Embed Size (px)
Citation preview

Infrared and Raman spectra, conformational stability, ab initio
calculations of structure, and vibrational assignment of 2-hexyneq
Stephen Bella, Xiaodong Zhub, Gamil A. Guirgisb, James R. Durigb,*
aDepartment of Chemistry, University of Dundee, Dundee, Scotland DD1 4HN, UKbDepartment of Chemistry and Geosciences, University of Missouri-Kansas City, Kansas City, MO 64110-2499, USA
Received 20 March 2002; revised 30 May 2002; accepted 30 May 2002
Abstract
The infrared spectra (3500–50 cm21) of the gas and solid and the Raman spectra (3500–50 cm21) of the liquid and solid
have been recorded for 2-hexyne, CH3–CxC–CH2CH2CH3. Variable temperature studies of the infrared spectrum (3500–
400 cm21) of 2-hexyne dissolved in liquid krypton have also been recorded. Utilizing four anti/gauche conformer pairs, the
anti(trans ) conformer is found to be the lower energy form with an enthalpy difference of 74 ^ 8 cm21 (0.88 ^ 0.10 kJ/mol)
determined from krypton solutions over the temperature range 2105 to 2150 8C. At room temperature it is estimated that there
is 42% of the anti conformer present. Equilibrium geometries and energies of the two conformers have been determined by ab
initio (HF and MP2) and hybrid DFT (B3LYP) methods using a number of basis sets. Only the HF and DFT methods predict the
anti conformer as the more stable form as found experimentally. A vibrational assignment is proposed based on the force
constants, relative intensities, depolarization ratios from the ab initio and DFT calculations and on rotational band contours
obtained using the calculated equilibrium geometries. From calculated energies it is shown that the CH3 group exhibits almost
completely free rotation which is in agreement with the observation of sub-band structure for the degenerate methyl vibrations
from which values of the Coriolis coupling constants, z, have been determined. The results are compared to similar properties of
some corresponding molecules. q 2002 Elsevier Science B.V. All rights reserved.
Keywords: Infrared spectra; Conformational stability; Ab initio calculations; 2-hexyne
1. Introduction
We have been studying the vibrational spectra
and structural parameters of several alkynes [1–4]
and some fluorine substituted alkynes [5–9]. The
infrared and Raman spectra of 1-pentyne, HCxC–
CH2CH2CH3 [2,4], presents an interesting problem
concerning which conformer has the lower energy,
the anti (trans) or gauche form. For this molecule,
the anti rotamer was found to have the lower
enthalpy by 50 ^ 6 cm21 (0.60 ^ 0.07 kJ/mol) and
45 ^ 4 cm21 (0.54 ^ 0.05 kJ/mol) by variable
low-temperature infrared spectra of liquid xenon
and krypton solutions, respectively [4]. In the
infrared spectrum of 2-pentyne, CH3 – CxC –
CH2CH3, in the gas phase [3], some interesting
‘fine’ structure was observed involving three
nearly degenerate antisymmetric modes of the
0022-2860/02/$ - see front matter q 2002 Elsevier Science B.V. All rights reserved.
PII: S0 02 2 -2 86 0 (0 2) 00 3 26 -5
Journal of Molecular Structure 616 (2002) 135–158
www.elsevier.com/locate/molstruc
q Taken in part from the thesis of X. Zhu which will be submitted
to the Department of Chemistry at the University of Missouri-
Kansas City in partial fulfillment of the PhD degree.* Corresponding author. Tel.: þ1-816-235-6038; fax: þ1-816-
235-5502.
E-mail address: [email protected] (J.R. Durig).

methyl group adjacent to the CC triple bond
which show strong–weak–weak–strong sub-bands
much like that observed in the spectra of
symmetric top molecules such as the methyl
halides. For 2-pentyne, the sub-band structure
was assigned as arising from Coriolis coupling
of the vibrational angular momenta with the
almost-free internal rotation of the methyl group
adjacent to the triple bond.
Both of these features are allowed in the 5-fluoro-
2-pentyne CH3–CxC–CH2CH2F molecule [9] which
is like 2-pentyne exhibiting sub-band structure
associated with the methyl antisymmetrical
vibrational modes. For this molecule, sub-bands are
observed for only two modes, namely the antisym-
metric stretch and antisymmetric deformation, but not
for the antisymmetric rock, perhaps due to the close
proximity of the C–F stretch. For 5-fluoro-2-pentyne,
the anti rotamer was determined [9] to be lower in
energy than the gauche form with an enthalpy
difference of 273 ^ 15 cm21.
The 2-hexyne molecule CH3–CxC–CH2CH2CH3
is also expected to exhibit both of these interesting
features in its infrared spectrum. It can exist in two
conformers, the anti and gauche forms, like 1-pentyne
[2,4], and it may also exhibit Coriolis sub-band
structure like 2-pentyne [3] due to the almost
symmetric methyl group adjacent to the CC triple
bond. It is isoelectronic and isostructural with 5-
fluoro-2-pentyne apart from a methyl replacing the F
atom at the 5-position. This methyl group should not
have a low barrier to internal rotation but it should
have a value much like that in ethane or 1-pentyne.
Additionally, 2-hexyne is another example in a
series of 1,2-disubstituted ethane molecules, XCH2-
CH2Y, whose conformational stabilities and struc-
tural parameters have been of interest to chemists
for many years. The 1,2-difluoroethane molecule
has the gauche conformer as the more stable form
where the DH value has been determined [10] to be
280 ^ 30 cm21 (3.35 ^ 0.36 kJ/mol). However, for
both 1,2-dichloroethane [11] and n-butane [12] the
anti conformer is the more stable rotamer with DH
values of 318 ^ 25 cm21 (3.81 ^ 0.30 kJ/mol) and
234 ^ 33 cm21 (2.80 ^ 0.39 kJ/mol), respectively.
However, both 1-fluoropropane [13] and 1-chlor-
opropane [14] have the gauche conformers as the
more stable form where the enthalpy differences
from the rare gas solutions have been determined
to be 104 ^ 6 cm21 (1.24 ^ 0.07 kJ/mol) and
52 ^ 3 cm21 (0.62 ^ 0.06 kJ/mol), respectively.
With the replacement of the methyl group of 1-
fluoropropane by the ethynyl group, CxCH, i.e.
FCH2CH2CxCH (4-fluoro-1-butyne), the confor-
mational stability changes from the gauche con-
former to the anti rotamer being the more stable
form [5] by 215 ^ 22 cm21 (2.57 ^ 0.26 kJ/mol) in
krypton. This is a large change in conformational
stability which was rather unexpected although the
CxCH and CN groups have recently been shown to
have a pronounced effect on the relative stabilities
of the conformers of n-butane when they replace
one of the methyl groups [2,15]. The results from
these studies indicate that the conformational
stability for the series FCH2CH2X has additional
factors to the electronegativity and size of the
substituent X determining the rotamer that will be
the most stable form.
As a continuation of our conformational studies
of 1,2-disubstituted ethane molecules, we initiated
a Raman and infrared vibrational investigation of
2-hexyne, CH3CxCCH2CH2CH3 where the methyl
group of n-butane has been replaced by the
CxCCH3 group to determine the effect on the
conformational stability by the propynyl group as
well as by the replacement of the hydrogen atom
on the ethynyl moiety by the methyl group for the
1-pentyne molecule [2]. Therefore, we have
recorded the Raman spectra of the liquid and
solid along with the infrared spectra of the gas,
krypton solutions with variable temperatures, and
solid 2-hexyne. We have also carried out ab initio
calculations employing the 6-31G(d) basis set at
the level of restricted Hartree–Fock (RHF) as well
as with electron correlation with the 6-31G(d)
basis set by the Moller–Plesset method to the
second order (MP2) to obtain equilibrium geome-
tries, force constants, vibrational frequencies,
infrared and Raman intensities, and conformation-
al stabilities. Structural parameters and confor-
mational stabilities have also been obtained from
the larger basis sets of 6-311G(d,p) and 6-
311G(2df,2p) at the MP2 level and by density
functional theory (DFT) by the B3LYP method.
The results of these spectroscopic and theoretical
studies are reported herein.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158136

2. Experimental
The sample of 2-hexyne was purchased from
Aldrich Chemical Corp., Milwaukee, WI with a
stated purity of 99%. Further purification was
performed with a low-temperature, low-pressure
fractionating column. The sample was collected at
255 8C and the purity was checked by infrared
spectroscopy [16]. The sample was stored under
vacuum at low temperature.
The mid-infrared spectrum of the gas (Fig. 1A) was
recorded using a Perkin–Elmer model 2000 Fourier
transform spectrometer equipped with a Ge/CsI
beamsplitter and DTGS detector. Atmospheric water
vapor was removed from the spectrometer housing by
purging with dry nitrogen. The spectrum of the gas
was obtained using a 10 cm cell fitted with CsI
windows. The infrared spectrum of the solid (Fig. 1D)
was obtained by condensing the sample on a CsI
substrate held at the temperature of boiling liquid
nitrogen and housed in a vacuum cell fitted with CsI
window. The sample was condensed as an amorphous
or glassy solid and repeatedly annealed until no
further changes were observed in the spectrum.
The mid-infrared spectra of the sample dissolved in
liquid krypton as a function of variable temperatures
were recorded on a Bruker model ISF-66 Fourier
transform spectrometer equipped with a globar
source, a Ge/KBr beamsplitter and a TGS detector.
In all case 100 interferograms were collected at
1.0 cm21 resolution, averaged and transformed with a
boxcar truncation function. A specially designed
cryostat cell was used to obtain the spectral data. It
consists of a copper cell with a path length of 4 cm
with wedged silicon windows sealed to the cell with
indium gaskets. This cell was cooled by boiling liquid
nitrogen and the temperature was monitored with two
Pt thermoresistors. The complete cell was connected
to a pressure manifold, which allowed the filling and
evacuation of the cell. After the cell had cooled to the
desired temperature, a small amount of the compound
was condensed into the cell. Next, the pressure
manifold and the cell were pressurized with the
noble gas, which immediately started to condense in
the cell, allowing the compound to dissolve.
The far infrared spectrum of the gas (Fig. 2) was
recorded on a Bomem model DA3.002 Fourier
transform spectrometer equipped with a vacuum
bench, 6.25 and 25 mm Mylar beamsplitters, and a
liquid helium-cooled Si bolometer. The spectrum was
obtained from the sample contained in a 1-m folded
path cell equipped with mirrors coated with gold, and
fitted with polyethylene windows with an effective
resolution of 0.1 cm21. The far infrared spectra of the
amorphous and crystalline solids (Fig. 2) were
obtained with the Perkin – Elmer model 2000
equipped with a metal grid beamsplitter and a
DTGS detector.
The Raman spectra were recorded on a SPEX
model 1403 spectrophotometer equipped with a
Spectra-Physics model 164 argon ion laser operating
on the 514.5 nm line. The laser power used was 0.5 W
with a spectral bandpass of 3 cm21. The spectrum of
the liquid was recorded with the sample sealed in a
Pyrex glass capillary held in a Miller–Harney
apparatus [17]. Depolarization ratio measurements
were checked by measuring the depolarization values
of the Raman bands of CCl4 immediately before
depolarization measurements were made on the liquid
sample. The Raman wavenumbers are expected to be
accurate to ^2 cm21 and typical spectra are shown in
Fig. 1. All of the observed bands with significant
intensity in both the infrared and Raman spectra,
along with the proposed assignments, are listed in
Table 1.
3. Conformational stability
For 2-hexyne, many of the fundamentals for the
two conformers have nearly the same frequency.
Nevertheless, there are significant differences among
lower frequencies as indicated in the spectrum of 2-
hexyne in liquid krypton which is shown in Fig. 3. The
assignment of fundamentals of the gauche conformer
is according to ab initio MP2/6-31G(d) predication
calculations and in agreement with the anti conformer
being the only conformer existing in the annealed
solid state. The conformer pairs at 478/513, 740/1074,
740/820, and 740/513 cm21, where the first frequen-
cies are due to the anti conformer, were used to
determine the enthalpy difference between the two
conformers. With the sample dissolved in liquid
krypton, the spectral changes with variation of the
temperature are shown in Fig. 4. It is evident that the
increase in the intensity of the band assigned to
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158 137

