Embed Size (px)
Citation preview

Naunyn-Schmiedeberg's Arch Pharmacol (1984) 328:69-75 Naunyn-Schrniedeberg's
Archivesof Pharmacology �9 Springer-Verlag 1984
Interactions between a "calcium channel agonist", Bay K 8644, and calcium antagonists differentiate calcium antagonist subgroups in K +- depolarized smooth muscle Michael Spedding and Christiane Berg Merrell Dow Research Institute, Strasbourg Center, 16 rue d'Ankara, F-67084 Strasbourg Cedex, France
Summary. 1. The proposal that calcium antagonists have different sites of action has been tested by attempting to reverse their inhibitory effects with a dihydropyridine which augments Ca 2+ entry into cells, Bay K 8644.
2. Bay K 8644 (1-1000 nmol/1) increased the sensitivity to Ca 2+ of K+-depolarized taenia preparations from the guinea-pig caecum. Thus Bay K 8644 augmented estab- lished submaximal Ca2+-induced contractions and also shifted cumulative concentration-response curves to Ca 2+ to the left, even in the presence of an optimal K +- depolarization.
3. The inhibitory effects of nifedipine (10nmol/1), verapamil (0.2 ~tmol/1), diltiazem (1 ~tmol/1) and diclo- furime (1 ~tmol/1) on Ca 2+ - induced contractions were reversed by Bay K 8644 (1-100 nmol/1). In contrast, Bay K 8644 did not reverse the effects of cinnarizine (1 ~tmol/1), flunarizine (1 ~tmol/1), fendiline (3 ~tmol/1), prenylamine (3~tmol/1), pimozide (l~tmol/1), bepridil (3~tmol/1), perhexiline (10 ~tmol/1) or the calmodulin antagonist W-7 (200 ~tmol/1).
4. Bay K8644 (1-100nmol/1) was less effective at reversing the effects of nisoldipine (10 nmol/1), a slowly dissociating dihydropyridine, than the effects of nifedipine. However, preincubation with Bay K 8644 (1 ~tmol/1) pro- tected the taenia from the inhibitory effects of nisoldipine (10 nmol/1). These findings are compatible with interac- tions of nisoldipine and Bay K 8644 at a common site.
5. Taenia preparations incubated with Bay K8644 (1 ~tmol/1) were protected from the inhibitory effects of nifedipine (10 nmol/1), nisoldipine (10 nmol/1) and to a lesser extent verapamil (0.2 ~tmol/1) and diltiazem (1 ~tmol/1). However, there was no protection against the inhibitory effects of cinnarizine (1 ~tmol/1), flunarizine (l~tmol/1), pimozide (1 ~tmol/1), fendiline (3~tmol/1), prenylamine (3 ~tmol/1) or W-7 (200 ~tmol/1). Thus, the lack of protection indicates that there is no interaction between the dihydropyridine site and the site for the latter drugs. These experiments show that Bay K 8644 defines subgroups of calcium antagonists which are the same as those previously reported from functional studies.
Key words: Dihydropyridines - Calcium - Calcium anta- gonists - Verapamil - Diltiazem
Send offprint requests to M. Spedding at the above address
Introduction
The diversity of the drugs classed as calcium antagonists is so great that this group may form the most chemically disparate class of therapeutic agents (Kazda et al. 1983). It is evident that a single molecular site could not accept such diverse compounds and therefore drugs inhibiting Ca ~+ slow channels may act at several site either in the channel or intracellularly. Several workers have proposed that different subclasses of calcium antagonist may exist, depending on their site of action (Ferry and Glossmann 1982; Glossmann et al. 1982) or on functional differences between the compounds (Fleckenstein 1981, Spedding 1981, 1982a, b; 1983a, b; 1984a and b; Rodeukirchen et al. 1982; Bou et al. 1983); the various subclassifications are listed in Table 1. Generally there appear to be three major classes of drugs: a) dihydropyridines which bind to a site in the channel, b) drugs such as diltiazem and verapamil which can positively or negatively regulate the dihydropyridine site by an allosteric mechanism and c) diphenylalkylamines which can be readily differentiated from the other agents in functional tests (Spedding 1984b).
However, a unitary model for calcium antagonists has recently been proposed, where the dihydropyridine binding site, associated with a selective cation binding site, is allosterically modified by a site which is common to verapamil, diltiazem and the diphenylalkylamines (Gould et al. 1983; Murphy et al. 1983). Thus in this model the drugs would only act at two sites and Murphy et al. (1983) demonstrated modulation of the inhibitory effects of diphenylalkylamines on dihydropyridine binding by dil- tiazem and by a verapamil derivative, D 600. However, these important experiments were performed in guinea-pig brain membranes: there is at present no functional cor- relate to this binding site and it cannot clearly be ascribed to the Ca ~+ channel (Triggle 1984).
Functional consequences of calcium antagonist inter- actions are not striking in smooth muscle (Spedding, 1983c), but the recent availability of a dihydropyridine which stimulates Ca 2+ entry via Ca 2+ channels and thus behaves as a "calcium channel agonist" (Bay K8644, Schramm et al. 1983 a and b) should allow more striking functional interactions. Bay K8644 has been shown to bind to the dihydropyridine binding site (Glossmann et al. 1983; Sarmiento et al. 1984) arid to reverse competitively the effects of the dihydropyridine calcium antagonist nife-
70
dipine (Schramm etat. 1983b; Schramm and Towart 1984).
This report describes reversal of the effects of some, but not all, calcium antagonists by Bay K 8644. The results support the hypothesis that there are distinct subgroups of calcium antagonists. K+-depolarized taenia preparations from the guinea-pig caecum were used as these prepara- tions are very sensitive to Ca 2+ and competitive kinetics can be used to describe the interactions between anta- gonists and Ca 2+ (Riemer et al. 1974; Spedding 1982 a).
