Embed Size (px)
Citation preview

In Vitro Activity and Resistance Profile of Samatasvir, a Novel NS5AReplication Inhibitor of Hepatitis C Virus
J. P. Bilello, L. B. Lallos, J. F. McCarville, M. La Colla, I. Serra, C. Chapron, J. M. Gillum, C. Pierra, D. N. Standring, M. Seifer
Idenix Pharmaceuticals, Inc., Cambridge, Massachusetts, USA
The hepatitis C virus (HCV) nonstructural 5A (NS5A) protein is a clinically validated target for drugs designed to treat chronicHCV infection. This study evaluated the in vitro activity, selectivity, and resistance profile of a novel anti-HCV compound, sa-matasvir (IDX719), alone and in combination with other antiviral agents. Samatasvir was effective and selective against infec-tious HCV and replicons, with 50% effective concentrations (EC50s) falling within a tight range of 2 to 24 pM in genotype 1through 5 replicons and with a 10-fold EC50 shift in the presence of 40% human serum in the genotype 1b replicon. The EC90/EC50 ratio was low (2.6). A 50% cytotoxic concentration (CC50) of >100 �M provided a selectivity index of >5 � 107. Resistanceselection experiments (with genotype 1a replicons) and testing against replicons bearing site-directed mutations (with genotype1a and 1b replicons) identified NS5A amino acids 28, 30, 31, 32, and 93 as potential resistance loci, suggesting that samatasviraffects NS5A function. Samatasvir demonstrated an overall additive effect when combined with interferon alfa (IFN-�), ribavi-rin, representative HCV protease, and nonnucleoside polymerase inhibitors or the nucleotide prodrug IDX184. Samatasvir re-tained full activity in the presence of HIV and hepatitis B virus (HBV) antivirals and was not cross-resistant with HCV protease,nucleotide, and nonnucleoside polymerase inhibitor classes. Thus, samatasvir is a selective low-picomolar inhibitor of HCV rep-lication in vitro and is a promising candidate for future combination therapies with other direct-acting antiviral drugs in HCV-infected patients.
Approximately 150 million people are infected with hepatitisC virus (HCV) worldwide (http://www.who.int/mediacentre
/factsheets/fs164/en). In the United States, �4 million people sufferfrom persistent HCV infection, and 10,000 people die annually fromHCV-related liver diseases, such as cirrhosis and hepatocellular car-cinoma. Morbidity and mortality rates from chronic HCV infec-tion are projected to double in this decade and may surpass thoseof human immunodeficiency virus (1). To date, three proteaseinhibitors and a nucleotide prodrug inhibitor of the HCV poly-merase have been approved for HCV treatment in combinationwith pegylated interferon and ribavirin. However, due to the pos-sible emergence of resistant viruses upon single-drug therapy andthe side effects related to treatment with protease inhibitors (2–5)(see http://www.jnj.com/news/all/OLYSIO-simeprevir-Receives-FDA-Approval-for-Combination-Treatment-of-Chronic-Hepatitis-C), additional potent and safe direct-acting antiviral agentsare needed to effectively combat this disease.
The HCV genome consists of approximately 9,600 nucleotidesof positive single-stranded RNA that encode a �3,033-amino acidpolyprotein. Upon cleavage by cellular and viral proteases, thepolyprotein is processed into 10 viral proteins. The four amino-terminal structural proteins function in the formation of viralparticles. The six carboxy-terminal nonstructural proteins processthe viral polyprotein, serve in host and viral regulatory roles, par-ticipate in the formation of the viral replication complex, and/orcontribute to replication of the viral genome (6).
The nonstructural 5A (NS5A) protein is involved in the repli-cation and maturation of HCV virions and has been shown tointeract with numerous host cell proteins (7). Although the exactfunctions of the NS5A protein are not fully understood, inhibitorsof NS5A have been identified through replicon screening and arein various stages of clinical development (6, 8–10). The first suchinhibitor, daclatasvir (BMS-790052), was active against the repli-con, with 50% effective concentrations (EC50s) ranging from 9 to
146 pM, depending upon the HCV genotype (8). The activity ofdaclatasvir is markedly lower against genotype 2 and 3 intergeno-typic replicons than against those of genotypes 1, 4, and 5 (8). TheNS5A inhibitor samatasvir (IDX719) was designed to inhibit HCVreplication with enhanced activity across genotypes, potentiallyaffording a once-daily single-pill dosing regimen for all genotypes.This study assesses the in vitro efficacy, specificity, and resistancephenotype of samatasvir, a novel HCV NS5A inhibitor, and dem-onstrates its role in a combination treatment regimen for HCV.
MATERIALS AND METHODS
Compounds. Samatasvir [carbamic acid, N-[(1R)-2-[(2S)-2-[5-[4-[6-[2-[(2S)-1-[(2S)-2-[(methoxycarbonyl)amino]-3-methyl-1-oxobutyl]-2-pyrrolidinyl]-1H-benzimidazol-6-yl]thieno[3,2-b]thien-3-yl]phenyl]-1H-imidazol-2-yl]-1-pyrrolidinyl]-2-oxo-1-phenylethyl]-, methyl ester](Fig. 1), daclatasvir, and IDX184 were synthesized by Idenix Pharmaceu-ticals, Inc. (Cambridge, MA). Intron A and ribavirin (RBV) (Rebetol)were obtained from Schering-Plough (Kenilworth, NJ). Doxorubicin hy-drochloride, diclofenac sodium salt, and alpha-1 acid glycoprotein (AAG)were obtained from Sigma (St. Louis, MO). Efavirenz (EFV), lamivudine(3TC), lopinavir (LPV), ritonavir (RTV), RBV, telbivudine (LdT), teno-fovir (TFV), and zidovudine (AZT) were obtained from Moravek Bio-chemicals and Radiochemicals (Brea, CA). Raltegravir was purchasedfrom Selleck Chemicals (Houston, TX) (11). Lopinavir and ritonavir weremixed in a 4:1 ratio to constitute Kaletra (KLT).
Received 20 December 2013 Returned for modification 28 January 2014Accepted 1 May 2014
Published ahead of print 27 May 2014
Address correspondence to J. P. Bilello, [email protected].
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/AAC.02777-13
August 2014 Volume 58 Number 8 Antimicrobial Agents and Chemotherapy p. 4431– 4442 aac.asm.org 4431
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

HCV replicons. ZS11-luc (genotype 1b, Con1) and 1a-luc (strainH77) are bicistronic HCV replicons encoding the nonstructural proteinsfrom NS3 to NS5B under the control of the encephalomyocarditis virus(EMCV) internal ribosomal entry site (IRES) promoter, as well as a fireflyluciferase-neomycin phosphotransferase fusion gene under the control ofthe HCV promoter. The ZS11-luc replicon contains adaptive mutationsencoding E1202G, S2204I, and D2254E, and the 1a-luc replicon containsadaptive mutations encoding Q1067R, P1496L, V1655I, K1691R,K2040R, and S2204I in the HCV polyprotein. SP1�S (genotype 1b, Con1)and H1a (genotype 1a, H77) are bicistronic HCV replicons that containnonstructural regions from NS3 to NS5B under the EMCV IRES pro-moter. SP1�S contains a deletion at S2204 in NS5A for enhanced replica-tion. The parental ZS11 (without luciferase) and SP1�S replicons werekindly provided by Christoph Seeger (Fox Chase Cancer Center, Philadel-phia, PA).
Site-directed mutations on the wild-type 1a-luc or ZS11-luc repliconswere generated using the QuikChange II XL site-directed mutagenesis kit(Stratagene/Agilent Technologies), as recommended by the manufac-turer, and were confirmed by sequencing. cDNAs were transcribed afterScaI linearization using the RiboMAX large-scale RNA production system(Promega Corporation).
The NS5A intergenotypic (IGT) replicons were derived from the ge-notype 1b ZS11-luc replicon by removing the genotype 1b NS5A regionand either substituting the first 100 amino acids of NS5A or amino acids
12 to 437 of NS5A from the desired genotype. The following genotypesand strains were used: genotype 1a (H77 strain), 2a (JFH-1 strain,GenBank accession no. AB047639), 3a (NZL-1 strain, D17763), 4a (F7157strain, DQ418788), and 5a (SA13 strain, AF064490). The preliminarycDNA templates of NS5a for genotypes 2a, 3a, 4a, and 5a were synthesizedby an outside vendor.
Viruses. The JFH-1 DNA template was derived synthetically usingsequence information from NCBI accession no. AB047639 (12). JFH-1RNA, produced by in vitro transcription, was used to generate infectiousvirus by transfection of hepatitis C-producing (HPC) cells using a proce-dure similar to those previously reported (12, 13).
A panel of 17 RNA and DNA viruses was obtained from the AmericanType Culture Collection (ATCC), the BEI Research Resource Repository,and the NIH AIDS Research and Reference Reagent Program (ARRRP)and propagated by standard methods. With the exception of dengue virus,which was grown in Vero E6 cells, the stock virus pools for each of theviruses were grown in the same cell lines used for antiviral evaluations.
Cells and media. The CAKI-1, CCRF-CEM, COLO-205, SJCRH30,and HepG2 cell lines, as well as those listed in Table 1, were obtained fromthe ATCC, MAGI-CCR5 cells were obtained from the NIH ARRRP (14),and the SNB-78 cell line was provided by the National Cancer Institute(NCI). All cell lines were maintained as suggested by the respective man-ufacturers. The Huh-7 (15) and HPC cell lines were kindly provided byChristoph Seeger (Fox Chase Cancer Center, Philadelphia, PA) and werepropagated in Huh-7 medium (Dulbecco’s modified Eagle’s medium[DMEM] containing glucose, L-glutamine, sodium pyruvate, 10% fetalbovine serum [FBS], 100 IU/ml penicillin, 100 �g/ml streptomycin, 2 mMGlutaMAX, and nonessential amino acids). The HepaRG cell line (LifeTechnologies) was maintained in the supplier’s proprietary medium.
The GS4.1 (16) (kindly provided by Christoph Seeger, Fox Chase Can-cer Center, Philadelphia, PA), Zluc, H1a, and H1a-luc cell lines stablypossess a bicistronic HCV genotype 1a or 1b replicon and were propa-gated in Huh-7 medium containing 0.25 to 0.5 mg/ml of G418 (Geneticin;Life Technologies). The NS5A intergenotypic (IGT) replicon cell linesstably possess bicistronic replicons containing the luciferase reporter geneand represent NS5A of genotype 1a, 2a, 3a, 4a, or 5a. These cells weremaintained in Huh-7 medium containing 0.5 mg/ml of G418.
FIG 1 Molecular structure of samatasvir.
