Embed Size (px)
Citation preview

JOURNAL OF MOLECULAR RECOGNITION, VOL. 9,595-606 (1996)
Intermolecular Chiral Recognition Probed by Enantiodifferential Excited-state Quenching Kinetics Tonya Gowl Stockman,* Cynthia A. Klevickis,? C. M. Grisham and F. S. Richardson* Department of Chemistry, University of Virginia, Charlottesville, VA 22901, USA
Time-resolved chiroptical luminescence (TR-CL) measurements are used to study the kinetics of chirality- dependemt excited-state quenching processes in aqueous solution. Experiments are carried out on samples that contain a racemic mixture of chiral luminophore molecules (L.) in solution with a small, optically active concentration of chiral quencher molecules (Q). The luminophores are excited with a pulse of unpolarized light to create an initially racemic excited-state population of AL* and AL* enantiomers, and TR-CL measurements are then used to monitor the differential decay kinetics of the AL* and AL* subpopulations. Observed differences between the AL* and AL* decay kinetics reflect differential rate processes and efficiencies for a * - Q versus AL*-Q quenching actions, and they are diagnostic of chiral discriminatory interactions between the luminophore and quencher molecules. In this study, the luminophores are either Eu(dpa):- or Tb(dpa):- coordination complexes (where dpa denotes a dipicolinate dianion ligand), and the quenchers are diastereomeric structures of Cr(H,O),(ATP), Rh(H,O),(ATP) and Rh(H,O),(ATP) (where ATP = adenosine triphosphate). The Ln(dpa):- (Ln=Eu3+ or Tb3+) complexes have three-bladed propeller-like structures of 4 symmetry, and in aqueous solution they exist as a racemic mixture of left-handed (A) and right-handed (A) con6gurational isomers (enantiomers). The results show that the chiral quencher molecules can distinguish between the A and A enantiomeric structures of the luminophores in their excited-state quenching actions. The degree and sense of enantiomeric preference in these quenching actions are governed by the electronic and stereochemical properties of the quencher molecules. Twenty-one Merent luminophore-quencher systems are examined in this study, and they exhibit interestingly diverse enantiodifferential quenching kinetics. The results reflect the extraordinary sensitivity of chiral recognition and discrimination procesws to relatively small, and somethimes subtle, changes in molecular electronic and stereochemical structure.
Keywords: chiral recognition; luminescence; excited-state quenching; enantioselective interactions
Introduction Chirality-dependent intermolecular recognition and dis- crimination phenomena are of widespread interest and profound importance in chemistry and biology. These phenomena are manifested in a wide variety of chemical processes that occur in nature, and they are exploited extensively in laboratory syntheses and separations of complex chemical systems. The electronic and stereochem- ical determinants of intermolecular chiral recognition and discrimination in chemical processes have been the subject of extensive experimental and theoretical studies, but their characterization in terms of detailed structural and mecha- nistic considerations has been elusive. This is due, in large part, to the difficulties involved in identifying and sepa- rately evaluating contributions made by the various kinds of structural chirality (electronic and stereochemical) typically found in chiral moleclar systems. For some systems, intermolecular chiral discrimination processes can be rationalized, at a phenomenological level, in terms of steric/
* Authors to whom correspondence should be addressed. t Present address: College of Integrated Science and Technology, James Madison University, Hankonburg, VA 22807, USA.
electrostatic ‘lock-and-key’ models in which the discriminatory parameters reflect relative molecular shapes, sizes and static charge distributions. These models have been used with some success in cases where the interacting ‘lock‘ and ‘key’ molecules have highly differentiated stereochemical structures, but they are much less useful for treating systems in which the interacting molecules have similar shapes, sizes and stereochemical details. Fur- thermore, they do not address contributions to chiral discrimination from dynamic electronic interactions of the kinds found, for example, in intermolecular electronic energy-transfer processes and in stereoselective electron- transfer reactions. Electronic chirality in dynamically interacting (or reacting) systems is notoriously difficult to model, and it is also elusive to direct measurement and analysis.
Recently, the authors have developed a technique for studying chiral discrimination in electronic energy-transfer processes between dissymmetric molecules in solution (Richardson et al., 1991; Richardson and Metcalf, 1994). This technique involves the measurement of enantiodiffer- ential excited-state quenching kinetics in systems that contain a racemic mixture of chiral luminophore molecules (L) (the energy donors) in solution with a small concentra-
CCC 0952-3499/96/060595-12 0 1996 by John Wiley & Sons, Ltd. Received 27 May 1995
Accepted (revised) 4 November 1995

596 T. GOWL STOCKMAN ETAL
tion of either fully or partially resolved chiral quencher molecules (Q) (the energy acceptors). The luminophores are excited with a pulse of unpolarized light to produce a racemic excited-state population (denoted here by rac-L*), and time-resolved chiroptical luminescence (TR-CL) spec- troscopy is then used to measure differences between the decay rates of the two enantiomers within this population (denoted here by AL* and AL*). Observed differences between the AL* and AL* decay rates reflect differential rate processes and efficiencies for AL* - Q versus AL* - Q quenching actions, and they are diagnostic of chiral discriminatory interactions between the L and Q molecules. In all of the systems studied so far, including those examined in the present study, quenching occurs via short- range electronic energy transfer processes in transient L-Q encounter complexes, and the bimolecular quenching rates are significantly slower than those predicted for diffusion- limited rate processes (Glover-Fischer et al., 1995; Metcalf et aZ., 1993; Rexwinkel et al., 1993). The enantiodifferential quenching kinetics observed for these systems will, in general, reflect a combination of (a) chiral recognition/ discrimination in the formation and dissociation of (AL* - Q) versus (AL* - Q) encounter complexes, (b) chirality-dependent differential geometries and dynamics in (AL* - Q) versus (AL* - Q) diastereomeric structures, and (c) chiral discriminations in the electronic interactions between the resonant donor (L*)-acceptor (Q) state mani- folds involved in L*-to-Q energy transfer.
In the present paper, the authors report enantiodifferential excited-state quenching studies on systems in which the chiral L are tris(dipico1inate) complexes of either Eu3+ or Tb3+, and the Q are diastereomeric structures of either M(H20),( ATP) or M(H20)4( ATP) complexes, where M=Rh(III) or Cr(III), and ATP denotes an adenosine triphosphate ligand with either tridentate or bidentate chelation to the metal ion in the respective complexes. Four different structural isomers of Rh(H,O),(ATP), four struc- tural isomers of Cr(H,),(ATP) and two structural isomers of Rh(H,O),(ATP) are examined as Q. Each of these Q complexes contains several kinds (or elements) of structural chirality, and each exhibits distinctive chiroptical properties that are diagnostic of its overall stereochemical structure. Under the conditions of these experiments, the Ln(dpa);- L complexes (where Ln = Eu3+ or Tb3+, and dpa= dipicoli- nate dianion ligand) are present in solution as a racemic mixture of two enantiomeric structures (i.e. optical antipo- des) designated here as A-Ln(dpa):- and A-Ln(dpa):-. These structural enantiomers have trigonal-dihedral (D3) symmetry, and their absolute configuration labels reflect either left-handed (A) or right-handed (A) structural chirality about their trigonal (C,) symmetry axis.
Theory
Enantioditrerential quenching kinetics
In the authors' experiments, the sample contains a racemic mixture of chiral L (AL and AL) in solution with a small concentration of chiral Q. The L are excited with a pulse of
unpolarized (racemic) light to produce an initially racemic excited-state population of AL* and AL* enantiometers. The decay kinetics of the AL* and AL* subpopulations of the excited state are then measured and compared, and any differences observed between AL* and AL* decay kinetics may be attributed to differential AL* - Q versus AL* - Q interactions. The rate processes relevant to a purely phenomenological analysis of the AL* and AL* decay kinetics are: (a) unimolecular racemization (enantiomer interconver- sion);
AL* C AL*, AL* C AL*
c = C E k , (b) radiative and non-radiative decay in the absence of Q actions
AL* AL+ E,+ E,,, AL* AL + E,+ E,,,
&y=gfb (where E, and En, denote radiative and non-radiative energies, respectively), and (c) De-excitation via bimolecular quenching events.
