Embed Size (px)
Citation preview

INSULIN GRANULES
Insulin secretory granules controlautophagy in pancreatic β cellsAlexander Goginashvili,1 Zhirong Zhang,1 Eric Erbs,1 Coralie Spiegelhalter,1
Pascal Kessler,1 Michael Mihlan,1 Adrien Pasquier,1 Ksenia Krupina,1 Nicole Schieber,2
Laura Cinque,3 Joëlle Morvan,1 Izabela Sumara,1 Yannick Schwab,2
Carmine Settembre,3,4 Romeo Ricci1,5*
Pancreatic β cells lower insulin release in response to nutrient depletion. The questionof whether starved β cells induce macroautophagy, a predominant mechanismmaintaining energy homeostasis, remains poorly explored. We found that, in contrastto many mammalian cells, macroautophagy in pancreatic β cells was suppressedupon starvation. Instead, starved β cells induced lysosomal degradation of nascentsecretory insulin granules, which was controlled by protein kinase D (PKD), a keyplayer in secretory granule biogenesis. Starvation-induced nascent granule degradationtriggered lysosomal recruitment and activation of mechanistic target of rapamycinthat suppressed macroautophagy. Switching from macroautophagy to insulin granuledegradation was important to keep insulin secretion low upon fasting. Thus, β cellsuse a PKD-dependent mechanism to adapt to nutrient availability and couple autophagyflux to secretory function.
Upon feeding, the pancreatic b cell catab-olizes nutrients to secrete insulin. Con-versely, insulin secretion decreases uponfasting (1). A shortage of nutrients also in-duces macroautophagy (hereafter referred
to as “autophagy”). During autophagy, cellularcomponents are sequestered into autophago-somes and degraded upon targeting to lysosomes(autolysosomes). Resulting catabolites maintaincells in a metabolically active state and ensurecell survival (2). However, catabolism of nutrientscould also trigger insulin release, which shouldbe avoided during fasting. Autophagy might thusbe a bad strategy for a b cell to cope with nutri-ent deprivation. Both induction and lack of in-duction of autophagy have been reported infasted b cells (3, 4). We thus wanted to assess theimportance of autophagy in fasted b cells.Microtubule-associated protein 1 light chain
3 B (LC3B) incorporates into membranes of auto-phagosomes (5). Unexpectedly, INS1 cells (a ratinsulinoma–derived b cell line) exogenously ex-pressing LC3B tagged with green fluorescentprotein (GFP) (LC3B-GFP) deprived of serumand amino acids or glucose (Glc), but not ofserum alone, decreased LC3B-GFP puncta (fig.S1, A to C). Conversely, starved human embryonickidney (HEK) 293 cells increased LC3B-GFP
puncta (fig. S1, D and E) (6). INS1 cells endoge-nously expressing LC3B-GFP (INS1LC3B-GFPendo
cells) (fig. S2, A to C) treated without or withbafilomycin A1 (BafA1), which blocks autolyso-somal degradation of LC3B (7), decreased LC3B-GFP puncta upon starvation (Fig. 1A and fig. S2,D to F). Tandem RFP-GFP–tagged LC3 (ptfLC3;RFP, red fluorescent protein) allows for discrim-ination of yellow fluorescent autophagosomesand red fluorescent autolysosomes (8). MultipleRFP-GFP puncta were converted into RFP-onlypuncta upon the onset of starvation. A reductionin RFP puncta and no reappearance of RFP-GFPpuncta were observed over time (fig. S3, A and B).Correlative light and electron microscopy (CLEM)confirmed their autophagic origin (Fig. 1B). More-over, lipidated autophagosomal LC3B (LC3B-II)decreased in starved INS1 cells in the absenceand presence of BafA1 (Fig. 1C). In contrast, star-vation increased LC3B-II in HEK293 cells (fig.S4). p62 binds polyubiquitinylated substrates fortargeting to autophagosomes (9). p62 was mod-erately increased during starvation, indepen-dent of BafA1 (Fig. 1C). p62 puncta clustered andincreased in size (fig. S5A). Accordingly, p62/LC3B-GFP colocalization was decreased, indicat-ing suppressed autophagy-dependent clearanceof p62. ATG16L1 binds to preautophagosomes(10). ATG16L1- and ATG16L1/LC3B-GFP–positivepuncta moderately decreased upon starvation(fig. S5B). Quantitative electron microscopy (QEM)confirmed reduced autophagic compartmentsin b cells of starved murine primary islets (fig.S6). Moreover, in LC3B-GFP–expressing mice(11), autophagosomes decreased in b cells uponfasting (Fig. 1D and fig. S7). What about othersecretory cells, in which nutrients do not act assecretagogues? In contrast to b cells, primary mu-rine plasma cells induced autophagy upon star-vation, whereas secretion of immunoglobulin G
