Embed Size (px)
Citation preview

of December 26, 2018.This information is current as
IFN-Mediated ISGylation of HCV-NS5AInhibition of Hepatitis C Virus Replication by
Min-Jung Kim and Joo-Yeon Yoo
http://www.jimmunol.org/content/185/7/4311doi: 10.4049/jimmunol.1000098September 2010;
2010; 185:4311-4318; Prepublished online 1J Immunol
MaterialSupplementary
8.DC1http://www.jimmunol.org/content/suppl/2010/09/01/jimmunol.100009
Referenceshttp://www.jimmunol.org/content/185/7/4311.full#ref-list-1
, 21 of which you can access for free at: cites 50 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2010 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on Decem
ber 26, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on D
ecember 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Inhibition of Hepatitis C Virus Replication by IFN-MediatedISGylation of HCV-NS5A
Min-Jung Kim and Joo-Yeon Yoo
ISG15 is a ubiquitin-like molecule whose expression is induced by type I IFN (IFN-a/b) or in response to virus or bacterial
infection. ISG15 or conjugation of ISG15 to target proteins was reported to play critical roles in the regulation of antiviral
responses. IFN restricts replication of hepatitis C virus (HCV). However, molecular mechanism of IFN-a/b that inhibits HCV
replication is not clear yet. In the current study, we demonstrated that replication of HCV was inhibited by overexpression of
ISG15 and ISG15-conjugation enzymes in the HCV subgenomic replicon cells. Among various nonstructural proteins of HCV,
NS5A was identified as the substrate for ISGylation. Furthermore, protein stability of NS5A was decreased by overexpression of
ISG15 or ISG15-conjugating enzymes. The inhibitory effect of ISG15 or ISGylation on NS5A was efficiently blocked by sub-
stitution of lysine at 379 residue to arginine within the C-terminal region, suggesting that ISGylation directly controls protein
stability of NS5A. Finally, the inhibitory effect of IFN-a/b on HCV replication was further enhanced by ISGylation, suggesting
ISG15 as a therapeutic tool for combined therapy with IFN against HCV. The Journal of Immunology, 2010, 185: 4311–4318.
Hepatitis C virus (HCV) is an enveloped RNA virus thatbelongs to the genus Hepacivirus, family Flaviviridae(1). The HCV genome is a single-stranded, 9.6-kb–long
RNA molecule. It encodes a 3000-aa polyprotein that undergoesposttranslational processing to yield at least 10 functionally dis-tinct viral proteins: core, envelope protein 1 (E1), envelope protein2 (E2), p7, and nonstructural protein (NS)2, NS3, NS4A, NS4B,NS5A, and NS5B (2–4). Unlike other HCV nonstructural proteins,NS5A does not have enzymatic activity. Instead, HCV-NS5A phys-ically interacts with various cellular proteins, such as p53, IFN-induced, dsRNA-activated protein kinase, or a novel transcriptionfactor SNF2-related CBP activator protein, to elicit modulator rolesin cell-cycle regulation, cellular transformation, and antiviral im-mune responses (5–7). HCV-NS5A is a membrane-associated pro-tein that uses its N-terminal amphipathic helix domain and is usuallyfound in the HCV RNA replication complex (8, 9). HCV-NS5A isindispensable for HCV RNA replication, because HCV genomereplicates in the lipid droplet-associated membrane of the endo-plasmic reticulum (ER) (10).Chronic infection of HCV is a leading cause of liver disease,
including chronic hepatitis, liver cirrhosis, and hepatocellular car-cinoma (11).With the absence of effective vaccines to prevent HCVinfection, combined treatment of pegylated IFN-a with ribavirinis the most favorable choice for therapy. However, resistance to
IFN is often observed in patients with HCV infection (12, 13). In-terestingly, mutations in HCV-NS5A were reported to correlatewith responsiveness to IFN therapy in patients, although its precisemechanism is not conclusive (14). To overcome IFN resistance inpatients with HCV infection, the development of chemicals thatinhibit HCV-specific enzymatic activities or antisense oligomersthat inhibit HCV replication has been undertaken (15–17).Pathogen-associated molecular patterns of replicating HCV RNA
genomes, such as 59-triphosphate RNA or dsRNA, are recognizedby the product of RIG-I in the cytoplasm. It triggers the activationof intracellular signaling pathways and transcription factors, suchas NF-kB and IFN regulatory factors (18–22). An outcome of theseevents is the production of type I IFN (IFN-a/b) (23, 24). SecretedIFN-a/b induces the activation of the IFN-stimulated gene factor 3complex, which translocates to the nucleus, where it binds to theIFN-stimulated response element regions of target genes to directthe expression of IFN-stimulated genes (ISGs). Among the genesinduced by IFN, IFN-induced, dsRNA-activated protein kinase,MxA, and ISG15 function as effector molecules in the host cellresponse to viral infection (24). Multiple studies have shown a clearabsence or low level of IFN-a/b in the hepatocytes of patients withchronic HCV infection. In addition, patients with chronic HCVinfection show no significant level of ISG induction upon treatmentwith IFN (25). Based on these findings, it has been suggested thatHCV viral proteins disturb the innate immune response of the hostand that altered hepatic ISG expression is associated with liverpathology.ISG15 is a type I IFN-inducible, ubiquitin-like protein. Most
components for ISG15 conjugation (ISGylation), including UbelL(E1, ISG15-activating enzyme), UbcH8 (E2, ISG15-conjugating en-zyme), and HERC (E3, ISG15 ligase), are highly responsive to IFN-a/b–induced signal transduction (26, 27). Expression of ISG15 andthe conjugation of ISG15 to target proteins are strongly promoted byIFN-a/b treatment, dsRNA, and viral or bacterial infection (28).Production of ISG15 or ISGylation is believed to play an importantrole in establishing the antiviral state of an infected cell. ISG15-deficient mice are highly susceptible to influenza, herpes, and Sind-bis viral infections (29), whereas the protective role of overexpressedISG15 in Sindbis virus-infected IFN-a/bR-deficient mice has beenreported (30). It was also reported that ISGylation of cellular proteins
Department of Life Sciences, Pohang University of Science and Technology, Pohang,Republic of Korea
Received for publication January 13, 2010. Accepted for publication July 29, 2010.
This work was supported by Grant A080084 from the Korea Healthcare TechnologyR&D Project.
