Embed Size (px)
Citation preview

— —< <
Integral Transformation with Low-OrderScaling for Large Local Second-OrderMøller]Plesset Calculations
GUNTRAM RAUHUT,1,U PETER PULAY,1 HANS-JOACHIM WERNER2
1 Department of Chemistry and Biochemistry, University of Arkansas, Fayetteville, Arkansas 727012 Institut fur Theoretische Chemie, Universitat Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart,¨ ¨Germany
Received 29 October 1997; accepted 5 March 1998
ABSTRACT: An algorithm is presented for the four-index transformation ofŽ .electron repulsion integrals to a localized molecular orbital MO basis. Unlike
in most programs, the first two indices are transformed in a single step. This andthe localization of the orbitals allows the efficient neglect of small contributionsat several points in the algorithm, leading to significant time savings. Thresholdsare applied to the following quantities: distant orbital pairs, the virtual spacebefore and after the orthogonalizing projection to the occupied space, and smallcontributions in the transformation. A series of calculations on medium-sizedmolecules has been used to determine appropriate thresholds that keep the
Ž .truncation errors small below 0.01% of the correlation energy in most cases .ŽBenchmarks for local second-order Møller]Plesset perturbation theory MP2;
.i.e., MP2 with a localized MO basis in the occupied subspace are presented forseveral large molecules with no symmetry, up to 975 contracted basis functions,and 60 atoms. These are among the largest MP2 calculations performed on a
Ž .single processor. The computational time with constant basis set scales with a
Correspondence to: G. RauhutU Present address: Institut fur Theoretische Chemie, Univer-¨
sitat Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany¨Contractrgrant sponsors: Deutsche Forschungsgemein-
schaft; Fonds der Chemischen IndustrieContractrgrant sponsor: Air Force Office for Scientific Re-
search; contractrgrant number: F49620-94-1Contractrgrant sponsor: National Science Foundation; con-
tractrgrant numbers: CHE-9319929, CHE-9707202Contractrgrant sponsor: European Union TMR; contractr
grant number: FMRX-CT96-088
( )Journal of Computational Chemistry, Vol. 19, No. 11, 1241]1254 1998Q 1998 John Wiley & Sons, Inc. CCC 0192-8651 / 98 / 111241-14

RAUHUT, PULAY, AND WERNER
somewhat lower than cubic power of the molecular size, and the memorydemand is moderate even for large molecules, making calculations that requirea supercomputer for the traditional MP2 feasible on workstations. Q 1998 JohnWiley & Sons, Inc. J Comput Chem 19: 1241]1254, 1998
Keywords: integral transformation; low-order scaling; second-orderMøller]Plesset calculations
Introduction
Žor low cost post-Hartree]Fock methods e.g.,F second-order Møller]Plesset perturbation.theory, MP2 , the transformation of electron repul-
Ž . Ž .sion integrals ERIs from an atomic orbital AOŽ .to molecular orbital MO basis is the most de-
manding step, in regard to both computing timeand disk space. Thus, there have been numeroussuggestions to improve this transformation.1] 16
Parallel implementations12 ] 16 have revived thetopic quite recently. For MP2, only exchange inte-
Ž < . Žgrals of the form ai jb are necessary i, j denote.occupied orbitals and a, b virtuals , and in the
following we will restrict ourselves to this type.Almost all proposed algorithms have in commonthat the four-index transformation is split into fourquarter transformations11:
Ž < . Ž .ai jb s C C C C mr N sn .Ý Ý Ý Ýma n b r i s jž /ž /ž /m n r s
Ž .1
This partitioning reduces the formal scaling fromŽ 2 2 4. Ž 2 6. Ž 4.O n V N f O n N to O nN . In the above,
C is the matrix of MO coefficients; m, n , r, and sdenote AO labels; N is the number of contractedbasis functions; V f N is the number of virtualorbitals; and n is the number of occupied orbitals.
Ž .As n g N ratios Nrn f 5]10 are common , mostof the computational effort is required in the first
Ž .quarter transformation, which requires nO I op-Ž .erations, where O I is proportional to the number
w Ž .of nonneglected AO integrals in practice O I is2 3 xusually between N and N . The main bottleneck
of the traditional algorithm is the memory require-ment. The fastest algorithm, which fully exploitsthe eight-fold permutational symmetry of the integ-rals,5 still requires the storage of all half-trans-
Ž 2 2formed integrals in high-speed memory n N r2. 2memory locations and needs in addition nSN
memory locations for intermediate storage of quar-
Žter transformed integrals. S is the maximum shell.size and is independent of the molecular size. The
storage for the quarter transformed integrals canbe reduced to nS2N at the cost of two integralevaluations, but because all transformed integralsmust be held in memory, the total memory stillscales with the fourth power of the molecular size.Alternatively, one can reduce the total memoryrequirement to n2S2, at the cost of four integralevaluations and a resorting of the half-transformedintegrals, which requires n2N 2r2 words of diskspace.7, 17 This approach is limited by disk spaceand is probably the best conventional method forcalculations up to about 1000 basis functions and100 occupied orbitals in C symmetry. However, it1is evident that for such large cases alternativeapproaches have to be investigated.
In this article we explore the simultaneoustransformation of two indices in the first half-transformation,
i jŽ . Ž < . Ž < . Ž .mnK s m i jn s C C mr sn . 2Ý r i s jrs
This method was analyzed earlier by Taylor,6 whonotes that the optimum memory for a two-index transformation is n2N 2r2 but the minimummemory demand is only N 2, although at the con-siderable cost of n2r2 integral evaluations. Thecomputational work in the simultaneous two-index transformation scales formally more steeplyw 2 Ž .xn O I than in a series of consecutive one-indextransformations. However, although few of theorbital coefficients are negligible, many of themare small and a significant fraction of their prod-ucts is then very small. Precomputing upper limitsfor these products allows an efficient prescreeningfor both the integral evaluation and the transfor-mation. As will be shown, this offsets the poorerformal scaling for large systems.
This method is particularly useful if localizedMOs are used. First, the correlation of electrons in
Ž .distant localized orbitals ‘‘distant pairs’’ is a weakdispersionlike interaction, and its leading term de-
VOL. 19, NO. 111242

INTEGRAL TRANSFORMATION FOR LARGE LOCAL MP2 CALCULATIONS
creases with the inverse sixth power of the dis-tance between the orbital centers. Depending onthis distance, very distant pairs can be neglected,calculated approximately,18 or even modeled em-pirically.19 This asymptotically reduces the num-
Ž . Ž . Ž 2 .ber of orbital pairs ij in eq. 2 from O n toŽ . 2 2 2O n , and the storage from n N r2 to f ? nN ,
where f ? n is the number of correlated orbitalŽ .pairs simply pairs in the following . For large
molecules f is constant, and thus f ? n shouldgrow linearly with the molecular size. Second, forsufficiently large molecules the number of AOsŽ . w Ž .xm, n see eq. 2 needed for an appropriate de-
Ž .scription of a given orbital pair ij becomes con-stant and independent of the molecular size aswell. This reduces both the computational workand the storage requirement. Denoting the averagesize of the local AO space per pair by L, thememory requirement is reduced to f ? nL2 ; asymp-totically it scales only linearly with the molecularsize, even for a single integral pass computation.