the anti conformer as the temperature decreases
confirms the ab initio prediction. In order to obtain
the enthalpy difference, spectral data at ten different
temperatures were obtained from these four pairs of
bands over the temperature range from 2105 to
2150 8C for the krypton solution as given in Table 2.
The enthalpy difference was calculated by the van’t
Hoff equation, 2ln K ¼ ðDH=RTÞ2 DS=R; with the
assumption that the value of DH is constant within the
temperature range utilized and It/Ig is substituted for
K. The value of DH for the four pairs of bands ranged
from a low value of 44 ^ 5 cm21 to a high value of
96 ^ 7 cm21 from the slope of the lines with the anti
conformer the more stable form. Using a least squares
Fig. 1. Vibrational spectra: (A) gas, infrared; (B) liquid, Raman; (C)
annealed solid, Raman; (D) annealed solid, infrared of 2-hexyne.
Fig. 2. Far infrared spectra of 2-hexyne: (A) gas phase and (B)
annealed solid.
Fig. 3. Mid-infrared spectrum (400–1100 cm21) of 2-hexyne
dissolved in liquid krypton at 2105 8C.
Fig. 4. Variation with temperature of the 740/1074 cm21 conformer
pair in liquid krypton.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158138

Table 1
Observed infrared and Raman wavenumbers (cm21) for 2-hexyne
Gas Rel. Int. Infrared Raman Assignment
Krypton Rel. Int. Solid Rel. Int. Liquid Rel.Int. and Depol. Solid Rel. Int. ni
0 Approximate description
3076 m
2972 ’ 2976 vs 2972 s 2 964 sh 2972 m n1pCH3 antisymmetric stretch
2965 vs 2962 s 2964 2965 m n26pCH3 antisymmetric stretch
2958 m 2955 m n27 CH2 antisymmetric stretch
2955 sh S 2955 S 2955 m 2953 sh n2 CH3 antisymmetric stretch
2942 max vs 2941 vs 2945 s 2938 s 2945 m n28 CH3 antisymmetric stretch
2937 vs 2937 s 2939 m n29pCH2 antisymmetric stretch
2931 sh s 2925 vs 2920 s 2922 s 2921 s n3 CH2 symmetric stretch
2919 QC s 2914 s 2911 s 2908 vs n4pCH3 symmetric stretch
2909 s 2903 s 2904 s 2908 vs n5 CH3 symmetric stretch
2880 max s 2877 s 2873 s 2875 m 2884 m n6pCH2 symmetric stretch
2730 max 2730 m 2738 w 2738 w
2302 s 2296 s
2236 vs 2228 vs n7 CxC stretch, Fermi doublet
1462 QC s 1467 s 1467 s 1460 w n8 CH2 scissors
1453QC s 1457 s 1458 s 1453 m n30 CH3 antisymmetric deformation
1447 QC 1450 m 1447 m 1449 m n9 CH3 antisymmetric deformation
1435 QC s 1436 s 1421 s 1435 m n10pCH2 scissors
1450 ’ m 1448 s 1443 s 1443 m 1444 m n31pCH3 antisymmetric deformation
1436 m 1441 m n11pCH3 antisymmetric deformation
1390 QC m 1381 s 1387 m 1381 m 1394 m n12pCH3 symmetric deformation
1384 QA m 1372 m 1371 w n13 CH3 symmetric deformation
1380 P
1348 QC m 1353 m 1352 m 1349 w 1348 w n14 CH2 wag
1342 QC m 1341 m 1332 m n 014
1337QC m 1333 s 1332 m n 015
1332 P
1296 w 1294 vw 1296 w 1296 m n32 CH2 twist
1287 R
1282 QA w 1280 m 1277 s 1279 w 1283 w n15pCH2 wag
1277 P
1261 w n 032
1237 R
1231 QA vw 1230 w 1240 w n33pCH2 twist
1225 P
1217 w 1228 w n 033
(continued on next page)
S.
Bell
eta
l./
Jou
rna
lo
fM
olecu
lar
Stru
cture
61
6(2
00
2)
13
5–
15
81
39

Table 1 (continued)
Gas Rel. Int. Infrared Raman Assignment
Krypton Rel. Int. Solid Rel. Int. Liquid Rel.Int. and Depol. Solid Rel. Int. ni
0 Approximate description
1135 max w 1147 w 1137 vw 1144 vw 1140 vw n16 C3C4 stretch
1103 R
1097 QC w 1101 w 1105 w 1095 m 1092 m n17 CH3 rock ip
1091 QC w 1096 m 1094 w n34pCH2 rock
1080 R
1074 QA w 1074 m 1071 w n 017
1067 P
1039 R
1036 QA w 1034 m 1037 w n18pCH3 rock ip
1032 QA w 1031 m 1034 w 1033 m 1035 m n19, n35 C5C6 stretch, pCH3 rock op
1028 P
1024 sh w 1024 w 1020 w n 018
1018 sh w 1013 w n 019
894 R
892 QC w 888 m 890 m 888 m 890 m n20 C4C5 stretch
889 QA w 879 w n 036
885 P
866 QC 865 w 866 w 866 vw n36 CH3 rock op
856 QC 855 w 855 m n 020
849 P
807 R
804 QA w 802 w 804 w n 037
800 P
740 w 754 w 755 vw 745 vw n21 C1C2 stretch
751 R
740 QC vw 739 m 730 w 734 vw 735 vw n37 CH2 rock
726 P
690 R
685 QC vw 686 w 687 w n 021
681 P
518 R
513 QA w 513 w 514 w n 022
508 P
483 R
478 ctr B vw 479 vw 475 w 479 w 476 w n22 C3C4C5 bend
474 P
399 R
S.
Bell
eta
l./
Jou
rna
lo
fM
olecu
lar
Stru
cture
61
6(2
00
2)
13
5–
15
81
40

Table 1 (continued)
Gas Rel. Int. Infrared Raman Assignment
Krypton Rel. Int. Solid Rel. Int. Liquid Rel.Int. and Depol. Solid Rel. Int. ni
0 Approximate description
392 ctr B vw 382 vw 374 s 384 m n38 C2xC3C4 bend op
385 P
368 max vw 368 w n 038
359 sh vw 348 w 343 w 349 w n23 C4C5C6 bend
288 R
286 QC w 289 w n 039
280 R
272 QA m 298 m 285 w 282 w n24 C1C2xC3 bend ip
268 P
210 max m 243 m n40 C1C2xC3 bend op
202 QC m n 040
129 R
120 QC w n 025
115 sh w 141 w 139 m n25 C2xC3C4 bend ip
90 R
88 QC w 103 w 111 w n41 C6C5C4C3 torsion
86 P
92
91
75 Lattice mode
74
47
Abbreviations used: s, strong; m, medium; w, weak; v, very; ctr, center; A, B, C, and ’ refer to infrared band envelopes; P, Q, and R refer to the rotational–vibrational
branches. n indicates anti mode and n0 indicates gauche mode.
S.
Bell
eta
l./
Jou
rna
lo
fM
olecu
lar
Stru
cture
61
6(2
00
2)
13
5–
15
81
41