Address correspondence and reprint requests to Dr. Joo-Yeon Yoo, Department ofLife Sciences, Pohang University of Science and Technology, Hyouja-dong 31, Po-hang 790-784, Republic of Korea. E-mail address: [email protected].
The online version of this article contains supplemental material.
Abbreviations used in this paper: E1, envelope protein 1; E2, envelope protein 2; ER,endoplasmic reticulum; HCV, hepatitis C virus; ISDR, interferon sensitivity-deter-mining region; ISG, IFN-stimulated gene; NS, nonstructural protein; poly-IC,polyinosinic-polycytidylic acid; POSTECH, Pohang University of Science and Tech-nology; WCE, whole-cell extracts; WT, wild-type.
Copyright� 2010 by The American Association of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1000098
by guest on Decem
ber 26, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

can be suppressed by infection with influenza B virus through theinteraction of ISG15 with the NS1B viral protein (31). In the currentstudy, we investigated the role of ISGylation on HCV-NS5A and itsimplication for viral replication in subgenomic HCV replicon cells.
Materials and MethodsCell culture and transfection
Huh-7 and COS7 cells were maintained in DMEM (Invitrogen, GrandIsland, NY), supplemented with 10% FBS (HyClone, Logan, UT) and 1%penicillin/streptomycin (Invitrogen). Huh-5-15 and Huh-luc/neo-ET HCV(genotype 1b) subgenomic replicon cells (gift of R. Bartenschlager, Uni-versity of Heidelberg, Heidelberg, Germany) were maintained in G418 (600mg/ml; Calbiochem, San Diego, CA)-containing media. Cells were treatedwith IFN-b (100 or 1500 U/ml; PBL Biomedical Laboratories, Piscataway,NJ) or polyinosinic-polycytidylic acid (poly-IC; 25 mg/ml; Amersham Bio-sciences, Piscataway, NJ) for 24 h. For transfection, equal quantities of ex-pression plasmids (2 mg plasmids/35-mm dish) were transfected using Lipo-fectamine 2000 (Invitrogen).
Plasmid construction
The coding regions forHCV-E2, -NS3, -NS4, and -NS5B ofHCVgenotype 1bwere obtained by PCR amplification using pGX10gDs4ST, pGX10-NS34,and pGX10-NS5 plasmids as templates, respectively (gift of Y.C. Sung,Pohang University of Science and Technology [POSTECH], Pohang, SouthKorea), and were cloned into pDEST mammalian expression vectors (Invi-trogen). For the HCV-NS5A expression vector, coding regions for NS5A ofHCV genotype 1b were amplified by PCR using pGX10-NS5 as the template(gift of Y.C. Sung of POSTECH), digested with KpnI and XhoI, and insertedinto the pcDNA3.1/myc or subsequently cloned into the pFlag/CMV plasmid.Deletionmutants of HCV-NS5Awere generated by PCR and were cloned intothe pFlag/CMV plasmid. Point mutations in NS5Awere generated using theQuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA),according to the manufacturer’s instructions. DNA sequences of the con-structed plasmids were verified by automated sequencing. The expressionvectors for HA-Ube1L and Flag-UbcM8 were provided by Dr. D.-E. Zhang(The Scripps Research Institute, La Jolla, CA), and HA-ubiquitin plasmid wasprovided by Chin Ha Chung (Seoul National University, Seoul, Korea). Myc-ISG15 plasmid was cloned, as described (32).
Generation of mutant HCV subgenomic replicon cells
The K379R point mutation was introduced into NS5A of NK5.1 plasmidconstruct (provided by S.K. Jang, POSTECH) using a QuickChange site-directed mutagenesis kit (Stratagene). Linear form of the NK5.1 mutantconstruct (NS5A-K379R) was used as a template for in vitro transcriptionusing T7 RNA polymerase (Ambion, Austin, TX). Huh-7 cells were electro-porated with 10 mg RNA, and stable transfectants were selected in thecomplete DMEM media with 0.6 mg/ml G418 (Calbiochem).
Immunoblot analysis
Cells were harvested in lysis buffer (50mMTris-HCl [pH 7.4], 150mMNaCl,1% Triton X-100, 0.5% deoxycholic acid, 0.1% SDS) containing 1 mMDTT,0.57 mM PMSF, 5 mg/ml leupeptin, 2 mg/ml pepstatin A, 5 mg/ml aprotinin,and 1mMbenzamidine. Total-cell lysates (15–40mg) were separated by SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA).For immunoblotting, commercially available anti-Myc (Roche, Basel, Swit-zerland), anti-Flag (Sigma-Aldrich, St. Louis,MO), anti-V5 (Invitrogen), anti-GAPDH (Chemicon International, Temecula, CA), anti-HA (Roche), andanti–HCV-NS5A Abs (Virogen, Watertown, MA) were used.
Immunoprecipitation
Cellswereharvested in lysis buffer, and1mg total-cell lysateswas preclearedwith normal IgG and protein A/G agarose beads. Lysates were incubated for12 h at 4˚C with the appropriate Ab, followed by an additional 2 h of in-cubation with protein A/G agarose beads.
Immunofluorescence staining
Cells on glass cover slips were fixed with 4% paraformaldehyde solutionand permeabilized with 0.2% Triton X-100. Cells were blocked with 3%goat serum/2.5% BSA and incubated with anti–HCV-NS5A (Virogen) oranti-FLAG (Santa Cruz Biotechnology, Santa Cruz, CA) Abs for 2 h. Cellswere then incubated with Alexa Fluor 568- or Alexa Fluor 488-conjugatedsecondary Abs (both from Invitrogen). Slides were mounted and analyzedusing a fluorescence microscope.
RNA isolation and analysis
Total RNAwas extracted using TRIzol reagent (Invitrogen) and subjected toreal-time RT-PCR. The primer sequences used for PCR were as follows:HCV-NS4, 59-ACAACAGGCAGCGTGGTCATT-39 and 59-TTCCACAT-GTGCTTTGCCCA-39 and actin, 59-TCATGAAGGTGACGTTGACATC-CGT-39 and 59-CCTAGAAGCATTTGCGGTGCACGATG-39.
Luciferase assay
Cells carrying the Huh-luc/neo-ET HCV subgenomic replicon construct weretransfected with empty vectors or expression vectors for the ISGylation-conjugation system. Cells were lysed 48 h posttransfection, and luciferaseactivity was measured using a dual-luciferase assay kit (Promega, Madison,WI). To ensure that equal amounts of cell lysates were used for luciferaseassay, protein quantification using the Bradford method was performed priorto the assay.
