Embed Size (px)
Citation preview


The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific Research (CNRS) on its electronic publishing platform I-Revues. Online and PDF versions of the Atlas of Genetics and Cytogenetics in Oncology and Haematology are hosted by INIST-CNRS.
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
Scope The Atlas of Genetics and Cytogenetics in Oncology and Haematology is a peer reviewed on-line journal in open access, devoted to genes, cytogenetics, clinical entities in cancer, and cancer-prone diseases. It presents structured review articles ("cards") on genes, leukaemias, solid tumours, cancer-prone diseases, more traditional review articles on these and also on surrounding topics ("deep insights"), case reports in hematology, and educational items in the various related topics for students in Medicine and in Sciences.
Editorial correspondance Jean-Loup Huret Genetics, Department of Medical Information, University Hospital F-86021 Poitiers, France tel +33 5 49 44 45 46 or +33 5 49 45 47 67 [email protected] or [email protected]
Staff Sylvie Yau Chun Wan - Senon
Database Director of the on-line version Philippe Dessen (Villejuif, France)
Chairman of the on-line version Alain Bernheim (Villejuif, France)
The Atlas of Genetics and Cytogenetics in Oncology and Haematology (ISSN 1768-3262) is published 4 times a year by ARMGHM, a non profit organisation.
http://AtlasGeneticsOncology.org
© ATLAS - ISSN 1768-3262

Atlas Genet Cytogenet Oncol Haematol. 2006;10(2)
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
Editor-in-Chief
Jean-Loup Huret (Poitiers, France)
Editorial Board Alessandro Beghini (Milan, Italy) Genes Section Anne von Bergh (Rotterdam, The Netherlands) Genes / Leukemia Sections Vasantha Brito-Babapulle (London, UK) Leukemia Section Charles Buys (Groningen, The Netherlands) Deep Insights Section Anne Marie Capodano (Marseille, France) Solid Tumors Section Fei Chen (Morgantown, West Virginia) Genes / Deep Insights Sections Antonio Cuneo (Ferrara, Italy) Leukemia Section Paola Dal Cin (Boston, Massachussetts) Genes / Solid Tumors Sections Louis Dallaire (Montreal, Canada) Education Section François Desangles (Paris, France) Leukemia / Solid Tumors Sections Gordon Dewald (Rochester, Minnesota) Leukemia / Deep Insights Sections Richard Gatti (Los Angeles, California) Cancer-Prone Diseases / Deep Insights Sections Oskar Haas (Vienna, Austria) Genes / Leukemia Sections Anne Hagemeijer (Leuven, Belgium) Deep Insights Section Nyla Heerema (Colombus, Ohio) Leukemia Section Jim Heighway (Liverpool, UK) Genes / Deep Insights Sections Sakari Knuutila (Helsinki, Finland) Deep Insights Section Lidia Larizza (Milano, Italy) Solid Tumors Section Lisa Lee-Jones (Newcastle, UK) Solid Tumors Section Edmond Ma (Hong Kong, China) Leukemia Section Cristina Mecucci (Perugia, Italy) Genes / Leukemia Sections Yasmin Mehraein (Homburg, Germany) Cancer-Prone Diseases Section Fredrik Mertens (Lund, Sweden) Solid Tumors Section Konstantin Miller (Hannover, Germany) Education Section Felix Mitelman (Lund, Sweden) Deep Insights Section Hossain Mossafa (Cergy Pontoise, France) Leukemia Section Florence Pedeutour (Nice, France) Genes / Solid Tumors Sections Susana Raimondi (Memphis, Tennesse) Genes / Leukemia Section Mariano Rocchi (Bari, Italy) Genes Section Alain Sarasin (Villejuif, France) Cancer-Prone Diseases Section Albert Schinzel (Schwerzenbach, Switzerland) Education Section Clelia Storlazzi (Bari, Italy) Genes Section Sabine Strehl (Vienna, Austria) Genes / Leukemia Sections Nancy Uhrhammer (Clermont Ferrand, France) Genes / Cancer-Prone Diseases Sections Dan Van Dyke (Rochester, Minnesota) Education Section Roberta Vanni (Montserrato, Italy) Solid Tumors Section Franck Viguié (Paris, France) Leukemia Section Thomas Wan (Hong Kong, China) Genes / Leukemia Sections Bernhard Weber (Würzburg, Germany) Education Section

Atlas Genet Cytogenet Oncol Haematol. 2006;10(2)
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
Volume 10, Number 2, April-June 2006
Table of contents
Gene Section MLLT10 (myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to, 10) 65 Cristina Morerio, Claudio Panarello
AF4p12 (ALL1 fused gene from chromosome 4p12) 68 Sandrine Hayette
CBFA2T3 (core-binding factor, runt domain, alpha subunit 2; translocated to, 3) 70 Anthony J Bais
CLDN4 (claudin-4) 77 Stefanie Ripka, Thomas M Gress
JAG1 (jagged 1 (Alagille syndrome)) 79 Michèle Meunier-Rotival, Catherine Driancourt, Julie Boyer-Di Ponio
MLL (myeloid/lymphoid or mixed lineage leukemia) 83 Jean-Loup Huret
MMP2 (matrix metallopeptidase 2 (gelatinase A, 72kDa gelatinase, 72kDa type IV collagenase) 88 Gopal Chandra Kundu, Pralhad Deepak Patil
PRRX2 (paired related homeobox 2) 91 Carine Gervais
SEL1L (sel-1 suppressor of lin-12-like (C. elegans)) 93 Ida Biunno, Monica Cattaneo
SIL (SCL/TAL1 interrupting locus) 97 Asher Castiel, Shai Izraeli
SIX1 (sine oculis homeobox homolog 1) (mammalian) 100 Heide L Ford, Aaron N Patrick, Marileila Varella-Garcia
STK4 (serine/threonine kinase 4) 103 Jonathan Chernoff
XRCC3 (X-ray repair complementing defective repair in Chinese hamster cells 3) 105 Ulla Vogel
Leukaemia Section Amplified NUP214/ABL1 107 Nathalie Nadal
dic(7;9)(p11-13;p11) 110 Sabine Strehl
dup(21q) amplified (RUNX1) 112 Anthony V Moorman, Christine J Harrison

Atlas Genet Cytogenet Oncol Haematol. 2006;10(2)
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
t(1;11)(q23;p15) 114 Jean-Loup Huret
t(2;9)(p11;p13) 115 Jean-Loup Huret
t(3;7)(q27;p12-13) 116 Jean-Loup Huret
t(3;16)(q27;p11) 118 Jean-Loup Huret
inv(7)(p15q34), t(7;7)(p15;q34) 120 Barbara Cauwelier, Nicole Dastugue, Anne Hagemeijer, Frank Speleman
t(9;22)(p24;q11.2) 123 Stefan K Bohlander
t(1;11)(q21;q23) 125 Marie-Agnès Collonge-Rame
Solid Tumour Section Angiomatoid fibrous histiocytoma (AFH) 127 Carolina Vicente-Dueñas, Isidro Sánchez-Garcîa
Cancer Prone Disease Section Alagille syndrome (AGS) 131 Michèle Meunier-Rotival, Michelle Hadchouel
Deep Insight Section Three-dimensional organization of the mammalian nucleus in normal and tumor cells 134 Sabine Mai

Gene Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 65
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
MLLT10 (myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to, 10) Cristina Morerio, Claudio Panarello
Dipartimento di Ematologia ed Oncologia Pediatrica, IRCCS Istituto Giannina Gaslini, Largo G. Gaslini 5, 16147 Genova, Italy
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/AF10.html DOI: 10.4267/2042/38285
This article is an update of: Huret JL. AF10 (ALL1 fused gene from chromosome 10). Atlas Genet Cytogenet Oncol Haematol.1997;1(2):51-52. This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: MLLT10 Other names: AF10 (ALL1 fused gene from chromosome 10) Location: 10p12
AF10 (10p12) - Courtesy Mariano Rocchi, Resources for Molecular Cytogenetics.
DNA/RNA Transcription 5' telomeric → 3' centromeric direction; 5.5 kb mRNA; coding sequence: 3.1 kb.
Protein Description 1027 amino acids; 109 KDa; N-term - LAP (leukemia associated protein)/PHD finger - Ext-LAP/PHD (Cys-rich region) - NLS (nuclear localisation signal) - AT-hook - LZ (leucine zipper) - Gln-rich domain - C-term.
Expression Mainly in the testis.
Localisation Nuclear.
Function Transcription factor.
Homology With AF17 and BR140.

MLLT10 (myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to, 10) Morerio C, Panarello C
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 66
Representation of the AF10 protein modified by Linder B et al. J Mol Biol 2000 and Jones LK et al. Leukemia 2001.
Implicated in
t(10;11)(p12;q23)/ANLL → MLL-AF10 Disease Mainly M4/M5 ANLL.
Cytogenetics Due to the opposite orientation of AF10 and MLL on their respective chromosome arms, MLL-AF10 gene fusion requires complex rearrangements with three or more breakpoints. An inversion of the 5'MLL or 3'AF10 is required in order to allow an in-frame MLL-AF10 fusion.
Hybrid/Mutated Gene 5' MLL - 3' AF10; breakpoints are at variable places along AF10.
Abnormal Protein The AF10 Ext-LAP/PHD is always deleted, as are the MLL PHD fingers. Both retain the C-term LZ domain necessary for malignant transformation.
inv ins(10;11)(p12;q23q12)/ANLL → MLL-AF10 Disease One case of pediatric M5 ANLL.
Hybrid/Mutated Gene 5' MLL - 3' AF10 and 5' AF10 - 3' HEAB, a gene at 11q12.
Abnormal Protein Only MLL-AF10 is expressed.
t(10;11)(p13;q14-21) → CALM -AF10 and/or AF10-CALM Disease Present both in myeloid and non myeloid acute leukemias: in T-cell ALL specific to TCRgd lineage; in myeloid leukemia described in FAB M0-AML, M1-AML, M5-AML, M7-AML.
Prognosis Poor.
Cytogenetics May well be confused with the above t(10;11)(p12;q23).
Hybrid/Mutated Gene 5' CALM - 3' AF10 and 5' AF10 - 3' CALM. In a 5' breakpoint cluster region (nucleotides 424 and 589), AF10 sequences retained the Ext-LAP/PHD domain. The presence of these kinds of sequences seems to be necessary for maturation toward the TCRgd lineage, whereas their absence leads to maturation arrest at a more immature stage.
Abnormal Protein Both CALM-AF10 and the reciprocal AF10-CALM are expressed. However, the CALM-AF10 contains most of the functional domains present in each of the two proteins.
Breakpoints Note: the breakpoint in the t(10;11)(p13;q14-21) is more in 5' of AF10.

MLLT10 (myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to, 10) Morerio C, Panarello C
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 67
References Bernard OA and Berger R. Molecular basis of 11q23 rearrangements in hematopoietic malignant proliferations. Genes Chromosomes Cancer 1995 Jun;13(2):75-85.
Chaplin T, Bernard O, Beverloo HB, Saha V, Hagemeijer A, Berger R, Young BD. The t(10;11) translocation in acute myeloid leukemia (M5) consistently fuses the leucine zipper motif of AF10 onto the HRX gene. Blood 1995 Sep 15;86(6):2073-2076.
Dreyling MH, Martinez-Climent JA, Zheng M, Mao J, Rowley JD, Bohlander SK. The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proc Natl Acad Sci USA 1996 May 14;93(10):4804-4809.
Rubnitz JE, et al. 11q23 rearrangements in acute leukemia. Leukemia 1996 Jan;10(1):74-82. (Review).
Young BD and Saha V. Chromosome abnormalities in leukaemia: the 11q23 paradigm. Cancer Surv 1996;28:225-245.
Kobayashi H, Hosoda F, Maseki N, Sakurai M, Imashuku S, Ohki M, Kaneko Y. Hematologic malignancies with the t(10;11) (p13;q21) have the same molecular event and a variety of morphologic or immunologic phenotypes. Genes Chromosomes Cancer 1997 Nov;20(3):253-259.
Dreyling MH, Schrader K, Fonatsch C, Schlegelberger B, Haase D, Schoch C, Ludwig WD, Löffler H, Büchner T, Wörmann B, Hiddemann W, Bohlander SK. MLL and CALM are fused to AF10 in morphologically distinct subsets of acute leukemia with translocation t(10;11): both rearrangements are associated with a poor prognosis. Blood 1998;12:4662-4667.
Lillington DM, Young BD, Berger R, Martineau M, Moorman AV, Secker-Walker LM. The t(10;11)(p12;q23) translocation in acute leukaemia: a cytogenetic and clinical study of 20 patients. Leukemia 1998;12:801-804.
Linder B, Jones LK, Chaplin T, Mohd-Sarip A, Heinlein UA, Young BD, Saha V. Expression pattern and cellular distribution of the murine homologue of AF10. Biochim Biophys Acta 1998;1443:285-96.
Bohlander SK, Muschinsky V, Schrader K, Siebert R, Schlegelberger B, Harder L, Schemmel V, Fonatsch C, Ludwig WD, Hiddemann W, Dreyling MH. Molecular analysis of the CALM/AF10 fusion: identical rearrangements in acute myeloid
leukemia, acute lymphoblastic leukemia and malignant lymphoma patients. Leukemia 2000;14:93-99.
Linder B, Newman R, Jones LK, Debernardi S, Young BD, Freemont P, Verrijzer CP, Saha V. Biochemical analyses of the AF10 protein: the extended LAP/PHD-finger mediates oligomerisation. J Mol Biol 2000;299:369-378.
Chaplin T, Jones L, Debernardi S, Hill AS, Lillington DM, Young BD. Molecular analysis of the genomic inversion and insertion of AF10 into MLL suggests a single-step event. Genes Chromosomes Cancer 2001;30:175-180.
Jones LK, Chaplin T, Shankar A, Neat M, Patel N, Samuel Dp, Hill AS, Debernardi S, Bassini A, Young BD, SahaV. Identification and molecular characterisation of a CALM-AF10 fusion in acute megakaryoblastic leukaemia. Leukemia 2001;15:910-914.
DiMartino JF, Ayton PM, Chen EH, Naftzger CC, Young BD, Cleary ML. The AF10 leucine zipper is required for leukemic transformation of myeloid progenitors by MLL-AF10. Blood 2002;99:3780-3785.
Van Limbergen H, Poppe B, Janssens A, De Bock R, De Paepe A, Noens L, Speleman F. Molecular cytogenetic analysis of 10;11 rearrangements in acute myeloid leukemia. Leukemia 2002;16:344-351.
Asnafi V, Radford-Weiss I, Dastugue N, Bayle C, Leboeuf D, Charrin C, Garand R, Lafage-Pochitaloff M, Delabesse E, Buzyn A, Troussard X, Macintyre E. CALM-AF10 is a common fusion transcript in T-ALL and is specific to the TCRgd lineage. Blood 2003;102:1000-1006.
Morerio C, Rapella A, Rosanda C, Lanino E, Lo Nigro L, Di Cataldo A, Maserati E, Pasquali F, Panarello C. MLL-MLLT10 fusion in acute monoblastic leukemia: variant complex rearrangements and 11q proximal breakpoint heterogeneity. Cancer Genet Cytogenet 2004;152:108-112.
Morerio C, Rapella A, Tassano E, Rosanda C, Panarello C. MLL-MLLT10 fusion gene in pediatric acute megakaryoblastic leukemia. Leukemia Res 2005;29:1223-1226.
This article should be referenced as such: Morerio C, Panarello C. MLLT10 (myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to, 10). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):65-67.

Gene Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 68
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
AF4p12 (ALL1 fused gene from chromosome 4p12) Sandrine Hayette
Laboratoire d'Hématologie et de cytogénétique, Hôpital Ed Herriot and INSERM U590, Lyon, France
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/AF4q12ID42970ch4p12.html DOI: 10.4267/2042/38286
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: FRYL Other names: DKFZp686E205; KIAA0826 Location: 4p12 Note: AF4p12 must be considered as a human ortholog of Drosophila Furry gene.
DNA/RNA Description The genomic size of the gene is about 185 kb and contains at least 61 exons.
Transcription mRNA size are about 11,42 kb with a large open reading frame of 9,318 kb. mRNA are expressed in a wide spectrum of normal tissues. The highest steady-state levels are in colon, placenta and brain.
Pseudogene No known pseudogene.
Protein Description The protein size is 3105 amino acids. It contains two potential leucine zipper domains (aa 1229-1250 and 2923-2944).
Expression See above the mRNA expression, protein expression has not been studied.
Localisation Not determined.
Function Not determined but displays transcriptional activation potential.
Homology AF4p12 shows about 60% identity to the human protein CAB42442. Two paralogs are found in human, rat and chicken, and one ortholog is found in Drosophila, C elegans, and Arabidopsis.
Implicated in t(4;11)(p12;q23)/Treatment-related acute lymphoblastic leukemia (t-ALL) → MLL-AF4p12
The t(4;11) translocation breakpoint between exon 6 from the MLL gene and exon 49 from AF4p12. Black bars, chromosome 11 DNA regions; grey bars, chromosome 4 DNA regions. MLL exons are indicated by black boxes, AF4p12 exons are indicated by grey boxes.
Schematic representation of the domain structures of MLL and of the MLL/AF4p12 fusion protein. MT, DNA methyltransferase homology domain; SET, SET domain; LZ, Leucine Zipper domain. Arrows show the fusion point. Numbers refer to the positions of amino acids in wild-type MLL or AF4p12. In the predicted chimeric MLL/AF4p12 fusion protein, the MLL zinc finger and the MLL SET domains have been replaced by the AF4p12 leucine zipper domain.

AF4p12 (ALL1 fused gene from chromosome 4p12) Hayette S
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 69
Disease B-ALL.
Prognosis Only one patient described, but she died one month after ALL diagnosis.
Cytogenetics Translocation t(4;11)(p12;q23).
Hybrid/Mutated Gene MLL-AF4p12.
Abnormal Protein MLL-AF4.
Oncogenesis The fusion domain of AF4p12 to the chimeric protein
MLL-AF4p12 displays transcriptional activation potential and the gain of transcriptional effector properties could contribute to the transformation of lymphoid progenitor by the fusion protein.
References Hayette S, Cornillet-Lefebvre P, Tigaud I, Struski S, Forissier S, Berchet A, Doll D, Gillot L, Brahim W, Delabesse E, Magaud JP, Rimokh R. AF4p12, a human homologue to the furry gene of Drosophila, as a novel MLL fusion partner. Cancer Res 2005 ;65:6521-6525.
This article should be referenced as such: Hayette S. AF4p12 (ALL1 fused gene from chromosome 4p12). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):68-69.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 70
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
CBFA2T3 (core-binding factor, runt domain, alpha subunit 2; translocated to, 3) Anthony J Bais
Department of Haematology and Genetic Pathology, Flinders University, Bedford Park, Adelaide, SA 5042, Australia
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/CBFA2T3ID428ch16q24.html DOI: 10.4267/2042/38287
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: CBFA2T3 Other names: MTG16; MTGR2; ZMYND4 Location: 16q24.3 Local order: telomere; centromeric to CDH15 and telomeric to GALNS.
DNA/RNA Description CBFA2T3 encodes two alternative transcripts, CBFA2T3A and CBFA2T3B, via the use of alternative start sites at exons 1A and 1B, respectively. CBFA2T3A is 4,265-bp in length, composed of 13 exons (1A and 2 to 12) spanning approximately 130-kb of genomic DNA, and has an ORF of 1,959-bp encoding a protein of 653 amino acids. CBFA2T3B is 4,034-bp in length, composed of 12 exons (1B to 12 splicing out exon 3) spanning approximately 50-kb of genomic DNA, and has an ORF of 1,701-bp encoding a protein of 567 amino acids. Additional CBFA2T3C and CBFA2T3D isoforms have been identified in leukemic and HEL cell lines. CBFA2T3C encodes a protein that lacks exons 2 and 3, and CBFA2T3D is a truncated protein with out-of-frame splicing of exon 1A to exon 5. The CBFA2T3A open-reading-frame (ORF) may include an additional 177 amino acids beyond the originally proposed methionine start codon. The CBFA2T3B isoform contains a high-density 1-kb CpG island within and five prime to the exon 1B start site. A hypothetical protein FLJ26728 located within and proximal to the CpG island transcribes antisense to
CBFA2T3. Another hypothetical protein FLJ23429 transcribes antisense starting from within the first alternative intron.
Pseudogene None identified.
Protein Description ETO proteins are composed of four evolutionarily conserved domains termed nervy homology regions (NHR1 to 4) and three proline-serine-threonine (PST) rich regions. The fourth NHR region is also referred to as the zinc-finger MYND (zf myeloid-nervy-DEAF-1) domain. NHR1 shares significant homology to human TATA-binding protein (TBP)-associated factor 130 (hTAF 130), hTAF 105, Drosophila accessory or activation factor TAF 110, and is often referred to as the TAF 110 domain. NHR2 is a small region containing homo and heterodimerization domains and a hydrophobic heptad repeat (HHR) unit. The sequence of NHR3 is unremarkable in homology and often referred to as the nervy domain. The C-terminal NHR4 domain exists in numerous human, murine, Caenorhabditis elegans and Drosophila proteins, and contains a MYND zinc-finger motif. The motif is composed of CXXC and two (C-H)XXXC regions which correspond to cysteine-histidine 'knuckle structures' that are the basic building blocks of many zinc-finger proteins. The zinc-finger is common to the developmental proteins rat programmed cell death (RP-8), the human homolog programmed cell death 2 (PDCD2), deformed epidermal autoregulatory factor-1

