Embed Size (px)
Citation preview

O P I N I O N
Intermediate phenotypes and genetic mechanisms of psychiatric disordersAndreas Meyer-Lindenberg and Daniel R. Weinberger
Abstract | Genes are major contributors to many psychiatric diseases, but their
mechanisms of action have long seemed elusive. The intermediate phenotype
concept represents a strategy for characterizing the neural systems affected by
risk gene variants to elucidate quantitative, mechanistic aspects of brain function
implicated in psychiatric disease. Using imaging genetics as an example, we
illustrate recent advances, challenges and implications of linking genes to structural
and functional variation in brain systems related to cognition and emotion.
Although the genomic revolution transforms all areas of medicine, psychiatry arguably stands to benefit the most. For one, the majority of common psychiatric disorders show high genetic familiality. Heritability (the proportion of total variance in a trait due to genetic variation) has been estimated from twin studies at 81% for schizophrenia1 and 37% for major depression2. As well as being debilitating for sufferers, these diseases impose enormous medical and economic burdens, making understanding the genetic mechanisms of mental disorders crucial. In addition, genetic research is expected to aid in the definition of psychiatric disease entities themselves, which are largely based on clinical phenomenology and lack biologi-cal validity. However, understanding the neural mechanisms by which genetic vari-ation increases risk has long been elusive. The genetic architecture of psychiatric risk is complex and is dominated by multiple inter-acting contributing factors. As genes do not encode for psychopathology, it is reasonable to expect that the association or penetrance of gene effects will be greater at the level of relatively more simple and biologically based phenotypes. Therefore, intermediate pheno-types were initially envisaged to be tools for gene discovery, improving the power of association studies by reducing phenotypic heterogeneity.
However, as we review here, the develop-ment of imaging genetics — a strategy for mapping neural structure and activity as a function of genotype in living humans — has encouraged a conceptual transformation by showing that the greater power of inter-mediate phenotypes lies in using genetic risk variants as tools for the discovery of the
mediating neural mechanisms that bridge the gap from DNA sequence to pathological behaviour. Other quantitative parameters from electrophysiology, neurobiochemistry and neuropsychology can also be powerful tools to index intermediate neurobiological processes that are influenced by genetic variation, but these are not in the scope of this article. After briefly recapitulating the development of the intermediate phenotype concept, we review recent advances in the characterization of prefrontal circuits in schizophrenia3–5 as target neural systems for mechanisms of genetic susceptibility and their epistatic and environmental interac-tions. Although we use schizophrenia as our example, most of the arguments equally apply to other complex psychiatric disorders such as depression6 (BOX 1), attention deficit hyperactivity disorder7, addictions8 and autism9. We conclude with an assessment of the methodological and conceptual challenges of the intermediate phenotype approach.
Intermediate phenotypes in psychiatry
When the tools for gene identification in simple Mendelian disorders — mainly link-age analysis followed by positional cloning — became available, they were applied with enthusiasm to psychiatry10. The success of linkage analysis is influenced by several assumptions, including genetic homoge-neity, moderate to major gene effects in families and a valid model of inheritance11. Given the likelihood that these assumptions do not hold in many complex disorders such as mental illness11, it is not surprising that the first wave of linkage studies in psychiatry was hindered by weak results
and non-replication10. Subsequent studies of larger and more carefully characterized samples were more successful and have identified several replicable linkage sites, but these probably reflect regions where multiple genes of small effect are found12. Indeed, the weight of the evidence indicates that risk for psychiatric disease is usually conferred by multiple small effect genetic variants interacting with one another and with the environment13, that is, psychiatric disorders are genetically complex, similar to other common conditions such as hyperten-sion, obesity and diabetes. This implies that no particular constellation of genes or environmental conditions will be character-istic of most ill individuals, and gene–gene and gene–environment interactions, both additive and epistatic, further complicate analysis14. The intricate genetic architecture of susceptibility also makes it more difficult to deal with problems such as pleiotropy or variable penetrance found in many, even Mendelian, genetic diseases15.
The rate-limiting factor in gene identifi-cation is often the effect size of a risk allele on phenotypic variance. Many factors contribute to the small effect size of genes in psychiatry. Few variants involve changes in protein structure or function. More often, aspects of gene regulation are implicated, which have relatively subtle biological effects. Importantly, genes do not encode for psychiatric phenomena (for example, hallucinations and panic attacks), and so, almost by definition, the more behavioural the phenotype, the less directly it will be predicted by a genotype (FIG. 1). This leads to the strategy of studying underlying quantitative traits that more directly index biology, analogous to moving from the study of cardiac insufficiency or stroke (complex diseases) to ventricular hypertrophy16 and cholesterol metabolism. This strategy offers several advantages for behavioural disorders: biological traits are expected to be closer to the genetic substrate, enhancing penetrance; the traits should be observable in geneti-cally at risk but behaviourally unaffected individuals; and, if the traits are sufficiently causally upstream to index a biological process that makes a separable contribution to disease, the genetic architecture should be simplified.
There are two key reasons why the intermediate phenotype concept has reson-ated strongly with psychiatry17. First, the uncertain and phenomenological nature of psychiatric diagnosis makes reference to a biological level of description attractive. Second, the complexity of the human brain
P E R S P E C T I V E S
818 | OCTOBER 2006 | VOLUME 7 www.nature.com/reviews/neuro
© 2006 Nature Publishing Group

1.7
1.9
2.2
2.5
2.8
3.1
3.3
c d
Dec
reas
e in
func
tion
al
conn
ecti
vity
wit
h am
ygda
la
1.9
2.3
2.7
3.1
3.4
3.8
4.2
Dec
reas
e in
gre
y m
atte
r
a b
virtually necessitates an effort to parse this problem into tractable biological subpro-cesses. Almost uniquely in the psychiatric arena, risk factors or intermediate biological phenotypes have come to be known as ‘endophenotypes’17, from the Greek word endos for interior, within. This usage dates back to a seminal paper by Gottesman and Shields14, who adopted it from a report on evolutionary biology. As used later by Gottesman and Gould17, and subsequently by the field, it was hoped that endopheno-types would assist both “in the identification of aberrant genes in the hypothesized polygenic systems conferring vulnerabilities to disorders”17 and in the decomposition of psychiatric diagnosis into biologically valid disease entities (FIG. 2a). Although the original use of the term did not focus on the biological mechanisms of gene effects, but more on psychological processes, it has come to be used in this context as well.
From the outset, it was stressed that using endophenotypes for gene discovery mandates that they be ‘sufficiently’ herita-ble17. Elaborating on this, several authors7,18,19 have specified that, as well as being herit-able, an endophenotype should: have good psychometric properties; be related to the disorder and its symptoms in the general population; be stable over time; show increased expression in unaffected relatives of probands; cosegregate with the disorder in families; and have common genetic influences with the disorder. It is clear that establishing these criteria for any given measure is an extensive and costly process, and published work suggests that few, if any, endophenotypes actually fulfil them7. This issue has become pressing with the applica-tion and, arguably, success of neuroimaging in characterizing neural system intermediate phenotypes in psychiatry. After revolution-izing our understanding of the neural under-pinnings of normal cognition and altered brain function in disease, neuroimaging has been used to study genetic variation, which has proved surprisingly penetrant at this level14–21. For the phenotype discussion, imaging provides, for each participant, an enormous amount of functional–structural data that can potentially characterize gene effects in the brain. However, this unprec-edented access to brain biology also carries unique problems: it is unclear, for example, how the issue of multiple testing should be handled in this context, how multimodal datasets can be related to genetic informa-tion of growing complexity in the compara-tively small populations currently studied, and how reliable and comparable across
laboratories and populations these data are. More importantly for some, heritability has not been conclusively demonstrated for many structural and most functional param-eters used. The traditional endophenotype model was a strategy for reducing genetic complexity and increasing genetic effect size for facilitating gene discovery. Imaging strategies might prove to be valuable in this respect, but large imaging datasets necessary for gene discovery are difficult to acquire.