Statistics
Data are represented as mean 6 SD, and differences between groups wereanalyzed by the t test.
ResultsReplication of HCV was inhibited by the overexpression ofISG15-conjugation components in the HCV subgenomicreplicon cells
ISG15 is an IFN-inducible gene that is strongly induced by virusinfection in immune and nonimmune cells. ISG15 was shown tonegatively regulate the replication, assembly, and budding of Sindbisvirus,HIV, andEbolavirus, respectively, which strongly suggests thatit functions as an antiviral agent (30, 33). We wondered whetherISG15 or ISG15 conjugation had similar inhibitory effect on thereplication of HCV. To explore the effect of ISGylation, we trans-fected Huh-luc/neo-ET replicon cells with ISG15, Ube1L, andUbcM8 expression plasmids and monitored the replication of sub-genomic HCV replicons. Huh-luc/neo-ET replicon cells harbor theluciferase reporter gene in the HCV replicon construct (34). There-fore, it can be used to quantitatively analyze the effect of ISG15conjugation on HCV replication (Fig. 1). Quite surprisingly, theoverexpression of ISG15 and ISG15-conjugation components sig-nificantly inhibited the replication of the HCV subgenomic repliconto up to 65% of that observed in the empty vector-transfectedcontrol cells.
FIGURE 1. Overexpression of the ISGylation system inhibits HCV
replication in the HCV subgenomic replicon cells. Huh-luc/neo-ET repli-
con construct used in this study (34) (top panel). Cells were transfected
with the indicated expression plasmids, and replication was measured by
luciferase assay (bottom panel). Graph shows mean 6 SD of triplicate
experiment set. Representative data repeated in at least three independent
experiments.
4312 INHIBITION OF HCV REPLICATION BY ISGylation
by guest on Decem
ber 26, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
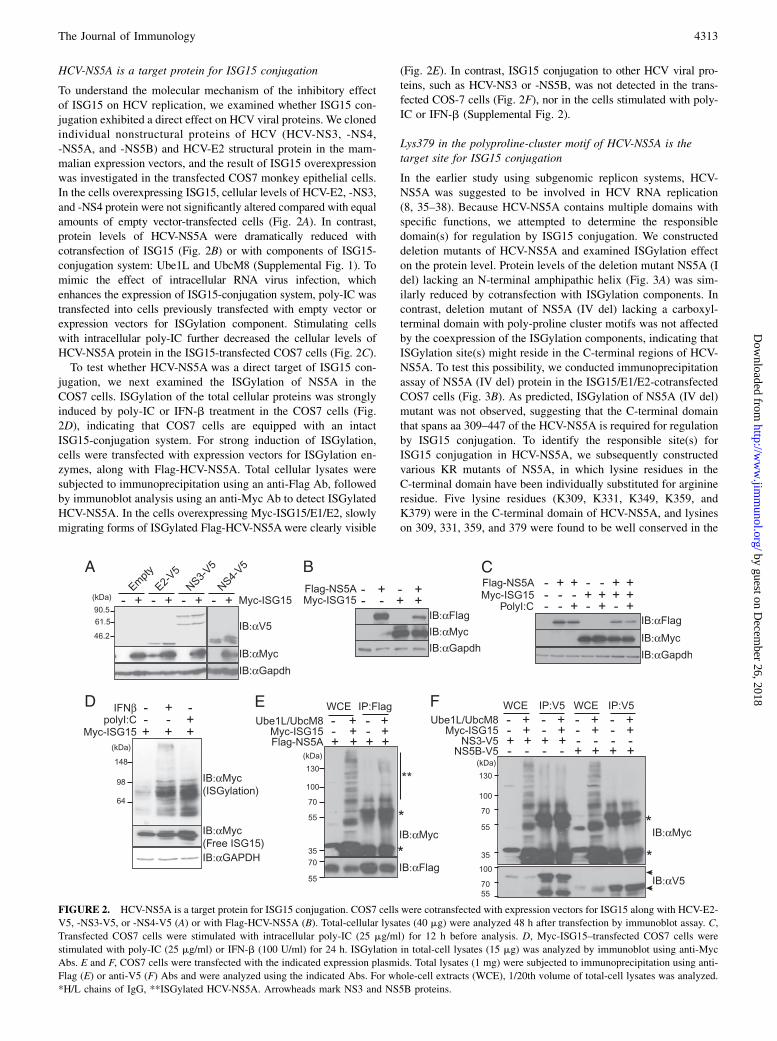
HCV-NS5A is a target protein for ISG15 conjugation
To understand the molecular mechanism of the inhibitory effectof ISG15 on HCV replication, we examined whether ISG15 con-jugation exhibited a direct effect on HCV viral proteins. We clonedindividual nonstructural proteins of HCV (HCV-NS3, -NS4,-NS5A, and -NS5B) and HCV-E2 structural protein in the mam-malian expression vectors, and the result of ISG15 overexpressionwas investigated in the transfected COS7 monkey epithelial cells.In the cells overexpressing ISG15, cellular levels of HCV-E2, -NS3,and -NS4 protein were not significantly altered compared with equalamounts of empty vector-transfected cells (Fig. 2A). In contrast,protein levels of HCV-NS5A were dramatically reduced withcotransfection of ISG15 (Fig. 2B) or with components of ISG15-conjugation system: Ube1L and UbcM8 (Supplemental Fig. 1). Tomimic the effect of intracellular RNA virus infection, whichenhances the expression of ISG15-conjugation system, poly-IC wastransfected into cells previously transfected with empty vector orexpression vectors for ISGylation component. Stimulating cellswith intracellular poly-IC further decreased the cellular levels ofHCV-NS5A protein in the ISG15-transfected COS7 cells (Fig. 2C).To test whether HCV-NS5A was a direct target of ISG15 con-
jugation, we next examined the ISGylation of NS5A in theCOS7 cells. ISGylation of the total cellular proteins was stronglyinduced by poly-IC or IFN-b treatment in the COS7 cells (Fig.2D), indicating that COS7 cells are equipped with an intactISG15-conjugation system. For strong induction of ISGylation,cells were transfected with expression vectors for ISGylation en-zymes, along with Flag-HCV-NS5A. Total cellular lysates weresubjected to immunoprecipitation using an anti-Flag Ab, followedby immunoblot analysis using an anti-Myc Ab to detect ISGylatedHCV-NS5A. In the cells overexpressing Myc-ISG15/E1/E2, slowlymigrating forms of ISGylated Flag-HCV-NS5Awere clearly visible
(Fig. 2E). In contrast, ISG15 conjugation to other HCV viral pro-teins, such as HCV-NS3 or -NS5B, was not detected in the trans-fected COS-7 cells (Fig. 2F), nor in the cells stimulated with poly-IC or IFN-b (Supplemental Fig. 2).