CBFA2T3 (core-binding factor, runt domain, alpha subunit 2; translocated to, 3) Bais AJ
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 71
(DEAF-1), suppressin (SPN), BLU or zinc-finger MYND domain containing 10 (ZMYND10), adenovirus 5 E1A binding protein (BS69), and CD8 beta opposite (BOP).
Expression CBFA2T3 is widely expressed in B-cells, blood, brain, breast, cervix, colon, eye, kidney tumor, lymph, marrow, muscle, pancreas, placenta and tonsil. CBFA2T3 exists predominantly as 4.5 and 4.2-kb transcripts along with several other minor RNAs in heart, brain, placenta, lung, liver, skeletal muscle, kidney, pancreas, spleen, thymus, prostate, testis, ovary, small intestine, colon and peripheral blood leukocyte.
Localisation All ETO members contain nuclear localization signals (NLS), some of which may be abrogated through five prime variations to enable extracellular targeting. For instance, CBFA2T1 has been detected in the cytoplasm of Purkinje cells in adult human brain, and both CBFA2T1 and CBFA2T3B have been detected in the Golgi apparatus, where they may function as cAMP-dependent protein kinase anchoring proteins. The majority of ETO proteins presumably remain in the nucleus for transcriptional regulation.
Function CBFA2T3 has been considered to function as a transcriptional repressor via interaction with corepressor complexes. The CBFA2T3A isoform oligomerizes and drags MTG8 and MTGR1 to the nucleus, oligomerizes with RUNX1-MTG16 fusion proteins in the nucleoplasm, interacts with nuclear HDACs 1 and 3, and when overexpressed accumulates at the periphery of nucleoli in characteristic rings. Because clustered arrays of inactive methylated ribosomal DNA (rDNA) repeats are also found at the nucleoli periphery, it has been speculated that CBFAT23A could be involved in methylation silencing of rDNA in the nucleolus. The CBFA2T3B isoform has been shown to function in T lymphocytes as a kinase anchorage protein, and interact with cyclic nucleotide phosphodiesterases, suggesting it may function in T cell activation and inflammatory response. CBFA2T3B has been shown to function as a transcriptional repressor when tethered to a GAL4 DNA-binding domain in gene reporter assays, and inhibit the growth of breast tumor cell lines with reduced expression when ectopically expressed using retrovirus. CBFA2T3 has been found to interact with a novel zinc finger protein KIA00924 to mediate potent transcriptional repression as determined by CAT
reporter gene assays. The presence of a zinc-finger motif common to developmental proteins suggests that CBFA2T3 might function in regulating differentiation and morphogenesis. The RP-8 and human homolog PDCD2 proteins assume a role in programmed cell death, a process essential for epithelial turnover. DEAF-1 is essential for early embryonic dorsal epidermal, eye and wing development in Drosophila. BOP encodes a muscle-restricted protein essential for cardiomyocyte differentiation and morphogenesis. BLU is a candidate tumor suppressor gene from the 3p21.3 LOH region in many human cancers, and SPN from rat functions as a potent tumor suppressor of leukemia, lymphoma and thymoma cells and tumor cells from the brain, breast, pituitary and adrenal glands. CBFA2T3 transcripts of CD34(+) progenitor cells have been shown to be rapidly reduced by cytokine-induced differentiation into myeloid or erythroid lineages, supporting suggestion that CBFA2T3 may function in hematopoietic differentiation. The CBFA2T3B CpG island contains several Specificity protein 1 (Sp1), homeotic, epidermal and insulin growth factor recognition sites. High conserved binding sites include GATA-1, CREB, F-1 and PKNOX1.
Homology The human gene CBFA2T3 is a member of the eight-twenty-one (ETO) family of proteins. The human ETO family consists of the ETO gene, also known as the myeloid translocation gene 8 (MTG8, CBFA2T1), the myeloid translocation gene related protein-1 (EHT, MTGR1, CBFA2T2), and the myeloid translocation gene 16 (MTG16, MTGR2, ZMYND4, HGNC:1537, CBFA2T3). Murine homologs of the ETO family include mETO (cbfa2t1h), ET0-2 (cbfa2t3h), and cbfa2t2h, the latter of which is uncharacterised. Chicken cETO and Drosophila nervy homologs have also been identified. The ETO protein family members and NHRs are highly homologous and conserved to each other. The CBFA2T3A and B isoforms share significant homology to MTG8 (67 and 75%, respectively) and MTGR1 (54 and 61%, respectively), and approximately 30% homology to nervy. CBFA2T3 shares 86% homology to the murine ETO-2 (cbfa2t3h), which in turn shares 75 and 60% homology to MTG8 and MTGR1, respectively, suggesting that ETO-2 is the murine homolog of CBFA2T3. MTG8 is approximately 99, 65 and 30% homologous to murine ETO, MTGR1 and nervy, respectively.
Mutations Note: None recorded.

CBFA2T3 (core-binding factor, runt domain, alpha subunit 2; translocated to, 3) Bais AJ
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 72
Implicated in t(16;21)(q24;q22) in therapy related acute myeloid leukemia (t-AML) → CBFA2T3/RUNX1 Note: CBFA2T3 or MTG16 was identified by molecular characterization of a second less common therapy-related AML translocation involving t(16;21). Characterization of the t(16;21) demonstrated that RUNX1-MTG16 fusion transcripts similar to RUNX1-ETO were generated.
Abnormal Protein The RUNX1-MTG8, and to a lesser extent the RUNX1-MTG16, fusion transcripts of the t(8;21)(q22;q22) and t(16;21) events are most recognized for their ability to function as transcriptional repressors of genes normally activated by RUNX1 through their interaction with corepressor complexes involving N-CoR, mSin3A, SMRT, and HDACs. The MTG8 or CBFA2T1 gene is the most studied member of the ETO family and is additionally recognized for its ability to function independently as a nuclear transcriptional repressor through nuclear corepressor complex interaction. Gene targeting experiments demonstrating that homozygous mutant mice with an inactivating insertion of LacZ in exon 2 of the MTG8 locus resulted in massive gastrointestinal defects, have also shown that MTG8 and perhaps ETO family members are essential for gastrointestinal development. Multiple NHRs of the ETO family of proteins are required and cooperate to mediate transcriptional repression. Most of the proteins that interact with NHRs have been assigned from studies of RUNX1-MTG8 and its interacting proteins. Studies originally demonstrated that MTG8s NHR2 and NHR4 were required for AML-MTG8 to inhibit RUNX1 mediated transcriptional activation and initiated the search for 'corepressor complexes' that bind to these regions. Several yeast two-hybrid systems using portions of MTG8 as bait demonstrated that specific portions of NHR2, NHR3 and NHR4 interact with the human nuclear receptor co-repressor (HuN-CoR)-mSin3A-HDAC1 corepressor complex. The N-CoR protein was originally described to interact with DNA-bound nuclear receptors to repress transcription of target genes through recruitment of HDAC containing complexes and was latter shown to form complexes with mammalian Sin3 to alter chromatin structure and mediate transcriptional repression via nuclear receptors and oncoregulatory proteins. A similar yeast two-hybrid approach also established that the corepressor silencing mediator of retinoic acid and thyroid hormone receptors (SMRT) interacts with
MTG8. The zinc-finger NHR4 is necessary but not sufficient for repression and interaction with SMRT and N-CoR in vitro. The direct physical association of MTG8 with corepressors is more complex. A 'core-repressor domain' containing NHR2 and the neighboring amino and carboxy terminal sequences was defined and found to be the strongest region of interaction with mSin3a and transcriptional repression. In the RUNX1-MTG8 translocation product associated with myeloid malignancies, the transactivation domain of the RUNX1 gene, which normally binds the transcriptional coactivators p300-CBP, is replaced by almost the entire MTG8 protein. The resultant fusion protein recruits a corepressor complex containing HDAC activity instead of the coactivators p300 - CBP to RUNX1 responsive genes giving rise to leukemia. The repression activity of the MTG8 protein was demonstrated from a GAL4 DNA binding domain (DBD) MTG8 fusion construct which mediated strong repression through multimerized GAL4 binding sites upstream of a minimal promoter driving a reporter gene. Consistent with a mechanism involving MTG8 and HDAC corepressor interactions, the repressive effect of MTG8 was partially overcome with the addition of the HDAC inhibitor trichostatin A (TSA). Mutational studies of RUNX1-MTG8 have shown that NHR4 is responsible for repression of the multidrug resistance 1 promoter. MTG8 has also been shown to interact with the Dentato-rubral and Pallido-luysian atrophy gene product, atrophin-1, in a yeast two-hybrid assay. Both Dentato-rubral and Pallido-luysian atrophies are neurodegenerative disorders caused by expansion of polyglutamine tracts. Several other transcriptional repressors have been shown to interact with MTG8. The promyelocytic leukemia zinc-finger (PLZF), a transcriptional repressor found in hematopoietic cells and down-regulated during differentiation of myeloid cell lines was shown to exhibit enhanced repressor activity when interacting with MTG8 in 293T cells and assayed using a reporter plasmid containing four copies of a high affinity PLZF binding site linked to firefly luciferase. The ability of MTG8 to enhance repression was abolished upon addition of HDAC inhibitors TSA and sodium butyrate, suggesting that the MTG8 enhanced repression by PLZF is also mediated through the recruitment of HDACs. The Growth factor independence-1 (Gfi-1), a HDAC interacting transcriptional repressor found in hematopoietic cells, was shown in vitro and in vivo to interact with NHR1 and NHR2 of MTG8. These interactions together with gene targeting experiments demonstrating MTG8s involvement in gastrointestinal development, the well established interactions with corepressor complexes, and the presence of a zinc-finger motif common to numerous developmental proteins, suggests that ETO family members including

CBFA2T3 (core-binding factor, runt domain, alpha subunit 2; translocated to, 3) Bais AJ
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 73
CBFA2T3 may play function in regulating cell cycle transcription during differentiation and proliferation of specific cell type-lineages, for example hematopoietic cells as indicated for MTG8.
Loss of heterozygosity (LOH) of 16q22-qter in breast cancer, prostate cancer, and several other cancers Note: This region is frequently deleted in several human cancers causing loss of heterozygosity. The 16q24.3 region including CBFA2T3 spans approximately 3-Mb from the marker D16S498 to the telomere and contains at least two smallest regions of overlap (SROs). These SROs are most frequently deleted in early and late stage breast cancer and in prostate cancer. Loss of normal function of CBFA2T3 may be a key event in the early stage of breast cancer. LOH on the whole 16q22-qter region is frequently detected in breast and prostate cancer. CBFA2T3B is a potential tumor suppressor gene affected by LOH, aberrant gene expression and promoter methylation in breast cancer. Quantitative gene expression analysis of 78 genes in the 16q24.3 region demonstrated that CBFA2T3 was the only gene with an aberrant expression profile distinctly similar to the known tumor suppressors SYK and CDKN2A. 68 genes displayed normal expression, six displayed mildly aberrant expression (DPEP1, CDH15, Hs.17074, Hs.189419, SLC7A5 and AA994450), one displayed excessively reduced expression (CA5A), and two displayed moderately aberrant expression (CYBA and FBX031). The CBFA2T3B promoter region displays aberrant hypo and hypermethylation in breast tumor cell lines and primary breast tumor samples in correlation with aberrant gene expression.
Disease 16q22-qter LOH is detected in bilateral breast cancer and ductal lavage, in rare inflammatory breast cancer, and in several other cancers, including central nervous system neuroectodermal and primary ependymomas, colorectal liver metastases, gastric tumor, head and neck squamous cell carcinoma, hepatocellular carcinoma, lung tumor, nasopharyngeal tumor, ovarian tumor, rhabdomyosarcoma, and Wilms' tumor. 16q22-qter LOH in ovarian, hepatocellular and particularly breast and prostate cancers, exhibit similar SROs, suggesting common molecular pathways are affected.
References Carter BS, Ewing CM, Ward WS, Treiger BF, Aalders TW, Schalken JA, Epstein JI, Isaacs WB. Allelic loss of chromosomes 16q and 10q in human prostate cancer. Proc Natl Acad Sci USA 1990;87:8751-8755.
Tsuda H, Zhang WD, Shimosato Y, Yokota J, Terada M, Sugimura T, Miyamura T, Hirohashi S. Allele loss on chromosome 16 associated with progression of human hepatocellular carcinoma. Proc Natl Acad Sci USA 1990;87:6791-6794.
Zhang WD, Hirohashi S, Tsuda H, Shimosato Y, Yokota J, Terada M, Sugimura T. Frequent loss of heterozygosity on chromosomes 16 and 4 in human hepatocellular carcinoma. Jpn J Cancer Res 1990;81:108-111.
Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci USA 1991;88:10431-10434.
Owens GP, Hahn WE, Cohen JJ. Identification of mRNAs associated with programmed cell death in immature thymocytes. Mol Cell Biol 1991;11:4177-4188.
Sato T, Akiyama F, Sakamoto G, Kasumi F, Nakamura Y. Accumulation of genetic alterations and progression of primary breast cancer. Cancer Res 1991;51:5794-5799.
Thomas GA, Raffel C. Loss of heterozygosity on 6q, 16q, and 17p in human central nervous system primitive neuroectodermal tumors. Cancer Res 1991;51:639-643.
Erickson P, Gao J, Chang KS, Look T, Whisenant E, Raimondi S, Lasher R, Trujillo J, Rowley J, Drabkin H. Identification of breakpoints in t(8;21) acute myelogenous leukemia and isolation of a fusion transcript, AML1/ETO, with similarity to Drosophila segmentation gene, runt. Blood 1992;80:1825-1831.
Maw MA, Grundy PE, Millow LJ, Eccles MR, Dunn RS, Smith PJ, Feinberg AP, Law DJ, Paterson MC, Telzerow PE, et al. A third Wilms' tumor locus on chromosome 16q. Cancer Res 1992;52:3094-3098.
Sakai K, Nagahara H, Abe K, Obata H. Loss of heterozygosity on chromosome 16 in hepatocellular carcinoma. J Gastroenterol Hepatol 1992;7:288-292.
Hoey T, Weinzierl RO, Gill G, Chen JL, Dynlacht BD, Tjian R. Molecular cloning and functional analysis of Drosophila TAF110 reveal properties expected of coactivators. Cell 1993;72:247-260.
Miyoshi H, Kozu T, Shimizu K, Enomoto K, Maseki N, Kaneko Y, Kamada N, Ohki M. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J 1993;12:2715-2721.
Erickson PF, Robinson M, Owens G, Drabkin HA. The ETO portion of acute myeloid leukemia t(8;21) fusion transcript encodes a highly evolutionarily conserved, putative transcription factor. Cancer Res 1994;54:1782-1786.
Schwabe JW, Klug A. Zinc mining for protein domains. Nat Struct Biol 1994;1:345-349.
Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 1995;377:454-457.
Feinstein PG, Kornfeld K, Hogness DS, Mann RS. Identification of homeotic target genes in Drosophila melanogaster including nervy, a proto-oncogene homologue. Genetics 1995;140:573-586.
Hörlein AJ, Näär AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Söderström M, Glass CK, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995;377:397-404.
Hwang I, Gottlieb PD. Bop: a new T-cell-restricted gene located upstream of and opposite to mouse CD8b. Immunogenetics 1995;42:353-361.
Lenny N, Meyers S, Hiebert SW. Functional domains of the t(8;21) fusion protein, AML-1/ETO. Oncogene 1995;11:1761-1769.
Meyers S, Hiebert SW. Indirect and direct disruption of transcriptional regulation in cancer: E2F and AML-1. Crit Rev Eukaryot Gene Expr 1995;5:365-383.

CBFA2T3 (core-binding factor, runt domain, alpha subunit 2; translocated to, 3) Bais AJ
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 74
Niwa-Kawakita M, Miyoshi H, Gotoh O, Matsushima Y, Nishimura M, Shisa H, Ohki M. Cloning and gene mapping of the mouse homologue of the CBFA2T1 gene associated with human acute myeloid leukemia. Genomics 1995;29:755-759.
Ross CA. When more is less: pathogenesis of glutamine repeat neurodegenerative diseases. Neuron 1995;15:493-496.
Grimes HL, Chan TO, Zweidler-McKay PA, Tong B, Tsichlis PN. The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol Cell Biol 1996;16:6263-6272.
Gross CT, McGinnis W. DEAF-1, a novel protein that binds an essential region in a Deformed response element. EMBO J 1996;15:1961-1970.
Suzuki H, Komiya A, Emi M, Kuramochi H, Shiraishi T, Yatani R, Shimazaki J. Three distinct commonly deleted regions of chromosome arm 16q in human primary and metastatic prostate cancers. Genes Chromosomes Cancer 1996;17:225-233.
Alland L, Muhle R, Hou H Jr, Potes J, Chin L, Schreiber-Agus N, DePinho RA. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature 1997;387:49-55.
Godfrey TE, Cher ML, Chhabra V, Jensen RH. Allelic imbalance mapping of chromosome 16 shows two regions of common deletion in prostate adenocarcinoma. Cancer Genet Cytogenet 1997;98:36-42.
Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature 1997;387:43-48.
Latil A, Cussenot O, Fournier G, Driouch K, Lidereau R. Loss of heterozygosity at chromosome 16q in prostate adenocarcinoma: identification of three independent regions. Cancer Res 1997;57:1058-1062.
Visser M, Sijmons C, Bras J, Arceci RJ, Godfried M, Valentijn LJ, Voûte PA, Baas F. Allelotype of pediatric rhabdomyosarcoma. Oncogene 1997;15:1309-1314.
Calabi F, Cilli V. CBFA2T1, a gene rearranged in human leukemia, is a member of a multigene family. Genomics 1998;52:332-341.
Chou YH, Chung KC, Jeng LB, Chen TC, Liaw YF. Frequent allelic loss on chromosomes 4q and 16q associated with human hepatocellular carcinoma in Taiwan. Cancer Lett 1998;123:1-6.
Fracchiolla NS, Colombo G, Finelli P, Maiolo AT, Neri A. EHT, a new member of the MTG8/ETO gene family, maps on 20q11 region and is deleted in acute myeloid leukemias. Blood 1998;92:3481-3484.
Gamou T, Kitamura E, Hosoda F, Shimizu K, Shinohara K, Hayashi Y, Nagase T, Yokoyama Y, Ohki M. The partner gene of AML1 in t(16;21) myeloid malignancies is a novel member of the MTG8(ETO) family. Blood 1998;91:4028-4037.
Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol 1998;18:7185-7191.
Kitabayashi I, Ida K, Morohoshi F, Yokoyama A, Mitsuhashi N, Shimizu K, Nomura N, Hayashi Y, Ohki M. The AML1-MTG8 leukemic fusion protein forms a complex with a novel member of the MTG8(ETO/CDR) family, MTGR1. Mol Cell Biol 1998;18:846-8458.
Lutterbach B, Westendorf JJ, Linggi B, Patten A, Moniwa M, Davie JR, Huynh KD, Bardwell VJ, Lavinsky RM, Rosenfeld
MG, Glass C, Seto E, Hiebert SW. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol 1998;18:7176-7184.
Lutterbach B, Sun D, Schuetz J, Hiebert SW. The MYND motif is required for repression of basal transcription from the multidrug resistance 1 promoter by the t(8;21) fusion protein. Mol Cell Biol 1998;18:3604-3611.
Sacchi N, Tamanini F, Willemsen R, Denis-Donini S, Campiglio S, Hoogeveen AT. Subcellular localization of the oncoprotein MTG8 (CDR/ETO) in neural cells. Oncogene 1998;16:2609-2615.
Sato M, Mori Y, Sakurada A, Fukushige S, Ishikawa Y, Tsuchiya E, Saito Y, Nukiwa T, Fujimura S, Horii A. Identification of a 910-kb region of common allelic loss in chromosome bands 16q24.1-q24.2 in human lung cancer. Genes Chromosomes Cancer 1998;22:1-8.
Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci USA 1998;95:10860-10865.
Davis JN, Williams BJ, Herron JT, Galiano FJ, Meyers S. ETO-2, a new member of the ETO-family of nuclear proteins. Oncogene 1999;18:1375-1383.
Mori Y, Matsunaga M, Abe T, Fukushige S, Miura K, Sunamura M, Shiiba K, Sato M, Nukiwa T, Horii A. Chromosome band 16q24 is frequently deleted in human gastric cancer. Br J Cancer 1999;80:556-562.
Wang X, Gleich L, Pavelic ZP, Li YQ, Gale N, Hunt S, Gluckman JL, Stambrook PJ. Cervical metastases of head and neck squamous cell carcinoma correlate with loss of heterozygosity on chromosome 16q. Int J Oncol 1999;14:557-561.
Wang J, Saunthararajah Y, Redner RL, Liu JM. Inhibitors of histone deacetylase relieve ETO-mediated repression and induce differentiation of AML1-ETO leukemia cells. Cancer Res 1999;59:2766-2769.
Akhmanova A, Verkerk T, Langeveld A, Grosveld F, Galjart N. Characterisation of transcriptionally active and inactive chromatin domains in neurons. J Cell Sci 2000;113:4463-4474.
Gramantieri L, Trerè D, Pession A, Piscaglia F, Masi L, Gaiani S, Mazziotti A, Bolondi L. Allelic imbalance on 16q in small, unifocal hepatocellular carcinoma: correlation with HBV and HCV infections and cellular proliferation rate. Dig Dis Sci 2000;45:306-311.
Ikonomov OC, Petrov T, Soden K, Shisheva A, Manji HK. Lithium treatment in ovo: effects on embryonic heart rate, natural death of ciliary ganglion neurons, and brain expression of a highly conserved chicken homolog of human MTG8/ETO. Brain Res Dev Brain Res 2000;123:13-24.
Launonen V, Mannermaa A, Stenbäck F, Kosma VM, Puistola U, Huusko P, Anttila M, Bloigu R, Saarikoski S, Kauppila A, Winqvist R. Loss of heterozygosity at chromosomes 3, 6, 8, 11, 16, and 17 in ovarian cancer: correlation to clinicopathological variables. Cancer Genet Cytogenet 2000;122:49-54.
Masselink H, Bernards R. The adenovirus E1A binding protein BS69 is a corepressor of transcription through recruitment of N-CoR. Oncogene 2000;19:1538-1546.
Melnick A, Carlile GW, McConnell MJ, Polinger A, Hiebert SW, Licht JD. AML-1/ETO fusion protein is a dominant negative inhibitor of transcriptional repression by the promyelocytic leukemia zinc finger protein. Blood 2000;96:3939-3947.
Pederson JA, LaFollette JW, Gross C, Veraksa A, McGinnis W, Mahaffey JW. Regulation by homeoproteins: a comparison of deformed-responsive elements. Genetics 2000;156:677-686.