Although these problems and possible solutions will be discussed, our primary perspective is that neuroimaging is bringing about a conceptual change in the way in
which biological intermediate phenotypes are viewed and pursued in psychiatry and behavioural genetics by enabling a previ-ously inaccessible level of biological charac-terization and validation of genetic effects (FIG. 2b). Imaging genetics can delineate neural systems that are affected by genetic variation, offering a way of ‘functionating’ polymorphisms beyond simple clinical statistical association. The assumption of the intermediate phenotype strategy is that gene effects at the level of the brain are a more direct effect of genetic variation than is complex behaviour, and will show associa-tion in carriers of risk alleles even if the
Box 1 | The application of imaging genetics to depression
The pathophysiology of depression is complex, affecting integrated pathways linking cortical, subcortical and limbic sites and their molecular mediators6,72,73. A useful example of the application of imaging genetics to understanding the neural mechanisms of genetic risk concerns a key node of this system, the subgenual cingulate (Brodmann’s area 25)6,74,75, which interacts with the amygdala76 in a functional feedback circuit that regulates amygdala processing of environmental adversity77,78. This circuit is closely linked to serotonergic (5-hydroxytryptamine; 5-HT) neurotransmission79. The serotonin transporter gene (SLC6A4) contains a variable number of tandem repeats variant in the 5′ promoter region (5-HTTLPR), with reduced transcription of the 5-HTTLPR short (S) allele in comparison to the long (L) allele. Individuals carrying the S allele tend to have increased anxiety-related temperamental traits80: this variant predicts risk for depression in conjunction with early environmental adversity81. Several studies have found that S allele carriers evince an exaggerated response during functional MRI70,82,83, suggesting that amygdala hyper-reactivity might be a neural substrate of trait anxiety predisposing to psychiatric disease. A reduction in grey matter was found in the subgenual cingulate region of healthy carriers of the S allele6 (a). Analyses of functional and structural connectivity confirmed close interactions of this region with the amygdala and suggested an inhibitory feedback circuit6. The S allele was associated with reduced coupling between the amygdala and the subgenual cingulate cortex (b), and the degree of that coupling predicted close to 30% of the variability of trait anxiety in these normal individuals6, suggesting that the psychiatric risk associated with 5-HTTLPR is mediated by a weakened circuit for the extinction of fear. Two studies have also shown that a frequent regulatory variant (844G>T) of tryptophan hydroxylase 2 biases the reactivity of the amygdala84,85. A common variable number of tandem repeats polymorphism86 in monoamine oxidase A (MAOA), which encodes a key enzyme for the catabolism of serotonin and other neurotransmitters during neurodevelopment, has been implicated in violence; the low expression variant also showed a pronounced gene–environment interaction in predicting violent offences in males with adverse early experience87. Multimodal imaging results88 have indicated a similar effect to that of 5-HTTLPR on the structure and function of the amygdala and perigenual cingulate cortex, suggesting a shared serotonergic mechanism of emotional regulation. However, MAOA showed more extensive effects in both structure (c, reductions in blue) and activation (d, reductions in yellow), notably affecting more caudal regions of the cingulate, which are associated with cognitive control — in agreement with previous findings89 — as well as the orbitofrontal cortex and hippocampus. Panels a and b reproduced, with permission, from Nature Neuroscience REF. 6 © (2005) Macmillan Publishers Ltd. Panels c and d modified, with permission, from REF. 88 © (2006) National Academy of Sciences.
P E R S P E C T I V E S
NATURE REVIEWS | NEUROSCIENCE VOLUME 7 | OCTOBER 2006 | 819
© 2006 Nature Publishing Group

Episodic memory
Working memory
RewardEmotional regulation
GRM3(7q21.1–q21.2)
DRD2(11q23)
COMT(22q11.2)
MAOA VNTR(Xp11.23)
5-HTTLPR/SLC6A4(17q11.1–q12)
PFCCaudate
Putamen
MB
PFC
OFC
BA 25 AM
HF
a
b
Schizophrenia
Depression
carriers show no clinical diagnostic charac-teristics. As illustrated below, many risk gene associations to brain-based phenotypes are observed even in healthy individuals. To the considerable degree that susceptibility genes contribute to psychiatric risk, this approach offers a powerful bottom-up strategy to discover biologically valid knowledge about previously unknown mechanisms. Imaging genetics therefore becomes a guide to the discovery of neural circuitry that translates genetic effects into behaviour, and endo-
phenotypes implicate endomechanisms. Although this approach does not depend on the demonstrated heritability of the imaging probe used, to increase the prior probability of observing biologically relevant neural activity, the chosen paradigm must activate brain systems that are plausibly related to the disease under study based on independent evidence. We outline such evidence below for neural systems related to schizophrenia.
We stress that although we use the term endophenotype in deference to an earlier
period in the evolution of thinking about this genetic strategy, there is nothing ‘hidden’ about the biological phenomena in the con-text of neurobiology and imaging. The term intermediate phenotype is preferred, both because it implies a biological trait that is in a predictable path from gene to behaviour and because the phenotypes and mecha-nisms are not secondary, but probably pri-mary. This is analogous to its usage in other areas of complex medical genetics. From this perspective, it might be more correct to refer
Figure 1 | The complex path from genes to behavioural and disease phenotype: mediation through brain circuitry. Multiple genetic risk
variants affect, through interaction with each other and the environment,
multiple neural systems linked to several neuropsychological and behav-
ioural domains that are impaired, in differing proportions, in psychiatric
diseases. No one-to-one mapping exists between genes and neural system
mechanisms, or between mechanisms and behaviour. As examples, the
following genetic variants are depicted (chromosomal variation in paren-
theses): GRM3 single nucleotide polymorphism 4 (REF. 57) (7q21.1–q21.2),
dopamine receptor D2 (DRD2) Taq 1a56 (11q23), catechol-O-methyltrans-
ferase (COMT) Val66Met4,46 (22q11.2), serotonin transporter length
polymorphism (5-HTTLPR/SLC6A4)6,70 (17q11.1–q12) and monoamine
oxidase A variable number tandem repeat (MAOA VNTR)92 (Xp11.23).
These are shown to affect a circuit that links the prefrontal cortex (PFC)
with the midbrain (MB) and striatum (caudate and putamen) (a), which is
relevant for schizophrenia, and a circuit that connects the amygdala (AM)
with regulatory cortical and limbic areas (b), which is implicated in depres-
sion and anxiety (BOX 1). These circuits, in turn, are shown to mediate risk
for schizophrenia and depression and various neuropsychological func-
tions. Although illustrative, the connections shown correspond to pub-
lished work and show that a given gene will influence a variety of neural
circuits, which in turn influence several behavioural and clinical
parameters. BA 25, Brodmann’s area 25; HF, hippocampal formation;
OFC, orbitofrontal cortex.
P E R S P E C T I V E S
820 | OCTOBER 2006 | VOLUME 7 www.nature.com/reviews/neuro
© 2006 Nature Publishing Group

Schizophrenia
Depression
Episodic memory
Working memory
RewardEmotional regulation
Schizophrenia
Depression
Episodic memory
Working memory
RewardEmotional regulation
Schizophrenia
Depression
Episodic memory
Working memory
RewardEmotional regulation
Schizophrenia
Depression
Episodic memory
Working memory
RewardEmotional regulation
PFC
PFC
OFC
BA 25 AM
HF
PFC
Caudate
PutamenMB
GRM3(7q21.1–q21.2)
COMT(22q11.2)
MAOA VNTR(Xp11.23)
MAOA VNTR(Xp11.23)
5-HTTLPR/SLC6A4
(17q11.1–q12)
COMT(22q11.2)
COMT(22q11.2)
a
b
BA 25 AM
HF
to the behavioural phenomena as emergent or exophenotypes.
Neural circuits as psychiatric phenotypes. Convergent molecular, cellular, clinical, neurophysiological, neuropsychological and imaging work clearly indicate dorsolateral prefrontal cortex (DLPFC) dysfunction as a key feature of schizophrenia3. A defining feature of the DLPFC is its extensive inter-connectedness with other brain regions20,21, making the characterization of these distributed functional networks impor-tant22,23. We focus on two circuits involving the DLPFC, one interlinking it with the hippo campus and the other with the stria-tum, as intermediate phenotype targets for genetic association.
In a topographically well-organized manner, the neostriatum receives excita-tory glutamatergic projections from the cortex and thalamus, integrates them with monoaminergic inputs and sends them via the globus pallidus and substantia nigra pars reticulata to the thalamus, which projects back to the prefrontal cortex (PFC)20. These parallel processing loops are crucial for the integration of sensorimotor, cognitive and emotional information20. Lesions to the neostriatal-prefrontal system impair prefrontal-dependent cognitive functions24 that are characteristic of the cognitive deficits found in schizophrenia25. Prefrontal-striatal interactions have been proposed to ‘filter’ different pieces of information that are competing for cortical processing26. This filtering could underlie cognitive symptoms and potential schizophrenia intermediate phenotypes such as the abnormal pre-pulse inhibition of the startle response26.
