Embed Size (px)
Citation preview

C A R B O N 4 9 ( 2 0 1 1 ) 4 0 4 0 – 4 0 4 9
. sc iencedi rec t .com
avai lab le at wwwjournal homepage: www.elsev ier .com/ locate /carbon
In vitro and in vivo behaviors of dextran functionalizedgraphene
Shuai Zhang 1, Kai Yang 1, Liangzhu Feng 1, Zhuang Liu *
Jiangsu Key Laboratory for Carbon-Based Functional Materials and Devices, Institute of Functional Nano and Soft Materials,
Soochow University, Suzhou, Jiangsu 215123, China
A R T I C L E I N F O
Article history:
Received 27 February 2011
Accepted 30 May 2011
Available online 2 June 2011
0008-6223/$ - see front matter � 2011 Elsevidoi:10.1016/j.carbon.2011.05.056
* Corresponding author: Fax: +86 512 6588284E-mail address: [email protected] (Z. Liu)
1 These authors contributed equally to this
A B S T R A C T
Development of biocompatible surface coating is critical to engineer various functional
nanomaterials for biomedical applications. Here, we present a new surface chemistry of
graphene by covalently conjugating graphene oxide (GO) with dextran (DEX), a biocompat-
ible polymer widely used for surface coating of biomaterials. Compared with GO, the
graphene–dextran (GO–DEX) conjugate shows reduced sheet sizes, increased thickness
and significantly improved stability in physiological solutions. Cellular experiments
uncover that DEX coating on GO offers remarkably reduced cell toxicity. We further label
GO–DEX with a radioactive isotope, 125I, for in vivo tracking in animal studies. It is found
that GO–DEX accumulates in the reticuloendothelial system (RES) including liver and
spleen after intravenous injection, and importantly, shows obvious clearance from the
mouse body within a week without causing noticeable short-term toxicity to the treated
animals. Our results suggest that this DEX coating method on GO may potentially be useful
to the further development of novel graphene-based bioconjugates for various biomedical
applications.
� 2011 Elsevier Ltd. All rights reserved.
1. Introduction
Graphene with unique two-dimensional structures, fascinat-
ing electronic, physical and chemical properties has shown
promise in a wide range of fields including electronic devices,
solar cells, nano-catalysts, as well as chemical and biological
sensors [1–11]. Recently, tremendous attentions have been
paid to the graphene based nanomedicine for biological
detection, drug delivery, and cancer therapies [12–15]. In the
past three years, numerous publications have reported
graphene-based novel biosensors by utilizing the interesting
chemical, optical, electrical, and electrochemical properties
of graphene for detection of various biomolecules with high
sensitivities [11,14,15]. Monolayer graphene with all atoms
exposed on its surface exhibits a huge surface area (theoreti-
cally 2600 m2/g), which can be utilized for ultra-efficient
er Ltd. All rights reserved
6..
work.
loading of aromatic molecules such as anti-cancer drugs,
useful for applications in drug and gene delivery [16–21]. In
a recent work by our group, polyethylene glycol (PEG) func-
tionalized nano-graphene with strong optical absorption in
the near-infrared region (NIR) and was used for in vivo photo-
thermal therapy of cancer, showing excellent tumor ablation
effect in a mouse model [7]. Although still at its infant stage,
the graphene-based nanomedicine appears to have great
potential in a number of different directions.
It is well known that the surface chemistry of nanomateri-
als is highly important to their behaviors and applications in
biological systems. Several latest reports uncovered that as-
prepared GO exhibited dose-dependent toxicity in vitro to
cells [21]. After intravenous injection, GO would dominantly
accumulated in lungs of mice over long periods of time,
inducing obvious pulmonary toxicity [22,23]. Coating of
.

C A R B O N 4 9 ( 2 0 1 1 ) 4 0 4 0 – 4 0 4 9 4041
biocompatible polymers on graphene may help to circumvent
these problems. Our recent work showed that PEGylated
nano-graphene sheets with sizes smaller than 50 nm (10–
30 nm) could be cleared out from the mouse body after intra-
venous injection, without rendering noticeable toxicity to the
treated animals. Although PEG has been used to functionalize
GO for several in vitro and in vivo studies [7,8,24,25], efforts
are still needed to develop new biocompatible surface coat-
ings of graphene for different applications in biomedicine.
DEX is a hydrophilic natural polymer extensively used to
functionalize many nanomaterials such as iron oxide, quan-
tum dots, single walled carbon nanotubes and nanodiamond,
to improve their stability, biocompatibility, pharmacokinetics,
and thus biomedical functions [26–31]. Different from PEG
with linear structure, DEX in aqueous phase exhibits spheri-
cal shape which may help to better coat nanomaterials. More-
over, DEX is a natural polymer that can be fully degraded in
living biological systems [32–35]. Herein, we report a new sur-
face coating of GO by functionalized DEX, and test the solubil-
ity of the yielded GO–DEX in physiological solutions in
comparison to non-functionalized GO. It is found that GO–
DEX is able to enter cells, without showing significant inter-
ference to the cell growth. Using radioactive 125I-labeled GO–
DEX (125I–GO–DEX), we further study the in vivo pharmacoki-
netics and biodistribution of GO–DEX in mice, observing high
uptake of GO–DEX in the mouse liver and spleen together
with neglectable accumulation in the lung. Both radiolabel-
ing-based biodistribution data and liver slice images indicate
the significant clearance of graphene from the mouse organs,
strongly favorable for future biomedical applications.
2. Experimental section
2.1. Chemical materials
Graphite powder was purchased from Hua Dong Graphite Man-
ufactory, China. Dextran (Mw = 8000–12,000 Da) was obtained
from SERVA. Sodium periodate, 3-(4,5-dimethylthiazol-2-yl)-
2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide
(DMSO), 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide
hydrochloride (EDC) and sodium cyanoborohydride were pur-
chased from Sigma–Aldrich. Cy5-NHS was purchased from
Fanbo Chemicals Co. Ltd. Ethanediamine and sodium azide
were obtained from Sinopharm Chemical Reagent Co. Ltd.
Na125I was obtained from Chengdu nuclear isotope Qualcomm
Inc. Ultra-filter tubes with 100 kD and 10 kD molecular weight
cut off (MWCO) were purchased from Millipore.
2.2. Synthesis of GO
GO was prepared from graphite following a modified Hum-
mers’ method [7,36]. The as-prepared material was stirred
in the presence of sodium hydroxide for 2 h at 50 �C. After
adjusting the solution pH to 1 by hydrochloric acid, the sus-
pension was centrifuged at 8000g for 5 min and washed by
distilled (DI) water several times to remove excess acid and
salts. The GO suspension became rather stable afterwards
and was then centrifuged at 10,000g for 5 min to remove
any insoluble aggregates, leaving based treated GO (GO-b) in
the supernatant for future use.