CBFA2T3 (core-binding factor, runt domain, alpha subunit 2; translocated to, 3) Bais AJ
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 75
Wood JD, Nucifora FC Jr, Duan K, Zhang C, Wang J, Kim Y, Schilling G, Sacchi N, Liu JM, Ross CA. Atrophin-1, the dentato-rubral and pallido-luysian atrophy gene product, interacts with ETO/MTG8 in the nuclear matrix and represses transcription. J Cell Biol 2000;150:939-948.
Zheng PP, Pang JC, Hui AB, Ng HK. Comparative genomic hybridization detects losses of chromosomes 22 and 16 as the most common recurrent genetic alterations in primary ependymomas. Cancer Genet Cytogenet 2000;122:18-25.
Amann JM, Nip J, Strom DK, Lutterbach B, Harada H, Lenny N, Downing JR, Meyers S, Hiebert SW. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol 2001;21:6470-6483.
Calabi F, Pannell R, Pavloska G. Gene targeting reveals a crucial role for MTG8 in the gut. Mol Cell Biol 2001;21:5658-5666.
Costoya JA, Pandolfi PP. The role of promyelocytic leukemia zinc finger and promyelocytic leukemia in leukemogenesis and development. Curr Opin Hematol 2001;8:212-217.
Fukuyama T, Sueoka E, Sugio Y, Otsuka T, Niho Y, Akagi K, Kozu T. MTG8 proto-oncoprotein interacts with the regulatory subunit of type II cyclic AMP-dependent protein kinase in lymphocytes. Oncogene 2001;20:6225-6232.
Heibert SW, Lutterbach B, Durst K, Wang L, Linggi B, Wu S, Wood L, Amann J, King D, Hou Y. Mechanisms of transcriptional repression by the t(8;21)-, t(12;21)-, and inv(16)-encoded fusion proteins. Cancer Chemother Pharmacol 2001;48:S31-34.
Hildebrand D, Tiefenbach J, Heinzel T, Grez M, Maurer AB. Multiple regions of ETO cooperate in transcriptional repression. J Biol Chem 2001;276:9889-9895.
Yakicier MC, Legoix P, Vaury C, Gressin L, Tubacher E, Capron F, Bayer J, Degott C, Balabaud C, Zucman-Rossi J. Identification of homozygous deletions at chromosome 16q23 in aflatoxin B1 exposed hepatocellular carcinoma. Oncogene 2001;20:5232-5238.
Yan J, Fang Y, Liang Q, Huang Y, Zeng Y. Novel chromosomal alterations detected in primary nasopharyngeal carcinoma by comparative genomic hybridization. Chin Med J 2001;114:418-421.
Zhang J, Hug BA, Huang EY, Chen CW, Gelmetti V, Maccarana M, Minucci S, Pelicci PG, Lazar MA. Oligomerization of ETO is obligatory for corepressor interaction. Mol Cell Biol 2001;21:156-163.
Ansieau S, Leutz A. The conserved Mynd domain of BS69 binds cellular and oncoviral proteins through a common PXLXP motif. J Biol Chem 2002;277:4906-4910.
Gottlieb PD, Pierce SA, Sims RJ, Yamagishi H, Weihe EK, Harriss JV, Maika SD, Kuziel WA, King HL, Olson EN, Nakagawa O, Srivastava D. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat Genet 2002;31:25-32.
Hansen LL, Jensen LL, Dimitrakakis C, Michalas S, Gilbert F, Barber HR, Overgaard J, Arzimanoglou II. Allelic imbalance in selected chromosomal regions in ovarian cancer. Cancer Genet Cytogenet 2002;139:1-8.
Hoogeveen AT, Rossetti S, Stoyanova V, Schonkeren J, Fenaroli A, Schiaffonati L, van Unen L, Sacchi N. The transcriptional corepressor MTG16a contains a novel nucleolar targeting sequence deranged in t (16; 21)-positive myeloid malignancies. Oncogene 2002;21:6703-6712.
Kochetkova M, McKenzie OLD, Bais AJ, Martin JM, Secker GA, Seshadri R, Powell JA, Hinze SJ, Gardner AE, Spendlove HE, O'Callaghan NJ, Cleton-Jansen A, Cornelisse C, Whitmore SA, Crawford J, Kremmidiotis G, Sutherland GR, Callen DF.
CBFA2T3 (MTG16) is a putative breast tumour suppressor from the breast cancer loss of heterozygosity region at 16q24.3. Cancer Res 2002;62:4599-4604.
Lerebours F, Bertheau P, Bieche I, Driouch K, De The H, Hacene K, Espie M, Marty M, Lidereau R. Evidence of chromosome regions and gene involvement in inflammatory breast cancer. Int J Cancer 2002;102:618-622.
Powell JA, Gardner AE, Bais AJ, Hinze SJ, Baker E, Whitmore S, Crawford J, Kochetkova M, Spendlove HE, Doggett NA, Sutherland GR, Callen DF, Kremmidiotis G. Sequencing, transcript identification, and quantitative gene expression profiling in the breast cancer loss of heterozygosity region 16q24.3 reveal three potential tumor-suppressor genes. Genomics 2002;80:303-310.
Scarr RB, Sharp PA. PDCD2 is a negative regulator of HCF-1 (C1). Oncogene 2002;21:5245-54.
Schillace RV, Andrews SF, Liberty GA, Davey MP, Carr DW. Identification and characterization of myeloid translocation gene 16b as a novel a kinase anchoring protein in T lymphocytes. J Immunol 2002;168:1590-1599.
Veraksa A, Kennison J, McGinnis W. DEAF-1 function is essential for the early embryonic development of Drosophila. Genesis 2002;33:67-76.
Agathanggelou A, Dallol A, Zöchbauer-Muller S, Morrissey C, Honorio S, Hesson L, Martinsson T, Fong KM, Kuo MJ, Yuen PW, Maher ER, Minna JD, Latif F. Epigenetic inactivation of the candidate 3p21.3 suppressor gene BLU in human cancers. Oncogene 2003;22:1580-1588.
Davis JN, McGhee L, Meyers S. The ETO (MTG8) gene family. Gene 2003;303:1-10.
Diep CB, Parada LA, Teixeira MR, Eknaes M, Nesland JM, Johansson B, Lothe RA. Genetic profiling of colorectal cancer liver metastases by combined comparative genomic hybridization and G-banding analysis. Genes Chromosomes Cancer 2003;36:189-197.
Matsuyama H, Pan Y, Yoshihiro S, Kudren D, Naito K, Bergerheim US, Ekman P. Clinical significance of chromosome 8p, 10q, and 16q deletions in prostate cancer. Prostate 2003;54:103-111.
McGhee L, Bryan J, Elliott L, Grimes HL, Kazanjian A, Davis JN, Meyers S. Gfi-1 attaches to the nuclear matrix, associates with ETO (MTG8) and histone deacetylase proteins, and represses transcription using a TSA-sensitive mechanism. J Cell Biochem 2003;89:1005-1018.
Safford SD, Goyeau D, Freemerman AJ, Bentley R, Everett ML, Grundy PE, Skinner MA. Fine mapping of Wilms' tumors with 16q loss of heterozygosity localizes the putative tumor suppressor gene to a region of 6.7 megabases. Ann Surg Oncol 2003;10:136-143.
Asirvatham AL, Galligan SG, Schillace RV, Davey MP, Vasta V, Beavo JA, Carr DW. A-kinase anchoring proteins interact with phosphodiesterases in T lymphocyte cell lines. J Immunol 2004;173:4806-4814.
Bais AJ, Gardner AE, McKenzie OLD, Callen DF, Sutherland GR, Kremmidiotis G. Aberrant CBFA2T3B gene promoter methylation in breast tumors. Mol Cancer 2004;3:22.
Ibanez V, Sharma A, Buonamici S, Verma A, Kalakonda S, Wang J, Kadkol S, Saunthararajah Y. AML1-ETO decreases ETO-2 (MTG16) interactions with nuclear receptor corepressor, an effect that impairs granulocyte differentiation. Cancer Res 2004;64:4547-4554.
Rossetti S, Hoogeveen AT, Sacchi N. The MTG proteins: chromatin repression players with a passion for networking. Genomics 2004;84:1-9.
Härkönen P, Kyllönen AP, Nordling S, Vihko P. Loss of heterozygosity in chromosomal region 16q24.3 associated with progression of prostate cancer. Prostate 2005;62:267-274.

CBFA2T3 (core-binding factor, runt domain, alpha subunit 2; translocated to, 3) Bais AJ
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 76
Lindberg SR, Olsson A, Persson AM, Olsson I. The Leukemia-associated ETO homologues are differently expressed during hematopoietic differentiation. Exp Hematol 2005;33:189-198.
Yonekura Y, Yamamoto D, Okugawa H, Tanaka K, Kamiyama Y. Loss of heterozygosity in ductal lavage for breast tumor and the contralateral breast. Oncol Rep 2005;13:739-743.
This article should be referenced as such: Bais AJ. CBFA2T3 (core-binding factor, runt domain, alpha subunit 2; translocated to, 3). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):70-76.

Gene Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 77
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
CLDN4 (claudin-4) Stefanie Ripka, Thomas M Gress
Abteilung Innere Medizin I, Universitätsklinikum Ulm, Robert Koch Str.8, 89081 Ulm, Germany
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/CLDN4ID42975ch7q11.html DOI: 10.4267/2042/38288
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: CLDN4 Other names: CPETR; CPETR1; WBSCR8 (Williams-Beuren syndrome chromosome region 8 protein); hCPE-R, CPE-R (Clostridium perfringens enterotoxin receptor) Location: 7q11.23
Probe(s) - Courtesy Mariano Rocchi, Resources for Molecular Cytogenetics.
DNA/RNA Description Intronless; one exon spanning 1.68 kb.
Transcription One transcript of 1.68 kb with 630 bp of coding sequence.
Protein Description The CLDN4 protein contains 209 amino acids and has a molecular weight of 22.1 kDa with four putative transmembrane segments. It directly interacts with TJP1/ZO-1, TJP2/ZO-2 and TJP3/ZO-3.
Expression Claudin-4 is expressed in many fetal and adult tissues, predominantly in lung, intestine and kidney. Overexpressed in pancreatic, breast, ovarian, and prostate cancer.
Localisation Integral membrane protein. Tight junction component.
Function CLDN4 plays a major role in tight junction-specific obliteration of the intercellular space.
Homology Belongs to the claudin family.
Implicated in Williams-Beuren syndrom Disease Williams-Beuren syndrom (WBS) includes supravalvular aortic stenosis (SVAS), multiple peripheral pulmonary arterial stenoses, elfin face, mental and statural deficiency, characteristic dental malformation, and infantile hypercalcemia. It is associated with an autosomal dominant contiguous gene deletion involving genes from chromosome band 7q11.23, including CLDN4, elastin and LIM-kinase1. Haploinsufficiency for CLDN4 may be the cause of certain cardiovascular and musculo-skeletal abnormalities observed in the context of this disease.
Gastric cancer Oncogenesis Downregulated in gastric cancer. Absence of CLDN4 may play a role in the disruption of cell-to-cell adhesion in diffuse type gastric cancer and in a loss of differentiation.
Pancreatic cancer Oncogenesis Overexpressed in pancreatic cancer. Overexpression is predominantly observed in well-differentiated tumors with decreased metastatic potencial.

CLDN4 (claudin-4) Ripka S, Gress TM
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 78
Breast cancer Oncogenesis Overexpressed in breast cancer and Paget's disease. Significance unclear. Ovarian cancer Oncogenesis CLDN4 is upregulated in ovarian tumors and cell lines and may represent a novel marker for this disease.
Squamous cell carcinoma and Bowen's disease Oncogenesis Expression of claudin-4 is associated with keratinization in SCC and BD.
Prostate cancer Oncogenesis Overexpressed in prostate cancer ephitelium. Significance unclear.
References Paperna T, Peoples R, Wang YK, Kaplan P, Francke U. Genes for the CPE receptor (CPETR1) and the human homolog of RVP1 (CPETR2) are localized within the Williams-Beuren syndrom deletion. Genomics 1998 Dec 15;54(3):453-459.
Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci USA 1999 Jan 19;96(2):511-516.
Long H, Crean CD, Lee WH, Cummings OW, Gabig TG. Expression of Clostridium perfringens enterotoxin receptors claudin-3 and claudin-4 in prostate cancer epithelium. Cancer Res 2001 Nov 1;61(21):7878-7881.
Michl P, Barth C, Buchholz M, Lerch MM, Rolke M, Holzmann KH, Menke A, Fensterer H, Giehl K, Löhr M, Leder G, Iwamura T, Adler G, Gress TM. Claudin-4 expression decreases invasiveness and metastatic potential of pancreatic cancer. Cancer Res 2003 Oct 1;63(19):6265-6271.
Nichols LS, Ashfaq R, Iacobuzio-Donahue CA. Claudin 4 protein expression in primary and metastatic pancreatic cancer: support for use as a therapeutic target. Am J Clin Pathol 2004 Feb;121(2):226-230.
Morita K, Tsukita S, Miyachi Y. Tight junction-associated proteins (occludin,ZO-1; claudin-1, claudin-4) in squamous cell carcinoma and Bowen's disease. Br J Dermatol 2004 Aug;151(2):328-334.
Nichols LS, Ashfaq R, Iacobuzio-Donahue CA. Claudin 4 protein expression in primary and metastatic pancreatic cancer: support for use as a therapeutic target. Am J Clin Pathol 2004 Feb;121(2):226-230.
Soini Y. Claudins 2, 3, 4, and 5 in Paget's disease and breast carcinoma. Hum Pathol 2004 Dec;35(12):1531-1536.
Lee SK, Moon J, Park SW, Song SY, Chung JB, Kang JK. Loss of the tight junction protein claudin 4 correlates with histological growth pattern and differentiation in advanced gastric adenocarcinoma. Oncol Rep 2005 Feb;13(2):193-199.
Tokes AM, Kulka J, Paku S, Szik A, Paska C, Novak PK, Szilak L, Kiss A, Bogi K, Schaff Z. Claudin-1, -3 and -4 proteins and mRNA expression in benign and malignant breast lesions: a research study. Breast Cancer Res 2005;7(2):R296-305.
This article should be referenced as such: Ripka S, Gress TM. CLDN4 (claudin-4). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):77-78.
Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 79
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
JAG1 (jagged 1 (Alagille syndrome)) Michèle Meunier-Rotival, Catherine Driancourt, Julie Boyer-Di Ponio
INSERM E0020, 80 rue du General Leclerc, F-94276 Le Kremlin-Bicêtre Cedex, France
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/JAG1ID41029ch20p12.html DOI: 10.4267/2042/38289
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: JAG1 Other names: JAGGED1; HJ1; hJ1; JAGL1 Location: 20p12.1-11.23 Local order: telomere PLCB1, PLCB4, PAK7, SNAP25, MKKS, JAG1 centromere.
DNA/RNA
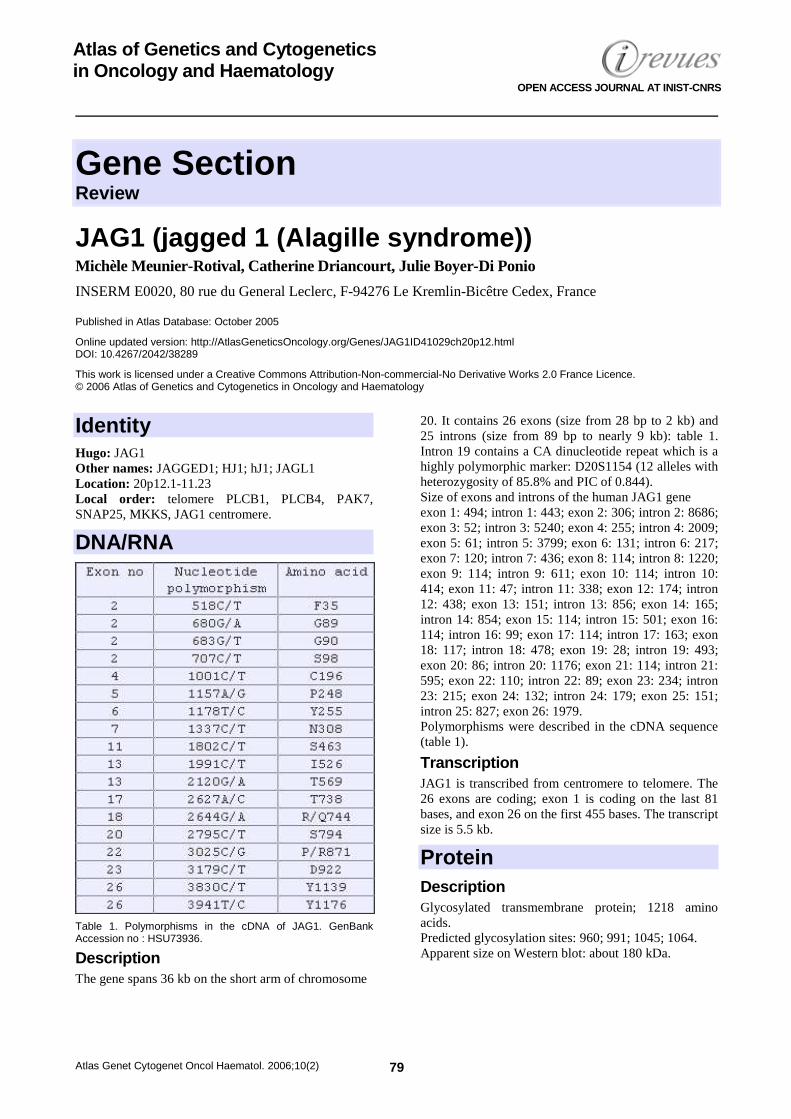
Table 1. Polymorphisms in the cDNA of JAG1. GenBank Accession no : HSU73936.
Description The gene spans 36 kb on the short arm of chromosome
20. It contains 26 exons (size from 28 bp to 2 kb) and 25 introns (size from 89 bp to nearly 9 kb): table 1. Intron 19 contains a CA dinucleotide repeat which is a highly polymorphic marker: D20S1154 (12 alleles with heterozygosity of 85.8% and PIC of 0.844). Size of exons and introns of the human JAG1 gene exon 1: 494; intron 1: 443; exon 2: 306; intron 2: 8686; exon 3: 52; intron 3: 5240; exon 4: 255; intron 4: 2009; exon 5: 61; intron 5: 3799; exon 6: 131; intron 6: 217; exon 7: 120; intron 7: 436; exon 8: 114; intron 8: 1220; exon 9: 114; intron 9: 611; exon 10: 114; intron 10: 414; exon 11: 47; intron 11: 338; exon 12: 174; intron 12: 438; exon 13: 151; intron 13: 856; exon 14: 165; intron 14: 854; exon 15: 114; intron 15: 501; exon 16: 114; intron 16: 99; exon 17: 114; intron 17: 163; exon 18: 117; intron 18: 478; exon 19: 28; intron 19: 493; exon 20: 86; intron 20: 1176; exon 21: 114; intron 21: 595; exon 22: 110; intron 22: 89; exon 23: 234; intron 23: 215; exon 24: 132; intron 24: 179; exon 25: 151; intron 25: 827; exon 26: 1979. Polymorphisms were described in the cDNA sequence (table 1).
Transcription JAG1 is transcribed from centromere to telomere. The 26 exons are coding; exon 1 is coding on the last 81 bases, and exon 26 on the first 455 bases. The transcript size is 5.5 kb.
Protein Description Glycosylated transmembrane protein; 1218 amino acids. Predicted glycosylation sites: 960; 991; 1045; 1064. Apparent size on Western blot: about 180 kDa.
JAG1 (jagged 1 (Alagille syndrome)) Meunier-Rotival M et al.
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 80
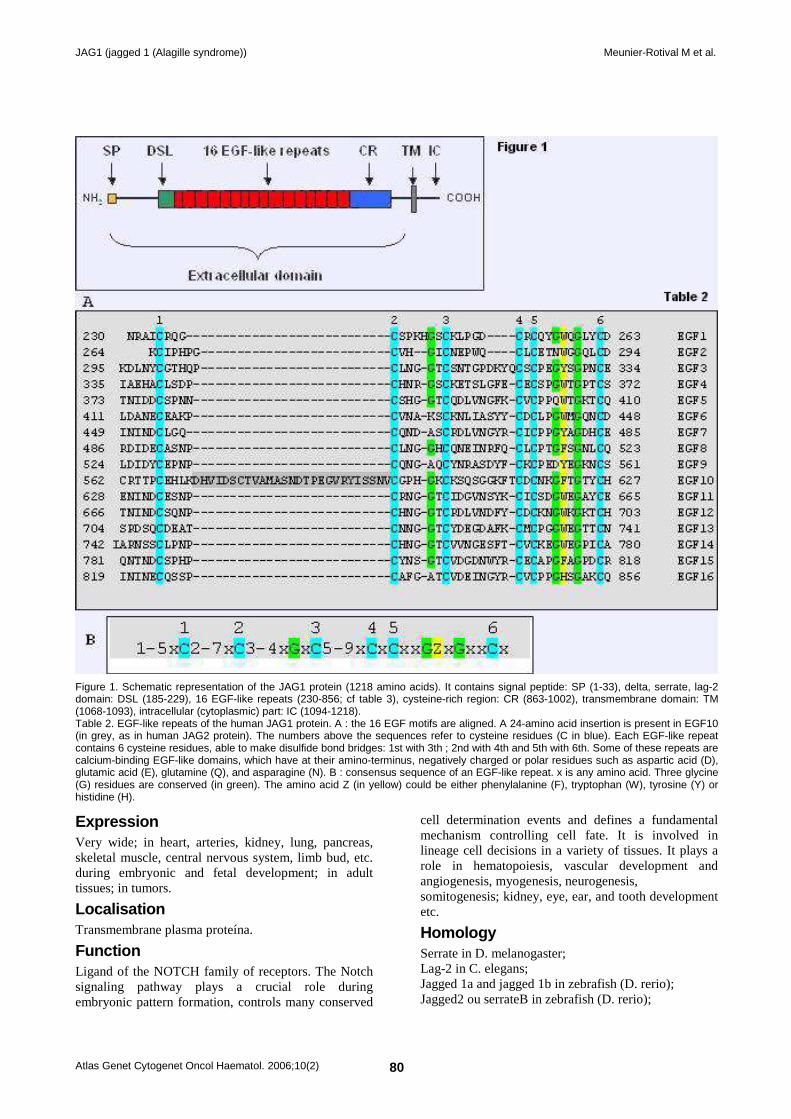
Figure 1. Schematic representation of the JAG1 protein (1218 amino acids). It contains signal peptide: SP (1-33), delta, serrate, lag-2 domain: DSL (185-229), 16 EGF-like repeats (230-856; cf table 3), cysteine-rich region: CR (863-1002), transmembrane domain: TM (1068-1093), intracellular (cytoplasmic) part: IC (1094-1218). Table 2. EGF-like repeats of the human JAG1 protein. A : the 16 EGF motifs are aligned. A 24-amino acid insertion is present in EGF10 (in grey, as in human JAG2 protein). The numbers above the sequences refer to cysteine residues (C in blue). Each EGF-like repeat contains 6 cysteine residues, able to make disulfide bond bridges: 1st with 3th ; 2nd with 4th and 5th with 6th. Some of these repeats are calcium-binding EGF-like domains, which have at their amino-terminus, negatively charged or polar residues such as aspartic acid (D), glutamic acid (E), glutamine (Q), and asparagine (N). B : consensus sequence of an EGF-like repeat. x is any amino acid. Three glycine (G) residues are conserved (in green). The amino acid Z (in yellow) could be either phenylalanine (F), tryptophan (W), tyrosine (Y) or histidine (H).
Expression Very wide; in heart, arteries, kidney, lung, pancreas, skeletal muscle, central nervous system, limb bud, etc. during embryonic and fetal development; in adult tissues; in tumors.
Localisation Transmembrane plasma proteína.
Function Ligand of the NOTCH family of receptors. The Notch signaling pathway plays a crucial role during embryonic pattern formation, controls many conserved
cell determination events and defines a fundamental mechanism controlling cell fate. It is involved in lineage cell decisions in a variety of tissues. It plays a role in hematopoiesis, vascular development and angiogenesis, myogenesis, neurogenesis, somitogenesis; kidney, eye, ear, and tooth development etc.
Homology Serrate in D. melanogaster; Lag-2 in C. elegans; Jagged 1a and jagged 1b in zebrafish (D. rerio); Jagged2 ou serrateB in zebrafish (D. rerio);
JAG1 (jagged 1 (Alagille syndrome)) Meunier-Rotival M et al.
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 81
X-serrate-1 in tadpole (Xenopus laevis); C-serrate-1 and C-serrate-2 in chicken (Gallus gallus); Jagged1 and jagged2 in mouse (Mus musculus); Jagged1 and jagged2 in rat (Rattus norvegicus); Jagged1 and jagged2 in dog (Canis familiaris); Partial jagged1 in Bos Taurus; JAGGED2 in Homo sapiens.
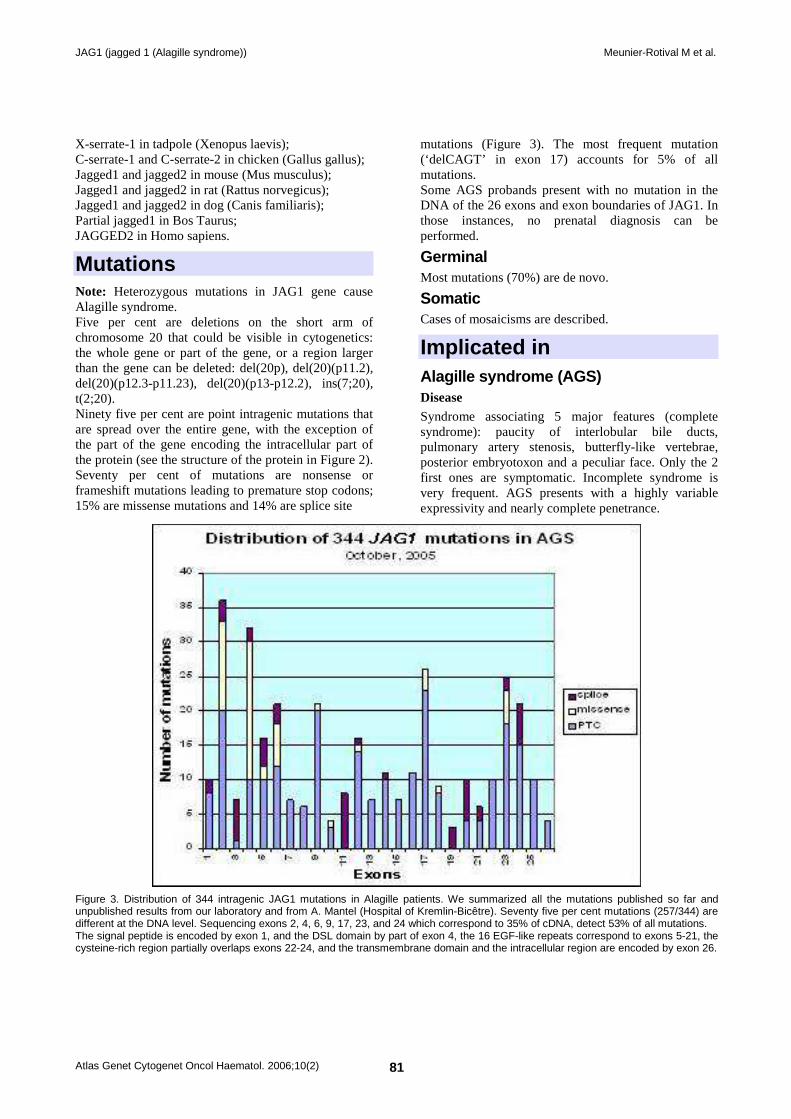
Mutations Note: Heterozygous mutations in JAG1 gene cause Alagille syndrome. Five per cent are deletions on the short arm of chromosome 20 that could be visible in cytogenetics: the whole gene or part of the gene, or a region larger than the gene can be deleted: del(20p), del(20)(p11.2), del(20)(p12.3-p11.23), del(20)(p13-p12.2), ins(7;20), t(2;20). Ninety five per cent are point intragenic mutations that are spread over the entire gene, with the exception of the part of the gene encoding the intracellular part of the protein (see the structure of the protein in Figure 2). Seventy per cent of mutations are nonsense or frameshift mutations leading to premature stop codons; 15% are missense mutations and 14% are splice site
mutations (Figure 3). The most frequent mutation (‘delCAGT’ in exon 17) accounts for 5% of all mutations. Some AGS probands present with no mutation in the DNA of the 26 exons and exon boundaries of JAG1. In those instances, no prenatal diagnosis can be performed.
Germinal Most mutations (70%) are de novo.
Somatic Cases of mosaicisms are described.
Implicated in Alagille syndrome (AGS) Disease Syndrome associating 5 major features (complete syndrome): paucity of interlobular bile ducts, pulmonary artery stenosis, butterfly-like vertebrae, posterior embryotoxon and a peculiar face. Only the 2 first ones are symptomatic. Incomplete syndrome is very frequent. AGS presents with a highly variable expressivity and nearly complete penetrance.
Figure 3. Distribution of 344 intragenic JAG1 mutations in Alagille patients. We summarized all the mutations published so far and unpublished results from our laboratory and from A. Mantel (Hospital of Kremlin-Bicêtre). Seventy five per cent mutations (257/344) are different at the DNA level. Sequencing exons 2, 4, 6, 9, 17, 23, and 24 which correspond to 35% of cDNA, detect 53% of all mutations. The signal peptide is encoded by exon 1, and the DSL domain by part of exon 4, the 16 EGF-like repeats correspond to exons 5-21, the cysteine-rich region partially overlaps exons 22-24, and the transmembrane domain and the intracellular region are encoded by exon 26.