The dopaminergic system, originating from the midbrain, is an important modula-tor of the neostriatal-prefrontal circuit27 and is the target of a large number of therapeutic and abused drugs. The DLPFC participates in the control of midbrain dopaminergic neurons28. These prefrontal–midbrain interactions are crucial for motivated behaviour, working memory29 and reward-related learning30. A large body of work has established an inverted u-shaped relation-ship between working-memory-related activation of DLPFC neurons and dopamin-ergic — especially dopamine D1 receptor — stimulation29, with dopaminergic tone essential for optimizing signal-to-noise ratio, or tuning. In schizophrenia, dopaminergic abnormalities are suggested by the fact that antipsychotic agents block D2 receptors — a cornerstone of the so-called ‘dopamine hypothesis’. Although a simple model of
Figure 2 | Intermediate phenotypes as tools for gene discovery versus neural mechanism characterization. Examples of two alternative approaches to the identification of genetic variants
linked to psychiatric disorders are illustrated, with the relevant genes, neural systems and behavioural
phenotypes highlighted in red, and arrows indicating the direction of research inference. a | In the gene discovery approach, behavioural or neural systems phenotypes are used to reduce
genetic complexity and increase penetrance to identify genes implicated in psychiatric disorders. For
example, deficiencies in the electrophysiological response to auditory stimulation were used to iden-
tify an association of schizophrenia with the α7 nicotinic cholinergic receptor93. In the figure, prefron-
tal cortex (PFC) dysfunction has been linked to catechol-O-methytransferase (COMT) and GRM3
genetic variation57,65, and emotional regulation has been linked to variation in COMT, monoamine
oxidase A (MAOA) and the serotonin transporter length polymorphism (5-HTTLPR/SLC6A4)6,83,88, and
so could have been hypothetically employed as a phenotype to identify these genes. b | In the neural
mechanism approach, genes known to be associated with psychiatric disorders or behavioural traits
are used to discover neural mechanisms mediating their complex emergent phenotypic associations,
implicating these mechanisms in the psychiatric disorders to which they have been linked. Examples
include the use of the COMT Val158Met polymorphism to characterize prefrontal function and
prefrontal–midbrain interactions linked to risk for schizophrenia4,46, and the delineation of cingulate
circuitry regulating amygdala (AM) function mediating risk for anxiety and depression through an
investigation of the MAOA variable number tandem repeat (VNTR)6,70. BA 25, Brodmann’s area 25;
HF, hippocampal formation; MB, midbrain; OFC, orbitofrontal cortex.
P E R S P E C T I V E S
NATURE REVIEWS | NEUROSCIENCE VOLUME 7 | OCTOBER 2006 | 821
© 2006 Nature Publishing Group
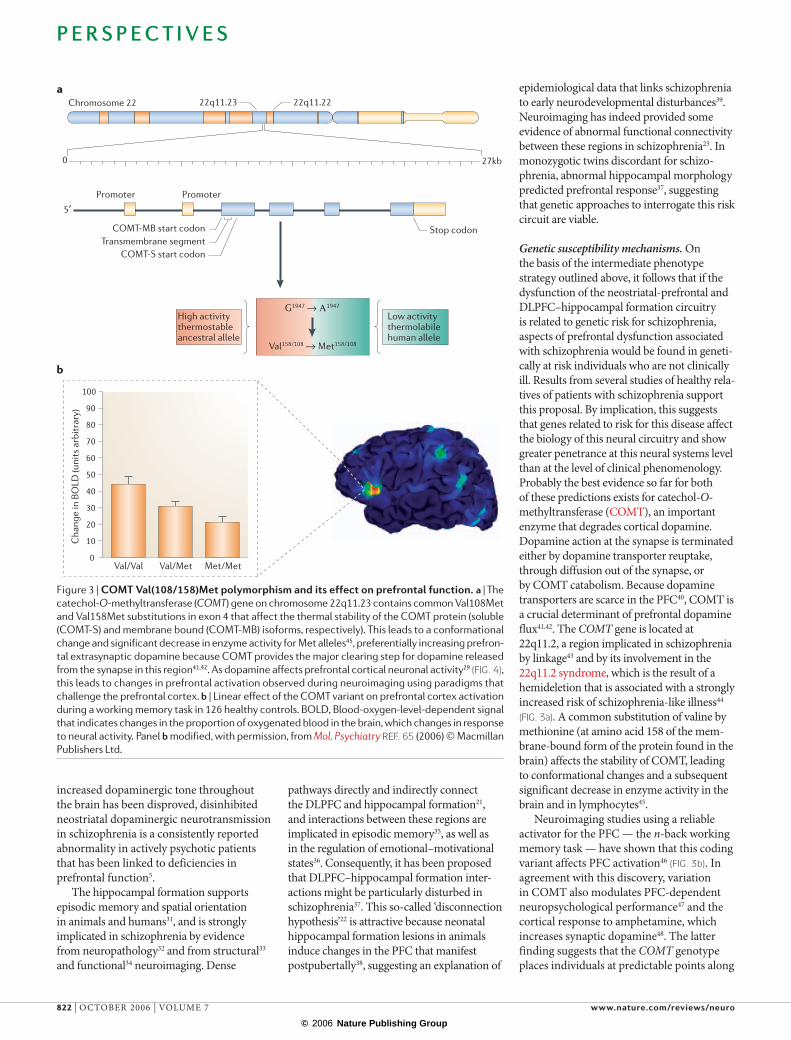
Chromosome 22 22q11.2222q11.23
0 27kb
Promoter Promoter
5′
Stop codonCOMT-MB start codonTransmembrane segment
COMT-S start codon
High activitythermostableancestral allele
G1947 → Α1947
Val158/108 → Met158/108
Low activitythermolabilehuman allele
a
b
0
10
20
30
40
50
60
70
80
90
100
Cha
nge
in B
OLD
(uni
ts a
rbit
rary
)
Val/Val Val/Met Met/Met
increased dopaminergic tone throughout the brain has been disproved, disinhibited neostriatal dopaminergic neurotransmission in schizophrenia is a consistently reported abnormality in actively psychotic patients that has been linked to deficiencies in prefrontal function5.
The hippocampal formation supports episodic memory and spatial orientation in animals and humans31, and is strongly implicated in schizophrenia by evidence from neuropathology32 and from structural33 and functional34 neuroimaging. Dense
pathways directly and indirectly connect the DLPFC and hippocampal formation21, and interactions between these regions are implicated in episodic memory35, as well as in the regulation of emotional–motivational states36. Consequently, it has been proposed that DLPFC–hippocampal formation inter-actions might be particularly disturbed in schizophrenia37. This so-called ‘disconnection hypothesis’22 is attractive because neonatal hippocampal formation lesions in animals induce changes in the PFC that manifest postpubertally38, suggesting an explanation of
epidemiological data that links schizophrenia to early neurodevelopmental disturbances39. Neuroimaging has indeed provided some evidence of abnormal functional connectivity between these regions in schizophrenia23. In monozygotic twins discordant for schizo-phrenia, abnormal hippocampal morphology predicted prefrontal response37, suggesting that genetic approaches to interrogate this risk circuit are viable.
Genetic susceptibility mechanisms. On the basis of the intermediate phenotype strategy outlined above, it follows that if the dysfunction of the neostriatal-prefrontal and DLPFC–hippocampal formation circuitry is related to genetic risk for schizophrenia, aspects of prefrontal dysfunction associated with schizophrenia would be found in geneti-cally at risk individuals who are not clinically ill. Results from several studies of healthy rela-tives of patients with schizophrenia support this proposal. By implication, this suggests that genes related to risk for this disease affect the biology of this neural circuitry and show greater penetrance at this neural systems level than at the level of clinical phenomenology. Probably the best evidence so far for both of these predictions exists for catechol-O-methyltransferase (COMT), an important enzyme that degrades cortical dopamine. Dopamine action at the synapse is terminated either by dopamine transporter reuptake, through diffusion out of the synapse, or by COMT catabolism. Because dopamine transporters are scarce in the PFC40, COMT is a crucial determinant of prefrontal dopamine flux41,42. The COMT gene is located at 22q11.2, a region implicated in schizophrenia by linkage43 and by its involvement in the 22q11.2 syndrome, which is the result of a hemideletion that is associated with a strongly increased risk of schizophrenia-like illness44
(FIG. 3a). A common substitution of valine by methionine (at amino acid 158 of the mem-brane-bound form of the protein found in the brain) affects the stability of COMT, leading to conformational changes and a subsequent significant decrease in enzyme activity in the brain and in lymphocytes45.
