Embed Size (px)
Citation preview

PEDIATRICS Volume 141, number s5, April 2018:e20170213CASE REPORT
Pediatric panniculitis occurs in a group of rare disorders characterized by inflammation of the subcutaneous fat.1, 2 Histopathological analysis is required to confirm the diagnosis because the lesions may present differently, often consisting of erythematous nodules or large infiltrated plaques. Panniculitis cases are classified as mostly lobular, mostly septal, or mixed, depending on the distribution of the dominant subcutaneous inflammatory infiltrates.3 Panniculitis can be a primary disease but is most often a secondary process related to many different underlying
disorders. Some types of panniculitis are specific to children, including those only seen in neonates such as subcutaneous fat necrosis of the newborn, or occur in a specific context such as postcorticosteroid panniculitis and cold panniculitis.1, 2 Other recognized etiologies described in adults may be observed in children but are less frequent or rare in this population, including: infectious, connective tissue disease–related, subcutaneous lymphoma–like, metabolic (alpha1-antitrypsin deficiency, pancreatitis), and drug- or trauma-induced panniculitis. In
Inherited Immunodeficiency: A New Association With Early-Onset Childhood PanniculitisBrigitte Bader-Meunier, MD, a, b Frédéric Rieux-Laucat, PhD, b Fabien Touzot, MD, PhD, c Marie-Louise Frémond, MD, a, b Isabelle André-Schmutz, PhD, b Sylvie Fraitag, MD, d Christine Bodemer, MD, PhDb, e, f
We report on 4 children who presented with aseptic panniculitis associated with inherited immunodeficiency. Three patients had a B-cell immunodeficiency resulting from mutations in the TRNT1 and NF-κb2 genes (no mutation was found in the third patient), and 1 had a T-cell deficiency (mutation in the LCK gene). Panniculitis occurred before the age of 2 years in the 4 patients and preceded the onset of recurrent infections because of immunodeficiency in 2. It presented either as nodules, which resolved spontaneously within 1 to 2 weeks (3 patients), or chronic ulcerative lesions (1 patient) associated with unexplained fever and elevated acute phase reactants, without evidence of infection or high-titer autoantibodies. Febrile nodules relapsed in 2 patients, and recurrent attacks of unexplained fever (without relapse of panniculitis) occurred in the third. Skin biopsy revealed predominantly lympho-histiocytic or septal neutrophilic panniculitis in 1 and 3 patients, respectively. Panniculitis was associated with dermal involvement in the 4 patients. Patients with B-cell deficiency received monthly intravenous immunoglobulin replacement. Two patients who underwent bone marrow transplant died of bone marrow transplant-related complications. The 2 remaining patients had persistent, mild autoinflammatory disease, which did not require specific treatment. In these cases, the need for careful immunologic evaluation of patients who present with unexplained panniculitis, especially early-onset panniculitis before the age of 2 years, is highlighted.
abstract
To cite: Bader-Meunier B, Rieux-Laucat F, Touzot F, et al. Inherited Immunodeficiency: A New Association With Early-Onset Childhood Panniculitis. Pediatrics. 2018;141(s5):e20170213
aDépartements d'Immunologie, Hématologie, et Rhumatologie Pédiatrique, dd'Anatomie Pathologique, and ede Dermatologie Pédiatrique, Hôpital Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris, Paris, France; bInstitut Imagine, INSERM U1163; fUniversité Paris Descartes, Sorbonne Cité, Paris, France; and cDépartement d’Immunologie et Rhumatologie Pédiatrique, Centre Hospitalier Universitaire Sainte-Justine, Université de Montréal, Quebec, Canada
Drs Bodemer, Fraitag, and Bader-Meunier conceptualized and designed the study, supervised the data collection, and drafted the initial manuscript; Drs Touzot and Frémond conducted the initial analyses and reviewed and revised the manuscript; Dr Fraitag was involved in the pathologic analysis; Drs André-Schmutz and Rieux-Laucat were involved in the genetic studies; and all authors approved the final manuscript as submitted.
