Embed Size (px)
Citation preview

INTESTINAL STRESS
Paneth cells secrete lysozyme viasecretory autophagy during bacterialinfection of the intestineShai Bel,1 Mihir Pendse,1 Yuhao Wang,1 Yun Li,1 Kelly A. Ruhn,1 Brian Hassell,1
Tess Leal,1 Sebastian E. Winter,2 Ramnik J. Xavier,3,4,5 Lora V. Hooper1,6*
Intestinal Paneth cells limit bacterial invasion by secreting antimicrobial proteins, includinglysozyme. However, invasive pathogens can disrupt the Golgi apparatus, interfering withsecretion and compromising intestinal antimicrobial defense. Here we show that duringbacterial infection, lysozyme is rerouted via secretory autophagy, an autophagy-basedalternative secretion pathway. Secretory autophagy was triggered in Paneth cells bybacteria-induced endoplasmic reticulum (ER) stress, required extrinsic signals from innatelymphoid cells, and limited bacterial dissemination. Secretory autophagy was disruptedin Paneth cells of mice harboring a mutation in autophagy gene Atg16L1 that confersincreased risk for Crohn’s disease in humans. Our findings identify a role for secretoryautophagy in intestinal defense and suggest why Crohn’s disease is associated withgenetic mutations that affect both the ER stress response and autophagy.
Themammalian intestine is home to a diversepopulation of bacteria,which includes patho-gens that can disrupt host cellular functions.The intestinal epithelium defends againstbacterial encroachment through multiple
mechanisms, including antimicrobial proteinsecretion and destruction of invading bacteriathrough autophagy (1). Paneth cells are special-ized intestinal epithelial cells that secrete abun-dant antimicrobial proteins, including lysozyme;thus, disrupting Paneth cell secretion can lead toinflammatory disease (2–4). Pathogenicmicrobescan trigger endoplasmic reticulum (ER) stressthat interferes with protein secretion (5, 6) andcompromises antimicrobial protein delivery, rais-ing the question of how Paneth cells preservetheir antimicrobial function during pathogen-induced stress.Invasive bacteria, including Salmonella enterica
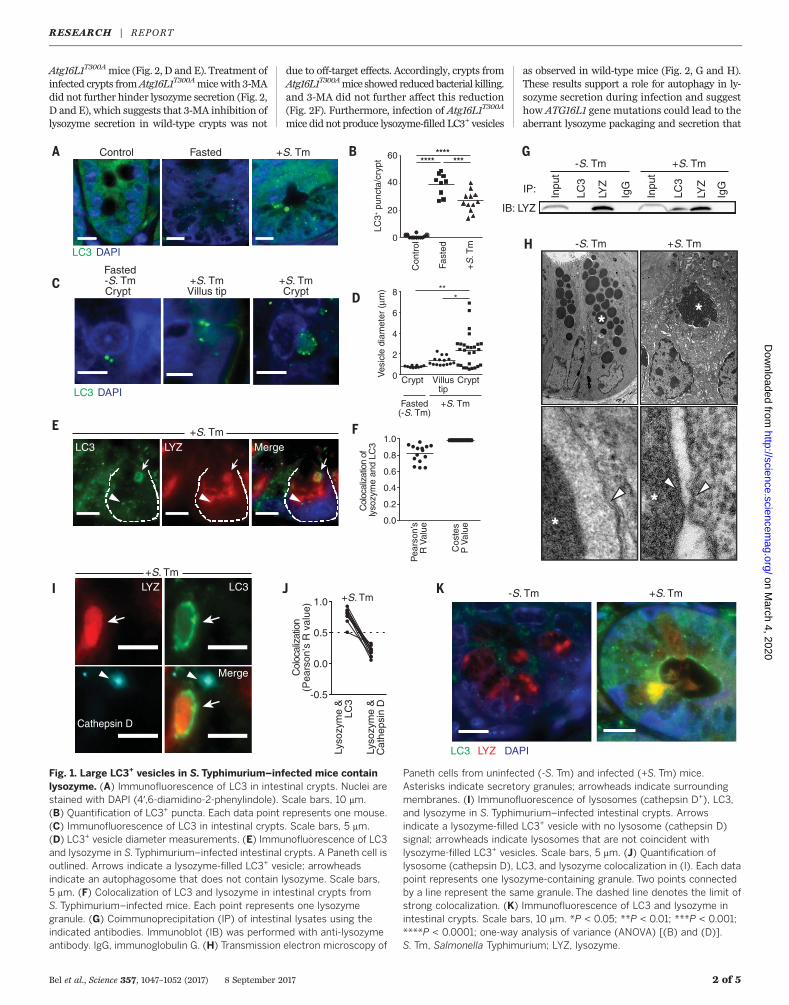
serovar Typhimurium (S. Typhimurium), triggerautophagy in intestinal enterocytes. This is indi-cated by abundant epithelial cell autophagosomes,marked by LC3 (microtubule-associated protein1 light chain 3), that capture and eliminate invad-ing bacteria (7). S. Typhimurium also invadedPaneth cells (fig. S1), and invasion was associatedwith elevated numbers of LC3+ puncta in Panethcells (Fig. 1, A and B). The puncta numbers were
comparable to those inmice subjected to fasting,a trigger of canonical autophagy (8) (Fig. 1, A andB). However, many of the LC3+ structures ininfected Paneth cells were markedly larger (0.2to 7 mm in diameter) than the LC3+ puncta inS. Typhimurium–infected enterocytes (~1 mm) orin Paneth cells of fasted (~0.5 mm) mice (Fig. 1, Cand D).To characterize the contents of the LC3+ vesicles,
we performed immunofluorescence, transmissionelectron microscopy, and coimmunoprecipitationassays. These assays revealed that the large LC3+
vesicles contained lysozyme (Fig. 1, E to G) andwere absent in Paneth cells of uninfected andfasted mice, where lysozyme was packaged intoLC3– vesicles (fig. S2). Ultrastructure analysisshowed that the large granules in Paneth cells ofinfected mice contained lysozyme (fig. S3) andwere surrounded by a double membrane (Fig.1H), a hallmark of autophagosomes (8). Gran-ules from uninfected mice were surrounded bya single membrane (Fig. 1H). The LC3+ vesiclesdid not contain bacteria (fig. S4, A and B) orthe antimicrobial proteins REG3g or cryptdin 5(fig. S5). Also, cryptdin 5 was not packaged insecretory granules and was excluded from theLC3+ vesicles in infected mice (fig. S6, A and B).This suggested that infection interferes withpackaging and secretion of Paneth cell antimicro-bial proteins and that lysozymemight be reroutedthrough an alternative secretion pathway involv-ing an LC3+ vesicle.Canonical autophagy targets the cargo in LC3+