Neuroimaging studies using a reliable activator for the PFC — the n-back working memory task — have shown that this coding variant affects PFC activation46 (FIG. 3b). In agreement with this discovery, variation in COMT also modulates PFC-dependent neuropsychological performance47 and the cortical response to amphetamine, which increases synaptic dopamine48. The latter finding suggests that the COMT genotype places individuals at predictable points along
Figure 3 | COMT Val(108/158)Met polymorphism and its effect on prefrontal function. a | The
catechol-O-methyltransferase (COMT) gene on chromosome 22q11.23 contains common Val108Met
and Val158Met substitutions in exon 4 that affect the thermal stability of the COMT protein (soluble
(COMT-S) and membrane bound (COMT-MB) isoforms, respectively). This leads to a conformational
change and significant decrease in enzyme activity for Met alleles45, preferentially increasing prefron-
tal extrasynaptic dopamine because COMT provides the major clearing step for dopamine released
from the synapse in this region41,42. As dopamine affects prefrontal cortical neuronal activity29 (FIG. 4),
this leads to changes in prefrontal activation observed during neuroimaging using paradigms that
challenge the prefrontal cortex. b | Linear effect of the COMT variant on prefrontal cortex activation
during a working memory task in 126 healthy controls. BOLD, Blood-oxygen-level-dependent signal
that indicates changes in the proportion of oxygenated blood in the brain, which changes in response
to neural activity. Panel b modified, with permission, from Mol. Psychiatry REF. 65 (2006) © Macmillan
Publishers Ltd.
P E R S P E C T I V E S
822 | OCTOBER 2006 | VOLUME 7 www.nature.com/reviews/neuro
© 2006 Nature Publishing Group
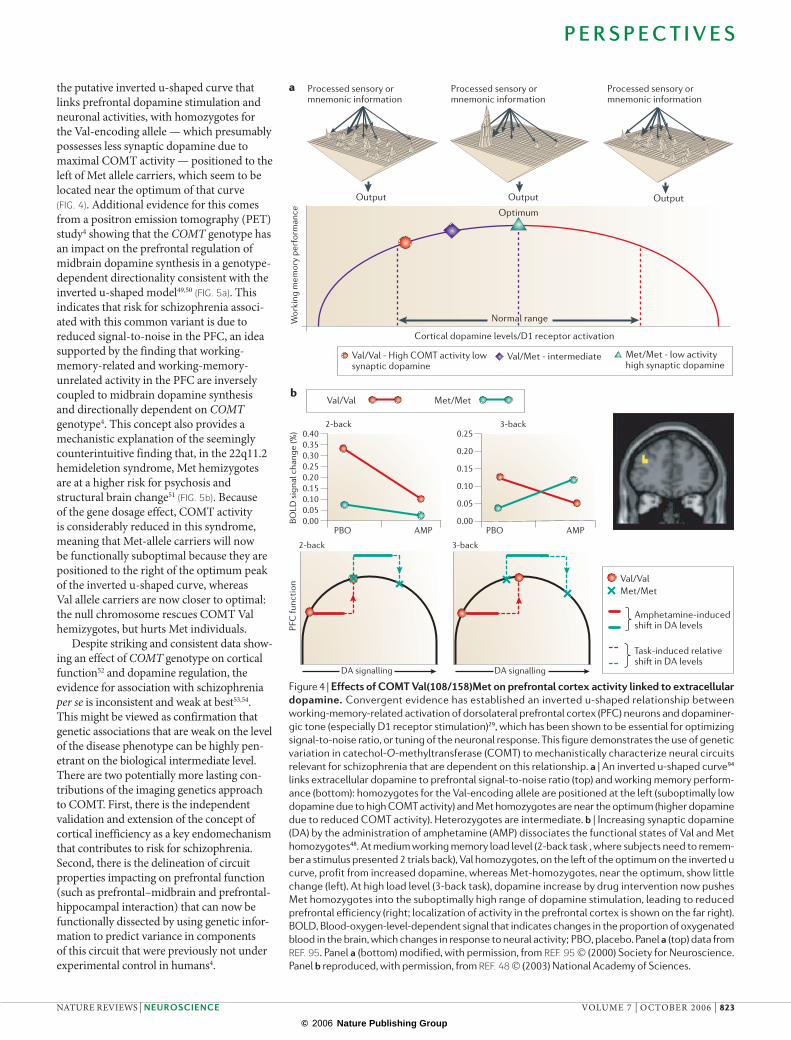
Processed sensory or mnemonic information
Output
Processed sensory or mnemonic information
Output
Val/Val - High COMT activity low synaptic dopamine
Val/Met - intermediate Met/Met - low activityhigh synaptic dopamine
Cortical dopamine levels/D1 receptor activation
Wor
king
mem
ory
perf
orm
ance
Normal range
PBO AMP
Val/Val Met/Metb
PBO AMP0.00 0.00
0.05
0.10
0.15
0.20
0.25
0.050.100.150.200.250.300.350.40
2-back
2-back
3-back
3-back
PFC
func
tion
DA signalling
Val/ValMet/Met
Amphetamine-inducedshi� in DA levels
Task-induced relativeshi� in DA levels
DA signalling
BOLD
sig
nal c
hang
e (%
)
Processed sensory or mnemonic information
Output
a
Optimum
the putative inverted u-shaped curve that links prefrontal dopamine stimulation and neuronal activities, with homozygotes for the Val-encoding allele — which presumably possesses less synaptic dopamine due to maximal COMT activity — positioned to the left of Met allele carriers, which seem to be located near the optimum of that curve (FIG. 4). Additional evidence for this comes from a positron emission tomography (PET) study4 showing that the COMT genotype has an impact on the prefrontal regulation of midbrain dopamine synthesis in a genotype-dependent directionality consistent with the inverted u-shaped model49,50 (FIG. 5a). This indicates that risk for schizophrenia associ-ated with this common variant is due to reduced signal-to-noise in the PFC, an idea supported by the finding that working-memory-related and working-memory-unrelated activity in the PFC are inversely coupled to midbrain dopamine synthesis and directionally dependent on COMT genotype4. This concept also provides a mechanistic explanation of the seemingly counterintuitive finding that, in the 22q11.2 hemideletion syndrome, Met hemizygotes are at a higher risk for psychosis and structural brain change51 (FIG. 5b). Because of the gene dosage effect, COMT activity is considerably reduced in this syndrome, meaning that Met-allele carriers will now be functionally suboptimal because they are positioned to the right of the optimum peak of the inverted u-shaped curve, whereas Val allele carriers are now closer to optimal: the null chromosome rescues COMT Val hemizygotes, but hurts Met individuals.
Despite striking and consistent data show-ing an effect of COMT genotype on cortical function52 and dopamine regulation, the evidence for association with schizophrenia per se is inconsistent and weak at best53,54. This might be viewed as confirma tion that genetic associations that are weak on the level of the disease phenotype can be highly pen-etrant on the biological intermediate level. There are two potentially more lasting con-tributions of the imaging genetics approach to COMT. First, there is the independent validation and extension of the concept of cortical inefficiency as a key endomechanism that contributes to risk for schizophrenia. Second, there is the delinea tion of circuit properties impacting on prefrontal function (such as prefrontal–midbrain and prefrontal-hippo campal interaction) that can now be functionally dissected by using genetic infor-mation to predict variance in components of this circuit that were previously not under experimental control in humans4.
Figure 4 | Effects of COMT Val(108/158)Met on prefrontal cortex activity linked to extracellular dopamine. Convergent evidence has established an inverted u-shaped relationship between
working-memory-related activation of dorsolateral prefrontal cortex (PFC) neurons and dopaminer-
gic tone (especially D1 receptor stimulation)29, which has been shown to be essential for optimizing
signal-to-noise ratio, or tuning of the neuronal response. This figure demonstrates the use of genetic
variation in catechol-O-methyltransferase (COMT) to mechanistically characterize neural circuits
relevant for schizophrenia that are dependent on this relationship. a | An inverted u-shaped curve94
links extracellular dopamine to prefrontal signal-to-noise ratio (top) and working memory perform-
ance (bottom): homozygotes for the Val-encoding allele are positioned at the left (suboptimally low
dopamine due to high COMT activity) and Met homozygotes are near the optimum (higher dopamine
due to reduced COMT activity). Heterozygotes are intermediate. b | Increasing synaptic dopamine
(DA) by the administration of amphetamine (AMP) dissociates the functional states of Val and Met
homozygotes48. At medium working memory load level (2-back task , where subjects need to remem-
ber a stimulus presented 2 trials back), Val homozygotes, on the left of the optimum on the inverted u
curve, profit from increased dopamine, whereas Met-homozygotes, near the optimum, show little
change (left). At high load level (3-back task), dopamine increase by drug intervention now pushes
Met homozygotes into the suboptimally high range of dopamine stimulation, leading to reduced
prefrontal efficiency (right; localization of activity in the prefrontal cortex is shown on the far right).
BOLD, Blood-oxygen-level-dependent signal that indicates changes in the proportion of oxygenated
blood in the brain, which changes in response to neural activity; PBO, placebo. Panel a (top) data from
REF. 95. Panel a (bottom) modified, with permission, from REF. 95 © (2000) Society for Neuroscience.