TABLE 1 Antiviral activity of samatasvir against 17 RNA and DNA virusesa
Genome type andvirus family Virus (strain) Cell line EC50 (�M)
RNAFlaviviridae Bovine viral diarrhea virus (NADL) MDBK 33.4
Dengue virus, serotype 2 (New Guinea C) Huh-7 �50West Nile virus (NY-99) Vero �50Yellow fever virus (17D) HeLa 16.5
Orthomyxoviridae Influenza A virus (A/Victoria/3/75 [H3N2]) MDCK �50Influenza B virus (Lee) MDCK �50
Paramyxoviridae Measles virus (Edmonston) HeLa �50Respiratory syncytial virus (Long) Vero �50
Picornaviridae Coxsackie B virus (B4) Vero �50Poliovirus (Chat) Vero �50Rhinovirus (1B) MRC-5 �50
Retroviridae Human immunodeficiency virus type 1 (Ba-L) MAGI-CCR5 �50Togaviridae Venezuelan equine encephalitis virus (Trinidad) Vero E6 �50Reoviridae Rotavirus (WI61) Vero �50
DNAHerpesviridae Herpes simplex virus 1 (HF) Vero �50Poxviridae Monkeypox virus (Zaire) Vero E6 �50
Vaccinia virus (WR-56) Vero E6 �50a The values are from a single data set. The samatasvir CC50 values for each cell line used were �50 �M.
Bilello et al.
4432 aac.asm.org Antimicrobial Agents and Chemotherapy
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

Cytotoxicity assays. The cells were seeded and treated as in the repli-con activity assay (described below). After 3 days of treatment, viabilitywas determined with the CellTiter-Blue cell viability assay solution (Pro-mega) in a Victor3 V 1420 multilabel counter (PerkinElmer), and 50%cytotoxic concentration (CC50) values were determined using the Mi-crosoft Excel and XLFit 4.1 softwares.
HepG2 cells (1 � 104 cells/well) were subjected to 3-day treatmentwith serial dilutions of compound, after which a 4-parameter In Cy-totox toxicity test system was used as suggested by the manufacturer(Xenometrics). This test system consecutively monitors different cy-totoxic endpoints, such as cell death (lactate dehydrogenase [LDH]assay), general physiological cell state (glucose consumption), meta-bolic activity (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetra-zolium-5-carboxanilide [XTT] assay), and lysosomal activity (acidphosphatase) in the same well.
The CAKI-1, SNB-78, CCRF-CEM, SJCRH30, and COLO-205 celllines were exposed to serial dilutions of samatasvir or daclatasvir for 3 daysand then assessed for cell viability via CellTiter-Glo (Promega), as de-scribed above. Doxorubicin (10 �M) was used as a toxicity control.
Long-term cytotoxicity assays were performed with HepaRG cells(1 � 105 cells/well) in collagen I-coated 96-well plates that were main-tained for 7 days in the recommended medium without drug to ensureterminal differentiation prior to drug exposure. The cells were treatedwith compound for 14 days and were refed every other day to ensureproper nutrition and exposure to the drug. At the end of treatment, cellviability was determined using the WST-1 cell proliferation solution(Clontech), and absorbance (450 nm) was measured in a Victor3 V 1420multilabel counter. CC50 values were determined using the XLFit 4.1 soft-ware.
Non-HCV antiviral activity assays. Depending on the virus, standardcytoprotection (cytoprotective effect [CPE]), reporter gene (HIV), orplaque reduction (vaccinia virus, monkeypox virus, or Venezuelan equineencephalitis [VEE] virus) assays were used to evaluate antiviral activity(17). The appropriate positive controls of virus inhibition were used foreach virus. For each assay, an aliquot of virus whose titer had been previ-ously determined was diluted into tissue culture medium such that theamount of virus added to each well was either the amount determined togive between 85 and 95% cell killing (CPE assays), approximately 10� the50% tissue culture infective dose (TCID50)/well (reporter gene), or therequired number of PFU per well (plaque reduction assays).
HCV replicon activity assay. Solid opaque 96-well culture plates wereseeded with Zluc, NS5A IGT (7.5 � 103 cells/well), or H1a-luc (1 � 104
cells/well) cells in Huh-7 medium. At least 4 h later, drug treatment wasinitiated and the cells incubated for 3 days at 37°C under 5% CO2. Lucif-erase activity was measured on a Victor3 V 1420 multilabel counter (1-sread time, 700-nm cutoff filter) using ONE-Glo luciferase assay reagent(Promega). The EC50s were calculated from dose-response curves fromthe resulting best-fit equations determined by the Microsoft Excel andXLFit 4.1 software programs.
HCV replicon combination assay. Duplicate 96-well culture plateswere seeded with Zluc and Huh-7 cells, respectively, at a density of 7.5 �103 cells per well in Huh-7 medium. The cells were incubated at 37°Cunder 5% CO2 for 3 to 4 h, and drug treatment was then initiated byadding drug dilutions in a 5-by-5 checkerboard design. The final concen-trations of both drugs ranged from 0.25� to 4� their respective EC50s.Cytotoxicity was measured in parallel using the CellTiter-Blue cell viabil-ity assay solution (Promega).
The activity data were analyzed for synergism or antagonism usingthree mathematical models, (i) the Bliss independence model (Mac-Synergy II program, version 1.0; M. N. Prichard, L. E. Prichard, and C.Shipman, Jr., University of Michigan, MI, USA) (18, 19), (ii) Loewe ad-ditivity model (CombiTool program, version 2.001; V. Dressler, G.Muller, and J. Suhnel, Institute of Molecular Biotechnology, Jena, Ger-many) (20), and (iii) the median-effect approach (combination index,CalcuSyn program, version 2.1; Biosoft, Cambridge, United Kingdom)
(19–22). MacSynergy II calculates a theoretical additive dose-responsesurface based on the dose-response curves for each individually titrateddrug. Peaks above the plane at 0% inhibition indicate areas of greater-than-additive interaction, or synergism. Conversely, peaks below theplane at 0% inhibition indicate areas of less-than-additive interaction, orantagonism. The data sets were assessed at the 99.9, 99, and 95% confi-dence levels; volumes of �25 �M2% (log volumes, �2) were consideredadditive, volumes of �25 but �50 �M2% (log volumes, �2 and �5,respectively) were considered weakly synergistic or antagonistic, volumesof �50 but �100 �M2% (log volumes, �5 and �9, respectively) wereconsidered moderately synergistic or antagonistic, and volumes of �100�M2% (log volumes, �9) were considered strongly synergistic or antag-onistic. CombiTool calculates the differences between the predicted ef-fects (according to the Loewe additivity model) and observed effects (ex-perimental data) (20). Deviations of �0.25 from the predicted effectswere considered significant. The CalcuSyn program, version 2.1 (Biosoft),was used to determine the nature of the drug-drug interaction based onthe combination index equation proposed by Chou and Talalay (22). Thecombination index at the calculated EC50 for the drug combination wasdetermined; the description of the degree of drug interaction based on thecombination index (CI) value is as recommended in the CalcuSyn forWindows software manual. Analyses were performed on the data derivedfrom at least five independent experiments.
For the protein binding experiments, Huh-7 medium containing 10 to50% human sera (HS) (Valley Biomedical), 10% FBS plus 1 mg/ml AAG,or 10% FBS alone was used. The fold change in antiviral activity for eachdrug was calculated by dividing the EC50 in the respective medium by thereference EC50 in 10% FBS. In addition, the EC50s obtained in 10, 20, 30,40, 45, and 50% HS for each drug were subjected to linear regressionanalysis using the XLFit 4.1 software to extrapolate the 100% serum-adjusted EC50. The cytotoxicity assays were run in parallel using MTS[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] reagent (Promega).
For drug-drug interaction studies, the replicon cells were treated withserial dilutions of samatasvir in the presence of a fixed concentration of ahepatitis B virus (HBV) or HIV agent and 45% HS. The fixed concentra-tions used were five times the reported maximum concentration of drugin serum (Cmax) value: 94 �M KLT, 28 �M AZT, 32.5 �M 3TC, 5 �MTFV, 65 �M LdT, 65 �M EFV, and 27 �M raltegravir (RLT) (11, 23–28).The cytotoxicity assays were run in parallel using MTS reagent.
Extended-treatment replicon assay. H1a cells were seeded into a6-well plate (3 � 105 cells per well) in medium containing compoundsand incubated at 37°C under 5% CO2. The cells were split every 3 to 4 daysin medium containing fresh compound. On the days indicated, RNA wasextracted using the High Pure RNA isolation kit (Roche) with DNase Itreatment, according to the manufacturer’s protocol. Replicon RNA wasmeasured by real-time quantitative reverse transcription-PCR (RT-PCR)in a StepOnePlus instrument using TaqMan One-Step RT-PCR mastermix (Life Technologies) containing 0.9 �M forward and reverse prim-ers and 0.25 �M probe specific for the 5=-untranslated region (UTR) ofthe replicon, and these were compared to an in vitro-transcribed stan-dard curve. The primers used were as follows: forward, 5=-AGCCATGGCGTTAGTATGAGTGT-3=; reverse, 5=-TTCCGCAGACCACTATGG-3=. The probe was 5=-6FAM-CCTCCAGGACCCCCCCTCCC-TAMRA-3= (6FAM, 6-carboxyfluorescein; TAMRA, tetramethylrhodamine).The replicon RNA values were normalized to human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) endogenous control (Life Technol-ogies), using TaqMan control total human RNA (Life Technologies) forthe standard curve. The log10 change from baseline of the HCV repliconfor each RNA extraction day (days 3, 7, 10, and 14) in a single experimentwas then calculated by subtracting the mean log10 HCV replicon cop-ies/ng RNA of the sample from the mean log10 HCV replicon copies/ngRNA of the untreated control at day 0. The mean log10 change frombaseline and standard deviation for each RNA extraction day were thencalculated across the experiments in the analysis.