DOI: https:// doi. org/ 10. 1542/ peds. 2017- 0213
Accepted for publication Aug 28, 2017
Address correspondence to Brigitte Bader-Meunier, MD, Hémato-Immunologie Pédiatrique, Hôpital Necker APHP, 149 rue de Sèvres, 75015 Paris, France. E-mail: [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275).
Copyright © 2018 by the American Academy of Pediatrics
FINANCIAL DISCLOSURE: The authors have indicated they have no financial relationships relevant to this article to disclose.
FUNDING: No external funding.
POTENTIAL CONFLICT OF INTEREST: The authors have indicated they have no potential conflicts of interest to disclose.
by guest on June 6, 2020www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 141, number s5, April 2018 S497
many pediatric patients, no classic, specific cause can be identified. Such cases are designated as idiopathic panniculitis. Here, we describe the cases of 4 patients who presented with early-onset panniculitis associated with inherited B- or T-cell immunodeficiency. We propose that inherited immunodeficiency is a new cause of panniculitis, underlying the need for careful immunologic evaluation for cases of unexplained pediatric panniculitis, particularly with onset before the age of 2 years.
CASE REPORTS
We identified 4 patients who displayed inherited immunodeficiency associated with panniculitis among 18 cases of pediatric idiopathic panniculitis (onset before the age of 16 years) recorded in the database of the Department of Dermatological Pathology of the Necker-Enfants Malades hospital from January 2004 to December 2014. Specific infectious causes for the panniculitis lesions were excluded by specific stains (periodic acid–Schiff, Grocott, Ziehl, and Gram) in cases of neutrophilic and/or macrophagic infiltrates, and Epstein-Barr encoding region staining was used for those composed of lymphocytes. Blood cultures and serological tests for Epstein-Barr virus, human herpesvirus 6, and cytomegalovirus were performed if an infection was suspected. Specific gene sequencing or whole exome sequencing (WES) was performed on DNA of the 4 patients after obtaining informed consent from the patients’ parents.
The 4 patients were referred to the Department of Pediatric Immunology and Rheumatology of the Necker-Enfants Malades hospital at the onset of disease. All were born to healthy, nonconsanguineous parents. Their demographic, biochemical, immunologic, pathologic, and genetic features are shown in Table 1.
Patient 1
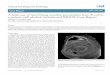
A 2-month-old girl was referred because of fever and an erythematous nodule on 1 toe lasting 2 weeks (Fig 1). C-reactive protein (CRP) was 42 mg/L. A skin biopsy revealed predominantly septal lympho-histiocytic panniculitis (Fig 1). The symptoms resolved spontaneously within 4 weeks. During the course of the disease, the patient presented with relapsing noninfectious panniculitis coinciding with febrile attacks without any other manifestations and elevated acute-phase reactants without any other manifestations. Two to 4 episodes per year occurred and resolved spontaneously within 1 to 2 weeks. Other findings included recurrent otitis and ataxia, and we performed immunologic tests that revealed hypogammaglobulinemia and CD27+ memory B-cell deficiency (Table 1). Replacement intravenous immunoglobulin (IVIg) therapy was started (400 mg/kg per day). At 8 years of age, she was normal-sized for her age, and no more infections had occurred. Relapsing panniculitis and a mild ataxia persisted. WES was performed but did not allow identification of the underlying genetic defect.
