Embed Size (px)
Citation preview

Surface Science 543 (2003) 75–86
www.elsevier.com/locate/susc
Interaction of chlorine radicals with polyethyleneand hydrocarbon thin films under vacuum
conditions––a comparison with atomic oxygen reactivity
Jessica Torres, C.C. Perry, A.J. Wagner, D. Howard Fairbrother *
Department of Chemistry, Johns Hopkins University, 3400 N. Charles Street, Baltimore, MD 21218, USA
Received 22 January 2003; accepted for publication 24 July 2003
Abstract
The surface reactions of atomic chlorine and oxygen with hydrocarbon-based polymers and organic thin films under
vacuum conditions have been investigated with in situ X-ray photoelectron spectroscopy (XPS). The interaction of
chlorine radicals (Cl(2P)) with polyethylene (PE) under vacuum conditions produces a partially chlorinated layer
containing both CCl and CCl2 groups whose concentration was maximized at the surface. Compared to higher-pressure
photochlorination experiments where the flux of chlorine atoms is higher, the maximum extent of PE chlorination as
measured by the C:Cl XPS ratio and the evolution of the C(1s) region was reduced in the present study while the surface
selectivity of the reaction was enhanced. This influence of chlorine atom flux on the extent of chlorination and surface
selectivity can be rationalized by a simple stochastic model of the PE chlorination process that incorporates steric effects
associated with the production of mono and dichlorinated carbon atoms as well as cross-linking reactions between
carbon-containing radicals. During the reaction of PE with atomic oxygen (O(3P)), a concentration gradient of oxygen-
containing carbon functionality is also observed in the near surface region. Experiments carried out on hydrocarbon
thin films based on self-assembled monolayers (SAMs) reveal that chlorination proceeds without erosion. In contrast,
the incorporation of new carbon containing-oxygen functionalities during reactions of hydrocarbon films with atomic
oxygen occurs in competition with carbon erosion.
� 2003 Elsevier B.V. All rights reserved.
Keywords: Surface chemical reaction; Chlorine; Self-assembly; Oxygen
1. Introduction
The interaction of atomic species (radicals) witha variety of different surfaces is important in a
number of technologically significant processes.
For example, plasmas are one of the most popular
* Corresponding author. Tel.: +1-410-516-4328; fax: +1-410-
516-8420.
E-mail address: [email protected] (D.H. Fairbrother).
0039-6028/$ - see front matter � 2003 Elsevier B.V. All rights reserv
doi:10.1016/S0039-6028(03)00992-0
methods of modifying a polymer�s chemical and
physical surface characteristics, such as wettability,
adhesion, biocompatibility [1] and gas permeability[2], without changing bulk properties. Within the
plasma, the concentration of reactive neutrals and
radicals are typically estimated to be 3–4 orders of
magnitude greater than their ion counterparts [3].
Chlorine plasmas for example, are used to modify a
polymer�s gas permeability [4,5] while in the mi-
croelectronics industry chlorine-based plasmas are
ed.

76 J. Torres et al. / Surface Science 543 (2003) 75–86
used to etch and clean semiconductors and metals
such as Si [6], GaAs, AlGaAs [7,8], Pt [9], and Al
[10]. The interaction between gas-phase radical
species and solid substrates during chemical vapor
deposition is also a central process in the growth of
a variety of semiconductor materials [11–14].Studies on the reactions of radical species with
surfaces have, however, been limited by the need to
generate clean controllable sources of neutral rad-
icals while maintaining the capability to monitor
changes in the physical and chemical composition
of the interface.
The interaction of atomic chlorine with hydro-
carbon surfaces has been studied by X-rayphotoelectron spectroscopy (XPS) during the
photochemical gas phase chlorination of polyeth-
ylene (PE) (–CH2–CH2–)n using Cl2 at pressures
between 10 and 500 Torr [15–18]. For example,
Elman et al. [17] showed that during chlorination
of PE under atmospheric pressures, CCl and CCl2species were produced with monochlorinated spe-
cies dominating, ultimately producing a CC/CH2:CCl:CCl2 ratio of 1:1:0.5. The uptake of
chlorine was found to slow drastically at longer
exposures, although angle resolved XPS measure-
ments indicated that chlorination was uniform
throughout the sampling depth (10–70 �AA). In a
later study, using a combination of XPS, attenu-
ated total reflectance (ATR) IR and gravimetric
measurements, McCarthy and co-workers [15]showed that the depth as well as the extent of
chlorination could be controlled by adjusting the
Cl2 pressure, photointensity and exposure. Em-
ploying a combination of ATR and UV–VIS
measurements, Vernekar and co-workers [18]
showed that gas-phase photochlorination of PE
was accompanied by simultaneous dehydrochlori-
nation. In related studies, Chidsey and co-workers[19] examined the surface functionalization of
methyl-terminated alkyl self-assembled monolay-
ers (SAM) on silicon during gas-phase photo-
chlorination (PCl2 � 0:2 Torr). Results from this
study indicated that chlorination of the monolayer
under these conditions was limited. More than
50% of the original carbon atoms remained unat-
tached to chlorine with a steric preference forchlorination at the methyl ends of the alkyl chains,
evidenced by ATR measurements.
In this study, we present results on the exposure
of atomic chlorine (generated from the thermal
dissociation of molecular chlorine) to hydrocar-
bon surfaces under vacuum conditions. A deter-
mination of the processes that accompany the
interaction of chlorine radicals with hydrocarbonsurfaces under these low-pressure conditions was
motivated by the need to better understand the
reactions of chlorine-based plasmas with poly-
meric substrates and also to explore the possibility
that this approach could be employed as a route
for selective surface functionalization.
Under the low-pressure conditions that char-
acterize our investigation, the exposure of PE tochlorine atoms produces a depth dependent con-
centration gradient of CCl and CCl2 species in the
near surface region. Compared to previous higher-
pressure photochlorination experiments on PE
[15,17], the maximum chlorine content obtained
was significantly reduced while the surface selec-
tivity was enhanced. A simple stochastic kinetic
model, which considered steric effects on thechlorination as well as cross-linking reactions be-
tween carbon containing radicals, was employed
to model the chlorination of PE. To develop a
more comprehensive understanding of radical/
polymer surface reaction dynamics, the reactivity
of chlorine atoms with hydrocarbon substrates has
also been compared to those associated with
atomic oxygen. During their interaction with PE,both atomic chlorine and atomic oxygen produced
a depth dependent concentration of new species in
the near surface region. Separate experiments
carried out on SAMs, used as models for poly-
meric interfaces, reveal that while chlorination
proceeds without significant substrate erosion, the
grafting of new oxygen-containing functionality
during reactions with atomic oxygen is accompa-nied by carbon loss from the overlayer.