Panel b reproduced, with permission, from REF. 48 © (2003) National Academy of Sciences.
P E R S P E C T I V E S
NATURE REVIEWS | NEUROSCIENCE VOLUME 7 | OCTOBER 2006 | 823
© 2006 Nature Publishing Group
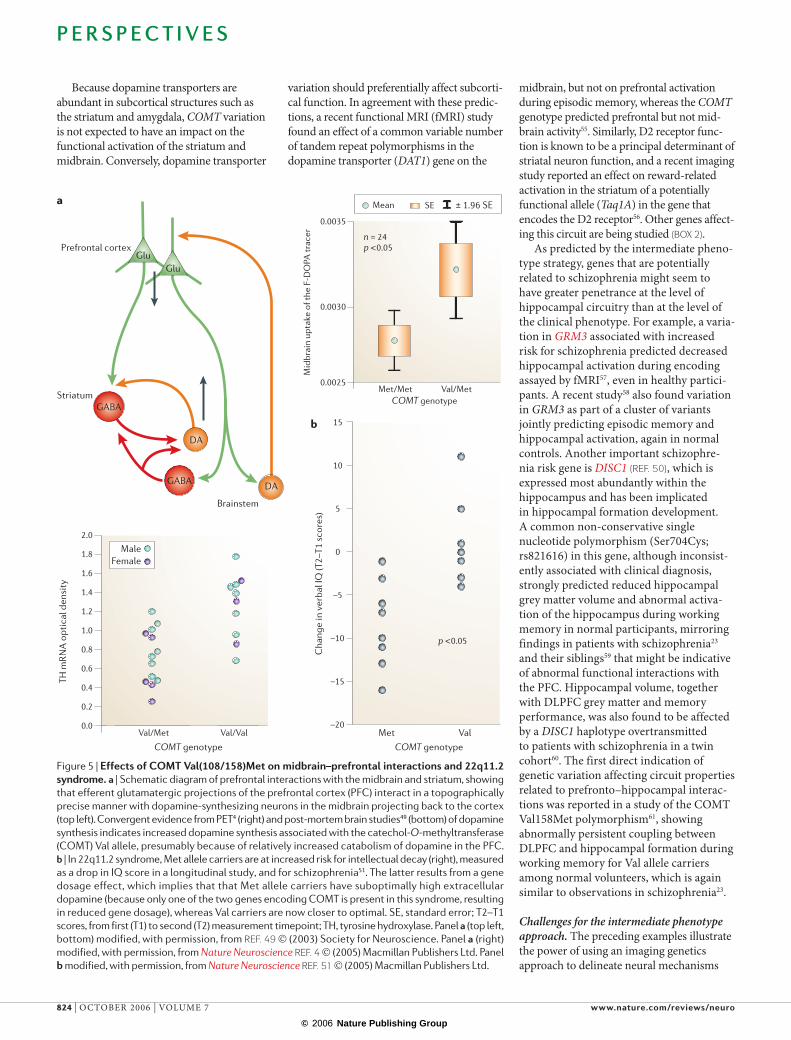
Prefrontal cortex
Striatum
Brainstem
GluGlu
DA
DA
GABA
GABA
0.0025
0.0030
0.0035
Mid
brai
n up
take
of t
he F
-DO
PA tr
acer n = 24
p <0.05
Mean SE
COMT genotypeMet/Met Val/Met
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
TH m
RN
A o
ptic
al d
ensi
ty
Val/Met Val/Val Met Val
Cha
nge
in v
erba
l IQ
(T2–
T1 s
core
s)
–20
–15
–10
–5
0
5
10
15
p <0.05
a
b
± 1.96 SE
MaleFemale
COMT genotype COMT genotype
Because dopamine transporters are abundant in subcortical structures such as the striatum and amygdala, COMT variation is not expected to have an impact on the functional activation of the striatum and midbrain. Conversely, dopamine transporter
variation should preferentially affect subcorti-cal function. In agreement with these predic-tions, a recent functional MRI (fMRI) study found an effect of a common variable number of tandem repeat polymorphisms in the dopamine transporter (DAT1) gene on the
midbrain, but not on prefrontal activation during episodic memory, whereas the COMT genotype predicted prefrontal but not mid-brain activity55. Similarly, D2 receptor func-tion is known to be a principal determinant of striatal neuron function, and a recent imaging study reported an effect on reward-related activation in the striatum of a potentially functional allele (Taq1A) in the gene that encodes the D2 receptor56. Other genes affect-ing this circuit are being studied (BOX 2).
As predicted by the intermediate pheno-type strategy, genes that are potentially related to schizophrenia might seem to have greater penetrance at the level of hippo campal circuitry than at the level of the clinical phenotype. For example, a varia-tion in GRM3 associated with increased risk for schizophrenia predicted decreased hippocampal activation during encoding assayed by fMRI57, even in healthy partici-pants. A recent study58 also found variation in GRM3 as part of a cluster of variants jointly predicting episodic memory and hippocampal activation, again in normal controls. Another important schizophre-nia risk gene is DISC1 (REF. 50), which is expressed most abundantly within the hippocampus and has been implicated in hippocampal formation development. A common non-conservative single nucleotide polymorphism (Ser704Cys; rs821616) in this gene, although inconsist-ently associated with clinical diagnosis, strongly predicted reduced hippocampal grey matter volume and abnormal activa-tion of the hippocampus during working memory in normal participants, mirroring findings in patients with schizophrenia23 and their siblings59 that might be indicative of abnormal functional interactions with the PFC. Hippocampal volume, together with DLPFC grey matter and memory performance, was also found to be affected by a DISC1 haplotype overtransmitted to patients with schizophrenia in a twin cohort60. The first direct indication of genetic variation affecting circuit properties related to prefronto–hippocampal interac-tions was reported in a study of the COMT Val158Met polymorphism61, showing abnormally persistent coupling between DLPFC and hippocampal formation during working memory for Val allele carriers among normal volunteers, which is again similar to observations in schizophrenia23.
Challenges for the intermediate phenotype approach. The preceding examples illustrate the power of using an imaging genetics approach to delineate neural mechanisms
Figure 5 | Effects of COMT Val(108/158)Met on midbrain–prefrontal interactions and 22q11.2 syndrome. a | Schematic diagram of prefrontal interactions with the midbrain and striatum, showing
that efferent glutamatergic projections of the prefrontal cortex (PFC) interact in a topographically
precise manner with dopamine-synthesizing neurons in the midbrain projecting back to the cortex
(top left). Convergent evidence from PET4 (right) and post-mortem brain studies49 (bottom) of dopamine
synthesis indicates increased dopamine synthesis associated with the catechol-O-methyltransferase
(COMT) Val allele, presumably because of relatively increased catabolism of dopamine in the PFC.
b | In 22q11.2 syndrome, Met allele carriers are at increased risk for intellectual decay (right), measured
as a drop in IQ score in a longitudinal study, and for schizophrenia51. The latter results from a gene
dosage effect, which implies that that Met allele carriers have suboptimally high extracellular
dopamine (because only one of the two genes encoding COMT is present in this syndrome, resulting
in reduced gene dosage), whereas Val carriers are now closer to optimal. SE, standard error; T2–T1
scores, from first (T1) to second (T2) measurement timepoint; TH, tyrosine hydroxylase. Panel a (top left,
bottom) modified, with permission, from REF. 49 © (2003) Society for Neuroscience. Panel a (right)
modified, with permission, from Nature Neuroscience REF. 4 © (2005) Macmillan Publishers Ltd. Panel
b modified, with permission, from Nature Neuroscience REF. 51 © (2005) Macmillan Publishers Ltd.
P E R S P E C T I V E S
824 | OCTOBER 2006 | VOLUME 7 www.nature.com/reviews/neuro
© 2006 Nature Publishing Group

for genetic risk in the context of single gene effects. However, dealing with genetically complex disorders requires going beyond this stage by dealing with multiple interact-ing genetic variants in a gene, between separate genes and gene–environment inter-actions, and with questions of polygenicity versus genetic specificity.