2.3. Synthesis of dextran amine (DEX-NH2)
DEX with a molecular weight of 8000–12,000 Da was dissolved
in DI water at a concentration of 0.2 g/ml. Sodium periodate
was then added in dark to break the glycol C–C bond to two
aldehyde groups. The mole ratios of sodium periodate to dex-
tran monomers were 2.5%, 5%, 10% and 20%. After dialysis
against water for 48 h to remove excess sodium periodate,
the oxidized dextran was frozen dried. To introduce amino
groups into dextran, oxidized dextran (0.2 g) in 15 mL DI water
was mixed with excessive ethanediamine (48 mg) and 0.5 mL
sodium cyanoborohydride (caution!) (5 mol/L) alkaline solu-
tion at pH 14 for 24 h. After removing small molecules by dial-
ysis, the final product was lyophilized and stored at �20 �C.
Oxidized dextran and DEX-NH2 were characterized to
determine the aldehyde and amino contents, respectively.
The content of aldehyde in oxidized dextran was measured
by a classical redox titration method. In this method, excess
sodium bisulfate was reacted with aldehyde groups on dex-
tran, with leftover free sodium bisulfate expended by iodime-
try. One gram of sodium bicarbonate was then added into the
solution to release bisulfate conjugated to aldehyde groups.
Iodimetry was used again to measure the concentration of so-
dium bisulfate, which was equivalent to that of aldehyde
groups. The nitrogen concentration of DEX-NH2 was mea-
sured by a standard Kjeldahl assay to determine the ratios
of amino groups in various DEX-NH2 samples [37,38].
2.4. Synthesis of GO–DEX
The protocol to synthesize GO–DEX was similar to that of
PEGylated GO synthesis reported earlier [7]. 1 mL GO solution
(2 mg/mL) dispersed in 4 mL DI water was first mixed with
�15 mg DEX-NH2 in 400 lL DI water and sonciated for 5 min.
1 mg EDC dissolved in 100 lL DI water was then added into
the suspension instantly. After 30 min reaction under sonica-
tion, another 2.5 mg EDC was added afterwards. After 6 h
reaction under stirring, the suspension was filtrated through
a MWCO 100 kDa centrifugal filter (Millipore) several times
to completely remove excess EDC and dextran amine. Finally,
the prepared GO–DEX was dispersed in DI water at the con-
centration of 2 mg/mL.
2.5. Fluorescent labeling of GO–DEX
To fluorescently label GO–DEX, 50 lg Cy5-NHS in 20 lL DMSO
was added into 500 lL GO–DEX (2 mg/mL) in phosphate buf-
fered saline (PBS) solution and stirred for a few seconds.
The amine reactive N-hydroxysuccinimide (NHS) ester in
Cy5-NHS would react with the leftover amino groups on the
DEX-NH2 coating of the GO–DEX conjugate. After 12 h reac-
tion in dark, excess fluorescent dyes were removed by filtra-
tion via 100 kDa centrifugal filters.
2.6. Cellular experiments
HeLa cells were cultured in standard Dulbecco’s modified ea-
gle medium (DMEM) supplemented with 10% fetal bovine ser-
um (FBS) and 1% penicillin–streptomycin solution under
37 �C. For cell proliferation measurement, HeLa cells were

4042 C A R B O N 4 9 ( 2 0 1 1 ) 4 0 4 0 – 4 0 4 9
seeded into 6-well plates at the density of 2 · 105 per well. Dif-
ferent concentrations (10 mg/L, 50 mg/L, 200 mg/L) of GO and
GO–DEX were then added into the cells. Doxorubicin (DOX) at
the concentration of 20 mg/L was used as the positive control.
Cells were detached from the plates and collected for cell
number counting after 24, 48, and 72 h of incubation. Dead
cells were excluded by trypan blue staining.
For confocal fluorescence imaging experiments, HeLa cells
cultured in 35 mm dishes were added with 10 lL Cy5 labeled
GO–DEX (2 mg/ml) and incubated at different temperature
(4 �C and 37 �C) for 2 h. After washing with PBS for several
times, cells were then imaged by a laser scanning confocal
fluorescence microscope (Leica TCS SP5). The excitation
wavelength was 633 nm and the emission band is between
660 and 680 nm. All the images were taken under the same
instrumental condition.
The Calcein AM/propidium iodide (PI) staining experiment
was carried out following a classic method. Briefly, Hela cells
were seeded in 35 mm culture dishes overnight. GO and GO–
DEX solutions at concentrations of 50 mg/L and 200 mg/L (fi-
nal concentrations in the cell medium) were added into cells.
Doxorubicin (DOX) at the concentration of 20 mg/L was added
as the positive control. After incubation for various periods of
time, the cells were washed with PBS and stained with 1 mL
PBS solution containing Calcein-AM (0.3 mg/L) and PI
(0.5 mg/L). Confocal fluorescence images were taken 20 min
after staining.
2.7. Histology examination
Healthy female Balb/c mice were injected with 200 ll of 2 mg/
ml GO–DEX per mouse (a dose of 20 mg/kg) and sacrificed at
various time points after injection (1 day, 3 and 7 days). Major
organs were then taken and fixed in 4% neutral buffered for-
malin, processed routinely into paraffin, sectioned at 8 lm,
stained with hematoxylin and eosin (H&E) and examined by
a digital microscope (Leica QWin). Examined tissues include
liver, spleen, kidney and lung.
2.8. Radioactive labeling of GO–DEX
The radioactive labeling of GO–DEX was carried out by a chlo-
ramine-T oxidation method [25,39]. 500 lL GO–DEX solution
(2 mg/mL), �800 lCi Na125I and 100 lL chloramine-T (4 mg/
mL) were mixed together in a phosphate buffer (pH �7.5). Ex-
cess Na125I and chloramine-Twere removed by filtration with
100 kDa centrifugal filters. The suspension was washed 4–6
times with DI water until no detectable gamma radioactivity
in the filtrate. The radiolabeling yield was about 40–45%. As
a control experiment, GO–DEX and Na125I were mixed to-
gether without chloramine-T. Almost no gamma radioactivity
signal could be detected in the GO–DEX suspension after the
same washing procedures.
2.9. Radiolabeling stability test
10 lL 125I labeled GO–DEX was added into 200 lL mouse plas-
ma (collected from a healthy Balb/c mouse) and incubated in
a 37 �C water bath. After incubation for different periods of
time, 10 lL of the mixture was added into 1 mL DI water.
The solution was filtered by a centrifugal filter (MWCO =
10 kD) and washed with water. The filtrate was collected
and measured by a Gamma counter (Science and Technology
Institute of China in Jia Branch Innovation Co., Ltd.) to detect
the released radioactive iodine.