2. Experimental
In situ XPS measurements were carried out in
the same ultra-high vacuum (UHV) chamber
(Pbase � 5� 10�9 Torr) as the chlorine radicalsource, as described previously [20]. XPS mea-
surements were performed with a Physical Elec-

J. Torres et al. / Surface Science 543 (2003) 75–86 77
tronics 04-500 dual anode source, using Mg Ka
(1253.6 eV) irradiation. All XP spectra were col-
lected at 15 kV and 300 W with a take off angle of
45� from the sample normal unless otherwise no-
ted. Samples were mounted in a carousel stage
with xyz translational and rotational capabilities.Binding energy scales were referenced to the CC/
CH2 peak in the C(1s) region at 284.6 eV [21]. The
C(1s) and Cl(2s) peaks were best fit using mixed
Gaussian–Lorentzian curves in conjunction with
Shirley background subtraction. The reliability of
the fitting protocol employed in Fig. 1 was verified
by comparing the Cl/C ratio obtained from the fits
in the C(1s) region shown in Fig. 1 against the Cl/C ratio calculated from the integrated C(1s) and
Cl(2s) XPS areas. 1 Results from this analysis re-
vealed a good qualitative agreement between
the Cl/C ratios obtained using these two methods.
The validity of the angle resolved XPS measure-
ments reported in the present study were veri-
fied by measurements on the polymer nylon-6
(NH(CH2)5(C@O))n, which revealed an angle in-dependent N(1s)/O(1s) XPS ratio and C(1s) spec-
tral envelope. ATR measurements were made with
a Spectra Tech Thunderdomee accessory using a
Mattson Infinity Series FTIR spectrometer. Sto-
chastic kinetic simulations were performed using a
software package [22] that utilizes the algorithm
developed by Gillespie [23,24].
2.1. Polyethylene sample preparation
A PE bar (McMaster Carr, high-density tech-
nical grade) was cut with a razor and washed
in 2,2,4-trimethylpentane to remove adventitious
carbon and oxygen impurities. Typical XPS sur-
veys showed <5% oxygen impurities in the native
PE samples.
2.2. Radical source
Atomic chlorine and oxygen were generated
using a Thermal Gas Cracker TC-50 (Oxford
1 This comparison is facilitated by the proximity of the C(1s)
and Cl(2s) binding energies (284.6 vs. 271 eV) that ensures the
relative surface sensitivity of these peak areas can be directly
compared.
Applied Research). This gas cracker works by
dissociating molecular gases such as chlorine or
oxygen into a stream of atomic, low-energy reac-
tive species to produce a mixture of Cl(2P) and Cl2,
or O(3P) and O2, hereafter referred to as atomic
Cl or atomic O respectively. A description of theradical source can be found in a previous publi-
cation [20]. The gas cracker was operated at 30 W
with a chlorine pressure of �5 · 10�8 Torr, and at
�7 · 10�7 Torr during oxygen exposure. Samples
were placed in line of sight with the effluent from
the gas cracker (target-to-sample distance �5 cm).
2.3. Preparation of self-assembled monolayers
Gold substrates were initially cleaned by 2 keV
Arþ ion sputtering and then dipped into a 5 mM
solution of hexadecane thiol (CH3(CH2)15SH) in
hexanes for 10–12 h to prepare an alkanethiolate
SAM (C16-SAM). Samples were then washed in
ethanol, water and hexanes and placed in the XPS
analysis chamber on a UHV carousel stage via afast entry load lock system. X-ray irradiation is
known to initiate electron-stimulated C–H, C–C
and S–Au bond breaking within alkanethiolate
SAMs [25–27] compromising the structural integ-
rity of the SAM and modifying the elemental
composition [25]. To minimize the effects of X-ray
irradiation in the present investigation, XPS
analysis was carried out on SAMs only after ex-posure to atomic chlorine and oxygen. Further-
more, individual SAMs were used for each atomic
chlorine/oxygen experiment and then discarded;
thus, the results shown in this investigation cor-
respond to single exposures of atomic chlorine/
oxygen to numerous C16-SAMs.
2.4. Atomic force microscopy (AFM) measure-
ments
Ex situ AFM experiments were carried out using
a Burleigh Metris 2000 NC instrument operating
in contact mode as described previously [28]. Sili-
con probes with a resonant frequency of 11–16
kHz (1:1 conical tip; 100 �AA) and a nominal stiff-
ness of 0.1–0.34 Nm�1 were used. A 6.62 · 5.83lm2 scan area was typically imaged. Consecu-
tive measurements on the same regions produced
C(1s)
Binding Energy (eV)282285288291
0 min
2 min
60 min
240 min
10 min
Cl(2s)
Binding Energy (eV)267270273276279
Cl/Cl2
ExposureTime
x 0.7
CC/CH2CHClCCl2 C(1s)
Satellites
C-Cl
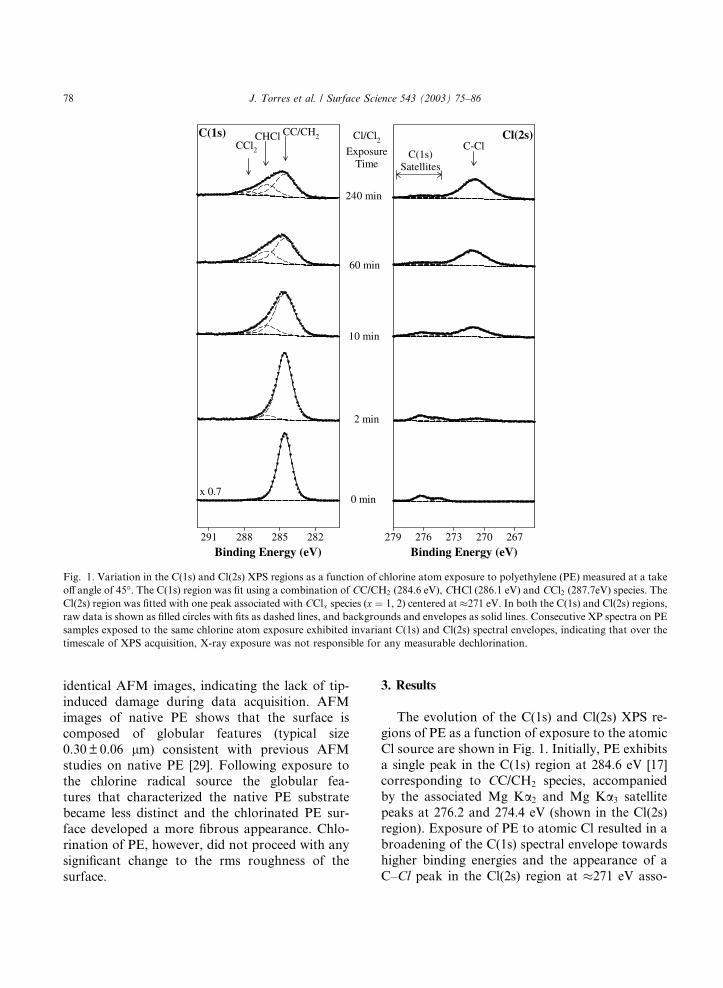
Fig. 1. Variation in the C(1s) and Cl(2s) XPS regions as a function of chlorine atom exposure to polyethylene (PE) measured at a take
off angle of 45�. The C(1s) region was fit using a combination of CC/CH2 (284.6 eV), CHCl (286.1 eV) and CCl2 (287.7eV) species. The
Cl(2s) region was fitted with one peak associated with CClx species (x ¼ 1, 2) centered at �271 eV. In both the C(1s) and Cl(2s) regions,
raw data is shown as filled circles with fits as dashed lines, and backgrounds and envelopes as solid lines. Consecutive XP spectra on PE
samples exposed to the same chlorine atom exposure exhibited invariant C(1s) and Cl(2s) spectral envelopes, indicating that over the
timescale of XPS acquisition, X-ray exposure was not responsible for any measurable dechlorination.
