Embed Size (px)
Citation preview

Kidney International, Vol. 48 (1995), pp. 1459—1468
Induction of plasminogen activator inhibitor type 1 in murine
lupus-like glomerulonephritisSOLANGE MOLL, PIERRE-ALAIN MENOUD, TFIIERRY FuLpIus, YVES PASTORE, SATORU TAKAHASHI,LILIANE F0ssATI, JEAN-DOMINIQUE VASSALLI, ANDRÉ-PASCAL SAPPIN0, JURG A. SCHIFFERLI,' and
SHozo Izui
Departments of Pathology, Morphology and Internal Medicine, University of Geneva Medical School, Geneva, Switzerland, and Department of Pathology,Tohoku University School of Medicine, Sendai, Japan
Induction of plasminogen activator inhibitor type 1 in murine lupus-like glomerulonephritis. Three major components of the plasminogenactivators (PA)/plasmin system are synthesized physiologically in glomer-uli, and can be involved in glomerular proteolysis and extracellular matrixmetabolism: tissue-type PA (tPA), urokinase (uPA) and PA inhibitor type1 (PAl-i). To explore the possible role of a dysregulation of the plasminprotease system in the development and progression of lupus-like glomer-ulonephritis, we studied the expression of the renal plasmin proteasecomponents during the course of the disease, either acute, induced byIgG3 monoclonal cryoglobulins, or chronic, occurring spontaneously inthree different lupus-prone mice: (NZBxNZW)FI, BXSB and MRL-lpr/lpr. RNase protection assays and in situ hybridizations revealed a markedglomerular induction of PAT-i mRNA abundance without any significantchanges in renal tPA and uPA mRNA levels in the two different types oflupus-like glomerulonephritis. The overexpression of PAT-i mRNA oc-curred in parallel with a significant decrease in glomerular tPA-catalyzedenzymatic activity as determined by zymographic analysis. In addition, aconcomitant increase in glomerular expression of transforming growthfactor /31 (TGF-131) mRNA was observed. The demonstration of a closecorrelation between the PAL-i and TGF-/31 mRNA levels and the severityof lupus-like glomerular lesions suggests that a perturbation of theglomerular PA/PA! balance, resulting from a marked TGF-j3i-mediatedinduction of PAl-i gene expression, plays an important role in theprogression of lupus-like glomerular lesions, leading to glomerulosclerosis.
Glomeruloscierosis is characterized by an accumulation ofextracellular matrix (ECM) as a result of an excessive productionand/or an insufficient degradation of ECM. This process isconsidered to be the final pathogenetic pathway leading to theprogressive loss of renal function in a number of glomerulopathiesincluding lupus nephritis. Although the molecular basis for thedevelopment of glomerulosclerosis remains unclear, it has beenproposed that a dysfunction in glomerular coagulation and pro-teolysis may play a role in this process [1—7]. Fibrin deposits maypromote glomerular injuries by occluding capillaries, by promot-ing inflammatory cell migration and glomerular cell proliferation,
and by a direct cytotoxic effect on mesangial cells [8]. This isfurther supported by the fact that anticoagulant treatment pre-vents the development of glomeruloscierosis in an experimentalmodel of glomerulonephritis [9].
It is now well documented that three major components of theplasminogen activators (PA)/plasmin system—tissue-type PA(tPA), urokinase (uPA) and PA inhibitor type I (PAI-1)—areexpressed in glomeruli under physiological and pathological con-ditions [10—13]. Since PAs and their inhibitors play an importantrole in fibrinolysis and in ECM degradation, a diminished synthe-sis of glomerular PA and/or an increased production of glomer-ular PAl following primary glomerular injuries may result in animbalance in the extracellular proteolytic process, thereby leadingto the generation of glomerular sclerotic lesions.
To explore the possible role of a dysregulation of the plasminprotease system in the development and progression of lupus-likeglomerulonephritis, we have studied the expression of the renalplasmin protease system during the course of acute lupus-likeglomerulonephritis induced by murine IgG3 monoclonal cryo-globulins [14—161 and of chronic glomerulonephritis occurringspontaneously in three different strains of lupus-prone mice:(NZBxNZW)F1, BXSB and MRL-lpr/lpr [17]. In parallel, wedetermined the renal mRNA expression of transforming growthfactor 131 (TGF-pI) for two reasons. First, it has been shown thatTGF-p1 decreases the synthesis of proteases, including tPA anduPA, and induces the production of PAT-i both in vitro and in vivo[18—25]. Second, TGF-p1 appears to play a crucial role in thedevelopment of glomerulosclerosis in experimental anti-Thy-iglomerulonephritis and diabetic nephropathy [26, 27]. Our resultsindicate that a marked induction of PAl-i and TGF-/31 geneexpression in glomeruli is associated with the progression ofglomerulonephritis in these different experimental models ofmurine lupus nephritis.
MethodsPresent address: Department of Medicine, University of Basel Medi-
cal School, Basel, Switzerland.
Received for publication April 14, 1995and in revised form June 12, 1995Accepted for publication June 12, 1995
© 1995 by the International Society of Nephrology
Mice
BALB/c and C57BL/6 mice were obtained from BomholtgardLtd. (Ry, Denmark). (NZBxNZW)F1, BXSB, MRL-lpr/lpr andMRL-+/+ mice were purchased from Harlan Olac Ltd. (Oxon,
1459
a e
a —
e
a— —
'._
rs
I
•,
S
.j p.
,'
1460 Moll et al: PAl-I induction in murine lupus nephritis
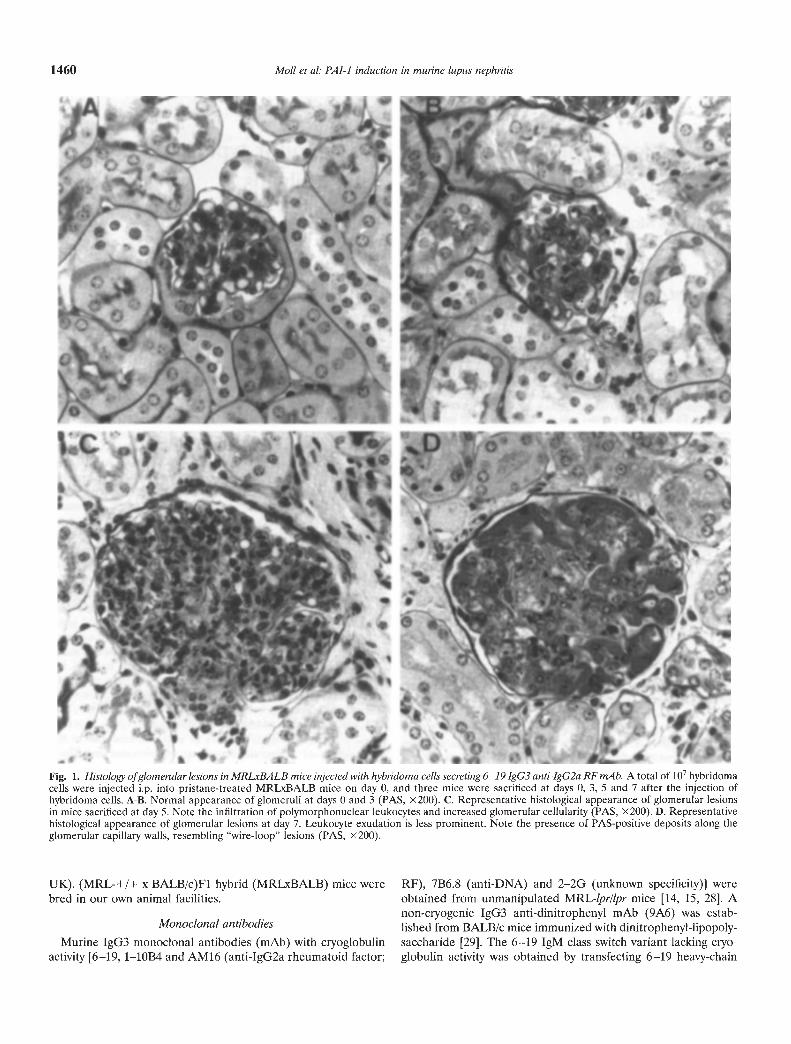
Fig. 1. Histology of glomerular lesions in MRLXBALB mice injected with hybridoma cells secreting 6—19 IgG3 anti-lgG2a RF mAb. A total of io hybridomacells were injected i.p. into pristane-treated MRLxBALB mice on day 0, and three mice were sacrificed at days 0, 3, 5 and 7 after the injection ofhybridoma cells. A-B. Normal appearance of glomeruli at days 0 and 3 (PAS, X200). C. Representative histological appearance of glomerular lesionsin mice sacrificed at day 5. Note the infiltration of polymorphonuclear leukocytes and increased glomerular cellularity (PAS, X200). D. Representativehistological appearance of glomerular lesions at day 7. Leukocyte exudation is less prominent. Note the presence of PAS-positive deposits along theglomerular capillary walls, resembling "wire-loop" lesions (PAS, )<200).