1 fast 1 fast Q Q
On the basis of the rate processes shown above in (a), (b) and (c). the differential rate equations for [AL*] and [AL*] decay may be written as:
--- d[AL*l'-(b+k,+e[Q])[AL*]' - k,[AL*]' (1) dt
--- d[AL* " - (b + k, + T[Q])[AL*]' - k,[AL*]' (2) dr
where [QJ denotes the ground-state Q concentration, and [AL*]' and [AL*]' denote the time-dependent concentrations of the AL* and AL* L enantiomers (after excitation at t=O) . Under the excitation conditions employed in the present study, the AL* and AL* enantiomers are prepared in initially equal concentrations, in which case [AL*Io= [AL*]O and Eqs (1) and (2) may be integrated to the following forms:
[AL*]'=- rAL*30 [(Z+ k@[Q] - 2k,)e'/'l 22

INTERMOLECULAR CHIRAL RECOGNITION 591
k q = ( q + e ) / 2 . These equations may be combined to obtain an expression for the time evolution of enantiomeric excess (q) in the L excited-state population:
[AL*]‘- [AL*]‘ - -k&[QI w(Zt12) ‘%*(‘)= [AL*]I+ [AL*]‘ - Z+2kc tanh(Zt/2)
where tanh denotes a hyperbolic tangent function. Among the rate parameters that appear in the equations
given above, ko and k, reflect properties of the L and are entirely independent of any Q properties (or quenching actions). All the information about quenching kinetics is contained in the enantiospec8c kb\* and e rate parameters, which in Eqs (3) and (4) have been combined to define a mean quenching constant, k q = ( q + e ) / 2 , and an enantio- diflerential quenching parameter, k&=e-kf. It is of further value to introduce yet another parameter that provides a simple and direct measure of both the degree and sense of enantioselectivity in the quenching processes. This enantioselectivity parameter is defined by
and it can have values between +1 (for purely AL* quenching) and - 1 (for purely AL* quenching).
Finally, the authors note that under conditions in which excited-state enantiomer interconversion rates are much slower than excited-to-ground-state decay rates, Eqs (3), (4) and ( 5 ) may be reduced to the following forms:
[AL*]‘= [&*lo e-‘IT1 (7) [AL*]’= [AL*]O e-‘Id (8)
%.(t) = - tanh(ka[Qlt/2) = tanh($ kq[Q]t) (9) with (l/rl)=ko+kb\‘[Q] and (1/r2)=$+e[Q].
TR-CL measurements
In TR-CL spectroscopy one measures sums, differences, and ratios of left (L)- and right (R)-circularly polarized emission intensities as a function of time after sample excitation. The quantities ,obtained from these measure- ments are generally expressed as follows:
where A’ denotes emission wavelength, t denotes time after excitation, and ZL and ZR denote L- and R-circularly polarized emission intensities. Following standard practice the authors refer to I as a total luminescence (TL) intensity, to AZ as a circularly polarized luminescence (CPL) intensity and to gem as an emission dissymmetry factor.
For a sample that contains a mixture of L enantiomers, AL* and AL*, the time dependence of I(A’, t ) reflects the time dependence of [AL*]‘+[AL*]‘, and the time depend- ence of AZ(A’, t) reflects the time dependence of
[AL*]I - [AL*]‘. It follows, therefore, that the time depend- ence of g,,(A’, t) will reflect the time evolution of enantiomeric excess ( qL,) in the L excited-state population. Within the context of the present study, the relevant expressions for [AL*]‘, [AL*]‘, and qL.(t) are given by Eqs (3), (4) and (5). respectively. Using these expressions and previously derived relationships between [AL*]‘* [AL*]‘, q,.(t), and the TR-CL observables, it may be written:
I(A’, f)=w [(Z- 2kc) e-‘IT1+(Z+2kc) e-‘”2] (13) 22
(15) where gti(A’) denotes the emission dissymmetry factor for pure AL* enantiomers at the emission wavelength A‘, and the remaining notation corresponds to that used in Eqs
All of the TR-CL measurements reported in this study were carried out under sample conditions in which L enantiomer interconversion processes (AL* ++ AL*) are at least two orders of magnitude slower than excited-to- ground-state decay rates. Under these conditions, the expressions for Z(A’, t ) , AZ(A’, t). and gem(A’, t) may be approximated by:
(3)-(5) and (10)-(12)
gem@‘, t)= - gti(A’) tanh r“:“’tl - (18) L -I
where ( 1 / rl) =$+ e [QI, (1 / r2) = ko+ e [ Q ] , and, therefore,
1 1 - + - = 2ko + 2kq[Q] 71 72
1 1 - - - = 2ka[Q] 71 72
Measurements performed on samples with [Q]=O yield AZ(A’, t ) = O and gem@’, t ) = O at all times, and they yield Z(A’ , t ) decay curves with a time dependence given by Z(A‘, t)=I(A’, 0) exp( - kot). These measurements permit direct evaluation of &, which may then be treated as a ‘known’ quantity in Eq. (19). Finally, the authors note that the quenching enantioselectivity parameter, Eq, may be evaluated from measured values of r,, r2, and k,, according
T. GOWL STOCKMAN ET AL.
22000 - 21000 -
2oooo-
19000 -
18000 - h d I 17000-
0 - 6000- h
5000 -
m-
3000-
2000-
1000 - 0-
E
t? 8 w
598
to:
c,
71 - 72 Eq = 71 + 72 - 2&7, 72
Under the sample conditions employed in the present study, $=610 s-' for the Eu(dpa);- L and k,,=478 s-' for the Tb(dpa):-L. These values of k,, correspond to 'unquen- ched' luminsescence lifetimes (ro=llk,,) of 1.64 ms for Eu(dpa):- and 2.09 ms for Tb(dpa):-.
Spectroscopic and Structural Properties of the Luminophores and Quenchers
Eu(dpa):- and Tb(dpa):- luminophores
The equilibrium structures of Eu(dpa):- and Tb(dpa):- in aqueous solution have trigonal dihedral (D,) symmetry, and the three bicyclic chelate rings in these structures form either a left-handed (A) or a right-handed (A) three-bladed propeller arrangement about the threefold axis. The individ- ual ligands and their chelate rings have achirul structures, so structural chirality in Eu(dpa):- and Tb(dpa):- com- plexes is determined entirely by the helical (propeller like) distribution of chelate rings about the metal atom. Follow- ing standard practice, the authors label the structural enantiomers of these complexes as either A or A, which refers to the helicity of the three bicyclic chelate rings about the trigonal axis. It is important to note, however that although the A enantiomer is defined to have left-handed chirality about its trigonal (C,) symmetry axis, it has right- handed chirality about each of its three digonal (C,) symmetry axes. Analogously, the A enantiomer, defined to have right-handed chirality about its trigonal axis, has kf- handed chirality about each of its digonal axes. The absolute configuration of a given enantiomer is uniquely specified by the label A or A, but to specify the 'handedness' of its structure one must also choose a particular reference axis. The chiroptical properties of this enantiomer are determined by its absolute configuration, irrespective of how one chooses to define its structural handedness. However, when considering interactions between this enantiomer and other chiral molecules, it is often useful (and sometimes essential) to specify the handedness of the enantiomer with respect to axes that are coincident with likely intermolecular interaction axes. Structural representations of A-Ln(dpa):- and A-Ln(dpa):- enantiomers are shown in Plate 1 as viewed along the trigonal (C,) axis.