The method described in the present work isbased on the local correlation approach introducedby Saebø and Pulay.20 ] 22 It was originally con-ceived for higher order correlation methods, andthus little attention was paid to the integral trans-formation. By contrast, the present article focuseson MP2 calculations and the associated integraltransformation when applied to large systems byusing an integral-direct approach.23 The local cor-relation method has been extended to the fullcoupled cluster singles and doubles level24 andcoupled with the pseudospectral formalism.25, 26
Analytical local MP2 gradients have been devel-oped recently.27 Other, somewhat different localcorrelation methods are also being pursued.28, 29
The weak orthogonality concept of Szalewicz et al.30
could be used within the framework of local MP2theory to avoid the explicit projection of the AObasis. We have not yet explored this possibility.An alternative approach to the application of thelocal correlation concept to MP2 theory is the useof Laplace transform orbitals as presented by Haser¨and Almlof.31 This method has recently been com-¨pared with the theory outlined above.32, 33 Approx-imating electron repulsion integrals can also beused to speed up MP2 calculations. This resolution
Ž .of identity RI approach was explored by variousresearchers.34 ] 37 Although this approach retains theformal fifth power scaling of the traditional MP2
Žmethod, it can lead to significant speedups up to.an order of magnitude , depending on the com-
pleteness of the auxiliary basis set used to approxi-mate the charge densities.
Local MP2 Theory
In the local MP2 method,20 the Fock matrix isnondiagonal; thus, the pair correlation coefficientsŽ . i jamplitudes T have to be determined from ap q
coupled set of linear equations. In a compact ma-trix formalism,38 using generator state spin adap-tation for closed shells,39 these equations are
R i j s K i j q FT i jS q ST i j Fk j i k Ž .y S f T q f T S s 0. 3Ý i k k j
k
Ž . i jHere i, j, k are internal occupied MO labels, Rare residuum matrices, and F and S are the Fockand overlap matrices, respectively. f is the Focki kmatrix element between orbitals i and k, and K i j
is the internal exchange matrix of the orbital pairij.
Ž .In eq. 3 all matrices are defined in a basis of< :projected AOs p , which are orthogonalized
against the occupied space but nonorthogonalamong themselves. In terms of the basis functionsŽ . < :AOs m the projected orbitals are defined as
< : < : Ž .p s P m . 4Ý m pm
The projection matrix given by
1 Ž .P s 1 y DS, 52
where D is the closed-shell density matrix and SŽ .the overlap matrix in the unprojected AO basis.
The exchange matrices K i j in the projected basisare defined as
i j i jŽ < . Ž .K s pi jq s P K P , 6Ýp q m p mn n qmn
i j Ž < .where K s m i jn are the half-transformed in-mn
Ž .tegrals defined in eq. 2 . The transformation of Fand S into the projected basis is analogous. In the
Ž .following, eq. 6 is denoted as the second half-transformation.
Due to the coupling terms introduced by thenondiagonal Fock matrix elements F and f , thep q i k
Ž .set of eq. 3 has to be solved iteratively. To achievefast convergence, it is necessary to transform theresiduum matrices R i j to pair-adapted intermedi-
JOURNAL OF COMPUTATIONAL CHEMISTRY 1243

RAUHUT, PULAY, AND WERNER
ate pseudocanonical MO basis sets, perform theupdates of the amplitude matrices T i j in this form,and transform them back to the original projectednonorthogonal AO basis.21 This procedure has beenrecently described in detail for the local coupledcluster method.24 Because the iteration is per-formed in the small projected basis and only theamplitudes have to reside readily available inmemory, there are essentially no memory limita-tions in this step. In our implementation of theiteration we used a dynamic update procedureŽi.e., each amplitude matrix is updated immedi-ately after the evaluation of its corresponding
.residue . This has the advantage that the residuummatrices do not have to be stored, and it alsoconverges significantly faster than static updatingin which all amplitudes are updated at once. DIISconvergence acceleration40 cannot be applied withdynamic updating, but sorting the pairs accordingto their coupling to other pairs accelerates conver-gence efficiently in a very simple manner. In mostcases 5]6 iterations are sufficient to achieve anaccuracy of 5 = 10y8 E , which is the default con-hvergence threshold applied in all calculations andtimings presented below.
Using the residual matrices R i j and amplitudesT i j one can compute the Hylleraas functional
i j i j ˜i j Ž .E s K q R T , 7Ž .Ý Ýcorr p q p q p qpqij
˜ i jwhere the contravariant coefficient matrices T aredefined as
Ti j i j ji ji i j˜ w x Ž .T s 2T y T with T s T . 8
The Hylleraas functional gives for any choice ofthe amplitudes an upper bound to the MP2 en-ergy. For the converged amplitudes T i j the residualmatrices R i j vanish and the MP2 correlation en-ergy is obtained simply as
2 i j ˜i jE s K TÝÝcorr p q p qpqij
2i j i j˜ Ž .s K T s e . 9Ý Ý Ýp q p q i j1 q di j pqiGj iGj
ŽWithout further approximations i.e., no restriction.of the virtual space, etc. , these formulas are ex-
actly equivalent to canonical MP2 theory; and thetotal correlation energies obtained with the localand canonical treatments agree within numericalaccuracy. The individual pair energies e are dif-i jferent, however.
In the local correlation approach two distinctŽ .approximations are introduced. First, for a pair ij
substitutions are limited to projected orbitals p, qthat lie in the vicinity of the two localized orbitalsi and j. This leads to very sparse amplitude matri-ces, because only those elements T i j are nonzerop q
w xfor which p, q belong to the pair domain ij . Be-cause the same restriction applies to the residual
i j Ž . i jmatrices R in eq. 3 , only elements K withp q p q
w xp, q g ij are needed. This fact can be exploited toreduce the memory requirements and the compu-tational effort in the integral transformation. Therestriction of the configuration space to projectedfunctions in direct neighborhood of the correlatedorbitals i and j may thus be considered as aphysically well-defined configuration selectionscheme. This truncation of the correlation space
Ž .causes a small loss 1]2% in the correlation en-ergy.22 The fraction of correlation energy loss de-creases with increasing basis set quality and can beat least partially interpreted as arising from re-moving the intramolecular basis set superpositionerror.41 Second, distant pairs can be neglected; theexchange matrices K i j need to be computed onlyp q
Ž .for a subset of orbital pairs ij . As will be dis-cussed in the next section, this leads to furthersavings in the integral transformation.
With these two approximations, the correlationenergy takes the form
2Ž2. i j i j˜ Ž .E s K T , 10Ý Ýcorr p q p q1 q di jŽ . w xij gP p , qg ij
Ž .where P denotes a list of pairs ij with i G j. Dueto its stationary property, the Hylleraas functionalcomputed with the reduced set of amplitudes isstill an upper bound to the full MP2 energy. Inaddition to these reductions of the configurationspace, which have been used and tested in previ-ous local correlation methods,20 ] 22, 24 we will in-vestigate in this article two additional approxima-tions of more numerical nature: truncations of thesummations in the first and second halves of the
Ž . Ž .integral transformation, eqs. 2 and 6 , respec-tively. These two approximations influence the ac-curacy of the exchange integrals K i j , and theyp qmay therefore violate the upper bound property ofthe Hylleraas functional. In fact, as will be dis-cussed in the following sections, the correlationenergy is quite sensitive to such approximationsand they must therefore be tested carefully. Theimplementation of these approximations will bedescribed in more detail in the next section.
VOL. 19, NO. 111244

INTEGRAL TRANSFORMATION FOR LARGE LOCAL MP2 CALCULATIONS
Transformation Algorithm
Our local MP2 program42 is based on the shell-driven integral package of the quantum chemistryprogram TX96 in which a shell is defined as a setof contracted functions built from the same set ofprimitive radial Gaussian functions. In Pople’s ba-sis sets, s and p functions sharing the same primi-tive Gaussian exponents are grouped in shells of LŽ .or sp type. In the numerical approximations dis-cussed below whole shells of AO are always con-sidered. This corresponds to the logic of integralcomputation and guarantees strict rotational in-variance. As outlined above, we use four differenttruncation schemes to reduce the step scaling ofthe traditional integral transformation algorithms.Those schemes are described in the following. Thethird and the fourth are new and essential to thepresent code.