UK). (MRL-+/+ x BALB/c)F1 hybrid (MRLxBALB) mice were RF), 7B6.8 (anti-DNA) and 2—2G (unknown specificity)] werebred in our own animal facilities, obtained from unmanipulated MRL-lprllpr mice [14, 15, 28].A
non-cryogenic IgG3 anti-dinitrophenyl mAb (9A6) was estab-Monoclonal antibodies lished from BALB/c mice immunized with dinitrophenyl-lipopoly-
Murine 1g03 monoclonal antibodies (mAb) with cryoglobulin saccharide [29]. The 6—19 1gM class switch variant lacking cryo-activity [6—19, 1—10B4 and AM16 (anti-lgG2a rheumatoid factor; globulin activity was obtained by transfecting 6—19 heavy-chain
00-
Dl
•'1
Moll et al: PAl-i induction in murine lupus nephritis 1461
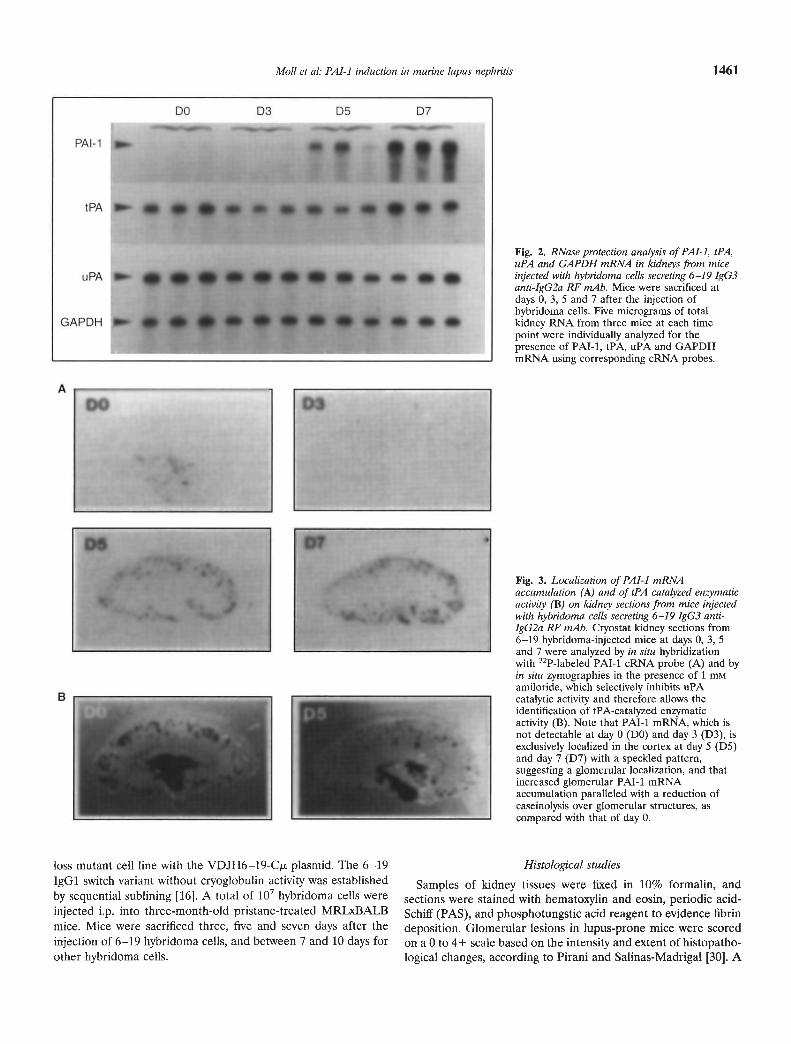
Fig. 2. RNase protection analysis of PA 1-1, IPA,uPA and GAPDH mRNA in kidneys from miceinjected with hybridoma cells secreting 6—19 IgG3anti-IgG2a RF mAb. Mice were sacrificed atdays 0, 3, 5 and 7 after the injection ofhybridoma cells. Five micrograms of totalkidney RNA from three mice at each timepoint were individually analyzed for thepresence of PAl-I, tPA, uPA and GAPDHmRNA using corresponding cRNA probes.
Fig. 3. Localization of PAl-i mRNAaccumulation (A) and of tPA -catalyzed enzymaticactivity (B) on kidney sections from mice injectedwith hybridoma cells secreting 6—19 IgG3 anti-IgG2a RF mAb. Cryostat kidney sections from6—19 hybridoma-injected mice at days 0, 3, 5and 7 were analyzed by in situ hybridizationwith 32P-labeled PAT-i cRNA probe (A) and byin situ zymographies in the presence of 1 mMamiloride, which selectively inhibits uPAcatalytic activity and therefore allows theidentification of tPA-catalyzed enzymaticactivity (B). Note that PAl-i mRNA, which isnot detectable at day 0 (DO) and day 3 (D3), isexclusively localized in the cortex at day 5 (D5)and day 7 (D7) with a speckled pattern,suggesting a glomerular localization, and thatincreased glomerular PAT-i mRNAaccumulation paralleled with a reduction ofcaseinolysis over glomerular structures, ascompared with that of day 0.
loss mutant cell line with the VDJH6—i9-C.r plasmid. The 6—19 Histological studiesIgGi switch variant without cryoglobulin activity was established Samples of kidney tissues were fixed in 10% formalin, andby sequential sublining [16]. A total of io hybridoma cells were sections were stained with hematoxylin and eosin, periodic acid-injected i.p. into three-month-old pristane-treated MRLxBALB Schiff (PAS), and phosphotungstic acid reagent to evidence fibrinmice. Mice were sacrificed three, five and seven days after the deposition. Glomerular lesions in lupus-prone mice were scoredinjection of 6—19 hybridoma cells, and between 7 and 10 days for on a 0 to 4+ scale based on the intensity and extent ofhistopatho-other hybridoma cells. logical changes, according to Pirani and Salinas-Madrigal [30]. A
1462 Mollet al: PAl-i induction in murine lupus nephritis
i 2.00
0IL0I-
0.5
0
t....••
. .. ... . .— -—_—? .