Lys379 in the polyproline-cluster motif of HCV-NS5A is thetarget site for ISG15 conjugation
In the earlier study using subgenomic replicon systems, HCV-NS5A was suggested to be involved in HCV RNA replication(8, 35–38). Because HCV-NS5A contains multiple domains withspecific functions, we attempted to determine the responsibledomain(s) for regulation by ISG15 conjugation. We constructeddeletion mutants of HCV-NS5A and examined ISGylation effecton the protein level. Protein levels of the deletion mutant NS5A (Idel) lacking an N-terminal amphipathic helix (Fig. 3A) was sim-ilarly reduced by cotransfection with ISGylation components. Incontrast, deletion mutant of NS5A (IV del) lacking a carboxyl-terminal domain with poly-proline cluster motifs was not affectedby the coexpression of the ISGylation components, indicating thatISGylation site(s) might reside in the C-terminal regions of HCV-NS5A. To test this possibility, we conducted immunoprecipitationassay of NS5A (IV del) protein in the ISG15/E1/E2-cotransfectedCOS7 cells (Fig. 3B). As predicted, ISGylation of NS5A (IV del)mutant was not observed, suggesting that the C-terminal domainthat spans aa 309–447 of the HCV-NS5A is required for regulationby ISG15 conjugation. To identify the responsible site(s) forISG15 conjugation in HCV-NS5A, we subsequently constructedvarious KR mutants of NS5A, in which lysine residues in theC-terminal domain have been individually substituted for arginineresidue. Five lysine residues (K309, K331, K349, K359, andK379) were in the C-terminal domain of HCV-NS5A, and lysineson 309, 331, 359, and 379 were found to be well conserved in the
FIGURE 2. HCV-NS5A is a target protein for ISG15 conjugation. COS7 cells were cotransfected with expression vectors for ISG15 along with HCV-E2-
V5, -NS3-V5, or -NS4-V5 (A) or with Flag-HCV-NS5A (B). Total-cellular lysates (40 mg) were analyzed 48 h after transfection by immunoblot assay. C,
Transfected COS7 cells were stimulated with intracellular poly-IC (25 mg/ml) for 12 h before analysis. D, Myc-ISG15–transfected COS7 cells were
stimulated with poly-IC (25 mg/ml) or IFN-b (100 U/ml) for 24 h. ISGylation in total-cell lysates (15 mg) was analyzed by immunoblot using anti-Myc
Abs. E and F, COS7 cells were transfected with the indicated expression plasmids. Total lysates (1 mg) were subjected to immunoprecipitation using anti-
Flag (E) or anti-V5 (F) Abs and were analyzed using the indicated Abs. For whole-cell extracts (WCE), 1/20th volume of total-cell lysates was analyzed.
*H/L chains of IgG, **ISGylated HCV-NS5A. Arrowheads mark NS3 and NS5B proteins.
The Journal of Immunology 4313
by guest on Decem
ber 26, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

diverse HCV isolates (Fig. 3C). Among the tested KR mutants ofHCV-NS5A, only the K379R mutant remained unchanged afterthe overexpression of ISGylation components (Fig. 3D). Further-more, ISGylation on HCV-NS5A was completely blocked in theK379R mutant compared with normal ISGylation in the wild-type(WT) or other KR mutants (Fig. 3E), suggesting that lysine 379 onHCV-NS5A is critical for protein regulation by ISGylation.
Reduction of HCV-NS5A protein by ISG15 conjugation in thesubgenomic HCV replicon cells
We demonstrated that HCV-NS5A was a target protein of ISG15conjugation, which led to decreased protein levels in COS7 cells.Based on these observations, we hypothesized that the reductionof HCV-NS5A by ISG15 conjugation may be responsible for thedecreased replication of HCV observed in the Huh-luc/neo-ETreplicon cells (Fig. 1). To investigate the molecular mechanismof HCV replication controlled by ISG15, we examined whetherNS5A was directly controlled by ISG15 conjugation in the HCVsubgenomic replicon cells. With good agreement with the pre-vious results, overexpression of ISG15/E1/E2 restrained proteinlevels of endogenous HCV-NS5A in the Huh-5-15 or Huh-luc/neo-ET replicon cells (Fig. 4A, 4B). The inhibitory effect ofISG15 conjugation on HCV-NS5A became evident when proteinlevels of NS5A were visualized by immunofluorescence stainingat the single-cell level. Immunoreactive HCV-NS5A proteins werediminished in a cell with high levels of UbcM8 (E2) but remainedunchanged in a nontransfected cell (Fig. 4C). Finally, we confirmed
that NS5A in the HCV replicon cells was subject to ISG15 con-jugation (Fig. 4D). Taken together, our results strongly suggest thatISG15 induced by type I IFNs may suppress replication of HCV,presumably because of the reduced cellular levels of HCV-NS5Aprotein mediated by ISG15 conjugation.