fit of all of these data the value of DH is 74 ^ 4 cm21
(0.88 ^ 0.04 kJ/mol). The uncertainty is the statistical
uncertainty and does not take into account possible
overtone or combination bands contributing to the
intensities of the bands used for the DH determination.
Thus, the probably error is expected to be at least ten
percent, so a value of 74 ^ 8 cm21 (0.88 ^ 0.10 kJ/
mol) is a more realistic one.
4. Ab Initio calculations
The electronic structure calculations were per-
formed with GAUSSIAN98 program [18] with the
Gaussian type basis functions. The energies of 2-
hexyne were obtained from restricted Hartree–Fock
(RHF) calculations, Møller–Plesset perturbation cal-
culations at the second order (MP2) as well as hybrid
density functional theory (B3LYP) with the 6-31G(d)
and 6-311G(2d,2p) basis sets, and the results are given
in Table 3. The geometrical parameters have been
optimized for all the conformers considered. For each
of the calculation methods, potential energy functions
have been obtained from these energies by least
squares fit. It is evident from Table 3 and Fig. 5 that
the gauche conformer is predicted to be lower in
energy than the anti rotamer with the MP2 method for
any size of basis set. However, at both the Hartree–
Fock and B3LYP levels of calculation the anti
conformer is more stable with a small conformer
energy difference as found experimentally.
The optimized values of the structural parameters
obtained by computation are given in Table 4 for the
anti and the gauche form. The parameter symbols
adopted are also given in this table and in Fig. 6. The
GAUSSIAN98 program was also employed in calculat-
ing the force field in Cartesian coordinates for each of
the computational methods. In order to carry out a
normal coordinate analysis and attempt a complete
assignment of vibrational frequencies for 2-hexyne,
the internal coordinates defined in Table 4 and shown
in Fig. 6 were used to form the symmetry coordinates
listed in Table 5. The B-matrix elements were used to
convert the ab initio force field from Cartesian
coordinates into the force field in internal coordinates
[19]. These force constants were used in a mass-
weighted Cartesian coordinate calculation to repro-
duce the ab initio vibrational frequencies and to
determine the potential energy distributions (PED),
which are given in Table 6 for anti 2-hexyne and in
Table 7 for the gauche rotamer. The diagonal and off-
diagonal elements of the force field in internal
coordinates were then modified with scaling factors
of 0.88 for the carbon–hydrogen stretches, 0.9 for the
heavy atoms stretch and carbon–hydrogen bends, 1.0
for the skeletal bends except the CxCC motions with
a scaling factor of 1.5. The off-diagonal elements were
scaled by the geometric mean of the scaling factors.
The calculation was repeated to obtain the fixed
scaled force field and scaled vibrational frequencies.
To aid in the vibrational assignment for each of the
conformers, we simulated the Raman spectra for each
form. The Raman scattering activities were obtained
from the output of the ab initio calculations. The
Raman scattering cross sections, ›sj/›V, which are
proportional to the Raman intensities, can be
calculated from the scattering activities and the
predicted wavenumbers for each normal mode
[20–23]. To obtain the polarized Raman cross
sections, the polarizabilities are incorporated into Sj
by Sj[(1 2 rj)/(1 þ rj)], where rj is the depolarization
ratio of the jth normal mode. The Raman scattering
cross sections and the predicted scaled wavenumbers
were used together with a Lorentzian function to
obtain the calculated spectra.
The predicted Raman spectra of the pure anti
conformer of 2-hexyne is shown in Fig.7D and that of
Table 2
Temperature and intensity ratios for the conformational study of 2-
hexyne dissolved in liquid krypton
T (8C) 1000/K I478/513 I740/1074 I740/820 I740/513
2105 5.9471 0.6681 1.0841 – 1.061
2110 6.1293 0.7035 1.1424 0.400 1.096
2115 6.3231 0.7112 1.1540 0.425 –
2120 6.5295 0.7210 1.1908 0.445 1.141
2125 6.7499 0.7469 1.2004 0.453 –
2130 6.9857 0.7515 1.2513 0.474 1.198
2135 7.2385 0.7517 – 0.486 –
2140 7.5103 0.7632 1.3297 0.507 1.272
2145 7.8033 0.7693 1.3759 0.520 1.307
2150 8.1202 0.7888 1.4202 0.534 1.359
DHa (cm21) 44 ^ 5 82 ^ 3 96 ^ 7 77 ^ 1
a Average value of DH is 74 ^ 4 cm21 (0.88 ^ 0.04 kJ/mol) with
the anti conformer the more stable form.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158142

pure gauche conformer in Fig. 7C. The predicated
Raman spectrum of the mixture of the two con-
formers, with an enthalpy difference of 74 cm21 with
the anti conformer the more stable rotamer at ambient
temperature, is shown in Fig. 7B. This result is
considered satisfactory when it is compared to the
experimental Raman spectrum (Fig. 7A) of the liquid.
One major difference is the doublet of the bands near
2250 cm21, which results from the Fermi resonance
of the CxC stretch with an overtone as in 2-pentyne
[3]. In general, the predicted Raman spectrum for the
lines in the region of 100–1500 cm21 showed very
Table 3
Calculated energies and energy difference for several conformations of 2-hexyne by ab initio and hybrid DFT methods
Method/basis Energy (Eh), Tse Energy differencesa (cm21)
Gse Cse Sse Teeb (V3 trans ) Geeb V3 gauche Tss
HF/3-21G 2231.692497 249.4 1752 1287 1069 1032 2.0
HF/6-31G 2232.889762 þ57.7 1826 1305 1026 1073 2.5
MP2(FC)/6-31G(d) 2233.747418 2145.8 1072 1171 3.8
MP2(full)/6-31G(d) 2233.776573 2148.3 1594 1292 1081 1179 3.8
MP2(full)/6-31G(d,p) 2233.859273 2142.6
MP2(full)/6-311G(2d,2p) 2234.088154 2153.9
B3LYP/6-31G(d) 2234.606622 þ26.3 1604 1217 1019 1086 4.2
B3LYP/6-31G(d,p) 2234.620509 þ27.0
B3LYP/6-31 þ G(d,p) 2234.629772 þ105.6
B3LYP/6-311G(2d,2p) 2234.683270 þ58.2
Conformation labels: capital letter refers to main skeletal torsional angle, C6C5C4C3, T ¼ trans or anti, G ¼ gauche, C ¼ cis or syn, and
S ¼ skew; lower case letters refer to methyl torsional angles, staggered or eclipsed, the first for the C6 methyl and the second for the C1 methyl.a Energies of conformations relative to Tse; A negative energy difference means gauche form is lower than trans conformer.b Tee energy is relative to Tse and Gee energy is relative to Gse to obtain methyl barriers.
Fig. 5. Potential energy function for torsion about the CH2–CH2 bond.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158 143

Table 4
Calculated structural parameters of the anti and gauche conformer of 2-hexyne
Parametersa HF 6-31G MP2(Full) 6-31G(d) B3LYP 6-31G(d) MP2(Full) 6-311G(d,p)
anti gauche anti gauche anti gauche anti gauche
C1–C2 S 1.4646 1.4646 1.4617 1.4639 1.4614 1.4658 1.4623 1.4624
C2xC3 T 1.1961 1.1963 1.2211 1.2215 1.2098 1.2101 1.2195 1.2198
C3–C4 U 1.4688 1.4700 1.4629 1.4618 1.4647 1.4615 1.4629 1.4641
C4–C5 V 1.5393 1.5406 1.5327 1.5339 1.5436 1.5456 1.5344 1.5359
C5–C6 X 1.5301 1.5296 1.5240 1.5228 1.5307 1.5303 1.526 1.5252
C1C2C3 z 180.06 180.12 180.47 180.40 180.38 180.43 179.27 179.32
C2C3C4 j 179.42 179.87 178.72 179.16 178.88 179.23 178.19 178.63
C3C4C5 u 113.05 113.55 112.70 112.67 113.37 113.73 112.66 112.51
C4C5C6 e 112.15 113.42 112.03 112.61 112.42 113.47 112.02 112.41
C3C4C5C6 t 180.0 64.08 180.0 61.47 180.0 62.62 180.0 61.78
C1H1 r1 1.0839 1.0838 1.0937 1.0937 1.0971 1.0971 1.0934 1.0934
C1H2 r2 1.0838 1.0838 1.0937 1.0937 1.0971 1.0971 1.0933 1.0934
C1H3 r3 1.0838 1.0838 1.0937 1.0937 1.0971 1.0971 1.0933 1.0934
C4H4 r4 1.0866 1.0867 1.0976 1.0978 1.1000 1.1001 1.0970 1.0973
C4H5 r5 1.0866 1.0859 1.0976 1.0968 1.1000 1.0990 1.0970 1.0960
C5H6 r6 1.0849 1.0864 1.0950 1.0964 1.0970 1.0979 1.0948 1.0959
C5H7 r7 1.0849 1.0850 1.0950 1.0952 1.0970 1.0972 1.0948 1.0951
C6H8 r8 1.0841 1.0845 1.0934 1.0935 1.0957 1.0958 1.0932 1.0934
C6H9 r9 1.0854 1.0854 1.0941 1.0944 1.0971 1.0973 1.0944 1.0947
C6H10 r10 1.0854 1.0832 1.0941 1.0926 1.0971 1.0952 1.0944 1.0929
C2C1H1 b1 110.98 111.01 111.02 111.04 111.36 111.37 110.86 110.85
C2C1H2 b2 111.03 110.99 111.04 111.02 111.40 111.36 110.84 110.87
C2C1H3 b3 111.03 111.06 111.04 111.05 111.40 111.44 110.84 110.84
C3C4H4 b4 108.99 108.03 108.84 108.41 108.83 107.95 109.52 109.33
C3C4H5 b5 108.99 108.98 108.84 108.79 108.83 108.78 109.52 109.26
C4C5H6 b6 109.27 109.22 109.67 109.44 109.62 109.43 108.65 108.46
C4C5H7 b7 109.27 108.92 109.67 109.43 109.62 109.33 108.65 108.69
C5C6H8 b8 111.05 110.92 111.23 111.18 111.18 111.11 111.25 110.87
C5C6H9 b9 111.20 110.99 110.97 110.77 111.29 111.13 110.77 110.64
C5C6H10 b10 111.20 111.09 110.97 110.65 111.29 110.86 110.77 110.55
H1C1C2C5 f1 0.0 22.81 0.0 12.85 0.0 16.68 0.0 8.62
H2C1C2C5 f2 119.98 142.76 119.98 132.80 119.98 136.61 119.99 128.80
H3C1C2C5 f3 2119.98 297.22 2119.98 2107.17 2119.98 2119.92 2119.99 2111.34
H4C4C3C5 f4 121.97 122.18 119.98 121.59 119.98 122.16 119.99 121.37
H5C4C3C5 f5 2121.97 2121.90 2121.89 2122.24 2122.15 2122.20 2121.71 2122.08
H6C5C4C6 f6 121.97 121.60 122.10 121.94 122.25 122.02 122.06 121.31
H7C5C4C6 f7 2121.97 2122.71 2122.10 2122.16 2122.25 2122.55 2122.06 2120.94
H8C6C5C4 f8 180.0 179.24 180.0 179.55 180.0 180.31 180.0 179.98
H9C6C5C4 f9 60.06 59.43 59.94 59.58 59.98 60.34 59.89 58.96
H10C6C5C4 f10 260.06 260.64 259.94 260.12 259.98 259.45 259.89 260.61
lmal 0.014 0.037 0.012 0.047 0.019 0.067 0.146 0.047
lmbl 0.142 0.070 0.170 0.090 0.137 0.069 0.087 0.090
lmcl 0.000 0.085 0.000 0.093 0.000 0.078 0.000 0.093
lmtl 0.143 0.116 0.170 0.138 0.139 0.123 0.170 0.138
A 18168 7876 17854 7735 17907 7755 17732 7721
B 1195 1586 1196 1616 1189 1588 1199 1618
C 1153 1402 1153 1421 1147 1400 1155 1423
k 20.9950 20.9431 20.9948 20.9382 20.9949 20.8407
F 6.0827 5.4956 5.9783 5.3940 5.9672 5.3809
F p 6.0858 5.6895 5.9783 5.5893 5.9731 5.5818
a Kinetic constants for methyl internal rotation: F applies to C6 methyl, F p to C1 methyl labeled pCH3.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158144