2.10. Blood circulation and biodistribution of radioactivelabeled GO–DEX
To study the blood circulation and biodistribution of GO–DEX,125I–GO–DEX was injected via tail veins into healthy female
Balb/c mice. Blood was drawn at different time points after
injection and measured by the Gamma Counter. For biodistri-
bution study, mice were sacrificed 4, 24, 72 and 168 h after
injection. Major organs were collected and wet weighted for
radioactivity measurement.
3. Results and discussion
The procedure of converting dextran to DEX-NH2 and conju-
gating GO to DEX-NH2 was shown in Fig. 1 To synthesize
DEX-NH2, the glycol bond in a dextran monomer was oxidized
by sodium periodate, giving two aldehyde groups which were
available for conjugation to ethanediamine by reductive ami-
nation. Excess ethanediamine was added into the solution of
oxidized dextran, forming a Schiff base between the amino
group in ethanediamine and the aldehyde group in oxidized
dextran. The Schiff base could then be reduced by a middling
reducing agent, sodium cyanoborohydride, transforming the
C@N bond into a more stable C–N bond. The measured aver-
age numbers of aldehyde and amino groups per oxidized dex-
tran and DEX-NH2, respectively, in the four samples were
presented in Table 1.
We then conjugated DEX-NH2 with different amine ratios
to GO. It was found that the synthesis yield of GO–DEX (after
reaction and the followed centrifugation to remove unstable
aggregates) increased as the raise of nitrogen contents in
DEX-NH2 (Fig. 2a), and reached to the optimized yield when
the DEX-NH2 with 1.20% N was used. Further increase in the
nitrogen content of DEX-NH2 resulted in slightly decreased
GO–DEX yield, likely due to the heavy oxidization that dam-
aged the dextran structure. The DEX-NH2 sample with 1.20%
nitrogen composition (4.28 amino groups per dextran) was
thus chosen for the following experiments. Compared to
non-coated GO which precipitated in the presence of salts
and was unstable in various biological solutions including sal-
ine, serum and cell medium, GO–DEX was rather stable in the
above solutions without obvious agglomeration (Fig. 2b). The
UV–vis absorption spectra of GO, base treated GO (GO-b),
and GO–DEX were recorded (Fig. 2c), showing a similar
absorption peak at 230–240 nm, which was originated from
the p-plasmon of carbon [40,41]. Interestingly, the optical den-
sity of GO-b and GO–DEX in the NIR region significantly in-
creased by 3.76 and 18.6 folds at 800 nm, compared to the
as-prepared GO. We attributed the increase of absorbance to
the base treatment and DEX conjugation, which would de-
crease the surface flaw by breaking epoxide and ester groups,
releasing the local strain and increasing the p-plasmon of car-
bon, that would raise the absorbance of GO-b and GO–DEX in
the visible-NIR range [8,42,43].
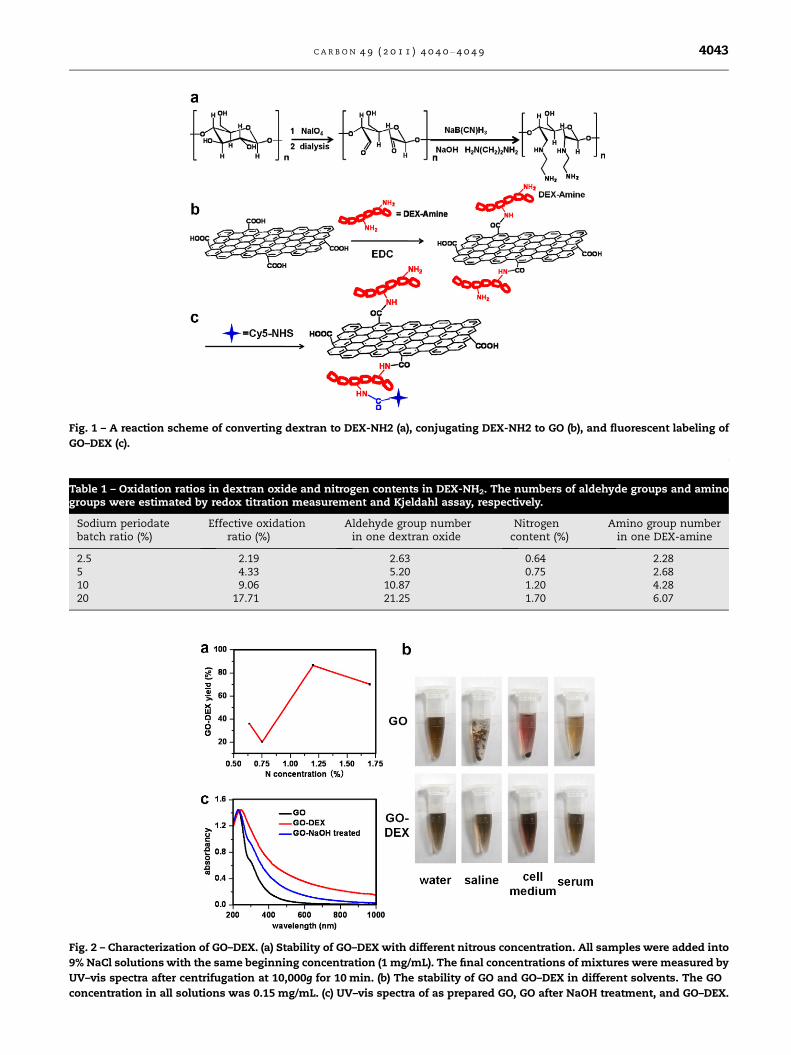
Fig. 1 – A reaction scheme of converting dextran to DEX-NH2 (a), conjugating DEX-NH2 to GO (b), and fluorescent labeling of
GO–DEX (c).
Table 1 – Oxidation ratios in dextran oxide and nitrogen contents in DEX-NH2. The numbers of aldehyde groups and aminogroups were estimated by redox titration measurement and Kjeldahl assay, respectively.
Sodium periodatebatch ratio (%)
Effective oxidationratio (%)
Aldehyde group numberin one dextran oxide
Nitrogencontent (%)
Amino group numberin one DEX-amine
2.5 2.19 2.63 0.64 2.285 4.33 5.20 0.75 2.6810 9.06 10.87 1.20 4.2820 17.71 21.25 1.70 6.07
Fig. 2 – Characterization of GO–DEX. (a) Stability of GO–DEX with different nitrous concentration. All samples were added into
9% NaCl solutions with the same beginning concentration (1 mg/mL). The final concentrations of mixtures were measured by
UV–vis spectra after centrifugation at 10,000g for 10 min. (b) The stability of GO and GO–DEX in different solvents. The GO
concentration in all solutions was 0.15 mg/mL. (c) UV–vis spectra of as prepared GO, GO after NaOH treatment, and GO–DEX.