Interacting genetic variants and epistasis. Interacting variants cause specific problems with regard to necessary sample size and mode of inference. An illustrative example is again provided by COMT, in which the evidence supports the existence of multiple variants in the gene. A haplotype combin-ing the Val/Met polymorphism (rs4680) with two common single nucleotide polymorphisms at other loci, one upstream in intron 1 (rs737865) and the other in a 3′ untranslated region (rs165599), was highly associated with schizophrenia in a large sample of Israelis of Ashkenazi descent62. This haplotype differentially affected the expression of rs4680 alleles in human brain tissue63, suggesting the presence of a cis-acting functional locus in COMT that interacts with Val/Met. A population study found that this three marker haplotype is markedly heterogeneous in populations worldwide64, despite the relatively constant prevalence of schizophrenia, and suggested the relevance of another possible cis-acting functional variant (rs2097603) linked upstream in the P2 promoter driving transcription of the predominant form of COMT in the brain (membrane-bound COMT). This variant also affects COMT activity in lymphocytes and post-mortem brain tissue45. So, COMT could contain at least three functional polymorphisms that differentially affect its biological actions and confound its clinical associations. The com-binatorial possibilities of diplotypes based on varying alleles at these three sites are difficult to model in preclinical systems, but imaging offers a unique potential to identify the functional effects of these combinations. Recent work in our laboratory using a method adapted from haplotype regression showed the interacting effects of these func-tional variants on prefrontal function65. The combined effects of these loci are not linear, which is consistent with predictions based on the inverted u-shaped function described above. Confirmatory convergent evidence comes from a study of executive cognition that found similar non-linear effects of these haplotypes on working memory perform-ance66. So, imaging genetics approaches offer strategies for functionating complex
genetic interactions at the level of brain function that might not be approachable with non-human models.
Methodological issues in the characteriza-tion of genetic neural mechanisms. As whole-genome scans of hundreds of thousands of genetic variants have become feasible, the selection of variants to study has become a pressing, and so far unsolved, problem. Few variants are well-defined functional polymorphisms. Indeed, many variants that are statistically associated with psychiatric disease are intronic and of no known functional consequence. Although many are likely to be characterized by advances in predicting novel exon, splice, transcription factor or microRNA binding sites, a translational approach will be essential in functionating them by demonstrating, for example, an effect of genetic variation on mRNA expression, protein levels or cellular physiology. It is our opinion that, given the absence of reliable information on the heritability and reliability of most imaging phenotypes cur-rently in use, a statistically significant result in neuroimaging is by itself not sufficient to establish that a given polymorphism is functional, and the complex nature of psychiatric disease predicts that the isolated genetics evidence for association will usually not be unequivocal for a given variant. This leads to a new kind of multiple comparison problem, this time over the number of studied genetic variants, that will need to be addressed by future research. Similarly, it is important to select neuroim-aging tasks that tap into neural systems plausibly related to the disease under study, such as working memory tasks that activate DLPFC-striatal systems in schizophrenia.
Given the small contribution of each individual genetic variant to complex pheno-types and the importance of gene–gene interactions, it is crucial to control for occult stratification effects that might confound the analysis of a target variant. This applies to demographic variables such as age, gender, IQ or socioeconomic status, and also to an assessment of genetic stratification that is necessary to investigate whether the studied groups, defined by one genotype, systematically differ in the distribution of other genetic variants. Genomic control or ancestral marker panels should be carried out67 to investigate this important source of potential confounders.
Further research is also necessary to determine which imaging designs are the most conducive to genetic research. It is clear that the assessment of genetic effects across participants crucially depends on a reliable and robust brain response. Researchers need to balance this require-ment against the wish to isolate cognitive subcomponents by subtle manipulation in the context of the limited time and budget available for each participant in these high-volume, long-term experiments. It is our view that a two-step approach is often useful and practical. The first line of research establishes genetic effects using well-tested tasks that reliably elicit strong activation in known functional networks, and more specific neural processes are then elucidated in a second step using specifically tailored experiments. Ideally, behavioural confound-ers in these tasks should be avoided by design (simplicity), analysis (for example, by using mixed event-related designs68 allowing for the inclusion of only correct responses) or matching. For the field as a whole, reaching consensus on questions of standardization
Box 2 | Characterization of additional risk genes in schizophrenia
Glutamate is the most important excitatory neurotransmitter in the cortex, and the excitability of glutamate neurons is regulated in part by dopamine35. GRM3, which encodes a metabotropic glutamate receptor responsible for modulating synaptic glutamate, is a candidate gene linked to schizophrenia57. A single nucleotide polymorphism in GRM3 was found to predict prefrontal activation analogous to catechol-O-methyltransferase (COMT)57. This functional convergence of a glutamate and a dopamine gene on a common cortical phenotype illustrates the impact that imaging genetics can have on illuminating core mechanisms behind clinical associations. Recent work in our laboratory has focused on DARPP32, which is expressed in regions receiving dopaminergic innervation, especially the neostriatum (caudate and putamen)90. This protein, a phosphatase encoded by PPP1R1B, acts as a central molecular switch in dopaminoceptive neurons90, integrating dopamine and glutamate signals. DARPP32 is a key node in a final common pathway of psychotomimetics in both the frontal cortex and striatum, making it an attractive candidate gene for schizophrenia90. We recently identified, through the resequencing of the gene in 298 chromosomes, a frequent haplotype in PPP1R1B associated with schizophrenia in a family-based sample91. Imaging genetics showed a pronounced and convergent effect on the structure and activation of the neostriatum, as well as on prefrontal–striatal interactions, indicating that PPP1R1B contributes to risk for schizophrenia by causing disturbed gating26 subsequent to impaired fronto-striatal function.
P E R S P E C T I V E S
NATURE REVIEWS | NEUROSCIENCE VOLUME 7 | OCTOBER 2006 | 825
© 2006 Nature Publishing Group

of tasks and comparability of data across centres69 will also allow more rapid progress towards acquisition of the large sample sizes necessary to investigate genetic variation of small penetrance, and gene–gene and gene–environment interactions.
Even for studies of a single established genetic variant, sample sizes must be con-sidered critically. Most studies published so far report significant effects in groups of around 20–40 participants. However, many of these results have not yet been replicated, and a publication bias against negative results in this newly developing field is likely. Moreover, even for the same genetic mechanism the effect size can vary widely depending on which imaging target measure (structural variation, functional activation or functional connections) is examined6,70. Further research, ideally contrasting genes of no known or likely function with known functional variants, is necessary to achieve a principled assessment of expected false posi-tive rates that can guide recommendations for statistical inference in this field.
Another area of interest is how to com-bine independent streams of information, such as structural and functional neuro-imaging together with clinical and neuro-psychological assessment, into a common analytical framework that can formalize the intuition that convergent information of this kind provides a stronger argument for the functional relevance of the studied genetic variations. It is possible that Bayesian approaches might be a fruitful71, although as yet unexplored in this domain, way to pursue this goal, as in principle they allow the quantitative consideration of sources of uncertainty relevant to the field, such as the prior probability of a given genetic variation or imaging phenotype being causally related to the disorder under study.
Conclusion
Using schizophrenia as an example, we have discussed recent insights gained from a translational approach to investigate and define neural mechanisms of psychiatric illness based on genetic risk. Although methodological problems in this fast-moving area of research exist and must be tackled in the interest of reliable and replicable results, we believe that this methodology of using genetic variation as a tool for the discovery of brain mechanisms will become a widely applied and fruitful research field in psychiatry and related disciplines. As the characterization of single genetic variants is rapidly proceeding, we predict that research will increasingly turn to dissecting gene–gene
and gene–environment interactions, leading to the next crucial step in the field — the identification of converging molecular pathways and their neuronal and systems-level targets, which would then constitute the genetically discovered, biologically valid core pathophysiology of the disorders under study. As these difficult questions posed by interacting genetic variation in complex disease are tackled in the future, we expect that the results mentioned here will then be seen as ‘low-hanging fruit’ harvested at the beginning of an effort that is ultimately expected to not only reform our view of the taxonomy and pathophysiology of psychiat-ric disease, but also to point the way to new treatment targets and more principled clinical management.
Andreas Meyer-Lindenberg is at the Unit for Systems Neuroscience in Psychiatry, Clinical Brain Disorders
Branch, Genes, Cognition and Psychosis Program, National Institute for Mental Health, National Institutes
of Health, Department of Health and Human Services, 9000 Rockville Pike, Bethesda, Maryland 20892, USA.
Daniel R. Weinberger is at the Clinical Brain Disorders Branch, Genes, Cognition and Psychosis Program,
National Institute for Mental Health, National Institutes of Health, Department of Health and Human Services,
9000 Rockville Pike, Bethesda, Maryland 20892, USA.
Correspondence to A.M.-L. or D.R.W. e-mails: [email protected]; [email protected]
doi:10:1938/nrn1993
1. Sullivan, P. F., Kendler, K. S. & Neale, M. C. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 60, 1187–1192 (2003).
2. Sullivan, P. F., Neale, M. C. & Kendler, K. S. Genetic epidemiology of major depression: review and meta-analysis. Am. J. Psychiatry 157, 1552–1562 (2000).