In Vitro Profile of Samatasvir, an HCV NS5A Inhibitor
August 2014 Volume 58 Number 8 aac.asm.org 4433
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
HCV in vitro infection core ELISA. For an enzyme-linked immu-nosorbent assay (ELISA), 96-well plates were seeded with HPC cells at adensity of 2.5 � 103 cells per well in Huh-7 medium. At least 4 h after theHPC cells were seeded, JFH-1 HCV virus stock and serial dilutions of drugin Huh-7 medium were added to each well. At 16 h after treatment andinfection, the virus inoculum and drug solution were removed by aspira-tion. The cultures were then treated at the same final concentrations ofdrug diluted in Huh-7 medium. The cells were incubated in the presenceof drug for 4 additional days at 37°C under 5% CO2, upon which the cellswere fixed with a 1:1 solution of acetone to methanol for 1 min, washedthree times with KPL wash solution (KPL, Inc., Gaithersburg, MD), andthen blocked with 10% FBS-TNE (50 mM Tris-HCl [pH 7.5], 100 mMNaCl, 1 mM EDTA, and 10% FBS) for 1 h at room temperature. The cellswere washed three times with KPL wash solution and incubated with ananti-hepatitis C virus core protein antibody (clone MA1-080; ThermoScientific, Rockford, IL) diluted in 10% FBS-TNE for 2 h at 37°C. The cellswere washed as described above and incubated with a horseradish perox-idase (HRP)-conjugated goat anti-mouse antibody (Invitrogen) dilutedin 10% FBS-TNE for 1 h at 37°C, and then washed. Color developmentwas initiated by adding o-phenylenediamine (OPD) solution. After 30min, the reaction was stopped with 2 N H2SO4 and the absorbance mea-sured at 490 nm on a Victor3 V 1420 multilabel counter. The EC50s werecalculated from dose-response curves from the resulting best-fit equa-tions determined by the Microsoft Excel and XLFit 4.1 software programs.
Resistance selection. H1a-luc cells were cultured in the presence of0.25 mg/ml of G418 and increasing concentrations of samatasvir for 90days to generate samatasvir-resistant cell lines in three independent ex-periments. Selection began with 2� the EC50 of samatasvir; untreatedreplicon cells were cultured in parallel. On day 0 and after selection, com-pound activity was evaluated in the replicon activity assay. Populationsequencing of the genomic region corresponding to the amino-terminal100 residues of NS5A was performed during selection. At the conclusionof the selection period, the NS3 to NS5b genomic sequence was deter-mined by population sequencing. Finally, clonal sequencing of the NS5Aamino-terminal region (encoding amino acids 1 to 100) from one sama-tasvir-resistant cell line (719R-A) and its matched untreated control rep-licon cell line was performed after selection.
Population and clonal sequencing. RNA was extracted from cell pel-lets as per the manufacturer’s protocol for the High Pure RNA isolation kit(Roche Diagnostics), including the optional DNase I digestion procedure.All RT-PCRs were carried out using the Easy-A one-tube RT-PCR system(Stratagene/Agilent Technologies). The entire replicon was sequenced us-ing primers that overlapped each nonstructural gene: NS3F, 5=-CCTCGGTGCACATGCTTTACATGTGTTT-3=; NS3R, 5=-GACAATCCTGCCCACTATGACCAC-3=; NS4F, 5=-CCCTGACGCACCCAATCACCAAAT-3=;NS4R, 5=-CGCGCTGGCAGGACACAAAG-3=; NS5AF, 5=-GCACTACGTGCCGGAGAGCG-3=; NS5AR, 5=-CTGGAAGACAGTGTAACACC-3=;NS5BF, 5=-CGGATCTCAGCGACGGGTCATGGTC-3=; and NS5BR, 5=-GGAAAAAAACAGGATGGCC-3=. The region encoding the first 100amino acids of NS5A was amplified using the following primers: forward,5=-GCACTACGTGCCGGAGAGCG-3=, and reverse, 5=-GATTGTCAGTAGTCATACCC-3=. The resulting cDNAs were then sequenced with over-lapping primers in both directions.
Clonal sequencing of the genomic region encoding the first 100 aminoacids of NS5A was carried out using RT-PCR products per the TOPO TACloning kit for sequencing (Invitrogen) protocol and electroporated intoOne Shot TOP10 Electrocomp cells. One hundred individual bacterialcolonies were expanded and purified using the Wizard SV 96 plasmidDNA purification system (Promega). The plasmids were sequenced withforward and reverse primers.
The sequences were assembled and compared against a wild-type rep-licon sequence from untreated H1a-luc cells using Sequencher 4.9 (GeneCodes Corporation) and aligned using GeneDoc (http://www.nrbsc.org/gfx/genedoc/ebinet.htm).
Transient-transfection assay. Four million HPC cells were electropo-rated with the wild-type or mutant replicon RNA and plated (3 � 104
cells/well) on a 96-well opaque white plate (29). Approximately 4 h later,serial dilutions of drug were added and the plates incubated at 37°C under5% CO2 for 4 days. The experiment was performed as described above forthe replicon activity assay to determine the EC50 of each drug against eachof the mutant replicons. The activity was expressed as the mean foldchange in EC50 relative to that of a wild-type genotype 1a, 1b, or interge-notypic 2a replicon.
To determine the replicative capacity of each mutant, two identicalplates of transfected cells were plated without treatment. Luciferase activ-ity was measured at 4 h and 4 days after plating for each mutant repliconand compared to that of the wild-type replicon. The replicative capacity ofeach mutant was expressed as a mean percentage of the wild-type replica-tion activity.
RESULTSSamatasvir is a selective inhibitor of HCV replication withbroad genotypic activity. Samatasvir inhibited genotype 1a and1b HCV replicons at picomolar concentrations, with a mean CC50
value of �100 �M in the Zluc genotype 1b replicon cell line (Ta-bles 2 and 3), giving rise to a selectivity index of �5 � 107. Whentested against replicons bearing NS5A sequences from other ge-notypes or a genotype 2a JFH-1 infectious virus, EC50s of 2 to 24pM were obtained. This represents a 12-fold change across thetested genotypes compared to an 84-fold change for daclatasvir(Table 2).
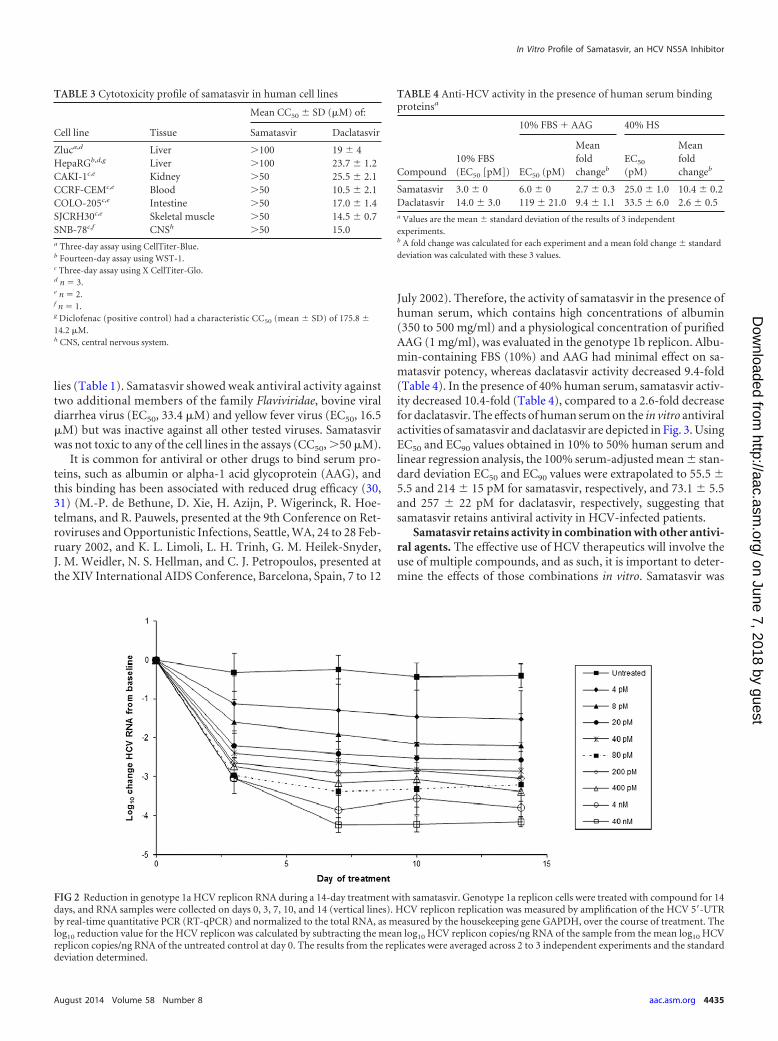
Samatasvir reduced genotype 1a replicon RNA in a dose-de-pendent manner over 14 days (Fig. 2). The maximum reductionswere 1.5 and 4.2 log10 for 4 pM (1� the EC50) and 40 nM(10,000� the EC50), respectively. Depending on the drug concen-tration, there was a 1.1- to 3.0-log10 reduction in replicon RNAafter 3 days of treatment, followed by a second lower rate of RNAreduction until day 7, when the RNA reductions reached a plateau.There was no evidence of replicon RNA rebound. Maximal sup-pression occurred at 7 to 14 days, depending on the dose.
The specificity of samatasvir antiviral activity was testedagainst 17 RNA and DNA viruses, spanning several viral fami-
TABLE 2 Antiviral potency against HCV genotypes 1 to 5
Genotype Virus/replicona
Mean EC50 � SD (pM) ofb:
Samatasvir Daclatasvir
1a H1a-luc 4.1 � 1.1 51 � 11100-IGT 8.5 � 1.7 84 � 27FL-IGT 6.2 � 1.7 80 � 33
1b Zluc 2.4 � 0.6 13 � 4
2a JFH-1 virus 21 � 4 158 � 50FL-IGT 24 � 4 176 � 1
3a 100-IGT 23 � 11 1,086 � 301FL-IGT 17 � 3 299 � 4
4a 100-IGT 6.0 � 0.1 72 � 66FL-IGT 2.0 � 0 19 � 3
5a 100-IGT 18 � 2 42 � 1a 100-IGT, ZS11-luc replicon containing intergenotypic substitutions in amino acids 1to 100 of NS5A; FL-IGT, ZS11-luc replicon containing intergenotypic substitutions inamino acids 12 to 437 of NS5A.b Data are from 3 to 5 independent experiments.
Bilello et al.
4434 aac.asm.org Antimicrobial Agents and Chemotherapy
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
lies (Table 1). Samatasvir showed weak antiviral activity againsttwo additional members of the family Flaviviridae, bovine viraldiarrhea virus (EC50, 33.4 �M) and yellow fever virus (EC50, 16.5�M) but was inactive against all other tested viruses. Samatasvirwas not toxic to any of the cell lines in the assays (CC50, �50 �M).