was reduced (fig. S8, A and B). Thus, nutrient-sensing b cells appear to use a distinct mechanismto overcome shortage of nutrients.Targeting of secretory granules (SGs) to lyso-
somes might be an alternative strategy (12). Lyso-somal Lamp1 and SG protein Phogrin colocalizedclose to the Golgi upon starvation in INS1 cells.Lysosomal inhibitors (LIs) increased their co-localization (fig. S9, A to C). QEM, immunogoldlabeling, and CLEM revealed abundant granule-containing lysosomes (GCLs) (fig. S10, A to D).We next used density gradients to purify GCLs(see supplementary materials and methods). Uponstarvation, Phogrin- and lysosomal Lamp2-positivesignals shifted to heavier fractions containingGCLs. LC3B was almost undetectable in shiftedfractions, suggesting independence of GCLs fromautophagy (Fig. 2A). Accordingly, starvation didnot increase LC3B-GFP/Phogrin colocalization(fig. S11). Moreover, inactivation of autophagy didnot change the amount of GCLs in starved cells(fig. S12, A and B).Secretory granules are generated at the Golgi.
Proinsulin, a marker for nascent SGs (13), marked-ly decreased upon starvation (Fig. 2B). Its reduc-tion was partially restored by LIs. AbundantGCLs were confirmed ex vivo by QEM of starvedprimary murine islets (Fig. 2C and fig. S13A). Infasted GFP-LC3B–expressing mice, Lamp2 andproinsulin/insulin [(Pro)insulin] colocalization in-creased, whereas there was no increase in LC3B-GFP/(Pro)insulin colocalization (Fig. 2D and fig.S13, B and C). Thus, b cells employ starvation-induced nascent granule degradation (SINGD)instead of autophagy during fasting.Lysosome-derived amino acids induce translo-
cation of mechanistic target of rapamycin (mTOR)complex 1 (mTORC1) to lysosomal membranes,mTORC1 activation (14, 15), and subsequent sup-pression of autophagy (16–18). The mTOR inhib-itors rapamycin and torin 1 increased the numberof LC3B-GFP puncta in starved INS1 cells (Fig. 3A).Moreover, starvation localized mTOR to Phogrin/Lamp1-positive puncta close to the Golgi (Fig. 3B).mTORC1 suppresses starvation-induced autophagythrough phosphorylation of Unc-51–like kinase1 (ULK1) (19). Indeed, phospho-ULK1 remainedhigh upon starvation and was abolished byrapamycin, whereas mTORC1-mediated phospho-rylation of S6K1 (20) diminished during starvation(Fig. 3C). Accordingly, S6K1 phosphorylation wasshown to require higher mTOR activity thanULK1 phosphorylation (21). Moreover, starvationled to formation of large phospho-ULK1 punctathat colocalized with Lamp1/Phogrin (fig. S14, Aand B). Thus, nutrient depletion leads to induc-tion of SINGD that locally activates mTOR tosuppress autophagy.Fasting suppresses both insulin secretion and
autophagy. Would insulin secretion increase ifautophagy is induced upon starvation? Keep-ing autophagy high by rapamycin precludedsuppression of insulin secretion in INS1 cells(fig. S15). We next used tat-beclin1 (22) to specif-ically trigger autophagy in b cells of murine pri-mary islets upon nonstimulatory Glc (fig. S16,A and B). Tat-beclin1 increased insulin secretion
878 20 FEBRUARY 2015 • VOL 347 ISSUE 6224 sciencemag.org SCIENCE
1Institut de Génétique et de Biologie Moléculaire et Cellulaire(IGBMC), INSERM, CNRS, Université de Strasbourg, 67404Illkirch, France. 2Cell Biology and Biophysics Unit, EuropeanMolecular Biology Laboratory (EMBL), 69117 Heidelberg,Germany. 3Dulbecco Telethon Institute and Telethon Instituteof Genetics and Medicine (TIGEM), 80131 Naples, Italy.4Medical Genetics, Department of Medical and TranslationalScience Unit, Federico II University, Via Pansini 5, 80131Naples, Italy. 5Nouvel Hôpital Civil, Laboratoire de Biochimieet de Biologie Moléculaire, Université de Strasbourg, 67091Strasbourg, France.*Corresponding author. E-mail: [email protected]
RESEARCH | REPORTSon N
ovember 16, 2017
http://science.sciencem
ag.org/D
ownloaded from