JAG1 (jagged 1 (Alagille syndrome)) Meunier-Rotival M et al.
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 82
Tetralogy of Fallot Disease The heterozygous mutation (G274D) in EGF2 of JAG1 has been reported in one family: affected family members also had characteristic facies.
Familial deafness, congenital heart defects, and posterior embryotoxon Disease The heterozygous mutation (C234Y) in EGF1 of JAG1 has been reported in one family.
References Alagille D, Estrada A, Hadchouel M, Gautier M, Odièvre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr 1987;110:195-200.
Anad F, Burn J, Matthews D, Cross I, Davison BC, Mueller R, Sands M, Lillington DM, Eastham E. Alagille syndrome and deletion of 20p. J Med Genet 1990;27:729-737. (Review).
Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 1997;16:243-251.
Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet 1997;16:235-242.
Oda T, Elkahloun AG, Meltzer PS, Chandrasekharappa SC. Identification and cloning of the human homolog (JAG1) of the rat Jagged1 gene from the Alagille syndrome critical region at 20p12. Genomics 1997;43:376-379.
Krantz ID, Colliton RP, Genin A, Rand EB, Li L, Piccoli DA, Spinner NB. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet 1998;62:1361-1369.
Crosnier C, Driancourt C, Raynaud N, Dhorne-Pollet S, Pollet N, Bernard O, Hadchouel M, Meunier-Rotival M. Mutations in JAGGED1 gene are predominantly sporadic in Alagille syndrome. Gastroenterology 1999;116:1141-1148.
Crosnier C, Lykavieris P, Meunier-Rotival M, Hadchouel M. Alagille syndrome. The widening spectrum of arteriohepatic dysplasia. Clin Liver Dis 2000;4:765-78. (Review).
Crosnier C, Attie-Bitach T, Encha-Razavi F, Audollent S, Soudy F, Hadchouel M, Meunier-Rotival M, Vekemans M. JAGGED1 gene expression during human embryogenesis elucidates the wide phenotypic spectrum of Alagille syndrome. Hepatology 2000;32:574-581.
Jones EA, Clement-Jones M, Wilson DI. JAGGED1 expression in human embryos: correlation with the Alagille syndrome phenotype. J Med Genet 2000;37:658-662.
Crosnier C, Driancourt C, Raynaud N, Hadchouel M, Meunier-Rotival M. Fifteen novel mutations in the JAGGED1 gene of patients with Alagille syndrome. Hum Mutat 2001;17:72-73.
Deloukas P et al. The DNA sequence and comparative analysis of human chromosome 20. Nature 2001;414:865-871.
Eldadah ZA, Hamosh A, Biery NJ, Montgomery RA, Duke M, Elkins R, Dietz HC. Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Hum Mol Genet 2001;10:163-169.
Giannakudis J, Röpke A, Kujat A, Krajewska-Walasek M, Hughes H, Fryns JP, Bankier A, Amor D, Schlicker M, Hansmann I. Parental mosaicism of JAG1 mutations in families with Alagille syndrome. Eur J Hum Genet 2001;9:209-216.
Morrissette JD, Colliton RP, Spinner NB. Defective intracellular transport and processing of JAG1 missense mutations in Alagille syndrome. Hum Mol Genet 2001;10:405-413.
Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in Alagille syndrome. Hum Mutat 2001;17:18-33. (Review).
Yuan ZR, Okaniwa M, Nagata I, Tazawa Y, Ito M, Kawarazaki H, Inomata Y, Okano S, Yoshida T, Kobayashi N, Kohsaka T. The DSL domain in mutant JAG1 ligand is essential for the severity of the liver defect in Alagille syndrome. Clin Genet 2001;59:330-337.
Heritage ML, MacMillan JC, Anderson GJ. DHPLC mutation analysis of Jagged1 (JAG1) reveals six novel mutations in Australian Alagille syndrome patients. Hum Mutat 2002;20:481.
Le Caignec C, Lefevre M, Schott JJ, Chaventre A, Gayet M, Calais C, Moisan JP. Familial deafness, congenital heart defects, and posterior embryotoxon caused by cysteine substitution in the first epidermal-growth-factor-like domain of jagged 1. Am J Hum Genet 2002;71:180-186.
Lu F, Morrissette JJ, Spinner NB. Conditional JAG1 mutation shows the developing heart is more sensitive than developing liver to JAG1 dosage. Am J Hum Genet 2003;72:1065-70.
Röpke A, Kujat A, Gräber M, Giannakudis J, Hansmann I. Identification of 36 novel Jagged1 (JAG1) mutations in patients with Alagille syndrome. Hum Mutat 2003;21:100.
Boyer J, Crosnier C, Driancourt C, Raynaud N, Gonzales M, Hadchouel M, Meunier-Rotival M. Expression of mutant JAGGED1 alleles in patients with Alagille syndrome. Hum Genet 2005;116:445-453.
Jurkiewicz D, Popowska E, Glaser C, Hansmann I, Krajewska-Walasek M. Twelve novel JAG1 gene mutations in polish Alagille syndrome patients. Hum Mutat 2005;25:321.
This article should be referenced as such: Meunier-Rotival M, Driancourt C, Boyer-Di Ponio J. JAG1 (jagged 1 (Alagille syndrome)). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):79-82.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 83
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
MLL (myeloid/lymphoid or mixed lineage leukemia) Jean-Loup Huret
Genetics, Dept Medical Information, UMR 8125 CNRS, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/MLL.html DOI: 10.4267/2042/38290
This article is an update of: Marschalek R. MLL (myeloid/lymphoid or mixed lineage leukemia). Atlas Genet Cytogenet Oncol Haematol.2003;7(1):16-18. Hess JL, Huret JL. MLL (myeloid/lymphoid or mixed lineage leukemia). Atlas Genet Cytogenet Oncol Haematol.2001;5(1):12-14. Huret JL. MLL (myeloid/lymphoid or mixed lineage leukemia). Atlas Genet Cytogenet Oncol Haematol.1997;1(2):68-69. This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
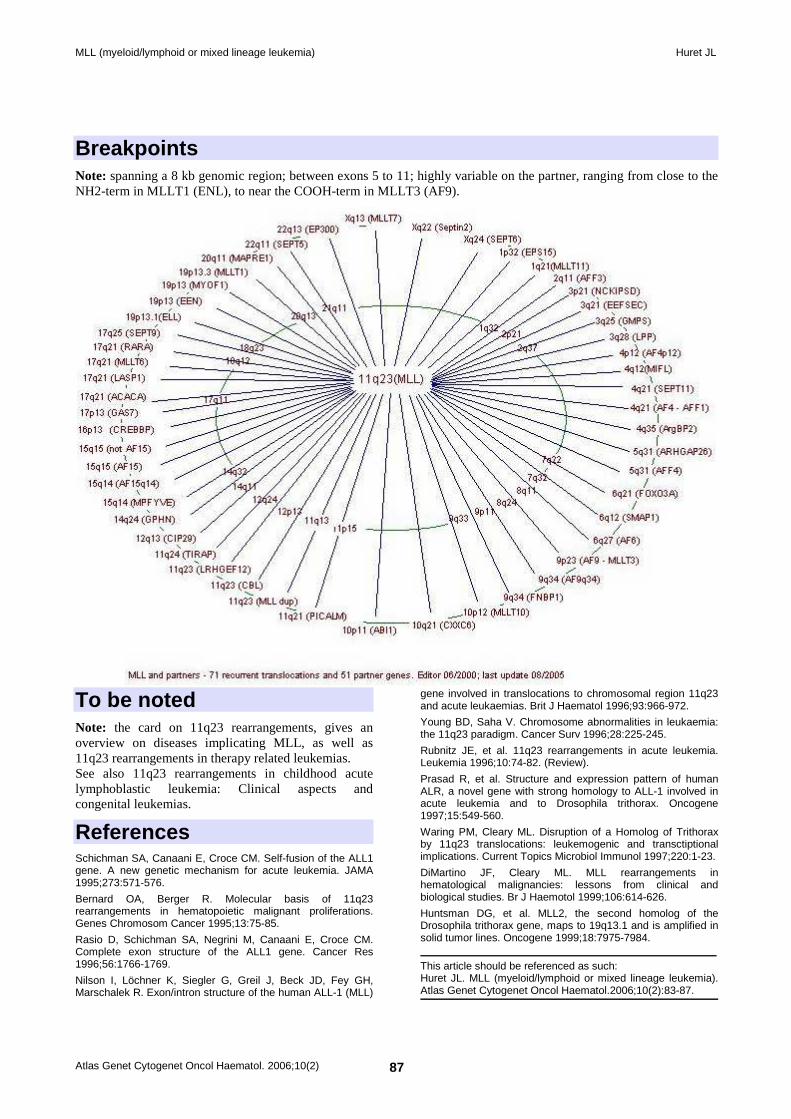
Identity Hugo: MLL Other names: ALL1, HRX, Htrx (human trithorax), TRX1 Location: 11q23 Local order: telomeric to PLZF, centromeric from RCK.
DNA/RNA
MLL (11q23) - Courtesy Mariano Rocchi, Resources for Molecular Cytogenetics.
Description 37 exons, spanning over 100 kb
Transcription In a centromeric to telomeric direction; 13 and 15 kb; coding sequence: 11.9 kb
Protein Description 3969 amino acids; 431 KDa; contains from N-term to C-term 3 AT hooks homologous to high mobility group proteins HMGA1 and HMGA2, binding to the minor grove of DNA; 2 speckled nuclear localisation signals; 2 repression domains RD1 and RD2: RD1 or CXXC: cystein methyl transferase, binds CpG rich DNA, has a transcriptional repression activity; RD2 recruits histone desacetylases HDAC1 and 2; 3 plant homeodomains (cystein rich zinc finger domains, with homodimerization properties), 1 bromodomain (may bind acetylated histones), and 1 plant homeodomain; these domains may be involved in protein-protein interaction; a FYRN and a FRYC domain; a transactivation domain which binds CBP; may acetylates H3 and H4 in the HOX area; a SET domain: methyltransferase; methyltates H3, including histones in the HOX area for allowing chromatin to be open to transcription. MLL is cleaved by taspase 1 into 2 proteins before entering the nucleus: a p300/320 N-term protein called MLL-N, and a p180 C-term protein, called MLL-C. The FYRN and a FRYC domains of native MLL associate MLL-N and MLL-C in a stable complex; they form a multiprotein complex with transcription factor TFIID.
Expression Wide; especially in: brain, kidney, thyroid; expressed in Taned B lymphocytes and myeloid cells.

MLL (myeloid/lymphoid or mixed lineage leukemia) Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 84
MLL partner genes - Rolf Marschalek Nov 2002.
Localisation Nuclear, in punctate spots.
Function Transcriptional regulatory factor; MLL may have yin-yang functions though actions of MLL-N and MLL-C (e.g. desacetylation/acetylation); MLL-N acts as a transcriptional repressor; MLL can be associated with more than 30 proteins, including the core components of the SWI/SNF chromatin remodeling complex and the transcription complex TFIID. MLL binds promotors of HOX genes through acetylation and methylation of histones. MLL is a major regulator of hematopoesis and embryonic development, through regulation of HOX genes expression regulation (HOXA9 in particular).
Homology Trithorax (Drosophila), ALR (human), MLL2 (human).
Mutations Note: MLL is implicated in at least 10 % of acute leukaemias (AL) of various types: acute lymphoblastic leukemias (ALL), acute non lymphocytic leukemias (ANLL), biphenotypic ALs, treatment related leukemias, infant leukemias; the prognosis is poor.
Implicated in
t(4;11)(q21;q23)/acute leukaemias → MLL-AFF1 (AF4) Disease Typically CD19+ CD10-precursor B-ALL, biphenotypic AL, at times ANLL (M4/M5); common in infants may be congenital; treatment related leukaemia (secondary to epipodophyllotoxins). Prognosis Median survival < 1 year Cytogenetics Additional chromosome anomalies are found in 1/4 of cases, one of which is the i(7q). Hybrid/Mutated Gene 5' MLL - 3' AF4; 12kb. Abnormal Protein 240 kDa protein with about 1400 aminoacids from NH2 MLL and 850 from COOH AF4 (variable breakpoints); the reciprocal may or may not be expressed.