It is common for antiviral or other drugs to bind serum pro-teins, such as albumin or alpha-1 acid glycoprotein (AAG), andthis binding has been associated with reduced drug efficacy (30,31) (M.-P. de Bethune, D. Xie, H. Azijn, P. Wigerinck, R. Hoe-telmans, and R. Pauwels, presented at the 9th Conference on Ret-roviruses and Opportunistic Infections, Seattle, WA, 24 to 28 Feb-ruary 2002, and K. L. Limoli, L. H. Trinh, G. M. Heilek-Snyder,J. M. Weidler, N. S. Hellman, and C. J. Petropoulos, presented atthe XIV International AIDS Conference, Barcelona, Spain, 7 to 12
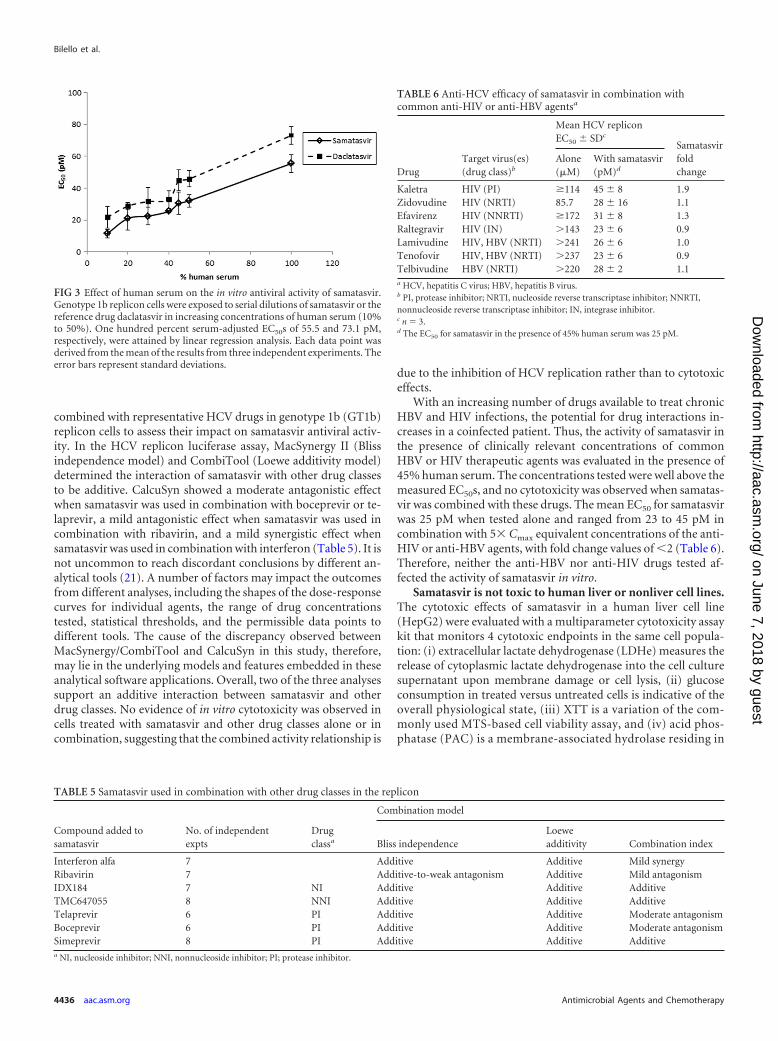
July 2002). Therefore, the activity of samatasvir in the presence ofhuman serum, which contains high concentrations of albumin(350 to 500 mg/ml) and a physiological concentration of purifiedAAG (1 mg/ml), was evaluated in the genotype 1b replicon. Albu-min-containing FBS (10%) and AAG had minimal effect on sa-matasvir potency, whereas daclatasvir activity decreased 9.4-fold(Table 4). In the presence of 40% human serum, samatasvir activ-ity decreased 10.4-fold (Table 4), compared to a 2.6-fold decreasefor daclatasvir. The effects of human serum on the in vitro antiviralactivities of samatasvir and daclatasvir are depicted in Fig. 3. UsingEC50 and EC90 values obtained in 10% to 50% human serum andlinear regression analysis, the 100% serum-adjusted mean � stan-dard deviation EC50 and EC90 values were extrapolated to 55.5 �5.5 and 214 � 15 pM for samatasvir, respectively, and 73.1 � 5.5and 257 � 22 pM for daclatasvir, respectively, suggesting thatsamatasvir retains antiviral activity in HCV-infected patients.
Samatasvir retains activity in combination with other antivi-ral agents. The effective use of HCV therapeutics will involve theuse of multiple compounds, and as such, it is important to deter-mine the effects of those combinations in vitro. Samatasvir was
TABLE 3 Cytotoxicity profile of samatasvir in human cell lines
Cell line Tissue
Mean CC50 � SD (�M) of:
Samatasvir Daclatasvir
Zluca,d Liver �100 19 � 4HepaRGb,d,g Liver �100 23.7 � 1.2CAKI-1c,e Kidney �50 25.5 � 2.1CCRF-CEMc,e Blood �50 10.5 � 2.1COLO-205c,e Intestine �50 17.0 � 1.4SJCRH30c,e Skeletal muscle �50 14.5 � 0.7SNB-78c,f CNSh �50 15.0a Three-day assay using CellTiter-Blue.b Fourteen-day assay using WST-1.c Three-day assay using X CellTiter-Glo.d n 3.e n 2.f n 1.g Diclofenac (positive control) had a characteristic CC50 (mean � SD) of 175.8 �14.2 �M.h CNS, central nervous system.
FIG 2 Reduction in genotype 1a HCV replicon RNA during a 14-day treatment with samatasvir. Genotype 1a replicon cells were treated with compound for 14days, and RNA samples were collected on days 0, 3, 7, 10, and 14 (vertical lines). HCV replicon replication was measured by amplification of the HCV 5=-UTRby real-time quantitative PCR (RT-qPCR) and normalized to the total RNA, as measured by the housekeeping gene GAPDH, over the course of treatment. Thelog10 reduction value for the HCV replicon was calculated by subtracting the mean log10 HCV replicon copies/ng RNA of the sample from the mean log10 HCVreplicon copies/ng RNA of the untreated control at day 0. The results from the replicates were averaged across 2 to 3 independent experiments and the standarddeviation determined.
TABLE 4 Anti-HCV activity in the presence of human serum bindingproteinsa
Compound10% FBS(EC50 [pM])
10% FBS AAG 40% HS
EC50 (pM)
Meanfoldchangeb
EC50
(pM)
Meanfoldchangeb
Samatasvir 3.0 � 0 6.0 � 0 2.7 � 0.3 25.0 � 1.0 10.4 � 0.2Daclatasvir 14.0 � 3.0 119 � 21.0 9.4 � 1.1 33.5 � 6.0 2.6 � 0.5a Values are the mean � standard deviation of the results of 3 independentexperiments.b A fold change was calculated for each experiment and a mean fold change � standarddeviation was calculated with these 3 values.
In Vitro Profile of Samatasvir, an HCV NS5A Inhibitor
August 2014 Volume 58 Number 8 aac.asm.org 4435
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
combined with representative HCV drugs in genotype 1b (GT1b)replicon cells to assess their impact on samatasvir antiviral activ-ity. In the HCV replicon luciferase assay, MacSynergy II (Blissindependence model) and CombiTool (Loewe additivity model)determined the interaction of samatasvir with other drug classesto be additive. CalcuSyn showed a moderate antagonistic effectwhen samatasvir was used in combination with boceprevir or te-laprevir, a mild antagonistic effect when samatasvir was used incombination with ribavirin, and a mild synergistic effect whensamatasvir was used in combination with interferon (Table 5). It isnot uncommon to reach discordant conclusions by different an-alytical tools (21). A number of factors may impact the outcomesfrom different analyses, including the shapes of the dose-responsecurves for individual agents, the range of drug concentrationstested, statistical thresholds, and the permissible data points todifferent tools. The cause of the discrepancy observed betweenMacSynergy/CombiTool and CalcuSyn in this study, therefore,may lie in the underlying models and features embedded in theseanalytical software applications. Overall, two of the three analysessupport an additive interaction between samatasvir and otherdrug classes. No evidence of in vitro cytotoxicity was observed incells treated with samatasvir and other drug classes alone or incombination, suggesting that the combined activity relationship is
due to the inhibition of HCV replication rather than to cytotoxiceffects.
With an increasing number of drugs available to treat chronicHBV and HIV infections, the potential for drug interactions in-creases in a coinfected patient. Thus, the activity of samatasvir inthe presence of clinically relevant concentrations of commonHBV or HIV therapeutic agents was evaluated in the presence of45% human serum. The concentrations tested were well above themeasured EC50s, and no cytotoxicity was observed when samatas-vir was combined with these drugs. The mean EC50 for samatasvirwas 25 pM when tested alone and ranged from 23 to 45 pM incombination with 5� Cmax equivalent concentrations of the anti-HIV or anti-HBV agents, with fold change values of �2 (Table 6).Therefore, neither the anti-HBV nor anti-HIV drugs tested af-fected the activity of samatasvir in vitro.
Samatasvir is not toxic to human liver or nonliver cell lines.The cytotoxic effects of samatasvir in a human liver cell line(HepG2) were evaluated with a multiparameter cytotoxicity assaykit that monitors 4 cytotoxic endpoints in the same cell popula-tion: (i) extracellular lactate dehydrogenase (LDHe) measures therelease of cytoplasmic lactate dehydrogenase into the cell culturesupernatant upon membrane damage or cell lysis, (ii) glucoseconsumption in treated versus untreated cells is indicative of theoverall physiological state, (iii) XTT is a variation of the com-monly used MTS-based cell viability assay, and (iv) acid phos-phatase (PAC) is a membrane-associated hydrolase residing in
FIG 3 Effect of human serum on the in vitro antiviral activity of samatasvir.Genotype 1b replicon cells were exposed to serial dilutions of samatasvir or thereference drug daclatasvir in increasing concentrations of human serum (10%to 50%). One hundred percent serum-adjusted EC50s of 55.5 and 73.1 pM,respectively, were attained by linear regression analysis. Each data point wasderived from the mean of the results from three independent experiments. Theerror bars represent standard deviations.
TABLE 5 Samatasvir used in combination with other drug classes in the replicon
Compound added tosamatasvir
No. of independentexpts
Drugclassa
Combination model
Bliss independenceLoeweadditivity Combination index
Interferon alfa 7 Additive Additive Mild synergyRibavirin 7 Additive-to-weak antagonism Additive Mild antagonismIDX184 7 NI Additive Additive AdditiveTMC647055 8 NNI Additive Additive AdditiveTelaprevir 6 PI Additive Additive Moderate antagonismBoceprevir 6 PI Additive Additive Moderate antagonismSimeprevir 8 PI Additive Additive Additivea NI, nucleoside inhibitor; NNI, nonnucleoside inhibitor; PI; protease inhibitor.