3. Weinberger, D. R. et al. Prefrontal neurons and the genetics of schizophrenia. Biol. Psychiatry 50, 825–844 (2001).
4. Meyer-Lindenberg, A. et al. Midbrain dopamine and prefrontal function in humans: interaction and modulation by COMT genotype. Nature Neurosci. 8, 594–596 (2005).
5. Meyer-Lindenberg, A. et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nature Neurosci. 5, 267–271 (2002).
6. Pezawas, L. et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nature Neurosci. 8, 828–834 (2005).
7. Waldman, I. D. Statistical approaches to complex phenotypes: evaluating neuropsychological endophenotypes for attention-deficit/hyperactivity disorder. Biol. Psychiatry 57, 1347–1356 (2005).
8. Goldman, D., Oroszi, G. & Ducci, F. The genetics of addictions: uncovering the genes. Nature Rev. Genet. 6, 521–532 (2005).
9. Belmonte, M. K. et al. Autism as a disorder of neural information processing: directions for research and targets for therapy. Mol. Psychiatry 9, 646–663 (2004).
10. Riley, B. P. & McGuffin, P. Linkage and associated studies of schizophrenia. Am. J. Med. Genet. 97, 23–44 (2000).
11. Menzel, S. Genetic and molecular analyses of complex metabolic disorders: genetic linkage. Ann. NY Acad. Sci. 967, 249–257 (2002).
12. Harrison, P. J. & Weinberger, D. R. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 10, 40–68 (2005).
13. Risch, N. Genetic linkage and complex diseases, with special reference to psychiatric disorders. Genet. Epidemiol. 7, 3–16; discussion 17–45 (1990).
14. Gottesman, I. I. & Shields, J. A polygenic theory of schizophrenia. Proc. Natl Acad. Sci. USA 58, 199–205 (1967).
15. Page, G. P., George, V., Go, R. C., Page, P. Z. & Allison, D. B. ‘Are we there yet?’: Deciding when one has demonstrated specific genetic causation in complex diseases and quantitative traits. Am. J. Hum. Genet. 73, 711–719 (2003).
16. Deschepper, C. F., Boutin-Ganache, I., Zahabi, A. & Jiang, Z. In search of cardiovascular candidate genes: interactions between phenotypes and genotypes. Hypertension 39, 332–336 (2002).
17. Gottesman, I. I. & Gould, T. D. The endophenotype concept in psychiatry: etymology and strategic intentions. Am. J. Psychiatry 160, 636–645 (2003).
18. Almasy, L. & Blangero, J. Endophenotypes as quantitative risk factors for psychiatric disease: rationale and study design. Am. J. Med. Genet. 105, 42–44 (2001).
19. Weinberger, D. R. Schizophrenia: new phenes and new genes. Biol. Psychiatry 46, 3–7 (1999).
20. Alexander, G. E., DeLong, M. R. & Strick, P. L. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu. Rev. Neurosci. 9, 357–381 (1986).
21. Goldman-Rakic, P. S., Selemon, L. D. & Schwartz, M. L. Dual pathways connecting the dorsolateral prefrontal cortex with the hippocampal formation and parahippocampal cortex in the rhesus monkey. Neuroscience 12, 719–743 (1984).
22. Friston, K. J. The disconnection hypothesis. Schizophr. Res. 30, 115–125 (1998).
23. Meyer-Lindenberg, A. S. et al. Regionally specific disturbance of dorsolateral prefrontal-hippocampal functional connectivity in schizophrenia. Arch. Gen. Psychiatry 62, 379–386 (2005).
24. Dunnett, S. B., Meldrum, A. & Muir, J. L. Frontal-striatal disconnection disrupts cognitive performance of the frontal-type in the rat. Neuroscience 135, 1055–1065 (2005).
25. Pantelis, C. et al. Frontal-striatal cognitive deficits in patients with chronic schizophrenia. Brain 120, 1823–1843 (1997).
26. Swerdlow, N. R., Geyer, M. A. & Braff, D. L. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl.) 156, 194–215 (2001).
27. Carlsson, A. A paradigm shift in brain research. Science 294, 1021–1024 (2001).
28. Jaskiw, G. E., Karoum, F. K. & Weinberger, D. R. Persistent elevations in dopamine and its metabolites in the nucleus accumbens after mild subchronic stress in rats with ibotenic acid lesions of the medial prefrontal cortex. Brain Res. 534, 321–323 (1990).
29. Williams, G. V. & Goldman-Rakic, P. S. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature 376, 572–575 (1995).
30. Schultz, W. Predictive reward signal of dopamine neurons. J. Neurophysiol. 80, 1–27 (1998).
31. Squire, L. R., Stark, C. E. & Clark, R. E. The medial temporal lobe. Annu. Rev. Neurosci. 27, 279–306 (2004).
32. Harrison, P. J. & Eastwood, S. L. Neuropathological studies of synaptic connectivity in the hippocampal formation in schizophrenia. Hippocampus 11, 508–519 (2001).
33. Heckers, S. Neuroimaging studies of the hippocampus in schizophrenia. Hippocampus 11, 520–528 (2001).
34. Heckers, S. et al. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nature Neurosci. 1, 318–323 (1998).
35. Simons, J. S. & Spiers, H. J. Prefrontal and medial temporal lobe interactions in long-term memory. Nature Rev. Neurosci. 4, 637–648 (2003).
36. Phillips, M. L., Drevets, W. C., Rauch, S. L. & Lane, R. Neurobiology of emotion perception I: the neural basis of normal emotion perception. Biol. Psychiatry 54, 504–514 (2003).
37. Weinberger, D. R., Berman, K. F., Suddath, R. & Torrey, E. F. Evidence of dysfunction of a prefrontal-limbic network in schizophrenia: a magnetic resonance imaging and regional cerebral blood flow study of discordant monozygotic twins. Am. J. Psychiatry 149, 890–897 (1992).
38. Bertolino, A. et al. Altered development of prefrontal neurons in rhesus monkeys with neonatal mesial temporo-limbic lesions: a proton magnetic resonance
P E R S P E C T I V E S
826 | OCTOBER 2006 | VOLUME 7 www.nature.com/reviews/neuro
© 2006 Nature Publishing Group

spectroscopic imaging study. Cereb. Cortex 7, 740–748 (1997).
39. Weinberger, D. R. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 44, 660–669 (1987).
40. Lewis, D. A. et al. Dopamine transporter immunoreactivity in monkey cerebral cortex: regional, laminar, and ultrastructural localization. J. Comp. Neurol. 432, 119–136 (2001).
41. Tunbridge, E. M., Bannerman, D. M., Sharp, T. & Harrison, P. J. Catechol-o-methyltransferase inhibition improves set-shifting performance and elevates stimulated dopamine release in the rat prefrontal cortex. J. Neurosci. 24, 5331–5335 (2004).
42. Tunbridge, E. M., Harrison, P. J. & Weinberger, D. R. Catechol-O-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol. Psychiatry 60, 141–151 (2006).
43. Owen, M. J., Williams, N. M. & O’Donovan, M. C. The molecular genetics of schizophrenia: new findings promise new insights. Mol. Psychiatry 9, 14–27 (2004).
44. Murphy, K. C. Schizophrenia and velo-cardio-facial syndrome. Lancet 359, 426–430 (2002).
45. Chen, J. et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am. J. Hum. Genet. 75, 807–821 (2004).
46. Egan, M. F. et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc. Natl Acad. Sci. USA 98, 6917–6922 (2001).
47. Goldberg, T. E. et al. Executive subprocesses in working memory: relationship to catechol-O-methyltransferase Val158Met genotype and schizophrenia. Arch. Gen. Psychiatry 60, 889–896 (2003).
48. Mattay, V. S. et al. Catechol O-methyltransferase val158-met genotype and individual variation in the brain response to amphetamine. Proc. Natl Acad. Sci. USA 100, 6186–6191 (2003).
49. Akil, M. et al. Catechol-O-methyltransferase genotype and dopamine regulation in the human brain. J. Neurosci. 23, 2008–2013 (2003).
50. Callicott, J. H. et al. Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc. Natl Acad. Sci. USA 102, 8627–8632 (2005).
51. Gothelf, D. et al. COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nature Neurosci. 8, 1500–1502 (2005).
52. Craddock, N., Owen, M. J. & O’Donovan, M. C. The catechol-O-methyl transferase (COMT) gene as a candidate for psychiatric phenotypes: evidence and lessons. Mol. Psychiatry 11, 446–458 (2006).
53. Fan, J. B. et al. Catechol-O-methyltransferase gene Val/Met functional polymorphism and risk of schizophrenia: a large-scale association study plus meta-analysis. Biol. Psychiatry 57, 139–144 (2005).