78 J. Torres et al. / Surface Science 543 (2003) 75–86
identical AFM images, indicating the lack of tip-
induced damage during data acquisition. AFM
images of native PE shows that the surface is
composed of globular features (typical size
0.30 ± 0.06 lm) consistent with previous AFM
studies on native PE [29]. Following exposure tothe chlorine radical source the globular fea-
tures that characterized the native PE substrate
became less distinct and the chlorinated PE sur-
face developed a more fibrous appearance. Chlo-
rination of PE, however, did not proceed with any
significant change to the rms roughness of the
surface.
3. Results
The evolution of the C(1s) and Cl(2s) XPS re-
gions of PE as a function of exposure to the atomic
Cl source are shown in Fig. 1. Initially, PE exhibits
a single peak in the C(1s) region at 284.6 eV [17]corresponding to CC/CH2 species, accompanied
by the associated Mg Ka2 and Mg Ka3 satellite
peaks at 276.2 and 274.4 eV (shown in the Cl(2s)
region). Exposure of PE to atomic Cl resulted in a
broadening of the C(1s) spectral envelope towards
higher binding energies and the appearance of a
C–Cl peak in the Cl(2s) region at �271 eV asso-
J. Torres et al. / Surface Science 543 (2003) 75–86 79
ciated with CClx (x ¼ 1, 2) species [17]. Fig. 1
shows that the evolution of the C(1s) region during
exposure to atomic chlorine could be well fitted to
a combination of CC/CH2 (284.6 eV), CHCl (286.1
eV) and CCl2 (287.4 eV) species [17,19]. It should
also be noted that in separate experiments whenPE was exposed to Cl2 alone at similar pressures to
those used in Fig. 1, no changes in the C(1s) or
Cl(2s) XPS peaks were observed over a period of
several hours.
Cl/C
Rat
io
0.0
0.2
0.4
0.6
0.8
Cl/Cl2 Exposu0 50 100
Cl/C
Rat
io
0.0
0.2
0.4
0.6
0.8
1.0
0
20
40
60
80
100
Per
cen
tC
ompo
siti
on
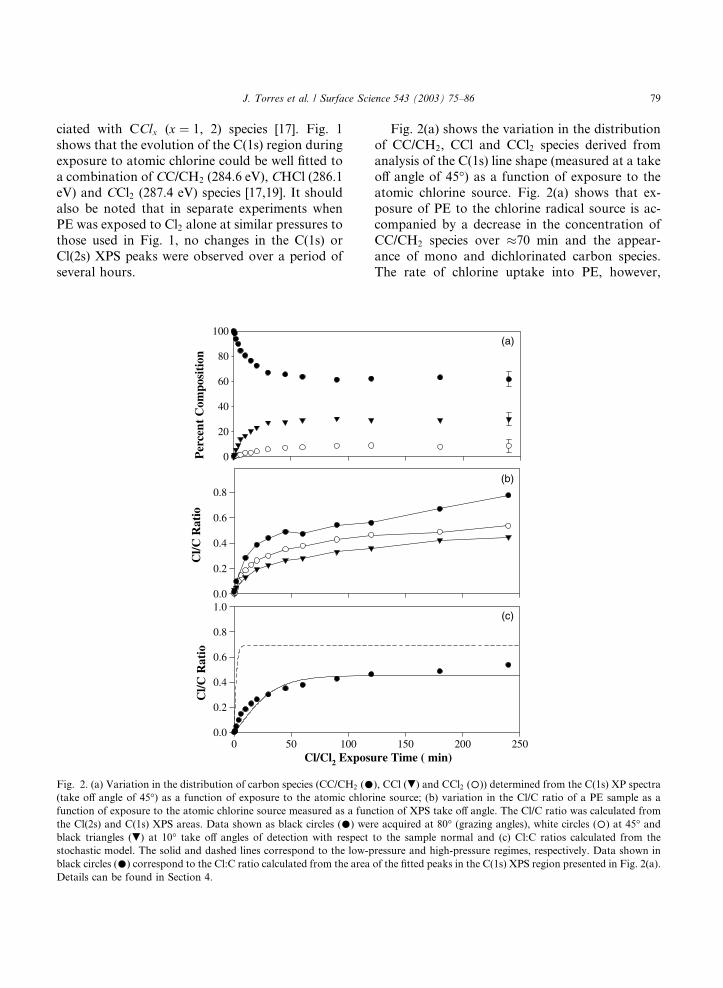
Fig. 2. (a) Variation in the distribution of carbon species (CC/CH2 (�(take off angle of 45�) as a function of exposure to the atomic chlor
function of exposure to the atomic chlorine source measured as a fun
the Cl(2s) and C(1s) XPS areas. Data shown as black circles (�) wer
black triangles (.) at 10� take off angles of detection with respect
stochastic model. The solid and dashed lines correspond to the low-p
black circles (�) correspond to the Cl:C ratio calculated from the area
Details can be found in Section 4.