TABLE 6 Anti-HCV efficacy of samatasvir in combination withcommon anti-HIV or anti-HBV agentsa
DrugTarget virus(es)(drug class)b
Mean HCV repliconEC50 � SDc
Samatasvirfoldchange
Alone(�M)
With samatasvir(pM)d
Kaletra HIV (PI) �114 45 � 8 1.9Zidovudine HIV (NRTI) 85.7 28 � 16 1.1Efavirenz HIV (NNRTI) �172 31 � 8 1.3Raltegravir HIV (IN) �143 23 � 6 0.9Lamivudine HIV, HBV (NRTI) �241 26 � 6 1.0Tenofovir HIV, HBV (NRTI) �237 23 � 6 0.9Telbivudine HBV (NRTI) �220 28 � 2 1.1a HCV, hepatitis C virus; HBV, hepatitis B virus.b PI, protease inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; NNRTI,nonnucleoside reverse transcriptase inhibitor; IN, integrase inhibitor.c n 3.d The EC50 for samatasvir in the presence of 45% human serum was 25 pM.
Bilello et al.
4436 aac.asm.org Antimicrobial Agents and Chemotherapy
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
the cellular Golgi apparatus and is a marker for lysosomal ac-tivity. No measurable in vitro cytotoxicity of samatasvir wasobserved after 3 days of treatment up to 100 �M; the meanCC50 values were �100 �M across all four assays (Table 7).Daclatasvir was moderately cytotoxic in HepG2 cells, withmean CC50 values of 14.9 to 17.7 �M.
The long-term effects of samatasvir were evaluated in the im-mortalized hepatic cell line HepaRG, which retains many charac-teristics of primary hepatocytes (32). HepaRG cells terminally dif-ferentiate into hepatocyte-like cells and express various liver-specific functions, including major cytochrome P450 enzymes.This cell line provides a novel in vitro tool for cytotoxicity studies(33). Diclofenac, a positive control of liver toxicity, exhibitedcharacteristic concentration-dependent cytotoxicity, with amean � standard deviation CC50 of 175.8 � 14.2 �M, which isconsistent with published results (34). Daclatasvir also showedconcentration-dependent cytotoxicity in HepaRG cells, with amean CC50 value of 23.7 �M. In contrast, no measurable in vitrocytotoxicity of samatasvir was observed in these cells after 14 daysof treatment at concentrations up to 100 �M (Table 3).
The off-target activity of samatasvir was evaluated in a panel offive commonly used human cell lines derived from renal, centralnervous system, blood, skeletal muscle, and colon tumors. As inthe Zluc replicon-bearing hepatic cells (CC50, �100 �M), no invitro cytotoxicity was apparent in any of the nonhepatic cell linesat the highest test concentration tested (50 �M) after 3 days ofsamatasvir exposure (Table 3). As in the hepatic lines, daclatasvirwas moderately toxic to all five nonhepatic cell lines, with CC50
values ranging from 10.5 to 25.5 �M.
Samatasvir resistance loci are limited to NS5A. As more ge-notype 1a- than 1b-infected subjects treated with daclatasvirmonotherapy exhibited viral breakthrough due to the selection ofresistant variants (35, 36), we selected genotype 1a replicon-bear-ing cells to determine the viral target of replicon inhibition bysamatasvir and its resistance selection profile. A genotype 1a rep-licon cell line was cultured in the presence of increasing concen-trations of samatasvir for 90 days, while untreated cells were cul-tured in parallel. Selection began at 2� the EC50 and increased in2-fold increments to a final concentration of 256� the EC50. Atthe end of selection, each samatasvir-treated cell line (719R-A,719R-B, and 719R-C) exhibited high resistance to samatasvir inthe replicon activity assays (1,100- to 3,100-fold change). Thesecell lines were also highly resistant to daclatasvir (630- to 2,200-fold change) but remained sensitive to the nucleotide polymeraseinhibitor IDX184 (0.81- to 1.6-fold change), suggesting that sa-matasvir may target NS5A (Table 8).
Population sequencing across the entire coding sequence of thereplicon was performed at the end of samatasvir selection. Dom-inant or minor substitutions appeared at 12 loci throughout thereplicon in at least one of the samatasvir-selected cell lines (Table9). Four minor substitutions (Y413H, L419P, and E420K in NS5Aand F574S in NS5B) were detected in only one samatasvir-selectedcell line. NS3 H593R was detected as a minor substitution in anuntreated and a single samatasvir-selected cell line. NS3 D518Nwas selected as a dominant or minor substitution in each of thesamatasvir-selected or untreated control cell lines. The low fre-quency of emergence and/or detection of these substitutions in the
TABLE 7 In vitro cytotoxicity in HepG2 cells after 3 days of treatment
Compound
Mean CC50 � SD (�M) fora:
LDHe Glucose XTT PAC
Samatasvir �100 �100 �100 �100Daclatasvir 17.4 � 0.3 14.9 � 3.7 17.7 � 3.1 17.6 � 1.6Doxorubicin 0.26 0.06 � 0.02 0.46 � 0.04 0.06 � 0.01a n 3. Cytotoxicity endpoints were membrane permeability for LDH3, physiologicalcell state for glucose, mitochondrial metabolism for XTT, and lysosomal activityfor PAC.
TABLE 8 Extent of resistance in samatasvir-selected genotype 1areplicon cell lines
Cell line
Mean fold change � SD witha:
Samatasvir Daclatasvir IDX184
719R-A 3,100 � 1,500 1,700 � 500 0.81 � 0.24719R-B 1,100 � 200 630 � 80 0.93 � 0.19719R-C 1,300 � 400 2,200 � 1,900 1.6 � 0.5a Mean fold changes � standard deviations are for the mean results from threeindependent experiments for each samatasvir-selected cell line compared to theirmatched untreated control cell line.
TABLE 9 Amino acid substitutions identified by population sequencing of untreated or samatasvir-selected genotype 1a replicon cellsa
Region Substitution 719R-AUntreatedcontrol A 719R-B
Untreatedcontrol B 719R-C
Untreatedcontrol C
NS3 T260A Dominant Minor Minor DominantD518N Dominant Dominant Minor Minor Dominant MinorH593R Minor Minor
NS4B E15G Dominant Minor Dominant Minor Dominant Minor
NS5A Y93H Dominant Dominant DominantE295G Dominant Minor DominantR318W Minor Minor DominantC404W Minor Minor MinorY413H MinorL419P MinorE420K Minor
NS5B F574S Minora The 719R-A/untreated control A, 719R-B/untreated control B, and 719R-C/untreated control C cell lines represent three independent experiments.
In Vitro Profile of Samatasvir, an HCV NS5A Inhibitor
August 2014 Volume 58 Number 8 aac.asm.org 4437
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
untreated cell lines suggests these were either random mutationsunrelated to samatasvir exposure or adaptive mutations acquiredto enhance the replicon replication.
NS3 T260A and NS4B E15G were observed as minor variantsin the untreated cell lines but emerged as dominant variants insamatasvir-selected cells (Table 9). Site-directed mutant repliconsbearing these individual substitutions significantly increased thereplication capacity of the replicon; however, their susceptibilityto samatasvir was comparable to that of the wild-type replicon.The NS3 T260A and NS4B E15G variants do not contribute tosamatasvir resistance but may confer a replicative/adaptive advan-tage (Table 10).
In NS5A, three dominant variants were observed (Y93H,E295G, and R318W), two of which (Y93H and R318W) were pres-ent in the samatasvir-selected cell lines but not in the control celllines. NS5A E295G was present as a minor variant in one controlcell line but became dominant in two samatasvir-treated cell lines.However, the activity of samatasvir against this substitution wassimilar to that in the wild type, indicating that this substitutiondoes not confer resistance to samatasvir (Table 10). The R318Wsubstitution dramatically reduced the fitness of the genotype 1areplicon to 1.8% of that of the wild type; the poor fitness of thisreplicon precluded measurement of an EC50 due to low luciferasesignal; therefore, its contribution to samatasvir resistance was notdetermined in the genotype 1a replicon. In contrast, the R318Wsubstitution in the genotype 1b replicon was replication compe-tent (79% of the wild type) and was susceptible to samatasvir(Table 10).
NS5A Y93H is an amino acid substitution that has been shownto confer significant resistance to NS5A inhibitors (10, 37–40),and when introduced into the genotype 1a replicon, this substitu-tion conferred a 4,400-fold shift in activity against samatasvir (Ta-ble 10). The emergence of Y93H was examined from RNA samples
collected at each cell passage. As the concentration of samatasvirincreased during selection, the Y93H substitution emerged as adominant variant in all three cell lines at approximately 128� theEC50. None of the three cell lines produced any other dominant(�50% penetrance) or minor (25 to 50% penetrance) variantwithin the first 100 amino acids of NS5A over the course of selec-tion (Table 9). An examination of the population sequences fromthe control cell lines showed no changes at Y93, suggesting that theobserved changes in the samatasvir-treated cell lines were aconsequence of selection pressure exerted by the drug. Thesedata strongly suggest that NS5A is a viral target of samatasvir,with Y93H being the predominant resistance-associated vari-ant identified following population sequencing of the genotype1a replicon.
Samatasvir resistance loci within NS5A. At the end of selec-tion, the cell line 719R-A was chosen for further investigation byclonal sequencing of the genomic region encoding the first 100amino acids of NS5A. All of the clones (100/100) selected fromthis cell line contained a substitution at one or more of the five lociM28, Q30, L31, P32, or Y93. In some clones, only a single substi-tution at one of these five loci was observed, suggesting that asingle substitution might be sufficient to promote resistance tosamatasvir. The clonal sequences obtained from the untreatedcontrol cell line A revealed only an M28I substitution at these loci,which was not identified in the 719R-A samatasvir-selected cellline; this suggests that M28I may be a random substitution. Theabsence of other substitutions at these loci in the control cell linesuggests that the substitutions observed in the 719R-A cell line arethe result of selection pressure from samatasvir.
Clonal sequencing of 719R-A confirmed that Y93 was the onlylocus with a dominant variant and the most highly mutated locus:58/100 clonal sequences contained a mutation at the Y93 locus,with 51/58 encoding Y93H variants, 4/58 encoding Y93C, and
TABLE 10 Activity of samatasvir against genotype 1a replicons bearing amino acid substitutions in NS3, NS4B, and NS5A
Region Substitution
Mean fold change � SD witha:Replicative capacity(% of WT � SD)aSamatasvir Daclatasvir IDX184
NS3 T260A 1.6 � 0.3 1.7 � 0.3 1.7 � 0.2 300 � 140NS4B E15G 1.5 � 0.1 1.6 � 0.2 1.6 � 0.1 264 � 18
NS5A K24E 1.8 � 0.5 1.8 � 0.4 1.6 � 0.2 79 � 25M28T 150 � 10 170 � 20 1.06 � 0.14 66 � 7Q30E 420 � 80 2,900 � 400 1.11 � 0.14 119 � 7Q30H 24 � 2 150 � 30 1.07 � 0.10 100 � 2Q30K 310 � 100 750 � 190 0.89 � 0.17 100 � 14Q30R 10 � 1 95 � 22 1.04 � 0.07 119 � 7L31F 68 � 14 43 � 9 1.06 � 0.31 85 � 4L31M 310 � 60 110 � 30 1.04 � 0.21 143 � 32L31V 420 � 80 900 � 180 1.33 � 0.10 210 � 8P32L 170 � 10 140 � 20 0.89 � 0.04 16 � 3Y93C 40 � 5 290 � 50 1.06 � 0.16 92 � 18Y93H 4,400 � 900 1,400 � 500 1.05 � 0.25 43 � 4Y93N 14,000 � 3,000 7,800 � 3,500 1.01 � 0.25 60 � 6E295G 1.6 � 0.2 1.6 � 0.2 1.3 � 0.1 132 � 36R318W (GT 1a)b RDc RD RD 1.8 � 0.8R318W (GT 1b)d 1.7 � 0.6 1.0 � 0.1 0.75 � 0.23 79 � 4
a n 3. WT, wild type.b NS5A R318W substitution in the genotype 1a replicon.c RD, replication deficient.d NS5A R318W substitution in the genotype 1b replicon.