autophagosomes for degradation in lysosomes(8). Immunofluorescence analysis indicated thatthe lysozyme-filled LC3+ vesicles do not fuse withlysosomes, as the vesicles did not colocalize withthe lysosomal marker cathepsin D (Fig. 1, I andJ), implying that lysozyme is not targeted fordegradation. The lysozyme-filled LC3+ vesiclesalso did not colocalize with p62/sequestesome
1 (SQSTM1), which selects autophagosome cargofor degradation (8). SQSTM1was associated withthe smaller LC3+ puncta in Paneth cells but wasnot associated with the larger LC3+ lysozyme+
vesicles in infected mice (fig. S7), implying thatlysozyme is not selected for degradation in theautolysosome through the canonical selective auto-phagy pathway. LC3 also accumulated at the apicalsurface ofPaneth cells from infectedmice, andLC3+
vesicles had fused with the apical surface and dis-charged lysozyme into the intestinal lumen (Fig. 1K).This was not observed in uninfected mice (fig. S7),which suggests that the LC3+ vesicles might beinvolved in lysozyme secretion.During conventional protein secretion, proteins
are transported through the ER–Golgi complex,packaged in secretory granules, and released tothe extracellular space. There are various alter-native secretory pathways, including one thatutilizes components of the autophagy pathwayand is known as secretory autophagy (9). In se-cretory autophagy, cargo is transported in anLC3+ vesicle and discharged at the plasmamem-brane, thus bypassing the ER-Golgi complex(9). Rab8a, a marker of secretory autophagy ves-icles (10), colocalized with the lysozyme-filledLC3+ vesicles. Rab8a also coimmunoprecipi-tated with LC3 only in infected mice but did notcolocalize with cryptdin-5 (fig. S8). This sug-gested that lysozymemight be selectively secretedthrough the secretory autophagy pathway duringinfection.To further test this idea, we isolated Paneth
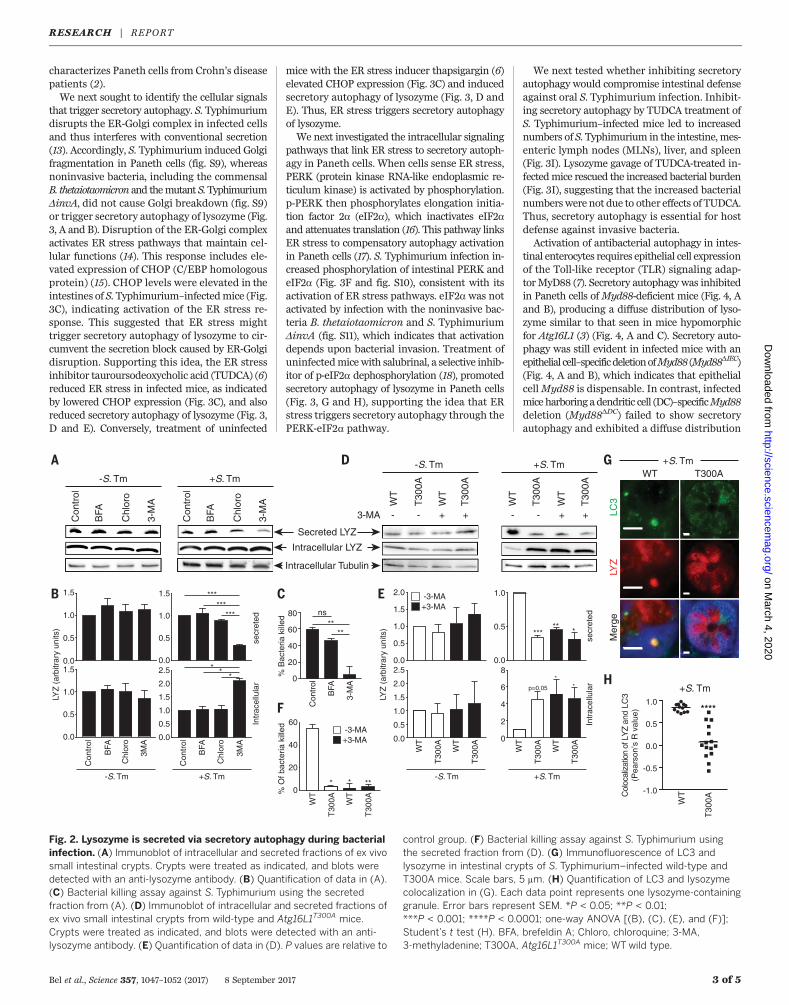
cell–containing crypts, infected them in vitrowhile treating with chemical inhibitors of con-ventional secretion and autophagy, and analyzedthe supernatants for lysozyme secretion. Inhibit-ing ER-Golgi trafficking with brefeldin A (BFA)did not affect lysozyme secretion in infected oruninfected crypts (Fig. 2, A and B), indicatingthat lysozyme secretion can bypass the ER-Golgipathway. Lysozyme secretion was also not alteredby treatment with chloroquine (Fig. 2, A and B),which prevents lysosome acidification (8), imply-ing that inhibiting autophagic degradation doesnot affect lysozyme secretion. However, treat-ment with 3-methyladenine (3-MA), which in-hibits autophagosome nucleation (8), impairedlysozyme secretion and caused an accumula-tion of intracellular lysozyme in infected crypts(Fig. 2, A and B). Accordingly, secretions fromBFA-treated but not 3-MA–treated crypts killedbacteria (Fig. 2C), indicating that secretory auto-phagy is essential for antibacterial defense ininfected crypts.We next studied mice in which autophagy is
perturbed by a mutation in the Atg16L1 (autoph-agy related 16–like 1) gene. A mutation in theAtg16L1 gene [Thr300→Ala300 (T300A)] confersan increased risk of developing Crohn’s diseasein humans (11). Mice harboring this mutation(Atg16L1T300A) exhibit decreased antibacterialautophagy and abnormal Paneth cell lysozymedistribution (12). Whereas crypts from uninfectedwild-type and Atg16L1T300A mice secreted similaramounts of lysozyme, lysozyme secretion was im-paired in crypts from S. Typhimurium–infected
RESEARCH
Bel et al., Science 357, 1047–1052 (2017) 8 September 2017 1 of 5