Methods
Strips of taenia ( 1 - 2 m m diameter, 1 .5-2cm relaxed length) from the caecum of male guinea-pigs (200-350 g) were set up in 10 ml isolated organ baths containing K +- Tyrode solution which was gassed with 95% 05:5% CO2 and maintained at 35 ~ The K+-Tyrode solution had the following composition (mmol/1): NaC1 97; KC140; NaHCO3 11.9; NaH2PO4 0.4; glucose 5.5. In some experi- ments the concentration of KC1 was changed with the appropriate change of NaC1 to keep the molarity constant. The preparations were washed in the Ca 2 +-free, 40 mmol/1 K+-Tyrode solution for 30 rain by which time they were fully relaxed. Contractile responses were measured under isotonic conditions (lg load) using Harvard or Bioscience isotonic transducers connected to Rikadenki potentio- metric recorders. Cumulative concentration-response curves to Ca 2+ (30-3000 ~tmol/1) were obtained by in- creasing the Ca 2+ concentration at 3-5 min intervals in logarithmic increments (Van Rossum 1963). When con- centration-response curves were repeated the curves were obtained at 40 rain intervals. Antagonists were prein- cubated for 25 rain. Concentration ratios were calculated as the ratios of the concentrations of Ca 2+ which produced a 50% maximal response (EC50) in the presence and in the absence of the antagonists. The slopes were calculated from the concentrations producing 80% and 20% maximal
effects (Stephenson 1956). Student's t-test was used for comparisons of mean values and the Mann-Whitney U test for comparison of % changes. Concentration ratios were calculated, and tested for statistical differences, in logarithmic units and the antilog,arithms are quoted in the test. SEMs are illustrated on percentage changes for representational purposes only. All concentrations refer to final bath concentrations of drugs.
Drugs. The following drugs were used: Bay K 8644, nife- dipine, nisoldipine (Bayer AG, Leverkusen, FRG), cinna- rizine tartrate, flunarizine hydrochloride, pimozide tartrate (Janssen, Dtisseldorf FRG), bepridil (Organon, France), dil- tiazem hydrochloride (Synthelabo, France), fendiline hydro- chloride (Dr. Thiemann GmbH), prenylamine (Segontin Hoechst AG), perhexiline hydrochloride (Men'ell Dow, USA), (__)-verapamil hydrochloride (Knoll AG, Ludwigshafen, FRG) N-(6-aminohexyl)-5-chloro- 1-naphthalenesulphon- amide (W-7) (Synthelabo, France). Dihydropyridines were dissolved in ethanol (1 retool/l), prior to diluting in distilled water. Stock solutions were kept at 0 ~ for up to 12 h and aqueous dilutions prepared immediately prior to use. All solutions were shielded from light. The other drugs were dissolved in distilled water.
Results
Effects of Bay K 8644
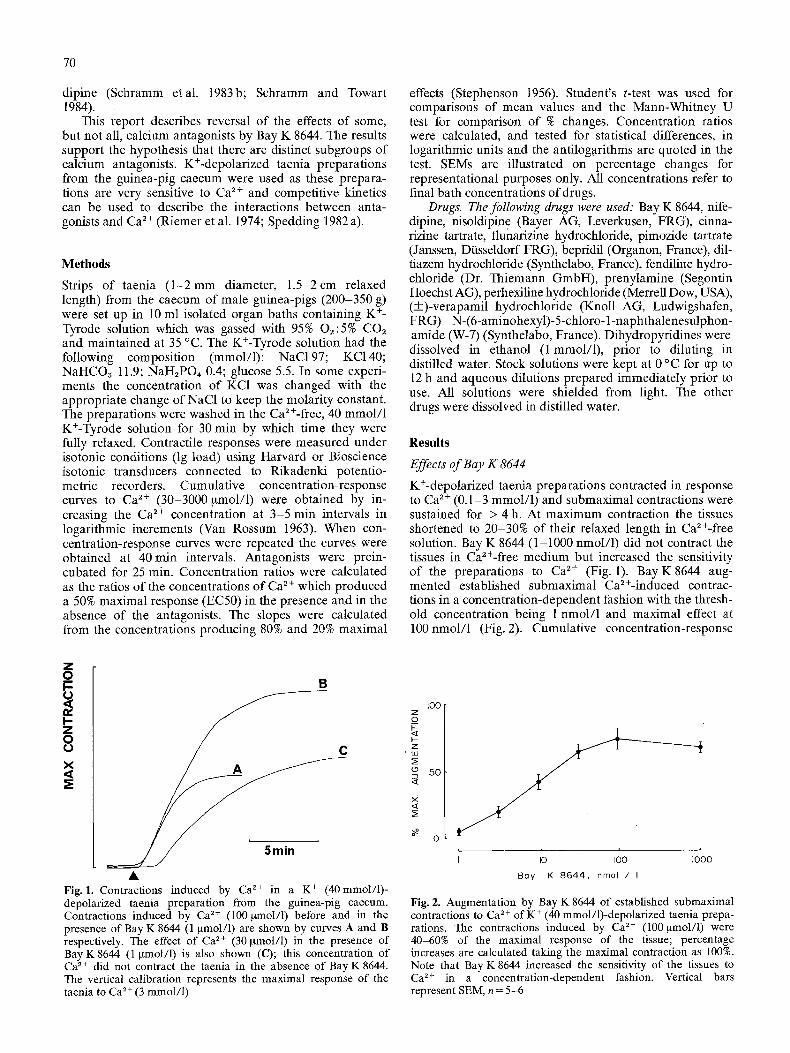
K+-depolarized taenia preparations contracted in response to Ca 2+ (0.1-3 mmol/1) and submaximal contractions were sustained for > 4 h. At maximum contraction the tissues shortened to 20-30% of their relaxed length in Ca2+-free solution. Bay K 8644 (1-1000 nmol/1) did not contract the tissues in Ca2+-free medium but increased the sensitivity of the preparations to Ca 2+ (Fig. 1). Bay K8644 aug- mented established submaximal Ca2+-induced contrac- tions in a concentration-dependent fashion with the thresh- old concentration being 1 nmol/1 and maximal effect at 100nmol/1 (Fig. 2). Cumulative concentration-response