C A R B O N 4 9 ( 2 0 1 1 ) 4 0 4 0 – 4 0 4 9 4043
4044 C A R B O N 4 9 ( 2 0 1 1 ) 4 0 4 0 – 4 0 4 9
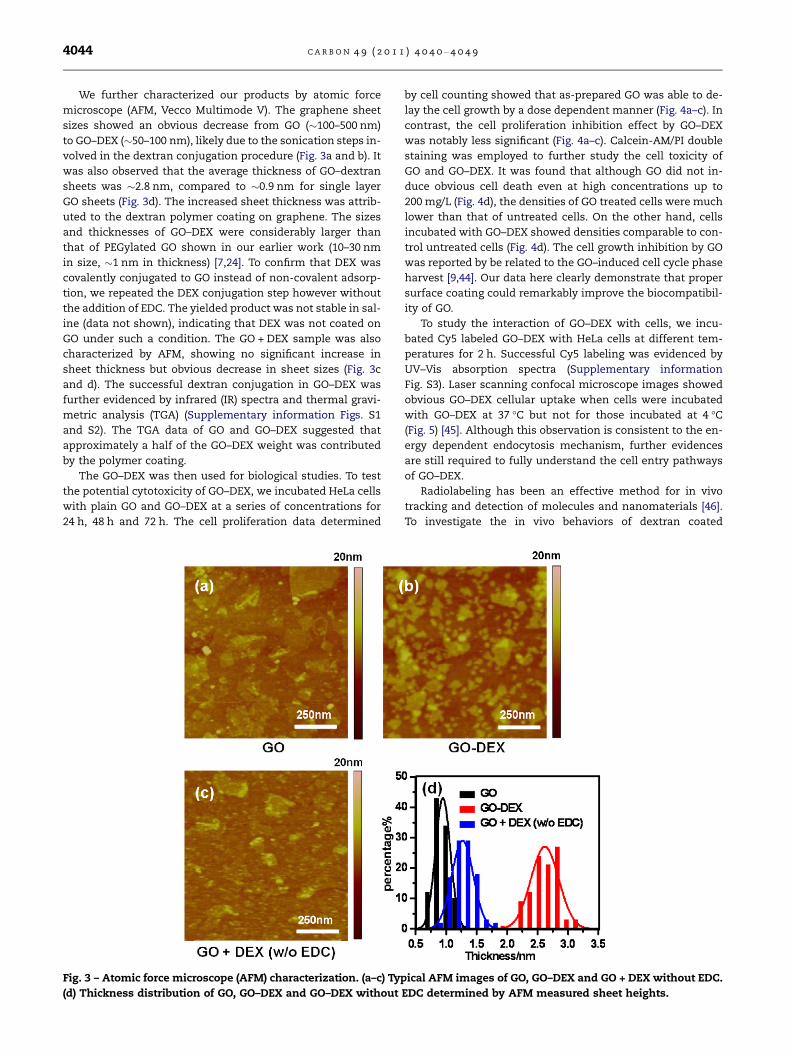
We further characterized our products by atomic force
microscope (AFM, Vecco Multimode V). The graphene sheet
sizes showed an obvious decrease from GO (�100–500 nm)
to GO–DEX (�50–100 nm), likely due to the sonication steps in-
volved in the dextran conjugation procedure (Fig. 3a and b). It
was also observed that the average thickness of GO–dextran
sheets was �2.8 nm, compared to �0.9 nm for single layer
GO sheets (Fig. 3d). The increased sheet thickness was attrib-
uted to the dextran polymer coating on graphene. The sizes
and thicknesses of GO–DEX were considerably larger than
that of PEGylated GO shown in our earlier work (10–30 nm
in size, �1 nm in thickness) [7,24]. To confirm that DEX was
covalently conjugated to GO instead of non-covalent adsorp-
tion, we repeated the DEX conjugation step however without
the addition of EDC. The yielded product was not stable in sal-
ine (data not shown), indicating that DEX was not coated on
GO under such a condition. The GO + DEX sample was also
characterized by AFM, showing no significant increase in
sheet thickness but obvious decrease in sheet sizes (Fig. 3c
and d). The successful dextran conjugation in GO–DEX was
further evidenced by infrared (IR) spectra and thermal gravi-
metric analysis (TGA) (Supplementary information Figs. S1
and S2). The TGA data of GO and GO–DEX suggested that
approximately a half of the GO–DEX weight was contributed
by the polymer coating.
The GO–DEX was then used for biological studies. To test
the potential cytotoxicity of GO–DEX, we incubated HeLa cells
with plain GO and GO–DEX at a series of concentrations for
24 h, 48 h and 72 h. The cell proliferation data determined
Fig. 3 – Atomic force microscope (AFM) characterization. (a–c) Typ
(d) Thickness distribution of GO, GO–DEX and GO–DEX without
by cell counting showed that as-prepared GO was able to de-
lay the cell growth by a dose dependent manner (Fig. 4a–c). In
contrast, the cell proliferation inhibition effect by GO–DEX
was notably less significant (Fig. 4a–c). Calcein-AM/PI double
staining was employed to further study the cell toxicity of
GO and GO–DEX. It was found that although GO did not in-
duce obvious cell death even at high concentrations up to
200 mg/L (Fig. 4d), the densities of GO treated cells were much
lower than that of untreated cells. On the other hand, cells
incubated with GO–DEX showed densities comparable to con-
trol untreated cells (Fig. 4d). The cell growth inhibition by GO
was reported by be related to the GO–induced cell cycle phase
harvest [9,44]. Our data here clearly demonstrate that proper
surface coating could remarkably improve the biocompatibil-
ity of GO.
To study the interaction of GO–DEX with cells, we incu-
bated Cy5 labeled GO–DEX with HeLa cells at different tem-
peratures for 2 h. Successful Cy5 labeling was evidenced by
UV–Vis absorption spectra (Supplementary information
Fig. S3). Laser scanning confocal microscope images showed
obvious GO–DEX cellular uptake when cells were incubated
with GO–DEX at 37 �C but not for those incubated at 4 �C(Fig. 5) [45]. Although this observation is consistent to the en-
ergy dependent endocytosis mechanism, further evidences
are still required to fully understand the cell entry pathways
of GO–DEX.
Radiolabeling has been an effective method for in vivo
tracking and detection of molecules and nanomaterials [46].
To investigate the in vivo behaviors of dextran coated
ical AFM images of GO, GO–DEX and GO + DEX without EDC.
EDC determined by AFM measured sheet heights.