6—19 y3 6—1911 6—19 1-10B4 AM16 766.8 2—2G 9A6 Control
S
S.I•%
SIi'
•:
••
. .'6—19y36—19y1 6—19i 1-1064 AM16 7B6.8 2—2G 9A6 Control
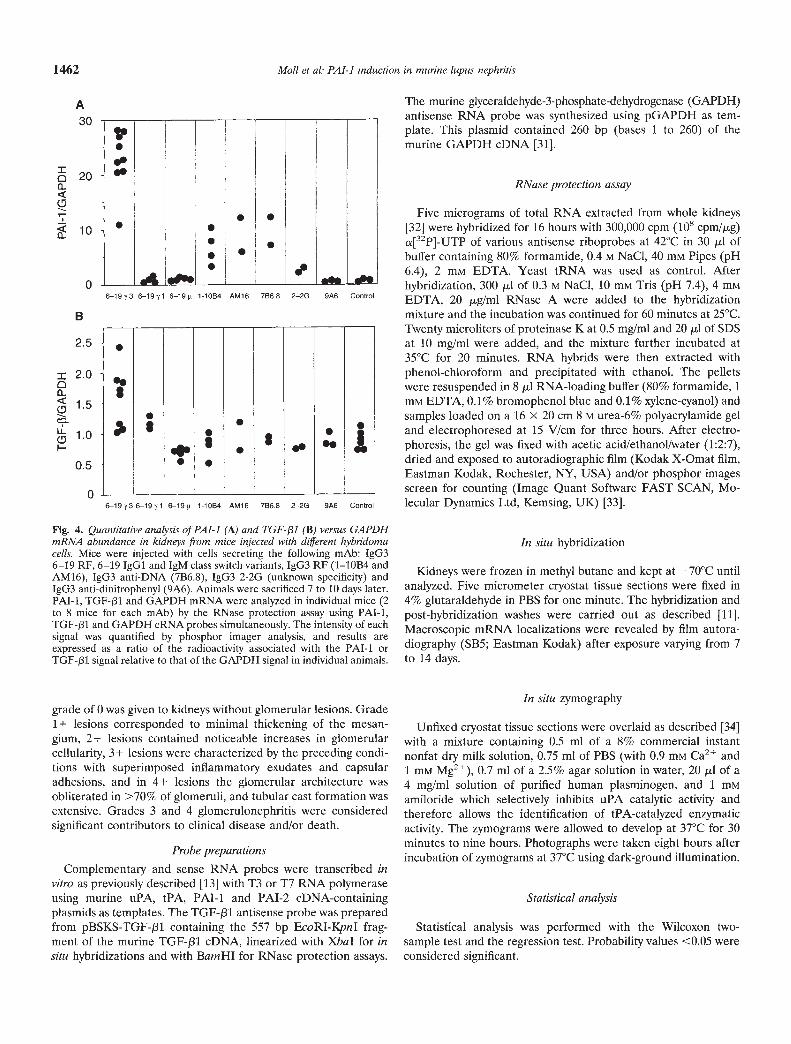
Fig. 4. Quantitative analysis of PAl-i (A) and TGF- j31 (B) versus GAPDHmRNA abundance in kidneys from mice injected with different hybridomacells. Mice were injected with cells secreting the following mAb: IgG36—19 RF, 6—19 IgGi and 1gM class switch variants, IgG3 RF (1—10B4 andAM16), IgG3 anti-DNA (7B6.8), IgG3 2-2G (unknown specificity) andIgG3 anti-dinitrophenyl (9A6). Animals were sacrificed 7 to 10 days later.PAT-i, TGF-131 and GAPDH mRNA were analyzed in individual mice (2to 8 mice for each mAb) by the RNase protection assay using PAl-i,TGF-f31 and GAPDH cRNA probes simultaneously. The intensity of eachsignal was quantified by phosphor imager analysis, and results areexpressed as a ratio of the radioactivity associated with the PAl-i orTGF-131 signal relative to that of the GAPDH signal in individual animals.
grade of 0 was given to kidneys without glomerular lesions. Grade1 + lesions corresponded to minimal thickening of the mesan-gium, 2 + lesions contained noticeable increases in glomerularcellularity, 3 + lesions were characterized by the preceding condi-tions with superimposed inflammatory exudates and capsularadhesions, and in 4+ lesions the glomerular architecture wasobliterated in >70% of glomeruli, and tubular cast formation wasextensive. Grades 3 and 4 glomerulonephritis were consideredsignificant contributors to clinical disease and/or death.
Probe preparationsComplementary and sense RNA probes were transcribed in
vitro as previously described [131 with T3 or T7 RNA polymeraseusing murine uPA, tPA, PAl-i and PAI-2 cDNA-containingplasmids as templates. The TGF-/31 antisense probe was preparedfrom pBSKS-TGF-/31 containing the 557 bp EcoRI-KpnI frag-ment of the murine TGF-pl eDNA, linearized with XbaI for insitu hybridizations and with BamHI for RNase protection assays.
The murine glyceraldehyde-3-phosphate-dehydrogenase (GAPDH)antisense RNA probe was synthesized using pGAPDH as tem-plate. This plasmid contained 260 bp (bases 1 to 260) of themurine GAPDH cDNA [311.
RNase protection assay
Five micrograms of total RNA extracted from whole kidneys[32] were hybridized for 16 hours with 300,000 cpm (108 cpm/jg)c432P]-UTP of various antisense riboprobes at 42°C in 30 itI ofbuffer containing 80% formamide, 0.4 M NaCl, 40 m Pipes (pH6.4), 2 mM EDTA. Yeast tRNA was used as control. Afterhybridization, 300 jtl of 0.3 M NaCl, 10 mM Tris (pH 7.4), 4 mMEDTA, 20 g/ml RNase A were added to the hybridizationmixture and the incubation was continued for 60 minutes at 25°C.Twenty microliters of proteinase K at 0.5 mg/ml and 20 d of SDSat 10 mg/mI were added, and the mixture further incubated at35°C for 20 minutes. RNA hybrids were then extracted withphenol-chloroform and precipitated with ethanol. The pelletswere resuspended in 8 jl RNA-loading buffer (80% formamide, 1mM EDTA, 0.1% bromophenol blue and 0.1% xyiene-cyanol) andsamples loaded on a 16 >< 20 cm 8 M urea-6% polyacrylamide geland electrophoresed at 15 V/cm for three hours. After electro-phoresis, the gel was fixed with acetic acid/ethanol/water (1:2:7),dried and exposed to autoradiographic film (Kodak X-Omat film,Eastman Kodak, Rochester, NY, USA) and/or phosphor imagesscreen for counting (Image Quant Software FAST SCAN, Mo-lecular Dynamics Ltd, Kemsing, UK) [33].
In situ hybridization
Kidneys were frozen in methyl butane and kept at —70°C untilanalyzed. Five micrometer cryostat tissue sections were fixed in4% glutaraldehyde in PBS for one minute. The hybridization andpost-hybridization washes were carried out as described [111.Macroscopic mRNA localizations were revealed by film autora-diography (SB5; Eastman Kodak) after exposure varying from 7to 14 days.
In situ zymography
Unfixed cryostat tissue sections were overlaid as described [31with a mixture containing 0.5 ml of a 8% commercial instantnonfat dry milk solution, 0.75 ml of PBS (with 0.9 mM Ca2 and1 mM Mg2), 0.7 ml of a 2.5% agar solution in water, 20 jil of a4 mg/mi solution of purified human plasminogen, and 1 miviamiloride which selectively inhibits uPA catalytic activity andtherefore allows the identification of tPA-catalyzed enzymaticactivity. The zymograms were allowed to develop at 37°C for 30minutes to nine hours. Photographs were taken eight hours afterincubation of zymograms at 37°C using dark-ground illumination.
Statistical analysis
Statistical analysis was performed with the Wilcoxon two-sample test and the regression test. Probability values <0.05 wereconsidered significant.