8
B
A C
Fig. 1. Contractions induced by Ca ~+ in a K + (40mmol/1)- depolarized taenia preparation from the guinea-pig caecum. Contractions induced by Ca 2+ (100 ~nol/1) before and in the presence of Bay K 8644 (1 gmol/1) are shown by curves A and B respectively. The effect of Ca ~+ (30 ~tmol/1) in the presence of Bay K8644 (1 Nnol/l) is also shown (C); this concentration of Ca 2+ did not contract the taenia in the absence of Bay K 8644. The vertical calibration represents the maximal response of the taenia to Ca 2+ (3 mmol/l)
z _o
I-- Z
' Ld
[00
5 0
I I 0 0 0 I0 I O0
B e y K 8 6 4 4 , nm01 /
Fig. 2. Augmentation by Bay K 8644 of estabhshed submaximal contractions to Ca 2+ of K + (40 mmol/1)-depolarized taenia prepa- rations. 1he contractions induced by Ca 2+ (t00 ~tmol/1) were 40-60% of the maximal response of the tissue; percentage increases are calculated taking the maximal contraction as 100%. Note that Bay K 8644 increased the sensitivity of the tissues to Ca 2+ in a concentration-dependent fashion. Vertical bars represent SEM, n = 5-6
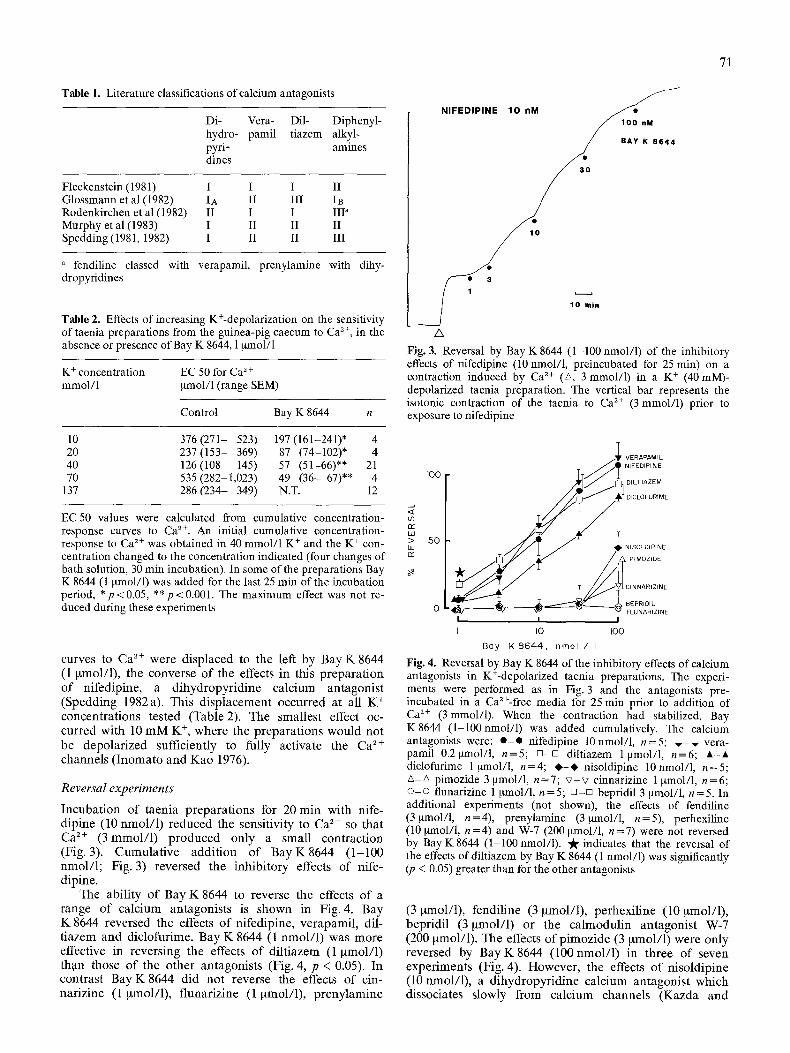
Table 1. Literature classifications of calcium antagonists
Di- Vera- Dil- hydro- pamil tiazem pyri- dines
Diphenyl- alkyl- ammes
Fleckenstein (1981) I I I II Glossmann et al (1982) IA II III IB Rodenkirchen et al (1982) II I I III a Murphy et al (1983) I II II II Spedding (1981, 1982) I II II III
a fendiline classed with verapamil, prenylamine with dihy- dropyridines
Table 2. Effects of increasing K+-depolarization on the sensitivity of taenia preparations from the guinea-pig caecum to Ca z+, in the absence or presence of Bay K 8644, 1 btmol/1
K + concentration EC 50 for Ca 2 + retool/1 btmol/1 (range SEM)
Control Bay K 8644 n
10 376 (271- 523) 197 (161-241)* 4 20 237(153- 369) 87 (74-102)* 4 40 126 (108- 145) 57 (51-66)** 21 70 535 (282-1,023) 49 (36- 67)** 4
137 286 (234- 349) N.T. 12
EC 50 values were calculated from cumulative concentration- response curves to Ca 2+. An initial cumulative concentration- response to Ca 2+ was obtained in 40 mmol/1 K + and the K + con- centration changed to the concentration indicated (four changes of bath solution, 30 min incubation). In some of the preparations Bay K 8644 (1 ~tmol/1) was added for the last 25 min of the incubation period, *p<0.05, **p<0.001. The maximum effect was not re- duced during these experiments
curves to Ca 2+ were displaced to the left by Bay K 8644 (1 btmol/1), the converse of the effects in this preparation of nifedipine, a dihydropyridine calcium antagonist (Spedding 1982a). This displacement occurred at all K + concentrations tested (Table 2). The smallest effect oc- curred with 10 m M K +, where the preparations would not be depolarized sufficiently to fully activate the Ca ~+ channels ( Inomato and Kao 1976).
Reversal experiments
Incubation of taenia preparations for 20 min with nife- dipine (10 nmol/1) reduced the sensitivity to Ca 2+ so that Ca ~+ (3retool/I) produced only a small contraction (Fig. 3). Cumulative addition of Bay K8644 (1-100 nmol/1; Fig. 3) reversed the inhibitory effects of nife- dipine.