Bilello et al.
4438 aac.asm.org Antimicrobial Agents and Chemotherapy
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

1/58 each of Y93R, Y93S, and Y93N (Table 11). Phenotypic anal-ysis of some of these substitutions indicated that similar to dacla-tasvir, these substitutions conferred moderate to high levels ofresistance in genotype 1 replicons (Table 10). Q30 was the secondmost highly mutated site (33/100) within the first 100 amino acidsof NS5A after selection. Unlike the Y93 locus that tended to mu-tate toward a single variant (Y93H), the substitutions at Q30 werenearly evenly distributed among four variants: Q30E (n 6),Q30H (n 6), Q30K (n 12), and Q30R (n 9) (Table 11).Substitutions at this locus conferred moderate to high levels ofresistance to samatasvir and high levels of resistance to daclatasvirin genotype 1a replicons (Table 10). Interestingly, the genotype 1bNS5A R30E substitution was not resistant to samatasvir (Table12). K24 was the third most highly mutated residue observed byclonal sequencing of the selected cell line (17/100). As with theQ30 locus, the substitutions at K24 were distributed among nu-merous variants: K24E (n 7), K24Q (n 3), K24R (n 3),K24T (n 3), and K24I (n 1) (Table 11); however, K24E did notconfer resistance to samatasvir or daclatasvir (Table 10). This,together with the fact that the substitutions at K24 were alwayslinked with at least one additional substitution shown to conferresistance to samatasvir (M28, Q30, or Y93 locus), suggests thatthe K24 locus may not confer resistance to samatasvir. Since theK24E substitution does not confer a replicative advantage (79% ofthe wild type), the contribution of this substitution to samatasvirresistance is unclear.
The two other loci identified with NS5A variants in the 719R-Acell line at some point during selection were M28 and L31. Byclonal sequencing, these loci show M28T (n 7), L31M (n 8),and L31P (n 1) substitutions (Table 11). M28T and L31M wereshown to confer high levels of resistance to samatasvir and dacla-tasvir (Table 10). The orthologous substitutions in genotype 1b,
L28T and L31M, conferred moderate and low resistance, respec-tively (Table 12). No single substitution at the K24, M28, Q30, andL31 loci appeared in �12 clones, but together, substitutions atthese loci accounted for 66 of the 189 variants observed in the 100clones from 719R-A. Ninety-six of the 100 clones contained atleast one mutation at the dominant variant locus Y93 or at one ofthe minor variant sites: K24, M28, Q30, or L31. The remainingclones (4/100) encoded a single P32L substitution, which con-ferred a high level of resistance to samatasvir (Table 10). Muta-tions in the K20 locus were identified in eight out of 100 clones,with 6/8 mutations encoding the K20T substitution. Because ofthe low penetrance of mutations at this site and the observationthat all substitutions at K20 were accompanied by substitutions atother loci, K20 was not considered a primary resistance locus. Twogenotype 1a substitutions reported in the literature but not ob-served in our resistance selection experiments conferred moderate(L31F; 68-fold) or high (L31V; 420-fold) resistance to samatasvir(Table 10) (37).
The resistance profile of samatasvir was further determined ingenotype 1b and 2a replicons bearing substitutions reported toconfer resistance to the NS5A inhibitor class (Table 12) (37, 41)(X. J. Zhou, B. Vince, J. Hill, E. Lawitz, W. O’Riordan, L. Webster,D. Gruener, R. S. Mofsen, A. Murillo, E. Donovan, J. Chen, J. Z.Sullivan-Bólyai, and D. Mayers, presented at the Asian Pacific As-sociation for the Study of the Liver, Suntec City, Singapore, 7 to 10March 2013). Substitutions at the L31 locus, as mentioned above,conferred 68- to 420-fold changes in genotype 1a but only 3.6- to15-fold changes in genotype 1b. The genotype 2a L31M polymor-phism confers approximately 90-fold resistance to samatasvir, inagreement with clinical results demonstrating a reduced responseto samatasvir in HCV genotype 2a-infected subjects bearing theNS5A M31 polymorphism (42; Zhou et al.). The genotype 1a P32Lsubstitution showed a 170-fold change in resistance, but in thecontext of genotype 1b, the change was only 6.7-fold. At the Y93locus, resistance ranged from 40- to 14,000-fold change with ge-notype 1a variants but 2.7- to 160-fold change in the genotype 1breplicon. Similar to previous studies, NS5A substitutions in thegenotype 1a replicon generally conferred higher resistance to sa-matasvir than those in the genotype 1b replicon (37, 38, 42).
Samatasvir is not cross-resistant with protease or polymer-ase inhibitors. The activity of samatasvir against substitutionsknown to confer resistance to different classes of HCV inhibitorswas evaluated. Three NS3 substitutions (R155K, A156T, and
TABLE 12 Activity of samatasvir against genotype 1b and 2a repliconsbearing amino acid substitutions in NS5A
Substitution Genotype
Mean fold change � SDa
Replicativecapacity(% of WT� SD)aSamatasvir Daclatasvir IDX184
L28T 1b 74 � 7 25 � 4 0.51 � 0.01 3 � 1R30E 1b 1.1 � 0.0 7.3 � 0.7 0.52 � 0.07 17 � 5L31F 1b 4.0 � 0.4 6.9 � 1.8 0.88 � 0.09 90 � 15L31M 1b 3.6 � 0.6 3.2 � 0.8 0.91 � 0.20 86 � 7L31M 2a 90 � 16 NDb ND 69 � 19L31V 1b 15 � 2 23 � 7 0.92 � 0.22 61 � 10P32L 1b 6.7 � 3.7 9.6 � 1.0 0.66 � 0.14 12 � 6Y93C 1b 2.7 � 0.3 2.1 � 0.1 0.73 � 0.14 33 � 8Y93H 1b 93 � 22 21 � 3 0.75 � 0.14 25 � 5Y93N 1b 160 � 30 35 � 5 0.69 � 0.14 23 � 6a n 3.b ND, not done.
TABLE 11 NS5A variants identified in the samatasvir-selected 719R-Acell linea
Locus Residue % penetrance
K20 M 1N 1T 6
K24b E 7I 1Q 3R 3T 3
M28 T 7
Q30 E 6H 6K 12R 9
L31 M 8P 1
P32 L 4
Y93 C 4H 51N 1R 1S 1
a Only variants with significance to samatasvir resistance are displayed.b All variants at K24 were linked to a substitution at either M28, Q30, or Y93.
In Vitro Profile of Samatasvir, an HCV NS5A Inhibitor
August 2014 Volume 58 Number 8 aac.asm.org 4439
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

D168V) and four NS5B substitutions (S282T, C316Y, M414T, andM423T) that confer resistance to protease or polymerase inhibi-tors, respectively, were tested (43–45) (J. P. Bilello, M. La Colla, I.Serra, J. M. Gillum, M. A. Soubasakos, C. Chapron, B. Li, A.Bonin, R. J. Panzo, L. B. Lallos, M. Seifer, and D. N. Standring,presented at the 16th International Symposium on Hepatitis CVirus and Related Viruses, Nice, France, 3 to 7 October 2009).Samatasvir exhibited similar activity against replicons bearingthese resistance variants and the wild-type replicon, indicatingthat samatasvir is not cross-resistant with protease, nucleoside, ornonnucleoside inhibitors (data not shown).
DISCUSSION
The current study evaluated the in vitro activity, cytotoxicity, andresistance profiles of samatasvir, a novel NS5A inhibitor of HCVreplication. As the in vitro activity of the initial NS5A inhibitors ofreplication varied significantly across HCV genotypes, our effortswere directed toward designing a pangenotypic NS5A replicationinhibitor with a narrow range of activity among the genotypes. Toassess this, we examined the activity of samatasvir against interge-notypic genotype 1b replicons bearing either the first 100 aminoacids or the full-length NS5A region from the orthologous regionsof genotypes 1 to 5. The in vitro activity of samatasvir is consistentamong all genotypes examined, with EC50s ranging from 2 to 24pM, which distinguishes it from other NS5A inhibitors, whoseactivity is reduced in genotypes beyond genotype 1b (8, 46). Fur-thermore, the in vitro activity of samatasvir against HCV repliconswas only mildly affected by human serum, such as AAG, and it wasnot reduced in the presence of commonly prescribed HBV or HIVantivirals. Samatasvir was effectively combined with other classesof direct-acting HCV antivirals, as well as interferon alfa (IFN-�)and ribavirin, demonstrating its suitability as part of a combina-tion treatment regimen in the clinical setting. Samatasvir was se-lective for HCV and was not active against other DNA and RNAviruses. Furthermore, samatasvir showed no evidence of in vitrocytotoxicity in an extensive panel of cell lines, affording a selectiv-ity index of �5 � 107.
Since NS5A does not have an enzymatic function, and a directassay to measure its inhibition is not available, we relied on indi-rect methods to verify the molecular target of inhibition by sama-tasvir. The only amino acid substitutions arising in selection ex-periments with samatasvir that were shown to confer resistance tosamatasvir were within NS5A, which suggests that samatasvir in-hibits the function(s) of NS5A.
Resistance selection with samatasvir generated a spectrum ofvariants in the genotype 1a replicon system within the first 100amino acids of NS5A, in concordance with previous results withdaclatasvir (8, 37, 47). The wide distribution of resistance variantswithin the first 100 amino acids of NS5A precluded their detectionby population sequencing because no single mutation achievedsufficient penetrance. The lone exception to this general observa-tion was the Y93 locus, for which the Y93H resistance substitutionwas clearly identified by population sequencing in all three sama-tasvir-resistant cell lines. NS5A Y93H was identified in 51 of the100 clonal sequences from the 719R-A resistant cell line. TheY93H substitution conferred 4,400-fold resistance to samatasvirwhen tested as a site-directed mutant in the genotype 1a replicon.An additional seven clonal sequences, representing four othervariants, were detected at the Y93 locus in the 719R-A resistant cellline. Thus, mutations at the Y93 locus were the predominant mu-
tations detected following selection with samatasvir in the geno-type 1a replicon.