Fig. 2(a) shows the variation in the distribution
of CC/CH2, CCl and CCl2 species derived from
analysis of the C(1s) line shape (measured at a take
off angle of 45�) as a function of exposure to the
atomic chlorine source. Fig. 2(a) shows that ex-
posure of PE to the chlorine radical source is ac-companied by a decrease in the concentration of
CC/CH2 species over �70 min and the appear-
ance of mono and dichlorinated carbon species.
The rate of chlorine uptake into PE, however,
re Time ( min)150 200 250
(a)
(b)
(c)
), CCl (.) and CCl2 (�)) determined from the C(1s) XP spectra
ine source; (b) variation in the Cl/C ratio of a PE sample as a
ction of XPS take off angle. The Cl/C ratio was calculated from
e acquired at 80� (grazing angles), white circles (�) at 45� andto the sample normal and (c) Cl:C ratios calculated from the
ressure and high-pressure regimes, respectively. Data shown in
of the fitted peaks in the C(1s) XPS region presented in Fig. 2(a).
80 J. Torres et al. / Surface Science 543 (2003) 75–86
decreases rapidly during exposure to the chlorine
atom source. Indeed, analysis of Fig. 1 indicates
that for exposure times greater than 70 min, the
composition of the film as measured by XPS re-
mained constant with 62.5% CC/CH2, 29.4%
CHCl and 8.1% CCl2 species. Fig. 2(b) shows theuptake of chlorine into the PE surface as a func-
tion of atomic Cl exposure measured by the vari-
ation in the Cl(2s)/C(1s) XPS ratios, recorded at
three different take off angles (80�, 45� and 10�)measured with respect to the sample normal. A
comparison of the angle resolved XPS data shows
that although the Cl(2s)/C(1s) ratio exhibits a
similar dependence on atomic Cl exposure for thedifferent take off angles, the Cl(2s)/C(1s) ratio was
always largest for data acquired at an 80� take off
angle and smallest for data acquired at 10�. Fig.2(c) shows a comparison of the calculated Cl(2s)/
C(1s) ratios in the case of low (solid line) and high
(dotted line) chlorine atom fluxes based on the
stochastic model developed in Section 4. Experi-
mental data on the variation in the Cl(2s)/C(1s)
10º
45º
80º
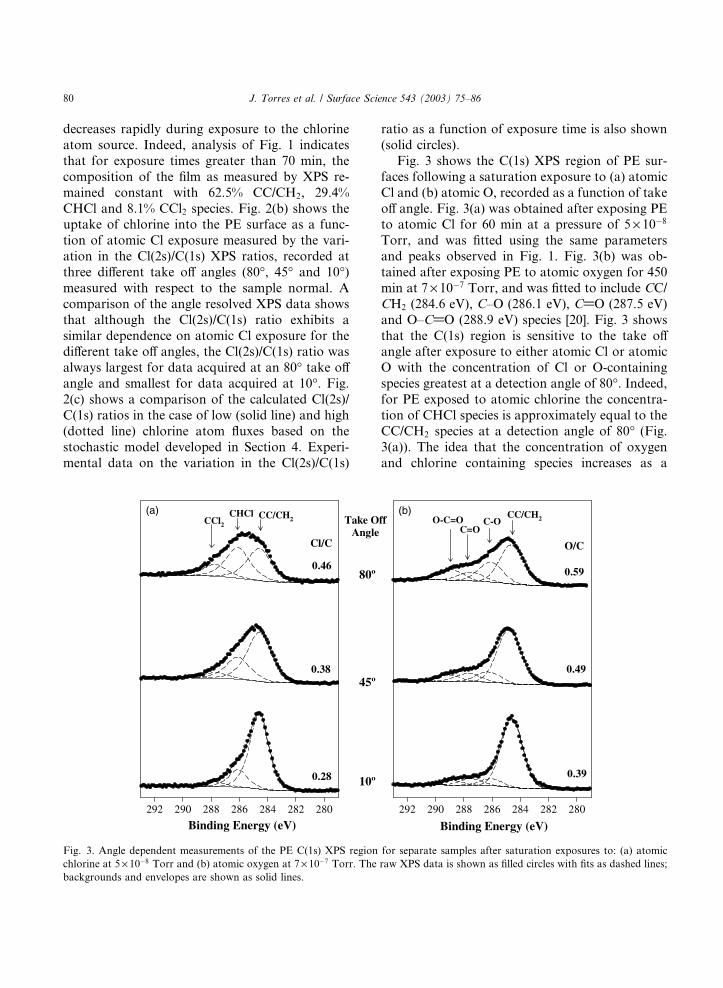
Binding Energy (eV)280282284286288290292
0.28
0.38
0.46
Cl/C
CC/CH2CHCl
CCl2 Take OAngle
(a)
Fig. 3. Angle dependent measurements of the PE C(1s) XPS region
chlorine at 5 · 10�8 Torr and (b) atomic oxygen at 7 · 10�7 Torr. The
backgrounds and envelopes are shown as solid lines.
ratio as a function of exposure time is also shown
(solid circles).
Fig. 3 shows the C(1s) XPS region of PE sur-
faces following a saturation exposure to (a) atomic
Cl and (b) atomic O, recorded as a function of take
off angle. Fig. 3(a) was obtained after exposing PEto atomic Cl for 60 min at a pressure of 5 · 10�8
Torr, and was fitted using the same parameters
and peaks observed in Fig. 1. Fig. 3(b) was ob-
tained after exposing PE to atomic oxygen for 450
min at 7 · 10�7 Torr, and was fitted to include CC/
CH2 (284.6 eV), C–O (286.1 eV), C@O (287.5 eV)
and O–C@O (288.9 eV) species [20]. Fig. 3 shows
that the C(1s) region is sensitive to the take offangle after exposure to either atomic Cl or atomic
O with the concentration of Cl or O-containing
species greatest at a detection angle of 80�. Indeed,for PE exposed to atomic chlorine the concentra-
tion of CHCl species is approximately equal to the
CC/CH2 species at a detection angle of 80� (Fig.
3(a)). The idea that the concentration of oxygen
and chlorine containing species increases as a
Binding Energy (eV)280282284286288290292
0.39
0.49
0.59
O/C
CC/CH2O-C=OC=O
C-Off(b)
for separate samples after saturation exposures to: (a) atomic
raw XPS data is shown as filled circles with fits as dashed lines;
J. Torres et al. / Surface Science 543 (2003) 75–86 81
function of increasing XPS take off angle is also
supported by the measured variation in the Cl(2s)/
C(1s) and O(1s)/C(1s) XPS ratios, shown in Fig.
3(a) and (b) respectively.
