002, 594, 41.
46. Conti, I.; Garavelli, M.; Orlandi, G. J Am Chem Soc 2008, 130,
5216; and references therein.
47. Briquet, I.; Vercauteren, D. P.; Andre, J.-M.; Perpete, E. A.; Jacque-
min, D. Chem Phys Lett 2007, 435, 257.
48. Yanai, T.; Tew, D. P.; Handy, N. C. Chem Phys Lett 2004, 393, 51.
49. Jacquemin, D.; Perpete, E. A.; Scuseria, G. E.; Ciofini, I.; Adamo,
C. J Chem Theory Comput 2008, 4, 123.
50. Maggini, M.; Scorrano, G.; Prato, M. J Am Chem Soc 1993, 115,
9798.
51. Stradins, J. P.; Glezer, V. T. In Encyclopedia of Electrochemistry of
the Elements; Organic Section; Bard, A. J.; Lund, H., Eds.; Marcel
Dekker, Inc.: New York, 1979; Vol. XIII, p. 163.
1194 Kumar and Patnaik • Vol. 31, No. 6 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc




![Quasi 3D polymerization in C60 bilayers in a fullerene solvate · 2017. 9. 25. · [16,17], and TDAE-C. 60 (TDAE: tetrakis-dimethylamino-ethylene) [18,19], etc. transformed to the](https://img.pdfslide.us/doc/110x75/60824ae0eb6af07ece3caf54/quasi-3d-polymerization-in-c60-bilayers-in-a-fullerene-2017-9-25-1617-and.jpg)

![Strong Electronic Polarization of the C Fullerene by the ... ion-molecular configurations of [MMIM][Cl], [MMIM][NO3], and [MMIM][PF6] with the C60 fullerene after the 10 ps long BOMD](https://img.pdfslide.us/doc/110x75/5aa8d0bb7f8b9a6c188bfdd3/strong-electronic-polarization-of-the-c-fullerene-by-the-ion-molecular-configurations.jpg)



![Inclusion of C Fullerene in a M L Subphthalocyanine Cage · S17 Complex [C60⊂2]•6PF6: [C60⊂2]•6PF6 was obtained by mixing 2•6PF6 and 5 equivalents C60 in acetone for five](https://img.pdfslide.us/doc/110x75/606126dbc2cd3f6b2e673f2e/inclusion-of-c-fullerene-in-a-m-l-subphthalocyanine-s17-complex-c60a2a6pf6.jpg)


![Division of Materials Chemistry – Polymer Controlled Synthesis · actions. We found that [10]CPP selectively encapsulated C60 forming the shortest fullerene-peapod, [10]CPP⊃C60,](https://img.pdfslide.us/doc/110x75/60a99b1b5c3efb00265d0c7b/division-of-materials-chemistry-a-polymer-controlled-synthesis-actions-we-found.jpg)