The ability of Bay K 8644 to reverse the effects of a range of calcium antagonists is shown in Fig. 4. Bay K 8644 reversed the effects o f nifedipine, verapamil, dil- tiazem and diclofurime. Bay K 8644 (1 nmol/1) was more effective in reversing the effects of diltiazem (1 ~tmol/1) th.an those of the other antagonists (Fig. 4, p < 0.05). In contrast Bay K 8644 did not reverse the effects of cin- narizine (1 ~tmol/1), flunarizine (1 ~tmol/1), prenylamine
71
NIFEDIPINE 10 nM ~ o 100 nM
BAY K 8 6 4 4
./,o 1 0 I l a i n
A
Fig. 3. Reversal by Bay K 8644 (1-100 nmol/1) of the inhibitory effects of nifedipine (10 nmol/1, preincubated for 25 min) on a contraction induced by Ca 2+ (A, 3 mmol/1) in a K + (40mM)- depolarized taenia preparation. The vertical bar represents the isotonic contraction of the taenia to Ca 2+ (3 mmol/1) prior to exposure to nifedipine
/•l? VERAPAMIL
,oo F~, DILTIAZEM
n -
/ / . . " , ~ q.CINNARIZ]NE
o L I I
I IQ IQQ
Bey K 8 6 4 4 , n m o l / I
Fig. 4. Reversal by Bay K 8644 of the inhibitory effects of calcium antagonists in K+-depolarized taenia preparations. The experi- ments were performed as in Fig. 3 and the antagonists pre- incubated in a Ca2+-free media for 25 min prior to addition of Ca 2+ (3retool/i). When the contraction had stabilized, Bay K8644 (1-100nmol/1) was added cumulatively. The calcium antagonists were: e - o nifedipine 10nmol/1, n=5; v - , , vera- pamil 0.2 btmol/1, n=5; []-D diltfazem 1 btmol/l, n=6; A-A diclofurime 1 ~tmol/1, n=4; ~ - ~ nisoldipine 10nmol/1, n=5; A-A pimozide 3 ~tmol/1, n=7; v - v cinnarizine 1 ~mol/1, n=6; o - o flunarizine 1 ~mol/1, n=5; []-~ bepridil 3 ~tmol/1, n---5. In additional experiments (not shown), the effects of fendiline (3~tmol/1, n=4), prenylamine (3~tmol/l, n=5), perhexiline (10 gmol/1, n=4) and W-7 (200 ~mol/1, n =7) were not reversed by Bay K8644 (1-100 nmol/1). "A" indicates that the reversal of the effects of diltiazem by Bay K 8644 (1 nmol/1) was significantly (p < 0.05) greater than for the other antagonists
(3 pmol/1), fendiline (3 gmol/1), perhexiline (10 ~tmol/1), bepridil (3 Nnol/1) or the calmodulin antagonist W-7 (200 ~tmol/1). The effects of pimozide (3 ~tmol/1) were only reversed by Bay K8644 (100 nmol/1) in three of seven experiments (Fig. 4). However, the effects of nisoldipine (10 nmol/1), a dihydropyridine calcium antagonist which dissociates slowly from calcium channels (Kazda and
72
~100 I.-- (J IZ F- Z 0 50 L)
X <Z =E
o
�9 /
oJ/ I I I I I I I
0.1 I I0
Ca~ (mmol / I )
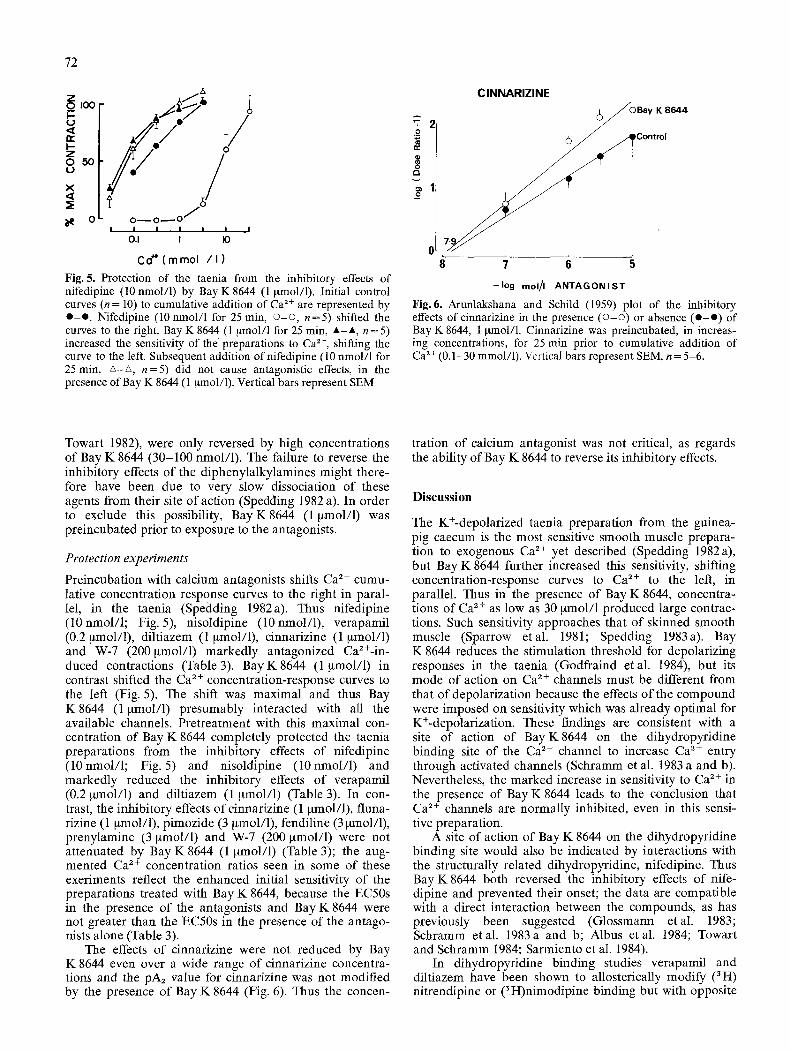
Fig. 5. Protection of the taenia from the inhibitory effects of nifedipine (10 nmol/1) by Bay K 8644 (1 ~tmol/1). Initial control curves (n = 10) to cumulative addition of Ca 2+ are represented by e - e . Nifedipine (10nmol/1 for 25 rain, �9169 n=5) shifted the curves to the right. Bay K 8644 (1 ~tmol/1 for 25 min, A-A, n=5) increased the sensitivity of the preparations to Ca 2§ shifting the curve to the left. Subsequent addition of nifedipine (10 nmol/1 for 25 min, zx-zx, n= 5) did not cause antagonistic effects, in the presence of Bay K 8644 (1 ~tmol/1). Vertical bars represent SEM
CINNARIZINE
~ / O B a y K 8644
._o Control r f
== O
- log mol/I ANTAGONIST
Fig. 6. Arunlakshana and Schild (1959) plot of the inhibitory effects of cinnarizine in the presence (o -o ) or absence ( e - e ) of Bay K 8644, 1 gmol/1. Cinnarizine was preincubated, in increas- ing concentrations, for 25 rain prior to cumulative addition of Ca 2+ (0.1-30 mmol/1). Vcrtical bars represent SEM, n = 5-6.
Towart 1982), were only reversed by high concentrations of Bay K 8644 (30-100 nmol/1). The failure to reverse the inhibitory effects of the diphenylalkylamines might there- fore have been due to very slow dissociation of these agents from their site of action (Spedding 1982 a). In order to exclude this possibility, Bay K8644 (1 ~mol/1) was preincubated prior to exposure to the antagonists.