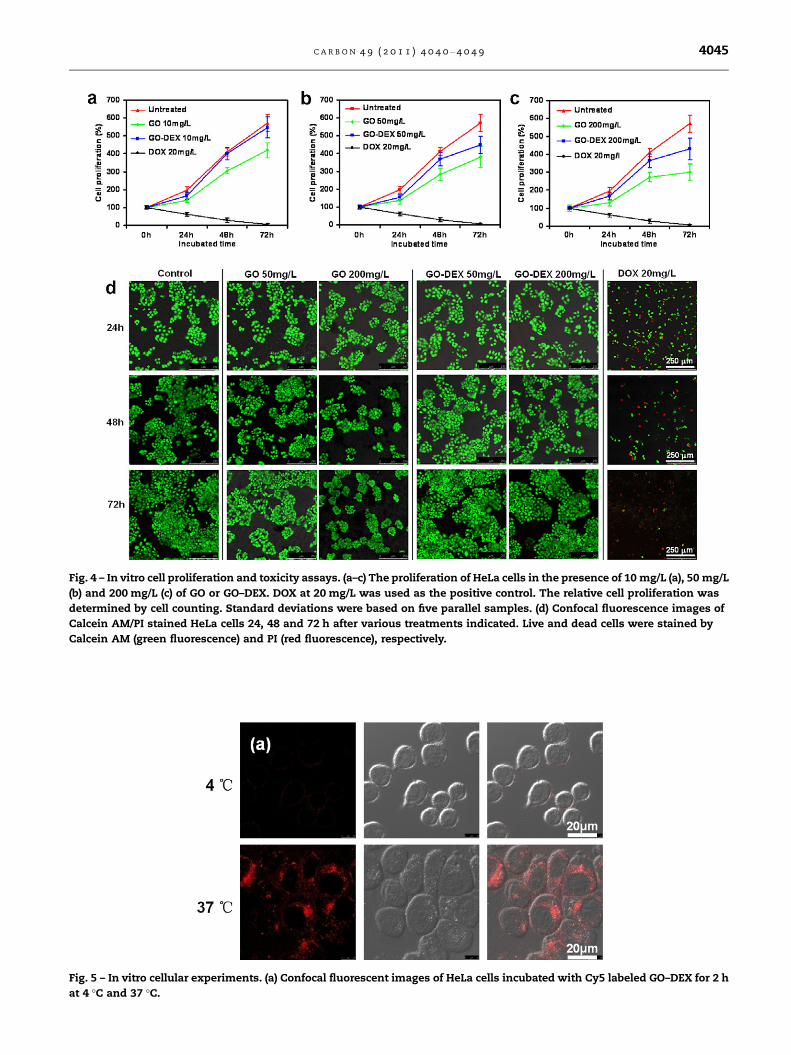
Fig. 4 – In vitro cell proliferation and toxicity assays. (a–c) The proliferation of HeLa cells in the presence of 10 mg/L (a), 50 mg/L
(b) and 200 mg/L (c) of GO or GO–DEX. DOX at 20 mg/L was used as the positive control. The relative cell proliferation was
determined by cell counting. Standard deviations were based on five parallel samples. (d) Confocal fluorescence images of
Calcein AM/PI stained HeLa cells 24, 48 and 72 h after various treatments indicated. Live and dead cells were stained by
Calcein AM (green fluorescence) and PI (red fluorescence), respectively.
Fig. 5 – In vitro cellular experiments. (a) Confocal fluorescent images of HeLa cells incubated with Cy5 labeled GO–DEX for 2 h
at 4 �C and 37 �C.
C A R B O N 4 9 ( 2 0 1 1 ) 4 0 4 0 – 4 0 4 9 4045
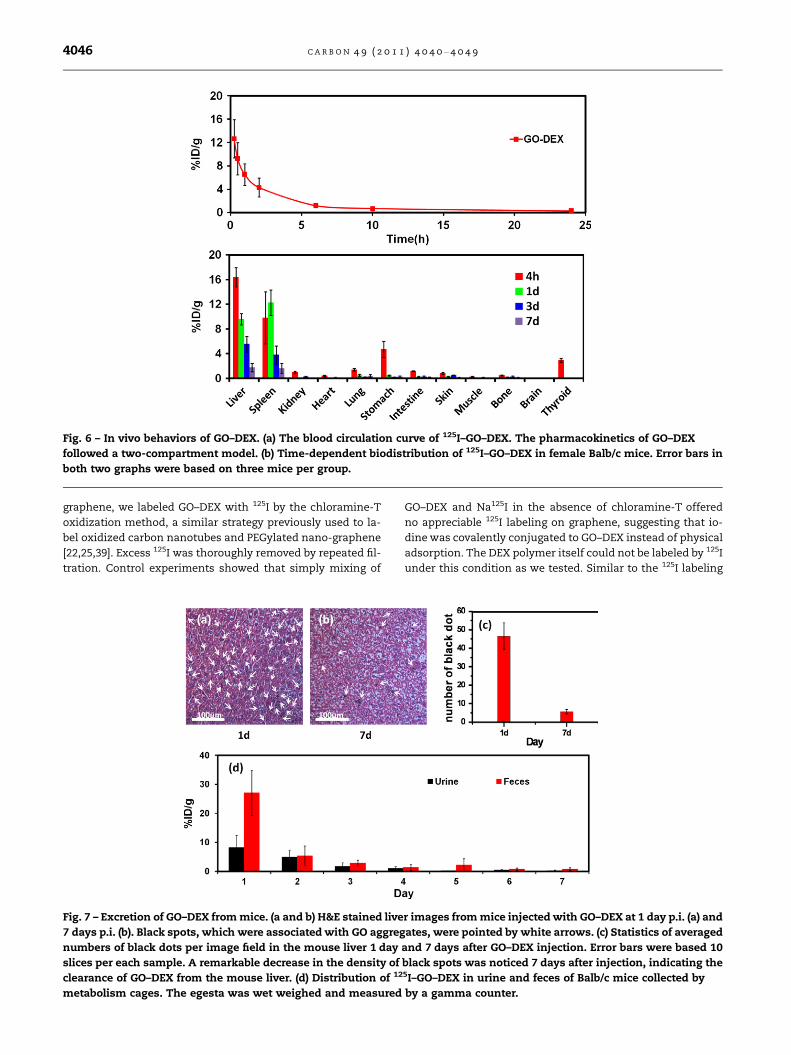
Fig. 6 – In vivo behaviors of GO–DEX. (a) The blood circulation curve of 125I–GO–DEX. The pharmacokinetics of GO–DEX
followed a two-compartment model. (b) Time-dependent biodistribution of 125I–GO–DEX in female Balb/c mice. Error bars in
both two graphs were based on three mice per group.