The optical excitation and emission properties of Eu(dpa):- and Tb(dpa):- complexes have been studied extensively in both solid-state and solution-phase samples. Analyses of these properties have produced reasonably good characterizations of the crystal-field energy-level structures within the 7F, (J=O-6) multiplet manifolds of
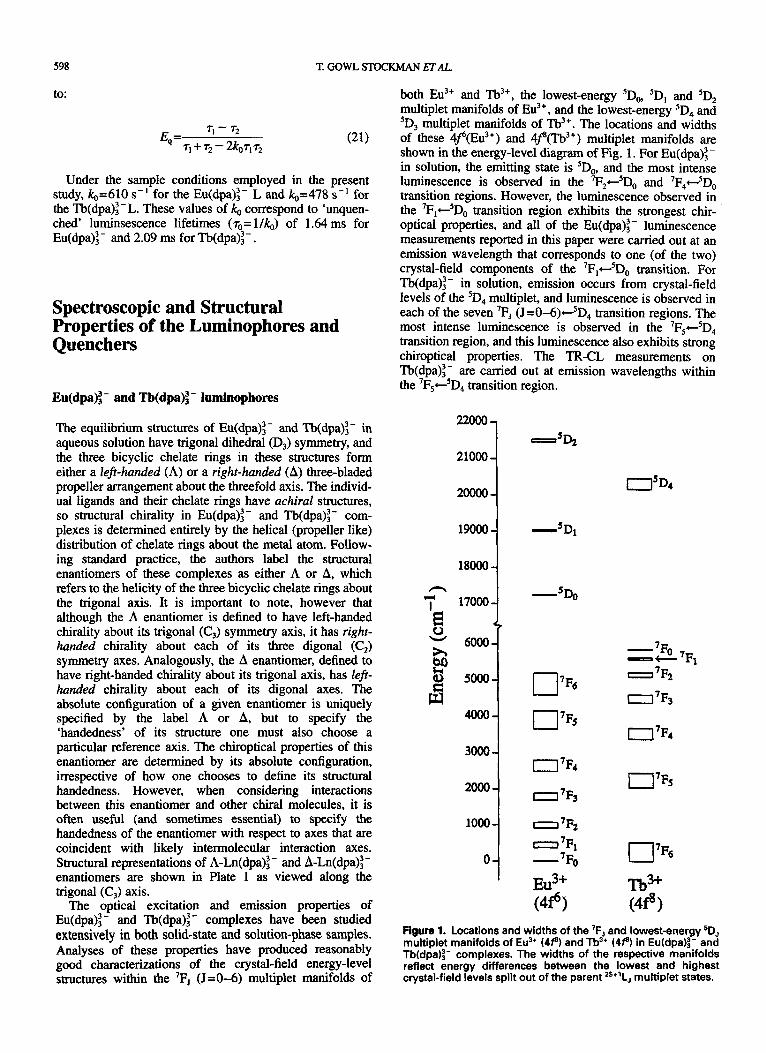
both Eu3+ and Tb3+, the lowest-energy 'Do, 'D, and 5D, multiplet manifolds of ELI,+, and the lowest-energy 'D, and 'D, multiplet manifolds of Tb3+. The locations and widths of these 4J6(Eu3+) and 4$(Tb3+) multiplet manifolds are shown in the energy-level diagram of Fig. 1. For Eu(dpa):- in solution, the emitting state is 'DO, and the most intense luminescence is observed in the 7F24-5D0 and 7F4+-5D0 transition regions. However, the luminescence observed in the 7F,-5Do transition region exhibits the strongest chir- optical properties, and all of the Eu(dpa):- luminescence measurements reported in this paper were carried out at an emission wavelength that corresponds to one (of the two) crystal-field components of the 7F,+5Do transition. For Tb(dpa):- in solution, emission occurs from crystal-field levels of the 'D, multiplet, and luminescence is observed in each of the seven 'F, (J =0-6)+'D4 transition regions. The most intense luminescence is observed in the 7Fs+SD, transition region, and this luminescence also exhibits strong chiroptical properties. The TR-CL measurements on Tb(dpa):- are carried out at emission wavelengths within the 7Fs+5D4 transition region.
Figure 1. Locations and widths of the 7F, and lowest-energy 5D, multiplet manifolds of Eu3+ (4p) and Tb3* (4 f9 in Eu(dpa):- and Tb(dpa):- complexes. The widths of the respective manifolds reflect energy differences between the lowest and highest crystal-field levels split out of the parent =+'LJ multiplet states.

INTERMOLECULAR CHIRAL RECOGNITION 599
Quenchers
The M(H,O),(ATP) and M(H,O),(ATP) transition-metal complexes employed as chiral quenchers in the present study are members of a larger class of M(H,O),(nucleotide), M(H,O),(nucleotide), and M(NH,),(nucleotide) complexes that have been used extensively as probes of the structural and biochemical properties of naturally occuring Mg(I1)- polyphosphate complexes (Cleland and Mildvan, 1979; Dunaway-Mariano and Cleland, 1980). Whereas the latter complexes are kinetically labile and have fluxional ster- eochemical structures in aqueous solution, the former are exchange inert and have reasonably well-defined ster- eochemical structures under similar solution conditions.
Four structural isomers (diastereomers) of Rh(H,O),(ATP), four of Cr(H,O),(ATP) and two of Rh(H,O),(ATP) were prepared and used as quenchers in the present study. In each case, the metal ion is in a M(II1) oxidation state and is coordinated to six oxygen atoms to form a nearly octahedral (0,) M(III)06 coordination core. In the M(H,O),(ATP) complexes, the ATP ligand is bound to the metal ion via tridentate chelation through three phosphate oxygen atoms (one each from the a, p and y phosphate groups of ATP). In the M(H,O),(ATP) com- plexes, the ATP ligand is bound to the metal ion via bidentate chelation through two phosphate oxygen atoms (one each from the p and y phosphate groups). General representations of the coordination modalities in the M(H,O),(ATP) and M(H,O),(ATP) complexes are shown in Fig. 2.
The authors' four structural isomers of Cr(H,O),(ATP) correspond exactly to those prepared, separated, charac- terized and described in previous work reported by
p,y-bidentate Metal-Adenosinetriphtmphate
-O--Pro--Pa-Adenosine "-'iT I I It 0 0 0
M3+ . *
.* I -*. : I '. -0'' 0- 0' I I I
II I I It -o--P~-o--Pp -0-Pa-Adenosine
0 0 0
Figure 2. Schematic representations of the M(H,O),(ATP) com- plexes used in this study.
Dunaway-Mariano and Cleland (1980). These isomers are distinguished entirely by structural features associated with the six-membered chelate ring formed by p, y-bidentate ATP coordination to Cr(1II). More specifically, they are distinguished by configurational chirality about the asym- metric P P atom of the chelate ring, and by the conformational state of the chelate ring. In two of the isomers, the absolute configuration at P P is R, and in the other two isomers the absolute configuration at P P is S (where the R and S labels for absolute configuration conform to the Cahn-Ingold-Prelog conventions) (Cahn et al., 1966). The two isomers within each of these R and S pairs are distinguished by twist-boat chelate ring conforma- tions that place the AMP substitutent on P P in either a pseudo-axial (a) or pseudo-equatorial (e) orientation rela- tive to the chelate ring. Cleland and co-workers have labeled the PP(R) and PP(S) isomers as A and A, respectively. Adopting this labeling scheme, the authors label their four Cr(H,O),(ATP) isomers as A@), A(a), A(e), and A(e), which correspond respectively to the isomers numbered as 1, 2, 3 and 4 by Dunaway-Mariano and Cleland (1980). The Cr(1II) d-d circular dichroism (CD) spectra of the A(a) and A(e) isomers exhibit signs opposite those observed in the CD spectra of the A(a) and A(e) isomers. Differences between the CD spectra of A(a) and A@), and between the CD spectra of A(a) and A@), are relatively small.
The structural isomers of Rh(H,O),(ATP) correspond to those described above for Cr(H,O),(ATP), but conforma- tional lability in the chelate ring of Rh(H,O),(ATP) precludes separation of the A(a) from A(e) and the A(a) from A(e) isomers. Therefore, the authors' Rh(H,O),(ATP) Qs consist of equilibirium mixtures of either A(a) + A(e) isomers or A(a)+A(e) isomers, which they label as A(ale) and A(ale), respectively.
The four structural isomers of Rh(H,O),(ATP) also correspond to previously published complexes (Lu ef al., 1988). These isomers possess the same asymmetric P P atom as the bidentate complexes described above, and as a result, the two structural isomers, A and A, are possible. For each of the two structural isomers arising from the asymmetric P P atom, there are two other structural possibilities created by the asymmetry of the a-P atom. The absolute configuration at the a-P atom can also be R or S using the Cahn-Ingold-Prelog convention. In order to remain consistent, however, the nomenclature established by Cleland which defines the absolute configuration of the a-P atom by describing the spatial relationship between the y P and the adenosine moiety is used. Cleland defines the structural isomer in which 'y-P and the adenosine are on the same face of the Rh-a, P chelate ring to be the endo isomer. When the y P and the adenosine are on opposite faces of the Rh-a, p chelate ring, the ex0 isomr is formed. Using Cahn- Ingold-Prelog nomenclature, the a-P(R) . configuration corresponds to the ex0 isomer and the a-P(S) configuration is the endo isomer. Combining the configurational possibil- ities of both chiral phosphorous atoms, four structural isomers arise and the authors label them as Aexo, Aexo, Aendo and Aendo. Similar to the bidentate complexes, the absolute configuration of the P P atom determines the sign of the CD spectra. Differences in the CD spectra between isomers with the same @P configuration and opposite a-P

600 T. GOWL STOCKMAN ETAL
configuration (Aexo relatively small.
and Aendo or Aexo and hendo) are
Experimental
Preparation of Eu(dpa)i- and Tb(dpa):- complexes Stock solutions of Eu(dpa):- and Tb(dpa):- were prepared by reaction of the appropriate lanthanide carbonate, Eu,(CO,)~ or Tb,(CO,),, with six equivalents of dipicolinic acid in water. The resulting solutions were neutralized with NaOH and diluted with water to bring the final lanthanide concentration to 50 m.