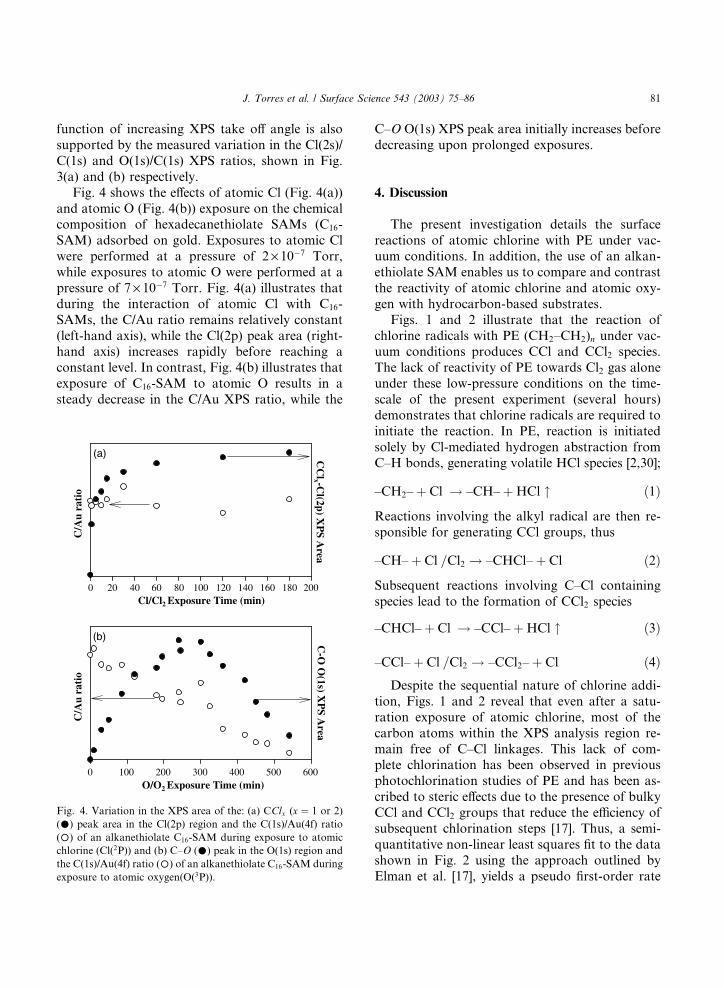
Fig. 4 shows the effects of atomic Cl (Fig. 4(a))
and atomic O (Fig. 4(b)) exposure on the chemicalcomposition of hexadecanethiolate SAMs (C16-
SAM) adsorbed on gold. Exposures to atomic Cl
were performed at a pressure of 2 · 10�7 Torr,
while exposures to atomic O were performed at a
pressure of 7 · 10�7 Torr. Fig. 4(a) illustrates that
during the interaction of atomic Cl with C16-
SAMs, the C/Au ratio remains relatively constant
(left-hand axis), while the Cl(2p) peak area (right-hand axis) increases rapidly before reaching a
constant level. In contrast, Fig. 4(b) illustrates that
exposure of C16-SAM to atomic O results in a
steady decrease in the C/Au XPS ratio, while the
(a)
Cl/Cl2 Exposure Time (min)200180160140120100806040200
C/A
u ra
tio
CC
lx -Cl(2p) X
PS A
rea
(b)
O/O2 Exposure Time (min)0 100 200 300 400 500 600
C/A
u ra
tio
C-O
O(1s) X
PS A
rea
Fig. 4. Variation in the XPS area of the: (a) CClx (x ¼ 1 or 2)
(�) peak area in the Cl(2p) region and the C(1s)/Au(4f) ratio
(�) of an alkanethiolate C16-SAM during exposure to atomic
chlorine (Cl(2P)) and (b) C–O (�) peak in the O(1s) region and
the C(1s)/Au(4f) ratio (�) of an alkanethiolate C16-SAM during
exposure to atomic oxygen(O(3P)).
C–O O(1s) XPS peak area initially increases before
decreasing upon prolonged exposures.
4. Discussion
The present investigation details the surface
reactions of atomic chlorine with PE under vac-
uum conditions. In addition, the use of an alkan-
ethiolate SAM enables us to compare and contrast
the reactivity of atomic chlorine and atomic oxy-
gen with hydrocarbon-based substrates.
Figs. 1 and 2 illustrate that the reaction of
chlorine radicals with PE (CH2–CH2)n under vac-uum conditions produces CCl and CCl2 species.
The lack of reactivity of PE towards Cl2 gas alone
under these low-pressure conditions on the time-
scale of the present experiment (several hours)
demonstrates that chlorine radicals are required to
initiate the reaction. In PE, reaction is initiated
solely by Cl-mediated hydrogen abstraction from
C–H bonds, generating volatile HCl species [2,30];
–CH2–þ Cl� ! –C�
H–þHCl " ð1Þ
Reactions involving the alkyl radical are then re-
sponsible for generating CCl groups, thus
–C�
H–þ Cl�=Cl2 ! –CHCl–þ Cl� ð2Þ
Subsequent reactions involving C–Cl containing
species lead to the formation of CCl2 species
–CHCl–þ Cl� ! –C�
Cl–þHCl " ð3Þ
–C�
Cl–þ Cl�=Cl2 ! –CCl2–þ Cl� ð4ÞDespite the sequential nature of chlorine addi-
tion, Figs. 1 and 2 reveal that even after a satu-
ration exposure of atomic chlorine, most of the
carbon atoms within the XPS analysis region re-
main free of C–Cl linkages. This lack of com-plete chlorination has been observed in previous
photochlorination studies of PE and has been as-
cribed to steric effects due to the presence of bulky
CCl and CCl2 groups that reduce the efficiency of
subsequent chlorination steps [17]. Thus, a semi-
quantitative non-linear least squares fit to the data
shown in Fig. 2 using the approach outlined by
Elman et al. [17], yields a pseudo first-order rate

82 J. Torres et al. / Surface Science 543 (2003) 75–86
constant for hydrogen abstraction from methylene
groups (reaction (1)) of 1.17 · 10�3 s�1. In contrast,
the pseudo first-order rate constant for the loss of
monochlorinated species (–CHCl–) as a result of
hydrogen abstraction (reaction (3)) has an upper
limit of 6 · 10�6 s�1.The most significant difference between the ex-
periments reported in the present study and
photochlorination experiments carried out at at-
mospheric pressures is the lower incident chlorine
atom flux, illustrated by the reduced rate of CH2
group loss during chlorination. Thus, under our
experimental conditions, the pseudo first-order
rate constant for the loss of methylene groups(1.17 · 10�3 s�1) is an order of magnitude less than
the reported value by Elman et al. (1.5 · 10�2 s�1)
[17]. In these higher-pressure chlorination studies
the maximum chlorine uptake was determined by
XPS to be CC/CH2:CCl:CCl2�1:1:0.5 [15] and the
C(1s) XPS profile was independent of the take off
angle. In contrast, Figs. 1–3(a) reveal that the
maximum chlorine uptake is reduced under ourlower-pressure conditions, along with an increased
surface selectivity, evidenced by the sensitivity of
the C(1s) spectral profiles to the XPS take off an-
gle. The surface selectivity of the chlorination
process under our experimental conditions was
further evidenced by the fact that ATR measure-
ments failed to detect any IR intensity between 600
and 800 cm�1 associated with C–Cl stretchingmodes.