1. TRUNCATION OF PROJECTEDVIRTUAL SPACE
Ž .Substitutions from an orbital pair ij are re-w xstricted to a pair domain ij of projected orbitals
w xp, q. The pair domains ij are chosen as the directw x w x 21sum of the orbital domains i and j , resulting in
square matrices K i j . An automatic method for thep q
selection of orbital domains was proposed byBoughton and Pulay,43 and this method has beenused in the present work throughout. The onlydifference from ref. 43 is that we do not restrict thedomains to four atoms. While most s bonds andlone pairs are localized on only one or two atoms,in extended p systems, like the azo-dye in our testsuite, a few orbitals may remain quite delocalized,which consequently leads to larger domains. Theselection threshold we used in the Boughton]Pulayalgorithm was taken to be the recommended valueof 0.02 and was not varied in this work, because itwas explored previously by us21, 24 and others.25, 26
2. NEGLECTING DISTANT ORBITAL PAIRS
Correlation between pairs of distant orbitalsŽ .weak and distant pairs can be neglected or calcu-lated approximately. For well-localized orbitals theleading term in the correlation energy betweendistant orbitals diminishes with the inverse sixthpower of the distance between orbital charge cen-ters and is usually below a few mE if the distanceh
˚of the orbital centroids exceeds 5 A. In our older
programs, which were directed toward higher levelcalculations, weak pairs were identified simply byestimating their correlation energy at the lowerMP2 level. Because our goal here is to avoid thecalculation of MP2 energies, we need a simplercriterion. For well-localized orbitals the distancebetween their charge centroids is a good indicatorof the pair energy and can be used to identifydistant pairs. However, for poorly localized or-bitals this criterion is not very reliable. Therefore,in all calculations presented in this work, we usedthe criterion suggested by Hampel and Werner.24
It is the minimum distance between any of thew xatoms comprising the orbital domain i and those
w xthat are part of the domain j . In the calculationspresented here, distant pairs are dropped if theminimum distance exceeds a threshold T . Ne-dglecting distant pairs contributes much to the effi-ciency of local MP2 for large systems with respectto memory and computation time requirements.
The question arises whether the effect of ne-glecting many weak pairs may accumulate to asignificant value and may thus lead to inaccuraterelative energies. This is possible if two structuresare compared that differ greatly in overall shape,
Žsuch as a compact and an extended structure e.g.,.the chain, bowl, and icosahedric structures of C .20
In most cases, however, the total energy contribu-tion of distant pairs is approximately constant overthe energy surface and thus cancels in relativeenergies. As shown recently,19 it is possible tomodel the correlation energies of such pairs empir-ically; this can account for the bulk of weak paircorrelation energy and at least indicate whetherweak pair contributions are important. Anotherpossibility is to use multipole expansions, and toapproximate the exchange matrices for distantpairs by products of one-electron operator matrixelements. This approach currently is being ex-plored.18
3. NEGLECTING SMALL CONTRIBUTIONS INFIRST HALF-TRANSFORMATION
The sparsity of the SCF coefficients can be usedto reduce the computational effort in the first half-
w Ž .xtransformation eq. 2 . A shell quartet of integralsŽ .is skipped for a given pair ij if the product of the
maximum integral value I and the largestmaxproduct of MO coefficients involving these shellsis smaller than a threshold T :1
< i j <D Imax max Ž .F T . 1112 2C Pmean mean
JOURNAL OF COMPUTATIONAL CHEMISTRY 1245

RAUHUT, PULAY, AND WERNER
Di j is the product of the two largest MO coeffi-maxŽ .cients for a given orbital pair ij and shell pair,
and C and P are mean values of a selectedmean meanset of MO coefficients of the occupied orbitals and
Žthe projected functions, respectively cf. the Ap-.pendix . These were introduced to eliminate basis
set dependencies of the criterion caused by Di j .maxThe Appendix gives a detailed description of thiscriterion and its rationale. This threshold leads tosignificant time savings and is essential in our
Ž 6.algorithm because it eliminates the formal O Nscaling. It does not affect the memory demand.
4. PAIR SPECIFIC TRUNCATION OF VIRTUALAO SPACE PRIOR TO PROJECTION
Truncations of the summations in the secondw Ž .xhalf-transformation eq. 6 limit the AO space in
i jwhich the exchange matrices K have to be com-puted. This benefits mainly the first half-transfor-mation, which has to be carried out for only a
Ž .limited number of AOs. Equation 6 is approxi-mated by
i j i jŽ < . Ž .K s pi jq s P K P , 12Ýp q m p mn n qw xmng ij AO
w x Ž .where ij denotes an AO domain for pair ijAOŽ w xin opposition to the usual pair domain ij in
.projected basis . This truncation reduces the com-putational effort in the first half-transformationfrom formally f ? nN 4 to f ? nN 2L2, where L is theaverage dimension of the AO domains and f ? n is
Ž . Žthe number of orbital pairs ij . The remainingfactor N 2 is reduced by the truncations described
.in the previous section. Furthermore, because theindices m, n of the half-transformed integrals isreduced from f ? nN 2 to f ? nL2. This is essential forthe present algorithm because it eliminates thefourth-order dependence of the memory require-ment, because L is significantly smaller than thefull AO basis for large systems and reaches anasymptotic limit that only depends on the basis setbut not on the molecular size. An example of theaverage size of L in comparison to the full AObasis and the average size of the pair domains inprojected AOs has been provided for a sequence ofglycine oligomers in Figure 1. Clearly, the accu-racy of the correlation energy is sensitive to thistruncation, and therefore L must be larger than thedomain of pair ij in projected orbitals but becomesconstant for extended systems. The reason for thissensitivity is twofold. First, limiting the summa-
Ž .tions in eq. 6 means approximating the projectedorbitals by subsets of AOs, and this may cause a
FIGURE 1. Comparison of the average size of the pairdomains in AOs and projected bases relative to thenumber of basis functions in an ascending sequence ofglycine oligomers. Results are given for a cc-pVDZ basis.
violation of the strong orthogonality condition.Second, the elements of the projection matrix maybe large if the projected functions are renormal-
i jized, and therefore neglecting small integrals Kmn
may lead to errors in the transformed integrals. Asalready mentioned, inaccuracies of the trans-formed integrals cause loss of the variational prop-erty of the Hylleraas functional, which is mini-mized in the iterative local MP2 procedure. Thecriteria for selecting the truncated AO space mustbe chosen carefully as shown in our tests.