1Department of Immunology, The University of TexasSouthwestern Medical Center, Dallas, TX 75390, USA.2Department of Microbiology, The University of TexasSouthwestern Medical Center, Dallas, TX 75390, USA.3Broad Institute, Cambridge, MA 02142, USA. 4Center forComputational and Integrative Biology, MassachusettsGeneral Hospital, Boston, MA 02114, USA. 5GastrointestinalUnit and Center for the Study of Inflammatory BowelDisease, Massachusetts General Hospital, Harvard MedicalSchool, Boston, MA 02142, USA. 6The Howard HughesMedical Institute, The University of Texas SouthwesternMedical Center, Dallas, TX 75390, USA.*Corresponding author. Email: [email protected]
on March 4, 2020
http://science.sciencem
ag.org/D
ownloaded from
Atg16L1T300Amice (Fig. 2, D and E). Treatment ofinfected crypts fromAtg16L1T300Amicewith 3-MAdid not further hinder lysozyme secretion (Fig. 2,D and E), which suggests that 3-MA inhibition oflysozyme secretion in wild-type crypts was not
due to off-target effects. Accordingly, crypts fromAtg16L1T300Amice showed reducedbacterial killing.and 3-MA did not further affect this reduction(Fig. 2F). Furthermore, infection of Atg16L1T300A
mice did not produce lysozyme-filled LC3+ vesicles
as observed in wild-type mice (Fig. 2, G and H).These results support a role for autophagy in ly-sozyme secretion during infection and suggesthow ATG16L1 gene mutations could lead to theaberrant lysozyme packaging and secretion that
Bel et al., Science 357, 1047–1052 (2017) 8 September 2017 2 of 5
0.0
0.2
0.4
0.6
0.8
1.0
Col
ocal
izat
ion
of
lyso
zym
e an
d LC
3
Pea
rson
’sR
Val
ue
Cos
tes
P V
alue
Control Fasted
LC3 DAPI
+S. Tm
LYZ+S. Tm
LC3
Cathepsin D
Merge
LC3 LYZ Merge+S. Tm
-0.5
0.0
0.5
1.0
Lyso
zym
e &
LC3
Lyso
zym
e &
Cat
heps
in D
+S. Tm
Col
ocal
izat
ion
(Pea
rson
’s R
val
ue)
+S. Tm-S. Tm
*
*
*
*
0
20
40
********
***
LC3+
pun
cta/
cryp
t
Con
trol
Fast
ed
+S
. Tm
60
0
2
4
6
8
Ves
icle
dia
met
er (
μm)
***
CryptVillustip
Fasted(-S. Tm)
Crypt
+S. Tm
+S. TmCrypt
+S. TmVillus tip
Fasted-S. Tm
LC3 DAPI
Crypt
LC3 LYZ DAPI
-S. Tm +S. Tm
IP: Inpu
t
LC3
LYZ
IgG
Inpu
t
LC3
LYZ
IgG
IB: LYZ
+S. Tm-S. Tm
Fig. 1. Large LC3+ vesicles in S.Typhimurium–infected mice containlysozyme. (A) Immunofluorescence of LC3 in intestinal crypts. Nuclei arestained with DAPI (4′,6-diamidino-2-phenylindole). Scale bars, 10 mm.(B) Quantification of LC3+ puncta. Each data point represents one mouse.(C) Immunofluorescence of LC3 in intestinal crypts. Scale bars, 5 mm.(D) LC3+ vesicle diameter measurements. (E) Immunofluorescence of LC3and lysozyme in S.Typhimurium–infected intestinal crypts. A Paneth cell isoutlined. Arrows indicate a lysozyme-filled LC3+ vesicle; arrowheadsindicate an autophagosome that does not contain lysozyme. Scale bars,5 mm. (F) Colocalization of LC3 and lysozyme in intestinal crypts fromS. Typhimurium–infected mice. Each point represents one lysozymegranule. (G) Coimmunoprecipitation (IP) of intestinal lysates using theindicated antibodies. Immunoblot (IB) was performed with anti-lysozymeantibody. IgG, immunoglobulin G. (H) Transmission electron microscopy of
Paneth cells from uninfected (-S. Tm) and infected (+S. Tm) mice.Asterisks indicate secretory granules; arrowheads indicate surroundingmembranes. (I) Immunofluorescence of lysosomes (cathepsin D+), LC3,and lysozyme in S. Typhimurium–infected intestinal crypts. Arrowsindicate a lysozyme-filled LC3+ vesicle with no lysosome (cathepsin D)signal; arrowheads indicate lysosomes that are not coincident withlysozyme-filled LC3+ vesicles. Scale bars, 5 mm. (J) Quantification oflysosome (cathepsin D), LC3, and lysozyme colocalization in (I). Each datapoint represents one lysozyme-containing granule. Two points connectedby a line represent the same granule. The dashed line denotes the limit ofstrong colocalization. (K) Immunofluorescence of LC3 and lysozyme inintestinal crypts. Scale bars, 10 mm. *P < 0.05; **P < 0.01; ***P < 0.001;****P < 0.0001; one-way analysis of variance (ANOVA) [(B) and (D)].S. Tm, Salmonella Typhimurium; LYZ, lysozyme.