good correspondence with the experimental spectrum
for 2-hexyne with the exception of the intensities of
the lines in the 250–380 cm21 region. The problem
here arises from the poor prediction of the out-of-
plane bend of the C2xC3–C4 group from the MP2/6-
31G(d) calculations. Without scaling this fundamental
which is the strongest predicted Raman line in this
region is predicted at 261 cm21 and with scaling at
299 cm21, whereas the band is actually observed
nearly 100 cm21 higher at 392 cm21. Excluding this
problem the predicted Raman spectrum is quite useful
for making the correct assignment of the observe
fundamentals for the two conformers.
Infrared intensities were calculated based on the
dipole moment derivatives with respect to the
Cartesian coordinates. The derivatives were taken
from the ab initio calculation transformed to normal
coordinates by
›mu
›Qi
� �¼X
j
›mu
›Xj
!Lij
where the Qi is the ith normal coordinate, Xj is the jth
Cartesian displacement coordinate, Lij is the trans-
formation matrix between the Cartesian displacement
coordinates and normal coordinates. The infrared
intensities were then calculated by
Ii ¼Np
3c2
›mx
›Qi
� �2
þ›my
›Qi
� �2
þ›mz
›Qi
� �2" #
In Fig. 8C and D are the predicated spectra of the pure
anti and gauche conformers, respectively. In Fig. 8B
the predicated spectrum of the mixture utilizing the
DH value of 74 cm21 is shown which is in good
agreement with the experimental spectrum of the
sample dissolved in liquid krypton solution (Fig. 8A)
at a temperature of 2105 8C. For example, it was
quite easy to identify the bands due to the anti and
gauche conformers in the region 1100–500 cm21
from the predicated spectrum (Fig. 3). Thus, the
predicted infrared spectrum made it relatively easy to
assign the fundamentals for both conformers.
5. Vibration assignment
The anti conformer of 2-hexyne has Cs symmetry
and the fundamental vibrations span the irreducible
representation 25A0 þ 17A00. The ab initio calculation
shows that the c principal axis of the anti conformer is
perpendicular to the symmetry plane (Table 4).
Therefore, the out-of-plane modes are expected to
give rise to C-type band contours in the infrared
spectrum, yield depolarized lines in the Raman spec-
trum and will not be observed in the Raman spectrum of
the gas, whereas the in-plane modes should give rise to
A-, B-, or A/B hybrid-type infrared contours. For the
gauche conformer, the infrared band contours can be A-,
B- and C-types and any hybrid of these three types
because it has only the trivial C1 symmetry. The
predicted vibrational–rotational infrared band contours
Fig. 6. Internal coordinates of 2-hexyne.
Fig. 7. Raman spectrum of 2-hexyne: (A) observed spectrum of the
liquid; (B) simulated spectrum of the mixture of anti and gauche
conformers with DH ¼ 74 cm21; (C) calculated for pure anti
conformer; (D) calculated for pure gauche conformer.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158 145

Table 5
Symmetry coordinates for vibrations of 2-hexyne
Description Symmetry coordinate
A0
pCH3 antisymmetric stretch S1 ¼ 2r1 2 r2 2 r3
CH3 antisymmetric stretch S2 ¼ 2r8 2 r9 2 r10
CH2 symmetric stretch S3 ¼ r6 þ r7pCH3 symmetric stretch S4 ¼ r1 þ r2 þ r3
CH3 symmetric stretch S5 ¼ r8 þ r9 þ r10pCH2 symmetric stretch S6 ¼ r4 þ r5
C2 xC3 stretch S7 ¼ T
CH2 scissors S8 ¼ (p
6 þ 2)d5 2 b6 2 b7 2 g6 2 g7 2 (p
6 2 2)u
CH3 antisymmetric deformation S9 ¼ 2a8 2 a9 2 a10pCH2 scissors S10 ¼ (
p6 þ 2)d4 2 b4 2 b5 2 g4 2 g5 2 (
p6 2 2)e
pCH3 antisymmetric deformation S11 ¼ 2a1 2 a2 2 a3pCH3 symmetric deformation S12 ¼ a1 þ a2 þ a3 2 b1 2 b2 2 b3
CH3 symmetric deformation S13 ¼ a8 þ a9 þ a10 2 b8 2 b9 2 b10
CH2 wag S14 ¼ b6 þ b7 2 g6 2 g7pCH2 wag S15 ¼ b4 þ b5 2 g4 2 g5
C3–C4 stretch S16 ¼ U
CH3 rock ip S17 ¼ 2b8 2 b9 2 b10pCH3 rock ip S18 ¼ 2b1 2 b2 2 b13
C5–C6 stretch S19 ¼ X
C4–C5 stretch S20 ¼ V
C1–C2 stretch S21 ¼ S
C3C4C5 bend S22 ¼ (p
6 þ 2)u 2 b4 2 b5 2 g4 2 g5 2 (p
6 2 2)d4
C4C5C6 bend S23 ¼ (p
6 þ 2)e 2 b6 2 b7 2 g6 2 g7 2 (p
6 2 2)d5
C1–C2xC3 bend ip S24 ¼ z
C2xC3–C4 bend ip S25 ¼ jpCH3 redundancy R1 ¼ a1 þ a2 þ a3 þ b1 þ b2 þ b3
CH3 redundancy R2 ¼ a8 þ a9 þ a10 þ b8 þ b9 þ b10pCH2 redundancy R3 ¼ e þ b4 þ b5 þ g4 þ g5 þ d4
CH2 redundancy R4 ¼ u þ b6 þ b7 þ g6 þ g7 þ d5
A00
pCH3 antisymmetric stretch S26 ¼ r2 2 r3
CH2 antisymmetric stretch S27 ¼ r6 2 r7
CH3 antisymmetric stretch S28 ¼ r9 2 r10pCH2 antisymmetric stretch S29 ¼ r4 2 r5
CH3 antisymmetric deformation S30 ¼ a9 2 a10pCH3 antisymmetric deformation S31 ¼ a2 2 a3
CH2 twist S32 ¼ b6 2 b7 2 g6 þ g7pCH2 twist S33 ¼ b4 2 b5 2 g4 þ g5pCH2 rock S34 ¼ b4 2 b5 þ g4 2 g5pCH3 rock op S35 ¼ b2 2 b3
CH3 rock op S36 ¼ b9 2 b10
CH2 rock S37 ¼ b6 2 b7 þ g6 2 g7
C2xC3–C4 bend op S38 ¼ j0
CH3 torsion S39 ¼ f
C1–C2xC3 bend op S40 ¼ z0
C6C5C4C3 torsion S41 ¼ tpCH3 torsion S42 ¼ f p
p Asterisks indicate the C atoms attached at both ends of the CxC bond.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158146

Table 6
Calculated and observed vibrational wavenumbers (cm21) of anti 2-hexyne
Vib. No. Approximate descriptiona HF 6-31G B3LYP 6-31G(d) MP2 6-31G(d) MP2 6-3lG(d) b IR Raman act.e Depol. ratio PEDf
Obs.c Int.d
A0
n1pCH3 antisymmetic stretch 3258 3115 3194 2997 2972 26.5 97.7 0.75 99S1
n2 CH3 antisymmetric stretch 3255 3095 3185 2988 (2955) 10.0 92.6 0.62 100S2
n3 CH2 symmetric stretch 3198 3052 3115 2922 2931 24.9 77.1 0.18 97S3
n4pCH3 symmetric stretch 3190 3042 3102 2910 2919 17.0 226.8 0.01 98S4
n5 CH3 symmetric stretch 3182 3035 3101 2909 (2903) 30.0 208.3 0.01 99S5
n6pCH2 Symmetric stretch 3177 3016 3081 2891 2880 18.0 29.8 0.32 98S6
n7 C2xC3 stretch 2547 2371 2309 2191 2188 0.0 134.5 0.34 81S7
n8 CH2 scissors 1665 1538 1571 1491 1462 5.2 2.4 0.55 53S8, 38S9
n9 CH3 antisymmetric deformation 1653 1522 1557 1479 1447 0.7 32.8 0.74 55S9, 40S8
n10pCH2 scissors 1644 1510 1547 1468 1435 6.6 16.8 0.70 93S10
n11pCH3 antisymmetric deformation 1642 1506 1542 1464 1450 1.0 27.6 0.75 91S11
n12pCH3 symmetric deformation 1589 1445 1472 1397 1390 5.5 33.6 0.51 61S12, 35S13
n13 CH3 symmetic deformation 1579 1442 1471 1396 1382 0.2 1.4 0.67 58S13, 36S12
n14 CH2 wag 1529 1404 1433 1361 1348 3.3 8.8 0.54 49S14, 31S15
n15pCH2 wag 1463 1323 1342 1276 1282 10.4 7.7 0.52 50S15, 29S14
n16 C3–C4 stretch 1240 1175 1203 1142 1135 0.6 0.4 0.51 41S16, 42S21
n17 CH3 rock in-plane 1225 1120 1151 1104 1097 3.0 8.3 0.15 38S17, 26S20, 11S23
n18pCH3 rock in-plane 1196 1070 1091 1036 1036 0.7 0.3 0.64 65S18, 22S20
n19 C5–C6 stretch 1115 1046 1074 1025 1032 0.9 12.1 0.62 90S19
n20 C4–C5 stretch 971 900 929 884 893 3.3 18.0 0.34 35S20, 34S17, 18S18
n21 C1–C2 stretch 824 772 778 746 739 0.3 2.4 0.39 35S21, 25S16, 14S7
n22 C3C4C5 bend 604 487 441 453 478 2.6 14.8 0.59 47S22, 21S25, 12S16
n23 C4C5C6 bend 387 338 325 328 359 0.7 6.6 0.43 72S23, 14S25
n24 C1–C2xC3 bend in-plane 320 279 212 257 272 3.8 1.2 0.73 100S24
n25 C2xC3–C4 bend in-plane 115 102 92 105 115 1.1 1.5 0.73 79S25, 25S22
A00
n26pCH3 antisymmetric stretch 3258 3110 3192 2995 2972 44.0 95.4 0.75 84S26, 16S28
n27 CH2 antisymmetric stretch 3258 3095 3186 2989 2995 9.9 22.8 0.75 100S27
n28 CH3 antisymmetric stretch 3230 3083 3165 2969 2942 8.9 51.4 0.75 78S28, 16S26
n29pCH2 antisymme tric stretch 3206 3042 3126 2932 (2937) 7.5 145.2 0.75 94S29
n30 CH3 antisymmetric deformation 1656 1530 1564 1484 1453 7.6 23.7 0.75 93S30
n31pCH3 antisymmetric deformation 1642 1510 1547 1469 1450 6.0 25.5 0.75 94S31
n32 CH2 twist 1454 1337 1364 1294 1294 0.0 24.9 0.75 64S32, 27S33
n33pCH2 twist 1397 1274 1303 1236 1231 0.2 0.6 0.75 39S33, 25S36, 20S37
n34pCH2 rock 1260 1138 1156 1101 1091 0.5 0.1 0.75 26S34, 29S33, 19S37
n35pCH3 rock out-of-plane 1196 1070 1075 1025 1032 1.2 0.2 0.75 88S35
(continued on next page)
S.
Bell
eta
l./
Jou
rna
lo
fM
olecu
lar
Stru
cture
61
6(2
00
2)
13
5–
15
81
47