4046 C A R B O N 4 9 ( 2 0 1 1 ) 4 0 4 0 – 4 0 4 9
graphene, we labeled GO–DEX with 125I by the chloramine-T
oxidization method, a similar strategy previously used to la-
bel oxidized carbon nanotubes and PEGylated nano-graphene
[22,25,39]. Excess 125I was thoroughly removed by repeated fil-
tration. Control experiments showed that simply mixing of
Fig. 7 – Excretion of GO–DEX from mice. (a and b) H&E stained live
7 days p.i. (b). Black spots, which were associated with GO aggreg
numbers of black dots per image field in the mouse liver 1 day
slices per each sample. A remarkable decrease in the density of
clearance of GO–DEX from the mouse liver. (d) Distribution of 12
metabolism cages. The egesta was wet weighed and measured
GO–DEX and Na125I in the absence of chloramine-T offered
no appreciable 125I labeling on graphene, suggesting that io-
dine was covalently conjugated to GO–DEX instead of physical
adsorption. The DEX polymer itself could not be labeled by 125I
under this condition as we tested. Similar to the 125I labeling
r images from mice injected with GO–DEX at 1 day p.i. (a) and
ates, were pointed by white arrows. (c) Statistics of averaged
and 7 days after GO–DEX injection. Error bars were based 10
black spots was noticed 7 days after injection, indicating the5I–GO–DEX in urine and feces of Balb/c mice collected by
by a gamma counter.
C A R B O N 4 9 ( 2 0 1 1 ) 4 0 4 0 – 4 0 4 9 4047
of oxidized carbon nanotubes [25,39], 125I was likely conju-
gated to the defect sites and dangling bonds of GO. The stabil-
ity of 125I labeled GO–DEX was tested in Balb/c mouse serum,
showing only a small degree of 125I detachment within 7 days
at 37 �C (Supplementary information Fig. S4).
To study the in vivo pharmacokinetics of GO–DEX, we
intravenously injected 100 ll of 125I–GO–DEX (4 mg/kg,
20 lCi) into mice. Blood was drawn at different time points
post injection (p.i.) and measured by a gamma counter.
(Fig. 6a) The blood circulation curve showed that the pharma-
cokinetics of GO–DEX followed a two-compartment model,
with first and second phases of half-lives at 0.19 ± 0.03 h
and 1.81 ± 0.17 h, respectively. Mice were sacrificed at 4, 24,
72 and 168 h for biodistribution study. It was found that125I–GO–DEX distributed in several organs including liver,
spleen, stomach, lung, kidney and intestine at 4 h, and
showed dominated RES uptake in liver and spleen at later
time points with neglectable lung accumulation (Fig. 6b). A
low thyroid uptake was observed only at 4 h and not detect-
able at later time points, suggesting the decent stability of
the 125I radiolabeling in the mouse body. Decreased signals
with elapsing time indicated the possible clearance of GO–
DEX in body. Interestingly, the excretion of GO–DEX appears
to be slightly faster compared to that of GO–PEG observed in
our previous study [25]. While �4% ID/g and �6% ID/g of
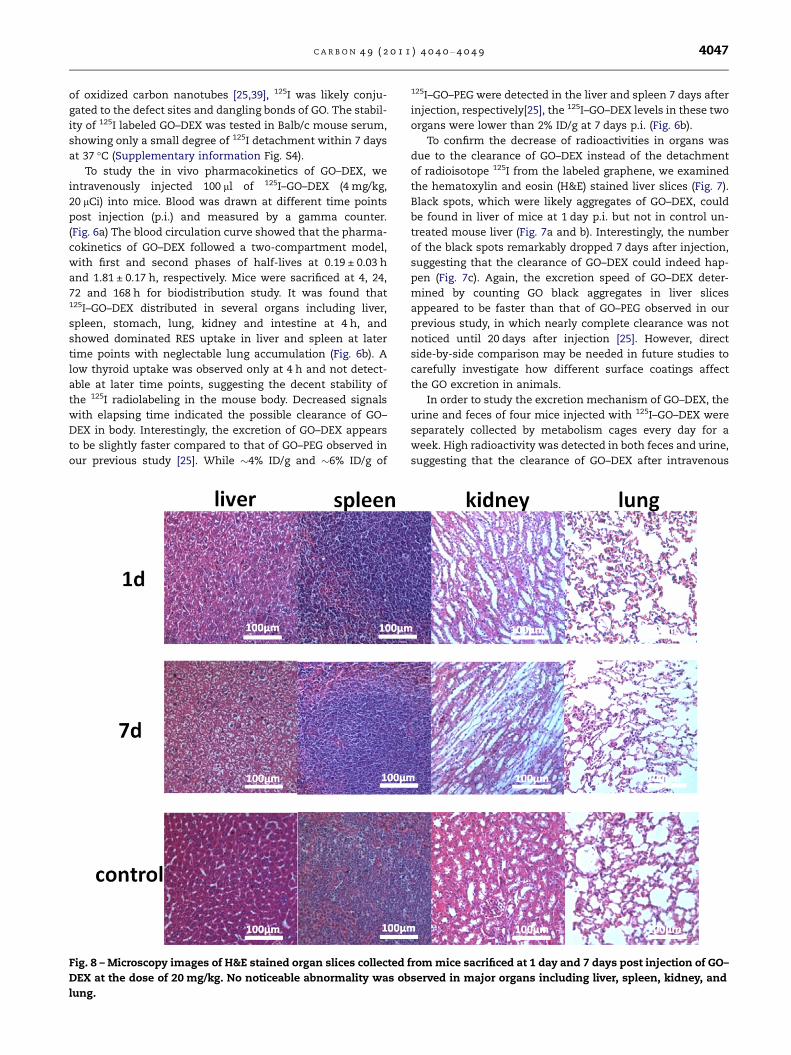
Fig. 8 – Microscopy images of H&E stained organ slices collected
DEX at the dose of 20 mg/kg. No noticeable abnormality was ob
lung.
125I–GO–PEG were detected in the liver and spleen 7 days after
injection, respectively[25], the 125I–GO–DEX levels in these two
organs were lower than 2% ID/g at 7 days p.i. (Fig. 6b).
To confirm the decrease of radioactivities in organs was
due to the clearance of GO–DEX instead of the detachment
of radioisotope 125I from the labeled graphene, we examined
the hematoxylin and eosin (H&E) stained liver slices (Fig. 7).
Black spots, which were likely aggregates of GO–DEX, could
be found in liver of mice at 1 day p.i. but not in control un-
treated mouse liver (Fig. 7a and b). Interestingly, the number
of the black spots remarkably dropped 7 days after injection,
suggesting that the clearance of GO–DEX could indeed hap-
pen (Fig. 7c). Again, the excretion speed of GO–DEX deter-
mined by counting GO black aggregates in liver slices
appeared to be faster than that of GO–PEG observed in our
previous study, in which nearly complete clearance was not
noticed until 20 days after injection [25]. However, direct
side-by-side comparison may be needed in future studies to
carefully investigate how different surface coatings affect
the GO excretion in animals.