RESEARCH | REPORTon M
arch 4, 2020
http://science.sciencemag.org/
Dow
nloaded from
characterizes Paneth cells from Crohn’s diseasepatients (2).We next sought to identify the cellular signals
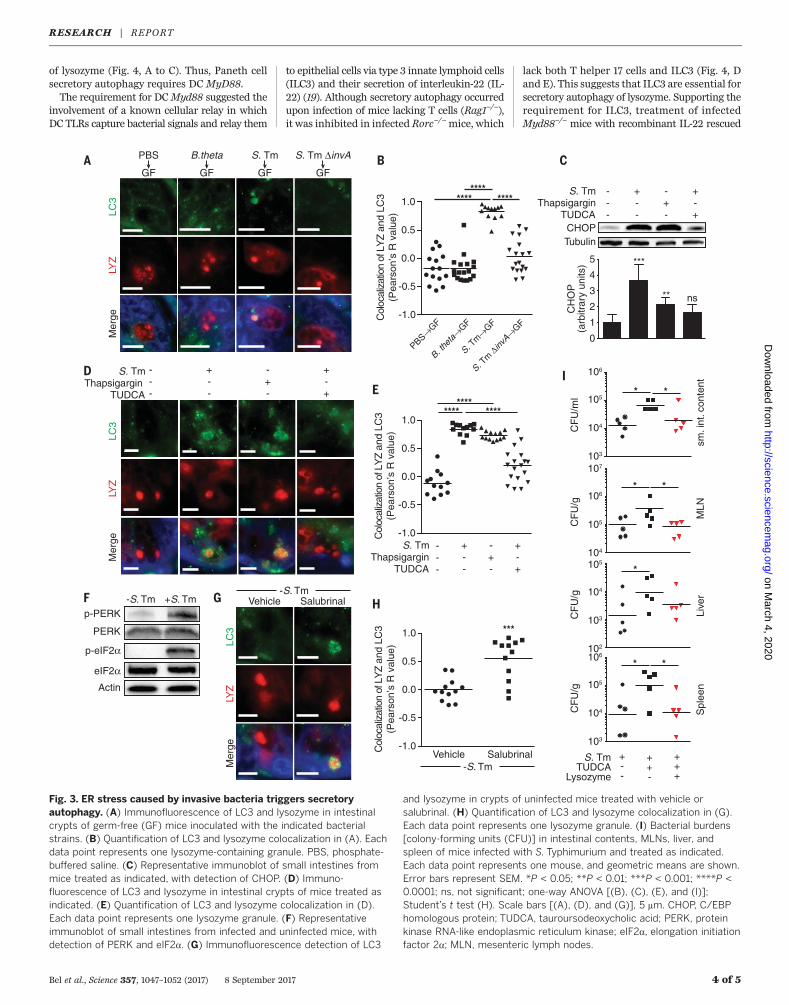
that trigger secretory autophagy. S. Typhimuriumdisrupts the ER-Golgi complex in infected cellsand thus interferes with conventional secretion(13). Accordingly, S. Typhimurium induced Golgifragmentation in Paneth cells (fig. S9), whereasnoninvasive bacteria, including the commensalB. thetaiotaomicron and themutant S.TyphimuriumDinvA, did not cause Golgi breakdown (fig. S9)or trigger secretory autophagy of lysozyme (Fig.3, A and B). Disruption of the ER-Golgi complexactivates ER stress pathways that maintain cel-lular functions (14). This response includes ele-vated expression of CHOP (C/EBP homologousprotein) (15). CHOP levels were elevated in theintestines of S. Typhimurium–infectedmice (Fig.3C), indicating activation of the ER stress re-sponse. This suggested that ER stress mighttrigger secretory autophagy of lysozyme to cir-cumvent the secretion block caused by ER-Golgidisruption. Supporting this idea, the ER stressinhibitor tauroursodeoxycholic acid (TUDCA) (6)reduced ER stress in infected mice, as indicatedby lowered CHOP expression (Fig. 3C), and alsoreduced secretory autophagy of lysozyme (Fig. 3,D and E). Conversely, treatment of uninfected
mice with the ER stress inducer thapsigargin (6)elevated CHOP expression (Fig. 3C) and inducedsecretory autophagy of lysozyme (Fig. 3, D andE). Thus, ER stress triggers secretory autophagyof lysozyme.We next investigated the intracellular signaling
pathways that link ER stress to secretory autoph-agy in Paneth cells. When cells sense ER stress,PERK (protein kinase RNA-like endoplasmic re-ticulum kinase) is activated by phosphorylation.p-PERK then phosphorylates elongation initia-tion factor 2a (eIF2a), which inactivates eIF2aand attenuates translation (16). This pathway linksER stress to compensatory autophagy activationin Paneth cells (17). S. Typhimurium infection in-creased phosphorylation of intestinal PERK andeIF2a (Fig. 3F and fig. S10), consistent with itsactivation of ER stress pathways. eIF2a was notactivated by infection with the noninvasive bac-teria B. thetaiotaomicron and S. TyphimuriumDinvA (fig. S11), which indicates that activationdepends upon bacterial invasion. Treatment ofuninfectedmicewith salubrinal, a selective inhib-itor of p-eIF2a dephosphorylation (18), promotedsecretory autophagy of lysozyme in Paneth cells(Fig. 3, G and H), supporting the idea that ERstress triggers secretory autophagy through thePERK-eIF2a pathway.