Protection experiments
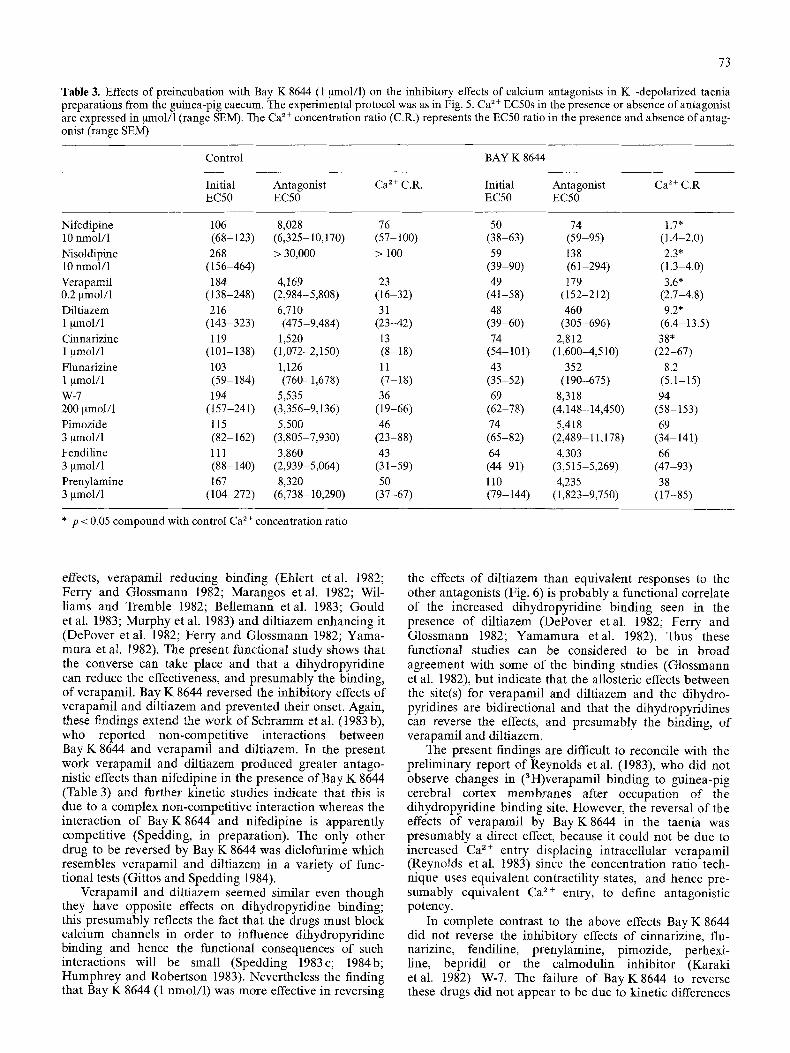
Preincubation with calcium antagonists shifts Ca ~+ cumu- lative concentration response curves to the fight in paral- lel, in the taenia (Spedding 1982a). Thus nifedipine (10 nmol/1; Fig. 5), nisoldipine (10 nmol/1), verapamil (0.2 ~tmol/1), diltiazem (1 ~tmol/1), cinnarizine (1 ~tmol/1) and W-7 (200gmol/1) markedly antagonized Ca2+-in - duced contractions (Table 3). Bay K8644 (1 ~mol/1) in contrast shifted the Ca 2+ concentration-response curves to the left (Fig. 5). The shift was maximal and thus Bay K8644 (1 ~tmol/1) presumably interacted with all the available channels. Pretreatment with this maximal con- centration of Bay K 8644 completely protected the taenia preparations from the inhibitory effects of nifedipine (10nmol/1; Fig. 5) and nisoldipine (10nmol/1) and markedly reduced the inhibitory effects of verapamil (0.2 ~mol/1) and diltiazem (1 gmol/1) (Table 3). In con- trast, the inhibitory effects ofcinnarizine (1 gmol/1), fluna- rizine (1 ~mol/1), pimozide (3 ~mol/1), fendiline (3 ~tmol/1), prenylamine (3 ~zmol/1) and W-7 (200 gmol/1) were not attenuated by Bay K 8644 (1 gmol/1) (Table 3); the aug- mented Ca 2+ concentration ratios seen in some of these exeriments reflect the enhanced initial sensitivity of the preparations treated with Bay K 8644, because the EC50s in the presence of the antagonists and Bay K 8644 were not greater than the EC50s in the presence of the antago- nists alone (Table 3).
The effects of cinnarizine were not reduced by Bay K 8644 even over a wide range of cinnarizine concentra- tions and the pA2 value for cinnarizine was not modified by the presence of Bay K 8644 (Fig. 6). Thus the concen-
tration of calcium antagonist was not critical, as regards the ability of Bay K 8644 to reverse its inhibitory effects.
Discussion
The K+-depolarized taenia preparation from the guinea- pig caecum is the most sensitive smooth muscle prepara- tion to exogenous Ca ~+ yet described (Spedding 1982a), but Bay K 8644 further increased this sensitivity, shifting concentration-response curves to Ca 2+ to the left, in parallel. Thus in the presence of Bay K 8644, concentra- tions of Ca 2+ as low as 30 9mol/1 produced large contrac- tions. Such sensitivity approaches that of skinned smooth muscle (Sparrow etal. 1981; Spedding 1983a). Bay K 8644 reduces the stimulation threshold for depolarizing responses in the taenia (Godfraind etal. 1984), but its mode of action on Ca 2+ channels must be different from that of depolarization because the effects of the compound were imposed on sensitivity which was already optimal for K+-depolarization. These findings are consistent with a site of action of Bay K8644 on the dihydropyridine binding site of the Ca 2+ channel to increase Ca 2+ entry through activated channels (Schramm et al. 1983 a and b). Nevertheless, the marked increase in sensitivity to Ca 2+ in the presence of Bay K 8644 leads to the conclusion that Ca 2+ channels are normally inhibited, even in this sensi- tive preparation.
A site of action of Bay K 8644 on the dihydropyridine binding site would also be indicated by interactions with the structurally related dihydropyridine, nifedipine. Thus Bay K 8644 both reversed the inhibitory effects of nife- dipine and prevented their onset; the data are compatible with a direct interaction between the compounds, as has previously been suggested (Glossmann et al. 1983; Schramm et al. 1983 a and b; Albus et al. 1984; Towart and Schramm 1984; Sarmiento et al. 1984).