In order to study the excretion mechanism of GO–DEX, the
urine and feces of four mice injected with 125I–GO–DEX were
separately collected by metabolism cages every day for a
week. High radioactivity was detected in both feces and urine,
suggesting that the clearance of GO–DEX after intravenous
from mice sacrificed at 1 day and 7 days post injection of GO–
served in major organs including liver, spleen, kidney, and

4048 C A R B O N 4 9 ( 2 0 1 1 ) 4 0 4 0 – 4 0 4 9
injection could take place in both renal and fecal pathways
(Fig. 7d). Since GO–DEX has a wide size distribution, a small
fraction of very small GO–DEX sheets may pass the glomeru-
lus for renal excretion, while the majority of GO–DEX
accumulated in RES organs could be cleared out in feces via
the biliary pathway. It worth noting that compared with
GO–PEG (sheet size 10–50 nm) in our previous work [25], nota-
bly higher portion of GO–DEX was excreted by feces than ur-
ine, likely owing to the increased sheet sizes of GO–DEX (50–
100 nm).
Mice injected GO–DEX at the dose of 20 mg/kg were sacri-
ficed at 1 day and 7 days p.i. Major organs were harvested and
sectioned into thin slices for H&E staining (Fig. 8). No obvious
abnormality was found in various organs including the lung
of the GO–DEX injected mice, in marked contrast to the obvi-
ous pulmonary toxicity induced by the as-prepared GO re-
cently observed in earlier studies [23]. Although further
careful long-term studies are needed to fully illustrate the
toxicology profiles of GO–DEX, our preliminary data here
showed that the dextran coating could offer GO improved bio-
compatibility. However, future experiments are still required
to understand the long-term fate, excretion pathways, and
toxicology of dextran functionalized GO in animals.
4. Conclusions
We develop a new graphene surface coating method using a
biocompatible polymer, dextran. After covalent conjugation
of dextran, the GO–DEX shows excellent stability in physio-
logical solutions. Compared to plain unfunctionalized GO,
GO–DEX exhibits remarkably improved in vitro biocompatibil-
ity with notably reduced cell growth inhibition effects. Radio-
labeling of GO–DEX by 125I enables tracking and detection of
graphene in animals. After being intravenously injected into
mice, GO–DEX is cleared from the circulating blood in a few
hours and accumulates in RES organs including liver and
spleen, in which the GO–DEX levels show a gradual decrease
over time with the majority excreted within a week. Different
from as-prepared GO, which is primarily trapped in the lung
after intravenous injection and causes obvious pulmonary
toxicity [22,23], neglectable lung uptake and no noticeable
in vivo short-term toxicity is found in GO–DEX treated mice.
Our work provides an alternative functionalization method
to produce biocompatible graphene bioconjugates for poten-
tial biomedical applications. However, the in vivo long-term
fate and toxicology of dextran coated graphene still require
further investigations. Using of GO–DEX for actual bio-appli-
cations such as drug delivery and bio-sensing is currently un-
der-going in our laboratory.
Acknowledgements
This work was partially supported by the National Natural
Science Foundation of China (51002100), a National ‘‘973’’ Pro-
gram of China (2011CB911002), and A Project Funded by the
Priority Academic Program Development of Jiangsu Higher
Education Institutions. We thank Dr. Jianmei Wan and Prof.
Youjiu Zhang in the Medical School of Soochow University
for their great help on radioactive experiments.
Appendix A. Supplementary data
Supplementary data associated with this article can be found,
in the online version, at doi:10.1016/j.carbon.2011.05.056.
R E F E R E N C E S
[1] Geim AK. Graphene: status and prospects. Science2009;324(5934):1530–4.
[2] Allen MJ, Tung VC, Kaner RB. Honeycomb carbon: a review ofgraphene. Chem Rev 2010;110(1):132–45.
[3] Hong WJ, Bai H, Xu YX, Yao ZY, Gu ZZ, Shi GQ. Preparation ofgold nanoparticle/graphene composites with controlledweight contents and their application in biosensors. J PhysChem C 2010;114(4):1822–6.
[4] Tang LAL, Wang JZ, Loh KP. Graphene-based SELDI probe withultrahigh extraction and sensitivity for DNA oligomer. J AmChem Soc 2010;132(32):10976–7.
[5] He QY, Sudibya HG, Yin ZY, Wu SX, Li H, Boey F, et al.Centimeter-long and large-scale micropatterns of reducedgraphene oxide films: fabrication and sensing applications.Acs Nano 2010;4(6):3201–8.
[6] Mao S, Lu GH, Yu KH, Bo Z, Chen JH. Specific protein detectionusing thermally reduced graphene oxide sheet decoratedwith gold nanoparticle-antibody conjugates. Adv Mater2010;22(32):3521–6.
[7] Yang K, Zhang SA, Zhang GX, Sun XM, Lee ST, Liu ZA.Graphene in mice: ultrahigh in vivo tumor uptake andefficient photothermal therapy. Nano Lett2010;10(9):3318–23.
[8] Sun XM, Liu Z, Welsher K, Robinson JT, Goodwin A, Zaric S,et al. Nano-graphene oxide for cellular imaging and drugdelivery. Nano Research 2008;1(3):203–12.
[9] Hu WB, Peng C, Luo WJ, Lv M, Li XM, Li D, et al. Graphene-based antibacterial paper. Acs Nano 2010;4(7):4317–23.
[10] Qi XY, Pu KY, Li H, Zhou XZ, Wu SX, Fan QL, et al.Amphiphilic graphene composites. Angew Chem Int Ed2010;49(49):9426–9.
[11] He SJ, Song B, Li D, Zhu CF, Qi WP, Wen YQ, et al. A graphenenanoprobe for rapid, sensitive, and multicolor fluorescentDNA analysis. Adv Func Mater 2010;20(3):453–9.
[12] Song YJ, Qu KG, Zhao C, Ren JS, Qu XG. Graphene oxide:intrinsic peroxidase catalytic activity and its application toglucose detection. Adv Mater 2010;22(19):2206–10.
[13] Wang Y, Li ZH, Hu DH, Lin CT, Li JH, Lin YH. Aptamer/graphene oxide nanocomplex for in situ molecular probing inliving cells. J Am Chem Soc 2010;132(27):9274–6.
[14] Mohanty N, Berry V. Graphene-based single-bacteriumresolution biodevice and DNA transistor: interfacinggraphene derivatives with nanoscale and microscalebiocomponents. Nano Lett 2008;8(12):4469–76.
[15] Gulbakan B, Yasun E, Shukoor MI, Zhu Z, You M, Tan X, et al.A dual platform for selective analyte enrichment andionization in mass spectrometry using aptamer-conjugatedgraphene oxide. J Am Chem Soc 2010;132(49):17408–10.
[16] Yang XY, Zhang XY, Liu ZF, Ma YF, Huang Y, Chen Y. High-efficiency loading and controlled release of doxorubicinhydrochloride on graphene oxide. J Phys Chem C2008;112(45):17554–8.
[17] Yang X, Wang Y, Huang X, Ma Y, Huang Y, Yang R, et al. Multi-functionalized graphene oxide based anticancer drug-carrierwith dual-targeting function and pH-sensitivity. J MaterChem 2011;21(10):3448–54.