w xThese AO pair domains ij are taken to beAOw x w xdirect sums of AO orbital domains i and j ,AO AO
which are selected as follows. Our first criterion isbased on the orthogonality condition
T² < : Ž . Ž .p ip i s P SC s 0. 13
The basis functions of a given shell S are includedŽ .in the AO domain of pair ij if
1<Ž . < Ž .m iSC G T , 14Ý 2nS mgS
where n is the number of basis functions in theSshell and S is the overlap matrix. However, thiscriterion is not sufficient, because the overlap can
Ž . Ž .be approximately zero if the local symmetries< : < :of the functions i and p are different. The
impact of such cases is minimized by taking thecriterion for shells of AOs; if any function in the
VOL. 19, NO. 111246

INTEGRAL TRANSFORMATION FOR LARGE LOCAL MP2 CALCULATIONS
shell has the right symmetry, the whole shell isretained. The criterion may still fail, e.g., for theoverlap of a p-type MO with an s function. Ide-
² < < < <:ally, we would use the absolute overlap m ¬ ibetween a localized orbital and a basis function,but this is difficult to calculate. It can be simu-lated, however, by the additional condition
v2 2 2 Ž .X q Y q Z G T , 15'Ý m i m i m i 2nS mgS
where, for example,
Ž i. Ž i.² < < : Ž . Ž . Ž .m i m iX s m x y x i s XC y x SC , 16ˆm i
and Y and Z are defined analogously. Here x ,̂m i m i
y, and z are the coordinate operators; x Ž i., yŽ i., z Ž i.ˆ ˆare the coordinates of the charge center of thelocalized MO i; and v is a scaling factor that wasempirically taken to be 0.12 auy1 without furtherrefinement. These dipole integrals give nonzerovalues for a s AO and a s MO if they spatiallyoverlap. We have found it advantageous to use a
Žlower thresholds T by about a half-order of mag-2. Žnitude for diagonal and close orbital pairs i.e.,
.pairs that share one atom , while all other pairscan be treated slightly less accurately. Poorly local-ized orbitals in large p systems also require asharper threshold. Useful ranges for threshold T2will be discussed later.
As is clear from the description of these criteria,they leave space for experimentation and furth-er improvements. Even though these thresholdsneeded adjustment for some critical cases as dis-cussed below, they work in most cases. The wholetransformation can be summarized as follows:
1. First we create a list of correlated orbitalŽ .pairs ij . Distant pairs are neglected accord-
ing to threshold T as described above. Thisdlist is further reduced by other criteria in thesubsequent steps.
Ž <2. The integrals in a given shell quartet MR. Ž X. X X X XSN are sorted into matrices I sm n , r s
Ž < .mr sn , where Greek indices denote indi-vidual AOs and capital letters refer to shellsŽ .m g M, n g N, etc. Here and in the follow-ing the primes indicates quantities whosedimensions are determined by the shell sizes.The absolute AO indices m are related to the
X Žshell indices m by shell offsets i.e., the abso-lute address of the first basis function in
X.shell m .
Ž3. Permutational symmetry is used fully i.e.,.each integral is used eight times , transform-
ing the indices belonging to the shells RS,RN, MS, and MN. Each of these cases gives
Ž .two contributions supermatrix symmetry .In the following we outline the algorithmonly for one of the cases, the one in whichthe shells RS are transformed to occupiedorbitals. The other cases are entirely similar.
Ž .4. We generate a list of all nondistant pairs ijŽ .according to the following criteria: 1 thresh-
w Ž .xold T cf. eq. 11 is exceeded for the shells1Ž . w Ž .R, S; and 2 threshold T cf. eqs. 14 and2
Ž .x15 is exceeded for the shells M, N. For thisreduced list of pairs we generate the matrices
Ž X . X X Ž .X Xr s , i jD s C C , 17r i s j
Ž X . X X Ž .X Xr s , jiD s C C . 18r j s i
Here ij and ji are to be considered as indicesof a reduced pair list.
w5. The actual first half-transformation cf. eq.Ž .x2 uses efficient matrix multiplications
X X X Ž .K s I ? D . 19
Shells with high angular momentum func-tions give rise to larger blocks and thereforeincrease the efficiency of the matrix multipli-cation.
6. In the last step the elements of the smallmatrices KX, whose dimension is determinedby the sizes of shells M and N, are scatteredto their final locations in the internal ex-
i jchange matrices K .
Xi j i j X XŽ . Ž . Ž . Ž .mn mn mn , i jK s K q K , 20Xi j i j X XŽ . Ž . Ž . Ž .nm nm mn , jiK s K q K . 21
After all integral shell quartets have been pro-cessed, the exchange matrices are transformed into
Ž .the projected basis according to eq. 6 . This re-quires just two matrix multiplications per pair,and the time for this step is quite negligible. Sub-sequently, the MP2 equations are solved itera-tively and the energy is computed.
Essentially all of the logic is required to gener-ate the effective pair list and to address the localinternal exchange matrices. The transformation ofthe shell blocks is free of time consuming logic. Ifthere is sufficient memory to store all local ex-
i jchange matrices K , then this algorithm requiresonly a single pass through the integrals. However,
JOURNAL OF COMPUTATIONAL CHEMISTRY 1247

RAUHUT, PULAY, AND WERNER
if this condition is not met, then it is possible tocalculate the internal exchange matrices for a sub-set of orbital pairs by projecting them and storingthe resulting small local exchange matrices on disk.This algorithm is essentially open ended but re-quires the repeated evaluation of the integrals foreach batch of pairs. Parallelizations of the algo-rithm appears feasible and will be explored in thefuture.
Effects of Thresholds
This section discusses the effects of the fourtruncation schemes on the results. The simplest is
Žthreshold T i.e., the neglect of pair correlationd.energies for distant pairs . The loss of correlation
energy caused by this grows with the number ofpairs neglected. The appropriate value of T can bed
estimated by using averaged pair energies for vari-ous distance ranges. Because of these simple char-acteristics, and because such tests have been per-formed in previous work, we did not test thistruncation explicitly. Unless otherwise noted, we
˚have used a threshold of 6.0 A for all moleculestested, which appears to be a safe choice. ThePipek]Mezey localization scheme44 has been usedin all calculations, even though it leads to largerpair domains for extended p-systems than the Boyslocalization.45
Truncation schemes T and T have been tested1 2Žwith several basis sets for lactic acid 18 correlated
. Ž .orbitals and glycine 15 correlated orbitals . Theresults are summarized in Table I. Two families ofbasis sets have been used; Dunning’s recent cc-pVDZ and cc-pVTZ bases,46 and 6]31G and6]311G sets augmented with polarization func-tions.47 The first family is generally contractedwhile the second has segmented contraction.
The error in the correlation energy caused byŽthreshold T truncations in the first half transfor-1
.mation , shown in column 8 of Table I, is smallŽ .around 0.01% for most basis sets. However, forthe cc-pVDZ we find large errors of up to 0.09%.This larger error is caused by a steep increase ofthe average magnitude of the MO coefficients forsmall bases, leading to a failure of the correction
wterm for basis set independency i.e., the denomi-Ž .xnator in eq. 11 . Refining this empirical correction
should resolve this problem. However, the error isbasis set specific and vanishes for larger basesŽ .cc-pVTZ . Particularly critical are basis sets con-
Ž .taining diffuse functions. Tests on 6]31qG d ,Ž .6]311qqG d, p , and aug-cc-pVDZ bases have
shown that criterion T is not yet able to handle1these functions properly. Considering a 6]311qqŽ .G d, p calculation on lactic acid, T introduces an1
error of 0.335% of the total correlation energy; T2leads to an inaccuracy of only 0.002%, which is in
Žthe usual range as shown in Table I thresholds for.this calculation are the same as given in Table I .
Thus, using this product criterion is not yet recom-mended for basis sets containing diffuse functions.
ŽThreshold T truncation of the virtual AO space2.prior to projection appears to be less basis set
dependent than T . Generally contracted bases per-1form slightly worse than segmented functions, butthe former behave more systematically. For mostbasis sets tested the numerical error introduced bythreshold T is less than 0.01% of the total correla-2tion energy. The combination of thresholds T and1T leads to about a 0.02% error in the correlation2
Ž .energy with the exceptions discussed above . Forall molecules and basis sets tested, using thresh-olds T and T yielded correlation energies that1 2are too low. This is characteristic of truncationsthat violate the variation theorem and leads to afortuitous error cancellation when invoking thresh-old T for distant pairs, as seen in the benchmarkdcalculations below.