Patient 2
A 20-month-old boy was referred because of undocumented lower respiratory tract infections, Salmonella infections, and failure to thrive. From the age of 10 months, he had a history of noninfectious, relapsing episodes of fever associated with multiple erythematous nodules (hands, feet, bottom) (Fig 1) and undocumented diarrhea. CRP levels rose substantially during these attacks (Table 1). A skin biopsy of 1 nodule revealed septal and lobular neutrophilic panniculitis (Fig 1). Immunologic investigation revealed a deficiency of serum immunoglobulin G (IgG), immunoglobulin A (IgA), and immunoglobulin M (IgM); poor vaccine responses; and abnormally
low levels of CD27+ B cells and switch memory B cells. IVIg was started (400 mg/kg per day). He developed further chronic fever, diarrhea, arthritis, failure to thrive, and multiple ulcerative plaques (Fig 1) that did not undergo biopsy. At onset, he responded to treatment with high doses of prednisone (2 mg/kg per day) and treatment with interleukin-1β inhibitors (anakinra, canakinumab), anti–tumor necrosis factor inhibitors (infliximab), and cyclophosphamide were not steroid-sparing. He underwent a bone marrow transplant but died of treatment-related complications within the first days of the procedure. WES identified a mutation in the NF-κB2 gene.
Patient 3
A 12-month-old girl was referred because of recurrent attacks of fever from the age of 2 months, with or without documented infections, and failure to thrive. CRP levels were elevated during each episode. Infections consisted of recurrent septicemias. Aseptic febrile manifestations were vulvitis, parotitis, adenitis, and the occurrence of 1 noninfectious erythematous nodule of the limb (at 10 months). A skin biopsy of the nodule revealed lobular and septal neutrophilic panniculitis. Other features included chronic microcytic anemia (from the age of 2 months), low levels of serum IgG, IgA, and IgM, and low levels of CD27+ B cells and memory B cells. IVIg was started (400 mg/kg per day). At 6 years of age, her height was normal, and no infections had occurred. She continued to have 3 to 4 febrile attacks (lasting for 1 to 2 days) each month, associated with elevated CRP levels. There were no further episodes of panniculitis or other inflammatory manifestations associated with fever, and the anemia resolved spontaneously. WES identified a compound heterozygous mutation in the TRNT1 gene.
by guest on June 6, 2020www.aappublications.org/newsDownloaded from

BADER-MEUNIER et alS498
Patient 4
A 22-month-old girl was referred because of daily spiking fever, accompanied by multiple ulcerative skin nodules and inflammation of the interphalangeal joints (Fig 1). No infection was found, and these manifestations persisted despite intravenous antibiotic therapy (ceftriaxone, gentamycin, vancomycin, clarithromycin, ciprofloxacin, rifampicin, isoniazid). From the age of 15 months, she had undocumented protracted diarrhea and recurrent undocumented upper and lower respiratory tract infections, resulting in failure to
thrive. A skin biopsy of a lesion revealed lobular neutrophilic panniculitis without granuloma (Fig 1). She had elevated CRP levels, and immunologic studies revealed CD4+ T-cell lymphopenia and low CD4 and CD8 expression. A LCK gene defect was identified. Given the severe T-cell deficiency, the patient underwent bone marrow transplant but died shortly after the procedure from treatment-related complications.
DISCUSSION