to levels comparable with those observed uponstimulatory Glc (fig. S16C). Potassium channelclosure that evokes calcium influx and exocyto-sis of SGs (1) was required, as the potassiumchannel activator diazoxide abolished this effect(fig. S16D). Insulin secretion was also potenti-ated by tat-beclin1 in murine islets at stimula-tory Glc, which was blocked by diazoxide (fig.S16, E and F). Tat-beclin1 also increased insulinsecretion from ex vivo fasted human islets (Fig.3D and fig. S17, A and B). Thus, SINGD-mediatedsuppression of autophagy is important to keepinsulin secretion low during fasting.Protein kinase D (PKD) controls insulin SG
biogenesis at the Golgi (23, 24). Thus, PKD in-activation could have an effect on the turnover ofnascent SGs. Indeed, inhibition of PKD decreasedthe amount of proinsulin (Fig. 4A). Proinsulinbiosynthesis was unchanged in PKD1-depletedcells (fig. S18, A and B). However, accumulationof newly formed insulin was decreased, suggest-ing enhanced degradation of de novo synthe-sized insulin (fig. S18C). QEM and immunogold
analyses revealed abundant GCLs in the Golgiarea (Fig. 4B and fig. S19, A and B), which wasfurther corroborated by subcellular fractiona-tion (fig. S19C). PKD inhibition increased Phogrin/Lamp1 colocalization, which was more prominentupon LI treatment (fig S19, D to F). Phogrin/LC3B-GFP colocalization remained unchanged (fig. S20).mTOR largely colocalized with Lamp1 (Fig. 4C),and phospho-ULK1 was increased (Fig. 4D) inPKD1-depleted cells. PKD1 knockdown decreasedaccumulation of LC3B-II in the presence of BafA1(fig. S21A). Moreover, PKD inhibition decreasedLC3B-GFP puncta in absence and presence ofBafA1 (fig. S21B).If PKD controls SINGD, its activity should be
regulated by nutrients. PKD activity at the Golgidecreased upon starvation (fig. S22, A and B).Loss of the p38d kinase leads to activation ofPKD in b cells (23). GCLs were markedly de-creased in b cells of ex vivo fasted p38d knockout(p38d−/−) islets (Fig. 4E and fig. S23A). Accord-ingly, (Pro)insulin/Lamp2 colocalized to a lesserextent in b cells of fasted p38d−/− mice (fig. S23,
C and D), suggesting that high PKD activity pre-vents SINGD. In contrast, autophagic compart-ments were increased in fasted p38d−/− b cells,indicating that SINGD-dependent suppressionof autophagy was diminished (Fig. 4E and fig.S23B). Thus, PKD is a major regulator of SINGDand autophagy in b cells.Altogether, inactivation of PKD evokes SINGD,
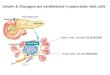
localized activation of mTOR, and suppressionof autophagy, which is critical to prevent insulinrelease during fasting. On the other hand, PKDactivity is required for SG biogenesis (Fig. 4F).Because the nascent SGs are preferentially se-creted (25), SINGD-mediated suppression of au-tophagy may represent an optimal strategy tocounteract insulin secretion, at the same timeproviding sufficient nutrients. Because SINGD-mediated suppression of autophagy depends onabundant nascent SGs, their depletion will prob-ably derepress autophagy without increasing in-sulin release. Timing when depletion occurs mayvary substantially depending on the models andprotocols used and may thus explain previously
SCIENCE sciencemag.org 20 FEBRUARY 2015 • VOL 347 ISSUE 6224 879
Fig. 1. Nutrient depletion suppresses autophagy in β cells. (A) LC3B-GFP puncta in INS1LC3B-GFPendo cells under growing culture (GC) and no aminoacids and fetal calf serum (AA/FCS) for 1 hour in the absence and presenceof BafA1 (10 nM). DAPI, 4′,6-diamidino-2-phenylindole. (B) CLEM of ptfLC3-expressing INS1 cells under GC and no AA/FCS for 2 hours. Regions of interest(ROI) are indicated with labeled dashed squares. Yellow and black asterisksindicate autophagosomes and autolysosomes, respectively. EM, electron
microscopy. (C) Immunoblot of LC3B and p62 using soluble and insolublefractions of lysates of INS1 cells under GC and no AA/FCS for 1.5 hours,nontreated or treated with 1 nM BafA1 for the last hour of incubation.Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loadingcontrol. (D) Immunofluorescence (IF) of LC3B-GFP puncta (white arrowheads)and insulin (red) in b cells in islets of fed and fasted LC3B-GFP–expressingmice. The nuclei were stained with DAPI.