of both the anti and gauche conformers are shown in
Figs. 9 and 10. They have been calculated by an
asymmetric top program using the rotational constants
given in Table 4 as obtained from the B3LYP
calculations.
The major difference of the band contours is for
the pure B and C bands of the anti conformer
where the spacing for the R and P branches is
significantly larger so the rotational fine structure
will be more discernible for this conformer
compared to the corresponding bands of the gauche
conformer. This information was particularly
important for distinguishing the bands in the low
wavenumber spectral region. In the ‘fingerprint’
spectral region most of the fundamentals could be
assigned based on the ab initio predicted frequen-
cies, infrared intensities, and Raman activities,
particularly once the bands were identified for the
gauche conformer after their disappearance from
the spectrum of the solid. A previous vibrational
assignment has been given [16] based on exper-
iment spectra and normal coordinate calculations
with transferred force constants but before theTab
le6
(co
nti
nued
)
Vib
.N
o.
Ap
pro
xim
ate
des
crip
tio
na
HF
6-3
1G
B3
LY
P6
-31
G(d
)M
P2
6-3
1G
(d)
MP
26
-3lG
(d)
bIR
Ram
anac
t.e
Dep
ol.
rati
oP
ED
f
Ob
s.c
Int.
d
n36
CH
3ro
cko
ut-
of-
pla
ne
98
08
84
90
18
57
86
60
.51
.40
.75
36
S36,4
0S
34,
16
S32
n37
CH
2ro
ck8
24
75
17
63
72
67
40
2.7
0.4
0.7
55
4S
37,
28
S34,
12
S36
n38
C2x
C3–
C4
ben
do
tlt-
Of-
pla
ne
53
93
80
26
12
99
39
20
.62
1.1
0.7
54
1S
38,4
3S
39,1
6S
41
n39
CH
3to
rsio
n2
50
24
52
42
24
92
38
0.5
0.0
0.7
55
5S
39,
40
S38
n40
C1–
C2x
C3
ben
do
ut-
of-
pla
ne
24
12
21
20
62
46
22
06
.40
.00
.75
10
0S
40
n41
C6C
5C
4C
3to
rsio
n8
88
48
38
78
80
.30
.60
.75
70
S41,
28
S38
n42
pC
H3
tors
ion
12
16
14
14
0.0
0.0
0.7
51
00
S42
aA
ster
isks
indic
ate
Cat
om
sat
tach
edto
both
ends
of
the
Cx
Cb
on
d.
bS
cale
fact
ors
for
forc
eco
nst
ants
:0
.9fo
ral
lC
Cst
retc
hes
,0.8
8fo
rC
Hst
retc
hes
,0.9
for
CC
Hd
efo
rmat
ion
s,1
.0fo
rh
eav
y-a
tom
ben
ds
exce
pt1
.5fo
rCx
C–
Cli
nea
rb
end
s,an
d1
.0
for
tors
ion
s.e
Cal
cula
ted
Ram
anac
tivit
ies
inA
4/a
mu
atth
eM
P2
/6-3
1G
(d)
lev
el.
fC
alcu
late
dat
the
MP
2/6
-31
G(d
)le
vel
;co
ntr
ibu
tio
ns
less
than
10
%ar
eo
mit
ted.
cO
bse
rved
freq
uen
cies
are
fro
mth
ein
frar
edsp
ectr
um
of
the
gas
exce
pt
tho
sew
ith
inb
rack
ets
are
fro
mth
esp
ectr
um
of
the
soli
d.
dC
alcu
late
din
frar
edin
ten
siti
esin
km
/mo
lat
the
MP
2/6
-31
G(d
)le
vel
.
Fig. 8. Mid-infrared spectrum of 2-hexyne: (A) observed spectrum
in the krypton solution (at 2105 8C); (B) simulated spectrum of the
mixture of anti and gauche conformers with DH ¼ 74 cm21; (C)
calculated for pure anti conformer; (D) calculated for pure gauche
conformer.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158148

Table 7
Calculated and observed vibrational wavenumbers (cm21) of gauche 2-hexyne
Vib. No. Approximate descriptiona HF 6-31G 3LYP 6-31G(d) MP2 6-31G(d) MP2 6-3lG(d)b IR Raman act.e Depol. ratio PEDf
Obs.c Int.d
n1pCH3 antisymmetric stretch 3258 3111 3191 2993 33.0 101.6 0.75 64S1,32S26
n2 CH3 antisymmetric stretch 3252 3095 3185 2988 9.6 69.2 0.64 57S2,43S27
n3 CH2 symmetric stretch 3190 3041 3103 2911 25.4 54.8 0.06 95S3
n4pCH3 symmetric stretch 3191 3046 3105 2913 19.4 448.1 0.04 99S4
n5 CH3 symmetric stretch 3183 3036 3101 2909 27.3 23.8 0.69 100S5
n6pCH2 symmetric stretch 3178 3020 3084 2893 19.6 51.4 0.30 98S6
n7 C2xC3 stretch 2545 2369 2306 2188 0.2 120.8 0.33 81S7
n8 CH2 scissors 1664 1538 1571 1491 5.4 2.8 0.75 72S8, 14S9
n9 CH3 antisymmetric deformation 1652 1518 1553 1474 1.1 34.6 0.74 79S9, 17S8
n10pCH2 scissors 1642 1510 1546 1467 6.3 19.3 0.74 92S10
n11pCH3 antisymmetric deformation 1642 1504 1539 1461 3.7 25.0 0.75 92S11
n12pCH3 symmetric deformation 1579 1442 1471 1396 4.7 2.4 0.60 86S12, 1013
n13 CH3 symmetric deformation 1589 1445 1474 1398 5.3 32.6 0.51 7813, 11S12
n14 CH2 wag 1532 1393 1418 1346 1342 3.2 2.3 0.60 70S14
n15pCH2 wag 1503 1380 1406 1334 1336 13.1 19.7 0.55 56S15, 24S32
n16 C3–C4 stretch 1259 1175 1203 1143 0.4 0.4 0.18 37S16, 37S21, 16S15
n17 CH3 rock in-plane 1202 1104 1133 1084 1073 2.2 5.2 0.71 29S17, 24S18, 15S34
n18pCH3 rock in-plane 1195 1070 1075 1026 1024 1.5 0.2 0.59 25S18, 20S20, 12S14
n19 C5–C6 stretch 1123 1043 1074 1021 1018 2.3 8.2 0.73 83S19
n20 C4–C5 stretch 928 867 896 853 856 0.2 16.3 0.28 20S20, 35S34, 289S17
n21 C1–C2 stretch 750 701 712 680 686 0.7 7.4 0.28 23S21, 27S16, 20S37
n22 C3C4C5 bend 622 523 505 512 513 1.2 10.8 0.59 34S22, 12S23, 25S25
n23 C4C5C6 bend 537 383 347 356 359 0.9 20.1 0.75 38S23, 26S18, 11S41
n24 C1–C2xC3 bend in-plane 261 243 221 255 210 4.5 0.0 0.58 61S24, 27S39
n25 C2xC3–C4 bend in-plane 140 127 123 137 120 1.6 0.9 0.69 40S25, 19S22, 24S41
n26pCH3 antisymmetric stretch 3270 3126 3205 3007 25.8 26.3 0.73 62S26, 36S1,
n27 CH2 antisymmetric stretch 3258 3095 3186 2988 9.7 99.8 0.75 57S27, 43S2
n28 CH3 antisymmetric stretch 3227 3080 3157 2961 19.7 66.6 0.74 88S28
n29pCH2 antisymmetric stretch 3215 3055 3132 2938 18.4 158.7 0.54 91S29
n30 CH3 antisymmetric deformation 1658 1529 1564 1484 5.6 12.3 0.64 77S30
n31pCH3 antisymmetric stretch 1642 1510 1547 1467 6.4 35.7 0.73 92S31
n32 CH2 twist 1427 1307 1332 1265 0.3 11.6 0.63 46S32, 17S36, 16S15
n33pCH2 twist 1389 1268 1291 1227 1217p 1.1 7.2 0.65 59S33
n34 CH2 rock 997 902 924 879 889 2.1 2.0 0.58 20S34, 27S17, 21S20
n35pCH3 rock out-of-plane 1196 1071 1077 1027 1.5 0.2 0.74 82S35
n36 CH3 rock out-of-plane 1244 1132 1157 1104 0.1 2.3 0.28 15S36, 12S33, 20S37, l2S33
n37 CH2 rock 897 816 830 793 804 3.4 1.3 0.41 31S37, 27S36
n38 C2xC3–C4 bend out-of-plane 440 382 289 323 368 1.0 6.7 0.74 41S38, 15S40, 15S23
(continued on next page)
S.
Bell
eta
l./
Jou
rna
lo
fM
olecu
lar
Stru
cture
61
6(2
00
2)
13
5–
15
81
49
widespread use of predicated frequencies from ab
initio calculations and spectra recorded with better
resolution or at low temperatures.
In the carbon–hydrogen stretching region there are
three modes from each of the two methyl groups and
two each for the two methylene groups for both
conformers, but we shall address the assignments for
these modes for the anti conformer first. Since thepCH3 rotor (methyl group attached to the triple bond)
is essentially a ‘free’ rotor, the two pCH3 antisym-
metric stretches are degenerate and readily assigned to
the vibrational–rotational fine structure which is
centered at 2972 cm21. For the CH3 group the two
antisymmetric stretches are split and assigned at 2953
and 2945 cm21 (n2 and n28) which are wavenumbers
from the Raman spectrum of the solid. However, it is
possible that the n2 fundamental is coincident with the
n1 fundamental at 2972 cm21 because the CH2
antisymmetric stretch is most reasonably assigned at
2955 cm21 or at 2972 cm21. The pCH2 antisymmetric
stretch is assigned at 2937 cm21 since it is predictedTab
le7
(co
nti
nued
)
Vib
.N
o.
Ap
pro
xim
ate
des
crip
tio
na
HF
6-3
1G
3L
YP
6-3
1G
(d)
MP
26
-31
G(d
)M
P2
6-3
lG(d
)bIR
Ram
anac
t.e
Dep
ol.
rati
oP
ED
f
Ob
s.c
Int.
d
n39
CH
3to
rsio
n3
30
24
32
55
28
22
86
0.1
1.5
0.4
13
3S
39,
23
S40,
36
S24
n40
C1–
C2x
C3
ben
do
ut-
of-
pla
ne
23
12
15
20
52
38
20
26
.10
.00
.46
51
S40,2
5S
39,
15
S38
n41
C6C
5C
4C
3to
rsio
n9
68
98
69
30
.51
.20
.72
47
S41,
24
S25
n42
pC
H3
tors
ion
11
31
11
0.0
0.0
0.6
11
00
S42
aA
ster
isks
indic
ate
Cat
om
sat
tach
edto
both
ends
of
the
Cx
Cb
on
d.
bS
cale
fact
ors
for
forc
eco
nst
ants
:0
.9fo
ral
lC
Cst
retc
hes
,0.8
8fo
rC
Hst
retc
hes
,0.9
for
CC
Hd
efo
rmat
ion
s,1
.0fo
rh
eav
y-a
tom
ben
ds
exce
pt1
.5fo
rCx
C–
Cli
nea
rb
end
s,an
d1
.0
for
tors
ion
s.e
Cal
cula
ted
Ram
anac
tivit
ies
inA
/am
uat
the
MP
2/6
-31
G(d
)le
vel
.f
Cal
cula
ted
atth
eM
P2
/6-3
1G
(d)
lev
elw
ith
the
seal
edfo
rce
con
stan
ts;
con
trib
uti
on
sle
ssth
an1
0%
are
om
itte
d.
cO
bse
rved
freq
uen
cies
are
fro
mth
ein
frar
edsp
ectr
um
of
the
gas
exce
pt
tho
sew
ith
aste
risk
are
fro
mth
esp
ectr
um
of
the
soli
d.
dC
alcu
late
din
frar
edin
ten
siti
esin
km
/mo
lat
the
MP
2/6
-31
G(d
)le
vel
.
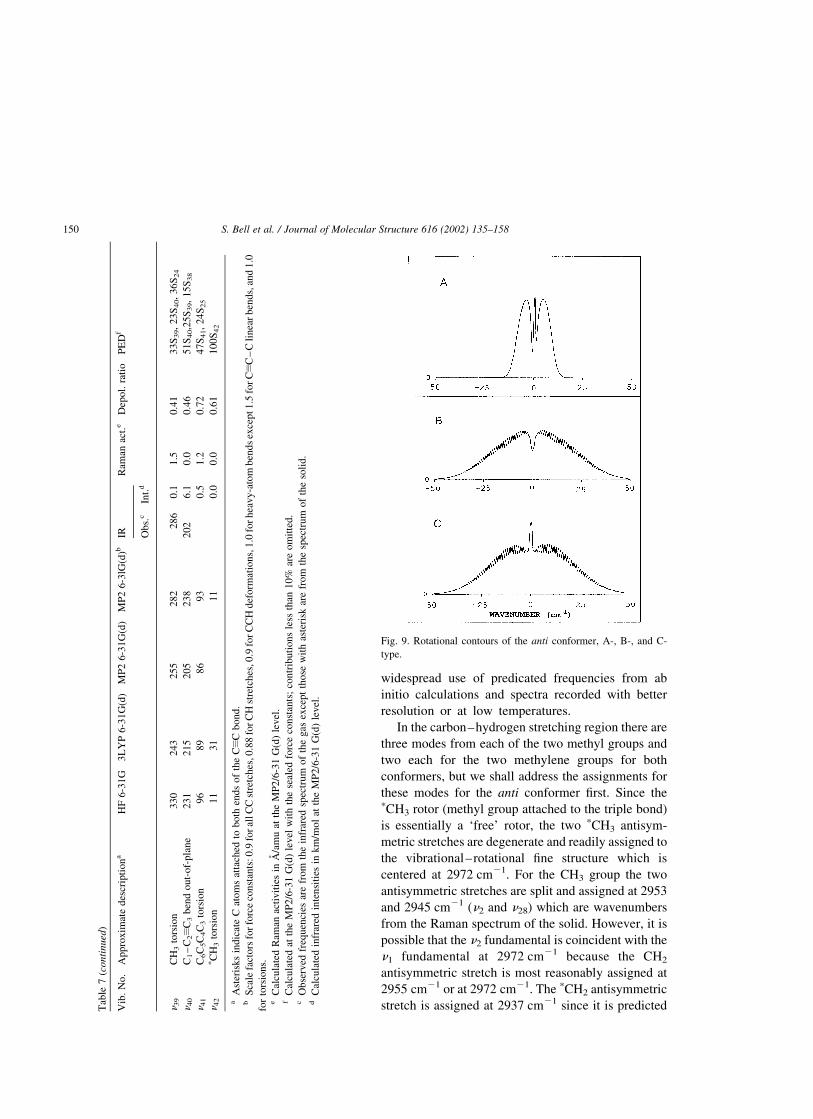
Fig. 9. Rotational contours of the anti conformer, A-, B-, and C-
type.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158150