We report on 4 patients with an inherited B- or T-cell deficiency that was associated with aseptic
panniculitis. Classic causes of panniculitis in children include infection, inflammatory rheumatic disorders, α1-antitrypsin deficiency, T-cell cytotoxic lymphoma, trauma- and cold-induced panniculitis, subcutaneous fat necrosis of the newborn, corticosteroid use, and erythema nodosum. Inherited immunodeficiency has not previously been considered as being associated with panniculitis.1, 2 Indeed, the association of aseptic panniculitis with inherited immunodeficiency has been reported only for GATA2 and CECR1 mutations. GATA2 mutations result in a broad phenotype that encompasses immunodeficiency,
TABLE 1 Demographic, Clinical, Biological, and Pathologic Features in 4 Patients With Panniculitis Associated With Immunodeficiency
Patient 1 Patient 2 Patient 3 Patient 4
Sex, ethnicity Female, white Male, white Female, white Female, whiteAge at onset of panniculitis,
mo (No. of infections)2 (36) 10 (20) 10 (2) 22 (15)
Clinical characteristics of panniculitis
Relapsing erythematous nodules coinciding with
febrile attacks (toes, abdomen, limb)
Relapsing erythematous nodules coinciding with
febrile attacks (hands, feet, bottom)
Unique episode of 1 aseptic erythematous nodule of the limb coinciding with
fever
Chronic multiple ulcerating skin nodules (PIP joints)
associated with fever
CRP at onset of panniculitis, mg/L (N: <6 mg/L)
42 60 90 77
Serum immunoglobulin (age, mo)
(37) (32) (4) (23)
IgG, g/L 3.5 (5.4–10.2) 3.26 (5.5–10.2) 1.14 (2.9–5.5) 8.7 (4.8–8.9) IgA, g/L 0.16 (0.4–1.4) <0.06 (0.4–1.4) <0.04 (0.1–0.4) 1.21 (0.3–1.2) IgM, g/L 0.16 (0.5–1.5) 0.18 (0.5–1.5) 0.07 (0.3–0.8) 3.06 (0.5–1.5)Lymphocytes/μL (age, mo) 2200 (90) 4200 (90) 5600 (15) 1300 (3600–8900) (29)T cells/μL CD3+ 1738 (1200–2600) 3822 (1200–2600) 2800 (1400–3700) 520 (2100–6200) CD4+ 1100 (650–1500) 3444 (650–1500) 1792 (700–2200) 156 (1300–3400) CD8+ 418 (370–1100) 380 (370–1100) 640 (490–1300) 338 (620–2000)B cells/μL CD19+ 286 (273–860) 250 (350–1100) 0 (390–1400) 559 (720–2600) CD27+/CD19+ (memory
B cells)11 (23–185) 42 (200–420) 0 (330–500) ND
Other immunologic tests Decrease of specific antibody responses to vaccines
Decrease of specific antibody responses to vaccines
Decrease of specific antibody responses to vaccines
Low levels of CD4 and CD8 expression at the cell
surface, lack of response to T-cell receptor activation
ANA — — — 1 out of 600 without specificityPathologic findings (age at
skin biopsy, mo)Lobular and septal
lymphohistiocytic panniculitis (2); CD3+ T cells++ (CD8+), CD20 ±, M++, N±, Eo±, dermis
involvement +
Lobular and septal neutrophilic panniculitis; (80); N+++, CD3+ T cells+
(CD8+), M+, CD20−, fat necrosis, dermis
involvement +
Lobular and septal neutrophilic panniculitis;
(10); N+++, CD3+ T cells++ (CD8+), CD20−, M+, dermis
involvement ±
Lobular and septal neutrophilic panniculitis; (24); N+++,
M++, CD3+ T cells, fat necrosis +, dermis involvement +++
Genetic analysis No mutation Mutation in NF-κB2 (c.2593/2594 del AG)
Compound heterozygous mutations in TRNT1
(c.1213G > A/c.1057–7C > G)
Homozygous mutations in LCK (c.1022T > C)
For immunoglobulin levels and T and B cells, normal values according to age are given in parentheses. Each cellular infiltrate was quantified as absent (−), weak (±), mild (+), moderate (++), or high (+++). ANA, antinuclear antibodies; Eo, eosinophils; PIP, proximal interphalangeal; M, macrophages; N, neutrophils; ND, not done; —, not applicable.