MLL (myeloid/lymphoid or mixed lineage leukemia) Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 85
t(6;11)(q27;q23)/ANLL → MLL-MLLT4 (AF6) Disease M5/M4 de novo and therapy related ANLL, T-cell ALL. Prognosis Poor.
t(9;11)(p22;q23) /ANLL → MLL-MLLT3 (AF9) Disease M5/M4 de novo and therapy related ANLL. Prognosis The prognosis may not be as poor as in other 11q23 leukaemias in de novo cases; very poor prognosis in secondary ANLL cases.
Cytogenetics May be overlooked; often as a sole anomaly.
Hybrid/Mutated Gene Variable breakpoints on both genes.
Abnormal Protein N-term - AT hook and DNA methyltransferase from MLL fused to the 192 C-term amino acids from AF9 (as breakpoints are variable, this is only an example).
t(10;11)(p12;q23)/ANLL → MLL-MLLT10 (AF10) Disease M4 or M5 ANLL; ALL at times; therapy related ANLL.
Prognosis Poor

MLL (myeloid/lymphoid or mixed lineage leukemia) Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 86
t(11;19)(q23;p13.1)/ANLL → MLL-ELL Disease Mainly M4/M5; treatment related leukemia; all ages.
Prognosis Very poor. Cytogenetics Detected with R banding.
Hybrid/Mutated Gene 5' MLL - 3' ELL.
Abnormal Protein AT hook and DNA methyltransferase from MLL fused to most of ELL.
Oncogenesis Potential transcription factor.
t(11;19)(q23;p13.3)/acute leukaemias → MLL-MLLT1 (ENL) Disease ALL (CD19+), biphenotypic AL, ANLL (M4/M5); mainly congenital; treatment-related leukaemia.
Prognosis Very poor, except in rare T-cell cases.
Cytogenetics Detected with G banding.
Hybrid/Mutated Gene 5' MLL - 3' ENL.
Abnormal Protein AT hook and DNA methyltransferase from MLL fused to, most often, the nearly entire ENL.
Other entities • t(X;11)(q13;q23)/ANLL, T-ALL → MLL - AFX1 • t(X;11)(q22;q23)/ANLL → MLL- Septin2 • t(1;11)(p32;q23)/ALL → MLL- EB15 (AF1p) • t(1;11)(q21;q23)/ANLL → MLL- MLLT11 (AF1q) • t(2;11)(q11;q23)/ALL → MLL- AFF3 (LAF4) • t(3;11)(p21;q23)/t-ANLL, ALL→ MLL- NCKIPSD (AF3p21) • t(3;11)(q21;q23)/ALL → MLL- EEFSEC (SELB) • t(3;11)(q25;q23)/t-ANLL → MLL - GMPS
• t(3;11)(q28;q23)/ANLL → MLL - LPP • t(4;11)(p12;q23) → MLL - AF4p12 • t(4;11)(q12;q23) → MLL - MIFL • t(4;11)(q21;q23)/atypical CML → MLL - SEPT11 • t(4;11)(q35;q23)→ MLL - ArgBP2 • t(5;11)(q31;q23)/ANLL, ALL → MLL - ARHGAP26 (GRAF) • ins(5;11)(q31;q13q23)/ALL → MLL - AFF4 (AF5q31) • t(6;11)(q12;q23)/ANLL → MLL - SMAP1 • t(6;11)(q21;q23)/ANLL → MLL - FOXO3A (AF6q21) • t(9;11)(q34;q23)/ANLL → MLL - DAB2IP (AF9q34) • t(10;11)(p11;q23)/ANLL → MLL - ABI1 • t(10;11)(q21;q23)/ANLL → MLL - CXXC6 (TET1) • t(11;11)(q21;q23)/ANLL → MLL - PICALM • trisomy 11/ANLL → MLL tandem duplication • t(11;11)(q23;q23)/ANLL → MLL - CBL • t(11;11)(q23;q23)/ANLL → MLL - ARHGEF12 (LARG) • t(11;11)(q23;q24)/ANLL → MLL - TIRAP • t(11;12)(q23;q13)/ANLL → MLL - CIP29 • t(11;14)(q23;q24)/ANLL, AUL → MLL - GPHN • t(11;15)(q23;q14)/ANLL, ALL → MLL - CASC5 (AF15q14) • t(11;15)(q23;q14) → MLL - MPFYVE • t(11;15)(q23;q15) → MLL - AF15 • t(11;16)(q23;p13)/MDS, ANLL, t-ANLL, ALL → MLL - CREBBP (CBP) • t(11;17)(q23;p13)/t-ANLL → MLL - GAS7 • t(11;17)(q23;q21)/ANLL → MLL - ACACA • t(11;17)(q23;q21)/ANLL → MLL - LASP1 • t(11;17)(q23;q21)/ANLL → MLL - MLLT6 (AF17) • t(11;17)(q23;q21)/ANLL → MLL - RARa • t(11;17)(q23;q25)/MDS, ANLL → MLL - SEPT9 (MSF1, AF17q25) • t(11;19)(q23;p13)/ANLL → MLL - SH3GLI1 (EEN) • t(11;19)(q23;p13)/ANLL → MLL - MYO1F • t(11;20)(q23;q11)/ALL → MLL - MAPRE1 (EB1) • t(11;22)(q23;q11.2)/ANLL → MLL - SEPT5 (hCDCRel) • t(11;22)(q23;q13)/ANLL → MLL - EP300 (P300)
MLL (myeloid/lymphoid or mixed lineage leukemia) Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 87
Breakpoints Note: spanning a 8 kb genomic region; between exons 5 to 11; highly variable on the partner, ranging from close to the NH2-term in MLLT1 (ENL), to near the COOH-term in MLLT3 (AF9).
To be noted Note: the card on 11q23 rearrangements, gives an overview on diseases implicating MLL, as well as 11q23 rearrangements in therapy related leukemias. See also 11q23 rearrangements in childhood acute lymphoblastic leukemia: Clinical aspects and congenital leukemias.
References Schichman SA, Canaani E, Croce CM. Self-fusion of the ALL1 gene. A new genetic mechanism for acute leukemia. JAMA 1995;273:571-576.
Bernard OA, Berger R. Molecular basis of 11q23 rearrangements in hematopoietic malignant proliferations. Genes Chromosom Cancer 1995;13:75-85.
Rasio D, Schichman SA, Negrini M, Canaani E, Croce CM. Complete exon structure of the ALL1 gene. Cancer Res 1996;56:1766-1769.
Nilson I, Löchner K, Siegler G, Greil J, Beck JD, Fey GH, Marschalek R. Exon/intron structure of the human ALL-1 (MLL)
gene involved in translocations to chromosomal region 11q23 and acute leukaemias. Brit J Haematol 1996;93:966-972.
Young BD, Saha V. Chromosome abnormalities in leukaemia: the 11q23 paradigm. Cancer Surv 1996;28:225-245.
Rubnitz JE, et al. 11q23 rearrangements in acute leukemia. Leukemia 1996;10:74-82. (Review).
Prasad R, et al. Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to Drosophila trithorax. Oncogene 1997;15:549-560.
Waring PM, Cleary ML. Disruption of a Homolog of Trithorax by 11q23 translocations: leukemogenic and transctiptional implications. Current Topics Microbiol Immunol 1997;220:1-23.
DiMartino JF, Cleary ML. MLL rearrangements in hematological malignancies: lessons from clinical and biological studies. Br J Haemotol 1999;106:614-626.
Huntsman DG, et al. MLL2, the second homolog of the Drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor lines. Oncogene 1999;18:7975-7984.
This article should be referenced as such: Huret JL. MLL (myeloid/lymphoid or mixed lineage leukemia). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):83-87.
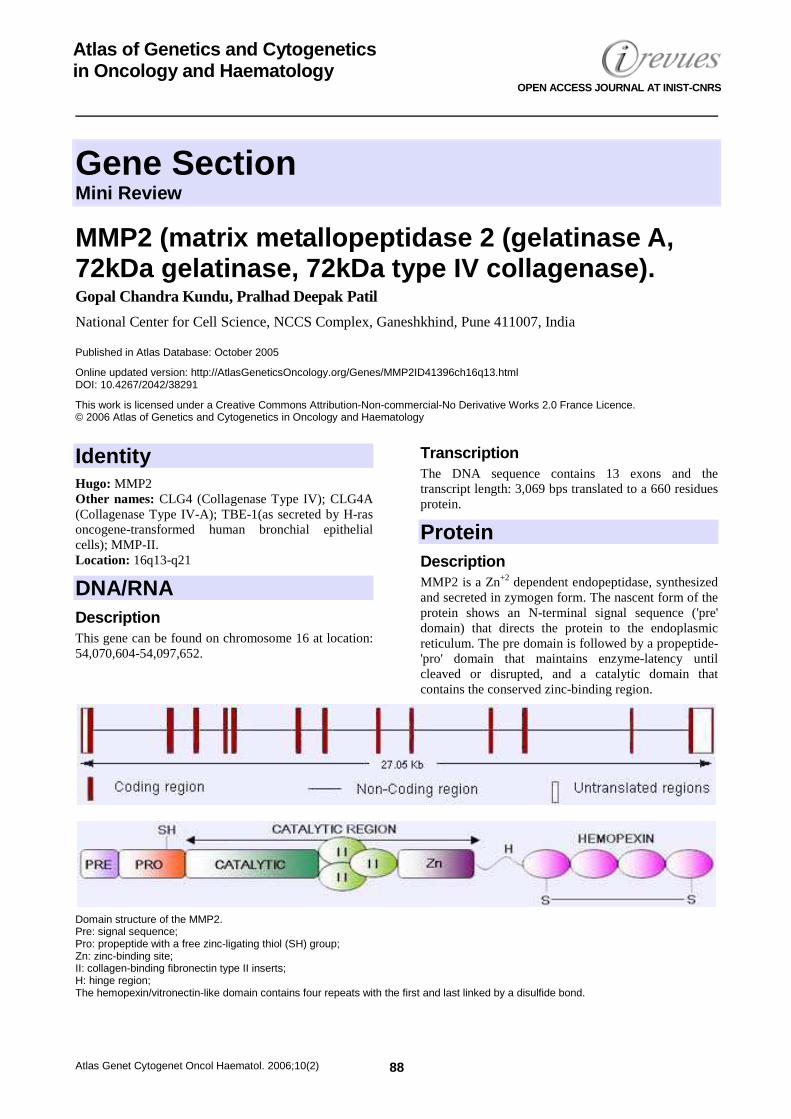
Gene Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 88
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
MMP2 (matrix metallopeptidase 2 (gelatinase A, 72kDa gelatinase, 72kDa type IV collagenase). Gopal Chandra Kundu, Pralhad Deepak Patil
National Center for Cell Science, NCCS Complex, Ganeshkhind, Pune 411007, India
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/MMP2ID41396ch16q13.html DOI: 10.4267/2042/38291
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: MMP2 Other names: CLG4 (Collagenase Type IV); CLG4A (Collagenase Type IV-A); TBE-1(as secreted by H-ras oncogene-transformed human bronchial epithelial cells); MMP-II. Location: 16q13-q21
DNA/RNA Description This gene can be found on chromosome 16 at location: 54,070,604-54,097,652.
Transcription The DNA sequence contains 13 exons and the transcript length: 3,069 bps translated to a 660 residues protein.
Protein Description MMP2 is a Zn+2 dependent endopeptidase, synthesized and secreted in zymogen form. The nascent form of the protein shows an N-terminal signal sequence ('pre' domain) that directs the protein to the endoplasmic reticulum. The pre domain is followed by a propeptide-'pro' domain that maintains enzyme-latency until cleaved or disrupted, and a catalytic domain that contains the conserved zinc-binding region.
Domain structure of the MMP2. Pre: signal sequence; Pro: propeptide with a free zinc-ligating thiol (SH) group; Zn: zinc-binding site; II: collagen-binding fibronectin type II inserts; H: hinge region; The hemopexin/vitronectin-like domain contains four repeats with the first and last linked by a disulfide bond.

MMP2 (matrix metallopeptidase 2 (gelatinase A, 72kDa gelatinase, 72kDa type IV collagenase) Kundu GC, Patil DP
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 89
A hemopexin/vitronectin-like domain is also seen, that is connected to the catalytic domain by a hinge or linker region. The hemopexin domain is involved in TIMP (Tissue Inhibitors of Metallo-Proteinases) binding, the binding of certain substrates, membrane activation, and some proteolytic activities. It also shows a series of three head-to-tail cysteine-rich repeats within its catalytic domain. These inserts resemble the collagen-binding type II repeats of fibronectin and are required to bind and cleave collagen and elastin. The regulation of MMP-2 activity occurs at many levels, of which regulation through TIMP-2 and its cell surface receptor, MT1-MMP (MMP14) is critically decisive. At higher levels of TIMP-2, MT1-MMP forms a ternary complex with MMP-2 through, leaving no free MT1-MMP receptors, thereby inhibiting the activation of pro-MMP-2 by MT1-MMP. But at lower levels of TIMP-2, due to availability of free MT1-MMP, MT1-MMP mediated activation of MMP-2 is observed. Further data also indicates that expression of TIMP-2, MMP-2 and MT1-MMP (MMP-14) is co-regulated transcriptionally, demonstrating an intricate network of regulation. Pro-MMP-2 activation is also seen by complex signaling induced by ECM proteins like osteopontin, various cytokines for example IL-8 in endothelial cells and other factors.
Expression MMP2 is tightly regulated at the transcriptional and post-transcriptional levels.
Localisation Peri/extracellular.
Function Primary function is degradation of proteins in the extracellular matrix. It proteolytically digests gelatin (denatured collagen), and types IV, V, VII, IX and X collagen. Physiologically, MMP-2 in coordination with other MMPs, play a role in normal tissue remodeling events such as embryonic development, angiogenesis, ovulation, mammary gland involution and wound healing. MMP2 is also involved in osteoblastic bone formation and/or inhibits osteoclastic bone resorption. Homology Homology in amino acid sequence is seen with the other members of Metalloproteinase family especially with MMP-9.
Mutations Germinal A G-to-A transition in codon 101 of exon 2 of MMP2 gene was detected in a Saudi family with idiopathic multicentric osteolysis. This mutation showed a replacement of an arginine by histidine (R101H) in the
prodomain, a region highly conserved across species and other members of the MMP gene family that is involved in autoproteolytic activation of MMP2. In a case of Winchester Syndrome, a homozygous 1210G-A transition in exon 8 of the MMP2 gene, leads to glu-to-lys (E404K) substitution in the catalytic domain of the protein. The glutamic acid at codon 404 is believed to be essential for the peptidase activity of all metalloproteinases, as its carboxyl group catalyzes 2 proton transfers, helps stabilize the transition state, and triggers the release of the products.
Implicated in Elevated expression of MMP-2, along with MMP-9 is usually seen in invasive and highly tumorigenic cancers such as colorectal tumors, gastric carcinoma, pancreatic carcinoma, breast cancer, oral cancer, melanoma, malignant gliomas, Chondrosarcoma, gastrointestinal adenocarcinoma. Levels are also increased in malignant astrocytomas, carcinomatous meningitis, and brain metastases.
Oncogenesis MMPs promote tumor progression and metastasis in invasive cancers by degradation of the ECM (ExtraCellular Matrix), which consists of two main components: Basement membranes and interstitial connective tissue. Though ECM comprises of many proteins (laminin-5, proteoglycans, entactin, osteonectin) collagen IV is the major element. MMP-2 & MMP-9 efficiently degrade collagen IV and laminin-5 thereby, assisting the metastatic cancerous cells to pass through the basement membrane. The degradation of ECM not only assists migration of metastatic cancerous cells, but also allows enhanced tumor growth by providing necessary space. Further, it is noteworthy that the ratio of active to latent form of MMP-2 increased with tumor progression in invasive cancers. MMP-2, with its family members also promotes angiogenesis (a critical process required for tumor cell survival) by degrading the vascular basement membrane and the interstitium.
Note: Arthritis, Autosomal recessive osteolysis disorder, Coronary Artery disease, pulmonary-emphysema and diabetic retinopathy.
References Hangauer D G, Monzingo AF and Matthews BW. An interactive computer graphics study of thermolysin-catalyzed peptide cleavage and inhibition by N-carboxymethyl dipeptides. Biochemistry 1984;23:5730-5741.
Davies B, Miles DW, Happerfield MLC et al. Activity of type IV collagenase in benign and malignant breast tissue. Br J Cancer 1993;67:1126-1131.
Morgunova E, Tuuttila A, Bergmann U, Isupov M, Lindqvist Y, Schneider G, and Tryggvason K. Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science 1999;284:1667-1670.

MMP2 (matrix metallopeptidase 2 (gelatinase A, 72kDa gelatinase, 72kDa type IV collagenase) Kundu GC, Patil DP
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 90
Stetler-Stevenenson W. Matrix metalloproteinases in angiogenesis: a moving target for therapeutic intervention. J Clin Invest 1999;103:1237-1241. (Review).
Zeng ZS, Cohen AM and Guillem JG. Loss of basement membrane type IV collagen is associated with increased expression of metalloproteinases 2 and 9 (MMP-2 and MMP-9) during human colorectal tumorigenesis. Carcinogenesis 1999;20(5):749-755.
Goldbach-Mansky R, Lee JM, Hoxworth JM, Smith D, Duray P, Schumacher HR, Yarboro CH, Klippel J, Kleiner D and El-Gabalawy HS. Active synovial matrix metalloproteinase-2 is associated with radiographic erosions in patients with early synovitis. Arthritis Res 2000, 2:145-153.
Woessner JF, Nagase H. Matrix Metalloproteinases and TIMPs. New York:Oxford Univ. Press 2000.
John A and Tuszynski G. The Role of Matrix Metalloproteinases in Tumor Angiogenesis and Tumor Metastasis. Path Onco Research 2001;7(1):14-23. (Review).
Martignetti JA, Al Aqeel A, Al Sewairi W, Boumah CE, Kambouris M, Al Mayouf S, Sheth KV, Al Eid W, Dowling O, Harris J, Glucksman MJ, Bahabri S, Meyer BF, Desnick RJ. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nature Genet 2001;28:261-265.
Philip S, Bulbule A and Kundu GC. Osteopontin Stimulates Tumor Growth and activation of Promatrix Metalloproteinase-2 through Nuclear Factor-kB-mediated Induction of Membrane Type 1 Matrix Metalloproteinase in Murine melanoma cells. J Biol Chem 2001;276(48):44926-44935.
Vu TH. Don't mess with the matrix. Nature Genet 2001;28:202-204.
Murphy G, Knauper V, Atkinson S,Butler G, English W, Hutton M, Stracke J and Clark I. Matrix metalloproteinases in arthritic disease. Arthritis Res 2002;4 (suppl 3):S39-S49. (Review).
Bloomston M, Zervos EE and Rosemurgy AS. Matrix Metalloproteinases and Their Role in Pancreatic Cancer: A Review of Preclinical Studies and Clinical Trials. Annals of Surgical Oncology 2002;9(7):668-674. (Review).
Matsuyama A, Sakai N, Ishigami M, Hiraoka H, Kashine S, Hirata A, Nakamura T, Yamashita S and Matsuzawa Y. Matrix metalloproteinases as novel disease markers in Takayasu arteritis. Circulation 2003;108:1469-1473.
Noda K, Ishida S, Inoue M, Obata K, Oguchi Y, Okada Y, Ikeda E. Production and activation of matrix metalloproteinase-2 in proliferative diabetic retinopathy. Invest Ophthal Vis Sci 2003;44:2163-2170.
Philip S and Kundu GC. Osteopontin Induces Nuclear Factor-kB-mediated Promatrix Metalloproteinase-2 activation through I-kBa/IKK Signaling Pathways, and curcumin (diferulolylmethane) Down-regulates these pathways. J Biol Chem 2003;278(16):14487-14497.
Philip S, Bulbule A and Kundu GC. Matrix metalloproteinase-2: Mechanism and regulation of NF-kB-mediated activation and its role in cell motility and ECM-invasion. Glycoconjugate Journal 2004;21:429-441.
Li A, Varney ML, Valasek J, Godfrey M, Dave BJ and Singh RK. Autocrine role of Interleukin-8 in induction of Endothelial cell proliferation, survival, migration and MMP-2 production and Angiogenesis. Angiogenesis 2005;8(1):63-71.
Zankl A, Bonafe L, Calcaterra V, Di Rocco M and Superti-Furga A. Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. Clin Genet 2005;67:261-266.
This article should be referenced as such: Kundu GC, Patil DP. MMP2 (matrix metallopeptidase 2 (gelatinase A, 72kDa gelatinase, 72kDa type IV collagenase).. Atlas Genet Cytogenet Oncol Haematol.2006;10(2):88-90.

Gene Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 91
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
PRRX2 (paired related homeobox 2) Carine Gervais
Laboratoire d'Hématologie et de Cytogénétique Onco-Hématologique, CHU de Hautepierre, Avenue Molière BP 49, 67098 Strasbourg cedex, France
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/PRRX2ID42897ch9q34.html DOI: 10.4267/2042/38292
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: PRRX2 Other names: PRX2; PMX2; MGC19843 Location: 9q34.1
DNA/RNA Description 57 kb, 4 exons.
Transcription 1327 bp mRNA.
Protein Note: Paired mesoderm homeobox protein 2, Paired related homeobox protein 2.
PRRX2 protein. HD = Homeodomain.
Description 253 amino acids, 27 kDa, contains an homeobox DNA-binding domain and an OAR domain.
Expression In embryon, higher levels of transcripts in heart, kidney, lung and skeletal muscle; lower levels in spleen and thymus; barely detecteble levels in brain and liver. In adult, higher levels in heart, lung, placenta and pancreas; moderate expression in kidney and skeletal muscle.
Localisation Nuclear.
Function Fetal skin development, cutaneous regeneration and possible role in cellular proliferation. Transcription factor activity.
Homology Member of the paired family of homeobox proteins. Murine Prrx2.
Implicated in
t(9;11)(q34;p15)/t-AML → NUP98-PPRX2 Disease One case of adult t-AML.
NUP98-PRRX2 fusion cDNA partial sequence.
Structure of the predicted chimeric NUP98-PRRX2 protein. FG = Phe-Gly repeats, GLEBS = RAE1 binding domain, HD = homeodomain.

PRRX2 (paired related homeobox 2) Gervais C
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 92
Cytogenetics No additional cytogenetic abnormality in this case.
Hybrid/Mutated Gene 5’ NUP98 - 3’ PRRX2
Abnormal Protein Fuses the GLFG repeat domains of NUP98 to the homeodomain of PRRX2.
References Stelnicki EJ, Arbeit J, Cass DL, Saner C, Harrison M, Largman C. Modulation of the human homeobox genes PRX-2 and HOXB13 in scarless fetal wounds. J Invest Dermatol 1998;111(1):57-63.
Nakamura T, Yamazaki Y, Hatano Y, Miura I. NUP98 is fused to PMX1 homeobox gene in human acute myelogenous leukemia with chromosome translocation t(1;11)(q23;p15). Blood 1999;94(2):741-747.
Norris RA, Scott KK, Moore CS, Stetten G, Brown CR, Jabs EW, Wulfsberg EA, Yu J, Kern MJ. Human PRRX1 and PRRX2 genes: cloning, expression, genomic localization, and exclusion as disease genes for Nager syndrome. Mamm Genome 2000;11(11):1000-1005.
Lam DH, Aplan PD. NUP98 gene fusions in hematologic malignancies. Leukemia 2001;15(11):1689-1695. (Review).
White P,Thomas DW,Fong S, Stelnicki E, Meijlink F, Largman C, Stephens P. Deletion of the homeobox gene PRX-2 affects fetal but not adult fibroblast wound healing responses. J Invest Dermatol 2003;120(1):135-144.
Gervais C, Mauvieux L, Perrusson N, Hélias C, Struski S, Leymarie V, Lioure B, Lessard M. A new translocation t(9;11)(q34;p15) fuses NUP98 to a novel homeobox partner gene, PRRX2, in a therapy-related acute myeloid leukemia. Leukemia 2005;19(1):145-148.
This article should be referenced as such: Gervais C. PRRX2 (paired related homeobox 2). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):91-92.