Preparation and separation of Cr(H,O),(ATP) isomers p, y-Bidentate Cr(H,O),(ATP) was synthesized according to the room-temperature procedure of Dunaway-Mariano and Cleland (1980). The four bidentate isomers were separated using a combination of known literature chromatographies. Unresolved Cr(H,O),(ATP) was concentrated by rotary evaporation and was loaded onto a cycloheptaamylose (CHpA) gel column equilibrated at 4°C in 1 O m Pipes buffer, pH 5.6. The Cr(H,O),(ATP) isomers eluted with buffer at a gravity-driven flow rate of 5 mlih. The 1.8 ml fractions were analyzed by HPLC; aliquots of 10-25 p1 were place on a 19 mmx 150 mm C,,-reverse phase HPLC preparative column in 10 m pH 2.5 methane sulfonic acid (MSA) buffer and eluted at a flow rate of 4.0 ml/min. The isomers were identified according to the order of elution from the HPLC column: (a) A-pseudo-axial, (b)A-pseudo- axial, (c) A-pseudo-equatorial, and (d) A-pseudo-equatorial (Gruys and Schuster, 1982). Fractions enriched in the four bidentate isomers were concentrated by rotary evaporation and rechromatographed on a second CHpA column. To prevent racemization, approximately three drops of 100 m MSA, pH 2.5, were added to each test tube prior to fraction collection. Fractions were analyzed by HPLC as described above. Because isomers b and c are somewhat difficult to separate by HPLC, the fractions enriched in b or a, the first two isomers off of the CHpA column, were combined and concentrated by rotary evaporation. Likewise, the third and fourth isomers eluting form the CHpA column, d and c, respectively, were combined and concentrated. Each con- centrated sample was injected onto the HPLC and individual isomers were collected by monitoring peak absorbances at 280 nm. Purified isomers were desalted by concentrating and loading onto a small Sephadex G10 column equilibrated in water. Fractions of each isomer having a pH of 3.5-4 were combined and used in CPL experiments. CD spectra recorded on a Jasco J-720 spectropolarimeter confirmed the isomeric assignments (Dunaway-Mariano and Cleland, 1980).
Preparation and separation of Rh(H,O),(ATP) and ~UI(H~O)~(ATP) isomers p, yBidentate Rh(H,O),ATP and a, p, ytridentate Rh(H,O),ATP were prepared by heating a mixture of 20 m N a A ” and 20 m [FUI(H,O)~I(C~O,), for 25 min at 80°C
amd pH 3 following the procedure of Lu et al. (1988). The resultant mixture of bidentate and tridentate RhATP was separated by size-exclusion chromatography at 5°C on a 180 cmx 0.7 cm Sephadex G10 column employing 1,4-bis(2-hydroxyethyl)-piperazine (pH 4.5) as the eluent according to the method of Shorter et al. (1987). The column fractions were monitored by CIS-reverse phase HPLC using 100 m MSA, pH 2.2, at a flow rate of 0.5 ml/ min. Because the bidentate isomers co-elute with two of the tridentate isomers on the HPLC, the fractions from the G10 column containing bidentate isomers were pooled and concentrated separately from the tridentate fractions. Fur- ther purification of the two bidentate Rh(H,O),(ATP) diastereomers as well as the tridentate isomers was achieved with a Waters C,,-reverse phase 19mmxlSOmm pre- parative HPLC column and a mobile phase of 10 m~ MSA, pH 2.2, at a flow rate of 4 mllmin). In the Rh(H,O),(ATP) fractions, the A isomer elutes before the A isomer from the HPLC column, whereas the tridentate isomers elute in the order Aexo, Aexo, Aendo and Aendo. CD spectra confirmed the isomeric assignments (Lu et al., 1988). Each isomer was concentrated by rotary evaporation and desalted as described for the Cr(H,O),(ATP) isomers.
Samples for spectroscopic measurements
All experiments were performed on aqueous solution samples that contained 100 m acetic acid buffer (pH 4.5) and a 1 O m concentration of either Eu(dpa):- or Tb(dpa):- complexes. The concentration of each of the resolved Rh(H,O),(ATP) isomers and of most of the Rh(H,O),(ATP) isomers was 75 p ~ . The resolved endo Rh(H,O),(ATP) isomer/Eu(dpa):- experiments were the only exception; in this case a 100 p~ concentration of the Q was used. The concentration of the resolved Cr(H,O),(ATP) isomers was 500 FM when in solution with Tb(dpa):- and 155 FM when used to quench Eu(dpa):-.
Spectroscopic measurements
Optical absorption spectra of the M(H,O),(ATP) and M(H,O),(ATP) complexes were obtained using a Cary Model 5E UV/Vis/NIR absorption spectrophotometer. CD measurements on these complexes were performed using a Jasco Model 5-720 CD/absorption spectrophotometer.
All TR-CL measurements were carried out using instru- mentation and techniques developed in the authors’ laboratory at the University of Virginia. In the TR-CL experiments on Eu(dpa):- L, excitation was at 465.8nm (with an argon ion laser), and emission intensity and polarization properties were monitored at 594.8 nm. In the TR-CL experiments on Tb(dpa):- L, excitation was at 488.0 nm (with an argon ion laser), and emission intensity and polarization properties were monitored at 543.7 nm. The data collected in these experiments were used to determine the time-dependent behavior of the Z(A’,f), AI(A’, t) and g,(A’, r ) quantitites defined by Eqs (10)-(12).