The greater surface selectivity and lower maxi-
mum chlorine uptake observed for PE chlorination
in the present investigation is in fact in agreement
with the previously observed effect of chlorine
pressure (50 vs. 760 Torr) on the photochlorina-
tion of PE [15]. Thus, the maximum chlorine up-
take determined by XPS (sensitive to the chlorinecontent on the nm length scale) was found to be
similar at both 50 and 760 Torr of Cl2, although
the maximum extent of chlorination measured by
ATR (sensitive to chlorination on the lm length
scale) and from gravimetric measurements was
significantly enhanced at 760 Torr [15]. Despite the
different means used to generate atomic chlorine in
the present study (thermal rather than photo-chemical), it therefore appears that the incident
chlorine radical flux is a significant factor in de-
termining both the maximum chlorine content and
surface selectivity of the reaction.
If steric effects alone were responsible for the
observed absence of complete chlorination in the
near surface region, the extent of chlorination
should be independent of the incident chlorineradical flux. Consequently, the influence of radical
flux on the chlorination process suggests that other
factors are operative. One possible explanation
that accounts for the influence of radical flux on
the chlorination process is the role of cross-linking
reactions (e.g. C�
Hþ C�
H ! CH–CH), since they
serve to limit the concentration of otherwise
available alkyl radicals for subsequent chlorina-tion. Evidence for the role of cross-linking reac-
tions in radical interactions with hydrocarbon
films can be found in a study by Kluth et al. [31] on
the reactions of H atoms with alkanethiolate
SAMs, where the observed decrease in contact
angle with increasing hydrogen atom exposure was
interpreted as indirect evidence for the presence of
alkyl radical cross-linking reactions. To test theidea that cross-linking reactions can play a role in
the chlorination process we developed a simple
stochastic model of PE chlorination assuming a
constant radical flux and a chlorination process
based on reactions (1)–(4). Steric effects were
simulated by assuming that hydrogen abstraction
from CHCl and chlorine addition to C�
Cl only
occurs when these species are adjacent to –CH2–groups. In addition, the production of CCl2 species
is assumed to sterically hinder reactions of adja-
cent CH2 groups as suggested previously [17].
Three types of cross-linking reactions were in-
cluded, specifically:
C .
H+ C
H
. C
H
C
Hð5Þ
C .
H
C
Cl
.+ C
H
C
Clð6Þ
C .
Cl
C
Cl
.+ C
Cl
C
Clð7Þ

Table 1
Rate constants used in the kinetic stochastic simulation
Reaction Rate constant
(1) 6.3 · 10�4 s�1
(2) 8.3 · 10�3 s�1
(3) 8.3 · 10�9 s�1
(4) 5 · 10�3 s�1
(5) 4.2 · 10�6 cm2 mol�1 s�1
(6) 4.2 · 10�6 cm2 mol�1 s�1
(7) 4.2 · 10�6 cm2 mol�1 s�1
J. Torres et al. / Surface Science 543 (2003) 75–86 83
The rate constants employed in this stochastic
model are shown in Table 1. The agreement be-
tween the Cl(2s)/C(1s) ratios measured experi-
mentally at a take off angle of 45� and those
calculated from this model were used as a guide tothe overall accuracy of the model. In addition, the
rate of hydrogen abstraction from methylene
groups (reaction (1)) was constrained to lie within a
factor of two of the value measured experimentally
from the loss of CC/CH2 groups during chlorina-
tion (Fig. 2(a)) [15]. Furthermore, chlorine addi-
tion reactions (steps (2) and (4)) were assumed to
have rate constants at least one order of magnitudehigher than the H abstraction reactions (steps (1)
and (3)). Reactions (1)–(4) that involve chlorine
atoms were treated as pseudo-first-order processes
due to the constant chlorine radical flux during
experiments, while reactions (5)–(7) were treated as
bimolecular second order processes. A comparison
of the Cl(2s)/C(1s) ratios measured experimentally
and those derived from the kinetic model outlinedin steps (1)–(7) is shown in Fig. 2(c).
Fig. 2(c) illustrates the qualitative agreement
between the Cl(2s)/C(1s) ratios measured experi-
mentally and those determined from the kinetic
model as a function of chlorine atom exposure
as well as the maximum extent of chlorination.
In addition, the limiting distribution of unreacted
carbon atoms (CH2/CC) as well as the mono(–CHCl–) and dichlorinated (–CCl2–) species can
be reproduced from this simulation. Thus, employ-
ing the rate constants listed in Table 1, the limiting
composition of the chlorinated overlayer was cal-
culated to be 65.2% CH2, 24.4% CHCl and 10.4%
CCl2, which compares favorably to the 62.5% CC/
CH2, 29.4% CHCl and 8.1% CCl2 obtained ex-
perimentally.
To simulate the effect of increasing chlorine
radical flux, the rate constants for those reactions
involving chlorine atoms were increased by a fac-
tor of 10 (reactions (1)–(4)). This value was cho-
sen to reflect the relative increase in the rate of
methylene group loss measured in higher-pressurephotochlorination experiments [15,17]. Under these
conditions of higher (fixed) incident chlorine rad-
ical flux, a more rapid rate and higher saturation
level of chlorination was observed (Fig. 2(c)),
consistent with previous photochlorination exper-
iments on PE [17]. In addition, the limiting com-
position of the chlorinated overlayer calculated
from our stochastic model changed to 46.6% CC/CH2, 37.7% CHCl and 15.7% CCl2 reflecting an
increase in the extent of chlorination as cross-
linking processes become less significant. This
change in the distribution of CC/CH2, CHCl and
CCl2 species is qualitatively similar to the limiting
values of 40% CC/CH2, 40% CHCl and 20% CCl2obtained by Elman et al. [17] in their higher-pres-
sure study. It should be noted that the chemicalcomposition of the film for both low- and high-
chlorine atom fluxes are derived from XPS mea-
surements of the C(1s) region on high-density PE
recorded at comparable take off angles.