Gene Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 93
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
SEL1L (sel-1 suppressor of lin-12-like (C. elegans)) Ida Biunno, Monica Cattaneo
Istituto di Tecnologie Biomediche CNR, V. Fratelli Cervi 93, Segrate 20090, Milan, Italy
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/SEL1LID42246ch14q24.html DOI: 10.4267/2042/38293
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: SEL1L Other names: IBD2; SEL1-like Location: 14q24.3-q31 Local order: SEL1L is located within a 'Gene Desert area' or 'Genome Deserts'; centromeric to FLRT2 (fibronectin leucine rich transmembrane protein 2) and telomeric to GTF2A1 (general transcription factor IIA) and TSHRq31 (thyroid stimulating hormone receptor). Note: SEL1L is the human ortholog of the C.Elegans
sel-1 (suppressor enhancer of lin-12) gene. It shows a high degree of cross-species conservation in its nucleotide and protein sequence.
DNA/RNA Description SEL1L genomic size is of 62,24 Kb localized from 81069886 to 81007646. 3’ the first exon lies the basal core of the promter, a TATA-less promoter containing four SP1 binding sites and a CAAT box. A CpG island
A graphical representation of SEL1L isorforms. The black numbered rectangles correspond to the exons, while the white rectangles correspond to the intronic sequence which is retained in the alternative isoforms. The SEL1L domains are indicated on the top of the isoforms. (FN2=fibronectin type II domain; I, II and III clusters of SEL-1 like repeats; Hrd3; TM=transmembrane; P= proline rich domain).

SEL1L (sel-1 suppressor of lin-12-like (C. elegans)) Biunno I, Cattaneo M
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 94
is located between -550bp and the start codon. SEL1L embryonic kidney cells. The C-terminal tail consists of over 5,0Kb untranslated sequences likely containing key regulatory elements.
Transcription The sequence is composed of 21 exons and produces at least five different alternative transcripts (A-E) which originate from alternative splicing and putative promoter usage. Exons 1-6 are common to forms A-B-C-E. Pseudogene No known pseudogenes
Protein Note: SEL1L is a multimodular protein consisting of several domains and signal sequences that confer the multifaceted specificities to the molecule.
Description SEL1L is not a member of a vast family of proteins but the several described isoforms (over 4) give the appearance of belonging to a multifamily of molecules having perhaps redundant functions.
Expression Ubiquitously expressed only in fetal and neoplastic tissues. In normal adult tissues is highly expressed in the acini and in the alpha cells of the pancreas; in general is highly represented in secretory cells such as plasma cells.
Localisation SEL1L protein can have a nuclear, cytoplasmic and nuclear-cytoplasmic location.
Function May have a role in the ER-associated protein degradation (ERAD) system (similarity with Hrd3). Negative regulator of the NOTCH pathway in C. Elegans. May play a role in TGF beta signalling. In breast and pancreatic tumor decrease tumor growth and aggressiveness, possibly involving cell-matrix interaction.
Homology Comparative sequence analysis across different regna, including metazoa, fungi, viridiplantae and bacteria, revealed the remarkable conservation of its primary sequence, although the gene structural complexity increased in evolution. Among mammals, SEL1L shares strict amino acid identity with chimpanzee (99%), dog (97%), hamster (92%), mouse (93%) and rat (92%). It also shows a good similarity with the model organisms such as xenopus (82%), chicken (83%), zebrafish (73%), Drosophila melanogaster (51%) and C. elegans (46%) (Table 1). Arabidopsis thaliana and Saccharomyces cerevisiae display lower similarity (34% and 28%, respectively).
Mutations Note: Neither causative nor functional mutations were found except for the presence of two base substitutions in the minimal promoter region in two well differentiated lung adenocarcinoma that led to a significant increase in the transcription. A polymorphic base substitution was reported in the fibronectin type II domain of the gene in children affected by persistent hyperinsulinemic hypoglycemia (insulinoma) of infancy which induces a major change in the amino acid composition.
SEL1L protein structure: SEL1L is a multimodular protein containing several structural and functional domains as well as signal sequences. The signal peptide (from 1 to 22 amino acid residues) and the Pest sequence (from 80 to 102 amino acid residues) are represented by red and pink rectangles. The fibronectin type II domain (from 120 to 168 residues) is symbolized by the hexagon (FNII), the SEL-1-like repeats are represented by rhombi and are distributed in tandem along the central portion of the protein in three large clusters (I cluster: 183-326; II cluster: 373-554 and III cluster: 664-675 residues). The Hrd3 like motif is located within the last SEL-1-like repeat (664-675 residues) and is represented by an circle. The transmembrane region (TM) (739-761 residues) and the proline-rich tail (770-793 residues) are symbolized by a blue rectangle. The N-linked glycosilation is also underlined.

SEL1L (sel-1 suppressor of lin-12-like (C. elegans)) Biunno I, Cattaneo M
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 95
Implicated in Considering the overall results published on SEL1L by various investigators working in different organisms, it can, perhaps, safely be deduced that this gene plays a fundamental role in eukaryotic intracellular protein degradation processes. Protein degradation is becoming a central theme in cancer biology and recently therapeutic approaches that use inhibitors of proteins belonging to ubiquitin-proteosoma pathway have been developed in solid tumors and haematological diseases. A survey of the expression of SEL1L mRNA as well as its encoded protein on a series of cancerous and pre-neoplastic lesion, revealed the role of SEL1L in cancer progression. Furthermore, its expression in breast cancer correlated with patient¹s survival. In vitro studies indicated that SEL1L protein affects those pathways which regulate signalling (cell-cell and or cell-matrix) interactions. Available data derived from several organisms indicate that it may function in the protein degradation processes through ubiquitin-proteosome system and perhaps in regulating important pathways such as Notch and TGF-beta. The fundamental question raised by the observation that SEL1L gets up-modulated during the early steps of tumor transformation is of paramount importance for early diagnosis. Currently it is only possible to hypothesize that the increased SEL1L levels are required in order to meet the advent of genetic and/or genomic structural alterations acquired during cancer initiation or to influence intra-cellular signalling. Its presence may be important in protecting cellular homeostasis from genetic mutations.
References Biunno I, Appierto V, Cattaneo M, Leone BE, Balzano G, Socci C, Saccone S, Letizia A, Della Valle G, Sgaramella V. Isolation of a pancreas-specific gene located on human chromosome 14q31: expression analysis in human pancreatic ductal carcinomas. Genomics 1997;46(2):284-286.
Donoviel DB, Donoviel MS, Fan E, Hadjantonakis A, Bernstein A. Cloning and characterization of Sel-1l, a murine homolog of the C. elegans sel-1 gene. Mech Dev 1998;78(1-2):203-207.
Donoviel DB, Bernstein A. SEL-1L maps to human chromosome 14, near the insulin-dependent diabetes mellitus locus 11. Genomics 1999;56(2):232-233.
Harada Y, Ozaki K, Suzuki M, Fujiwara T, Takahashi E, Nakamura Y, Tanigami A. Complete cDNA sequence and genomic organization of a human pancreas-specific gene homologous to Caenorhabditis elegans sel-1. J Hum Genet 1999;44(5):330-336.
Biunno I, Bernard L, Dear P, Cattaneo M, Volorio S, Zannini L, Bankier A, Zollo M. SEL1L, the human homolog of C. elegans sel-1: refined physical mapping, gene structure and identification of polymorphic markers. Hum Genet 2000;106(2):227-235.
Cattaneo M, Orlandi R, Ronchini C, Granelli P, Malferrari G, Menard S, Biunno I. The expression of SEL1L and TAN-1 in normal and neoplastic cells. Int J Biol Markers 2000;15(1):26-32.
Cattaneo M, Sorio C, Malferrari G, Rogozin IB, Bernard L, Scarpa A, Zollo M, Biunno I. Cloning and functional analysis of SEL1L promoter region, a pancreas-specific gene. DNA Cell Biol 2001;20(1):1-9.
Ban Y, Taniyama M, Tozaki T, Yanagawa T, Tomita M, Ban Y. SEL1L microsatellite polymorphism in Japanese patients with autoimmune thyroid diseases. Thyroid 2001;11(4):335-338.
Cattaneo M, Zollo M, Malferrari G, Orlandi R, D'Angelo A, Menard S, Biunno I. Allelic polymorphisms in the transcriptional regulatory region of human SEL1L. Mutat Res 2001;458(3-4):71-76.
Larsen ZM, Angelo AD, Cattaneo M, Nerup J, Biunno I, Zollo M, Pociot F. Complete mutation scanning of the human SEL 1L gene: a candidate gene for type 1 diabetes. Acta Diabetol 2001;38(4):191-192.
Pociot F, Larsen ZM, Zavattari P, Deidda E, Nerup J, Cattaneo M, Chiaramonte R, Comi P, Sabbadini M, Zollo M, Biunno I, Cucca F. No evidence for SEL1L as a candidate gene for IDDM11-conferred susceptibility. Diabetes Metab Res Rev 2001;17(4):292-295.
Biunno I, Castiglioni B, Rogozin IB, DeBellis G, Malferrari G, Cattaneo M. Cross-species conservation of SEL1L, a human pancreas-specific expressing gene. OMICS 2002;6(2):187-198.
Chiaramonte R, Calzavara E, Basile A, Comi P, Sherbet GV. Notch signal transduction is not regulated by SEL1L in leukaemia and lymphoma cells in culture. Anticancer Res 2002;22(6C):4211-4214.
Chiaramonte R, Sabbadini M, Balordi F, Comi P, Sherbet GV. Allele frequency of two intragenic microsatellite loci of SEL1L gene in Northern Italian population. Mol Cell Biochem 2002;232(1-2):159-161.
Orlandi R, Cattaneo M, Troglio F, Campiglio M, Biunno I, Ménard S. Production of a monoclonal antibody directed against the recombinant SEL1L protein. Int J Biol Markers 2002;17(2):104-111.
Orlandi R, Cattaneo M, Troglio F, Casalini P, Ronchini C, Menard S, Biunno I. SEL1L expression decreases breast tumor cell aggressiveness in vivo and in vitro. Cancer Res 2002;62(2):567-574.
Cattaneo M, Orlandini S, Beghelli S, Moore PS, Sorio C, Bonora A, Bassi C, Talamini G, Zamboni G, Orlandi R, Menard S, Bernardi LR, Biunno I, Scarpa A. SEL1L expression in pancreatic adenocarcinoma parallels SMAD4 expression and delays tumor growth in vitro and in vivo. Oncogene 2003;22(41):6359-6368.
Kaneko M, Nomura Y. ER signaling in unfolded protein response. Life Sci 2003;74(2-3):199-205.
Kim SH, Ma X, Klupa T, Powers C, Pezzolesi M, Warram JH, Rich SS, Krolewski AS, Doria A. Genetic modifiers of the age at diagnosis of diabetes (MODY3) in carriers of hepatocyte nuclear factor-1alpha mutations map to chromosomes 5p15, 9q22, and 14q24. Diabetes 2003;52(8):2182-2186.
Mathlouthi R, Aberle S, Schug N, Küpper JH, Schröder K, Seitz G, Blin N. Assessing optimal promoter activity for constructs in gastrointestinal gene. Anticancer Res 2003;23(5A):4011-4015.
Aberle S, Schug N, Mathlouthi R, Seitz G, Küpper JH, Schröder K, Blin N. Promoter selection for the cytosine deaminase suicide gene constructs in gastric cancer. Eur J Gastroenterol Hepatol 2004;16(1):63-67.
Cattaneo M, Canton C, Albertini A, Biunno I. Identification of a region within SEL1L protein required for tumour growth inhibition. Gene 2004;326:149-156.
Diaferia G, Cattaneo M, Saltini G, Proverbio MC, Monferini E, Malferrari G, Albertini A, Biunno I. RNA-mediated interference

SEL1L (sel-1 suppressor of lin-12-like (C. elegans)) Biunno I, Cattaneo M
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 96
indicates that SEL1L plays a role in pancreatic beta-cell growth. DNA Cell Biol 2004;23(8):510-518.
Granelli P, Cattaneo M, Ferrero S, Bottiglieri L, Bosari S, Fichera G, Biunno I. SEL1L and squamous cell carcinoma of the esophagus. Clin Cancer Res 2004;10(17):5857-5861.
Saltini G, Proverbio MC, Malferrari G, Biagiotti L, Boettcher P, Dominici R, Monferini E, Lorenzini E, Cattaneo M, Antonello D, Moore PS, Zamproni I, Viscardi M, Chiumello G, Biunno I. Identification of a novel polymorphism in the fibronectin type II domain of the SEL1L gene and possible relation to the persistent hyperinsulinemic hypoglycemia of infancy. Mutat Res 2004;554(1-2):159-163.
Bianchi L, Canton C, Bini L, Orlandi R, Ménard S, Armini A, Cattaneo M, Pallini V, Bernardi LR, Biunno I. Protein profile changes in the human breast cancer cell line MCF-7 in response to SEL1L gene induction. Proteomics 2005;5(9):2433-2442.
This article should be referenced as such: Biunno I, Cattaneo M. SEL1L (sel-1 suppressor of lin-12-like (C. elegans)). Atlas Genet Cytogenet Oncol Haematol.2006; 10(2):93-96.

Gene Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 97
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
SIL (SCL/TAL1 interrupting locus) Asher Castiel, Shai Izraeli
Department of Pediatric Hemato-Oncology, Cancer Research Center, Sheba Medical Center, Tel-Hashomer, Ramat Gan, Israel
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/SILID524ch1p32.html DOI: 10.4267/2042/38294
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: STIL Other names: TAL1 (SCL) interrupting locus Location: 1p32 Local order: UMP-CMPK (47511608-47556531); SIL (47427869-47491842); TAL1 (47393984-47407363). Note: 47,427,869 bp from pter; End: 47,491,842 bp from pter; Size: 63,973 bases; Orientation: minus strand.
DNA/RNA Description 18 exons distributed over 70 kb. 5' portion of the gene demonstrating alternate exon utilization.
Protein Description 1287 amino acids, 148 KDa protein which is highly conserved in vertebrates only. No homologies to known protein motifs except for conserved Serin/Threonine phosporylation sites.
Expression SIL is an immediate early gene, with ubiquitous expression in proliferating cells and during early embryonic development. SIL protein levels peak during mitosis and are degraded on transition to G1. SIL is phosphorylated in mitosis. It is expressed in multiple cancers. In lung cancer its expression correlates with the expression of mitotic checkpoint genes.
Localisation Cytosolic protein.
Function SIL knockout mouse embryos die in utero displaying holopresencephaly, randomized left/right asymmetry and marked apoptosis of the neural folds. Genetic evidence showed that SIL is required for the Sonic Hedgehog response pathway. SIL phosphorylation and interactions with PIN1 is required for maintenance of the mitotic checkpoint.
Homology There is no homology to other known proteins.
Genomic structure of SIL. EcoRI sites (R) are indicated. Exons are as shown; the smaller exons are not drawn to scale.

SIL (SCL/TAL1 interrupting locus) Castiel A, Izraeli S
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 98
Schematic representation of SIL/SCL fusion mRNA. The germ line SIL (solid boxes) and SCL (open boxes) genomic structures are shown. The deletion breakpoints are indicated with arrows. The SIL/SCL genomic rearrangement is indicated below. The SIL/SCL fusion mRNA is formed by SIL exon 1 (solid box) splicing to SCL exon 3 (open box) in a head-to-tail fashion.
Mutations Note: No mutations were found in families with hereditary holoprosencephaly.
Implicated in SIL- TAL1 (SCL) fusion Note: A submicroscopic deletions fuses the promoter of SIL to TAL1 to induce an abnormal expression of TAL1.
Disease T-cell ALL. This TAL1-SIL fusion transcript is found in approximately 25% of T-ALL patients.
Cytogenetics Normal karyotype.
Hybrid/Mutated Gene The promoter region of the SCL gene, a hematopoietic transcription factor, and the coding region of the SIL gene are deleted. The molecular result of this SIL/SCL rearrangement is an interstitial deletion on chromosome 1 that juxtaposes the 5' portion of the SIL gene to the coding region of the SCL gene. A SIL/SCL fusion mRNA is produced, with SIL exon 1 splicing to SCL exon 3 in a head-to-tail fashion. Because these are both 5' untranslated region (UTR) exons, the net result is that SIL promoter and enhancer elements drive the expression of a full length SCL gene product.
SIL overexpression in lung cancer Note: SIL is also overexpressed in various solid tumors (melanoma, lymphoma, ovary cancer, breast cancer colon cancer lung and prostate cancer) and leukemic
cell lines (Dami-acute megakaryocytic, and K562- erythroid blast crisis of chronic myeloid leukemia).
Disease High expression in non-small cell lung cancer (NSCLC). In addition, high expression levels in lung adenocarcinoma, lung squamous carcinoma and lung small cell carcinoma.
Prognosis SIL expression is associated with cell proliferation. In lung cancer, SIL overexpression is correlated with high mitotic activity.
References Aplan PD, Lombardi DP, Ginsberg AM, Cossman J, Bertness VL, Kirsch IR. Disruption of the human SCL locus by 'illegitimate' V-(D)-J recombinase activity. Science 1990 Dec 7;250(4986):1426-1429.
Peter D. Aplan, Donald P. Lombardi, Ilan R. Kirsch. Structural Characterization of SIL, a Gene Frequently Disrupted in T-Cell Acute Lymphoblastic Leukemia. Mol Cell Biol 1991 Nov;11(11):5462-5469.
Aplan PD, Lombardi DP, Reaman GH, Sather HN, Hammond GD, Kirsch IR. Involvement of the putative hematopoietic transcription factor SCL in T-cell acute lymphoblastic leukemia. Blood 1992;79(5):1327-1333.
Izraeli S, Colaizzo-Anas T, Bertness VL, Mani K, Aplan PD, Kirsch IR. Expression of the SIL gene is correlated with growth induction and cellular proliferation. Cell Growth Differ 1997;8(11):1171-1179.
Izraeli S, Lowe LA, Bertness VL, Good DJ, Dorward DW, Kirsch IR, Kuehn MR. The SIL gene is required for mouse embryonic axial development and left-right specification. Nature 1999 Jun 17;399(6737):691-694.
Izraeli S, Lowe LA, Bertness VL, Campaner S, Hahn H, Kirsch IR, Kuehn MR. Genetic evidence that Sil is required for the Sonic Hedgehog response pathway. Genesis 2001;31(2):72-77.

SIL (SCL/TAL1 interrupting locus) Castiel A, Izraeli S
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 99
Karkera JD, Izraeli S, Roessler E, Dutra A, Kirsch I, Muenke M. The genomic structure, chromosomal localization, and analysis of SIL as a candidate gene for holoprosencephaly. Cytogenet Genome Res 2002;97(1-2):62-67.
Colaizzo-Anas T, P. D. Aplan. Cloning and characterization of the SIL promoter. Biochim Biophys Acta - Gene Structure and Expression 2003;1625(2):207-213.
Erez A, Perelman M, Hewitt SM, Cojacaru G, Goldberg I, Shahar I, Yaron P, Muler I, Campaner S, Amariglio N, Rechavi G, Kirsch IR, Krupsky M, Kaminski N, Izraeli S. Sil
overexpression in lung cancer characterizes tumors with increased mitotic activity. Oncogene 2004 Jul 8;23(31):5371-5377.
Campaner S, Kaldis P, Izraeli S, Kirsch IR. Sil phosphorylation in a Pin1 binding domain affects the duration of the spindle checkpoint. Mol Cell Biol 2005 Aug;25(15):6660-6672.
This article should be referenced as such: Castiel A, Izraeli S. SIL (SCL/TAL1 interrupting locus). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):97-99.

Gene Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 100
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
SIX1 (sine oculis homeobox homolog 1) (mammalian) Heide L Ford, Aaron N Patrick, Marileila Varella-Ga rcia
Departments of Obstetrics and Gynecology and Biochemistry and Molecular Genetics, University of Colorado Health Sciences Center, Fitzsimons Campus, Mail stop 8309, P.O. Box 6511, Aurora, CO 80045, USA
Published in Atlas Database: November 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/SIX1ID42302ch14q23.html DOI: 10.4267/2042/38295
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: SIX1 Other names: HSIX1 (human SIX1); Sine oculis (Drosophila homolog) Location: 14q23
DNA/RNA
View of the SIX1 gene which is composed of 2 exons that are 833 and 543 bps respectively, and a single 2,052 bp intron. Start and stop codon positions are shown.
Description The gene is composed of 2 exons and one intron and can be found on chromosome 14 at location 60,182,506 - 60,185,933.
Transcription The transcript length is 1,376 base pairs and the start of the transcript is located in Contig AL049874.3.1.193047. The transcript can be detected in a variety of tissues during mouse development (see below for protein expression) and this has also been confirmed at the protein level (using reporter genes). The transcript is further detected throughout normal mouse mammary gland development with levels being
the highest in the embryonic mammary gland and decreasing as the mammary gland differentiates in pregnancy and lactation. In the adult human, it is detected in skeletal muscle, pituitary gland, salivary gland, kidney, lung, and trachea.
Pseudogene No.
Protein
View of the Six1 protein (total length 284 amino acids) that contains an N-terminal 115 amino acid Six domain (SD), and a 60 amino acid six-type homeodomain (HD). The SD is important for the interaction of Six1 with cofactors and also contributes to DNA binding along with the homeodomain.
Description Six1 belongs to the Six family of homeoproteins. Amino acids: 284. Predicted Molecular Weight: 32210 Dalton. It exists as a phosphoprotein and is hyperphosphorylated in mitosis.
Expression During mouse development, Six1 is expressed in otic vesicles, nasal placodes, branchial arches, Rathke¹s pouch, dorsal root ganglion, proximal cranial ganglia, somites, cranial mesenchyme, nephrogenic cords, and limb mesenchyme. Six1 expression in muscles is present throughout myogenesis and into adulthood.
Localisation Nuclear.