by guest on June 6, 2020www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 141, number s5, April 2018 S499
myelodysplastic syndrome, pulmonary disease, and vascular and/or lymphatic dysfunction.3 CECR1 mutations result in adenosine deaminase 2 deficiency associated with a spectrum of vascular and inflammatory phenotypes, ranging from early-onset recurrent stroke to systemic vasculopathy or vasculitis, and is associated with hypogammaglobulinemia in some patients.4
Panniculitis occurred before 2 years of age in the 4 patients and preceded the manifestation of immunodeficiency (infections) in 2. At the onset of the disease, panniculitis presented as relapsing nodules that coincided with febrile attacks in 2 patients who had a B-cell
deficiency and as chronic ulcerative plaques associated with fever in the patient with a T-cell deficiency. In the fourth patient, a single episode of febrile aseptic neutrophilic panniculitis occurred in addition to recurrent noninfectious febrile attacks associated with systemic inflammation. These episodes of panniculitis that are associated with an unexplained fever and systemic inflammation, without evidence of high-titer autoantibodies or autoantigen-specific T cells, are suggestive of autoinflammatory manifestations. Until now, early-onset panniculitis associated with systemic inflammation had been reported only in 4 autoinflammatory diseases that did not display an immunodeficiency phenotype:
(1) chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature, characterized by early-onset, recurrent fever, skin lesions, lipodystrophy, and multisystemic inflammatory manifestations, resulting from autosomal recessive mutations in proteasome genes5; (2) familial Mediterranean fever, which classically consists of short, recurrent febrile episodes, serositis, and arthritis resulting from mutations in marenostrin-encoding fever gene6; (3) otulipenia, which presents as neonatal-onset fever, neutrophilic dermatitis and/or panniculitis, and failure to thrive because of mutations in OTULIN, encoding a deubiquitinase7; and (4) granulomatous panniculitis in infants
FIGURE 1Clinical presentation and biopsy specimens from skin lesions stained with hematoxylin eosin saffron of panniculitis. A1, Erythematous plaque of the toe (patient 1). A2, Erythematous nodules and ulcerative plaque of the leg (patient 2). A3, Ulcerating skin nodules of the fingers (patient 4). B1, Lobular lymphocytic panniculitis with a slight lymphocytic infiltrate localized around the adipocytes (original magnification ×25) (patient 1). B2, Inflammation of the deep dermis and the subcutis, located in the septa and the lobules and composed of mixed cells with a predominance of neutrophils (original magnification ×200) (patient 2). B3, Lobular panniculitis with dense neutrophilic infiltrate replacing part of the subcutis (original magnification ×200) (patient 4).
by guest on June 6, 2020www.aappublications.org/newsDownloaded from

BADER-MEUNIER et alS500
who present with infantile-onset of widespread lobular panniculitis, panuveitis, arthropathy, and severe systemic illness.8
Skin biopsy revealed predominantly lympho-histiocytic or septal neutrophilic panniculitis in 1 and 3 patients, respectively. A histologic classification scheme has been proposed, dividing the cases into mostly lobular, septal, or mixed panniculitis, depending on the distribution of the dominant subcutaneous inflammatory infiltrate, with or without vasculitis.2 This classification is helpful to guide the etiological investigation in most of the cases. In our series, none of the 4 patients displayed a “pure” panniculitis without dermal involvement, and we could not identify a clear correlation between the nature of inflammatory infiltrate and the diagnosis of primary immunodeficiency because the panniculitis was either mostly neutrophilic or mostly lymphocytic in the 3 patients who presented with a B-cell deficiency.
Mutations were identified in 3 patients, whereas we failed to identify any mutation in the fourth patient using WES. Mutations were found in the LCK, TRNT1, and NF-κB2 genes. Panniculitis has been previously reported in 1 patient who had a mutation in LCK 9 but not in those with mutations in the TRNT1 or NF-κB2 genes. The LCK gene defect is a T-cell immunodeficiency characterized by lymphopenia, low levels of CD4 and CD8 expression at the cell surface, and a lack of response to T-cell receptor activation.9 Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency (B-cell lymphopenia), periodic fevers, and developmental delay.10 Mutations in
the NF-κB2 gene cause the recently defined clinical syndrome, common variable immunodeficiency, with central adrenal insufficiency.11 Thus, the occurrence of autoinflammatory panniculitis expands the phenotypes associated with mutations in TRNT1 and NF-κB2.
CONCLUSIONS
In these cases, we demonstrate that early-onset childhood panniculitis may reveal inherited immunodeficiency, and we highlight the need for careful immunologic evaluation of patients who present with unexplained panniculitis, even before clinical manifestations of immunodeficiency have emerged.
ACKNOWLEDGMENT
We thank Pr Capucine Picard for studying immunophenotyping of the patients.