to be more than 50 cm21 lower than the
corresponding mode for the CH2 group. The
symmetric methyl and methylene stretches (n3
through n6) are assigned consistent with the order
predicted from the ab initio calculations along
with the predicted infrared intensities and Raman
activities. These assignments must be considered
somewhat tentative and could be made only more
confidently if selected deuteration were made on
each carbon atom. Some of the wavenumbers
listed for these modes in Table 1 from the spectra
of the gas, krypton, or liquid, could be due to the
corresponding vibrations for the gauche conformer
which is in greater abundance in the fluid states even
though the anti conformer is the more stable form.
The predicted infrared intensity for the CxC
stretch is essentially zero and without the Raman
data it could not be assigned. The propyl group on one
side of the triple bond is not sufficiently different from
the methyl group on the other end of the triple bond to
give enough asymmetry for an observable dipole
change for the vibration. In fact, the band
(2259 cm21) readily observed in the infrared spectra
in this region is a combination band that falls between
the Fermi doublet at 2302 and 2236 cm21 for the CxC
stretch which is observed in the Raman spectrum.
Fig. 10. Rotational contours of the gauche conformer, A-, B-, and C-type.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158 151

There is also some ambiguity on the assignments
for two of the four methyl antisymmetric defor-
mations and the two methylene scissors. The twopCH3 antisymmetric deformations appear as a degen-
erate mode centered at 1450 cm21 with the typical
vibrational–rotational fine structure of a perpendicu-
lar symmetric top mode. The other four fundamentals
in this spectral region were assigned for the anti
conformer based on the scaled MP2/6-31G(d) ab
initio predicted wavenumbers and the observed bands
in the infrared and Raman spectra of the solid. The
remaining carbon–hydrogen bends, as well as the
heavy atom stretches, can be confidently assigned
from the ab initio predicted wavenumbers, infrared
intensities, Raman activities, and band contours.
However, the low frequency skeletal bends are better
predicted from the DFT calculations by the B3LPY
method than from the scaled ab initio calculations,
even with the scaling factor of 1.5 for the triple bond
bends. Nevertheless, there is little question concern-
ing their assignment for the anti conformer utilizing
the low wavenumber data from the infrared and
Raman spectra of the solid (Figs. 1 and 2).
For the gauche conformer, most of the funda-
mentals lower in wavenumber than the carbon–
hydrogen deformations could be assigned from their
disappearance from the spectra of the fluid phases
when the sample crystallizes to the solid phase. The
fundamental for this conformer in the higher wave-
number range, i.e. carbon–hydrogen stretches, CxC
stretch, and carbon–hydrogen deformations, are
expected to be in near coincident with the correspond-
ing modes of the anti conformer. For example, the
CxC stretch is predicted to only differ by three
wavenumbers for the two conformers. The complete
assignment of the normal modes of both anti and
gauche conformers are reported in Tables 6 and 7,
respectively.
6. Coriolis interaction
As in 2-pentyne [3] and 5-fluoro-2-pentyne [9],
the infrared spectrum of the gas exhibits sub-band
structure with strong–weak–weak–strong intensity
alternation associated with the antisymmetric pCH3
stretching vibrations and the antisymmetric pCH3
deformation modes (Figs. 11 and 12). This band
structure is typical of perpendicular bands of
symmetric top molecules such as the methyl
halides and 1-halo-propynes but in these molecules,
it is caused by the Coriolis interaction of the
overall rotational motion of the molecule with the
angular momentum of the E vibrations of
the methyl group. Similar sub-band structure has
been observed in the corresponding bands in the
infrared spectra of the 1-halo-2-butynes [7,24]
which are not symmetric top molecules. The Q
branches of the different sub-bands agree well with
the formula nsub0 ¼ c0 ^ c1m þ c2m2:
For a perpendicular band of a symmetric-top
molecule such as the degenerate fundamentals of a
methyl group, the formula for the sub-band structure
with the type of intensity alternation already men-
tioned has been given [25,26] by
nsub0 ¼ n0 þ ½A0ð1 2 zÞ2 2 B0�^ 2½A0ð1 2 zÞ2 B0�K
þ ½ðA0 2 B0Þ2 ðA00 2 B00Þ�K2
where z is the Coriolis coupling constant and the
factor (1 2 z )2 is used [25,26] rather than (1 2 2z ) as
in Herzberg [27] and Nyquist [28]. The single prime
refers to the upper state and double prime to the lower
state.
However, in the case of the 1-halo-2-butynes [7,
24], 2-pentyne [3], 5-fluoro-2-pentyne [9] and 2-
hexyne, the sub-band structure is due to the
interaction of the vibrational angular momentum of
the pseudo-degenerate vibrations with the internal
rotational angular momentum of the almost freely
rotating internal top and not with the overall rotation
of the molecule. From the energies of several ab initio
calculations of eclipsed and staggered configurations
of the C1 methyl of 2-hexyne (Table 3), it is evident
that the barrier to internal rotation of the pCH3 group
is very small, being only a few cm21 for all
calculation models used.
A quadratic equation can be derived from the first
principles using the appropriate Hamiltonian, but in
order to retain the z notation, the following equation
can be derived from the sub-band formula for overall
rotation of symmetric top molecules by elimination of
the kinetic constants for overall rotation, B or
(B þ C )/2, and by replacing the A constants with F1
which is h=8p2cIredt ; the inverse reduced moment of
inertia for internal rotation of the methyl top in the
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158152