SIX1 (sine oculis homeobox homolog 1) (mammalian) Ford HL et al.
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 101
I
Diagram of chromosome 14 (A) showing location of the SIX1 gene at 14q23 (clone RP11-1042B17, labeled in SpectrumRed). Clone RPMI-324B11 (labeled in SpectrumGreen) mapped at 14q11.2 was used as control. Metaphase spread (B) and interphase nuclei (C) of the non-malignant, immortalized MCF10A cell line showing two normal copies of chromosome 14 with signals generated by the SIX1 and control FISH probes. Metaphase spread of the mammary carcinoma cell line, 21MT2 (D), showing a normal copy of chromosome 14 with SIX1 (red) and control probe (green), two derivative chromosomes carrying only the SIX1 or the SIX1 and control signals and a copy of the derivative 14 chromosome with SIX1 gene amplification. Interphase nuclei from 21MT2 cells with multiple copies of red and green signals, including a cluster of SIX1 signals representing gene amplification (E).
Function Six1 is a transcription factor that is known to play a role in the proliferation and survival of precursor cells during normal development in numerous tissues including, amongst others, the kidney, inner ear, and muscle. It is also demonstrated to play a role in the proliferation of cancer cells and in cancer metastasis. It is known to activate several target genes, including cyclin A1, c-MYC, GDNF, and SLC12A2.
Mutations Germinal Germline mutations of SIX1 are observed in branchio-oto-renal sydrome, an autosomal dominant developmental disorder that is characterized by kidney and urinary tract malformations and hearing loss. The three SIX1 mutations identified to date all interfere with the ability of the Six1 protein to interact with its Eya1 cofactor, and two of the identified mutations additionally affect Six1-DNA binding. Somatic The SIX1 gene is amplified in about 5% of breast cancers (infiltrating ductal carcinomas).Overexpression of Six1 has been found in breast cancer, in Wilms¹tumors, and in rhabdomyosarcoma.
Implicated in Six1 is implicated in a number of cancers including breast cancer, rhabdomyosarcomas, and Wilms’ tumors. It has also been implicated in branchio-oto-renal syndrome. Note: Six1 is overexpressed in approximately 50% of primary breast cancers and 90% of metastatic lesions. Its overexpression in breast cancer has been linked to increased proliferation of breast cancer cells. Six1 is
also a critical mediator of metastasis in a mouse rhabdomyosarcoma model.
Disease Breast cancer, wilms’ tumor, rhabdomyosarcoma, branchio-oto-renal syndrome,
Cytogenetics The SIX1 gene is amplified in human breast cancer
Oncogenesis SIX1 is overexpressed in several tumor types, including breast cancer, rhabdomyosarcomas, and Wilms’ tumors. It has been implicated in both the proliferation and metastasis of tumor cells.
References Oliver G, Wehr R, Jenkins NA, Copeland NG, Cheyette BN, Hartenstein V, et al. Homeobox genes and connective tissue patterning. Development 1995;121(3):693-705.
Boucher CA, Carey N, Edwards YH, Siciliano MJ, Johnson KJ. Cloning of the human SIX1 gene and its assignment to chromosome 14. Genomics 1996;33(1):140-142.
Ford HL, Kabingu EN, Bump EA, Mutter GL, Pardee AB. Abrogation of the G2 cell cycle checkpoint associated with overexpression of HSIX1: a possible mechanism of breast carcinogenesis. Proc Natl Acad Sci USA 1998;95(21):12608-12613.
Spitz F, Demignon J, Porteu A, Kahn A, Concordet JP, Daegelen D, et al. Expression of myogenin during embryogenesis is controlled by Six/sine oculis homeoproteins through a conserved MEF3 binding site. Proc Natl Acad Sci USA 1998;95(24):14220-14225.
Khan J, Bittner ML, Saal LH, Teichmann U, Azorsa DO, Gooden GC, et al. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci USA 1999;96(23):13264-13269.
Gallardo ME, López-Ríos J, Fernaud-Espinosa I, Granadino B, Sanz R, Ramos C, et al. Genomic cloning and characterization of the human homeobox gene SIX6 reveals a cluster of SIX genes in chromosome 14 and associates SIX6 hemizygosity with bilateral anophthalmia and pituitary anomalies. Genomics 1999;61(1):82-91.

SIX1 (sine oculis homeobox homolog 1) (mammalian) Ford HL et al.
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 102
Relaix F, Buckingham M. From insect eye to vertebrate muscle: redeployment of a regulatory network. Genes Dev 1999;13(24):3171-3178. (Review).
Heanue TA, Reshef R, Davis RJ, Mardon G, Oliver G, Tomarev S, et al. Synergistic regulation of vertebrate muscle development by Dach2, Eya2, and Six1, homologs of genes required for Drosophila eye formation. Genes Dev 1999;13(24):3231-3243.
Ford HL, Landesman-Bollag E, Dacwag CS, Stukenberg PT, Pardee AB, Seldin DC. Cell cycle-regulated phosphorylation of the human SIX1 homeodomain protein. J Biol Chem 2000;275(29):22245-22254.
Kawakami K, Sato S, Ozaki H, Ikeda K. Six family genes- structure and function as transcription factors and their roles in development. Bioessays 2000;22(7):616-626. (Review).
Pandur PD, Moody SA. Xenopus SIX1 gene is expressed in neurogenic cranial placodes and maintained in the differentiating lateral lines. Mech Dev 2000;96(2):253-257.
Rodríguez de Córdoba S, Gallardo ME, López-Ríos J, Bovolenta P. The Human Six family of homeobox genes. Current Genomics 2001;2:231-242. (Review).
Li CM, Guo M, Borczuk A, Powell CA, Wei M, Thaker HM, et al. Gene expression in Wilms' tumor mimics the earliest committed stage in the metanephric mesenchymal-epithelial transition. Am J Pathol 2002;160(6):2181-2190.
Ikeda K, Watanabe Y, Ohto H, Kawakami K. Molecular interaction and synergistic activation of a promoter by Six, Eya, and Dach proteins mediated through CREB binding protein. Mol Cell Biol 2002;22(19):6759-6766.
Fougerousse F, Durand M, Lopez S, Suel L, Demignon J, Thornton C, et al. Six and Eya expression during human somitogenesis and MyoD gene family activation. J Muscle Res Cell Motil 2002;23(3):255-264.
Laclef C, Hamard G, Demignon J, Souil E, Houbron C, Maire P. Altered myogenesis in Six1-deficient mice. Development 2003;130(10):2239-2252.
Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 2003;426(6964):247-254.
Epstein JA, Neel BG. Signal transduction: an eye on organ development. Nature 2003;426(6964):238-239. (Review).
Xu PX, Zheng W, Huang L, Maire P, Laclef C, Silvius D. Six1 is required for the early organogenesis of mammalian kidney. Development 2003;130(14):3085-3094.
Zheng W, Huang L, Wei ZB, Silvius D, Tang B, Xu PX. The role of Six1 in mammalian auditory system development. Development 2003;130(17):3989-4000.
Laclef C, Souil E, Demignon J, Maire P. Thymus, kidney and craniofacial abnormalities in Six 1 deficient mice. Mech Dev 2003;120(6):669-679.
Bessarab DA, Chong SW, Korzh V. Expression of zebrafish six1 during sensory organ development and myogenesis. Dev Dyn 2004;230(4):781-786.
Yu Y, Khan J, Khanna C, Helman L, Meltzer PS, Merlino G. Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat Med 2004;10(2):175-181.
Zou D, Silvius D, Fritzsch B, Xu PX. Eya1 and Six1 are essential for early steps of sensory neurogenesis in mammalian cranial placodes. Development 2004;131(22):5561-5572.
Brodbeck S, Englert C. Genetic determination of nephrogenesis: the Pax/Eya/Six gene network. Pediatr Nephrol 2004;19(3):249-255. (Review).
Grifone R, Laclef C, Spitz F, Lopez S, Demignon J, Guidotti JE, et al. Six1 and Eya1 expression can reprogram adult muscle from the slow-twitch phenotype into the fast-twitch phenotype. Mol Cell Biol 2004;24(14):6253-6267.
Ozaki H, Nakamura K, Funahashi J, Ikeda K, Yamada G, Tokano H, et al. Six1 controls patterning of the mouse otic vesicle. Development 2004;131(3):551-562.
Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1. Proc Natl Acad Sci USA 2004;101(21):8090-8095.
Brugmann SA, Pandur PD, Kenyon KL, Pignoni F, Moody SA. Six1 promotes a placodal fate within the lateral neurogenic ectoderm by functioning as both a transcriptional activator and repressor. Development 2004;131(23):5871-5881.
Coletta RD, Christensen K, Reichenberger KJ, Lamb J, Micomonaco D, Huang L, et al. The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci USA 2004;101(17):6478-6483.
Reichenberger KJ, Coletta RD, Schulte AP, Varella-Garcia M, Ford HL. Gene amplification is a mechanism of SIX1 overexpression in breast cancer. Cancer Res 2005;65(7):2668-2675.
Gehring WJ. New perspectives on eye development and the evolution of eyes and photoreceptors. J Hered 2005;96(3):171-184. (Review).
Grifone R, Demignon J, Houbron C, Souil E, Niro C, Seller MJ, et al. Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development 2005;132(9):2235-2249.
Bonnin MA, Laclef C, Blaise R, Eloy-Trinquet S, Relaix F, Maire P, et al. Six1 is not involved in limb tendon development, but is expressed in limb connective tissue under Shh regulation. Mech Dev 2005;122(4):573-585.
Ando Z, Sato S, Ikeda K, Kawakami K. Slc12a2 is a direct target of two closely related homeobox proteins, Six1 and Six4. Febs J 2005;272(12):3026-3041.
This article should be referenced as such: Ford HL, Patrick AN, Varella-Garcia M. SIX1 (sine oculis homeobox homolog 1) (mammalian). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):100-102.

Gene Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 103
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
STK4 (serine/threonine kinase 4) Jonathan Chernoff
Fox Chase Cancer Center, 333 Cottman Ave, Philadelphia, PA 19111, USA
Published in Atlas Database: November 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/STK4ID42440ch20q11.html DOI: 10.4267/2042/38296
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: STK4 Other names: MST1; KRS2 Location: 20q11.2-q13.2 Local order: centromere - YWHAB - TOMM34 - STK4 - MATN4 - RBPSUHL - telomere. Note: STK4 encodes a serine/threonine specific protein kinase that is a member of the GC kinase branch of the STE20 family. STK4 plays a role in apoptosis and may have tumor suppressor function.
DNA/RNA Description The STK4 gene contains 11 exons. The sizes of the exons 1-11 are 68, 81, 129, 115, 165, 168, 138, 129, 192, 154, and 585 bps. Exon 1 contains the 5' untranslated region and the translation initation ATG, and a few additional codons. Exon 11 contains the stop codon and the 3' untranslated region. Other features of the STK4 gene, such as promoters or enhancer elements, have not been described.
Transcription A 7 kb transcript is detected in many tissues with
highest steady state levels in the thymus amd bone marrow. The predominant human STK4 mRNA encodes an open reading frame of 1883 bases, resulting in a predicted proteins of 487 amino acids.
Protein Description STK4 is a member of the GC kinase group of the STE20 family of serine/threonine protein kinases. STK4 homodimerizes through a C-terminal motif, and removal of the C terminus results in marked activation of the kinase. STK4 is cleaved by caspases during apotosis, releasing an active 34 kD kinase fragment. STK4 associates with the WW-domain protein Salvadore, which may link STK4 to the LATS tumor suppressor pathway.
Expression Widely expressed in both embryonic and adult tissues.
Localisation Nucleus and cytoplasm. In the nucleus, STK4 phosphorylates Histone 2B at Ser 14, a modification associated with chromosome condensation in apoptotic cells.
The alignment of STK4 mRNA to its genomic sequence.

STK4 (serine/threonine kinase 4) Chernoff J
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 104
Structure of STK4 protein. The catalytic (protein kinase) domain occupies the N-terminal half of STK4. The regulatory domain inhibits kinase activity and also contains a dimerization motif. A caspase-sensitive cleavage site is located between these two domains.
Function STK4 plays a role in promoting apotosis, in particular, in chromosome condensation during programmed cell death. STK4 is cleaved by caspase 3 during apoptosis, releasing the highly active N-terminal kinase domain. This active protein promotes apoptosis by activating JNK and also by further caspase activation. STK4, and/or the highly related protein STK3, may act as tumor suppressors, acting downstream of Raf.
Homology STK3 (a.k.a. MST2, KRS1).
References Creasy CL, Chernoff J. Cloning and characterization of a human protein kinase with homology to Ste20. J Biol Chem 1995;270:21695-21700.
Taylor LK, Wang HCR, Erikson RL. Newly identified stress-responsive protein kinases, Krs-1 and Krs-2. Proc Nat Acad Sci 1996;93:10099-10104.
Graves JD, Gotoh Y, Draves KE, Ambrose D, Han DKN, Wright M, Chernoff J, Clark EA, Krebs EG. Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J 1998;17:2224-2234.
Lee KK, Yonehara S. Phosphorylation and dimerization regulate nucleocytoplasmic shuttling of mammalian STE20-like kinase (MST). J Biol Chem 2002;277:12351-12358.
Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, Beeser A, Etkin LD, Chernoff J, Earnshaw WC, Allis CD. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 2003;113:507-517.
Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003, 114:457-467.
Praskova M, Khoklatchev A, Ortiz-Vega S, Avruch J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem J 2004;381:453-462.
O'Neill EE, Matallanas D, Kolch W. Mammalian sterile 20-like kinases in tumor suppression: an emerging pathway. Cancer Res 2005;65:5485-5487.
This article should be referenced as such: Chernoff J. STK4 (serine/threonine kinase 4). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):103-104.

Gene Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 105
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
XRCC3 (X-ray repair complementing defective repair in Chinese hamster cells 3) Ulla Vogel
National Research Centre for the Working Environment, Lerso Parkalle 105, DK-2100 Copenhagen O, Denmark
Published in Atlas Database: November 2005
Online updated version: http://AtlasGeneticsOncology.org/Genes/XRCC3ID335ch14q32.html DOI: 10.4267/2042/38297
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Hugo: XRCC3 Location: 14q32.3
DNA/RNA Description 17 800 bp, 9 exons.
Transcription 2,504 bp.
Protein Description 346 amino acids.
Function XRCC3 is required for efficient repair of double strand breaks via homologous recombinational repair (link), for correct chromosomal segregation and for repair of DNA cross links. Inactivation of XRCC3 in CHO cells results in increased radiation and cisplatin sensitivity. In human cells, XRCC3 forms a complex with Rad51C which is recruited early to DNA damage. Inactivation of XRCC3 in human cells leads to a two-fold sensitivity to DNA cross-linking agents, impaired Rad51 focus formation, elevated chromosome aberrations and five to seven-fold increased endoreduplication.
Homology XRCC3 is a paralog to rad51.
Implicated in No human disease has been linked to inactivation of XRCC3. However, polymorphisms in XRCC3 may be associated with increased cancer risk (see below). Note: Genetic Epidemiology: The most frequent polymorphism in XRCC3 is XRCC3 C18067T which results in a Thr to Met amino acid substitution at codon 241. Carriers of the variant T-allele of XRCC3 T241M have higher DNA adduct levels in lymphocyte DNA compared to homozygous C-allele carriers, indicating that the polymorphism is associated with lowered DNA repair capacity. The variant allele of XRCC3 T241M polymorphism has been associated with increased risk of squamous cell carcinoma of the head and neck in one study while another study found no association. No association has previously been found with colon cancer, non-melanoma skin cancer, prostate cancer, gastric cancer, ovarian cancer. Conflicting results have been published on the association with breast cancer, bladder cancer, malignant melanoma and lung cancer. Two frequent SNP are upstream of XRCC3 T241M have also been studied, namely XRCC3 A4541G and XRCC3 A17897G. Neither polymorphism gives rise to amino acid changes. The variant allele of A17897G has been associated with decreased risk of breast cancer and ovarian cancer. The same tendency, but no significant associations was found for lung cancer. XRCC3 A4541G was not associated to risk of breast cancer or lung cancer. However, homozygote carriers of the variant allele had lower risk of serous epithelial ovarian cancer.

XRCC3 (X-ray repair complementing defective repair in Chinese hamster cells 3) Vogel U
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 106
References Liu N, Lamerdin JE, Tebbs RS, Schild D, Tucker JD, Shen MR, Brookman KW, Siciliano MJ, Walter CA, Fan W, Narayana LS, Zhou ZQ, Adamson AW, Sorensen KJ, Chen DJ, Jones NJ, Thompson LH. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell 1998;1(6):783-793.
Griffin CS, Simpson PJ, Wilson CR, Thacker J. Mammalian recombination-repair genes XRCC2 and XRCC3 promote correct chromosome segregation. Nat Cell Biol 2000;2(10):757-761.
Winsey SL, Haldar NA, Marsh HP, Bunce M, Marshall SE, Harris AL, Wojnarowska F, Welsh KI. A variant within the DNA repair gene XRCC3 is associated with the development of melanoma skin cancer. Cancer Res 2000;60(20):5612-5616.
David-Beabes GL, Lunn RM, London SJ. No Association between the XPD (Lys751G1n) Polymorphism or the XRCC3 (Thr241Met) Polymorphism and Lung Cancer Risk. Cancer Epidemiol Biomarkers Prev 2001;10(8):911-912.
Matullo G, Guarrera S, Carturan S, Peluso M, Malaveille C, Davico L, Piazza A, Vineis P. DNA repair gene polymorphisms, bulky DNA adducts in white blood cells and bladder cancer in a case-control study. Int J Cancer 2001a;92(4):562-567.
Matullo G, Palli D, Peluso M, Guarrera S, Carturan S, Celentano E, Krogh V, Munnia A, Tumino R, Polidoro S, Piazza A, Vineis P. XRCC1, XRCC3, XPD gene polymorphisms, smoking and (32)P-DNA adducts in a sample of healthy subjects. Carcinogenesis 2001b;22(9):1437-1445.
Duan Z, Shen H, Lee JE, Gershenwald JE, Ross MI, Mansfield PF, Duvic M, Strom SS, Spitz MR, Wei Q. DNA repair gene XRCC3 241Met variant is not associated with risk of cutaneous malignant melanoma. Cancer Epidemiol Biomarkers Prev 2002, 11(10 Pt 1):1142-1143.
Kuschel B, Auranen A, McBride S, Novik KL, Antoniou A, Lipscombe JM, Day NE, Easton DF, Ponder BA, Pharoah PD, Dunning A. Variants in DNA double-strand break repair genes and breast cancer susceptibility. Hum Mol Genet 2002;11(12):1399-1407.
Shen H, Sturgis EM, Dahlstrom KR, Zheng Y, Spitz MR, Wei Q. A variant of the DNA repair gene XRCC3 and risk of squamous cell carcinoma of the head and neck: a case-control analysis. Int J Cancer 2002;99(6):869-872.
Stern MC, Umbach DM, Lunn RM, Taylor JA. DNA repair gene XRCC3 codon 241 polymorphism, its interaction with smoking and XRCC1 polymorphisms, and bladder cancer risk. Cancer Epidemiol Biomarkers Prev 2002;11(9):939-943.
Han J, Hankinson SE, Ranu H, De V, I, Hunter DJ. Polymorphisms in DNA double strand break repair genes and breast cancer risk in the nurses' health study. Carcinogenesis 2004;25(2):189-195.
Jacobsen NR, Nexø BA, Olsen A, Overvad K, Wallin H, Tjønneland A, Vogel U. No association between the DNA repair gene XRCC3 T241M polymorphism and risk of skin cancer and breast cancer. Cancer Epidemiol Biomarkers Prev 2003;12(6):584-585.
Mort R, Mo L, McEwan C, Melton DW. Lack of involvement of nucleotide excision repair gene polymorphisms in colorectal cancer. Br J Cancer 2003;89(2):333-337.
Sanyal S, Festa F, Sakano S, Zhang Z, Steineck G, Norming U, Wijkström H, Larsson P, Kumar R, Hemminki K. Polymorphisms in DNA repair and metabolic genes in bladder cancer. Carcinogenesis 2004;25(5):729-734.
Shen M, Hung RJ, Brennan P, Malaveille C, Donato F, Placidi D, Carta A, Hautefeuille A, Boffetta P, Porru S. Polymorphisms
of the DNA repair genes XRCC1, XRCC3, XPD, interaction with environmental exposures, and bladder cancer risk in a case-control study in northern Italy. Cancer Epidemiol Biomarkers Prev 2003;12(11 Pt 1):1234-1240.
Smith TR, Miller MS, Lohman K, Lange EM, Case LD, Mohrenweiser HW, Hu JJ. Polymorphisms of XRCC1 and XRCC3 genes and susceptibility to breast cancer. Cancer Lett 2003;190(2):183-190.
Forget AL, Bennett BT, Knight KL. Xrcc3 is recruited to DNA double strand breaks early and independent of Rad51. J Cell Biochem 2004;93(3):429-436.
Jacobsen NR, Raaschou-Nielsen O, Nexø B, Wallin H, Overvad K, Tjønneland A, Vogel U. XRCC3 polymorphisms and risk of lung cancer. Cancer Lett 2004;213(1):67-72.
Popanda O, Schattenberg T, Phong CT, Butkiewicz D, Risch A, Edler L, Kayser K, Dienemann H, Schulz V, Drings P, Bartsch H, Schmezer P. Specific combinations of DNA repair gene variants and increased risk for non-small cell lung cancer. Carcinogenesis 2004;25(12):2433-2441.
Yoshihara T, Ishida M, Kinomura A, Katsura M, Tsuruga T, Tashiro S, Asahara T, Miyagawa K. XRCC3 deficiency results in a defect in recombination and increased endoreduplication in human cells. EMBO J 2004;23(3):670-680.
Auranen A, Song H, Waterfall C, Dicioccio RA, Kuschel B, Kjaer SK, Hogdall E, Hogdall C, Stratton J, Whittemore AS, Easton DF, Ponder BA, Novik KL, Dunning AM, Gayther S, Pharoah PD. Polymorphisms in DNA repair genes and epithelial ovarian cancer risk. Int J Cancer 2005;117(4):611-618.
Festa F, Kumar R, Sanyal S, Undén B, Nordfors L, Lindholm B, Snellman E, Schalling M, Försti A, Hemminki K. Basal cell carcinoma and variants in genes coding for immune response, DNA repair, folate and iron metabolism. Mutat Res 2005;574(1-2):105-111.
Huang WY, Chow WH, Rothman N, Lissowska J, Llaca V, Yeager M, Zatonski W, Hayes RB. Selected DNA repair polymorphisms and gastric cancer in Poland. Carcinogenesis 2005a;26(8):1354-1359.
Huang WY, Olshan AF, Schwartz SM, Berndt SI, Chen C, Llaca V, Chanock SJ, Fraumeni JF, Jr., Hayes RB. Selected genetic polymorphisms in MGMT, XRCC1, XPD, and XRCC3 and risk of head and neck cancer: a pooled analysis. Cancer Epidemiol Biomarkers Prev 2005b;14(7):1747-1753.
Raaphorst GP, Leblanc M, Li LF. A comparison of response to cisplatin, radiation and combined treatment for cells deficient in recombination repair pathways. Anticancer Res 2005;25(1A):53-58.
Ritchey JD, Huang WY, Chokkalingam AP, Gao YT, Deng J, Levine P, Stanczyk FZ, Hsing AW. Genetic variants of DNA repair genes and prostate cancer: a population-based study. Cancer Epidemiol Biomarkers Prev 2005;14(7):1703-1709.
Webb PM, Hopper JL, Newman B, Chen X, Kelemen L, Giles GG, Southey MC, Chenevix-Trench G, Spurdle AB. Double-strand break repair gene polymorphisms and risk of breast or ovarian cancer. Cancer Epidemiol Biomarkers Prev 2005;14(2):319-323.
Yeh CC, Sung FC, Tang R, Chang-Chieh CR, Hsieh LL. Polymorphisms of the XRCC1, XRCC3, & XPD genes, and colorectal cancer risk: a case-control study in Taiwan. BMC Cancer 2005;5(1):12.
This article should be referenced as such: Vogel U. XRCC3 (X-ray repair complementing defective repair in Chinese hamster cells 3). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):105-106.