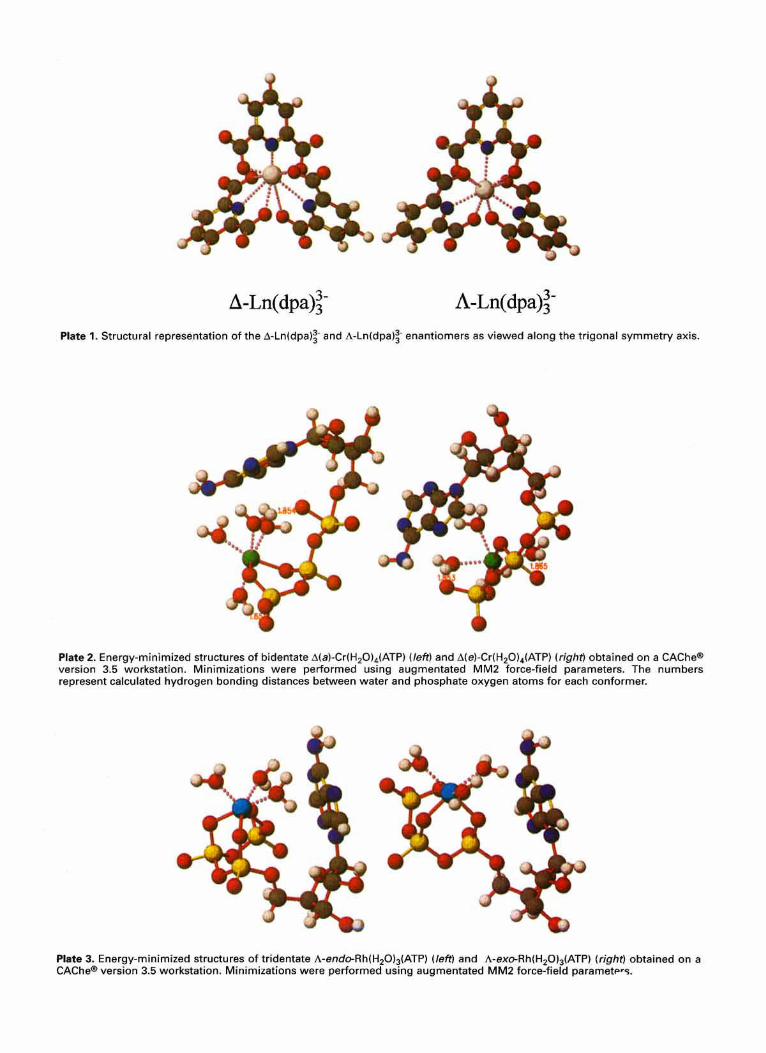

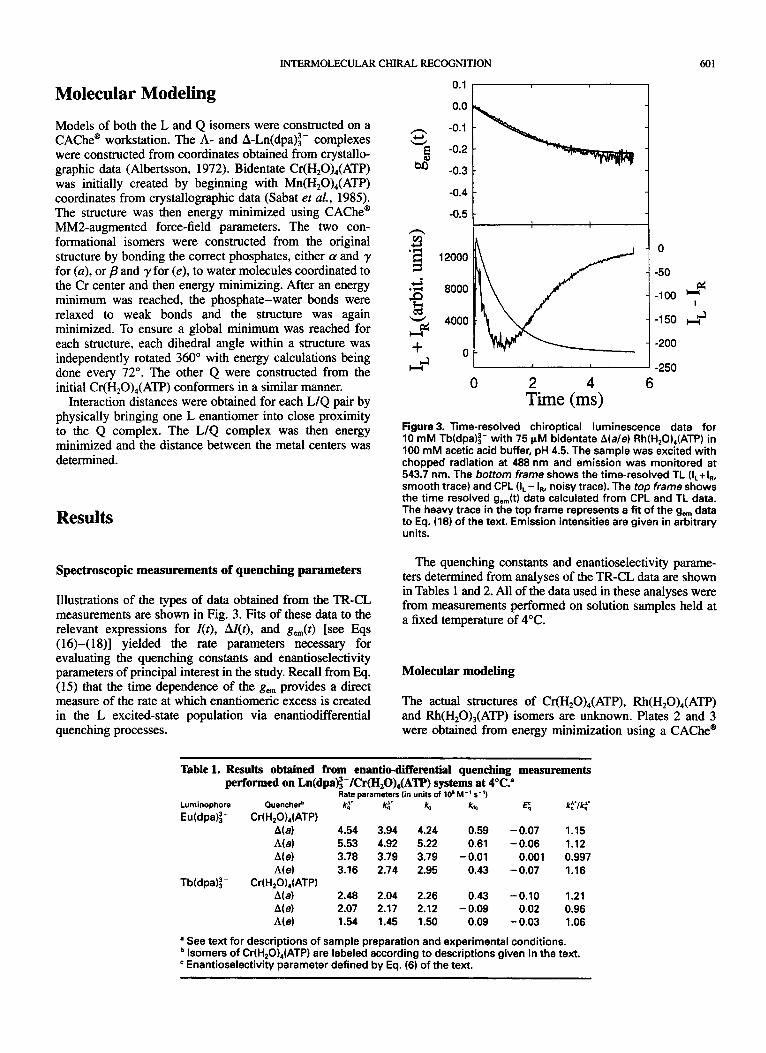
INTERMOLECULAR CHIRAL RECOGNITION 601
0.0 O.' 7 Molecular Modeling Models of both the L and Q isomers were constructed on a CAChe" workstation. The A- and A-Ln(dpa):- complexes were constructed from coordinates obtained from crystallo- graphic data (Albertsson, 1972). Bidentate Cr(H,O),(ATP) was initially created by beginning with Mn(H,O),(ATP) coordinates from crystallographic data (Sabat et al., 1985). The structure was then energy minimized using CAChe" MMZaugmented force-field parameters. The two con- formational isomers were constructed from the original structure by bonding the correct phosphates, either (Y and y for (a), or p and y for (e), to water molecules coordinated to the Cr center and then energy minimizing. After an energy minimum was reached, the phosphate-water bonds were relaxed to weak bonds and the structure was again minimized. To ensure a global minimum was reached for each structure, each dihedral angle within a structure was independently rotated 360" with energy calculations being done every 72". The other Q were constructed from the initial Cr(H,O),(ATP) conformers in a similar manner.
Interaction distances were obtained for each L/Q pair by physically bringing one L enantiomer into close proximity to the Q complex. The L/Q complex was then energy minimized and the distance between the metal centers was determined.
Results
Spectroscopic measurements of quenching parameters
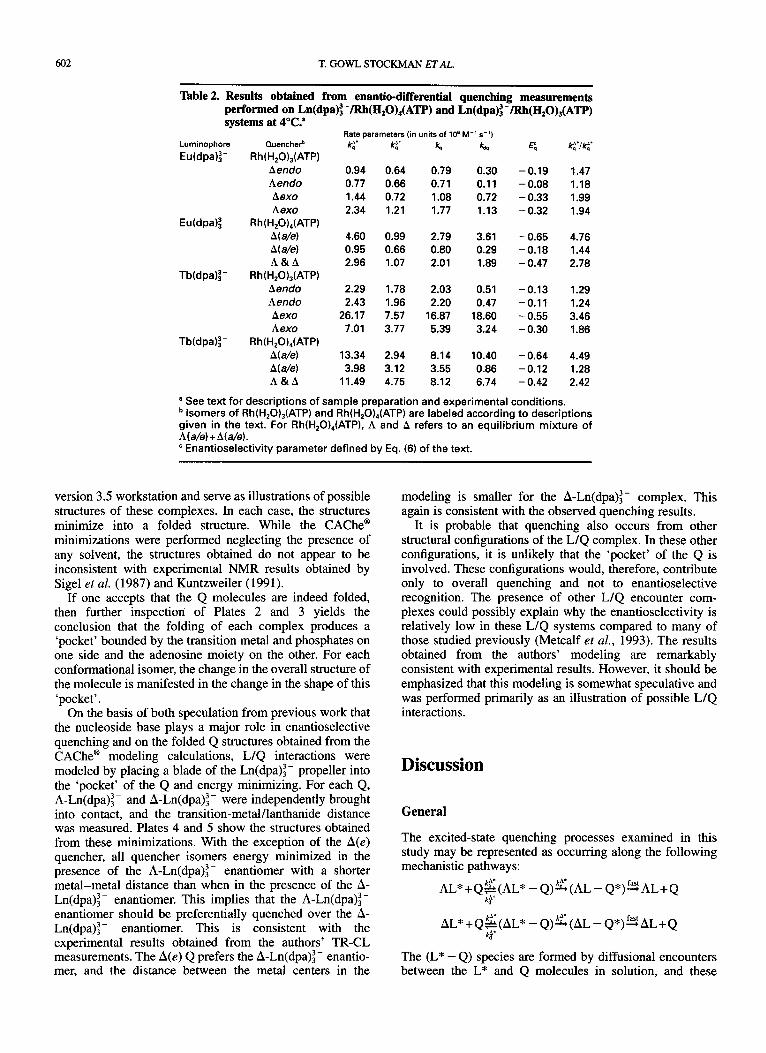
Illustrations of the types of data obtained from the TR-CL measurements are shown in Fig. 3. Fits of these data to the relevant expressions for Z(t), AZ(r), and g,,(t) [see Eqs (16)-(18)] yielded the rate parameters necessary for evaluating the quenching constants and enantioselectivity parameters of principal interest in the study. Recall from Eq. (15) that the time dependence of the g, provides a direct measure of the rate at which enantiomeric excess is created in the L excited-state population via enantiodifferential quenching processes.
'a'
3 4
-5 12000
8000 - 4000
3 2
+ 0
& U
J -50 /
' -250 4 CI
0 2 4 6 Time (ms)
Figure 3. Time-resolved chiroptical luminescence data for 10 mM Tb(dpa):- with 75 pM bidentate A(a/e) Rh(H,O),(ATP) in 100 mM acetic acid buffer, pH 4.5. The sample was excited with chopped radiation at 488 nm and emission was monitored at 543.7 nm. The bottom frame shows the time-resolved TL (lL+lR, smooth trace) and CPL (IL- IR, noisy trace). The fop frame shows the time resolved g,,(t) data calculated from CPL and TL data. The heavy trace in the top frame represents a fit of the gem data to Eq. (18) of the text. Emission intensities are given in arbitrary units.
The quenching constants and enantioselectivity parame- ters determined from analyses of the TR-CL data are shown in Tables 1 and 2. All of the data used in these analyses were from measurements performed on solution samples held at a fixed temperature of 4°C.