RESEARCH | REPORTSon N
ovember 16, 2017
http://science.sciencem
ag.org/D
ownloaded from

880 20 FEBRUARY 2015 • VOL 347 ISSUE 6224 sciencemag.org SCIENCE
Fig. 2. Nutrient depletion induces SINGD in β cells. (A) Immunoblot of Lamp2, Phogrin, and LC3B using lysates of indicated fractions from INS1 cellsunder GC and no AA/FCS for 30 min. EM of GCLs in shifted fractions is indicated by red dashed boxes. (B) Immunoblot of proinsulin using lysates of INS1cells treated or not with LIs under GC and no AA/FCS for 6 hours. GAPDH was served a loading control. (C) EM of Golgi areas in primary murine isletsunder GC and no AA/FCS for 2 hours. Yellow asterisks indicate GCLs. ROI are indicated with dashed squares. (D) IF of (Pro)insulin and Lamp2 (left) and(Pro)insulin and LC3B-GFP (right) in b cells in islets of fed and fasted LC3B-GFP expressing mice. The nuclei were stained with DAPI.
Fig. 3. SINGD suppresses autophagy in a mTOR-dependent manner to prevent insulin release. (A) IF of LC3B-GFP puncta (white arrowheads) in INS1 cells under GC and no AA/FCS for 2 hours treated or not with 100 nMrapamycin or 250 nM torin 1, as indicated. Quantification of LC3B-GFP puncta per cell (error bars denote mean T SEM).**P < 0.01. (B) IF of mTOR in INS1 cells coexpressing Phogrin-GFP and Lamp1-RFP under GC and no AA/FCSfor 2 hours. White arrowheads in ROI (dashed squares) indicate colocalization of Phogrin-GFP with mTOR andPhogrin-GFP with Lamp1-RFP. A Golgi marker (Giantin) was used. (C) Immunoblot of indicated proteins using lysatesof INS1 cells treated or not with 100 nM rapamycin under GC, no AA/FCS, or no Glc/FCS for 2 hours. GAPDH served asa loading control. DMSO, dimethyl sulfoxide. (D) Insulin in supernatants of human islets treated as indicated. Insulinconcentrations are expressed as a percentage of insulin upon 16.7 mM stimulatory Glc (mean T SEM). ***P < 0.001.
RESEARCH | REPORTSon N
ovember 16, 2017
http://science.sciencem
ag.org/D
ownloaded from

described inconsistencies (3). Accordingly, pro-longed fasting of mice can induce autophagy inb cells (26). Our model could also explain theregulation of autophagy despite constitutivelyhigh adenosine monophosphate–activated pro-tein kinase activity in b cells (27).Our data show that triggering autophagy re-
sults in increased secretion of insulin. Althoughthis should be avoided during fasting, it may bebeneficial when insulin demands are high; forexample, after a meal or in diabetes (28). Thepositive correlation between autophagy and in-sulin secretion may suggest an involvement ofautophagy in postprandial insulin release, prob-
ably going beyond the widely established house-keeping role of autophagy.
REFERENCES AND NOTES
1. P. Rorsman, M. Braun, Annu. Rev. Physiol. 75, 155–179(2013).
2. J. D. Rabinowitz, E. White, Science 330, 1344–1348(2010).
3. C. Ebato et al., Cell Metab. 8, 325–332 (2008).4. R. Singh, A. M. Cuervo, Cell Metab. 13, 495–504
(2011).5. I. Tanida, T. Ueno, E. Kominami, Int. J. Biochem. Cell Biol. 36,
2503–2518 (2004).6. Y. Kabeya et al., EMBO J. 19, 5720–5728 (2000).7. A. Yamamoto et al., Cell Struct. Funct. 23, 33–42
(1998).