We next tested whether inhibiting secretoryautophagy would compromise intestinal defenseagainst oral S. Typhimurium infection. Inhibit-ing secretory autophagy by TUDCA treatment ofS. Typhimurium–infected mice led to increasednumbers of S. Typhimurium in the intestine,mes-enteric lymph nodes (MLNs), liver, and spleen(Fig. 3I). Lysozyme gavage of TUDCA-treated in-fectedmice rescued the increased bacterial burden(Fig. 3I), suggesting that the increased bacterialnumberswere not due to other effects of TUDCA.Thus, secretory autophagy is essential for hostdefense against invasive bacteria.Activation of antibacterial autophagy in intes-
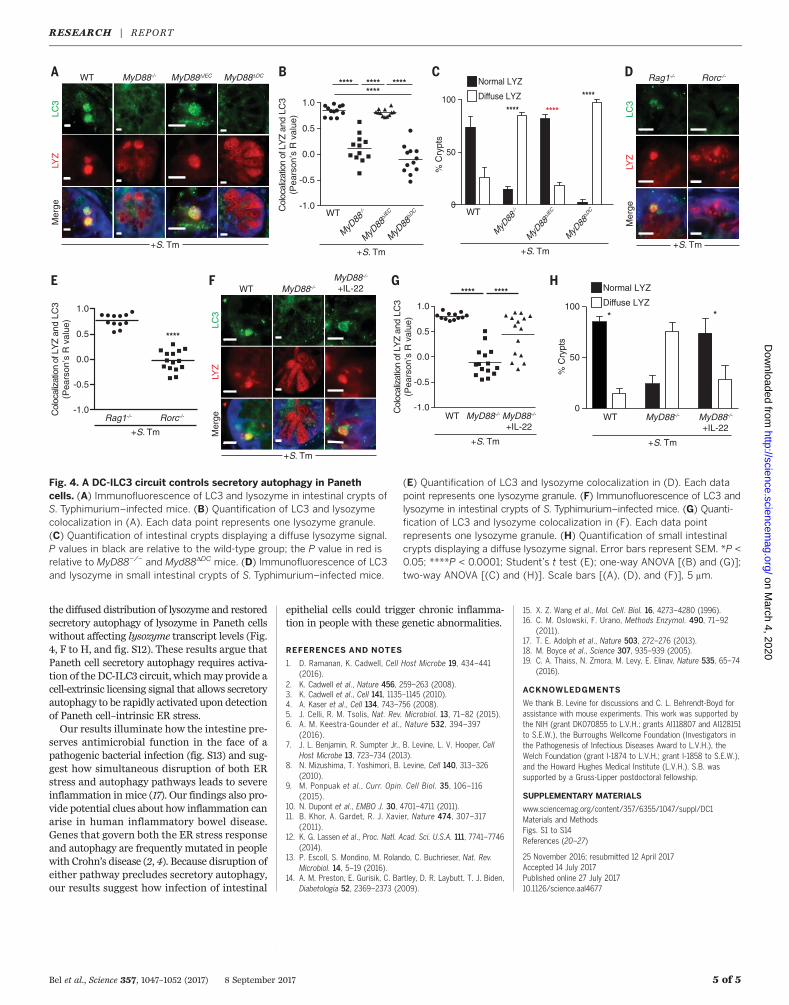
tinal enterocytes requires epithelial cell expressionof the Toll-like receptor (TLR) signaling adap-torMyD88 (7). Secretory autophagy was inhibitedin Paneth cells ofMyd88-deficient mice (Fig. 4, Aand B), producing a diffuse distribution of lyso-zyme similar to that seen in mice hypomorphicfor Atg16L1 (3) (Fig. 4, A and C). Secretory auto-phagy was still evident in infected mice with anepithelial cell–specificdeletionofMyd88 (Myd88DIEC)(Fig. 4, A and B), which indicates that epithelialcellMyd88 is dispensable. In contrast, infectedmice harboring adendritic cell (DC)–specificMyd88deletion (Myd88DDC) failed to show secretoryautophagy and exhibited a diffuse distribution
Bel et al., Science 357, 1047–1052 (2017) 8 September 2017 3 of 5
WT+S. Tm
LC3
LYZ
Mer
ge
T300A
Secreted LYZ
Intracellular LYZ
Intracellular Tubulin
+S. Tm-S. Tm
Con
trol
BFA
Chl
oro
3MA
Con
trol
BFA
Chl
oro
3MA
LYZ
(ar
bitr
ary
units
)
secr
eted
Intr
acel
lula
r
+S. Tm-S. Tm
0.0
0.5
1.0
1.5
0.0
0.5
1.0
1.5
0.0
0.5
1.0
1.5
2.0
2.5
0.0
0.5
1.0
1.5
Con
trol
BFA
Chl
oro
3-M
A
Con
trol
BFA
Chl
oro
3-M
A
+S. Tm-S. Tm
0
2
4
8
6
0.0
0.5
1.0
+S. Tm-S. Tm
secr
eted
Intr
acel
lula
r
LYZ
(ar
bitr
ary
units
)
0.0
0.5
1.0
1.5
2.0
0.0
0.5
1.0
1.5
2.0
2.5
WT
T30
0A WT
T30
0A WT
T30
0A WT
T30
0A
+3-MA-3-MA
WT
T30
0A
WT
T30
0A
WT
T30
0A
WT
T30
0A
3-MA - - + + - - + +
******
***
* **
*****
*
p=0.05
**
% B
acte
ria k
illed
0
20
40
60
80
Con
trol
BFA
3-M
A
****
0
20
40
60
% O
f bac
teria
kill
ed
+3-MA-3-MA
WT
T30
0A WT
T30
0A
* * **-1.0
-0.5
0.0
0.5
1.0
Col
ocal
izat
ion
of L
YZ
and
LC3
(Pea
rson
’s R
val
ue)
+S. Tm
****
WT
T30
0A
ns
Fig. 2. Lysozyme is secreted via secretory autophagy during bacterialinfection. (A) Immunoblot of intracellular and secreted fractions of ex vivosmall intestinal crypts. Crypts were treated as indicated, and blots weredetected with an anti-lysozyme antibody. (B) Quantification of data in (A).(C) Bacterial killing assay against S. Typhimurium using the secretedfraction from (A). (D) Immunoblot of intracellular and secreted fractions ofex vivo small intestinal crypts from wild-type and Atg16L1T300A mice.Crypts were treated as indicated, and blots were detected with an anti-lysozyme antibody. (E) Quantification of data in (D). P values are relative to
control group. (F) Bacterial killing assay against S. Typhimurium usingthe secreted fraction from (D). (G) Immunofluorescence of LC3 andlysozyme in intestinal crypts of S. Typhimurium–infected wild-type andT300A mice. Scale bars, 5 mm. (H) Quantification of LC3 and lysozymecolocalization in (G). Each data point represents one lysozyme-containinggranule. Error bars represent SEM. *P < 0.05; **P < 0.01;***P < 0.001; ****P < 0.0001; one-way ANOVA [(B), (C), (E), and (F)];Student’s t test (H). BFA, brefeldin A; Chloro, chloroquine; 3-MA,3-methyladenine; T300A, Atg16L1T300A mice; WT wild type.