Molecular modeling
The actual structures of Cr(H,O),(ATP), Rh(H,O),(ATP) and Rh(H,O),(ATP) isomers are unknown. Plates 2 and 3 were obtained from energy minimization using a CAChe"
Table 1. Results obtained from enantio-differential quenching measurements performed on Ln(dpa):-/Cr(H,O),(ATP) systems at 4°C.'
Rate parameters (in units of 1V W ' s - ' ) Lurninophore QuencheP kg ' kf 4 t E: k r / q Eu(dpa):- Cr(H,O),(ATP)
A(a) 4.54 3.94 4.24 0.59 -0.07 1.15 A(a) 5.53 4.92 5.22 0.61 -0.06 1.12 A( e) 3.78 3.79 3.79 -0.01 0.001 0.997 Ate) 3.16 2.74 2.95 0.43 -0.07 1.16
A(a) 2.48 2.04 2.26 0.43 -0.10 1.21 A(e) 2.07 2.17 2.12 -0.09 0.02 0.96 M e ) 1.54 1.45 1.50 0.09 -0.03 1.06
Tb(dpa)f Cr(H,O),(ATP)
a See text for descriptions of sample preparation and experimental conditions. Isomers of Cr(H,O),(ATP) are labeled according to descriptions given in the text. Enantioselectivity parameter defined by Eq. (6) of the text.
602 T. GOWL STOCKMAN ETAL.
Table 2. Results obtained from enantio-differential quenching measurements performed on L~(~~~):-/RII(H~O)~(AT€’) and Ln(dpa):-/Rh(H,O),(ATP) systems at 4°C.’
Rate parameters (in units of los M-’ s-’) Luminophore QuencheP ky k r k t E i kyikr
Aendo 0.94 0.64 0.79 0.30 -0.19 1.47 Aendo 0.77 0.66 0.71 0.11 -0.08 1.18 Aexo 1.44 0.72 1.08 0.72 -0.33 1.99 Aexo 2.34 1.21 1.77 1.13 -0.32 1.94
A(a/e) 4.60 0.99 2.79 3.61 -0.65 4.76 A(a/e) 0.95 0.66 0.80 0.29 -0.18 1.44 h & A 2.96 1.07 2.01 1.89 -0.47 2.78
Aendo 2.29 1.78 2.03 0.51 -0.13 1.29 Aendo 2.43 1.96 2.20 0.47 -0.11 1.24 Aexo 26.17 7.57 16.87 18.60 -0.55 3.46 Aexo 7.01 3.77 5.39 3.24 -0.30 1.86
A(a/e) 13.34 2.94 8.14 10.40 -0.64 4.49 A(a/e) 3.98 3.12 3.55 0.86 -0.12 1.28 A&A 11.49 4.75 8.12 6.74 -0.42 2.42
Eu(dpa):- Rh(H,O),(ATP)
Eu(dpa):- Rh(H,O),(ATP)
Tb(dpa):- Rh(H,O),(ATP)
Tb(dpa):- Rh(H,O),(ATP)
a See text for descriptions of sample preparation and experimental conditions. Isomers of Rh(H,O),(ATP) and Rh(H,O),(ATP) are labeled according to descriptions
given in the text. For Rh(H,O),(ATP), A and A refers to an equilibrium mixture of A(a/e) + A(a/e).
Enantioselectivity parameter defined by Eq. (6) of the text.
version 3.5 workstation and serve as illustrations of possible structures of these complexes. In each case, the structures minimize into a folded structure. While the CAChe@ minimizations were performed neglecting the presence of any solvent, the structures obtained do not appear to be inconsistent with experimental NMR results obtained by Sigel et al. (1987) and Kuntzweiler (1991).
If one accepts that the Q molecules are indeed folded, then further inspection of Plates 2 and 3 yields the conclusion that the folding of each complex produces a ‘pocket’ bounded by the transition metal and phosphates on one side and the adenosine moiety on the other. For each conformational isomer, the change in the overall structure of the molecule is manifested in the change in the shape of this ‘pocket’.
On the basis of both speculation from previous work that the nucleoside base plays a major role in enantioselective quenching and on the folded Q structures obtained from the CAChe@ modeling calculations, L/Q interactions were modeled by placing a blade of the Ln(dpa):- propeller into the ‘pocket’ of the Q and energy minimizing. For each Q, R-Ln(dpa):- and A-Ln(dpa):- were independently brought into contact, and the transition-metal/lanthanide distance was measured. Plates 4 and 5 show the structures obtained from these minimizations. With the exception of the A(e) quencher, all quencher isomers energy minimized in the presence of the h-Ln(dpa)z- enantiomer with a shorter metal-metal distance than when in the presence of the A- Ln(dpa): - enantiomer. This implies that the A-Ln(dpa): - enantiomer should be preferentially quenched over the A- Ln(dpa):- enantiomer. This is consistent with the experimental results obtained from the authors’ TR-CL measurements. The A(e) Q prefers the A-Ln(dpa):- enantio- mer, and the distance between the metal centers in the
modeling is smaller for the A-Ln(dpa):- complex. This again is consistent with the observed quenching results.
It is probable that quenching also occurs from other structural configurations of the L/Q complex. In these other configurations, it is unlikely that the ‘pocket’ of the Q is involved. These configurations would, therefore, contribute only to overall quenching and not to enantioselective recognition. The presence of other L/Q encounter com- plexes could possibly explain why the enantioselectivity is relatively low in these L/Q systems compared to many of those studied previously (Metcalf et al., 1993). The results obtained from the authors’ modeling are remarkably consistent with experimental results. However, it should be emphasized that this modeling is somewhat speculative and was performed primarily as an illustration of possible L/Q interactions.
Discussion
General
The excited-state quenching processes examined in this study may be represented as occurring along the following mechanistic pathways:
A L * + Q ~ ( A L * -Q$(AL-Q*)%L+Q
A L * + Q ~ ( A L * - Q$(AL-Q*)%AL+Q
k i
ky
The (L* - Q) species are formed by diffusional encounters between the L* and Q molecules in solution, and these

INTERMOLECULAR CHIRAL RECOGNITION 603
species may then either dissociate back to free L* and Q molecules or undergo a transformation to (L - Q*) species via L*-to-Q electronic energy transfer. On the basis of the mechanistic pathways shown here, and application of the steady-state approximation to the concentrations of (AL* - Q) and [AL* - Q) species, the quenching rates for AL* and AL* L may be expressed as:
where and kf correspond to the enantiospecgc quenching constants determined from the authors' TR-CL measurements. From these expresssions the following relationships between the various rate constants can be obtained:
where u= A* or A*, and K:=kf/kl. For all of the L/Q systems examined in this study, the
observed quenching rates are at least two orders of magnitude slower than those expected for diffusion-limited kinetics. This implies that (k,"k,")<.l, in which case Eq. (24) may be approximated by
Under the conditions of the experiments, the Kf and Kf parameters may be considered quasithermodynamic equilib- rium constants that reflect the 'stabilities' of the (AL* - Q) and (AL* - Q) complexes. The C and parameters are unimolecular rate constants associated with L*-to-Q elec- tronic energy-transfer processes within the (AL* - Q) and (AL* - Q) complexes. The Kzparameters are expected to be particularly sensitive to the relative charges, sizes and shapes of the AL*, AL*, and Q molecules, insofar as each of these properties may be expected to influence the formation and relative stabilities of the (AL* -Q) and (AL* - Q) complexes. For the L* - Q systems examined in the present study, the k," parameters are expected to be most strongly influenced by the relative electronic state structures and spatial proximity of the donor and acceptor metal ions in the (AL* - Q) and (AL* - Q) complexes.
The molecular modeling calculations performed in this study were used primarily to determine the most sterically compatible (and compact) structures of AL*-Q and AL* - Q contact pairs. The results obtained from these calculations provide information about the minimum allowed distances between the donor and acceptor metal centers in the (AL* - Q) and (AL* - Q) complexes, and, therefore, they are most germane to the k," parameters of Eq. (25).
ki= Kzk," (25)
Differences between Eu* and Tb* quenching results
The Eu(dpa)i- and Tb(dpa):- complexes are essentially identical with respect to size, shape, electrostatic charge distributions and detailed stereochemical properties. How-
ever, the 4.(Ln3+) electronic energy-level structures in these complexes are quite different (see Fig. 2 for relevant comparisons). Given these comparative properties of Eu(dpa):- and Tb(dpa):-, and with reference to the formulation and discussion of Eq. (25) in the preceding section, one may predict identical Kz factors but different y rate constants for Eu*-Q versus Tb*-Q quenching processes (with any given Q). It follows lthat for any given Q, the ratio of k,"(Eu*) to F(Tb*) should correspond to k,"(Eu*)/k,"(Tb*).