8. S. Kimura, T. Noda, T. Yoshimori, Autophagy 3, 452–460(2007).
9. G. Bjørkøy et al., J. Cell Biol. 171, 603–614 (2005).10. N. Fujita et al., Mol. Biol. Cell 19, 2092–2100 (2008).11. N. Mizushima, A. Yamamoto, M. Matsui, T. Yoshimori, Y. Ohsumi,
Mol. Biol. Cell 15, 1101–1111 (2004).12. R. E. Smith, M. G. Farquhar, J. Cell Biol. 31, 319–347 (1966).13. D. F. Steiner, D. Cunningham, L. Spigelman, B. Aten, Science
157, 697–700 (1967).14. R. Zoncu et al., Science 334, 678–683 (2011).15. Y. Sancak et al., Science 320, 1496–1501 (2008).16. L. Yu et al., Nature 465, 942–946 (2010).17. C. Betz, M. N. Hall, J. Cell Biol. 203, 563–574 (2013).18. S. G. Kim, G. R. Buel, J. Blenis, Mol. Cells 35, 463–473
(2013).19. J. Kim, M. Kundu, B. Viollet, K. L. Guan, Nat. Cell Biol. 13,
132–141 (2011).
SCIENCE sciencemag.org 20 FEBRUARY 2015 • VOL 347 ISSUE 6224 881
Fig. 4. PKD controls SINGD. (A) Immunoblot of proinsulin using lysates of INS1cells treated with CID755673 for the indicated times. GAPDH served as a loadingcontrol. (B) EM of Golgi areas in nonsilenced (NS) and PKD1-depleted (shPKD1)INS1 cells. Yellow asterisks indicate GCLs. ROI are indicated with dashed squares.(C) IF of mTOR and Lamp1 in nonsilenced and PKD1-depleted INS1 cells. Whitearrowheads indicate colocalization of mTOR with Lamp1. Nuclei were stained with
DAPI. (D) Immunoblot of indicated proteins using lysates of nonsilenced and PKD1-depleted INS1 cells.GAPDH served as a loading control. (E) (Top) EM of Golgi areasof fasted b cells in primary islets of p38d+/+ and p38d−/− mice. Yellow asterisksindicate GCLs. (Bottom) EM of cytoplasm of fasted b cells in primary islets ofp38d+/+ and p38d−/− mice. The yellow asterisk indicates an autophagosome.(F) Model linking SINGD, secretion, and autophagy. PM, plasma membrane.
RESEARCH | REPORTSon N
ovember 16, 2017
http://science.sciencem
ag.org/D
ownloaded from

20. R. B. Pearson et al., EMBO J. 14, 5279–5287 (1995).21. S. A. Kang et al., Science 341, 1236566 (2013).22. S. Shoji-Kawata et al., Nature 494, 201–206 (2013).23. G. Sumara et al., Cell 136, 235–248 (2009).24. H. Gehart et al., Dev. Cell 23, 756–768 (2012).25. G. Gold, M. L. Gishizky, G. M. Grodsky, Science 218, 56–58
(1982).26. K. Fujimoto et al., J. Biol. Chem. 284, 27664–27673 (2009).27. G. A. Rutter, G. Da Silva Xavier, I. Leclerc, Biochem. J. 375,
1–16 (2003).28. M.-S. Lee, Trends Endocrinol. Metab. 25, 620–627 (2014).