In dihydropyridine binding studies verapamil and diltiazem have been shown to allosterically modify (3H) nitrendipine or (3H)nimodipine binding but with opposite
73
Table 3. Effects of preincubation with Bay K 8644 (1 gmol/1) on the inhibitory effects of calcium antagonists in K+-depolarized taenia preparations from the guinea-pig caecum. The experimental protocol was as in Fig. 5. Ca 2 + EC50s in the presence or absence of antagonist are expressed in Nnol/1 (range SEM). The Ca 2+ concentration ratio (C.R.) represents the EC50 ratio in the presence and absence of antag- onist (range SEM)
Control BAY K 8644
Initial Antagonist Ca 2 + C.R. Initial Antagonist Ca 2 + C.R EC50 EC50 EC50 EC50
Nifedipine 106 8,028 76 50 74 1.7 * 10 nmol/1 (68-123) (6,325-10,170) (57-100) (38-63) (59-95) (1.4-2.0) Nisoldipine 268 > 30,000 > 100 59 138 2.3* 10 nmol/1 (156-464) (39-90) (61-294) (1.3-4.0) Verapamil 184 4,169 23 49 179 3.6* 0.2 ~tmol/l (138-248) (2,984-5,808) (16-32) (41-58) (152-212) (2.7-4.8) Diltiazem 216 6,710 31 48 460 9.2* 1 ~tmol/1 (143-323) (475-9,484) (23-42) (39-60) (305-696) (6.4-13.5) Cinnarizine 119 1,520 13 74 2,812 38" 1 ~mol/l (101-138) (1,072-2,150) (8-18) (54-101) (1,600-4,510) (22-67) Flunarizine 103 1,126 11 43 352 8.2 1 ~tmol/1 (59-184) (760-1,678) (7-18) (35-52) (190-675) (5.1-15) W-7 194 5,535 36 69 8,318 94 200 ~tmol/1 (157-241) (3,356-9,136) (19-66) (62-78) (4,148-14,450) (58-153) Pimozide 115 5,500 46 74 5,418 69 3 ~mol/l (82-162) (3,805-7,930) (23-88) (65-82) (2,489-11,178) (34-141) Fendiline 111 3,860 43 64 4,303 66 3 ~xmol/1 (88-140) (2,939-5,064) (31-59) (44-91) (3,515-5,269) (47-93) Prenylamine 167 8,320 50 110 4,235 38 3 ~tmol/1 (104-272) (6,738-10,290) (37-67) (79-144) (1,823-9,750) (17-85)
* p < 0.05 compound with control Ca 2+ concentration ratio
effects, verapamil reducing binding (Ehlert etal. 1982; Ferry and Glossmann 1982; Marangos et al. 1982; Wil- liams and Tremble 1982; Bellemann et al. 1983; Gould et al. 1983; Murphy et al. 1983) and diltiazem enhancing it (DePover et al. 1982; Ferry and Glossmann 1982; Yama- mura et al. 1982). The present functional study shows that the converse can take place and that a dihydropyfidine can reduce the effectiveness, and presumably the binding, of verapamil. Bay K 8644 reversed the inhibitory effects of verapamil and diltiazem and prevented their onset. Again, these findings extend the work of Schramm et al. (1983 b), who reported non-competitive interactions between Bay K 8644 and verapamil and diltiazem. In the present work verapamil and diltiazem produced greater antago- nistic effects than nifedipine in the presence of Bay K 8644 (Table 3) and further kinetic studies indicate that this is due to a complex non-competitive interaction whereas the interaction of Bay K 8644 and nifedipine is apparently competitive (Spedding, in preparation). The only other drug to be reversed by Bay K 8644 was diclofurime which resembles verapamil and diltiazem in a variety of func- tional tests (Gittos and Spedding 1984).
Verapamil and diltiazem seemed similar even though they have opposite effects on dihydropyridine binding; this presumably reflects the fact that the drugs must block calcium channels in order to influence dihydropyridine binding and hence the functional consequences of such interactions will be small (Spedding 1983c; 1984b; Humphrey and Robertson 1983). Nevertheless the finding that Bay K 8644 (1 nmol/1) was more effective in reversing
the effects of diltiazem than equivalent responses to the other antagonists (Fig. 6) is probably a functional correlate of the increased dihydropyfidine binding seen in the presence of dilfiazem (DePover etal. 1982; Ferry and Glossmann 1982; Yamamura etal. 1982). Thus these functional studies can be considered to be in broad agreement with some of the binding studies (Glossmann et al. 1982), but indicate that the allosteric effects between the site(s) for verapamil and diltiazem and the dihydro- pyridines are bidirectional and that the dihydropyfidines can reverse the effects, and presumably the binding, of verapamil and dilfiazem.
The present findings are difficult to reconcile with the preliminary report of Reynolds et al. (1983), who did not observe changes in (3H)verapamil binding to guinea-pig cerebral cortex membranes after occupation of the dihydropyridine binding site. However, the reversal of the effects of verapamil by Bay K 8644 in the taenia was presumably a direct effect, because it could not be due to increased Ca a+ entry displacing intracellular verapamil (Reynolds et al. 1983) since the concentration ratio tech- nique uses equivalent contractility states, and hence pre- sumably equivalent Ca 2+ entry, to define antagonistic potency.
In complete contrast to the above effects Bay K 8644 did not reverse the inhibitory effects of cinnarizine, flu- narizine, fendiline, prenylamine, pimozide, perhexi- line, bepridil or the calmodulin inhibitor (Karaki etal. 1982) W-7. The failure of Bay K8644 to reverse these drugs did not appear to be due to kinetic differences

74
between these agents and nifedipine, because Bay K 8644 did not protect the taenia from the inhibitory effects of these drugs even when pre-incubated in very high concen- trations. Kinetic differences explain the failure of Bay K 8644 to reverse the inhibitory effects of nisoldipine. Nisoldipine dissociates very slowly from its site of action (over 10 h; Kazda and Towart 1982) and thus there are presumably few dihydropyridine sites available for Bay K 8644 to interact with. In contrast, when Bay K 8644 is ,preincubated with the tissue prior to addition of nisold- ipine it can protect the taenia from the antagonistic effects of nisoldipine because both drugs presumably interact with the dihydropyridine site. Thus the failure of Bay K 8644 to protect the taenia from the inhibitory effects of cinnarizine and the other diphenylalkylamines means that the site of action of these drugs is independent of the dihydropyridine site, and consequently also independent of the site(s) for verapamil and diltiazem, the latter site(s) being allosterically linked to the dihydropyridine site.