Protein stability of HCV-NS5A was affected by ISGylation andubiquitination
To understand the underlying mechanism of HCV-NS5A down-regulation by ISGylation, we examined whether ISGylated NS5Acan be degraded by 26S proteasome. For this purpose, we measuredISGylation of HCV-NS5A in the cells treated with 26S proteasomeinhibitor MG132 (Fig. 5A). As previously reported (32), ISGylationwas strongly enhanced in the MG132-treated total-cell lysates, andISG15 conjugation to HCV-NS5A was correspondingly increased.Consequently, cellular levels of unconjugated NS5A protein werefurther decreased by MG132 treatment. Intriguingly, migrating pat-terns of ISGylated HCV-NS5A in SDS-PAGE were different fromthe untreated control, indicating that NS5A can be subject to furthermodification, such as ubiquitination. To prove this idea, ubiquiti-nation of HCV-NS5A protein was separately assessed by immuno-precipitation in the ubiquitin-overexpressing COS7 cells (Fig. 5B).As expected, ubiquitin-conjugated forms of HCV-NS5A were ob-served in the MG132-treated cells. Interestingly, the ubiquitinatedform of NS5A was also detected in the absence of MG132, sug-gesting that HCV-NS5A protein is a heavily ubiquitinated protein. Itis noteworthy that zinc mesoporphyrin, which suppresses HCV
FIGURE 3. K379 on HCV-NS5A is the ISG15-conjugation site. A, Diagram of the mutant forms of HCV-NS5A used in the study (top panel). COS7 cells
were transfected with indicated expression vectors, and total-cell lysates were analyzed by immunoblot assay (bottom panel). Arrowheads indicate NS5A
(IVdel) and NS5A (I del) proteins. B and E, COS7 cells were transfected with the indicated expression plasmids, and total lysates (1 mg) were subjected to
immunoprecipitation using anti-Flag Abs. *H/L chains of IgG; **ISGylated HCV-NS5A. Arrowheads indicate NS5A (WT) and NS5A (IV del) proteins. C,
Conserved lysine residues in the C-terminal region of HCV-NS5A from various genotype. D, COS7 cells were transfected with expression vectors for
various KR mutant and protein levels of WT, and mutant forms of HCV-NS5A were examined by immunoblot assay.
4314 INHIBITION OF HCV REPLICATION BY ISGylation
by guest on Decem
ber 26, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

replication, exhibited enhanced ubiquitination of HCV-NS5A (39).To determine whether ISGylation of NS5A affects ubiquitination ofNS5A, which leads to the protein degradation via 26S proteasome,we examined ubiquitination of HCV-NS5A (K379R) mutant protein.
Although basal ubiquitination of HCV-NS5A (K379R) was dimin-ished, ubiquitination of WT and K379R mutant NS5A protein wassimilarly increased when 26S proteasome activity was blocked byMG132, indicating that HCV-NS5A might be ubiquitinated on thelysine residue other than K379 (Fig. 5C, Supplemental Fig. 3). How-ever, despite MG132’s effect on the ubiquitination of NS5A, it isnoteworthy that cellular levels of HCV-NS5A and NS5A (K379R)proteins were not changed (Fig. 5B, 5C). We speculated that theprotein-synthesis rate of NS5A might be fast enough to counterbal-ance the degradation rate. To prove this idea, cells were treatedwith cycloheximide to block new protein synthesis, and the effect ofMG132 on protein degradation was examined (Fig. 5D, SupplementalFig. 4). Interestingly, expression of ubiquitin alone increased cellu-lar levels of HCV-NS5A and NS5A (K379R) proteins. However,the cellular levels of the proteins were not affected by MG132 treat-ment, in the absence or presence of cycloheximide treatment. Theseresults indicate that protein stability of HCV-NS5A is subject tocontrol that involves ISGylation and ubiquitination.
ISG15-conjugation system as a potential combined therapywith type I IFNs
Finally, we examined whether ISG15 or the ISG15-conjugationsystem could be used as combined treatment with type I IFN. Inthe Flag-HCV-NS5A–transfected COS7 cells, cellular levels ofHCV-NS5A protein were slightly suppressed by treatment withIFN-b, which was dramatically reduced by cotreatment of ISG15/E1/E2 overexpression (Fig. 6A). In the HCV replicon cells, IFN-b–mediated inhibition of HCV replication was further suppressedby cotransfection of ISGylation components (Fig. 6B). In addition,equivalent levels of the inhibitory effect of IFN-b (100 U/ml) onHCV replication were obtained using combined treatment witha lower dose of IFN-b (50 U/ml) and ISG15/E1/E2 overexpressionin the HCV subgenomic replicon cells (Fig. 6C). In contrast,ISGylation-mediated inhibition of HCV replication was not ob-served in the mutant HCV replicon cells with NS5A (K379R),demonstrating that HCV-NS5A might be the only target for ISG15conjugation (Fig. 6D). In conclusion, effective control of HCVreplication can be achieved by ISG15 or ISG15-conjugation sys-tem, in combination with IFN-b.
FIGURE 4. ISGylation-mediated downregulation of HCV-NS5A protein
in the HCV replicon cells. A, The replicon construct (I389/NS3-3/WT5-15)
(top panel) (36). Huh-5-15 replicon cells (bottom panel) or Huh-luc/neo-
ET replicon cells (B) were transfected with indicated expression vectors.
Endogenous HCV-NS5A protein levels were examined by immunoblot
assay. C, Huh-5-15 replicon cells were transfected with Myc-ISG15 and
Flag-UbcM8/HA-Ube1L plasmids and were analyzed by immunofluores-
cence staining using anti-Flag (green) and anti-HCV-NS5A (red) Abs and
DAPI (blue). D, Huh-5-15 replicon cells were transfected with expression
vectors for the ISG15-conjugation system, and ISGylation of the endog-
enous HCV-NS5A was examined by immunoprecipitation using anti–
HCV-NS5A Abs, followed by immunoblot analysis.
FIGURE 5. HCV-NS5A protein regulation by
ISGylation and ubiquitination. A–C, Transfected
COS7 cells were treated with MG132 (5 mM) for
12 h before analysis. Total lysates (1 mg) were
subjected to immunoprecipitation using anti-Flag
Abs. **ISGylated HCV-NS5A; #ubiquitinated HCV-
NS5A. D, COS-7 cells were transfected with in-
dicated expression plasmids and were treated with
cycloheximide (30 mg/ml) for 6 h before analysis.
