Embed Size (px)
Citation preview

Interactive tools and programming environments
for sequence analysis
Bernardo Barbiellini
Northeastern University
TATACATAAAGACCCAAATGGAACTGTTCTAGATGATACACTAGCATTAAGAGAAAAATTCGAAGAATCAGTCGATAAATACAAACTTCATTTTACTGGATTAATCGCTGACAAAATTGCAAAAGAAAAACTGAATACTTACGTCCTCACTTATAAAAAAGCAGACGAAGCTATGCCTGCAGACGAAGCTATGCCAACTGATGTACCTAGTACTTCTGTTACTGGATCAACAATGGCAAAC………………….

Overview
• Matlab and Darwin – bioinformatics tools
• Dotplot and Statistical signifance of alignments
• Scoring Matrices from Evolution Model
• Evolutionary Distances and Phylogenetic Trees.
• Unified approach for the sequence alignment and structure prediction

Matlab toolbox and Darwin• Computer language appropriate for bioinformatics
• A workbench to automate repetitive tasks
• Based on Linear Algebra & Statistics
• Matlab toolbox developed by Mathworks
• Darwin developed by Gaston Gonnet (ETHZ)

Extra features
• Loading of and retrieval in sequence databases• Fast searching for sequence fragments• Sequence alignment• Generation of random sequences, distributions and
mutations• Creation of Phylogenetic trees• Plotting functions - matrix and vector arithmetic• I/O comunicate with other programs

Calling Bioperl functions in MATLAB Documentation by Brian Madsen (NU and coop at the Mathworks)
>> help perl
PERL calls perl script using appropriate operating system PERL(PERLFILE) calls perl script specified by the file PERLFILE using appropriate perl executable.
PERL(PERLFILE,ARG1,ARG2,...) passes the arguments ARG1,ARG2,... to the perl script file PERLFILE, and calls it by using appropriate perl executable. RESULT=PERL(...) outputs the result of attempted perl call.

Visual Tool: Dotplot (1)
Pairwise sequence comparison

Visual Tool: Dotplot (2)
Filtered Image
The best alignment is achieved with dynamic programming . A score is obtained

Quantitative Tools To CheckStatistical Significance
Simulation with random sequences
Score in bits
extreme value distribution.

PAM Evolution Model
PAM means Accepted Point Mutation
The score of a paiwise alignment is obtained by using a scoring matrix.
We need a model to build scoring matrices.
This model is based on evolution in order to calculate evolution distances between species.

Step1: Order of the Amino-Acids

Step 2: Mutation Matrices
Markov Model pamX=(pam1)^X Stochastic matrices

Step 3: Distribution of Amino Acids
Eigenvector of the mutation matrix (eigenvalue 1)

Step 4: Evolutionary time vs. sequences differences

Step 5: Scoring Matrix
The Dayhoff scoring matrix is symmetric
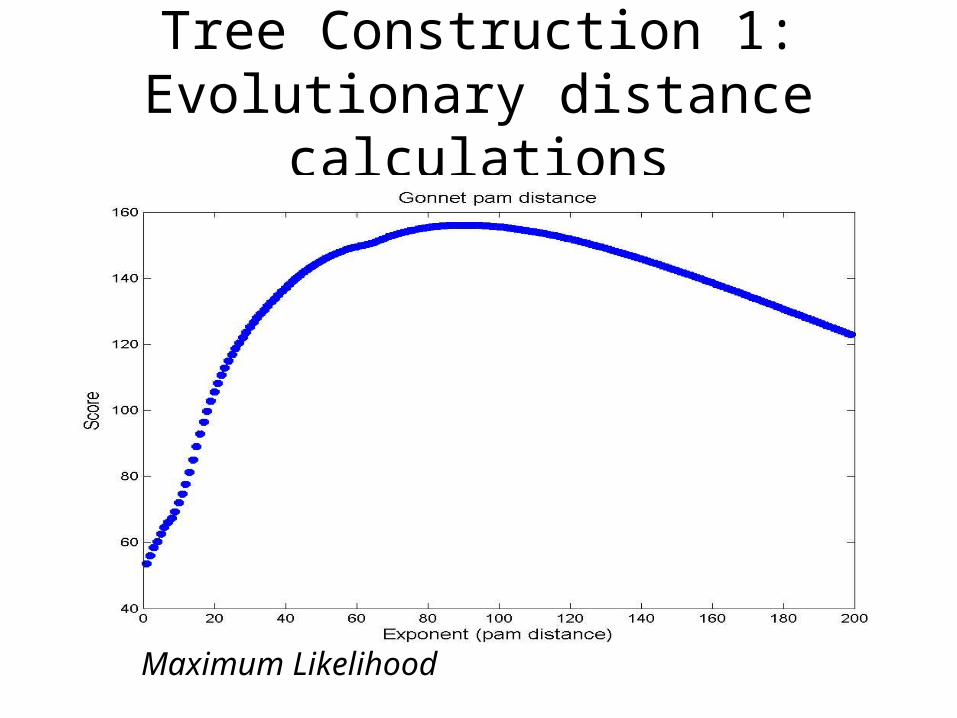
Tree Construction 1:Evolutionary distance calculations
Maximum Likelihood