These experiments can be considered as confirmatory evidence that calcium antagonist subgroups exist. The drugs which were not reversed by Bay K 8644 form a distinct subgroup of calcium antagonists: all the drugs are very lipophilic and most have a diphenylalkylamine moiety (Spedding 1984b). Their effects can be readily distinguished from the dihydropyridines, verapamil and diltiazem in that they are much less selective for calcium channels (Fleckenstein 1981; Bayer etal. 1982; Roden- kirchen etal. 1982; Anno etal. 1984), have time- and calcium-dependent effects in smooth muscle (Spedding 1982a; 1984b; Wadsworth and Moss 1983) and, like calmodulin inhibitors, can interact with the contractile proteins in high concentrations (Spedding 1983 a). Bepri- dil, flunarizine (Lugnier et al. 1984), pimozide (Levin and Weiss 1979), fendiline and prenylamine (Johnson and Fugman 1983) bind directly to calmodulin in concentra- tions similar to, or only slightly higher than, those used in this study. Furthermore, changing surface charge with salicylate increases the effectiveness of this group of antagonists in the taenia whereas the effectiveness of verapamil and diltiazem is reduced and that of nifedipine unchanged (Spedding 1983b and 1984a). There is thus considerable evidence that these very lipophilic calcium antagonists behave differently from the dihydropyridines, and from verapamil and diltiazem.
The results from these functional experiments cannot be reconciled with a unitary mechanism of action for calcium antagonists, proposed by Gould et al. (1983) and Murphy et al. (1983). These workers claim that verapamil, diltiazem and the diphenylalkylamines interact at a com- mon site which modifies the dihydropyridine binding site. The functional consequences of this model would be that cinnarizine should resemble verapamil. Why is this not the case? Cinnarizine and the diphenylalkylamines at the relevant pharmacological concentrations have certainly been shown to reduce dihydropyridine binding in mem- branes from all tissues studied, including smooth muscle (Bolger et al. 1983). However, the relevance of this phe- nomenon is questionable, because this effect is directly paralleled by the ability to bind to calmodulin (Sarmiento et al. 1983), which in turn parallels non-specific mem- branal interactions (Landry et al. 1981). Thus the effect of these lipophilic drugs on (3H)nitrendipine binding most likely reflects non-specific effects (see Spedding 1984b).
The results with Bay K 8644 in the present study indicate that the effects of cinnarizine would be independent of whether the dihydropyridine site was occupied or not. An effect of cinnarizine on events subsequent to Ca 2+ entry, similar to those demonstrated for W-7 (Karaki et al. 1982), cannot be ruled out.
In conclusion, differential reversal of the effects of calcium antagonists by Bay K 8644 confirm the proposal that the subgroups of calcium antagonists act at different sites (Spedding 1981 and 1982a; 1983 a and c; 1984a and b). Bay K 8644 reverses the effects of dihydropyridines such as nifedipine competitively (Schramm and Towart 1984), whereas the interaction of Bay K 8644 with vera- pamil and diltiazem is non-competitive (Schramm and Towart, 1984). Bay K 8644 does not reverse the effects of the lipophilic diphenylalkylamines. Actions at different sites would account for the disparate profiles of the subgroups in in vivo experimental models (Spedding 1982 b; Hof 1984) and in man (Opie 1980).
Acknowledgements. We are very grateful to Dr. M. Schramm (Bayer AG) for the gift of Bay K 8644 and to Janssen Pharma- ceutica, Synthelabo, Hoechst, Dr. Thiemann GmbH for gifts of drugs.
References
Albus U, Habermann E, Ferry DR, Glossmann H (1984) Novel 1,4-dihydropyridine (Bay K8644) facilitates calcium-depen- dent (~H)noradrenaline release from PC12 cells. J Neurochem 42:1186-1189
Anno T, Furuta T, Itoh M, Kodama I, Toyama J, Yamada K (1984) Electromechanical effects of bepridil on rabbit isolated hearts. Br J Pharmacol 81:41-47
Arunlakshana O, Schild HO (1959) Some quantitative uses of drug antagonists. Br J Pharmacol Chemother 14:48-58
Bayer R, Kaufmann R, Mannhold R, Rodenkirchen R (1982) ~Ihe action of specific Ca antagonists on cardiac electrical activity. Progr Pharmacol 5: 53-85
Bellemann P, Schade A, Towart R (1983) Dihydropyridine recep- tor in rat brain labelled with (3H)nimodipine. Proc Natl Acad Sci USA 80:2356-2360
Bolger GT, Gengo P, Klockowski E, Luchowski E, Siegel H, Janis RA, Triggle AM, Triggle DJ (1983) Characterization of binding of the Ca 2+ channel antagonist, (3H)nitrendipine, to guinea-pig ileal smooth muscle. J Pharmacol Exp Ther 225: 291-309
Bou J, Llenas J, Massingham R (1983) Calcium entry blocking drugs, "calcium antagonists" and vascular smooth muscle function. J Auton Pharmacol 3: 219-232
DePover A, Matlib MA, Lee SW, Dub6 GP, Grupp IL, Grupp G, Schwartz A (1982) Specific binding of (3H)nitrendipine to membranes from coronary arteries and heart in relation to pharmacological effects. Paradoxical stimulation by diltiazem. Biochem Biophys Res Comm 108:110-117
Ehlert FJ, Itoga E, Roeske WR, Yamamura HI (1982) The interaction of (~H)nitrendipine with receptors for calcium antagonists in the cerebral cortex and heart of rats. Bioehem Biophys Res Comm 104:937-943
Ferry DR, Glossmann H (1982) Evidence for multiple receptor sites within the putative calcium channel. Naunyn-Schmiede- berg's Arch Pharmaco1321:80-83
Fleckenstein A (1981) Fundamental actions of calcium antago- nists on myocardial and cardiac pacemaker cell membranes. In: Weiss GB (ed) New perspectives on calcium antagonists. Am Physiol Soc Baltimore, pp 59-81
Gittos M, Spedding M (1984) Comparison of cis and trans diclofurime with verapamil as calcium antagonists. Br J Pharmacol 81: 81P

75
Glossmann H, Ferry DR, Lt~bbecke F, Mewes R, Hofmann F (1982) Calcium channels: direct identification with radio- ligand binding studies. Trends Pharmacol Sci 3:431-437
Glossmann H, Linn T, Rombusch M, Ferry DR (1983) Tempera- ture-dependent regulation of d-cis-(3H)diltiazem binding to Ca 2+ channels by 1,4-dihydropyridine channel agonists and antagonists. FEBS Lett 160:226-232