The Journal of Immunology 4315
by guest on Decem
ber 26, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

DiscussionIn the current study, we demonstrated that the overexpression ofISG15 and ISGylation components induces ISGylation in the C-terminal regions of HCV-NS5A, reduces cellular levels of NS5Aprotein, and, hence, inhibits replication in the HCV subgenomicreplicon cells.The antiviral potential of ISG15 and ISGylation has been
reported in various viral systems. Overexpression of ISG15 wasshown to inhibit viral packing and replication of HIV, Ebola virus,and Sindbis virus in type I IFNR-deficient mice (30, 33, 40). Micewith a defect in the production of ISG15 are hyperresponsive toviral infection by influenza A and B, herpes, and Sindbis viruses(29, 41). Viral proteins that specifically target ISG15 to inhibitISGylation have been reported (31). Among the viruses that causeacute and chronic infection, HCV is notorious for evading host-immune mechanisms and developing resistance to current IFN-based therapy tools. An internal domain that determines sensi-tivity to IFN has been identified in HCV-NS5A proteins, althoughthe exact mechanism for regulation is not clear (14). Althoughthere is strong evidence that ISG15 can function as an antiviralmediator, its role in regulating HCV replication has not been ex-tensively explored. Silencing UBP43, which specifically cleavesISGylated residues, potentiates antiviral activity of IFN againstHCV replication (42), indicating that ISG15 or ISGylation mayplay a critical role in regulating anti-HCV replication.We showed that treating cells with intracellular poly-IC and
overexpressed ISG15 synergistically suppressed the cellular levelsof HCV-NS5A protein, suggesting that IFN-induced ISG15 conjuga-tion may be involved in the negative regulation of HCV replication.Although ISGylation does not directly target substrates for degra-dation by 26S proteasome complex (43), it inhibits normal functionsof target protein by reducing the amounts of unconjugated forms(32). Alternatively, it is possible that ISG15 conjugation could lead toaltered subcellular localization of target proteins, thereby inhibitingtheir physiological functions. HCV viral genomes are replicated inthe vicinity of ER membranes and form an lipid droplet-associatedreplication complex (10). ERmembrane association mediated by theN-terminal amphipathic helix domain of HCV-NS5A is a critical step
in HCV RNA replication, and a mutant form of HCV-NS5A lackingthe N-terminal domain does not support HCV viral replication (8).Recently, it was reported that E2-conjugating enzymes for ISGyla-tion suppress ubiquitination of cellar protein (44), presenting a com-petition mechanism between ISGylation and ubiquitination or cross-talk between ubiquitination and ISGylation (45). Therefore, futurestudies addressing the regulatory role of ubiquitinated NS5A and itscross-talk to ISGylation will be a key to our understanding of theunderlying mechanism of ISGylation-mediated regulation of HCV-NS5A protein.HCV-NS5A is composed of four functional regions: an N-terminal
amphipathic helix domain, a hyperphosphorylation cluster, the in-terferon sensitivity-determining region (ISDR), and a polyprolinecluster in the C-terminal region (46). Although internal ISDR in theHCV-NS5A is known to determine sensitivity to IFN therapy (14),mutant forms of NS5A lacking ISDR exhibited similar levels ofISGylation, indicating that IFN-dependent ISGylation of NS5A doesnot require ISDR region (Supplemental Fig. 5). Instead, we identifieda lysine residue, K379, in the polyproline cluster of HCV-NS5A asthe target site for ISGylation. Because K379 residue on HCV-NS5Ais conserved in most HCV genotypes, it is likely that the negativeeffect of ISG15 conjugation on HCV-NS5A may be universal. Al-though we now know that ISG15 conjugation of HCV-NS5A at Lys379 is critical for ISGylation-mediated negative control of HCV-NS5A, the molecular link between ISGylation and its effect on tar-get protein regulation is still elusive.We did not observe a similar inhibitory effect of HCV replica-
tion by ISG15 or ISGylation in the Huh-7.5 cells infected with theJFH strain of HCV (Supplemental Fig. 6). Similarly, ISG15 wasrecently reported to promote HCV replication in the HCV J6/JFH1-infected Huh-7.5 cells (47). There are several possible explan-ations for this difference. First, the effect of ISG15 or ISGylationon HCV replication may be specific to the genotypes. To confirmthe inhibitory effect of ISGylation, we examined two HCV sub-genomic replicon constructs, with all of the HCV genome se-quence derived from genotype 1b, whereas the HCV J6/JFH1strain used for cellular infection was derived from genotype 2a.HCV-NS5A from genotypes 1b and 2a showed only 64% amino
FIGURE 6. Effect of the combined treatment con-
sisting of ISG15 conjugation plus IFN-b. A, Trans-
fected COS7 cells were stimulated with IFN-b (1500
U/ml) for 24 h or left untreated. Protein levels of HCV-
NS5A were analyzed by immunoblot assay. Huh-5-15
replicon cells (B) and Huh-NS5A-K379R replicon
cells (D) were transfected with ISG15-conjugating
enzymes and stimulated with IFN-b (100 U/ml) for
the indicated times. Amplification of HCV replicons
was investigated by real-time RT-PCR using PCR
primers specific for HCV-NS4. Graphs show mean 6SD obtained from three separate experiments. C, Huh-
5-15 replicon cells were transfected, as indicated, and
amplification of the HCV replicon was examined by
conventional RT-PCR after treatment with IFN-b (50
or 100 U/ml) for 24 h.
4316 INHIBITION OF HCV REPLICATION BY ISGylation
by guest on Decem
ber 26, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

acid identity. In addition, it is possible that accumulated mutationsin the HCV J6/JFH1 RNA genome during replication providesan evasion mechanism against ISGylation. Finally, the effect ofISG15 or ISGylation on HCV-NS5A may be compromised byHCV viral proteins that are not present in the replicon construct.The HCV subgenomic replicon used in this study contained codingsequences covering NS3, NS4A/B, NS5A, and NS5B. Therefore, itlacked core, E1, E2, p7, and NS2 proteins.Currently available treatment of HCV infection is restricted to
pegylated IFN-a and ribavirin (48, 49). However, limited efficacyand side effects in patients treated with IFN-a demand the de-velopment of new therapeutic tools. Based on the focal replicationof HCV in the liver of patients, focal treatment has been suggestedto substitute for the classical systematic management of IFNs andto increase HCV clearance with fewer side effects (50). In thisstudy, combined treatment, consisting of enhanced ISG15 conju-gation and IFN-b, exhibited improved efficacy in the clearanceof HCV in the HCV subgenomic replicon cells. It would be in-teresting to see whether this combined therapy could limit HCVreplication efficiently in primates. For this purpose, future studiesaimed at the effective delivery of ISG15-conjugation system to thevirus-infected regions in the liver are required.
AcknowledgmentsWe thank Drs. D.-E. Zhang, Y.C. Sung, S.K. Jang, and R. Bartenschlager for
plasmids and HCV replicon cells. We also thank members of Dr. S.K. Jang’s
laboratory (S.J. Guam, M.H. Jee, and J.H. Jung; POSTECH) for technical
support.
DisclosuresThe authors have no financial conflicts of interest.
References1. Lindenbach, B. D., and C. M. Rice. 2005. Unravelling hepatitis C virus repli-
cation from genome to function. Nature 436: 933–938.2. Choo, Q. L., G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, and
M. Houghton. 1989. Isolation of a cDNA clone derived from a blood-bornenon-A, non-B viral hepatitis genome. Science 244: 359–362.