I0a-
0
A30
20
10
0
B
2.5
V
- ir03
07
Moll et al: PAl-i induction in murine lupus nephritis 1463
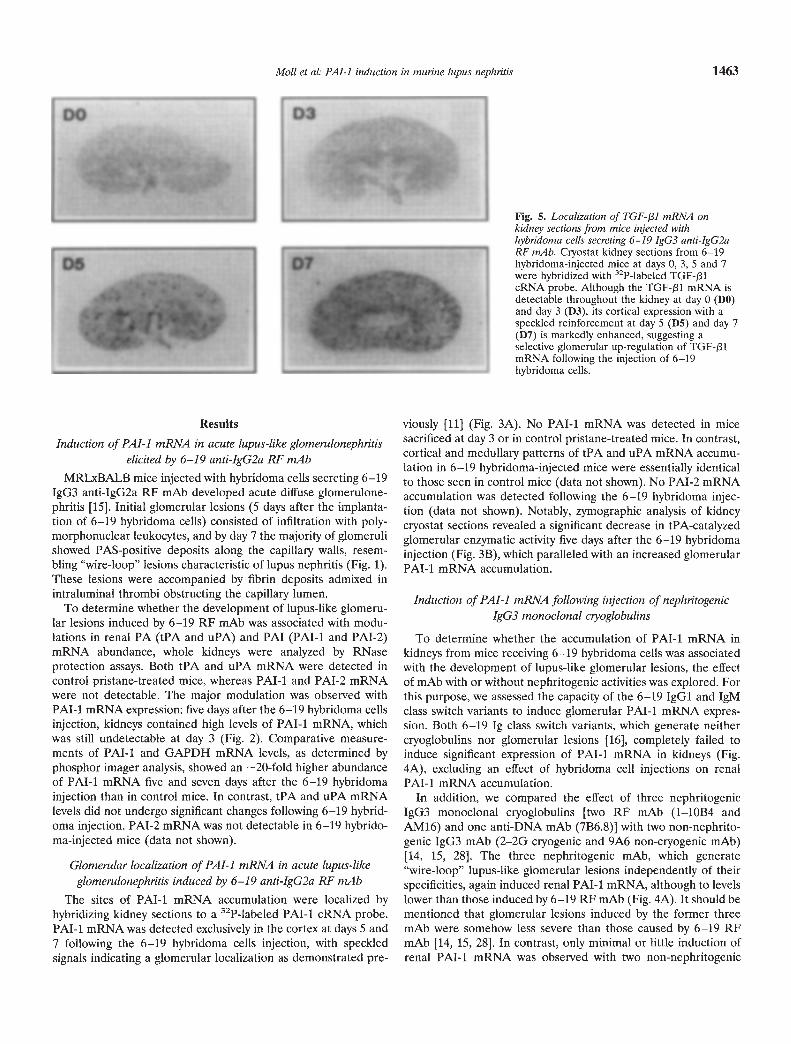
Fig. 5. Localization of TGF-131 mRNA onkidney sections from mice injected withhybridoma cells secreting 6—19 JgG3 anti-IgG2aRF mAb. Cryostat kidney sections from 6—19hybridoma-injected mice at days 0, 3, 5 and 7were hybridized with 32P-labeled TGF-131cRNA probe. Although the TGF-131 mRNA isdetectable throughout the kidney at day 0 (DO)and day 3 (D3), its cortical expression with aspeckled reinforcement at day 5 (D5) and day 7(D7) is markedly enhanced, suggesting aselective glomerular up-regulation of TGF-plmRNA following the injection of 6—19hybridoma cells.
Results
Induction of PAl-i mRNA in acute lupus-like glomerulonephritiselicited by 6—19 anti-IgG2a RF mAb
MRLXBALB mice injected with hybridoma cells secreting 6—19IgG3 anti-IgG2a RF mAb developed acute diffuse glomerulone-phritis [15]. Initial glomerular lesions (5 days after the implanta-tion of 6—19 hybridoma cells) consisted of infiltration with poly-morphonuclear leukocytes, and by day 7 the majority of glomerulishowed PAS-positive deposits along the capillary walls, resem-bling "wire-loop" lesions characteristic of lupus nephritis (Fig. 1).These lesions were accompanied by fibrin deposits admixed inintraluminal thrombi obstructing the capillary lumen.
To determine whether the development of lupus-like glomeru-lar lesions induced by 6—19 RF mAb was associated with modu-lations in renal PA (tPA and uPA) and PAl (PAl-i and PAI-2)mRNA abundance, whole kidneys were analyzed by RNaseprotection assays. Both tPA and uPA mRNA were detected incontrol pristane-treated mice, whereas PAl-i and PAI-2 mRNAwere not detectable. The major modulation was observed withPAl-i mRNA expression: five days after the 6—19 hybridoma cellsinjection, kidneys contained high levels of PAl-i mRNA, whichwas still undetectable at day 3 (Fig. 2). Comparative measure-ments of PM-i and GAPDH mRNA levels, as determined byphosphor imager analysis, showed an —20-fold higher abundanceof PAT-i mRNA five and seven days after the 6—19 hybridomainjection than in control mice. In contrast, tPA and uPA mRNAlevels did not undergo significant changes following 6—19 hybrid-oma injection. PAI-2 mRNA was not detectable in 6—19 hybrido-ma-injected mice (data not shown).
Glomerular localization of PAl-i mRNA in acute lupus-likeglomerulonephritis induced by 6—19 anti-IgG2a RF mAb
The sites of PAl-i mRNA accumulation were localized byhybridizing kidney sections to a 32P-labeled PAT-i cRNA probe.PAl-i mRNA was detected exclusively in the cortex at days 5 and7 following the 6—19 hybridoma cells injection, with speckledsignals indicating a glomerular localization as demonstrated pre-
viously [ii] (Fig. 3A). No PAl-i mRNA was detected in micesacrificed at day 3 or in control pristane-treated mice. In contrast,cortical and medullary patterns of tPA and uPA mRNA accumu-lation in 6—19 hybridoma-injected mice were essentially identicalto those seen in control mice (data not shown). No PAI-2 mRNAaccumulation was detected following the 6—19 hybridoma injec-tion (data not shown). Notably, zymographic analysis of kidneycryostat sections revealed a significant decrease in tPA-catalyzedglomerular enzymatic activity five days after the 6—19 hybridomainjection (Fig. 3B), which paralleled with an increased glomerularPAl-i mRNA accumulation.
Induction of PAl-i mRNA following injection of nephritogenicIgG3 monoclonal ciyoglobulins
To determine whether the accumulation of PAl-i mRNA inkidneys from mice receiving 6—19 hybridoma cells was associatedwith the development of lupus-like glomerular lesions, the effectof mAb with or without nephritogenic activities was explored. Forthis purpose, we assessed the capacity of the 6—19 IgGi and 1gMclass switch variants to induce glomerular PAl-i mRNA expres-sion. Both 6—19 Ig class switch variants, which generate neithercryoglobulins nor glomerular lesions [16], completely failed toinduce significant expression of PAl-i mRNA in kidneys (Fig.4A), excluding an effect of hybridoma cell injections on renalPAl-i mRNA accumulation.