slightly asymmetric top molecule
nsub0 ¼ ½n0 þ F0
1ð1 2 zÞ2� þ 2F01ð1 2 zÞm
þ ðF01 2 F00
1Þm2:
In 2-hexyne, the anti conformer is shown to be lower;
a complete vibrational analysis is given above, and it
is also more nearly a symmetric top than the gauche
conformer. The sub-band structure probably arises
from the near degeneracy of n1/n26 and n11/n31 of the
anti form. Values for kinetic constant F001 in the ground
vibrational state are obtained from ab initio optimized
geometric structures of the anti conformer given in
Table 4.
The measurements of the observed sub-bands shown
in Figs. 11 and 12 are given in Table 8. Fits to the sub-
band peaks have been made by the least-squares method
to the formula nsub0 ¼ c0 ^ c1m þ c2m2 and the coeffi-
cients are given in Table 8. For each of the bands, the
quadratic coefficient c2 is small; it is the change in the
internal rotational constant, F, between the ground and
excited states. Taking F00 ¼ 5:9731 cm21 from the
B3LYP geometric structure calculation, for the C–H
nearly degenerate stretching fundamental, n1/n26, we
obtain z ¼ 0:166 ^ 0:002 from the coefficient of the
linear term, 2F0ð1 2 zÞ; and from n0 þ F0ð1 2 zÞ2 the
band center, n0, is 2971.7 cm21. This value of
the antisymmetric pCH3 stretching fundamental is
very near that obtained for 2-pentyne [3].
The sub-band series is more difficult to discern for
the deformation fundamental, n11/n31 as the series is
interrupted in the middle with an interval of 22 cm21
instead of 14 cm21 and only the more obvious part is
used in a fit to obtain z ¼ 20:178 ^ 0:02 from the
linear term and the origin n0 is 1450.0 cm21.
However, this value for the deformational funda-
mental is in very good agreement with the value in 2-
pentyne and indeed it is within the range of
1451 ^ 2 cm21 for molecules considered with 1-
fluoro-2-butyne [7].
7. Discussion
The experimentally determined enthalpy differ-
ence of 74 ^ 8 cm21 for 2-hexyne is slightly lower
than the value of 50 ^ 6 cm21 obtained for 1-pentyne
[4] with the anti form the more stable conformer in
each case. Thus, the methyl group attached to the
carbon–carbon triple bond does not significantly
affect the conformational stability of 2-hexyne.
However, it should be noted that the ab initio MP2
calculations incorrectly predict the conformer stab-
ility and there is little difference in the predicted
energy difference with a small basis set of 6-31G(d)
with frozen core (146 cm21) and the much larger
basis set of 6-311G(2d,2p) with full electron corre-
lation (154 cm21). However, it should be noted that
all of the DFT calculations predict the correct
conformer stability. The similar problem exists for
Fig. 11. Sub-band structure in the degenerate CH3 antisymmetric
stretching band due to Coriolis interaction.
Fig. 12. Sub-band structure in the degenerate CH3 antisymmetric
deformation band due to Coriolis interaction.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158 153

1-pentyne where the ab initio MP2 calculations with
the 6-31G(d) basis set also incorrectly predicted the
gauche conformer as the more stable form by
122 cm21. Also, the MP2/g-311 þ G(d,p) calcu-
lations predicted the gauche conformer as the more
stable rotamer by 103 cm21. Because the ab initio
MP2 calculations predict such a long CxC bond, the
relative predicted energies for the two conformers is
in error for both molecules.
Apart from the frequency for the torsion of the
methyl group on the CxC (which is essentially free)
bond, the lowest frequency (Tables 6 and 7)
calculated for each of the conformers is the asym-
metric skeletal torsion: anti, 84.6, 83.2, and
86.8 cm21 from the B3LYP, MP2, MP2 scaled
calculations, respectively; and gauche, 89.4, 86.2,
and 93.9 cm21 from the B3LYP, MP2, MP2 scaled
calculations, respectively. From a calculation of the F
series for the kinetic part of the torsional problem and
from the potential function obtained from the B3LPY
energies of the four conformers in Table 3 and plotted
in Fig. 5, the torsional fundamentals are predicted as
83.5 and 86.0 cm21 for the anti and gauche
conformers, respectively. The absorption hump
around 90 cm21 is clearly to be identified with
asymmetric torsion. In view of the fact that the
gauche torsional fundamental is calculated as higher
than the anti fundamental, the small peak at 91 cm21
is assigned to the anti fundamental and the shoulder at
87 cm21 to the gauche fundamental. The value of DH
obtained from temperature-dependent infrared spectra
of 74 cm21 (Table 2) is used as the 0 þ (g ) ˆ 0(t )
transition energy. Using these torsional intervals, the
torsional angle of 62.68 of the gauche conformer, and
the F series from the B3LYP parameters an
experimental potential energy function is obtained
Table 8
Coriolis structure of nearly degenerate CH3 antisymmetric stretch and deformation bands of 2-hexyne
m CH3 antisymmetric stretch CH3 antisymmetric deformation
nobs nobs 2 ncalc nobs nobs 2 ncalc
13 3097.40 20.46
12 3089.19 0.24
11 3080.16 0.20 1605.10 20.27
10 3071.04 0.16 1592.63 0.04
9 3061.62 20.12 1580.32 0.63
8 3052.64 0.13 1566.69 0.02
7 3043.30 0.10 1553.05 20.48
6 3033.99 0.17 1540.30 0.03
5 3024.26 20.10 1526.70 20.20
4 3014.44 20.37 1513.60 0.19
3 3005.20 0.01 1499.83 0.03
2 2995.54 0.04 1477.56
1 2985.80 0.08 1464.91
0 2975.10 20.76 1448.02
21 2966.60 0.67 1434.59
22 1419.11
23 1404.98
24 1391.76
25 1377.85
26 1363.26
St. dev. 0.36 0.36
c0 2975.86 ^ 0.19 1458.3 ^ 0.9
c1 9.8947 ^ 0.0716 14.020 ^ 0.292
c2 20.0392 ^ 0.0057 20.059 ^ 0.021
z 0.166 ^ 0.002 20.178 ^ 0.024
n0 2971.74 ^ 0.2 1450.0 ^ 0.9
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158154

by fit. The constants are V1 ¼ 218; V2 ¼ 220; V3 ¼
1475; V4 ¼ 2104 cm21 with the DH of 72.2 cm21.
This function is also plotted in Fig. 5.
The predicted structural parameters for the two
conformers do not differ significantly with the bond
distances having variations of usually 0.001–0.002 A.
There are only slightly more variations in the angles
with the most apparent ones the dihedral angles.
However, such small changes are not expected to give
rise to large variations in the force constants. This is
exactly what is found with the variations usually less
than 2%. The exceptions are for several of the angle
bends.
The major difference in force constant values,
excluding those for the torsional modes, is found for
the C3C4C5 and C4C5C6 angle bends which are 11.8
and 8% smaller, respectively, for the anti conformer
compared to the corresponding ones for the gauche
form, i.e. 0.635 versus 0.710 and 0.710 versus
0.767 mdyn/A. Smaller differences of 4.9 and 3.2%
are obtained for the C2C3C4 and C1C2C3 in-plane
bends with those for the anti conformer again having
the lower values (0.389 versus 0.408 and 0.373 versus
0.385 mdyn/A). Another noteworthy difference is the
C6C5H7 bending force constant which is 3.4% lower
for the anti conformer (0.562 versus 0.581 mdyn/A).
Major differences are also predicted for the force
constants for the asymmetric and CH3 torsions with
values of 37 and 8.9% lower for the anti conformer.
Therefore, most of the significant differences in
frequencies for the normal modes are due to the
differences in the PEDs rather than differences in the
force constants.
The PEDs are reasonably pure for the anti
conformer with major mixing of the pCH2, CH2,pCH3 and CH3 rocks, along with some of the carbon–
carbon stretches. Several of these fundamentals have
only 35–41% contributions of the symmetry coordi-
nate for which the band is assigned. For most of these
vibrations there are significant contributions from
three symmetry coordinates. For the gauche con-
former which has no symmetry elements, the mixing
is much more extensive. For example, n17 and n18
which are methyl rocks and n20 and n21 which are
carbon–carbon stretches have only 20–29% of those
indicated symmetry coordinates contributing to these
bands. Similarly, n36 which is designed as the CH3
rock, has only 15% of S36 contributing to this mode,
with the other three contributions greater than 10%
being only 12, 12 and 20%. Therefore, the approxi-
mate descriptions given for the fundamentals of the
gauche conformer are much less an indication of the
motions than those for the anti conformer.
Assuming the internal rotation of the C5 methyl
group as an independent coordinate and from the
methyl barriers given in Table 3 and the F value given
in Table 4, the methyl torsional fundamental is
predicted to be at 220 cm21 in the anti conformer
and at 216 cm21 in the gauche form. These are a little
lower than predicted by the harmonic vibrational
force field but the symmetry coordinates are con-
siderably mixed as shown in the PED.
Taking the heavy atom torsional motion as a
separate coordinate and from a F series determined
from the geometrical structures of both anti and
gauche forms, the fundamental transitions for
C3C4C5C6 torsion are predicted to be at 111 cm21
for the anti conformer and at 98 cm21 in the gauche
form. These fundamental frequencies are a little
higher than indicated by the harmonic force field but
more in agreement with the magnitudes of the
observed and assigned far infrared bands.
In the earlier proposed assignment [15] many of
the fundamentals for the anti conformer were
assigned as being accidentally degenerate since only
the infrared spectra of the liquid and solid and the
Raman spectrum of the liquid were utilized for the
identification of the fundamentals. For example, in
the carbon–hydrogen stretch region only five of the
ten fundamentals were identified and the carbon–
hydrogen deformations only three individual bands
were assigned for the eight fundamentals. Similar dual
assignments were given for several of the funda-
mentals in the fingerprint region whereas others were
predicated form normal coordination calculations but
left unassigned, i.e. predicated fundamentals at 1102,
1019, and 911 cm21 which we observed at 1091,
1032, and 892 cm21, respectively.
Similar multiple assignments were previously
given [15] for some of the carbon–hydrogen bending
or carbon–carbon stretching modes for the gauche
conformer. However, from the spectral data from the
krypton solution it was possible to assign most of the
fundamentals in the fingerprint region to the gauche
conformer to individual bands that were not present in
the spectra of the solid. However, it should be noted
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158 155