The dependence of the total Cl uptake on the
incident chlorine radical flux results from the fact
that the –C�
H– group never reaches a steady-state
concentration. Under these conditions, the productpartitioning between –C
�
H– group chlorination
(reaction (2)) and bimolecular cross-linking pro-
cesses (reactions (5)–(7)) is sensitive to the incident
chlorine radical flux, effectively limiting the maxi-
mum chlorine content at lower incident fluxes. This
fact is illustrated in Fig. 2(c), where the Cl/C ratio
calculated from the model is shown in the low-
pressure limit (solid line) and the high-pressure limit(dashed line). Despite the inherent simplicity of our
model, the results clearly point to the role that
cross-linking reactions can play in limiting the ex-
tent of chlorination at lower incident radical fluxes.
During chlorination the effective flux of chlo-
rine atoms will in fact decrease as a function of
increasing distance below the PE surface. Thus,
the constant flux of chlorine radicals assumed inthe previous section should be regarded as an av-
erage flux within the PE substrate. As a result of

84 J. Torres et al. / Surface Science 543 (2003) 75–86
this depth dependence on the chlorine flux, the
extent of chlorination is also expected to decrease
below the surface as the significance of cross-
linking reactions increases, producing a concen-
tration gradient of CClx (x ¼ 1, 2) species. For the
low chlorine fluxes that characterize the presentstudy, this concentration gradient of CClx (x ¼ 1,
2) species is sufficiently localized in the near sur-
face region that it can be detected by angle re-
solved XPS measurements (Figs. 2 and 3). In
contrast, the C(1s) XPS profile was found to be
independent of the take off angle for higher-pres-
sure (flux) photochlorination experiments on PE.
The greater surface selectively of PE chlorinationunder our conditions of lower-chlorine radical flux
is also consistent with the lack of observable ATR
intensity associated with C–Cl incorporation
into the film, in contrast to ATR results obtained
under higher-pressure photochlorination condi-
tions [15]. It should be noted, however, that the
lower incident kinetic energy anticipated for chlo-
rine radicals produced from thermal rather thanphotochemical dissociation of Cl2 may also con-
tribute to the enhanced surface selectivity and
lower maximum chlorine uptake observed in the
present investigation.
In general, results from the present investiga-
tion suggest that free radical reactions with poly-
mers under vacuum conditions, where the flux of
reactive species is low, may offer a general route tomodify polymer surfaces with an extremely high
degree of surface selectivity. In polymer surface
modification processes, the ability to control sur-
face selectivity is important in the development
of optimal treatment strategies for a number of
properties, including gas permeability. Similarly,
the reaction of chlorine atoms with hydrocarbon
films under these low-pressure conditions may of-fer a means to selectively chlorinate the very top-
most layers of ordered molecular assemblies such
as SAMs as a convenient way to produce func-
tionalized monolayers.
4.1. A comparison of chlorine and oxygen radical
interactions with hydrocarbon thin films
Fig. 3 shows that the interaction of atomic
chlorine and atomic oxygen with PE substrates
both lead to a concentration gradient of chlorine
and oxygen containing species within the near
surface region. Despite these similarities, experi-
ments carried out on SAMs indicate that distinctly
different reaction pathways characterize the inter-
action of atomic chlorine and atomic oxygen withhydrocarbon surfaces.
In this investigation, SAMs are employed as
models for polymeric interfaces [20], where we
have exploited the fact that changes in the film�sthickness can be conveniently followed by moni-
toring the C(1s)/Au(4f) ratio, an advantage not
possible in experiments on bulk polymeric sub-
strates. Furthermore, the packing density of al-kanethiolate SAMs is comparable to PE [32]. In
the present study, Fig. 4(a) indicates that the re-
action with Cl radicals proceeds in the absence
of C–C bond cleavage. Chlorine radical-induced
carbon-carbon bond cleavage would produce a
decrease in the C(1s)/Au(4f) ratio due to the pro-
duction of volatile carbon-containing fragments
that were no longer tethered to the underlyingsubstrate [26]. This behavior is inconsistent with
the essentially constant C(1s)/Au(4f) ratio ob-
served during the interaction of atomic chlorine
with the SAM (Fig. 4(a)). The absence of –CCl3group production in this and other related studies
[15,17] is also consistent with the idea that chlorine
radicals can activate C–H bond cleavage via hy-
drogen atom abstraction but are unable to cleaveC–C bonds. The inability of atomic chlorine to
induce efficient C–C bond cleavage is also similar
to the results obtained on the interaction of fluo-
rine atoms with alkanethiolate SAMs [33] and in-
dicates that during chlorine and fluorine plasma
treatments, the removal of hydrocarbon contami-
nation is a result of ion bombardment.
In contrast to the reaction of atomic Cl withhydrocarbon films, Fig. 4(b) shows that the C(1s)/
Au(4f) ratio decreases monotonically during the
interaction of C16-SAMs with atomic oxygen, in-
dicating the presence of substrate erosion. This is
also reflected by the variation in the C–O(1s) XPS
area during exposure to atomic oxygen. Initially
the C–O(1s) signal increases as oxygen-containing
functional groups are formed at the hydrocarbonsurface, while upon prolonged exposure to atomic
oxygen the C–O(1s) signal decreases as volatile

J. Torres et al. / Surface Science 543 (2003) 75–86 85
carbon containing oxygen species (e.g. CO2) de-
sorb from the film [20]. The ability of atomic ox-
ygen to etch the hydrocarbon surface is related to
its ability to form linkages such as R–O–(C@O)–R
that can then react further via decarboxylation to
produce volatile carbon containing species, re-sulting in the formation of a steady-state etch
front. In contrast, the reactions of atomic chlorine
with hydrocarbon films are limited to those illus-
trated by reactions (1)–(4). Thus, the thin layer of
carbon-containing oxygen functionality that de-
velops during the interaction of atomic oxygen
with hydrocarbon films is at least in part a con-
sequence of the continuous etching of the film. Incontrast, the surface selectivity during chlorination
is postulated to derive from a combination of
steric and kinetic factors associated with the
chlorination process.
Based upon the reactivity of atomic chlorine
and oxygen with the prototypical C16-SAMs, a
model of radical interaction with PE can be de-
veloped; this is shown schematically in Fig. 5. Inthe case of atomic chlorine reactions with PE, a
(a)
(b)
Fig. 5. Schematic depiction of PE surface reactions during interaction
The evolution of the PE surface is depicted following specified radical
been depicted in an ordered array for ease of visualization.
partially chlorinated overlayer is produced con-
taining both CCl and CCl2 groups in the absence
of substrate erosion, with the extent of chlorina-
tion greatest at the vacuum/film interface (Fig.