54. Munafo, M. R., Bowes, L., Clark, T. G. & Flint, J. Lack of association of the COMT (Val158/108 Met) gene and schizophrenia: a meta-analysis of case-control studies. Mol. Psychiatry 10, 765–770 (2005).
55. Schott, B. H. et al. The dopaminergic midbrain participates in human episodic memory formation: evidence from genetic imaging. J. Neurosci. 26, 1407–1417 (2006).
56. Cohen, M. X., Young, J., Baek, J. M., Kessler, C. & Ranganath, C. Individual differences in extraversion and dopamine genetics predict neural reward responses. Brain Res. Cogn Brain Res. 25, 851–861 (2005).
57. Egan, M. F. et al. Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc. Natl Acad. Sci. USA 101, 12604–12609 (2004).
58. de Quervain, D. J. & Papassotiropoulos, A. Identification of a genetic cluster influencing memory performance and hippocampal activity in humans. Proc. Natl Acad. Sci. USA 103, 4270–4274 (2006).
59. Callicott, J. H. et al. Abnormal fMRI response of the dorsolateral prefrontal cortex in cognitively intact siblings of patients with schizophrenia. Am. J. Psychiatry 160, 709–719 (2003).
60. Cannon, T. D. et al. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch. Gen. Psychiatry 62, 1205–1213 (2005).
61. Bertolino, A. et al. Prefrontal-hippocampal coupling during declarative memory is modulated by COMT Val158Met genotype. Biol. Psychiatry 4 Sept 2006 (doi:10.1016/j.biopsych.2006.03.078).
62. Shifman, S. et al. A highly significant association between a COMT haplotype and schizophrenia. Am. J. Hum. Genet. 71, 1296–1302 (2002).
63. Bray, N. J. et al. A haplotype implicated in schizophrenia susceptibility is associated with reduced COMT expression in human brain. Am. J. Hum. Genet. 73, 152–161 (2003).
64. Palmatier, M. A. et al. COMT haplotypes suggest P2 promoter region relevance for schizophrenia. Mol. Psychiatry 9, 859–870 (2004).
65. Meyer-Lindenberg, A. et al. Impact of complex genetic variation in COMT on human brain function. Mol. Psychiatry 20 June 2006 (doi:10.1038/sj.mp.4001860).
66. Diaz-Asper, C. M. Weinberger, D. R. & Goldberg, T. E. Catechol-O-methyltransferase polymorphisms and some implications for cognitive therapeutics. NeuroRX 3, 97–105 (2006).
67. Devlin, B., Roeder, K. & Wasserman, L. Genomic control, a new approach to genetic-based association studies. Theor. Popul. Biol. 60, 155–166 (2001).
68. Laurienti, P. J., Burdette, J. H. & Maldjian, J. A. Separating neural processes using mixed event-related and epoch-based fMRI paradigms. J. Neurosci. Methods 131, 41–50 (2003).
69. Friedman, L. & Glover, G. H. Report on a multicenter fMRI quality assurance protocol. J. Magn. Reson. Imaging 23, 827–839 (2006).
70. Hariri, A. R. et al. Serotonin transporter genetic variation and the response of the human amygdala. Science 297, 400–403 (2002).
71. Friston, K. J. et al. Classical and Bayesian inference in neuroimaging: theory. Neuroimage 16, 465–483 (2002).
72. Manji, H. K., Drevets, W. C. & Charney, D. S. The cellular neurobiology of depression. Nature Med. 7, 541–547 (2001).
73. Mayberg, H. S. Limbic-cortical dysregulation: a proposed model of depression. J. Neuropsychiatry Clin. Neurosci. 9, 471–481 (1997).
74. Drevets, W. C. et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386, 824–827 (1997).
75. Mayberg, H. S. et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am. J. Psychiatry 156, 675–682 (1999).
76. Paus, T. Primate anterior cingulate cortex: where motor control, drive and cognition interface. Nature Rev. Neurosci. 2, 417–424 (2001).
77. Stefanacci, L. & Amaral, D. G. Some observations on cortical inputs to the macaque monkey amygdala: an anterograde tracing study. J. Comp. Neurol. 451, 301–323 (2002).
78. Sotres-Bayon, F., Bush, D. E. & LeDoux, J. E. Emotional perseveration: an update on prefrontal-amygdala interactions in fear extinction. Learn. Mem. 11, 525–535 (2004).
79. Nemeroff, C. B. & Owens, M. J. Treatment of mood disorders. Nature Neurosci. 5, S1068–S1070 (2002).
80. Lesch, K. P. et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 274, 1527–1531 (1996).
81. Caspi, A. et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301, 386–389 (2003).
82. Hariri, A. R. et al. A susceptibility gene for affective disorders and the response of the human amygdala. Arch. Gen. Psychiatry 62, 146–152 (2005).
83. Heinz, A. et al. Amygdala-prefrontal coupling depends on a genetic variation of the serotonin transporter. Nature Neurosci. 8, 20–21 (2005).
84. Brown, S. M. et al. A regulatory variant of the human tryptophan hydroxylase-2 gene biases amygdala reactivity. Mol. Psychiatry 10, 884–888, 805 (2005).
85. Canli, T., Congdon, E., Gutknecht, L., Constable, R. T. & Lesch, K. P. Amygdala responsiveness is modulated by tryptophan hydroxylase-2 gene variation. J. Neural Transm. 112, 1479–1485 (2005).
86. Sabol, S. Z., Hu, S. & Hamer, D. A functional polymorphism in the monoamine oxidase A gene promoter. Hum. Genet. 103, 273–279 (1998).
87. Caspi, A. et al. Role of genotype in the cycle of violence in maltreated children. Science 297, 851–854 (2002).
88. Meyer-Lindenberg, A. et al. Neural mechanisms of genetic risk for impulsivity and violence in humans. Proc. Natl Acad. Sci. USA 103, 6269–6274 (2006).
89. Fan, J., Fossella, J., Sommer, T., Wu, Y. & Posner, M. I. Mapping the genetic variation of executive attention onto brain activity. Proc. Natl Acad. Sci. USA 100, 7406–7411 (2003).
90. Svenningsson, P. et al. DARPP-32: an integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 44, 269–296 (2004).
91. Weinberger, D. R. et al. Variation in PPP1R1B predicts risk for schizophrenia, cognitive function, and gene expression in brain. Am. J. Med. Genet. B Neuropsychiatr. Genet. 138B, 139–140 (2005).
92. Meyer-Lindenberg, A. et al. Neural mechanisms of genetic risk for impulsivity and violence in humans. Proc. Natl Acad. Sci. USA 103, 6269–6274 (2006).
93. Freedman, R. et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc. Natl Acad. Sci. USA 94, 587–592 (1997).
94. Goldman-Rakic, P. S., Muly, E. C. & Williams, G. V. D1 receptors in prefrontal cells and circuits. Brain Res. Brain Res. Rev. 31, 295–301 (2000).
95. Seamans, J. K., Gorelova, N., Durstewitz, D. & Yang, C. R. Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J. Neurosci. 21, 3628–3638 (2001).
AcknowledgementsThis work was supported by the National Institute of Mental Health’s Intramural Research Program. We thank C. Rainey for help with figure preparation.
Competing interests statementThe authors declare no competing financial interests.
DATABASESThe following terms in this article are linked online to:Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.
fcgi?db=gene
COMT | DISC1 | GRM3 | PPP1R1B
OMIM: http://www.ncbi.nlm.nih.gov/entrez/query.
fcgi?db=OMIM
22q11.2 syndrome | attention deficit hyperactivity disorder |
autism | major depression | schizophrenia
FURTHER INFORMATIONUnit for Systems Neuroscience in Psychiatry: http://snp.nimh.nih.gov/
Access to this links box is available online.
ONLINE CORRESPONDENCE Nature Reviews Neuroscience publishes items of correspondence online. Such contributions are published at the discretion of the Editors and can be subject to peer review. Correspondence should be no longer than 500 words with up to 15 references and should represent a scholarly attempt to comment on a specific Review or Perspective article that has been published in the journal. To view correspondence, please go to our homepage at: http://www.nature.com/reviews/neuro and follow the link from the Review article by Hidehiko Komatsu.
The following correspondence has recently been published:
What gets filled-in during filling-in?Frans W. Cornelissen and Tony Vladusich
This correspondence relates to the article:
The neural mechanisms of perceptual filling-in Hidehiko Komatsu Nature Reviews Neuroscience 7, 220–231 (2006)
P E R S P E C T I V E S
NATURE REVIEWS | NEUROSCIENCE VOLUME 7 | OCTOBER 2006 | 827
© 2006 Nature Publishing Group





