In addition, we compared the effect of three nephritogenicIgG3 monoclonal cryoglobulins [two RF mAb (1—10B4 andAM16) and one anti-DNA mAb (7B6.8)] with two non-nephrito-genie IgG3 mAb (2—2G cryogenic and 9A6 non-cryogenic mAb)[14, 15, 28]. The three nephritogenic mAb, which generate"wire-loop" lupus-like glomerular lesions independently of theirspecificities, again induced renal PAl-i mRNA, although to levelslower than those induced by 6—19 RF mAb (Fig. 4A). It should bementioned that glomerular lesions induced by the former threemAb were somehow less severe than those caused by 6—19 RFmAb [14, 15, 28]. In contrast, only minimal or little induction ofrenal PAT-i mRNA was observed with two non-nephritogenic
1464 Mo/I et al. PAl-i induction in murine lupus nephritis
NZB x NZW BXSB
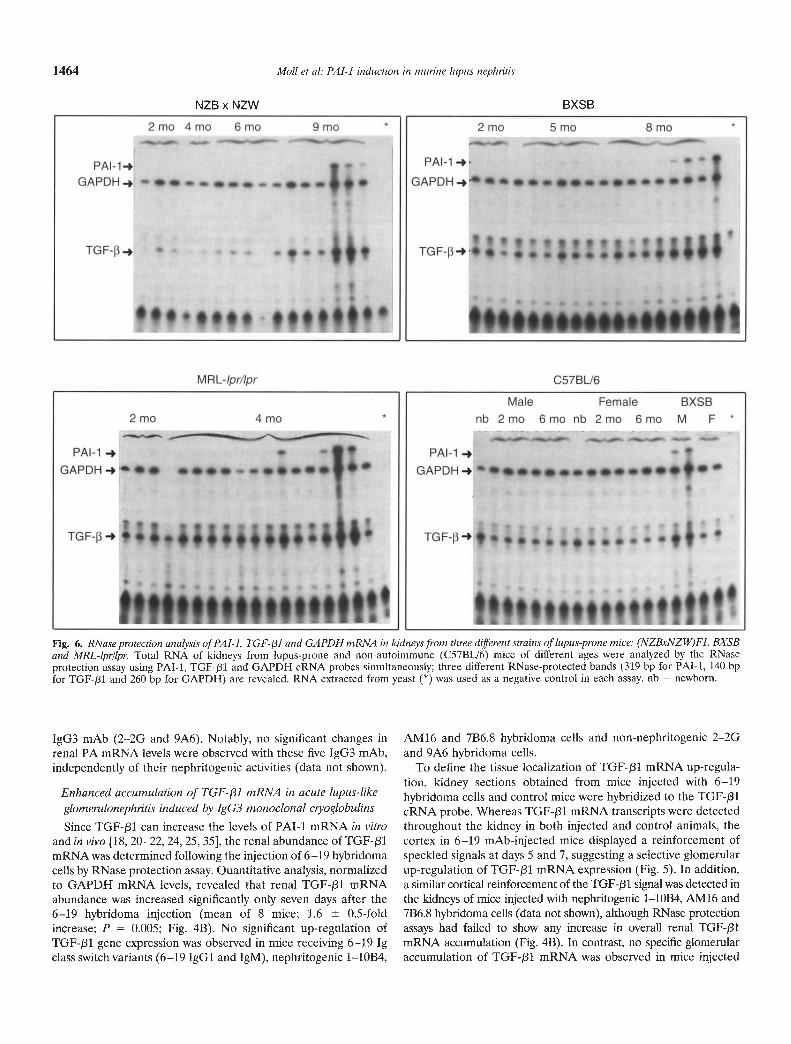
Fig. 6. RNase protection analysis of PAl-I, TGF-3I and GAPDH mRNA in kidneys from three different strains of lupus-prone mice: (NZBxNZW)Fi, BXSBand MRL-lpr/lpr. Total RNA of kidneys from lupus-prone and non-autoimmune (C57BL/6) mice of different ages were analyzed by the RNaseprotection assay using PAT-i, TGF-131 and GAPDH cRNA probes simultaneously; three different RNase-protected bands (319 bp for PAl-i, 140 bpfor TGF-/31 and 260 bp for GAPDH) are revealed. RNA extracted from yeast (*) was used as a negative control in each assay. nb = newborn.
IgG3 mAb (2—2G arid 9A6). Notably, no significant changes inrenal PA mRNA levels were observed with these five IgG3 mAb,independently of their nephritogeriic activities (data not shown).
Enhanced accumulation of TGF-/31 mRNA in acute lupus-like
glomenilonephritis induced by IgG3 monoclonal ciyoglobulinsSince TGF-J31 can increase the levels of PAl-I mRNA in vitro
and in vivo 18, 20—22, 24, 25, 35], the renal abundance of TGF-131mRNA was determined following the injection of 6—19 hybridomacells by RNase protection assay. Quantitative analysis, normalizedto GAPDH mRNA levels, revealed that renal TGF-131 mRNAabundance was increased significantly only seven days after the6—19 hybridoma injection (mean of 8 mice; 1.6 0.5-foldincrease; P = 0.005; Fig. 4B). No significant up-regulation ofTGF-/31 gene expression was observed in mice receiving 6—19 Igclass switch variants (6—19 IgGi and 1gM), nephritogenic 1—10B4,
AM16 and 7B6.8 hybridoma cells and non-nephritogenic 2—2Gand 9A6 hybridoma cells.
To define the tissue localization of TGF-131 mRNA up-regula-tion, kidney sections obtained from mice injected with 6—19hybridoma cells and control mice were hybridized to the TGF-J31cRNA probe. Whereas TGF-/31 mRNA transcripts were detectedthroughout the kidney in both injected and control animals, thecortex in 6—19 mAb-injected mice displayed a reinforcement ofspeckled signals at days 5 and 7, suggesting a selective glomerularup-regulation of TGF-131 mRNA expression (Fig. 5). In addition,a similar cortical reinforcement of the TGF-f31 signal was detected inthe kidneys of mice injected with nephritogenic 1—10B4, AMI6 and7B6.8 hybridoma cells (data not shown), although RNase protectionassays had failed to show any increase in overall renal TGF-f31mRNA accumulation (Fig. 4B). In contrast, no specific glomerularaccumulation of TGF-pl mRNA was observed in mice injected
80
60>1Na-z<>< 40
N-Z 20
0
50
40I030
(I)><—
= 20
10
0
50
40LI
10
0
Moll et a!: PAl-I induction in murine lupus nephritis 1465
I00CD
U-CDI-
I02a-CD
CDH
with 2—2G and 9A6 mAb lacking nephritogenic activities (data not
shown).
Up-regulation of PAl-i and TGF-/31 mRNA expression inspontaneous chronic lupus nephritis
Using three different strains of lupus-prone mice [(NZBxN-ZW)F1, BXSB and MRL-lprllpr] renal expression of PAT-i andTGF-pl mRNA was assessed in relation to the development oftheir glomerular lesions. For this purpose, total RNA was ex-tracted from kidneys of lupus-prone and non-autoimmuneC57BL/6 mice of various ages, and PAl-i, TGF-/31 and GAPDHmRNA abundance was simultaneously analyzed by RNase pro-tection assay, in which three different sizes of RNase-protectedbands (319 bp for PAl-i, 140 bp for TGF-pl and 260 bp forGAPDH) were revealed. All three strains of lupus-prone miceexhibited an increased abundance of both PAl-i and TGF-f31mRNA concomitantly with the development of severe glomerularlesions, that is, at eight to nine months for (NZBxNZW)F1
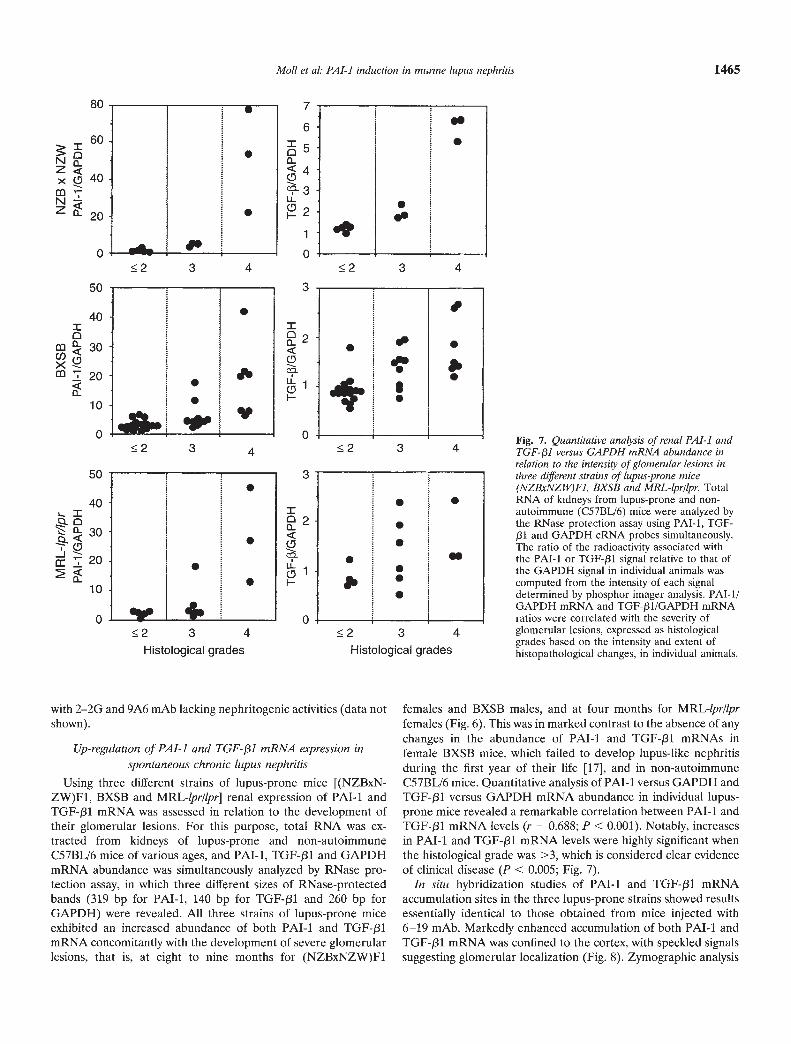
females and BXSB males, and at four months for MRL-lpr/lprfemales (Fig. 6). This was in marked contrast to the absence of anychanges in the abundance of PAl-I and TGF-131 mRNAs infemale BXSB mice, which failed to develop lupus-like nephritisduring the first year of their life [17], and in non-autoimmuneC57BL/6 mice. Quantitative analysis of PAl-i versus GAPDH andTGF-pl versus GAPDH mRNA abundance in individual lupus-prone mice revealed a remarkable correlation between PAl-i andTGF-f31 mRNA levels (r = 0.688; P < 0.001). Notably, increasesin PAl-i and TGF-131 mRNA levels were highly significant whenthe histological grade was >3, which is considered clear evidenceof clinical disease (P < 0.005; Fig. 7).