[18] Feng L, Zhang S, Liu Z. Graphene based gene transfection.Nanoscale 2011;3(3):1252–7.

C A R B O N 4 9 ( 2 0 1 1 ) 4 0 4 0 – 4 0 4 9 4049
[19] Zhang LM, Xia JG, Zhao QH, Liu LW, Zhang ZJ. Functionalgraphene oxide as a nanocarrier for controlled loading andtargeted delivery of mixed anticancer drugs. Small2010;6(4):537–44.
[20] Zhang L, Lu Z, Zhao Q, Huang J, Shen H, Zhang Z. Enhancedchemotherapy efficacy by sequential delivery of siRNA andanticancer drugs using PEI-grafted graphene oxide. Small2011;7(4):460–4.
[21] S A, Z ZX, Ye F, He QY, Chen GCK, Soo J, et al. Interfacing livecells with nanocarbon substrates. Langmuir2010;26(4):2244–7.
[22] Wang K, Ruan J, Song H, Zhang J, Wo Y, Guo S, et al.Biocompatibility of graphene oxide. Nanoscale Res Lett2010;6(1):8.
[23] Zhang XY, Yin JL, Peng C, Hu WQ, Zhu ZY, Li WX, et al.Distribution and biocompatibility studies of graphene oxidein mice after intravenous administration. Carbon2010;49(3):986–95.
[24] Liu Z, Robinson JT, Sun XM, Dai HJ. PEGylated nanographeneoxide for delivery of water-insoluble cancer drugs. J AmChem Soc 2008;130(33):10876–7.
[25] Yang K, Wan JM, Zhang S, Zhang YJ, Lee ST, Liu Z. Invivo pharmacokinetics, long-term biodistribution andtoxicology of PEGylated graphene in mice. ACS Nano2011;5(1):516–22.
[26] Goodwin AP, Tabakman SM, Welsher K, Sherlock SP, PrencipeG, Dai HJ. Phospholipid–dextran with a single coupling point:a useful amphiphile for functionalization of nanomaterials. JAm Chem Soc 2009;131(1):289–96.
[27] Hogemann D, Josephson L, Weissleder R, Basilion JP.Improvement of MRI probes to allow efficient detection ofgene expression. Bioconjugate Chem 2000;11(6):941–6.
[28] Harris LA, Goff JD, Carmichael AY, Riffle JS, Harburn JJ, PierreSt TG, et al. Magnetite nanoparticle dispersions stabilizedwith triblock copolymers. Chem Mater 2003;15(6):1367–77.
[29] Moore A, Weissleder R, Bogdanov A. Uptake of dextran-coated monocrystalline iron oxides in tumor cells andmacrophages. Jmri-Journal Magnetic Reson Imaging1997;7(6):1140–5.
[30] Sonvico F, Mornet S, Vasseur S, Dubernet C, Jaillard D,Degrouard J, et al. Folate-conjugated iron oxidenanoparticles for solid tumor targeting as potential specificmagnetic hyperthermia mediators: synthesis,physicochemical characterization, and in vitro experiments.Bioconjugate Chem 2005;16(5):1181–8.
[31] Wilson R, Spiller DG, Beckett A, Prior IA, See V. Highly stabledextran-coated quantum dots for biomolecular detection andcellular imaging. Chem Mater 2010;22(23):6361–9.
[32] Van Tomme SR, Hennink WE. Biodegradable dextranhydrogels for protein delivery applications. Expert Rev MedDevic 2007;4(2):147–64.
[33] Hiemstra C, van der Aa LJ, Zhong ZY, Dijkstra PJ, Feijen J.Rapidly in situ-forming degradable hydrogels from dextranthiols through michael addition. Biomacromolecules2007;8(5):1548–56.
[34] Bachelder EM, Beaudette TT, Broaders KE, Dashe J, FrechetJMJ. Acetal-derivatized dextran: an acid-responsivebiodegradable material for therapeutic applications. J AmChem Soc 2008;130(32):10494–5.
[35] Perez JM, Asati A, Nath S, Kaittanis C. Synthesis ofbiocompatible dextran-coated nanoceria with pH-dependentantioxidant properties. Small 2008;4(5):552–6.
[36] Hummers WS, Offeman RE. Preparation of graphitic oxide. JAm Chem Soc 1958;80:1339.
[37] Silverstein RM, Robert perthel J. Kjeldahlmicrodetermination. Anal Chem 1950;22(7):949–50.
[38] Concon JM, Soltess D. Rapid micro Kjeldahl digestion ofcereal grains and other biological materials. Anal Biochem1973;53(1):35–41.
[39] Wang HF, Wang J, Deng XY, Sun HF, Shi ZJ, Gu ZN, et al.Biodistribution of carbon single-wall carbon nanotubes inmice. J Nanosci Nanotechnol 2004;4(8):1019–24.
[40] Attal S, Thiruvengadathan R, Regev O. Determination of theconcentration of single-walled carbon nanotubes in aqueousdispersions using UV–visible absorption spectroscopy. AnalChem 2006;78(23):8098–104.
[41] Reed BW, Sarikaya M. Electronic properties of carbonnanotubes by transmission electron energy-lossspectroscopy. Phys Rev B 2001;64(19):195404.
[42] Li D, Muller MB, Gilje S, Kaner RB, Wallace GG. Processableaqueous dispersions of graphene nanosheets. NatureNanotechnol 2008;3(2):101–5.
[43] Stankovich S, Dikin DA, Dommett GHB, Kohlhaas KM,Zimney EJ, Stach EA, et al. Graphene-based compositematerials. Nature 2006;442(7100):282–6.
[44] Zhang YB, Ali SF, Dervishi E, Xu Y, Li ZR, Casciano D, et al.Cytotoxicity effects of graphene and single-wall carbonnanotubes in neural phaeochromocytoma-derived PC12cells. Acs Nano 2010;4(6):3181–6.
[45] Kam NWS, Liu ZA, Dai HJ. Carbon nanotubes as intracellulartransporters for proteins and DNA: an investigation of theuptake mechanism and pathway. Angew Chem Int Ed2006;45(4):577–81.
[46] Liu Z, Tabakman SM, Chen Z, Dai HJ. Preparation of carbonnanotube bioconjugates for biomedical applications. NatureProtocols 2009;4(9):1372–82.


![Graphene-Oxide Functionalized with 2-Ureido-4[1H]- pyrimidinone … · 2020. 10. 21. · 1 Graphene-Oxide Functionalized with 2-Ureido-4[1H]-pyrimidinone for Production of Nacre-Like](https://img.pdfslide.us/doc/110x75/61126fc5c8ab861af070f35f/graphene-oxide-functionalized-with-2-ureido-41h-pyrimidinone-2020-10-21.jpg)


