3. Clarke, B. 1997. Molecular virology of hepatitis C virus. J. Gen. Virol. 78: 2397–2410.
4. Hijikata, M., N. Kato, Y. Ootsuyama, M. Nakagawa, and K. Shimotohno. 1991.Gene mapping of the putative structural region of the hepatitis C virus genomeby in vitro processing analysis. Proc. Natl. Acad. Sci. USA 88: 5547–5551.
5. Gale, M., Jr., B. Kwieciszewski, M. Dossett, H. Nakao, and M. G. Katze. 1999.Antiapoptotic and oncogenic potentials of hepatitis C virus are linked to in-terferon resistance by viral repression of the PKR protein kinase. J. Virol. 73:6506–6516.
6. Ghosh, A. K., M. Majumder, R. Steele, P. Yaciuk, J. Chrivia, R. Ray, andR. B. Ray. 2000. Hepatitis C virus NS5A protein modulates transcription througha novel cellular transcription factor SRCAP. J. Biol. Chem. 275: 7184–7188.
7. Lan, K. H., M. L. Sheu, S. J. Hwang, S. H. Yen, S. Y. Chen, J. C. Wu, Y. J. Wang,N. Kato, M. Omata, F. Y. Chang, and S. D. Lee. 2002. HCV NS5A interacts withp53 and inhibits p53-mediated apoptosis. Oncogene 21: 4801–4811.
8. Brass, V., E. Bieck, R. Montserret, B. Wolk, J. A. Hellings, H. E. Blum, F. Penin,and D. Moradpour. 2002. An amino-terminal amphipathic alpha-helix mediatesmembrane association of the hepatitis C virus nonstructural protein 5A. J. Biol.Chem. 277: 8130–8139.
9. Lundin, M., H. Lindstrom, C. Gronwall, and M. A. Persson. 2006. Dual topologyof the processed hepatitis C virus protein NS4B is influenced by the NS5Aprotein. J. Gen. Virol. 87: 3263–3272.
10. Miyanari, Y., K. Atsuzawa, N. Usuda, K. Watashi, T. Hishiki, M. Zayas,R. Bartenschlager, T. Wakita, M. Hijikata, and K. Shimotohno. 2007. The lipiddroplet is an important organelle for hepatitis C virus production. Nat. Cell Biol.9: 1089–1097.
11. Di Bisceglie, A. M. 1998. Hepatitis C. Lancet 351: 351–355.12. De Francesco, R., and G. Migliaccio. 2005. Challenges and successes in de-
veloping new therapies for hepatitis C. Nature 436: 953–960.13. Feld, J. J., and J. H. Hoofnagle. 2005. Mechanism of action of interferon and
ribavirin in treatment of hepatitis C. Nature 436: 967–972.14. Enomoto, N., I. Sakuma, Y. Asahina, M. Kurosaki, T. Murakami, C. Yamamoto,
Y. Ogura, N. Izumi, F. Marumo, and C. Sato. 1996. Mutations in the non-structural protein 5A gene and response to interferon in patients with chronichepatitis C virus 1b infection. N. Engl. J. Med. 334: 77–81.
15. Forestier, N., H. W. Reesink, C. J. Weegink, L. McNair, T. L. Kieffer, H. M. Chu,S. Purdy, P. L. Jansen, and S. Zeuzem. 2007. Antiviral activity of telaprevir(VX-950) and peginterferon alfa-2a in patients with hepatitis C. Hepatology 46:640–648.
16. Modi, A. A., and J. H. Hoofnagle. 2007. New therapies for hepatitis C. Hep-atology 46: 615–617.
17. Sarrazin, C., R. Rouzier, F. Wagner, N. Forestier, D. Larrey, S. K. Gupta,M. Hussain, A. Shah, D. Cutler, J. Zhang, and S. Zeuzem. 2007. SCH 503034,a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2bfor genotype 1 nonresponders. Gastroenterology 132: 1270–1278.
18. Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innateimmunity. Cell 124: 783–801.
19. Hornung, V., J. Ellegast, S. Kim, K. Brzozka, A. Jung, H. Kato, H. Poeck,S. Akira, K. K. Conzelmann, M. Schlee, et al. 2006. 59-Triphosphate RNA is theligand for RIG-I. Science 314: 994–997.
20. Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, andC. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-strandedRNA bearing 59-phosphates. Science 314: 997–1001.
21. Schroder, M., and A. G. Bowie. 2005. TLR3 in antiviral immunity: key player orbystander? Trends Immunol. 26: 462–468.
22. Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi,M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-Ihas an essential function in double-stranded RNA-induced innate antiviralresponses. Nat. Immunol. 5: 730–737.
23. Kawai, T., and S. Akira. 2006. Innate immune recognition of viral infection. Nat.Immunol. 7: 131–137.
24. Sen, G. C. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55: 255–281.25. Smith, M. W., Z. N. Yue, M. J. Korth, H. A. Do, L. Boix, N. Fausto, J. Bruix,
R. L. Carithers, Jr., and M. G. Katze. 2003. Hepatitis C virus and liver disease:global transcriptional profiling and identification of potential markers. Hep-atology 38: 1458–1467.
26. Kim, K. I., N. V. Giannakopoulos, H. W. Virgin, and D. E. Zhang. 2004.Interferon-inducible ubiquitin E2, Ubc8, is a conjugating enzyme for proteinISGylation. Mol. Cell. Biol. 24: 9592–9600.
27. Kim, K. I., M. Yan, O. Malakhova, J. K. Luo, M. F. Shen, W. Zou, J. C. de laTorre, and D. E. Zhang. 2006. Ube1L and protein ISGylation are not essential foralpha/beta interferon signaling. Mol. Cell. Biol. 26: 472–479.
28. Loeb, K. R., and A. L. Haas. 1992. The interferon-inducible 15-kDa ubiq-uitin homolog conjugates to intracellular proteins. J. Biol. Chem. 267: 7806–7813.
29. Lenschow, D. J., C. Lai, N. Frias-Staheli, N. V. Giannakopoulos, A. Lutz,T. Wolff, A. Osiak, B. Levine, R. E. Schmidt, A. Garcia-Sastre, et al. 2007. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza,herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA. 104: 1371–1376.