ACKNOWLEDGMENTS
We thank H. de F. Magliarelli, H. Gehart, O. Sumara, G. Sumara,N. Djouder, E. Hafen, R. J. Loewith, J. Klumperman, E. Polishchuk,
R. Polishchuk, and all current members of the Ricci laboratoryfor critical scientific inputs. We thank A. Hausser for providingG-PKDrep-live, G. Rutter for providing Phogrin-GFP, J. Hutton(deceased) and H. Davidson for an antibody against Phogrin,and J. Klumperman for an antibody against Lamp1. We thankM. Koch, N. Messaddeq, and C. Ruhlmann for support in imaging;N. Mizushima for providing GFP-LC3 transgenic mice; andP. Halban and J.-I. Miyazaki for sharing the MIN6B1 cell line. Thiswork was supported by a European Research Council (ERC)starting grant (ERC-2011-StG, 281271-STRESSMETABOL) and aEuropean Foundation for the Study of Diabetes/Lilly EuropeanDiabetes Research Programme grant. A.G. and Z.Z. were supportedby the ERC grant, M.M. was supported by a fellowship of theDeutsche Forschungsgemeinschaft, A.P. was supported by anIGBMC Ph.D. fellowship, K.K. was supported by a fellowship
(Bourse Régionale de Recherche from region Alsace and INSERM),I.S. was supported by the Action Thématique et Incitative surProgramme (ATIP)–Avenir program, L.C. and C. Spiegelhalter weresupported by a Telethon Grant, and N.S. and Y.S. were supported byEMBL internal funding. A.G. and R.R. have applied for a patent thatprotects autophagy-inducing molecules to increase insulin release.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/347/6224/878/suppl/DC1Materials and MethodsFigs. S1 to S23References (29–36)
9 November 2014; accepted 5 January 201510.1126/science.aaa2628
KINASE DYNAMICS
Using ancient protein kinases tounravel a modern cancerdrug’s mechanismC. Wilson,1* R. V. Agafonov,1* M. Hoemberger,1 S. Kutter,1 A. Zorba,1 J. Halpin,1
V. Buosi,1 R. Otten,1 D. Waterman,1 D. L. Theobald,2 D. Kern1†
Macromolecular function is rooted in energy landscapes, where sequence determines not asingle structure but an ensemble of conformations. Hence, evolution modifies a protein’sfunction by altering its energy landscape. Here, we recreate the evolutionary pathway betweentwo modern human oncogenes, Src and Abl, by reconstructing their common ancestors. Ourevolutionary reconstruction combined with x-ray structures of the common ancestor andpre–steady-state kinetics reveals a detailed atomistic mechanism for selectivity of thesuccessful cancer drug Gleevec. Gleevec affinity is gained during the evolutionary trajectorytoward Abl and lost toward Src, primarily by shifting an induced-fit equilibrium that is alsodisrupted in the clinical T315I resistancemutation.This work reveals themechanism of Gleevecspecificity while offering insights into how energy landscapes evolve.
The evolution of protein kinases is a keyevent in the origin of multicellularity (1),which enabled the development of morecomplex signaling cascades essential forthe evolution of higher organisms. The
key role of protein kinases in the cell cycle hasplaced them at the center of cancer drug re-search. Despite an explosion in diversity in thekinome (2), the catalytic kinase domains havemaintained nearly identical structures (2–5). Itis therefore surprising that the clinically suc-cessful cancer drug Gleevec has such strongselectivity toward Abl versus other tyrosine ki-nases, including the closely related Src. This ispuzzling because the Gleevec-bound structuresof Abl and Src are nearly identical, including theN- and C-terminal lobes and the drug-bindingpocket (Fig. 1A) (3). Extensive work led to a fre-quently cited but controversial model whereGleevec selectivity is rooted in a pre-existing equi-librium between two alternative conformationsof the DFG-motif (for Asp-Phe-Gly), a conserved
segment of the activation loop (3, 6–12). A num-ber of x-ray structures have revealed the sam-pling of a Gleevec-binding–competent DFG-outposition and a binding-incompetent DFG-inposition in free kinases (Figs. 1A and 2A). Re-cently reported data rule out the predominantrole of the DFG-in/out equilibrium (conforma-tional selection) in Gleevec selectivity. It led to adifferent binding scheme (Scheme 1) that ac-counts for the 3000-fold difference in Gleevecaffinity between Src and Abl (11) due to a globalconformational change after drug binding (in-duced fit, Scheme 1).However, the molecular mechanism of this se-
lectivity remains unknown. Based on x-ray crys-tal structures, specific point mutations have beenmade in attempts to convert Abl to Src-like spec-ificity and vice versa. Despite two decades ofefforts, such sequence swaps in modern kinaseshave failed to illuminate the atomistic determi-nants of selectivity (3, 6). The differences be-tween Src, Abl, and other homologous kinaseshave evolved over a billion years from their com-mon ancestor—not via residue swaps from onemodern kinase to another. Sequence-swap exper-iments using modern enzymes have a funda-mental shortcoming by neglecting epistasis (theeffect of the surrounding amino acid background)