Stability with respect to molecular size has beenŽ .tested using the 6]31G d basis and a set of eight
test molecules given in Table II. The moleculeschosen show a variety of different functionalgroups and p systems of varying size. The aver-age numerical error for all molecules tested is lessthan 0.01% for the parameter values used. Wecould not find systematic tendencies with respectto the molecules size for either T or T . Individ-1 2ual characteristics of the molecules seem to have amuch stronger impact on the quality of the resultsthan just the molecular size. The somewhat largererror for coffein introduced by T can probably be2attributed to poor localization of the p system,
w x Ž .which indeed leads to pair domains ij up to 10 !atoms. In most cases pair domains include lessthan 6 atoms. However, the azo-dye51 given inTable II also has a very delocalized p system withpair domains up to 11 atoms and shows muchsmaller errors. Systems like these are stringenttests for the AO domain selection and certainlyshow the limits of our proposed criterion. Eventhough one can eliminate the error in coffein easilyby using a lower threshold, it would be desirableto change the criterion in such a way that a con-
VOL. 19, NO. 111248

INTEGRAL TRANSFORMATION FOR LARGE LOCAL MP2 CALCULATIONS
TABLE I.( y2 )LMP2 Correlation Energies and Numerical Errors in 10 % as Introduced by Thresholds Dependent on Basis
Set Used.
Test for T Test for T2 1
Ref.E E ELMP2 LMP2 LMP2
a 3CF T = 0.000 T = 0.000 T = 9.101 1 1b b cBasis Set No. T = 0.000 T = 0.004 Error T = 0.004 Error Error Ref.2 2 2
Lactic Acid
( )6-31G d 102 y0.89899090 y0.89899182 0.010 y0.89903361 0.475 0.476 47( )6-31G d, p 120 y0.93234490 y0.93244470 1.070 y0.93247581 1.404 0.334 47
( )6-311G d, p 144 y1.00426821 y1.00426179 0.064 y1.00428220 0.139 0.203 48( )6-311G 2d, p 174 y1.06923087 y1.06928618 0.517 y1.06933294 0.955 0.437 48( )6-311G 2d, 2p 192 y1.07547426 y1.07553284 0.545 y1.07557146 0.904 0.359 48( )6-311G 2df, 2pd 264 y1.17906340 y1.17922405 1.363 y1.17926544 1.714 0.351 48
[ ]cc-pVDZ 3s2p1d / 2s 96 y0.90681822 y0.90685535 0.409 y0.90761378 8.773 8.363 49[ ]cc-pVDZ 3s2p1d / 2s1p 114 y0.94204484 y0.94207054 0.273 y0.94252658 5.114 4.841 49[ ]cc-pVTZ 4s3p2d / 3s2p 192 y1.08878731 y1.08884136 0.496 y1.08888215 0.871 0.375 49[ ]cc-pVTZ 4s3p2d1f / 3s2p 234 y1.18243173 y1.18249497 0.543 y1.18252672 0.803 0.260 49[ ]cc-pVTZ 4s3p2d1f / 3s2p1d 264 y1.19065272 y1.19071300 0.506 y1.19075638 0.871 0.364 49
Glycine
( )6-31G d 85 y0.77338040 y0.77339406 0.177 y0.77340267 0.288 0.111 47( )6-31G d, p 100 y0.79907901 y0.79909218 0.165 y0.79910863 0.371 0.206 47
( )6-311G d, p 120 y0.85487988 y0.85487783 0.024 y0.85489175 0.139 0.163 48( )6-311G 2d, p 145 y0.90847054 y0.90848927 0.206 y0.90853979 0.762 0.556 48( )6-311G 2d, 2p 160 y0.91184823 y0.91186435 0.177 y0.91195605 1.182 1.006 48( )6-311G 2df, 2pd 220 y0.99718143 y0.99720337 0.220 y0.99736787 1.870 1.650 48
[ ]cc-pVDZ 3s2p1d / 2s 80 y0.77874478 y0.77881010 0.839 y0.77942891 8.785 7.946 49[ ]cc-pVDZ 3s2p1d / 2s1p 95 y0.80348198 y0.80353506 0.661 y0.80394824 5.803 5.142 49[ ]cc-pVTZ 4s3p2d / 3s2p 160 y0.92426394 y0.92430537 0.448 y0.92441831 1.670 1.222 49[ ]cc-pVTZ 4s3p2d1f / 3s2p 195 y1.00200186 y1.00205215 0.502 y1.00217422 1.720 1.218 49[ ]cc-pVTZ 4s3p2d1f / 3s2p1d 220 y1.00836828 y1.00841593 0.473 y1.00855113 1.813 1.341 49
Distant pairs have not been neglected, and the criterion of 0.02 for the virtual space selection as suggested by Boughton and Pulayhas been used throughout.43
a Number of contracted functions.b Relative to column 3.c Relative to column 4.
stant threshold would work in all cases. On thebasis of our tests, we recommend a threshold ofabout T s 5 ? 103 for the product criterion and1T s 0.004 for the AO space truncation. For gener-2ally contracted basis sets, somewhat sharperthresholds should be chosen.
Benchmark Calculations
For testing the performance of our code, wechose a sequence of glycine oligomers up to the
Žw x .hexamer C H NO ; x s 1]6 . The geometries3 5 2 xwere taken from an extended crystallographic unit
52 Ž .cell Fig. 2 . This sequence shows the scaling ofthe method directly, because the number of corre-lated orbitals and the number of contracted func-tions increase linearly with the number ofmonomer units. It is also useful to explore theeffect of molecular shape on the scaling. Up to thetrimer the shape of the cluster is essentially 2-
Ž .dimensional 2-D , while higher oligomers are 3-D.Ž .Lower dimension linear and planar structures are
expected to scale better than 3-D ones.Table III shows the memory requirements and
CPU times for two different basis sets: Dunning’scc-pVDZ and cc-pVTZ bases,46 the latter without dfunctions on the hydrogens. Two sets of thresholds
JOURNAL OF COMPUTATIONAL CHEMISTRY 1249
RAUHUT, PULAY, AND WERNER
TABLE II.( y2 )Correlation Energies and Numerical Errors in 10 % as Introduced by Thresholds Dependent on the
Molecular Size.
Test for T Test for T2 3
Ref.E E ELMP2 LMP2 LMP2
3No. No. T = 0.000 T = 0.000 T = 4 ? 101 1 1a b c c dMolecule CF CE T = 0.000 T = 0.004 Error T = 0.004 Error Error2 2 2
Acetone 72 24 y0.55480941 y0.55481146 0.037 y0.55481207 0.048 0.011Glycine 85 30 y0.77338040 y0.77339406 0.177 y0.77339973 0.250 0.073Lactic acid 102 36 y0.89899090 y0.89899182 0.010 y0.89902119 0.337 0.327Uracil 128 42 y1.15300124 y1.15301682 0.135 y1.15307166 0.611 0.476Adenine 160 50 y1.41041784 y1.41049751 0.565 y1.41054240 0.883 0.318Glycine dimer 170 60 y1.54524311 y1.54527699 0.219 y1.54528279 0.257 0.038Menthol 205 66 y1.45089391 y1.45089604 0.015 y1.45094701 0.366 0.351Coffein 230 74 y1.97181493 y1.97208814 1.386 y1.97222908 1.647 0.715
eHPAMP 262 80 y2.12201892 y2.12203600 0.080 y2.12211215 0.439 0.359
( )All calculations refer to a 6]31G d basis. Distant pairs have not been neglected, and the criterion of 0.02 for the virtual spaceselection as suggested by Boughton and Pulay has been used throughout.43
a Number of contracted functions.b Number of correlated electrons.c Relative to column 4.d Relative to column 5.e ( [( X ) ]4- 4 -hydroxyphenyl azo -N-methylpyridine.