5(a)). The reactions of atomic oxygen (O3P) with
hydrocarbon films in contrast, results in oxygenincorporation into the surface, followed by sub-
strate erosion with the ejection of volatile carbon
containing oxygen species (e.g. CO2) (Fig. 5(b)).
Thus, while the uptake of chlorine atoms from the
gas phase only occurs up until a point when the Cl/
C ratio reaches its limiting value, oxygen atoms
react continuously with the hydrocarbon film as
erosion proceeds.
5. Conclusions
Exposure of PE to atomic chlorine (Cl(2P))
under vacuum conditions leads to the production
of CCl and CCl2 groups. Compared to higher-
pressure studies on PE photochlorination, themaximum extent of chlorination is reduced while
with: (a) atomic chlorine (Cl(2P)) and (b) atomic oxygen (O(3P)).
exposures. It should be noted that the hydrocarbon chains have

86 J. Torres et al. / Surface Science 543 (2003) 75–86
the surface selectivity is enhanced. These results
have been rationalized on the basis of a simple
stochastic model of the PE chlorination process
that incorporates steric effects associated with the
production of mono and dichlorinated carbon at-
oms as well as cross-linking reactions betweencarbon-containing radicals. The surface selectivity
of the chlorination process is manifested by angle
resolved XPS measurements that illustrate the in-
homogeneous distribution of CCl and CCl2 groups
within the near surface region. A similar concen-
tration gradient is observed during the reaction of
atomic oxygen (O(3P)) with PE. Differences in the
reactivity of atomic chlorine and oxygen with hy-drocarbon films are manifested, however, in ex-
periments carried out on SAMs, used as models
for polymeric interfaces. Results from these studies
reveal that chlorination proceeds without sub-
strate erosion while the incorporation of new oxy-
gen functionality during reactions with atomic
oxygen competes with carbon loss from the film.
Acknowledgements
Support for this research was provided by a
National Science Foundation CAREER award (#
9985372) and a grant from the Petroleum Research
Fund (PRF # 35281–G5, G6) administered
through the American Chemical Society.
References
[1] F. Garbassi, M. Morra, E. Occhiello, Polymer Surfaces
from Physics to Technology, Revised and Updated Edi-
tion, John Wiley and Sons, Chichester, 1998.
[2] D.E. Bergbreiter, Prog. Polym. Sci. 19 (1994) 529.
[3] M.-C. Cahn, T.-M. Ko, H. Hiraoka, Surf. Sci. Rep. 24
(1996) 1.
[4] T. Nakagawa, S. Yamada, J. Appl. Pol. Sci. 16 (1972)
1997.
[5] J. Csernica, D.B. Rhodes, J. Polym. Eng. 19 (1999) 1.
[6] M.Y. Jung, S.S. Choi, J.W. Kim, D.W. Kim, Surf. Sci.
482–485 (2001) 1119.
[7] C.R.J. Eddy, D. Leonhardt, S.R. Douglass, B.D. Thoms,
V.A. Shamamian, J.E. Butler, J. Vac. Sci. Technol. A 17
(1999) 38.
[8] J.W. Coburn, H.F. Winters, J. Appl. Phys. 50 (1979) 3189.
[9] J.H. Kim, S.I. Woo, Appl. Surf. Sci. 156 (2000) 9.
[10] A. Ricard, Reactive Plasmas, Socite Francaise du Vide,
Paris, 1996.
[11] M. Kondow, K. Uomi, K. Hosomi, T. Mozume, Jpn.
J. Appl. Phys. 33 (1994) L1056.
[12] H. Ikeda, D. Matsushita, S. Naito, K. Ohmori, A. Sakai,
S. Zaima, Y. Yasuda, Jpn. J. Appl. Phys. 41 (2002) 2463.
[13] C.J. Chu, M.P. D�Evelyn, R.H. Hauge, J.L. Margrave,
J. Appl. Phys. 70 (1991) 1695.
[14] H.L. Duan, G.A. Zaharias, S.F. Bent, Thin Solid Films
395 (2001) 36.
[15] E.M. Cross, T.J. McCarthy, Macromolecules 25 (1992)
2603.
[16] M.B. Levitskii, Y.I. Kuryatnikov, R.V. Dzhagatspanyan,
Khim. Vysok. Energ. 20 (1986) 159.
[17] J.F. Elman, L.J. Gerenser, K.E. Goppert-Berarducci, J.M.
Pochan, Macromolecules 23 (1990) 3922.
[18] S. Balamurugan, A.B. Mandale, S. Badrinarayanan, S.P.
Vernekar, Polymer 42 (2001) 2501.
[19] M.R. Linford, C.E.D. Chidsey, Langmuir 18 (2002) 6217.
[20] J. Torres, C.C. Perry, S.J. Bransfield, D.H. Fairbrother,
J. Phys. Chem. B 106 (2002) 6265.
[21] The Handbook of X-ray Photoelectron Spectroscopy,
Perkin Elmer Corporation, 1979.
[22] IBM Corporation, Chemical Kinetics Simulator, Available
from <http://www.almaden.ibm.com/st/computational_
science/ck/msim/>, 1996.
[23] D.T. Gillespie, Comput. Phys. 22 (1976) 403.
[24] D.T. Gillespie, J. Phys. Chem. 81 (1977) 2340.
[25] A.J. Wagner, S. Carlo, C. Vecitis, D.H. Fairbrother,
Langmuir 18 (2002) 1542.
[26] A.J. Wagner, K. Han, A.L. Vaught, D.H. Fairbrother,
J. Phys. Chem. B 104 (2000) 3291.
[27] M. Wirde, U. Gelius, T. Dunbar, D.L. Allara, Nucl.
Instrum. Methods Phys. Res. 131 (1997) 245.
[28] S.R. Carlo, A. Wagner, D.H. Fairbrother, J. Phys. Chem.
B 104 (2000) 6633.
[29] G.J. Vancso, T.D. Allston, I. Chun, L.-S. Johansson, G.
Liu, P.F. Smith, Int. J. Polym. Anal. Character. 3 (1996)
89.
[30] D.J. Garton, T.K. Minton, M. Alagia, N. Balucani,
P. Casavecchia, G.G. Volpi, J. Chem. Phys. 112 (2000)
5975.
[31] G.J. Kluth, M.M. Sung, R. Maboudian, Langmuir 13
(1997) 6491.
[32] X. Qin, T. Tzvetkov, D.C. Jacobs, Nucl. Instrum. Methods
B 203 (2003) 130.
[33] G.N. Robinson, A. Freedman, Langmuir 11 (1995) 2600.