Besides the Y93 locus, no other minor or dominant mutationswithin the genomic region encoding the first 100 amino acids ofNS5A were identified by population sequencing. However, clonalsequencing analysis revealed a more nuanced resistance profile forsamatasvir. All clonal sequences examined from the 719R-A resis-tant cell line encoded at least one substitution at one of the follow-ing five loci: M28, Q30, L31, P32, or Y93. The P32L substitutionwas observed infrequently and was not linked with any other sub-stitution among the clonal sequences examined. Substitutions atthe other four loci were observed as either single or linked substi-tutions within the region examined. Whether a substitution wasobserved to occur alone or linked to another was determined atleast in part by the degree of resistance conferred by the aminoacid substitution. The second most mutated locus following sa-matasvir selection was at position Q30, distributed among fourvariants. The Q30E substitution, which conferred 420-fold resis-tance to samatasvir, was not observed to be linked to any othersubstitution within the first 100 amino acids of NS5A. In contrast,the Q30H substitution conferred only 24-fold resistance to sama-tasvir and was always found to be linked to a substitution at theY93 locus. The Q30K substitution (310-fold change in EC50) wassometimes observed as the sole substitution within the first 100amino acids of NS5A, and at other times, it was linked to anothersubstitution. However, no Q30K substitution was seen linked to amutation at Y93 in any clonal sequence. Thus, a possible correla-tion exists between the degree of resistance conferred by a substi-tution at Q30 and whether the substitution was observed to occuralone or linked to another substitution.
Unlike substitutions at M28, Q30, L31, or Y93, which werefound both linked and not linked to another substitution, the K24substitutions were always linked to another substitution withinthe first 100 amino acids of NS5A. The K24 substitutions weredistributed among five different variants (K24E, K24I, K24Q,K24R, and K24T), with the K24E substitution being the most fre-quent, and they were always linked with another substitution atone or more of the M28, Q30, and Y93 loci. The K24E substitu-tion, which did not confer resistance to samatasvir by itself, wasnever accompanied by a substitution at Y93 but was always linkedto a substitution at Q30. A possible explanation for the emergenceof K24E is that it functions as a compensatory substitution torestore replicon fitness. Yet, all genotype 1a mutant replicons con-taining K24E-linked substitutions at the Q30 locus demonstratedfitness at least as robust as that of the wild-type replicon. Thebenefit of substitutions at the K24 locus to the replicon whentreated with samatasvir remains unclear.
Four dominant amino acid substitutions outside the first 100amino acid region of NS5A were identified by population se-quencing of the replicons from samatasvir-resistant cell lines.These included NS5A E295G and R318W, NS3 T260A, and NS4BE15G. These substitutions were either replication deficient or didnot confer resistance to samatasvir when examined individually inthe genotype 1a replicon. Thus, all substitutions that conferredresistance to samatasvir were found within the first 100 aminoacids of the NS5A protein.
Genotype 1a substitutions generally conferred higher levels ofresistance to samatasvir in cell culture, which correlates with bothin vitro and clinical observations of other NS5A inhibitors of HCVreplication (35–38). Consistent with the in vitro resistance profile
Bilello et al.
4440 aac.asm.org Antimicrobial Agents and Chemotherapy
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

for samatasvir presented here, the most common treatment-emergent variants observed following 3-day monotherapy withsamatasvir in the clinic were at the NS5A M28, L31, and Y93 loci(48). However, as samatasvir is not cross-resistant to other classesof direct-acting antivirals in vitro, clinical resistance is expected tobe controlled by combination therapy.
In clinical studies, samatasvir was initially evaluated at doses of1 to 100 mg daily for up to 7 days in healthy volunteers and 3 daysin HCV-infected subjects. The pharmacokinetics was dose pro-portional, with a terminal half-life of approximately 24 h, sup-porting once-a-day dosing. Following 3 daily doses of 25 to 100mg samatasvir, viral load reductions of up to 4 log10 were observedin subjects with HCV genotypes 1 through 4. A reduced antiviralresponse was observed in HCV genotype 2-infected subjects whohad the NS5A M31 polymorphism at baseline. Currently, HCV-infected patients with genotype 1 or 4 are being treated with com-bination regimens, including the FDA-approved protease inhibi-tor simeprevir and samatasvir, for 12 weeks. Samatasvir has beengenerally safe and well tolerated in all clinical trials to date. For thedaily all-oral regimen containing 150 mg simeprevir, 50 mg sama-tasvir, and weight-based ribavirin, the sustained viral responserate at 4 weeks posttreatment (SVR4) was 85% (42; Zhou et al.).
The low-picomolar multigenotypic activity of samatasvir invitro has been confirmed clinically in patients infected with HCVgenotypes 1 through 4 (42; Zhou et al.). The lack of cross-resis-tance to other classes of HCV direct-acting antivirals and additivedirect-acting antiviral combination data in vitro support the on-going phase II studies of samatasvir as part of all-oral interferon-free direct-acting antiviral regimens for the treatment of HCV.
ACKNOWLEDGMENTS
The MAGI-CCR5 cell line was obtained through the NIH AIDS ReagentProgram, Division of AIDS, NIAID, NIH, and HeLa-CD4-LTR-�-gal wasobtained from Julie Overbaugh.
We thank Idenix personnel Bianca Heinrich and Alice Blouet for ex-amining the activity of samatasvir against the R318W substitution in thegenotype 1b replicon, and we thank Teresa Dahlman for assistance inmanuscript preparation.
REFERENCES1. Burke KP, Cox AL. 2010. Hepatitis C virus evasion of adaptive immune
responses: a model for viral persistence. Immunol. Res. 47:216 –227. http://dx.doi.org/10.1007/s12026-009-8152-3.
2. Vertex Pharmaceuticals, Inc. 2011. Incivek (telaprevir) package insert.Vertex Pharmaceuticals, Cambridge, MA. http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/201917lbl.pdf.
3. Zeuzem S, Berg T, Moeller B, Hinrichsen H, Mauss S, Wedemeyer H,Sarrazin C, Hueppe D, Zehnter E, Manns MP. 2009. Expert opinion onthe treatment of patients with chronic hepatitis C. J. Viral Hepat. 16:75–90. http://dx.doi.org/10.1111/j.1365-2893.2008.01012.x.
4. Lange CM, Sarrazin C, Zeuzem S. 2010. Review article: specificallytargeted anti-viral therapy for hepatitis C–a new era in therapy. Aliment.Pharmacol. Ther. 32:14 –28. http://dx.doi.org/10.1111/j.1365-2036.2010.04317.x.
5. Gilead Sciences, Inc. 2013. Sovaldi (sofosbuvir) package insert. GileadSciences, Inc., Foster City, CA. http://www.gilead.com/�/media/Files/pdfs/medicines/liver-disease/sovaldi/sovaldi_pi.pdf.
6. Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. 2004.Structural biology of hepatitis C virus. Hepatology 39:5–19. http://dx.doi.org/10.1002/hep.20032.
7. He Y, Staschke KA, Tan SL. 2006. HCV NS5A: a multifunctional regu-lator of cellular pathways and virus replication, p 267–229. In Tan SL (ed),Hepatitis C virus: genomes and molecular biology. Horizon Bioscience,Norfolk, United Kingdom.
8. Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA,
Serrano-Wu MH, Langley DR, Sun JH, O’Boyle DR, Jr, Lemm JA,Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA,Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5Ainhibitor with a potent clinical effect. Nature 465:96 –100. http://dx.doi.org/10.1038/nature08960.
9. Yang PL, Gao M, Lin K, Liu Q, Villareal VA. 2011. Anti-HCV drugs inthe pipeline. Curr. Opin. Virol. 1:607– 616. http://dx.doi.org/10.1016/j.coviro.2011.10.019.
10. Lawitz EJ, Gruener D, Hill JM, Marbury T, Moorehead L, Mathias A,Cheng G, Link JO, Wong KA, Mo H, McHutchison JG, Brainard DM.2012. A phase 1, randomized, placebo-controlled, 3-day, dose-rangingstudy of GS-5885, an NS5A inhibitor, in patients with genotype 1 hepatitisC. J. Hepatol. 57:24 –31. http://dx.doi.org/10.1016/j.jhep.2011.12.029.
11. Merck and Co., Inc. 2007. Isentress (raltegravir) package insert. MerckSharpe & Dohme, Whitehouse Station, NJ. http://www.merck.com/product/usa/pi_circulars/i/isentress/isentress_pi.pdf.
12. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z,Murthy K, Habermann A, Kräusslich HG, Mizokami M, BartenschlagerR, Liang TJ. 2005. Production of infectious hepatitis C virus in tissueculture from a cloned viral genome. Nat. Med. 11:791–796. http://dx.doi.org/10.1038/nm1268.
13. Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR,Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitisC virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294 –9299.http://dx.doi.org/10.1073/pnas.0503596102.
14. Chackerian B, Long EM, Luciw PA, Overbaugh J. 1997. Human immu-nodeficiency virus type 1 coreceptors participate in postentry stages in thevirus replication cycle and function in simian immunodeficiency virusinfection. J. Virol. 71:3932–3939.
15. Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growthof human hepatoma cell lines with differentiated functions in chemicallydefined medium. Cancer Res. 42:3858 –3863.
16. Zhu Q, Guo JT, Seeger C. 2003. Replication of hepatitis C virus subge-nomes in nonhepatic epithelial and mouse hepatoma cells. J. Virol. 77:9204 –9210. http://dx.doi.org/10.1128/JVI.77.17.9204-9210.2003.
17. Buckwold VE, Wilson RJH, Nalca A, Beer BB, Voss TG, Turpin JA,Buckheit RW, III, Wei J, Wenzel-Mathers M, Walton EV, Smith RJ,Pannansch M, Ward P, Wells J, Chuvala L, Sloane S, Paulman R,Russell J, Hartman T, Ptak R. 2004. Antiviral activity of hop constituentsagainst a series of DNA and RNA viruses. Antiviral Res. 61:57– 62. http://dx.doi.org/10.1016/S0166-3542(03)00155-4.
18. Prichard MN, Prichard LE, Shipman C, Jr. 1993. Strategic design andthree-dimensional analysis of antiviral drug combinations. Antimicrob.Agents Chemother. 37:540–545. http://dx.doi.org/10.1128/AAC.37.3.540.