have been chosen for the cc-pVDZ calculations toshow the variation of computing times and mem-ory requirements with the accuracy of the calcula-tions. The average dimension of the AO domainsw xij is also given. Even though the AO domainsAO
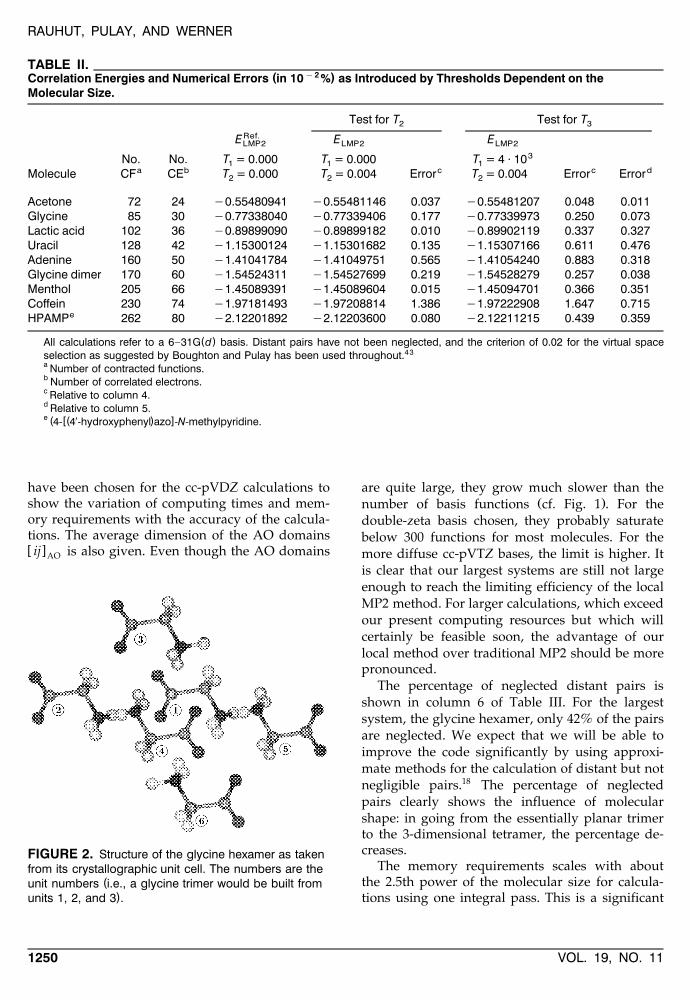
FIGURE 2. Structure of the glycine hexamer as takenfrom its crystallographic unit cell. The numbers are the
(unit numbers i.e., a glycine trimer would be built from)units 1, 2, and 3 .
are quite large, they grow much slower than theŽ .number of basis functions cf. Fig. 1 . For the
double-zeta basis chosen, they probably saturatebelow 300 functions for most molecules. For themore diffuse cc-pVTZ bases, the limit is higher. Itis clear that our largest systems are still not largeenough to reach the limiting efficiency of the localMP2 method. For larger calculations, which exceedour present computing resources but which willcertainly be feasible soon, the advantage of ourlocal method over traditional MP2 should be morepronounced.
The percentage of neglected distant pairs isshown in column 6 of Table III. For the largestsystem, the glycine hexamer, only 42% of the pairsare neglected. We expect that we will be able toimprove the code significantly by using approxi-mate methods for the calculation of distant but notnegligible pairs.18 The percentage of neglectedpairs clearly shows the influence of molecularshape: in going from the essentially planar trimerto the 3-dimensional tetramer, the percentage de-creases.
The memory requirements scales with aboutthe 2.5th power of the molecular size for calcula-tions using one integral pass. This is a significant
VOL. 19, NO. 111250

INTEGRAL TRANSFORMATION FOR LARGE LOCAL MP2 CALCULATIONS
TABLE III.Benchmark Calculations for Sequence of Glycine Oligomers with Structures Taken from Crystallographic Data.
Memory CPU time Correlation energiescAVa b d eMolecule Basis CF No. CE NP Trans. LMP2 Disk Trans. ERIs Total E ELMP2 Ref
Monomer cc-pVDZ 95 90 30 0.0 1.3 0.7 0.0 6 1 8 y0.80369515 y0.8034822889 1.3 6 1 8 y0.80379915
Dimer cc-pVDZ 190 130 60 17.2 8.0 2.5 0.0 50 4 56 y1.60590621 y1.60577350126 7.5 43 4 49 y1.60599622
Trimer cc-pVDZ 285 154 90 27.7 21.1 5.3 0.0 154 10 171 y2.41633266 y2.41637237148 19.6 131 10 141 y2.41643988
Tetramer cc-pVDZ 380 203 120 24.2 64.5 9.8 0.0 753 25 801 y3.22048798 y3.22062184193 58.6 612 25 661 y3.22056705
Pentamer cc-pVDZ 475 215 150 36.5 92.0 13.2 5.7 1408 42 1495 y4.02948436 }202 81.3 996 42 1083 y4.02957512
fHexamer cc-pVDZ 570 236 180 42.1 68.5 8.6 23.2 2306 131 2538 y4.84085174 }218 68.5 1633 129 1863 y4.84089500
Monomer cc-pVTZ 195 184 30 0.0 5.5 2.9 0.0 65 9 77 y1.00219536 y1.00200204Dimer cc-pVTZ 390 267 60 17.4 32.8 9.8 0.0 616 34 668 y2.00448738 y2.00441442Trimer cc-pVTZ 585 322 90 27.8 84.5 10.8 20.8 2397 87 2549 y3.01566041 }
gTetramer cc-pVTZ 780 424 120 24.2 78.5 16.5 39.9 11790 836 12828 y4.02324470 }hPentamer cc-pVTZ 975 454 150 36.6 90.5 22.7 52.1 20639 2064 23118 y5.03310598 }
CPU times are given in minutes and refer to an SGI Power Challenge R10000 / 194 MHz with 1.5 GB memory and 2 MB secondarycache. Memory and disk space requirements are given in MW, correlation energies in E . Thresholds used for the first set ofh
˚ 3 ˚cc-pVDZ calculations: T = 6.0 A, T = 0.005, T = 10 ; for the second set of cc-pVDZ calculations: T = 6.0 A, T = 0.006,d 2 1 d 23 ˚ 3T = 4 ? 10 . Thresholds used for cc-pVTZ benchmarks: T = 6.0 A, T = 0.005, T = 9 ? 10 . The criterion of 0.02 for the virtual1 d 2 1
space selection as suggested by Boughton and Pulay has been used throughout.43
a [ ]Dunning’s cc-pVTZ basis has been truncated for hydrogens 4s3p2d1f / 3s2p .b Number of contracted functions.c [ ]Average dimension of an AO pair domain ij .AOd Number of correlated electrons.e Percentage of neglected orbital pairs.f Two integral passes.g Four integral passes.h Six integral passes.
reduction relative to the n2N 2 scaling for fullconventional in-core transformations. Disk spacerequirements are easily satisfied by today’s largeŽ .multi-gigabyte disks. Loosening the thresholds asgiven in the lower set of numbers of Table III leadsto about a 10% memory savings. The bottleneckwith respect to memory requirements is still thetransformation step, because the AO pair domainsw x w xij have to be significantly larger than the ijAOdomains in projected space. Savings in CPU timedue to looser thresholds amount to about 30%.