ABBREVIATIONS
CRP: C-reactive proteinIgA: immunoglobulin AIgG: immunoglobulin GIgM: immunoglobulin MIVIg: intravenous
immunoglobulinWES: whole exome sequencing
REFERENCES
1. Torrelo A, Hernández A. Panniculitis in children. Dermatol Clin. 2008;26(4):491–500, vii
2. Polcari IC, Stein SL. Panniculitis in childhood. Dermatol Ther. 2010;23(4):356–367
3. Spinner MA, Sanchez LA, Hsu AP, et al. GATA2 deficiency: a protean disorder
of hematopoiesis, lymphatics, and immunity. Blood. 2014;123(6):809–821
4. Zhou Q, Yang D, Ombrello AK, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370(10):911–920
5. Liu Y, Ramot Y, Torrelo A, et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012;64(3):895–907
6. Leiva-Salinas M, Betlloch I, Arribas MP, Francés L, Pascual JC. Neutrophilic lobular panniculitis as an expression of a widened spectrum of familial Mediterranean fever. JAMA Dermatol. 2014;150(2):213–214
7. Zhou Q, Yu X, Demirkaya E, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci USA. 2016;113(36):10127–10132
8. Wouters CH, Martin TM, Stichweh D, et al. Infantile onset panniculitis with uveitis and systemic granulomatosis: a new clinicopathologic entity. J Pediatr. 2007;151(6):707–709
9. Hauck F, Randriamampita C, Martin E, et al. Primary T-cell immunodeficiency with immunodysregulation caused by autosomal recessive LCK deficiency. J Allergy Clin Immunol. 2012;130(5):1144–1152.e11
10. Chakraborty PK, Schmitz-Abe K, Kennedy EK, et al. Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood. 2014;124(18):2867–2871
11. Shi C, Wang F, Tong A, et al. NFKB2 mutation in common variable immunodeficiency and isolated adrenocorticotropic hormone deficiency: a case report and review of literature. Medicine (Baltimore). 2016;95(40):e5081
by guest on June 6, 2020www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2017-02132018;141;S496Pediatrics
Frémond, Isabelle André-Schmutz, Sylvie Fraitag and Christine BodemerBrigitte Bader-Meunier, Frédéric Rieux-Laucat, Fabien Touzot, Marie-Louise
PanniculitisInherited Immunodeficiency: A New Association With Early-Onset Childhood
ServicesUpdated Information &
http://pediatrics.aappublications.org/content/141/Supplement_5/S496including high resolution figures, can be found at:
References
#BIBLhttp://pediatrics.aappublications.org/content/141/Supplement_5/S496This article cites 11 articles, 3 of which you can access for free at:
Subspecialty Collections
s_subhttp://www.aappublications.org/cgi/collection/immunologic_disorderImmunologic Disordersubhttp://www.aappublications.org/cgi/collection/allergy:immunology_sAllergy/Immunologyhttp://www.aappublications.org/cgi/collection/dermatology_subDermatologyfollowing collection(s): This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtmlin its entirety can be found online at: Information about reproducing this article in parts (figures, tables) or
Reprintshttp://www.aappublications.org/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
by guest on June 6, 2020www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2017-02132018;141;S496Pediatrics
Frémond, Isabelle André-Schmutz, Sylvie Fraitag and Christine BodemerBrigitte Bader-Meunier, Frédéric Rieux-Laucat, Fabien Touzot, Marie-Louise
PanniculitisInherited Immunodeficiency: A New Association With Early-Onset Childhood
http://pediatrics.aappublications.org/content/141/Supplement_5/S496located on the World Wide Web at:
The online version of this article, along with updated information and services, is
1073-0397. ISSN:60007. Copyright © 2018 by the American Academy of Pediatrics. All rights reserved. Print
the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elk Grove Village, Illinois,has been published continuously since 1948. Pediatrics is owned, published, and trademarked by Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
by guest on June 6, 2020www.aappublications.org/newsDownloaded from