RESEARCH | REPORTon M
arch 4, 2020
http://science.sciencemag.org/
Dow
nloaded from
of lysozyme (Fig. 4, A to C). Thus, Paneth cellsecretory autophagy requires DC MyD88.The requirement for DCMyd88 suggested the
involvement of a known cellular relay in whichDC TLRs capture bacterial signals and relay them
to epithelial cells via type 3 innate lymphoid cells(ILC3) and their secretion of interleukin-22 (IL-22) (19). Although secretory autophagy occurredupon infection of mice lacking T cells (Rag1−/−),it was inhibited in infected Rorc−/−mice, which
lack both T helper 17 cells and ILC3 (Fig. 4, Dand E). This suggests that ILC3 are essential forsecretory autophagy of lysozyme. Supporting therequirement for ILC3, treatment of infectedMyd88−/− mice with recombinant IL-22 rescued
Bel et al., Science 357, 1047–1052 (2017) 8 September 2017 4 of 5
S. Tm
ΔinvA→
GF
S. Tm
→GF
B. the
ta→
GF
PBS→GF
-1.0
-0.5
0.0
0.5
1.0
CH
OP
(arb
itrar
y un
its)
S. Tm
TUDCAThapsigargin
+--
---
-+-
+-+
CHOP
0
1
2
3
4
5 ***
**
Tubulin
ns
**** ********
Col
ocal
izat
ion
of L
YZ
and
LC3
(Pea
rson
’s R
val
ue)
B.theta
GF
S. Tm ΔinvA
GF
PBS
GF
S. Tm
GF
LC3
LYZ
Mer
ge
-1.0
-0.5
0.0
0.5
1.0
S. TmThapsigargin
TUDCA
+--
---
-+-
+-+
********
Col
ocal
izat
ion
of L
YZ
and
LC3
(Pea
rson
’s R
val
ue)LC
3LY
ZM
erge
S. Tm
TUDCAThapsigargin
---
+--
-+-
+-+
p-PERK
PERK
p-eIF2α
eIF2α
Actin
-S. Tm +S. Tm
S. Tm
LysozymeTUDCA
+--
++-
+++
sm. i
nt. c
onte
nt
*
MLN
Live
rS
plee
n
CF
U/m
l
106
105
104
103
CF
U/g
107
106
105
104
CF
U/g
105
104
103
102
CF
U/g
106
105
104
103
*
*
*
*
*
*
****
-1.0
-0.5
0.0
0.5
1.0
SalubrinalVehicle
***
Col
ocal
izat
ion
of L
YZ
and
LC3
(Pea
rson
’s R
val
ue)
-S. Tm
SalubrinalVehicle
LC3
LYZ
Mer
ge
-S. Tm
Fig. 3. ER stress caused by invasive bacteria triggers secretoryautophagy. (A) Immunofluorescence of LC3 and lysozyme in intestinalcrypts of germ-free (GF) mice inoculated with the indicated bacterialstrains. (B) Quantification of LC3 and lysozyme colocalization in (A). Eachdata point represents one lysozyme-containing granule. PBS, phosphate-buffered saline. (C) Representative immunoblot of small intestines frommice treated as indicated, with detection of CHOP. (D) Immuno-fluorescence of LC3 and lysozyme in intestinal crypts of mice treated asindicated. (E) Quantification of LC3 and lysozyme colocalization in (D).Each data point represents one lysozyme granule. (F) Representativeimmunoblot of small intestines from infected and uninfected mice, withdetection of PERK and eIF2a. (G) Immunofluorescence detection of LC3
and lysozyme in crypts of uninfected mice treated with vehicle orsalubrinal. (H) Quantification of LC3 and lysozyme colocalization in (G).Each data point represents one lysozyme granule. (I) Bacterial burdens[colony-forming units (CFU)] in intestinal contents, MLNs, liver, andspleen of mice infected with S. Typhimurium and treated as indicated.Each data point represents one mouse, and geometric means are shown.Error bars represent SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P <0.0001; ns, not significant; one-way ANOVA [(B), (C), (E), and (I)];Student’s t test (H). Scale bars [(A), (D), and (G)], 5 mm. CHOP, C/EBPhomologous protein; TUDCA, tauroursodeoxycholic acid; PERK, proteinkinase RNA-like endoplasmic reticulum kinase; eIF2a, elongation initiationfactor 2a; MLN, mesenteric lymph nodes.