From the results shown in Tables 1 and 2 the authors note that the k,"(Eu*)/F(Tb*) ratios are >1 for the Cr(H,O),(ATP) Q, but are el for each of the Rh(H,O),(ATP) and Rh(H,O),(ATP) Q. These results imply significant differences between the donor (Ln*)-acceptor (M) interactions responsible for (Ln* - M)+(Ln - M*) electronic energy-transfer processes in the respective L-Q systems (where Ln* = Eu* or Tb* and M=Cr or Rh). The results also give clear evidence that Cr is a better acceptor than Rh in the (Eu* - M)-(Eu - M*) processes, whereas Rh is a better acceptor than Cr in the (Tb* - M+(Tb - M*) processes. A detailed interpretation of these findings would require considerably more informa- tion than is currently available about the energy-level structures of the Cr and Rh complexes examined in this study. The relevant energy-level structures of the Eu* and Tb* L are well characterized, but those of the Cr and Rh Q complexes are not. This precludes making any detailed comparisons between the donor-acceptor level resonances that might contribute to energy-transfer processes in the four different types of donor (Ln*)-acceptor (M) pairs.
The enantioselectivity (E,) parameters are of identical sign for Eu* and Tb* quenching, both within and among the different sets of Q. Rather remarkably, all except one of the Q exhibit a preference for ALn* (over ALn*) enantiomers in their quenching actions. The one exception is the A(e) isomer of Cr(H,O),(ATP), which exhibits a slight prefer- ence for ALn* enantiomers.
Quencher structure and enantioselectivity
Quencher differentiation between L enantiomers can occur in both the 'pre-equilibrium' and energy-transfer steps of the quenching process. In the pre-equilibrium step, this differentiation is reflected in the relative values of the KY and Kf 'stability' Parameters [defined according to Eqs (24) and (25)], and in the energy-transfer step, the differentiation is reflected in the relative values of the C and kf rate parameters.
For the L and Q examined in this study, it may be assumed that electronic energy transfer is induced by resonance interactions between f-f transition densities localized on the metal center (Ln*=Eu* or Tb*) of the L and d-d transition densities localized on the metal center (M=Cr or Rh) of the Q. The mechanistic details of these interactions will be identical for ALn* - M and ALn* - M pairs, and, to a very good approximation, the interactions may be presumed to be spatially isotropic. Therefore, the relative strengths of these interactions in ALn* - M versus ALn*-M pairs will depend only on scalar distance parameters that reflect the relative separation distances

604 T. GOWL STOCKMAN ETAL.
between the metal centers in ALn* - M versus ALn* - M. It follows that for any L*-Q system, the ratio of rate constants can be rationalized largely in terms of the relative metal-metal separation distances that exist in the (AL* - Q) versus (AL* - Q) encounter complexes. In previous work, the authors have derived functional forms for the e/kf ratios, based on both multipole-multipole and electron-exchange energy-transfer mechanisms (Glover- Fischer et al., 1995; Richardson et al., 1991). In each case, these ratios are predicted to exhibit an exponential depend- ence on the diflerence between the metal-metal separation distances in (AL* - Q) versus (AL* - Q) structures. More explicitly, the relevant expressions may be written as:
to
e k f -=exp[ - p(dA* - dA')]
where dA* and h' denote the metal-metal separation distances in (AL* - Q) and (AL* - Q) structures, respec- tively, and p is a parameter that depends on interaction details assumed to be identical in these two structures.
For any given L*-Q system, the ratio of KF to Kf 'stability' parameters may be expected to reflect the relative degree of compactness and/or surface contact that can be achieved in (AL* - Q) versus (AL* - Q) structures. This expectation is based on the assumption that compactness and surface contacts are closely correlated with the interactions that retard (L* - Q)-L*+Q dissociation proc- esses. If the authors adopt this view and further assume that the metal-metal distance parameters in Eq. (26) are inversely proportional to structural compactness (i.e. these distances are smaller in more compact structures and larger in less compact structures), then both e/e and KYlKf are predicted to be > 1 if (AL* - Q) stuctures are more compact than (AL* - Q) structures, and < 1 if (AL* - Q) structures are more compact than (AL* - Q) structures.
The connections, made earlier, between metal-metal distance, degrees of structural compactness and the relative stabilities (and lifetimes) of (AL* - Q) versus (AL* - Q) complexes provide a useful, though somewhat speculative and imprecise, basis for relating the present experimental results to the results obtained from molecular modeling calculations. The latter focused primarily on identifying the energetically favored structures of the (AL*-Q) and (AL* - Q) complexes and then determining the distances, 6' and dA*, between the metal centers in these structures. If these minimum energy structures are assumed to be the main precursors to energy-transfer events, then for dA*>dA' one may predict c<hf and Eq>O, whereas for d"'cdA* one may predict c>hf and E c0. These predicted qualitative correlations between dA'/h', TI?, and the sign of Eq are in substantial agreement with the combination of results obtained from the authors' modeling calculations and quenching experiments. Recall that for all except two of the L*-Q systems examined in this study, the sign of Eq is observed to be negative, and for each of these systems the modeling calculations show dA'<dA'. The two exceptions are those in which the A(e) isomer of Cr(H,O),(ATP) was used as the Q, and in these cases the sign of Eq is observed to be positive and the modeling calculations show 6' >dA*.
The /3 parameter in Eq. (26) depends on details of the interacting donor (Ln*) and acceptor (M) electronic transi-
tion densities that contribute to the (Ln* - M+(Ln - M*) energy-transfer processes. The value of this parameter will be different for Eu*-Cr, Eu*-Rh, Tb*-Cr, and Tb* - Rh donor-acceptor pairs, but in writing Eq. (26) the authors have assumed that p is essentially free of any chiral discriminatory effects (and, therefore, p= p ' r p"' for any given donor-acceptor pair). Support for this assumption comes from the observation that neither the signs nor the magnitudes of the quenching enantioselectivities (E,) show any correlations with the relative signs and magnitudes of the L* and Q chiroptical properties measured in the f-f electronic transition regions of Ln*=Eu* or Tb* CPL/ emission spectra and in the d-d electronic transition regions of M=Cr or Rh CDIabsorption spectra. The observed chiroptical properties show that the relevant f-f and d-d transition densities are strongly influenced by structural chirality in the respective L* and Q complexes, but the chirality dependence of these transition densities appears not to play any significant role in the interactions responsi- ble for (Ln* - M+(Ln - M*) energy transfer.
To illustrate the points made in the preceding paragraph, the authors consider the results obtained for Eu* quenching by the four structural isomers of Rh(H,O),(ATP). In Table 2 these isomers are labeled according to the prescriptions of Cleland (1979), in which A and A correlate, respectively, with S and R configurational chiralities at the asymmetric p P atom in the Rh-ATP chelate structure, and endo and ex0 correlate, respectively, with S and R configurational chiral- ities at the asymmetric a-P atom in this structure. Each of these isomers preferentially quenches AEu* (over AEu*) enantiomers; however, with respect to overall quenching efficiency and degree of enantioselectivity, there are major distinctions between the endo and ex0 isomeric structures, but only small distinctions between the A and A isomeric structures. By contrast, the CD spectra of the Rh(H,O),(ATP) isomers show major distinctions between the A and A isomeric structures, but only small distinctions between the endo and ex0 structures (Lu et al., 1988). In fact, in the Rh(II1) d-d transition regions, the signs observed in the CD spectra of the Aendo and Aexo isomers are the opposite of those observed for the Aendo and Aexo isomers. These results show that whereas the d-d chir- optical properties of Rh(H,O),(ATP) are governed largely by configurational chirality at the p P atom of the Rh-ATP chelate structure, the quenching properties are most sensi- tive to stereochemical structure dictated by configurational chirality at the a-P atom. A change from S to R absolute configuration at a-P involves an interchange of the exocyclic 0x0 and adenosine substituents on a-P. The resulting change in the spatial disposition of the adenosine moiety, relative to the remaining parts of the Rh(H,O),(ATP) structure, appears to have a significant effect on interactions between Rh(H,O),(ATP) and the A and A enantiomeric structures of the Ln(dpa):- L com- plexes.