30. Lenschow, D. J., N. V. Giannakopoulos, L. J. Gunn, C. Johnston, A. K. O’Guin,R. E. Schmidt, B. Levine, and H. W. Virgin, IV. 2005. Identification ofinterferon-stimulated gene 15 as an antiviral molecule during Sindbis virus in-fection in vivo. J. Virol. 79: 13974–13983.
31. Yuan, W., and R. M. Krug. 2001. Influenza B virus NS1 protein inhibits con-jugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J.20: 362–371.
32. Kim, M. J., S. Y. Hwang, T. Imaizumi, and J. Y. Yoo. 2008. Negative feedbackregulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15conjugation. J. Virol. 82: 1474–1483.
33. Okumura, A., G. Lu, I. Pitha-Rowe, and P. M. Pitha. 2006. Innate antiviral re-sponse targets HIV-1 release by the induction of ubiquitin-like protein ISG15.Proc. Natl. Acad. Sci. USA 103: 1440–1445.
34. Vrolijk, J. M., A. Kaul, B. E. Hansen, V. Lohmann, B. L. Haagmans,S. W. Schalm, and R. Bartenschlager. 2003. A replicon-based bioassay for themeasurement of interferons in patients with chronic hepatitis C. J. Virol. Methods110: 201–209.
35. Hughes, M., S. Gretton, H. Shelton, D. D. Brown, C. J. McCormick,A. G. Angus, A. H. Patel, S. Griffin, and M. Harris. 2009. A conserved prolinebetween domains II and III of hepatitis C virus NS5A influences both RNAreplication and virus assembly. J. Virol. 83: 10788–10796.
36. Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, andR. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs ina hepatoma cell line. Science 285: 110–113.
37. Moradpour, D., M. J. Evans, R. Gosert, Z. Yuan, H. E. Blum, S. P. Goff,B. D. Lindenbach, and C. M. Rice. 2004. Insertion of green fluorescent proteininto nonstructural protein 5A allows direct visualization of functional hepatitis Cvirus replication complexes. J. Virol. 78: 7400–7409.
38. Sauter, D., K. Himmelsbach, M. Kriegs, M. Carvajal Yepes, and E. Hildt. 2009.Localization determines function: N-terminally truncated NS5A fragmentsaccumulate in the nucleus and impair HCV replication. J. Hepatol. 50: 861–871.
39. Hou, W., Q. Tian, J. Zheng, and H. L. Bonkovsky. 2010. Zinc mesoporphyrininduces rapid proteasomal degradation of hepatitis C nonstructural 5A protein inhuman hepatoma cells. Gastroenterology 138: 1909–1919.
40. Okumura, A., P. M. Pitha, and R. N. Harty. 2008. ISG15 inhibits Ebola VP40VLP budding in an L-domain-dependent manner by blocking Nedd4ligase activity. Proc. Natl. Acad. Sci. USA 105: 3974–3979.
41. Osiak, A., O. Utermohlen, S. Niendorf, I. Horak, and K. P. Knobeloch. 2005.ISG15, an interferon-stimulated ubiquitin-like protein, is not essential for STAT1signaling and responses against vesicular stomatitis and lymphocytic chorio-meningitis virus. Mol. Cell. Biol. 25: 6338–6345.
The Journal of Immunology 4317
by guest on Decem
ber 26, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

42. Randall, G., L. Chen, M. Panis, A. K. Fischer, B. D. Lindenbach, J. Sun,J. Heathcote, C. M. Rice, A. M. Edwards, and I. D. McGilvray. 2006. Silencingof USP18 potentiates the antiviral activity of interferon against hepatitis C virusinfection. Gastroenterology 131: 1584–1591.
43. Liu, M., X. L. Li, and B. A. Hassel. 2003. Proteasomes modulate conjugation tothe ubiquitin-like protein, ISG15. J. Biol. Chem. 278: 1594–1602.
44. Arimoto, K., H. Konishi, and K. Shimotohno. 2008. UbcH8 regulates ubiquitinand ISG15 conjugation to RIG-I. Mol. Immunol. 45: 1078–1084.
45. Liu, Y. C., J. Penninger, and M. Karin. 2005. Immunity by ubiquitylation: a re-versible process of modification. Nat. Rev. Immunol. 5: 941–952.
46. Macdonald, A., and M. Harris. 2004. Hepatitis C virus NS5A: tales of a pro-miscuous protein. J. Gen. Virol. 85: 2485–2502.
47. Chen, L., J. Sun, L. Meng, J. Heathcote, A. Edwards, and I. McGilvray. 2010.ISG15, a ubiquitin-like interferon stimulated gene, promotes Hepatitis C Virus
production in vitro: Implications for chronic infection and response to treatment.
J. Gen. Virol. 91: 382–388.48. McHutchison, J. G., E. J. Lawitz, M. L. Shiffman, A. J. Muir, G. W. Galler,
J. McCone, L. M. Nyberg, W. M. Lee, R. H. Ghalib, E. R. Schiff, et al; IDEAL
Study Team. 2009. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment
of hepatitis C infection. N. Engl. J. Med. 361: 580–593.49. Watanabe, S., N. Enomoto, K. Koike, N. Izumi, H. Takikawa, E. Hashimoto,
F. Moriyasu, H. Kumada, and M. Imawari. 2010. Prolonged treatment with
pegylated interferon alpha 2b plus ribavirin improves sustained virological re-
sponse in chronic hepatitis C genotype 1 patients with late response in a clinical
real-life setting in Japan. Hepatol. Res. 40: 135–144.50. Stiffler, J. D., M. Nguyen, J. A. Sohn, C. Liu, D. Kaplan, and C. Seeger. 2009.
Focal distribution of hepatitis C virus RNA in infected livers. PLoS ONE 4: e6661.
4318 INHIBITION OF HCV REPLICATION BY ISGylation
by guest on Decem
ber 26, 2018http://w
ww
.jimm
unol.org/D
ownloaded from













![Why do I include peg-IFN in Hepatitis C treatment - genotype 1 Bendor July 2014 [Lecture seule].pdfInternational Liver Congress 2013, Amsterdam Treatment With Sofosbuvir+ Peginterferon+](https://img.pdfslide.us/doc/110x75/5e7c22806e17124ba231c001/why-do-i-include-peg-ifn-in-hepatitis-c-treatment-genotype-1-bendor-july-2014.jpg)