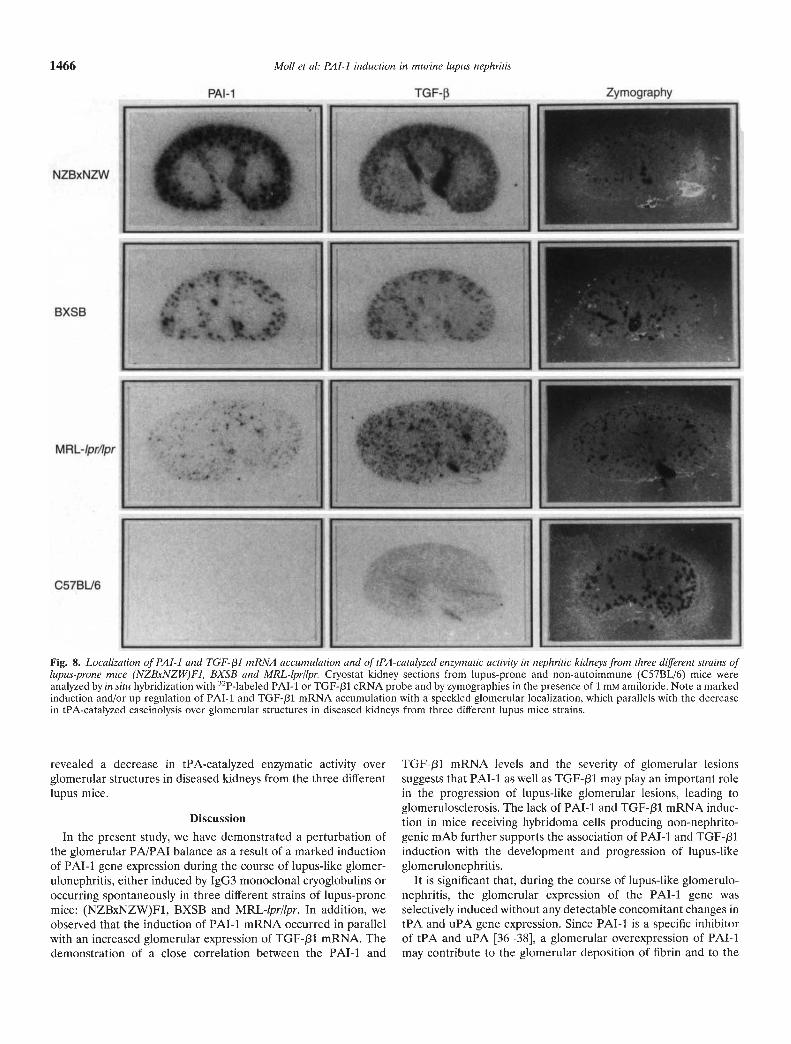
In situ hybridization studies of PAl-I and TGF-f31 mRNAaccumulation sites in the three lupus-prone strains showed resultsessentially identical to those obtained from mice injected with6—19 mAb. Markedly enhanced accumulation of both PAl-i andTGF-f31 mRNA was confined to the cortex, with speckled signalssuggesting glomerular localization (Fig. 8). Zymographic analysis
7
6I0a-<4CD
22
0
3
.
.
.-.-
3 4
S
S*,.
S
. .--
S...
3 4
'I. . S' S+! S
3 4
. .
.S
SS
S •.
0
3
3 4
Histological grades
0
Fig. 7. Quantitative analysis of renal PAl-i andTGF-/31 versus GAPDH mRNA abundance inrelation to the intensity of glomerular lesions inthree different strains of lupus-prone mice(NZBxNZW)Fi, BXSB and MRL-lpr/lpr. TotalRNA of kidneys from lupus-prone and non-autoimmune (C57BL/6) mice were analyzed bythe RNase protection assay using PAT-i, TGF-J31 and GAPDH cRNA probes simultaneously.The ratio of the radioactivity associated withthe PAT-I or TGF-pl signal relative to that ofthe GAPDH signal in individual animals wascomputed from the intensity of each signaldetermined by phosphor imager analysis. PAT-l/GAPDH mRNA and TGF-/31/GAPDH mRNAratios were correlated with the severity ofglomerular lesions, expressed as histologicalgrades based on the intensity and extent ofhistopathological changes, in individual animals.
3 4Histological grades
PAl-i TGF- Zymography
NZBxNZW
BXSB
MRL-/pr/!pr
C576L/6
1466 Mo!! et a!: PAl-i induction in murine !upus nephritis
Fig. 8. Loca!ization of PAl-I and TGF- /31 mRNA accumu!ation and of tPA-cata!yzed enzymatic activity in nephritic kidneys from three different strains of!upus-prone mice (NZBxNZW)Fi, BXSB and MRL-!pr/!pr. Ciyostat kidney sections from lupus-prone and non-autoimmune (C57BL/6) mice wereanalyzed by in situ hybridization with 32P-labeled PAl-i or TGF-/31 cRNA probe and by zymographies in the presence of 1 m'vi amiloride. Note a markedinduction and/or up-regulation of PAT-i and TGF-/31 mRNA accumulation with a speckled glomerular localization, which parallels with the decreasein tPA-catalyzed caseinolysis over glomerular structures in diseased kidneys from three different lupus mice Strains.
revealed a decrease in tPA-catalyzed enzymatic activity overglomerular structures in diseased kidneys from the three differentlupus mice.
Discussion
In the present study, we have demonstrated a perturbation ofthe glomerular PAJPAI balance as a result of a marked inductionof PAl-i gene expression during the course of lupus-like glomer-ulonephritis, either induced by IgG3 monoclonal cryoglobulins oroccurring spontaneously in three different strains of lupus-pronemice: (NZBxNZW)F1, BXSB and MRL-!pr/lpr. In addition, weobserved that the induction of PAT-i mRNA occurred in parallelwith an increased glomerular expression of TGF-/31 mRNA. Thedemonstration of a close correlation between the PAl-I and
TGF-/31 mRNA levels and the severity of glomerular lesionssuggests that PAl-i as well as TGF-f31 may play an important rolein the progression of lupus-like glomerular lesions, leading toglomerulosclerosis. The lack of PAl-i and TGF-/31 mRNA induc-tion in mice receiving hybridoma cells producing non-nephrito-genic mAb further supports the association of PAl-i and TGF-J31induction with the development and progression of lupus-likeglomerulonephritis.