(13). However, evolution has already navigatedthe complex epistatic protein space by producingfunctional proteins at each stage despite largenumbers of accumulated mutations. We there-fore examined the evolution of Src and Abl alongtheir phylogenetic branches using ancestral re-construction to understand differences not onlyin their equilibrium structures but also in theirenergy landscapes.Ancestral reconstruction has provided a way
to achieve mechanistic insight into protein func-tion (14–19). Here, we elucidate the basis of mod-ern kinase specificity toward Gleevec with atomicresolution by recapitulating the evolution of theSrc and Abl catalytic domain from their last com-mon ancestor. Analysis of the ancestral kinasesallows us to track the evolution of the proteinenergy landscape (20, 21). We define “energy land-scape” as a set of free energy and kinetic param-eters linking kinetically distinct states that arerelevant to biological processes.Seventy-six modern-day sequences spanning
the cytosolic tyrosine kinase family (Src/Abl/Tecfamilies) were used in a Bayesian phylogeneticanalysis with receptor tyrosine kinases as theout-group (Fig. 1B). Because the quality of the an-cestral reconstruction strongly depends on thealignment, we estimated the tree and alignmentssimultaneously. The most probable sequenceswere inferred for four key ancestral proteinsbetween modern Src and Abl and their last com-mon ancestor (Fig. 1B and figs. S1 and S2), andtheir corresponding proteins were expressed,purified, and characterized.We denote the reconstructed protein corre-
sponding to the last common ancestor of Src andAbl as ANC-AS. Similarly, on the lineage leadingfrom ANC-AS to modern Abl, ANC-A1 representsthe common ancestor between humans and co-lonial choanoflagellates, andANC-A2 correspondsto the common ancestor between humans andCaenorhabditis elegans. On the lineage leading tomodern Src, ANC-S1 is the last common ancestorbetween humans and colonial choanoflagellates/sponges. Despite the fact that ANC-AS differs by96 residues from any modern cytosolic tyrosinekinase, all ancestral kinases reconstructed hereare fully active and thermostable (Fig. 1C andfig. S3). We evaluated the specificity of Gleevectoward the ancestral kinases by measuring inhi-bition (Fig. 1D) and dissociation constants (fig. S4).The inhibition ofANC-AS is intermediate between
882 20 FEBRUARY 2015 • VOL 347 ISSUE 6224 sciencemag.org SCIENCE
1Howard Hughes Medical Institute and Department ofBiochemistry, Brandeis University, Waltham, MA 02452, USA.2Department of Biochemistry, Brandeis University, Waltham,MA 02452, USA.*These authors contributed equally to the work. †Correspondingauthor. E-mail: [email protected]
RESEARCH | REPORTSon N
ovember 16, 2017
http://science.sciencem
ag.org/D
ownloaded from

cellsβInsulin secretory granules control autophagy in pancreatic
Romeo RicciKsenia Krupina, Nicole Schieber, Laura Cinque, Joëlle Morvan, Izabela Sumara, Yannick Schwab, Carmine Settembre and Alexander Goginashvili, Zhirong Zhang, Eric Erbs, Coralie Spiegelhalter, Pascal Kessler, Michael Mihlan, Adrien Pasquier,
DOI: 10.1126/science.aaa2628 (6224), 878-882.347Science
, this issue p. 878; see also p. 826Sciencestrategy of choice in beta cells to adapt to starvation.controls this granule degradation in response to nutrient availability. Thus, unlike most other cells, autophagy is not the a process by which cells ''eat'' their own constituents. Protein kinase D, a major regulator of insulin granule biogenesis,
−−nutrient sensor mTOR is recruited to these lysosomes, leading to its local activation and the suppression of autophagy granules through their fusion with lysosomes, the cell's garbage disposal units (see the Perspective by Rutter). The
found that starvation of beta cells induced selective degradation of newly formed insulin et al.depletion. Goginashvili Pancreatic beta cells, the source of insulin in response to food, employ an unusual mechanism to adapt to nutrient
Too hungry to eat, too hungry not to eat
ARTICLE TOOLS http://science.sciencemag.org/content/347/6224/878
MATERIALSSUPPLEMENTARY http://science.sciencemag.org/content/suppl/2015/02/18/347.6224.878.DC1
CONTENTRELATED http://science.sciencemag.org/content/sci/347/6224/826.full
REFERENCES
http://science.sciencemag.org/content/347/6224/878#BIBLThis article cites 36 articles, 16 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.Sciencelicensee American Association for the Advancement of Science. No claim to original U.S. Government Works. The title Science, 1200 New York Avenue NW, Washington, DC 20005. 2017 © The Authors, some rights reserved; exclusive
(print ISSN 0036-8075; online ISSN 1095-9203) is published by the American Association for the Advancement ofScience
on Novem
ber 16, 2017
http://science.sciencemag.org/
Dow
nloaded from