Godfraind T, Hardy JP, Morel N (1984) The action of the spas- mogenic dihydropyridine Bay K 8644 on the mechanical and the electrical activity of guinea-pig taenia coli. Arch int Pharmacodyn Ther 268:167-168
Gould RJ, Murphy KMM, Reynolds IJ, Snyder SH (1983_) Antischizophrenic drugs of the diphenylbutylpiperidine type act as calcium channel antagonists. Proc Natl Acad Sci USA 80:5122-5125
Hof RP (1984) Selective effects of different calcium antagonists on the peripheral circulation. Trends Pharmacol Sci 5:100-103
Humphrey PPA, Robertson MJ (1983) Functional characteristics of verapamil and nitrendipine interactions at calcium channels in smooth muscle. Br J Pharmacol 80: 502P
Inomata H, Kao CY (1976) Ionic currents in the guinea-pig taenia coli. J Physio1255:347-378
Johnson JD, Fugman DA (1983) Calcium and calmodulin antag- onists binding to calmodulin and relaxation of coronary segments. J Pharmacol Exp Ther 226:330-334
Karaki H, Murakami K, Nakagawa H, Ozaki H, Urakawa N (1982) Effects of calmodulin antagonists on tension and cellular calcium content in depolarized vascular and intestinal smooth muscles. Br J Pharmaco177:661-666
Kazda S, Towart R (1982) The duration of action of calcium antagonists in vitro: a comparison of nifedipine and nisoldi- pine (Bay K 5552). Br J Pharmaco176:255P
Kazda S, Knorr A, Towart R (1983) Common properties and differences between various calcium antagonists. Progr Pharmacol 5: 84-116
I.andry Y, Amellal M, Ruckstnhl M (1981) Can calmodulin inhibitors be used to probe calmodulin effects? Biochem Pharmacol 30:2031-2032
Levin RM, Weiss GB (1979) Selective binding of antipsychotics and other psychoactive agents to the calcium-dependent activation of cyclic nucleotide phosphodiesterase. J Pharma- col Exp Ther 208:454-459
Lugnier C, Follenius A, Gerard D, Stoclet JC (1984) Bepridil and flunarizine as calmodulin inhibitors. Eur J Pharmacol 98: 157-158
Marangos PJ, Patel J, Miller C, Martino AM (1982) Specific calcium antagonist binding sites in brain. Life Sci 31: 1575-1585
Murphy KMM, Gould RJ, Largent BL, Snyder SH (1983) A unitary mechanism of calcium antagonist drug action. Proc Natl Acad Sci USA 80:860-864
Opie LH (1980) Calcium antagonists. Lancet 1:806-810 Reynolds I J, Gould R J, Snyder SH (1983) (sH)Verapamil binding
sites in brain and skeletal muscle: regulation by calcium. Eur J Pharmacol 95: 315-321
Riemer J, D/3rfler F, Mayer C-J, Ulbrecht G (1974) Calcium antagonistic effects on the spontaneous activity of guinea-pig taenia coli. Pflt~ger's Arch ges Physiol 351:33-37
Rodenkirchen R, Bayer R, Mannhold R (1982) Specific and non- specific Ca antagonists. A structure-activity analysis of cardio- depressive drugs. Progr Pharmacol 5:9-23
Sarmiento JG, Janis RA, Maurer SC, Katz AM, Triggle D J, Luchowski E (1983) Comparison of high affinity binding sites for Ca 2+ channel inhibitors in membranes for various tissues. Is calmodulin at the binding site for Ca ~+ channel inhibitors? Pharmacologist 25: 202
Sarmiento JG, Janis RA, Rampe D, Triggle DJ (1984) (3H) Bay K 8644, a calcium channel agonist, binds to high and low affinity sites on membranes from cardiac muscle and brain. Fed Proc 43: 448
Schramm M, Thomas G, Towart R, Franckowiak G (1983a) Novel dihydropyridines with positive inotropic action through activation of Ca 2+ channels. Nature 309:535-537
Schramm M, Thomas G, Towart R, Franckowiak G (1983b) Activation of calcium channels by novel 1,4-dihydropyridines. A new mechanism for positive inotropics or smooth muscle stimulants. Arzneim-Forsch 33:1268-1272
Schramm M, Towart R (1984) Modulation of calcium channel function with 1,4-dihydropyridine structure. J Physiol 349: 53P
Sparrow MP, Mrwa U, Hofmann F, Rt~egg JC (1981) Calmodulin is essential for smooth muscle contractions. FEBS Lett 125: 141-145
Spedding M (1981) Marked differences in the effects of "Ca 2+ antagonists" on CaC12-induced contractions in K+-depolarized smooth muscle. Br J Pharmacol 72: 144P
Spedding M (1982 a) Assessment of"Ca 2 +-antagonist" effects of drugs in K+-depolarized smooth muscle. Differentiation of antago- nist subgroups. Naunyn-Schmiedeberg's Arch Pharmacol 318: 234-240
Spedding M (1982b) Differences between the effects of calcium antagonists in the pithed rat preparation. J Cardiovase Phar- macol 4:973-979
Spedding M (1983a) Direct inhibitory effects of some "calcium antagonists" and trifluoperazine on the contractile proteins in smooth muscle. Br J Pharmaco179:225-231
Spedding M (1983 b) The effects of changing surface charge with salicylate on the potency of calcium antagonists in smooth muscle. Br J Pharmaco178:36 P
Spedding M (1983c) Functional interactions of calcium antago- nists in K+-depolarized smooth muscle. Br J Pharmacol 80: 485-488
Spedding M (1984a) Changing surface charge with salicylate differentiates between subgroups of calcium antagonists. Br J Pharmacol, in press
Spedding M (1984b) Calcium antagonist subgroups. Trends Pharmacol Sci: in press
Stephenson RP (1956) A modification of receptor theory. Br J Pharmacol Chemother 11 : 379-393
Towart R, Schramm M (1984) Recent advances in the pharmaco- logy of the calcium channel. Trends Pharmacol Sci 5:111-113
Triggle DJ (1984) Ca 2+ channels revisited: problems and prom- ises. Trends Pharmaeol Sci 5:4-5
Van Rossum JM (1963) Cumulative dose-response curves II. Technique for the making of dose-response curves in isolated organs and the evaluation of drug parameters. Arch Int Pharmacodyn 143:299-330
Wadsworth RM, Moss JP (1983) Time course of the effect of flunarizine on rabbit isolated vascular muscle. Eur J Pharma- col 85: 207-209
Williams LT, Tremble P (1982) Binding of a calcium antagonist, (3H)nitrendipine, to high affinity sites in bovine aortic smooth muscle and canine cardiac membranes. J Clin Inv 70:209-212
Yamamura HI, Schoemaker H, Boles RG, Roeske WR (1982) Diltiazem enhancement of (3H)nitrendipine binding to calcium channel associated drug receptor sites in rat brain synaptosomes. Biochem Biophys Res Comm 108:640-646
Received May 7, 1984/Accepted July 24, 1984




