19. Delaney WE, IV, Yang H, Miller MD, Gibbs CS, Xiong S. 2004.Combinations of adefovir with nucleoside analogs produce additive anti-viral effects against hepatitis B virus in vitro. Antimicrob. Agents Che-mother. 48:3702–3710. http://dx.doi.org/10.1128/AAC.48.10.3702-3710.2004.
20. Dressler V, Müller G, Sühnel J. 1999. CombiTool–a new computerprogram for analyzing combination experiments with biologically activeagents. Comput. Biomed. Res. 32:145–160. http://dx.doi.org/10.1006/cbmr.1999.1509.
21. Feng JY, Ly JK, Myrick F, Goodman D, White KL, Svarovskaia ES,Borroto-Esoda K, Miller MD. 2009. The triple combination of tenofovir,emtricitabine and efavirenz shows synergistic anti-HIV-1 activity in vitro:a mechanism of action study. Retrovirology 6:44 –59. http://dx.doi.org/10.1186/1742-4690-6-44.
22. Chou TC, Talalay P. 1984. Quantitative analysis of dose-effect relationships:the combined effects of multiple drugs or enzyme inhibitors. Adv. EnzymeRegul. 22:27–55. http://dx.doi.org/10.1016/0065-2571(84)90007-4.
23. AbbVie, Inc. 2000. Kaletra (lopinavir/ritonavir) package insert. AbbVie,Inc., North Chicago, IL. http://www.rxabbvie.com/pdf/kaletratabpi.pdf.
24. Bristol-Myers Squibb Company. 1998. Sustiva (efavirenz) package insert.Bristol-Myers Squibb Company, Princeton, NJ. http://packageinserts.bms.com/pi/pi_sustiva.pdf.
25. Gilead Sciences, Inc. 2001. Viread (tenofovir) package insert. Gilead Sci-ences, Inc., Foster City, CA. http://www.gilead.com/�/media/Files/pdfs/medicines/liver-disease/viread/viread_pi.pdf.
26. GlaxoSmithKline. 1995. Epivir-HBV (lamivudine) package insert. Glaxo-SmithKline, Research Triangle Park, NC. http://us.gsk.com/products/assets/us_epivir_hbv.pdf.
In Vitro Profile of Samatasvir, an HCV NS5A Inhibitor
August 2014 Volume 58 Number 8 aac.asm.org 4441
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

27. GlaxoSmithKline. 1987. Retrovir (zidovudine) package insert. Glaxo-SmithKline, Research Triangle Park, NC. http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/019910s033lbl.pdf.
28. Novartis Pharmaceuticals Corporation. 2006. Tyzeka (telbivudine)package insert. Novartis Pharmaceuticals Corporation, East Hanover,NJ. http://www.pharma.us.novartis.com/cs/www.pharma.us.novartis.com/product/pi/pdf/tyzeka.pdf.
29. van den Hoff M, Moorman A, Lamers W. 1992. Electroporation in‘intracellular’ buffer increases cell survival. Nucleic Acids Res. 20:2902.http://dx.doi.org/10.1093/nar/20.11.2902.
30. Molla A, Vasavanonda S, Kumar G, Sham HL, Johnson M, GrabowskiB, Denissen JF, Kohlbrenner W, Plattner JJ, Leonard JM, Norbeck DW,Kempf DJ. 1998. Human serum attenuates the activity of protease inhib-itors toward wild-type and mutant human immunodeficiency virus. Vi-rology 250:255–262. http://dx.doi.org/10.1006/viro.1998.9383.
31. Benet LZ, Hoener BA. 2002. Changes in plasma protein binding havelittle clinical relevance. Clin. Pharmacol. Ther. 71:115–121. http://dx.doi.org/10.1067/mcp.2002.121829.
32. Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, GuyomardC, Lucas J, Trepo C, Guguen-Guillouzo C. 2002. Infection of a humanhepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A.99:15655–15660. http://dx.doi.org/10.1073/pnas.232137699.
33. Jossé RM, Aninat C, Glaise D, Dumont J, Fessard V, Morel F, Poul JM,Guguen-Guillouza C, Guillouza A. 2008. Long-term functional stabilityof human HepaRG hepatocytes and use for chronic toxicity and genotox-icity studies. Drug Metab. Dispos. 36:1111–1118. http://dx.doi.org/10.1124/dmd.107.019901.
34. Bort R, Ponsoda X, Jover R, Gómez-Lechón MJ, Castell JV. 1998.Diclofenac toxicity to hepatocytes: a role for drug metabolism in cell tox-icity. J. Pharmacol. Exp. Ther. 288:65–72.
35. Nettles RE, Gao M, Bifano M, Chung E, Persson A, Marbury TC,Goldwater R, DeMicco MP, Rodriguez-Torres M, Vutikullird A, Fuen-tes E, Lawitz E, Lopez-Talavera JC, Grasela DM. 2011. Multiple ascend-ing dose study of BMS-790052, a nonstructural protein 5A replicationcomplex inhibitor, in patients infected with hepatitis C virus genotype 1.Hepatology 54:1956 –1965. http://dx.doi.org/10.1002/hep.24609.
36. Wang C, Sun JH, O’Boyle DR, Jr, Nower P, Valera L, Roberts S, FridellRA, Gao M. 2013. Persistence of resistant variants in hepatitis C virus-infected patients treated with the NS5A replication complex inhibitor da-clatasvir. Antimicrob. Agents Chemother. 57:2054 –2065. http://dx.doi.org/10.1128/AAC.02494-12.
37. Fridell R, Qiu D, Wang C, Valera L, Gao M. 2010. Resistance analysis ofthe hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro repliconsystem. Antimicrob. Agents Chemother. 54:3641–3650. http://dx.doi.org/10.1128/AAC.00556-10.
38. Fridell RA, Wang C, Sun JH, O’Boyle DR, Jr, Nower P, Valera L, QiuD, Roberts S, Huang X, Kienzle B, Bifano M, Nettles RE, Gao M. 2011.Genotypic and phenotypic analysis of variants resistant to hepatitis C virusnonstructural protein 5A replication complex inhibitor BMS-790052 inhumans: in vitro and in vivo correlations. Hepatology 54:1924 –1935. http://dx.doi.org/10.1002/hep.24594.
39. Lin HM, Wang JC, Hu HS, Wu PS, Yang CC, Wu CP, Pu SY, Hsu TA,Jiaang WT, Chao YS, Chern JH, Yeh TK, Yueh A. 2011. Resistanceanalysis and characterization of a thiazole analogue, BP008, as a potenthepatitis C virus NS5A inhibitor. Antimicrob. Agents Chemother. 56:44 –53. http://dx.doi.org/10.1128/AAC.00599-11.
40. Lin HM, Wang JC, Hu HS, Wu PS, Wang WH, Wu SY, Yang CC, YehTK, Hsu TA, Jiaang WT, Chao YS, Chern JH, Yueh A. 2012. Resistancestudies of a dithiazol analogue, DBPR110, as a potential hepatitis C virusNS5A inhibitor in replicon systems. Antimicrob. Agents Chemother. 57:723–733. http://dx.doi.org/10.1128/AAC.01403-12.
41. Vince B, Hill JM, Lawitz EJ, O’Riordan W, Webster LR, Gruener DM,Mofsen RS, Murillo A, Donovan E, Chen J, McCarville JF, Sullivan-Bólyai JZ, Mayers D, Zhou XJ. 2014. A randomized, double-blind,multiple-dose study of the pan-genotypic NS5A inhibitor samatasvir inpatients infected with hepatitis C virus genotype 1, 2, 3 or 4. J. Hepatol.60:920 –927. http://dx.doi.org/10.1016/j.jhep.2014.01.003.
42. Liu R, Kong R, Mann P, Ingravallo P, Zhai Y, Xia E, Meinke P, Olsen D,Ludmerer S, Kozlowski J, Coburn C. 2012. In vitro resistance analysis ofHCV NS5A inhibitor: MK-8742 demonstrates increased potency against clin-ical resistance variants and higher resistance profile. J. Hepatol. 56(Suppl 2):S334–S335. http://dx.doi.org/10.1016/S0168-8278(12)60870-8.
43. Lallos LB, Bilello JP, Soubasakos MA, Gillum J, La Colla M, Serra I,Luizzi M, McCarville J, Seifer M, Good SS, Larsson M, Selden J, ParsyC, Surleraux D, Standring DN. 2009. Preclinical profiles of IDX136 andIDX316, two novel macrocyclic HCV protease inhibitors. J. Hepatol.50(Suppl 1):S132. http://dx.doi.org/10.1016/S0168-8278(09)60346-9.
44. Cretton-Scott E, Perigaud C, Peyrottes S, Licklider L, Camire M,Larsson M, La Colla M, Hildebrand E, Lallos LB, Bilello JP, McCarvilleJ, Seifer M, Liuzzi M, Pierra C, Badaroux E, Gosselin G, Surleraux D,Standring DN. 2008. In vitro antiviral activity and pharmacology ofIDX184, a novel and potent inhibitor of HCV replication. J. Hepatol.48(Suppl 2):S220. http://dx.doi.org/10.1016/S0168-8278(08)60590-5.
45. Nicolas O, Boivin I, St-Denis C, Bedard J. 2008. Genotypic analysis of HCVNS5B variants selected from patients treated with VCH-759. J. Hepatol.48(Suppl 2):S317. http://dx.doi.org/10.1016/S0168-8278(08)60846-6.
46. Cheng G, Peng B, Corsa A, Yu M, Nash M, Lee YJ, Xu Y, Kirschberg T,Tian Y, Taylor J, Link J, Delaney W, IV. 2012. Antiviral activity andresistance profile of the novel HCV NS5A inhibitor GS-5885. J. Hepatol.56(Suppl 2):S464. http://dx.doi.org/10.1016/S0168-8278(12)61184-2.
47. Wang C, Huang H, Valera L, Sun JH, O’Boyle DR, Jr, Nower PT, Jia L,Qiu D, Huang X, Altaf A, Gao M, Fridell RA. 2012. Hepatitis C virusRNA elimination and development of resistance in replicon cells treatedwith BMS-790052. Antimicrob. Agents Chemother. 56:1350 –1358. http://dx.doi.org/10.1128/AAC.05977-11.
48. McCarville J, Seifer M, Standring D, Mayers D. 2013. Treatment-emergent variants following 3 days of monotherapy with IDX719, a po-tent, pan-genotypic NS5A inhibitor, in subjects infected with HCV geno-types 1– 4. J. Hepatol. 58(Suppl 1):S491–S492. http://dx.doi.org/10.1016/S0168-8278(13)61210-6.
Bilello et al.
4442 aac.asm.org Antimicrobial Agents and Chemotherapy
on June 7, 2018 by guesthttp://aac.asm
.org/D
ownloaded from