In a previous study of Co(NH,),(nucleotide) complexes as enantioselective excited-state Q of Ln(dpa):- L, it was found that both the quenching efficiencies and enantiose- lectivities showed a strong dependence on nucleoside structure (i.e. structure external to the Co-phosphate chelate ring system) (Metcalf et al., 1992). For example, Co(NH,),(GTP) amd Co(NH,),(ITP) complexes exhibited

INTERMOLECULAR CHIRAL RECOGNITION 605
enantioselectivities (E,) opposite in sign to those observed for Co(NH,),(ATP), Co(NH,),(CTP), Co(NH,),(UTP), and Co(NH,),(’ITP) complexes. All of the complexes used in this previous study consisted of unresolved mixtures of diastereomeric structures, so the results cannot be compared directly to those reported here. However, both sets of results are consistent with the conjecture that the nucleoside moiety of the nucleotide ligand plays a major (if not dominant) role in determining quenching behavior.
Summary TR-CL spectroscopy is a technique developed in the authors’ laboratory to study excited-state energy transfer between dissymmetric metal complexes in solution. In an effort to begin to understand enantioselective recognition in biological systems, they have examined a system that utilizes a racemic lanthanide tris-dipicolinate L complex (Eu(dpa)i- or Tb(dpa):-) with resolved diastereomers of both bidentate and tridentate Cr(H,O),(ATP) and Rh(H,O),(ATP) as chiral Q. All of these complexes have been commonly used as chemical probes of enzyme active sites.
The general mechanism for enantioselective quenching has been described. Twenty-one L-Q combinations have been investigated, and several trends can be noted. Each Q species generally shows the same degree and sense of enantioselective quenching for either lanthanide lumino- phore (either Tb or Eu) employed. When the quenching efficiencies of the two different transition metals are compared, Eu L are more strongly quenched by the chromium complexes than by the rhodium complexes, while quenching of the Tb species occurs more efficiently by the rhodium diastereomers than by the chromium diastereomers. The effect of Q structure on enantioselective recognition has also been investigated, and the observed trends are the most unanticipated results presented in this article. Subtle differences in the configuration and con- formation of these nucleotide structures result in substantial differences in the degree and sense of the enantioselective
quenching observed, but these trends do not correspond to the configuration about the Pphosphate (the factor deter- mining the sign of the CD signal). Additionally, molecular modeling is presented as an illustration of possible L/Q interactions and seems to correlate with experimental data.
This work represents the first time that differences in the molecular recognition properties among individual diaster- eomeric structures of transition metal nucleotide complexes have been investigated with the TR-CL technique. Bio- logical systems are usually relatively unstable and are, consequently, difficult to investigate. The Q employed in this study are no exception, but conditions were adjusted so that the authors could apply their technique to study recognition differences between the nucleotide diaster- eomers. The success achieved in this project of obtaining new and interesting recognition results encourages the authors to use their technique to study other biological systems. The TR-CL method has the advantage over other methods in that molecular recognition is determined from emission measurements. Consequently, studies can be performed using small amounts of material where molecular interactions approach recognition between single mole- cules. Having a system that measures single molecule interactions is especially important when considering energy transfer because the properties of a bulk solution can be very different from single molecular interactions. As more investigations are performed on systems similar to the ones studied here, a better understanding of chiral discrim- ination interactions, particularly in energy transfer processes, will be gained.
Acknowledgements
This work was supported by the US National Science Foundation (NSF Grant CHE-9213473 to F.S.R.) and by the NIH Training Program in Molecular Biophysics (5-T32 GMO 8323 to C.A.K.). T.G.S. gratefully acknowledges a fellowship from the BurgerlLutz fellowship endowment from the University of Virginia. The authors also gratefully acknowledge many helpful discussions with Dr David H. Metcalf (University of Virginia) who has been an important contrihuter to the theory and measurement of enantioselective quenching kinetics.
References
Albertsson, J. (1972). Structural studies on the rare earth carboxylates. Acta Chem. Scand. 26,1005-1017.
Cahn, R. S., Ingold, C. and Prelog, V. (1966). Specification of molecular chirality. Angew. Chem. Int .Ed. Engl. 5,385-415.
Cleland, W. W. and Mildvan, A. S. (1979). Chromium(ll1) and cobalt(ll1) nucleotides as biological probes. In Advances in Inorganic Biochemistry, ed. by G. Eichhorn and L. Marzilli, pp. 163-191. Elsevier/North Holland, New York.
Dunaway-Mariano, D. and Cleland, W. W. (1980). Preparation and properties of chromium(ll1) adenosine 5’-triphosphate, chromium (111) adenosine 5’-diphosphate, and related chro- mium (111) complexes. Biochemistry 19, 1496-1505.
Glover-Fischer, D., Metcalf, D., Bolender, J. and Richardson, F. (1995). Chiral discrimination in electronic energy-transfer processes in solution. Effects of temperature and solution properties on chirality-dependent rate parameters. Chem.
Gruys, K. J. and Schuster, S. M. (1982). Separation of p,? bidentate CrATP diastereomers by reverse-phase high performance liquid chromatography. Anal. Biochem. 125,
PhyS. 198,207-235.
66-73. Kuntzweiler, T. (1991). NMR studies on CaATPase intermediates
(Ph.D. dissertation). Lu, Z., Shorter, A. L., Lin, 1. and Dunaway-Mariano, D. (1988).
Preparation and configurational analysis of the stereo- isomers of p,ybidentate Rh(H,O),ATP and @,?tridentate Rh(H,O)dTP. A new class of enzyme active site probes. Inorg. Chem. 27,4135-4139.
Metcalf, D. H., Stewart, J. M., Snyder, S. W., Grisham, C. M. and Richardson, F. S. (1992). Chiral recognition between dissym- metric Ln(dpa):- and Co(lll)-nucleotide complexes in aqueous solution. Enantioselective luminescence quench- ing as a probe of intermolecular chiral discrimination. lnorg. Chem. 31,2445-2455.
Metcalf, D. H., Bolender, J. P., Driver, M. S. and Richardson, F. S. (1993). Chiral discrimination in electronic energy transfer between dissymmetric lanthanide (111) and cobalt(ll1) com- plexes in solution. Effects of ligand size, shape, and configuration in the acceptor complexes. J. fhys. Chem. 97, 553-564.

606 T. GOWL STOCKMAN ETAL
Rexwinkel, R. B., Meskers, S. C. J., Riehl, J. P. and Dekkers, H. P. J. M. (1993). Thermodynamics of the enantioselective quenching of Tb(2,B-pyridinedicarboxyIate):- luminescence by resolved Ru(1,lO-phenanthroline):'. J. Phys. Chem. 97,
Richardson, F. S. and Metcalf, D. H. (1994). Chiroptical lumines- cence spectroscopy as a probe of electronic excited-state structure and dynamics in dissymmetric metal complexes. In Circular Dichroism: Principles and Applications, ed. by K. Nakanishi, N. Berova and R. Woody, 153-178. VCH Pub- lishers, New York.
Richardson, F. S., Metcalf, D. H. and Glover, D. P. (1991) Intermolecular chiral recognition probed by enantiose- lective quenching kinetics. A mechanistic model for dissymmetric metal complexes in solution. J. Phys. Chem.
3857-3868.
95,6249-6260.
Sabat, M., Cini, R., Haromy, T. and Sundaralingam, M. (1985). Crystal structure of the a,p,ytridentate manganese complex of adenosine 5'-triphosphate cocrystallized with 2,2'-Dipyr- idylamine. Biochemistry, 24,7827-7883.
Shorter, A. L., Harmony, T., Scalzo-Brush, T., Knight, W., Dunaway-Mariano, D. and Sundaralingham, M. (1987). Structural and biochemical properties of bidentate tetra- aquarhodium(ll1) complexes of inorganic pyrophosDhate and adenosine 5'idiphosphate. Biochemistry, . 26, 2060-2066.
Sigel, H., Tribolet, R., Malini-Balakrishnan, R. and Martin, R. 6. (1987). Comparison of the stabilities of monomeric metal ion complexes formed with adenosine 5'4riphosphate (ATP) and pyrimidine-nucleoside 5'-triphosphates (CTP, UTP, TTP) and evaluation of the isomeric equilibria in the complexes of ATP and CTF? Inorg. Chem. 26,2149-2157.





