RESEARCH | REPORTon M
arch 4, 2020
http://science.sciencemag.org/
Dow
nloaded from
the diffused distribution of lysozyme and restoredsecretory autophagy of lysozyme in Paneth cellswithout affecting lysozyme transcript levels (Fig.4, F to H, and fig. S12). These results argue thatPaneth cell secretory autophagy requires activa-tion of the DC-ILC3 circuit, whichmay provide acell-extrinsic licensing signal that allows secretoryautophagy to be rapidly activated upon detectionof Paneth cell–intrinsic ER stress.Our results illuminate how the intestine pre-
serves antimicrobial function in the face of apathogenic bacterial infection (fig. S13) and sug-gest how simultaneous disruption of both ERstress and autophagy pathways leads to severeinflammation in mice (17). Our findings also pro-vide potential clues about how inflammation canarise in human inflammatory bowel disease.Genes that govern both the ER stress responseand autophagy are frequently mutated in peoplewith Crohn’s disease (2, 4). Because disruption ofeither pathway precludes secretory autophagy,our results suggest how infection of intestinal
epithelial cells could trigger chronic inflamma-tion in people with these genetic abnormalities.
REFERENCES AND NOTES
1. D. Ramanan, K. Cadwell, Cell Host Microbe 19, 434–441(2016).
2. K. Cadwell et al., Nature 456, 259–263 (2008).3. K. Cadwell et al., Cell 141, 1135–1145 (2010).4. A. Kaser et al., Cell 134, 743–756 (2008).5. J. Celli, R. M. Tsolis, Nat. Rev. Microbiol. 13, 71–82 (2015).6. A. M. Keestra-Gounder et al., Nature 532, 394–397
(2016).7. J. L. Benjamin, R. Sumpter Jr., B. Levine, L. V. Hooper, Cell
Host Microbe 13, 723–734 (2013).8. N. Mizushima, T. Yoshimori, B. Levine, Cell 140, 313–326
(2010).9. M. Ponpuak et al., Curr. Opin. Cell Biol. 35, 106–116
(2015).10. N. Dupont et al., EMBO J. 30, 4701–4711 (2011).11. B. Khor, A. Gardet, R. J. Xavier, Nature 474, 307–317
(2011).12. K. G. Lassen et al., Proc. Natl. Acad. Sci. U.S.A. 111, 7741–7746
(2014).13. P. Escoll, S. Mondino, M. Rolando, C. Buchrieser, Nat. Rev.
Microbiol. 14, 5–19 (2016).14. A. M. Preston, E. Gurisik, C. Bartley, D. R. Laybutt, T. J. Biden,
Diabetologia 52, 2369–2373 (2009).
15. X. Z. Wang et al., Mol. Cell. Biol. 16, 4273–4280 (1996).16. C. M. Oslowski, F. Urano, Methods Enzymol. 490, 71–92
(2011).17. T. E. Adolph et al., Nature 503, 272–276 (2013).18. M. Boyce et al., Science 307, 935–939 (2005).19. C. A. Thaiss, N. Zmora, M. Levy, E. Elinav, Nature 535, 65–74
(2016).