It is significant that, during the course of lupus-like glomerulo-nephritis, the glomerular expression of the PAl-i gene wasselectively induced without any detectable concomitant changes intPA and uPA gene expression. Since PAT-i is a specific inhibitorof tPA and uPA [36—381, a glomerular overexpression of PAl-imay contribute to the glomerular deposition of fibrin and to the

Molt et at: PAl-i induction in murine lupus nephritis 1467
progressive pathological accumulation of ECM occurring in lupusnephritis, leading to renal failure. Enzymatic analysis furtherconfirmed a decrease of glomerular tPA-catalyzed enzymaticactivity both in acute lupus-like glomerular lesions induced byIgG3 monoclonal cryoglobulins and in lupus-prone mice exhibit-ing severe glomerular lesions.
The glomerular localization of PAl-i mRNA, as indicated by insitu hybridization experiments, suggests that PAl-I may be syn-thesized by inflammatory cells infiltrating glomeruli or by glomer-ular mesangial cells [12] and/or endothelial cells as demonstratedrecently by Keeton et a! in MRL-lpr/lpr mice [39]. Since PAl-iexpression is not detectable in glomeruli under physiologicalconditions, if resident glomerular cells in nephritic lesions are thesites of PAT-i mRNA accumulation, this could be mediated bycytokines locally released by infiltrating PMN and other inflam-matory cells and/or by activated glomerular cells. In this regard,the concomitant increase in glomerular expression of TGF-131 isnoteworthy, since this cytokine is a potent inducer of PAT-ibiosynthesis in vitro and in vivo [22, 24, 25]. It should beemphasized that an increased glomerular expression of TGF-f31mRNA in diseased glomeruli could be detected by in situ hybrid-ization, even when RNase protection assays on total kidney RNAfailed to demonstrate any significant up-regulation in overall renalTGF-j31 mRNA accumulation. This is, for instance, the case formice injected with 1—10B4, AM16 and 7B6.8 hybridoma cells andfor mice sacrificed five days after injection with 6—19 anti-IgG2aRF hybridoma cells. This discrepancy is likely to be related to thefact that glomeruli contain only a minor portion of the overallrenal TGF-/31 mRNA.
That the levels of PAT-i and TGF-J31 mRNAs in mice receiving1—10B4, AMI6 and 7B6.8 mAb are lower than those in 6—19mAb-injected mice is likely to be related to the fact that theformer mAb induce milder glomerular lesions than the 6—19 mAb[14, 15, 28]. This is consistent with the finding that the increases inPAl-i and TGF-131 mRNA levels correlated very well with theseverity of glomerular lesions spontaneously occurring in thethree different lupus-prone mice. Taken together, our resultssuggest a model in which early events in the pathogenetic cascadeof lupus nephritis cause an increase in glomerular TGF-131expression, the cytokine in turn inducing a dramatic local increasein PAl-i production; this would result in an inhibition of prote-olysis and an accumulation of ECM, thereby actively participatingin the process of glomerulosclerosis. This is consistent withobservations from other laboratories in two different experimentalmodels of glomerulonephritis (anti-Thy-I antibody-induced me-sangial proliferative glomerulonephritis and anti-glomerular base-ment membrane antibody-induced glomerulonephritis) [40, 411.
In conclusion, we suggest that an increased expression ofTGF-/31 and PAl-i may be involved in the progression ofglomerular lesions during the course of acute and chronic lupus-like glomerulonephritis. The remarkable induction of PAl-imRNA in association with the development and progression oflupus-like glomerulonephritis does not necessarily imply thatPAl-i is a major factor in the process of generating glomerularsclerotic lesions. The participation of other proteases and/or theirinhibitors cannot be excluded in this process. This question can beanswered in studies on mice deficient in PAl-i [42], which are inprogress in our laboratory. The relative contribution of TGF-pI toglomerulosclerosis and the identification of the cellular sources ofTGF-f31 and PAT-i are other important questions that need to be
answered. Obviously, further elucidation of cellular and molecularevents involved in the progression of glomerulonephritis wouldhelp design new therapeutic approaches for lupus nephritis andother glomerular pathologies.
Acknowledgments
This work was supported by grants from the Swiss National Foundationfor Scientific Research. We thank Ms. Genevieve Leyvraz for histologicalprocessing of kidneys, and Mr. J.C. Rumbeli and Mr. E. Denkinger forphotograph work.
Reprint requests to Shozo Izui, M.D., Department of Pathology, C.M. U.,1211 Geneva 4, Switzerland.
Appendix
Abbreviations are: ECM, extracellular matrix; PA, plasminogen activa-tor; tPA, tissue-type PA; uPA, urokinase; PAT-i, PA inhibitor type 1;TGF-1, transforming growth factor 1; mAb, monoclonal antibody; RF,rheumatoid factor; PAS, periodic acid-Schiff; GAPDH, glyceraldehyde-3-phosphate-dehydrogenase.
References
1. TIPPING PG, DOWLING JP, HOLDSWORTH SR: Glomerular procoagu-lant activity in human proliferative glomerulonephritis. J Clin Invest81:119—125, 1988
2. WIGGINS RC, GLAFFELTER A, BRUCKMAN J: Procoagulant activity inglomeruli and urine of rabbits with nephrotoxic nephritis. Lab Invest53:156—165, 1985
3. BENTJENS JR: Glomerular procoagulant activity and glomerulonephri-tis. Lab Invest 57:107—111, 1987
4. GLAS-GREENWALT P, KANT KS, ALLEN C, POLLAK VE: Fibrinolysis inhealth and disease: Severe abnormalities in systemic lupus erythem-atosus. J Lab Clin Med 104:962—976, 1984
5. AYA N, YOSHIOKA K, MURAKAMI K, HIN0 S, OKADA K, MA-rsuo 0,MAKI 5: Tissue-type plasminogen activator and its inhibitor in humanglomerulonephritis. J Pathol 166:289—295, i992
6. RONDEAU E, MOUGENOT B, LACAVE R, PERALDI MN, KRuITFIOFEKO, SRAER JD: Plasminogen activator inhibitor 1 in renal fibrindeposits of human nephropathies. Clin Nephrol 33:55—60, 1990
7. TOMOOKA 5, BORDER WA, MARSHALL BC, NOBLE NA: Glomerularmatrix accumulation is linked to inhibition of the plasmin proteasesystem. Kidney mt 42:1462—1469, 1992
8. T5UMAGARI T, TANAKA K: Effects of fibrinogen degradation productson glomerular mesangial cells in culture. Kidney mt 26:712—718, 1984
9. VASSALLI P, MCCLUSKEY RT: The pathogenic role of the coagulationin rabbit Masugi nephritis. Am J Pathol 45:653—677, 1964
10. KEETON M, EGUCHI Y, SAWDEY M, AnN C, LOSKUTOFF DJ: Cellularlocalization of type 1 plasminogen activator inhibitor messenger RNAand protein in murine renal tissue. Am J Pathol 142:59—70, 1993
11. SAPPINO AP, HUARTE J, VASSALLI JD, BELIN D: Sites of synthesis ofurokinase and tissue-type plasminogen activators in the murine kid-ney. J Clin Invest 87:962—970, 1991