Ž 2.6.However, the overall CPU scaling of about O NŽ 2.9.to O N is not affected significantly by choosing
different parameters. This is evident in Figure 3that shows the CPU times of the integral transfor-mation as a function of the number of glycinemonomers for two choices of thresholds. The rela-tively steep increase in CPU time between thetrimer and the tetramer corresponds to the transi-tion from 2-D to 3-D clusters. Estimating the CPU
time for a full SCF calculation as 10 full integralevaluations, a local MP2 calculation takes roughlytwice as long as the preceding SCF step. The re-sults for cc-pVTZ calculations resemble those forcc-pVDZ benchmarks. The fraction of CPU timespent in the integral calculation becomes moresignificant for the cc-pVTZ benchmarks. This isdue to the memory limitations that require severalintegral passes for larger clusters. The efficiency ofthe thresholds is shown by the reduction of the
Ž 6. Ž 3.formal O N scaling to the observed fO N orw Ž 2.5. xlower O N in the best case . The benign scal-
ing of our method makes it feasible to treat largemolecules, which could so far be treated at the SCFor density functional theory level only, with MP2on single-processor workstations. However, therather large prefactor caused by the logic, as wellas the scatter and gather operations, makes themethod only competitive for large systems. Thecrossover point relative to conventional MP2
JOURNAL OF COMPUTATIONAL CHEMISTRY 1251

RAUHUT, PULAY, AND WERNER
FIGURE 3. Performance of the transformation step fora sequence of glycine units. The set labeling and thevalues presented refer to Table III.
should be around 400 basis functions for cc-pVDZbasis sets. For those oligomers for which local
Žreference calculations could be performed see.Table III , the numerical error of the total correla-
tion energy varies from 0.073 to 0.317 mE . Forhsmall clusters the absolute value of the correlationenergy is too high; it is too low for larger clusters.This effect is probably caused by neglecting dis-tant orbital pairs that accumulate for larger sys-tems and could be eliminated by an approximatetreatment of long-range correlation. Because thenumber of neglected pairs is related to the shape
Ž .of the molecule see above , the deviations causedby this threshold are as well.
Conclusions
A single step two index transformation of elec-tron repulsion integrals to localized MOs offers,
Ž 6.despite a formally unfavorable O N scaling, twoadvantages. First, it allows an effective use ofprescreening techniques, which dramatically re-duce the number of arithmetic operations and thus
Ž 3.the scaling to about O N . Second, because thenumber of half-transformed integrals is less than15% of the value in the traditional method, thebottleneck of storing huge arrays of transformedintegrals is largely eliminated. The algorithm
presently allows a memory efficient treatment ofmolecules up to 1000 basis functions on customaryworkstations, opening the use of more correlationmethods for larger molecules than previously pos-sible. The largest savings are expected for MP2,but all correlation methods could profit from thisnew transformation. A disadvantage is that it re-quires the orbital-invariant formulation of MP2,which is somewhat more costly than the canonicalformation. The actual scaling of our local MP2method is slightly lower than ;N 3, making thisapproach competitive with other nontraditionalmethods, for instance the pseudospectral LMP2 ofMurphy et al.25 Compared to traditional MP2, themethod offers significant advantages only for largesystems with around 400 basis functions or more.However, because LMP2 calculations needingmore than 600 basis functions are still quite expen-sive when using the algorithm presented, a smallwindow remains between 400 and 600 basis func-tions for routine applications at present. But, witha moderate future perspective of an increase infloating point operations by a factor of 10 withinthe next 5 years, our largest calculation can beperformed as a standard application easily by then.Moreover, making use of modern multiprocessorworkstations or ven large MPP supercomputersfor this algorithm, which is currently being ex-plored by one of us, will lead to an additionalspeedup and therefore we anticipate routine appli-cations of up to more than 1500 basis functionssoon.
The use of truncations introduces a small errorof about 0.01% of the correlation energy relative toa local MP2 calculation with full numerical accu-racy. For very large molecules, this amount maycome close to the accuracy desired for reactionpaths or rotational barriers. For example, if thetotal correlation energy amounts to 10 E , the errorh
may exceed 0.5 kcalrmol. However, it is expectedthat much of this error largely cancels out in calcu-lating relative energies.53 At any rate, it can bereduced easily by using tighter thresholds if higheraccuracy is needed. Friesner’s analytically cor-rected pseudospectral LMP2 also shows numerical
wdeviations in the same range reported to be typi-cally within 0.2 kcalrmol, e.g., 0.09 kcalrmolŽ UU . x 256]31G for the glycine moner . Weigend andHaser recently presented a systematic study on¨RI-MP2 energies for a large set of molecules.37 Forlarge systems, as considered in this study, theyobtain deviations in the range of about 0.05%
VOL. 19, NO. 111252

INTEGRAL TRANSFORMATION FOR LARGE LOCAL MP2 CALCULATIONS
wrelative to MP2 correlation energies benzene,Ž . Ž0.01% 0.07 kcalrmol ; porphyrin, 0.07% 1.45.xkcalrmol , but they are also able to show that
most of these deviations cancel out when consider-ing relative reaction energies. We expect the samefor the local approach.
All these absolute errors are insignificant com-pared to systematic errors in conventional MP2calculations, which are caused by the MP2 approx-imation itself, basis set superposition errors, andthe fact that for medium size basis sets typicallyonly ;80% of the MP2 correlation energy will berecovered. Further refinements of the local methodcan be expected from using mathematically local-ized orbitals, which minimize the number of sig-nificant SCF coefficients, and the approximatetreatment of distant pairs. Moreover, the ratherlarge prefactor, which is responsible for placingthe crossover point to about 400 basis functions
Ž .and 1000 pairs cc-pVDZ , should decrease by abetter addressing scheme that takes into accountthe size of the primary cache in dealing with thecompressed internal exchange matrices.
Appendix: Derivation for Criterion toExploit MO Coefficient Sparsity
The threshold introduced that neglects insignifi-Ž .cant coefficient products T acts on a shell level.1
ŽWe have generated a matrix shells = valence or-.bitals that contains the largest absolute LMO coef-
ficient per shell and orbital, C max. Likewise, weR isearch for the largest absolute electron repulsionintegral within a given shell quartet. Therefore,early after obtaining shell indices for a block oftwo electron integrals, we can decide about thesignificance of contributions to internal exchangematrices according to
maxi j max max †< < < Ž < . <D I s C C MR SN F Tmax max R i S j 1
Ž .M , N , R , S s shell indices. A.1
† ŽA useful range for threshold T which is different1.in magnitude than the finally used threshold T is1
10y7. Unfortunately, this simple criterion appearsto be basis set dependent, because the averagemagnitude of the MO coefficients strongly de-pends on the basis set chosen. In order to get abasis set independent criterion, the expression in
Ž .eq. A.1 has been divided by the square of theaverage magnitude of the largest shell coefficients
of the localized MOs and of the projected AOs:
< i j <D Imax max Ž .F T A.212 2C Pmean mean
where
1max< < Ž .C s C A.3Ý Ýmean T knT k k < <Tg k AO
is the average of all C max with the restriction thatT kw xthe AO shell T contributes to MO domain k .AO
1max< < Ž .P s P A.4Ý Ýmean T qnT q k w xT , qg k AO
max Žis in analogy the average of all P being de-T qmax .fined as C where T comprises all AO shells ofT k
w xorbital domain k and q runs over all projectedAOfunctions within this domain. Strictly, one would
w xhave to run q over all elements in k , but thisleads to a too small selection of significant ele-ments of the projection matrix. This correction of
Ž .eq. A.1 leads to a coupling of the criterion for thevirtual space selection and the criterion for smallcontributions. This coupling is necessary to restrictthe averaging to significant coefficients, becausethe bulk of coefficients is negligible for localizedMOs.