ACKNOWLEDGMENTS
We thank B. Levine for discussions and C. L. Behrendt-Boyd forassistance with mouse experiments. This work was supported bythe NIH (grant DK070855 to L.V.H.; grants AI118807 and AI128151to S.E.W.), the Burroughs Wellcome Foundation (Investigators inthe Pathogenesis of Infectious Diseases Award to L.V.H.), theWelch Foundation (grant I-1874 to L.V.H.; grant I-1858 to S.E.W.),and the Howard Hughes Medical Institute (L.V.H.). S.B. wassupported by a Gruss-Lipper postdoctoral fellowship.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/357/6355/1047/suppl/DC1Materials and MethodsFigs. S1 to S14References (20–27)
25 November 2016; resubmitted 12 April 2017Accepted 14 July 2017Published online 27 July 201710.1126/science.aal4677
Bel et al., Science 357, 1047–1052 (2017) 8 September 2017 5 of 5
WT MyD88-/- MyD88ΔIEC MyD88ΔDC
LC3
LYZ
Mer
ge
+S. TmM
yD88
ΔIEC
MyD
88ΔDCWT
MyD
88-/-
-1.0
-0.5
0.0
0.5
1.0
********
+S. Tm
Col
ocal
izat
ion
of L
YZ
and
LC3
(Pea
rson
’s R
val
ue)
Rag1-/- Rorc-/-
****
-1.0
-0.5
0.0
0.5
1.0
+S. Tm
Col
ocal
izat
ion
of L
YZ
and
LC3
(Pea
rson
’s R
val
ue)
Rag1-/- Rorc-/-
LC3
LYZ
Mer
ge
+S. Tm
**** ****
****
0
50
100
% C
rypt
s
MyD
88ΔIE
C
MyD
88ΔDCWT
MyD
88-/-
+S. Tm
Normal LYZ
Diffuse LYZ
LC3
LYZ
Mer
ge
WT MyD88-/-
MyD88-/-
+IL-22
+S. Tm
-1.0
-0.5
0.0
0.5
1.0
**** ****
Col
ocal
izat
ion
of L
YZ
and
LC3
(Pea
rson
’s R
val
ue)
WT MyD88-/- MyD88-/-
+IL-22
+S. Tm
0
50
100* *
% C
rypt
s
WT MyD88-/- MyD88-/-
+IL-22
+S. Tm
Normal LYZ
Diffuse LYZ
**** ****
Fig. 4. A DC-ILC3 circuit controls secretory autophagy in Panethcells. (A) Immunofluorescence of LC3 and lysozyme in intestinal crypts ofS. Typhimurium–infected mice. (B) Quantification of LC3 and lysozymecolocalization in (A). Each data point represents one lysozyme granule.(C) Quantification of intestinal crypts displaying a diffuse lysozyme signal.P values in black are relative to the wild-type group; the P value in red isrelative to MyD88−/− and Myd88DDC mice. (D) Immunofluorescence of LC3and lysozyme in small intestinal crypts of S. Typhimurium–infected mice.
(E) Quantification of LC3 and lysozyme colocalization in (D). Each datapoint represents one lysozyme granule. (F) Immunofluorescence of LC3 andlysozyme in intestinal crypts of S. Typhimurium–infected mice. (G) Quanti-fication of LC3 and lysozyme colocalization in (F). Each data pointrepresents one lysozyme granule. (H) Quantification of small intestinalcrypts displaying a diffuse lysozyme signal. Error bars represent SEM. *P <0.05; ****P < 0.0001; Student’s t test (E); one-way ANOVA [(B) and (G)];two-way ANOVA [(C) and (H)]. Scale bars [(A), (D), and (F)], 5 mm.
RESEARCH | REPORTon M
arch 4, 2020
http://science.sciencemag.org/
Dow
nloaded from

intestinePaneth cells secrete lysozyme via secretory autophagy during bacterial infection of the
and Lora V. HooperShai Bel, Mihir Pendse, Yuhao Wang, Yun Li, Kelly A. Ruhn, Brian Hassell, Tess Leal, Sebastian E. Winter, Ramnik J. Xavier
originally published online July 27, 2017DOI: 10.1126/science.aal4677 (6355), 1047-1052.357Science
, this issue p. 1047; see also p. 976Scienceto gut pathogens co-opts autophagy in intestinal immune defense.endoplasmic reticulum stress and required signals from type 3 innate lymphoid cells. Thus, the innate immune responsedelivery to the gut lumen, which protects against further bacterial invasion. Secretory autophagy was triggered by alternative autophagy-based secretion pathway (see the Perspective by Kaser and Blumberg). This ensures lysozymeepithelial cells sense an invading bacterial pathogen, they ''reroute'' the antimicrobial protein lysozyme through an
show that when intestinalet al.necessary for the delivery of antimicrobial proteins that kill pathogenic bacteria. Bel Intestinal pathogens can invade host cells and disrupt critical cellular functions, including secretion. Secretion is
Foiling bad bugs' sneaky tricks
ARTICLE TOOLS http://science.sciencemag.org/content/357/6355/1047
MATERIALSSUPPLEMENTARY http://science.sciencemag.org/content/suppl/2017/07/26/science.aal4677.DC1
CONTENTRELATED
http://stm.sciencemag.org/content/scitransmed/4/158/158ra144.fullhttp://stm.sciencemag.org/content/scitransmed/6/233/233ra53.fullhttp://stm.sciencemag.org/content/scitransmed/7/300/300ra128.fullhttp://stm.sciencemag.org/content/scitransmed/9/376/eaaf9655.fullhttp://science.sciencemag.org/content/sci/357/6355/976.full
REFERENCES
http://science.sciencemag.org/content/357/6355/1047#BIBLThis article cites 27 articles, 7 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.ScienceScience, 1200 New York Avenue NW, Washington, DC 20005. The title (print ISSN 0036-8075; online ISSN 1095-9203) is published by the American Association for the Advancement ofScience
Science. No claim to original U.S. Government WorksCopyright © 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of
on March 4, 2020
http://science.sciencem
ag.org/D
ownloaded from







![Paneth Colloquium 2008 () 1€¦ · Paneth Colloquium 2008 () 1 [PC2008 #001] Glass spherules related to the El’gygytgyn impact crater (Siberia) Adolph, L.*, Deutsch, A](https://img.pdfslide.us/doc/110x75/5f06df747e708231d41a28bc/paneth-colloquium-2008-1-paneth-colloquium-2008-1-pc2008-001-glass-spherules.jpg)





