12. HAGEGE J, PERALDI MN, RONDEAU E, ADIDA C, DELARUE F,MEDCALF R, SCHLEUNING WD, SRAER JD: Plasminogen activatorinhibitor-i deposition in the extracellular matrix of cultured humanmesangial cells. Am J Pathol 141:117—128, 1992
13. MOLL 5, SCHIFFERLI JA, HUARTE J, LEMOINE R, VASSALLI JD,SAPPING AP: LPS induces major changes in the extracellular proteo-lytic balance in the murine kidney. Kidney mt 45:500—508, 1994
14. BERNEY T, FULPIUS T, SHIBATA T, REININGER L, VAN SNICK J, SHANH, WEIGERT M, MARSHAK-ROTHSTEIN A, IzUl 5: Selective pathoge-nicity of murine rheumatoid factors of the clyoprecipitable IgG3subclass. mt Immunol 4:93—99, 1992
15. LEMOINE R, BERNEY T, SHIBATA T, FULPIUS T, GYOTOKU Y, SHIMADAH, SAWADA S, IzUl 5: Induction of "wire-loop" lesions by murinemonoclonal TgG3 cryoglobulins. Kidney mt 41:65—72, 1992

1468 Moll et al: PAl-i induction in murine lupus nephritis
16. FULPIUS T, SPERTINI F, REININGER L, Izui S: Immunoglobulin heavychain constant region determines the pathogenicity and the antigen-binding activity of rheumatoid factor. Proc Nati Acad Sci USA90:2345—2349, 1993
17. ANDREWS BS, EISENBERG RA, THEoFLLor'ouLos AN, Lzui S, WIlsoNCB, MCCONAHEY PJ, MURPHY ED, ROTHS JB, DIXON FJ: Spontane-ous murine lupus-like syndromes. Clinical and immunopathologicalmanifestations in several strains. J Exp Med 148:1198—1215, 1978
18. SHLEEF RR, BEVILACQUA MP, SAWDEY M, GIMBRONE MA JR,LOSKUTOFF DJ: Cytokine activation of vascular endothelium: Effectson tissue-type plasminogen activator and type 1 plasminogen activatorinhibitor. J Biol Chem 263:5797—5803, 1988
19. WESTERHAUSEN DR JR, HOPKINS WE, BILLADELLO JJ: Multipletransforming growth factor-il inducible elements regulate expressionof the plasminogen activator inhibitor type-i gene in Hep G2 cells. JBiol Chem 266:1092—1100, 1991
20. KESKI-OJA J, RAGHOW R, SAWDEW M, LOSKIJTOFF DJ, POSTLE-THWAITE AE, KANG AH, MOSES HL: Regulation of mRNAs for type-iplasminogen activator inhibitor, fibronectin, and type 1 procollagen bytransforming growth factor-fl. J Biol Chem 263:3111—3115, 1988
21. LUND LR, Riccio A, ANDREASEN AP, NIELSEN LS, KRISTENSEN P,LAIHO M, BLASI F, DANO K: Transforming growth factor-fl is a strongand fast acting positive regulator of the level of type-i plasminogenactivator inhibitor in mRNA in WI-38 human lung fibroblasts. EMBOJ 6:1281—1286, 1987
22. LLAIHO M, SAKSELA 0, ANDREASEN PA, KESKI-OJA J: Enhancedproduction and extracellular deposition of the endothelial-type plas-minogen activator inhibitor in cultured human lung fibroblasts bytransforming growth factor-fl. J Cell Biol 103:2403—2410, 1986
23. EDWARDS DR, GILLIAN M, REYNOLDS JJ, WHITMAN SE, DOCHERTYAJP, ANGEL P, HEATH JK: Transforming growth factor beta modu-lates the expression of collagenase and metalloproteinase inhibitor.EMBO J 6:1899—1904, 1987
24. GERWIN BL, KESKI-OJA J, SEDDON M, LECHNER JF, HARRIS CC:TGF-31 modulation of urokinase and PAl-i expression in humanbronchial epithelial cells. Am J Physiol 259:L262-L269, 1990
25. NEWMAN Mi, LANE EA, IANNOTrI AM, NUGENT MA, PEPINSKY RB,KESKI-OJA J: Characterization and purification of a secreted plasmin-ogen activator inhibitor (PAl-i) induced by transforming growthfactor-fl in normal rat kidney (NRK) cells: Decreased PAT-i expres-sion in transformed NRK cells. Endocrinology 126:2936—2946, 1990
26. BORDER WA, OKUDA S, LANGUINO L, SPORN MB, RUOSLAUTI E:Suppression of experimental glomerulonephritis by antiserum againsttransforming growth factor-fl. Nature 346:371—374, 1990
27. YAMAMOTO T, NAKAMURA T, NOBLE NA, RUOSLAHTI E, BORDERWA: Expression of transforming growth factor f3 is elevated in humanand experimental diabetic nephropathy. Proc Natl Acad Sci USA90:1814—1818, 1993
28. TAKAHASHI 5, ITOH J, 0o M, YAMAMOTO T, NOSE M, KYOGOKU M:eDNA sequence analysis of nephritogenic IgG3 monoclonal antibod-
ies derived from an MRL!lpr lupus mouse. Mol Immunol 30:177—182,1993
29. SPERTINI F, COULIE PG, VAN SNICK i, DAVIDSON E, LAMBERT PH,IzUl S: Inhibition of cryoprecipitation of murine IgG3 anti-dinitro-phenyl (DNP) monoclonal antibodies by anionic DNP-amino acidconjugates. Eur J Immunol 19:273—278, 1989
30. PIRANI CL, SALINAS-MADRIGAL L: Evaluation of percutaneous renalbiopsy, in Pathology Annual, edited by SOMMERS SC, New York,Appelton-Century-Crofts, 1968, p 249
31. SABATH DE, BROOME HE, PRYSTOWSKY MB: Glyceraldehyde-3-phosphate dehydrogenase mRNA is a major interleukin 2-inducedtranscript in a cloned T-helper lymphocyte. Gene 91:185—191, 1990
32. SAPPINO AP, BUSSO N, BELIN D, VASSALLI iD: Increase of urokinase-type plasminogen activator gene expression in human lung and breastcarcinomas. Cancer Res 47:4043—4046, 1987
33. BEUN D, WOHLWEND A, SCHLEUNING WD, KRUITHOF EKO, VAS-SALLI iD: Facultative popypeptide translocation allows a singlemRNA to encode the secreted and cytosolic forms of plasminogenactivator inhibitor 2. EMBO J 8:3287—3294, 1989
34. SAPPINO AP, MADANI R, HUARTE i, BELIN D, KISS JZ, WOHLWEND A,VASSALLI JD: Extracellular proteolysis in the adult murine brain. JClin Invest 92:679—685, 1993
35. SAWDEY MS, LOSKUTOFF DJ: Regulation of murine type 1 plasmino-gen activator inhibitor gene expression in vivo. Tissue specificity andinduction by lipopolysaccharide, tumor necrosis factor-a, and trans-forming growth factor-fl. J Clin Invest 88:1346—1353, 1991
36. DANO K, ANDREASEN PA, GRONDHAL-HANSEN i, KRISTENSEN P,NIELSEN LS, SKRIVER L: Plasminogen activators, tissue degradation,and cancer. Adv Cancer Res 44:139—266, 1985
37. HEKMAN CM, LOSKUTOFF Di: Kinetic analysis of the interactionsbetween plasminogen activator inhibitor 1 and both urokinase andtissue plasminogen activator. Arch Biochem Biophys 262:199—210,1988
38. VASSALLI JD, SAPPING AP, BELIN D: The plasminogen activator!plasmin system. J Clin Invest 88:1067—1072, 1988
39. KEETON M, AHN C, EGUCHI Y, BURLINGAME R, LOSKUTOFF Di:Expression of type 1 plasminogen activator inhibitor in renal tissue inmurine lupus nephritis. Kidney mt 47:148—157, 1995
40. OKUDA 5, LANGUINO LR, RUOSLAHTI E, BORDER WA: Elevatedexpression of transforming growth factor-fl and proteoglycan produc-tion in experimental glomerulonephritis. J Clin Invest 86:453—462,1990
41. FENG L, TANG WW, LOSKUTOFF Di, WILSON CB: Dysfunction ofglomerular fibrinolysis in experimental antiglomerular basementmembrane antibody glomerulonephritis. JASN 3:1753—1764, 1993
42. CARMELIET P, KIECKENS L, SCHOONJANS L, REAM B, VAN NUFFELENA, PAENDERGAST G, COLE M, BRONSON R, COLEN D, MULLIGAN RC:Plasminogen activator inhibitor-i gene-deficient mice. J Clin Invest92:2746—2755, 1993





