Acknowledgments
This work was supported by the DeutscheŽ .Forschungsgemeinschaft DFG in the Schwer-
punktsprogramm Molekulare Cluster. It was alsosupported in part by a grant of the Air Force Officeof Scientific Research to P. P. and the NationalScience Foundation under Grant CHE-9319929.P. P. thanks the Alexander von Humboldt Founda-tion for a Senior Scientist Award, the NationalScience Foundation for support under Grant CHE-9707202, and the Air Force Office for support un-der Grant F49620-94-1. H. J. W. acknowledges theEU support in the TMR network FMRX-CT96-088and the Fonds der Chemischen Industrie.
References
Ž .1. K. C. Tang and C. Edmiston, J. Chem. Phys., 52, 997 1970 .2. S. T. Elbert, In Numerical Algorithms in Chemistry: Algebraic
Methods, Reports of NRCC Workshop 1978, LBL-8158, 1978,p. 128.
3. M. Yoshimine, Report RJ-555 IBM Research Laboratory, SanJose, CA, 1969.
JOURNAL OF COMPUTATIONAL CHEMISTRY 1253

RAUHUT, PULAY, AND WERNER
Ž .4. H. J. Werner and W. Meyer, J. Chem. Phys., 73, 2342 1980 .5. V. R. Saunders and J. H. van Lenthe, Mol. Phys., 48, 923
Ž .1983 .Ž .6. P. R. Taylor, Int. J. Quantum Chem., 31, 521 1987 .
Ž .7. S. Saebø and J. Almlof, Chem. Phys. Lett., 154, 83 1989 .¨8. M. Head]Gordon, J. A. Pople, and M. J. Frisch, Chem. Phys.
Ž .Lett., 153, 503 1988 .9. M. J. Frisch, M. Head]Gordon, and J. A. Pople, Chem. Phys.
Ž .Lett., 166, 281 1990 .10. S. Wilson, In Methods in Computational Chemistry, S. Wilson,
Ed., Plenum Press, New York, 1987, p. 251.11. I. Shavitt, In Methods of Molecular Electronic Structure Theory,
H. F. Schaefer, ed., Plenum Press, New York, 1977, p. 189.12. A. M. Marquez and M. Dupuis, J. Comput. Chem., 16, 395´
Ž .1995 .13. I. M. B Nielsen and E. T. Seidl, J. Comput. Chem., 16, 1301
Ž .1995 .14. D. E. Bernholdt and R. J. Harrison, J. Comput. Chem., 102,
Ž .9582 1995 .15. A. T. Wong, R. J. Harrison, and A. P. Rendell, Theor. Chim.
Ž .Acta, 93, 317 1996 .Ž .16. M. Schutz and R. Lindh, Theor. Chim. Acta. 95, 13 1997 .¨
17. M. Schutz, R. Lindh, and H.-J. Werner, unpublished data.¨18. G. Hetzer, P. Pulay, and H.-J Werner, Chem. Phys. Lett., in
press.19. G. Rauhut, J. W. Boughton, and P. Pulay, J. Chem. Phys.,
Ž .103, 5662 1995 .Ž .20. P. Pulay and S. Seabø, Theor. Chim. Acta, 69, 357 1986 .
Ž . Ž . Ž .21. a S. Seabø and P. Pulay, J. Chem. Phys., 86, 914 1987 ; bŽ .S. Saebø and P. Pulay, J. Chem. Phys., 88, 1884 1988 .
22. S. Seabø and P. Pulay, Annu. Rev. Phys. Chem., 44, 213Ž .1993 .
23. J. Almlof, K. Faegri, and K. Korsell, J. Comput. Chem., 3, 385¨Ž .1982 .
24. C. Hampel and H.-J. Werner, J. Chem. Phys., 104, 6286Ž .1996 .
25. R. B. Murphy, M. D. Beachy, R. A. Friesner, and M. N.Ž .Rignalda, J. Chem. Phys., 103, 1481 1995 .
26. G. Reynolds, T. J. Martinez, and E. A. Carter, J. Chem. Phys.,Ž .105, 6455 1996 .
27. A. El Azhary, G. Rauhut, P. Pulay, and H.-J. Wener, J.Ž .Chem. Phys., 108, 5185 1998 .
28. R. Knab, W. Forner, J. Cizek, and J. Ladik, J. Mol. Struct.¨Ž . Ž .Theochem. , 366, 11 1996 .
Ž .29. E. Kapuy and C. Kozmutza, J. Chem. Phys., 94, 5565 1991 .30. K. Szalewicz, B. Jeziorski, H. J. Monkhorst, and J. G.
Ž .Zabolitzky, Chem. Phys. Lett., 91, 169 1982 .Ž .31. M. Haser and J. Almlof, J. Chem. Phys., 96, 489 1992 .¨ ¨
Ž .32. G. Rauhut and P. Pulay, Chem. Phys. Lett., 248, 223 1996 .Ž .33. A. K. Wilson and J. Almlof, Theor. Chim. Acta, 95, 49 1997 .¨
34. A. Komornicki and G. Fitzgerald, J. Chem. Phys., 98, 1398Ž .1993 .
35. M. Feyereisen, G. Fitzgerald, and A. Komornicki, Chem.Ž .Phys. Lett., 208, 359 1993 .
36. D. E. Bernholdt and R. J. Harrison, Chem. Phys. Lett., 250,Ž .477 1996 .
Ž .37. F. Weigend and M. Haser, Theor. Chem. Acc., 97, 331 1997 .¨Ž .38. W. Meyer, J. Chem. Phys., 64, 2901 1976 .
39. P. Pulay, S. Saebø, and W. Meyer, J. Chem. Phys., 81, 1901Ž .1984 .
Ž .40. P. Pulay, Chem. Phys. Lett., 73, 393 1980 .41. S. Saebø, W. Tong, and P. Pulay, J. Chem. Phys., 98, 2170
Ž .1993 .42. G. Rauhut and P. Pulay, LMP2, University of Arkansas,
1995. LMP2 is part of TEXAS-95, P. Pulay and K. Wolinski,University of Arkansas, 1995.
43. J. W. Boughton and P. Pulay, J. Comput. Chem., 14, 736Ž .1993 .
Ž .44. J. Pipek and P. G. Mezey, J. Chem. Phys., 90, 4916 1989 .45. S. F. Boys, In Quantum Theory of Atoms, Molecules and the
Solid State, P. O. Lowdin, Ed., Academic Press, New York,¨1966, p. 253.
Ž .46. T. H. Dunning, J. Chem. Phys., 90, 1007 1989 .47. P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 28, 213
Ž .1973 .48. R. Krishnan, J. S. Binkley, R. Seeger, and J. A. Pople, J.
Ž .Chem. Phys., 72, 650 1980 .Ž .49. T. Dunning, J. Chem. Phys., 90, 1007 1989 .
50. P. Pulay and G. Rauhut, Molecular Quantum Mechanics:w xMethods and Applications conference contribution , Cam-
bridge, U.K., 1995.Ž .51. E. Buncel and S. Rajagopal, J. Org. Chem., 54, 798 1989 .
52. P. G. Josson and A. Kvick, Acta Crystallogr., B28, 1827¨Ž .1972 .
53. G. Rauhut, unpublished data.
VOL. 19, NO. 111254





