that many of these fundamentals were predicated
reasonably well from the earlier [15] normal coordi-
nate calculation which was obtained from transferred
force constants from 1- and 2-butyne and 2-pentyne.
Nevertheless, many of the fundamentals for the two
conformers were predicated to be nearly degenerate,
particularly those associated with the two methyl
groups.
There does not appear to have been a structural
determination of 2-hexyne by the electron diffraction
Table 9
Comparison of the structural parameters (bond distances in A, angles in degrees, rotational constants in MHz, dipole moments in Debye, and
energies in Hartree) for anti and gauche 1-pentyne (all columns except the last two are for 1-pentyne) and 2-hexyne from experimental values
and ab initio calculations
MP2/6-311 þ G(d,p) EDa MWb ro-adjustedc 2-hexyne (MP2/6-311G(d,p))
anti gauche anti gacuhe anti gauche anti gauche
r(C2–C3) 1.219 1.219 1.2220(7) 1.210 1.210 1.209 1.209 1.220(1.210) 1.220(1.210)
r(C3–C4) 1.462 1.464 1.4604(30) 1.460 1.460 1.462 1.463 1.463 1.464
r(C4–C5) 1.535 1.536 1.5372(15) 1.544 1.544 1.536 1.537 1.534 1.536
r(C5–C6) 1.526 1.525 1.5262 1.536 1.536 1.526 1.525 1.526 1.525
r(C4–H4) 1.096 1.095 1.1072(9) 1.094 1.094 1.096 1.095 1.097 1.100
r(C4–H5) 1.096 1.097 1.1072(9) 1.094 1.094 1.096 1.097 1.097 1.099
r(C5–H6) 1.095 1.096 1.095 1.096 1.095 1.098
r(C5–H7) 1.095 1.095 1.095 1.095 1.095 1.097
r(C6–H9) 1.094 1.095 1.094 1.095 1.094 1.097
r(C6–H10) 1.094 1.093 1.094 1.093 1.094 1.095
r(C6–H8) 1.094 1.093 1.094 1.093 1.093 1.096
/C2C3C4 178.3 178.9 180.0 180.0 180.0 178.3 178.9 178.2 179.2
/C3C4C5 112.6 112.4 113.5(4) 111.5 111.5 112.3 112.1 112.7 112.5
/C4C5C6 111.9 112.4 111.7(5) 111.5 112.4 112.9 113.5 112.0 112.4
/C3C4H4 109.2 109.0 110.0(5) 109.5 109.5 109.4 109.1 109.5 109.3
/C3C4H5 109.2 109.1 109.5 109.5 109.4 109.2 109.5 109.3
/H3C4H5 106.8 106.9 106.8 106.9 106.6 106.8
/C4C5H6 108.7 108.2 108.2 107.7 108.7 108.5
/C4C5H7 108.7 108.9 108.2 108.3 108.7 108.7
/H6C5H7 106.9 107.2 106.9 107.2 106.8 107.2
/H6C5C6 110.3 110.0 109.5 109.5 110.3 110.0 110.3 110.0
/H7C5C6 110.3 110.0 109.5 109.5 110.3 110.0 110.3 110.0
/C5C6H9 111.1 110.6 110.14 110.14 111.1 110.6 1.094 110.6
/C5C6H10 110.8 110.8 110.14 110.14 110.8 110.8 1.094 110.6
/C5C6H8 110.8 111.1 110.14 110.14 110.8 111.1 111.2 111.2
/H9C6H10 107.9 108.0 107.9 108.0 108.0 108.0
/H9C6H8 107.9 108.0 107.9 108.0 108.0 108.1
/H10C6H8 108.0 108.3 108.0 108.3 108.0 108.3
/C3C4C5C6 180.0 61.4 64.9(1.1) 180 65(3) 180.0 63.5 180.0 67.8
A 23393 9817 23340.0 9921.1 23340.2 9921.7
B 2226 3209 2230.6 3172.8 2229.6 3172.0
C 2113 2651 2116.4 2634.0 2115.2 2636.3
lmal 0.902 20.761
lmbl 0.068 0.329
lmcl 0.000 20.002
lmtl 0.905 0.829
2 (E þ 194) 0.8023933 0.8032195
DE (cm21 181
a Ref. [29].b Ref. [30].c Ref. [4].
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158156

technique or from a microwave study. Since the
dipole moment is so small (predicted value of 0.17D
for the anti form and 0.13D for gauche conformer) for
the 2-hexyne molecule it would be a very difficult
microwave problem. However, an electron diffraction
study [29] and the microwave spectrum has been
reported [30] for 1-pentyne which has larger dipole
moments of 0.92 and 0.82D for the anti and gauche
conformers, respectively. It is expected that the
determined structural parameters for 1-pentyne will
be very nearly the same as those for 2-hexyne.
Therefore, we have listed (Table 9) the previously
reported structural parameters for 1-pentyne along
with those predicted from MP2/6-311G(d,p) ab initio
calculations for both 1-pentyne and 2-hexyne. The
major difference in the r0 adjusted parameters for 1-
pentyne and those from the electron diffraction study
[29] or the ab initio predictions is the triple bond
distance of 1.209 A which does not differ significantly
with difference substitutes on the CxC triple bond.
Therefore, the predicted structural parameters from
the MP2/6-311G(d,p) calculations for 2-hexyne
should provide C–H distances within 0.003 A,
whereas all of the C–C distances except for the triple
bond are expected to be a little too short. The CxC
bond distance is predicted too long from MP2
calculations by 0.010–0.012 A, so the actual value
is expected to be near the value of 1.210 A which is
the distance predicted from the B3LYP/6-31G(d)
calculations. Making these adjustments to the car-
bon–carbon distances, it is expected that the predicted
values for the distances relative to the actual values
will be 0.005 A for the carbon–carbon distances and
0.003 A for the carbon–hydrogen distances. These
adjusted ab initio parameters are expected to be as
accurate as they can be experimentally measured. The
uncertainty for all of the angles is 0.58 except for the
gauche dihedral angle, C3C4C5C6, which is expected
to be 1.58.
Acknowledgments
J.R. Durig acknowledges the University of Kansas
City Trustees for a Faculty Fellowship award for
partial financial support of this research.
References
[1] G.A. Guirgis, J.R. Durig, S. Bell, J. Mol. Struct. 196 (1989)
101.
[2] S. Bell, G.A. Guirgis, Y. Li, J.R. Durig, J. Phys. Chem., A 101
(1997) 5987.
[3] S. Bell, G.A. Guirgis, S.W. Hur, J.R. Durig, Spectrochim. Acta
A 55 (1999) 2361.
[4] J.R. Durig, B.R. Drew, J. Mol. Struct. 560 (2001) 247.
[5] G.A. Guirgis, S. Bell, J.R. Durig, Spectrochim. Acta A 52
(1996) 1861.
[6] G.A. Guirgis, X. Zhu, S. Bell, J.R. Durig, J. Phys. Chem. A
105 (2001) 363.
[7] G.A. Guirgis, S. Bell, J.R. Durig, Spectrochim. Acta 57 (2001)
1235.
[8] G.A. Guirgis, S. Bell, J.R. Durig, J. Raman Spectrosc. 31
(2000) 987.
[9] S. Bell, X. Zhu, G.A. Guirgis, J.R. Durig, Phys. Chem. Chem.
Phys. 3 (2001) 776.
[10] J.R. Durig, J. Liu, T.S. Little, J. Phys. Chem. 96 (1992)
8224.
[11] W.A. Herrebout, B.J. van der Veken, J. Phys. Chem. 100
(1996) 9671.
[12] W.A. Herrebout, B.J. van der Veken, A. Wang, J.R. Durig,
J. Phys. Chem. 99 (1995) 578.
[13] G.A. Guirgis, X. Zhu, J.R. Durig, Struct. Chem. 10 (1999)
445.
[14] J.R. Durig, X. Zhu, S. Shen, J. Mol. Struct. 570 (2001) 1.
[15] J.R. Durig, B.R. Drew, A. Koomer, S. Bell, Phys. Chem.
Chem. Phys. 3 (2001) 766.
[16] G.A. Crowder, P. Blankenship, J. Mol. Struct. 196 (1989)
125.
[17] F.A. Miller, B.M. Harney, Appl. Spectrosc. 24 (1970) 291.
[18] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A.
Robb, J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery,
Jr., R.E. Stratmann, J.C. Burant, S. Dapprich, J.M. Millam,
A.D. Daniels, K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi,
V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C.
Adamo, S. Clifford, J. Ochterski, G.A. Petersson, P.Y. Ayala,
Q. Cui, K. Morokuma, D.K. Malick, A.D. Rabuck,
K. Raghavachari, J.B. Foresman, J. Cioslowski, J.V. Ortiz,
B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi,
R. Gomperts, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham,
C.Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe,
P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, J.L. Andres,
C. Gonzalez, M. Head-Gordon, E.S. Replogle, J.A. Pople,
GAUSSIAN98 (Revision A.5), Gaussian Inc., Pittsburgh, PA,
1998.
[19] G. Guirgis, X. Zhu, Z. Yu, J.R. Durig, J. Phys. Chem. A 104
(2000) 4383.
[20] M.J. Frisch, Y. Yamagushi, J.F. Gaw, J.F. Schaefer III, J.S.
Binkley, J. Chem. Phys. 84 (1986) 531.
[21] R.D. Amos, Chem. Phys. Lett. 124 (1986) 376.
[22] P.L. Polavarapu, J. Phys. Chem. 94 (1990) 8106.
[23] G.W. Chantry, in: A. Anderson (Ed.), (1971) The Raman
Effect, vol. 1, Chapter 2, Marcel Dekker, New York, NY.
[24] R.D. McLachlan, Spectrochim. Acta A 23 (1967) 1793.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158 157

[25] D.W. Davidson, H.J. Bernstein, Can. J. Chem. 33 (1955) 1226.
[26] E.W. Jones, R.J.L. Popplewell, H.W. Thomson, Spectrochim.
Acta 22 (1966) 639.
[27] G. Herzberg, Molecular Spectra and Molecular Structure. II.
Infrared and Raman Spectra of Polyatomic Molecules, van
Nostrand, Princeton, 1945, p. 429.
[28] R.A. Nyquist, Spectrochim. Acta 21 (1965) 1245.
[29] M. Traetteberg, P. Bakken, H. Hopt, J. Mol. Struct. 509 (1999)
213.
[30] F.J. Wodarczyk, E.B. Wilson, J. Chem. Phys. 56 (1972)
166.
S. Bell et al. / Journal of Molecular Structure 616 (2002) 135–158158





