Tree Construction 2:Table of distances
PAM Spinach Rice Mosquito Monkey Human
Spinach 0.0 84.9 105.6 90.8 86.3
Rice 84.9 0.0 117.8 122.4 122.6
Mosquito 105.6 117.8 0.0 84.7 80.8
Monkey 90.8 122.4 84.7 0.0 3.3
Human 86.3 122.6 80.8 3.3 0.0
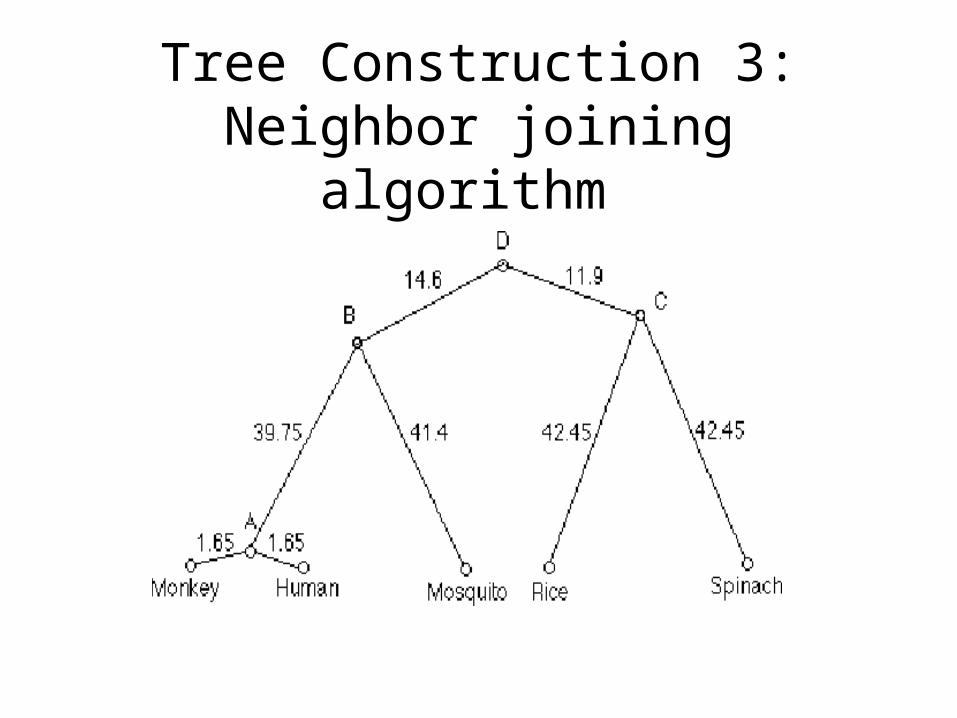
Tree Construction 3:Neighbor joining algorithm

Unified approach for the sequence alignment and structure prediction
Protein
Protein
Protein
Optimizationwith DynamicProgramming approach
Needleman-Wunsch Algorithm
orSmith-Waterman
Algorithm
Query
Subject Protein (letter of amino acids)
Scoring Matrix
Log (Aij/pi)
Penalties Gaps
Protein
Structure
Viterbi AlgorithmHMM
Protein
Structure (, , coil)
Log (P(im)/pi)
Transition from structure to another

Conclusions
• The highly efficient dynamic programming algorithms, used in this integrated environment, are particularly suitable for the high performance computers.
• Trees constructed using optimal PAM distances are better than the routinesingle distance scores obtained using a single scoring matrix.
• The unified approach for the sequence alignment and structure prediction provides a powerful formalism for biologists.

ASCC Northeastern University

Northeastern University (NU)/Hewlett-Packard (HP) Company Collaborative Research Program on Bioinformatics
Bernardo Barbiellini, Assoc. Director, ASCC
Arun Bansil, Professor of Physics & Director ASCC.
Bill Detrich, Prof. Biochem. & Marine Biology, Director Bioinformatics M. S.
Kostia Bergman, Prof. Biology
Mike Malioutov, Stone Professor of Applied Statistics
Mary Jo Ondrechen, Professor of Chemistry
Nagarajan Sankrithi, graduate student NU
Imtiaz Khan, graduate student NU
Alper Uzun, graduate student NU
Larry Weissman, staff HP/Compaq
Barry Latham, staff HP/Compaq
Bob Morgan, staff HP/Compaq

Other Bioinformatics activities at ASCC
• BIO3580: DNA and Protein Sequence Analysis (2001, 2002)
• MATLAB BIOINFORMATICS TOOL presentation (Robert Henson)
• Summer Institute of Mathematical Studies on Bioinformatics (2002) (Professor Mike Malioutov)
• Student projects proposed by Dr. Matteo Pellegrini, (Proteinpathways/UCLA).