Leukaemia Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 107
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
Amplified NUP214/ABL1 Nathalie Nadal
Laboratoire d'hématologie (niveau 1), Pavillon de Biologie, CHU Hôpital Nord, 42055 St Etienne Cedex 2, France
Published in Atlas Database: September 2005
Online updated version: http://AtlasGeneticsOncology.org/Anomalies/ampNUP214ABL1ID1397.html DOI: 10.4267/2042/38298
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: NUP214/ABL1 fusion gene on amplified episomes.
Episomal amplification of ABL detected by FISH with the commercial probe LSI BCR-ABL ES.
Clinics and pathology Disease T-cell acute lymphoblastic leukemia (T-ALL).
Phenotype / cell stem origin Immature T-cell leukemia (CD3+, CD2+ and CD7+). Not seen in B-cell ALL.
Epidemiology In about 6% of T-ALL. Mainly observed in T-ALL associated with the mutually exclusive overexpression of the oncogenes HOX11 and HOX11L2. Found in pediatric T-ALL and adults T-ALL.
Cytology Lymphoblasts.

Amplified NUP214/ABL1 Nadal N
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 108
Prognosis Present data suggests that NUP214-ABL1 fusion gene amplification in T-ALL is associated with poor outcome. NUP214-ABL1 is sensitive to the tyrosine kinase inhibitor imatinib mesylate. Targeting therapies may improve outcome.
Cytogenetics Note: Mechanism of gene amplification. The main hypothesis for genomic amplification mechanism is that it is a dynamic process. Molecular chronology of genomic amplification has been schematically described as follows. The first step is the production of submicroscopic, acentric, circular, extrachromosomal DNA molecules which replicate autonomously, called episomes. These DNA molecules are made of amplified genes. 2 mechanisms for the formation of episomes have been proposed: Conservative which preserves the original DNA sequence at the native chromosomal locus and non conservative which leads to the deletion of the original sequence at the native locus. The second step corresponds to an increase in copy number resulting from unequal mitotic segregation and an increase in size. They enlarge over time to form progressively heterogeneously sized structures, microscopically visible, called double minutes (dmin). In a later step they may integrate into chromosomes to generate intrachromosomally amplified structures (HSR). In some cases dmins or HSRs may form directly without precursors.
Cytogenetics morphological Cryptic extrachromosomal amplification of the fusion NUP214-ABL. Rearrangement invisible by conventional cytogenetics. No dmins observed.
Cytogenetics molecular FISH using commercially available ABL1 probe shows multiple extrachromosomal sites on metaphases and multiple signals in interphase nuclei. The extrachromosomal amplification of ABL1 appears to be pathognomonique for the presence of NUP214-ABL1 fusion in T-ALL. There may be a corresponding deletion of the ABL1 probe on one of the chromosomes 9 (see note below concerning mechanisms of gene amplification).
Additional anomalies Associated with an apparently normal karyotype with banding techniques or variable additional abnormalities.
Genes involved and Proteins ABL1 Location: 9q34.1
DNA / RNA Alternate splicing. mRNA of 6 and 7 kb.
Protein 145 kDa; Localization: nuclear and cytoplasmic; Tyrosine kinase; Ubiquitously expressed.
NUP214 (nuclear pore complex protein 241 kDa) or CAN, CAIN, Nucleoporin Location: 9q34.3, more telomeric than ABL1.
DNA / RNA 7.5 kb mRNA.
Protein 214 kDa; dimerization domains (2 leucine zippers) and a repeated motif; forms homodimers. Nuclear membrane localisation. Component of the Nuclear Pore Complex. Mediate nucleocytoplasmic transport.
Results of the chromosomal anomaly Hybrid gene Description Molecular analysis delineated the amplicon as a 500 kb region from chromosome band 9q34 containing the genes ABL1, LAMC3 and NUP214. The genomic region from ABL1 to NUP214 circularizes to generate the NUP214-ABL1 fusion gene = New mechanism for generation of a fusion gene. The breakpoint within ABL1 occurs in intron1 (coincides with ABL1 breakpoint in the Philadelphia chromosome). Whilst the breakpoint in NUP214 is variable (ranging from intron 23 to intron 34).
Detection protocole RT-PCR
Fusion protein Description The NUP214-ABL transcript encodes a 239-333 kDa protein. NUP214-ABL retains the N-terminal region of NUP 214 which includes the predicted coiled-coil domains that serve as oligomerisation motifs. NUP214-ABL contains SH3, SH2 and kinase domains of ABL1. NUP214-ABL is a constitutively activated tyrosine kinase that activates similar pathways as BCR-ABL1.
References Wahl GM. The importance of circular DNA in mammalian gene amplification. Cancer Res 1989 Mar 15;49(6):1333-1340.
Barber KE, Martineau M, Harewood L, Stewart M, Cameron E, Strefford JC, Rutherford S, Allen TD, Broadfield ZJ, Cheung KL, Harris RL, Jalali GR, Moorman AV, Robinson HM, Harrison CJ. Amplification of the ABL gene in T-cell acute lymphoblastic leukemia. Leukemia 2004 Jun;18(6):1153-1156.
Graux C, Cools J, Melotte C, Quentmeier H, Ferrando A, Levine R, Vermeesch JR, Stul M, Dutta B, Boeckx N, Bosly A,

Amplified NUP214/ABL1 Nadal N
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 109
Heimann P, Uyttebroeck A, Mentens N, Somers R, MacLeod RA, Drexler HG, Look AT, Gilliland DG, Michaux L, Vandenberghe P, Wlodarska I, Marynen P, Hagemeijer A. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat Genet 2004 Oct;36(10):1084-1089.
Ballerini P, Busson M, Fasola S, van den Akker J, Lapillonne H, Romana SP, Marynen P, Bernard OA, Landman-Parker J, Berger R. NUP214-ABL1 amplification in t(5;14)/HOX11L2-positive ALL present with several forms and may have a prognostic significance. Leukemia 2005 Mar;19(3):468-470.
De Keersmaecker K, Graux C, Odero MD, Mentens N, Somers R, Maertens J, Wlodarska I, Vandenberghe P, Hagemeijer A,
Marynen P, Cools J. Fusion of EML1 to ABL1 in T-cell acute lymphoblastic leukemia with cryptic t(9;14)(q34;q32). Blood 2005 Jun 15;105(12):4849-4852.
Stergianou K, Fox C, Russell NH. Fusion of NUP214 to ABL1 on amplified episomes in T-ALL - implications for treatment. Leukemia 2005 Sep;19(9):1680-1681.
This article should be referenced as such: Nadal N. Amplified NUP214/ABL1. Atlas Genet Cytogenet Oncol Haematol.2006;10(2):107-109.
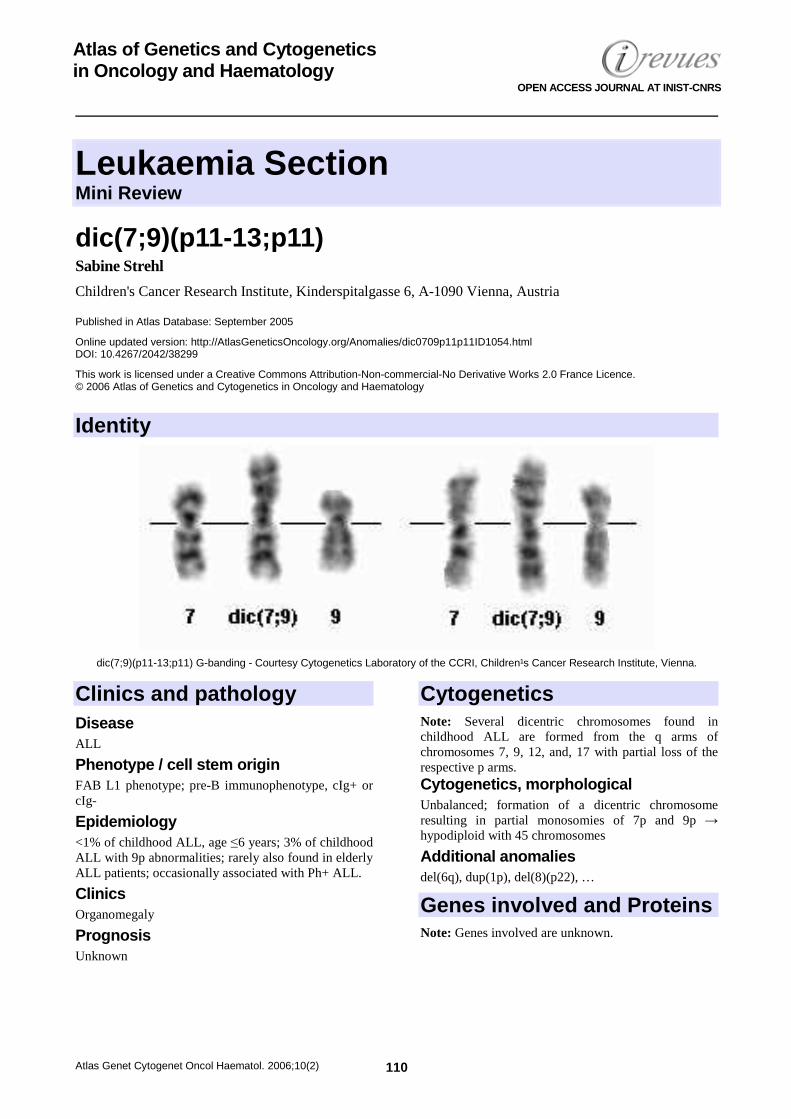
Leukaemia Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 110
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
dic(7;9)(p11-13;p11) Sabine Strehl
Children's Cancer Research Institute, Kinderspitalgasse 6, A-1090 Vienna, Austria
Published in Atlas Database: September 2005
Online updated version: http://AtlasGeneticsOncology.org/Anomalies/dic0709p11p11ID1054.html DOI: 10.4267/2042/38299
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity
dic(7;9)(p11-13;p11) G-banding - Courtesy Cytogenetics Laboratory of the CCRI, Children¹s Cancer Research Institute, Vienna.
Clinics and pathology Disease ALL
Phenotype / cell stem origin FAB L1 phenotype; pre-B immunophenotype, cIg+ or cIg-
Epidemiology <1% of childhood ALL, age ≤6 years; 3% of childhood ALL with 9p abnormalities; rarely also found in elderly ALL patients; occasionally associated with Ph+ ALL.
Clinics Organomegaly
Prognosis Unknown
Cytogenetics Note: Several dicentric chromosomes found in childhood ALL are formed from the q arms of chromosomes 7, 9, 12, and, 17 with partial loss of the respective p arms. Cytogenetics, morphological Unbalanced; formation of a dicentric chromosome resulting in partial monosomies of 7p and 9p → hypodiploid with 45 chromosomes
Additional anomalies del(6q), dup(1p), del(8)(p22), …
Genes involved and Proteins Note: Genes involved are unknown.

dic(7;9)(p11-13;p11) Strehl S
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 111
References Diaz MO, Rubin CM, Harden A, Ziemin S, Larson RA, Le Beau MM, Rowley JD. Deletions of interferon genes in acute lymphoblastic leukemia. N Engl J Med.1990;322:77-82.
Raimondi SC, Behm FG, Roberson PK, Williams DL, Pui CH, Crist WM, Look AT, Rivera GK. Cytogenetics of pre-B-cell acute lymphoblastic leukemia with emphasis on prognostic implications of the t(1;19). J Clin Oncol 1990;8:1380-1388.
Raimondi SC, Privitera E, Williams DL, Look AT, Behm F, Rivera GK, Crist WM, Pui CH. New recurring chromosomal translocations in childhood acute lymphoblastic leukemia. Blood 1991;77:2016-2022.
Collaborative study of karyotypes in childhood acute lymphoblastic leukemias. Groupe Français de Cytogénétique Hématologique. Leukemia 1993;7:10-19.
Uckun FM, Nachman JB, Sather HN, Sensel MG, Kraft P, Steinherz PG, Lange B, Hutchinson R, Reaman GH, Gaynon PS, Heerema NA. Clinical significance of Philadelphia chromosome positive pediatric acute lymphoblastic leukemia in the context of contemporary intensive therapies: a report from the Children's Cancer Group. Cancer 1998;83:2030-2039.
Wong N, Chen SJ, Cao Q, Su XY, Niu C, Wu QW, Leung TW, Wickham N, Johnson PJ, Chen Z. Detection of chromosome over- and underrepresentations in hyperdiploid acute lymphoblastic leukemia by comparative genomic hybridization. Cancer Genet Cytogenet 1998;103:20-24.
Heerema NA, Sather HN, Sensel MG, Liu-Mares W, Lange BJ, Bostrom BC, Nachman JB, Steinherz PG, Hutchinson R, Gaynon PS, Arthur DC, Uckun FM. Association of
chromosome arm 9p abnormalities with adverse risk in childhood acute lymphoblastic leukemia: A report from the Children's Cancer Group. Blood 1999;94:1537-1544.
Nacheva EP, Gribble S, Andrews K, Wienberg J, Grace CD. Screening for specific chromosome involvement in hematological malignancies using a set of seven chromosome painting probes. An alternative approach for chromosome analysis using standard FISH instrumentation. Cancer Genet Cytogenet 2000;122:65-72.
Thomas X, Olteanu N, Charrin C, Lhéritier V, Magaud JP, Fiere D. Acute lymphoblastic leukemia in the elderly: The Edouard Herriot Hospital experience. Am J Hematol 2001;67:73-83.
Raimondi SC, Zhou Y, Mathew S, Shurtleff SA, Sandlund JT, Rivera GK, Behm FG, Pui CH. Reassessment of the prognostic significance of hypodiploidy in pediatric patients with acute lymphoblastic leukemia. Cancer 2003;98:2715-2722.
Heerema NA, Nachman JB, Sather HN, La MK, Hutchinson R, Lange BJ, Bostrom B, Steinherz PG, Gaynon PS, Uckun FM. Deletion of 7p or monosomy 7 in pediatric acute lymphoblastic leukemia is an adverse prognostic factor: a report from the Children's Cancer Group. Leukemia 2004;18:939-947.
This article should be referenced as such: Strehl S. dic(7;9)(p11-13;p11). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):110-111.

Leukaemia Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 112
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
dup(21q) amplified (RUNX1) Anthony V Moorman, Christine J Harrison
Leukaemia Research Cytogenetics Group, Cancer Sciences Division, University of Southampton, MP822, Duthie Building, Southampton General Hospital, Tremona Road, Southampton, SO16 6YD, UK
Published in Atlas Database: September 2005
Online updated version: http://AtlasGeneticsOncology.org/Anomalies/dup21qID1382.html DOI: 10.4267/2042/38300
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Note: This molecular cytogenetic subgroup of ALL is characterised by the presence of multiple copies of the RUNX1 (AML1) gene on a duplicated chromosome 21q (Figure 1). Using metaphase FISH multiple RUNX1 signals are seen along the length of the
duplicated 21q (Figure 2), while in interphase the signals are clustered together (Figure 3). G-banded cytogenetic analysis shows that the morphology of the duplicated chromosome 21q is heterogeneous between cases (Figure 4). Currently, FISH with probes directed to RUNX1 is the only reliable method of detection.
Fig1 : Left: A metaphase showing an abnormal chromosome (whole chromosome paint 21) with multiple RUNX1 (red) and two normal ETV6 (TEL) signals. Middle: A metaphase showing multiple RUNX1 exon signals (red) along the length of an abnormal chromosome 21 Right: Interphase cells showing clustering of the red RUNX1 and the two normal green ETV6 signals, using the LSI TEL-AML1 translocation probe (Vysis).
Partial G-banded karyograms showing heterogeneity in the morphology of the duplicated 21q. The normal chromosome 21 is always on the left.

dup(21q) amplified (RUNX1) Moorman AV, Harrison CJ
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 113
Clinics and pathology Disease Acute lymphoblastic leukaemia: B-lineage immunophenotype (mostly common or Pre-B), FAB L1/L2.
Epidemiology The estimated incidence in childhood ALL is 1-3%. The majority of patients tend to be older children or adolescents with nearly three-quarters of reported patients aged between 6 and 14 years old. A few young adults have also been reported but too few to estimate its incidence in this age group. No gender bias has been observed.
Clinics Patients with this abnormality typically present with a low white cell count of <20x109L
Prognosis The three year event free survival of the 25 patients treated on the UK MRC ALL97 trial was just 51% (95% C.I. 27%-71%) and hence represent a poor risk cytogenetic feature in childhood ALL.
Cytogenetics Note: Although FISH with a probe to the RUNX1 (AML1) gene is required to accurately identify this abnormality, the duplicated chromosome 21 is easily visible using conventional cytogenetics. The morphology of the abnormal chromosome 21 is highly heterogeneous presenting as a metacentric, acrocentric or ring chromosome (Figure 4). Prior to the identify of this abnormality by FISH many karyotypes were described as -21,+mar. The duplicated 21q is rarely the sole chromosomal abnormality. The karyotype is frequently complex, although no recurrent secondary abnormality has yet emerged. This abnormality does not occur with other primary chromosomal abnormalities in ALL e.g. t(12;21)(p13;q22) / ETV6-RUNX1, t(9;22)(q34;q11), t(1;19)(q23;p13) etc. However, a few cases of high hyperdiploidy have been reported. FISH has been instrumental in defining this abnormality and is essential for its accurate detection.
Virtually all cases reported to date have been identified using the LSI TEL-AML1 translocation probe. However, any FISH probe directed to RUNX1 could be used. The identification of metaphases with multiple RUNX1 signals on a single chromosome 21 is the most accurate detection method. However, in the absence of metaphases the presence of multiple clustered RUNX1 signals is also reliable. The current published definitions of amplification within the context of this abnormality are as follows: only included cases in which the abnormality had been visualised in metaphases and three or more RUNX1 signals were seen on a single abnormal chromosome 21, while others used the same definition when the abnormality was seen in metaphase. Additionally, they included cases which revealed only interphases with five or more RUNX1 signals.
Genes involved and Proteins RUNX1 Note: By definition the RUNX1 (AML1) gene is amplified in all these patients. Over-expression of RUNX1 or any of the other genes within the amplified region has yet to be established.
References Harewood L, Robinson H, Harris R, Al Obaidi MJ, Jalali GR, Martineau M, Moorman AV, Sumption N, Richards S, Mitchell C, Harrison CJ. Amplification of AML1 on a duplicated chromosome 21 in acute lymphoblastic leukemia: a study of 20 cases. Leukemia 2003;17:547-553.
Soulier J, Trakhtenbrot L, Najfeld V, Lipton JM, Mathew S, Avet-Loiseau H, De Braekeleer M, Salem S, Baruchel A, Raimondi SC, Raynaud SD. Amplification of band q22 of chromosome 21, including AML1, in older children with acute lymphoblastic leukemia: an emerging molecular cytogenetic subgroup. Leukemia 2003;17:1679-1682.
Robinson HM, Broadfield ZJ, Cheung KL, Harewood L, Harris RL, Jalali GR, Martineau M, Moorman AV, Taylor KE, Richards S, Mitchell C, Harrison CJ. Amplification of AML1 in acute lymphoblastic leukemia is associated with a poor outcome. Leukemia 2003;17:2249-2250.
This article should be referenced as such: Moorman AV, Harrison CJ. dup(21q) amplified (RUNX1). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):112-113.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 114
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
t(1;11)(q23;p15) Jean-Loup Huret
Genetics, Dept Medical Information, UMR 8125 CNRS, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France
Published in Atlas Database: September 2005
Online updated version: http://AtlasGeneticsOncology.org/Anomalies/t0111q23p15ID1169.html DOI: 10.4267/2042/38301
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics and pathology Disease Acute non lymphocytic leukemia (ANLL), M2-ANLL type; this leukemia case is likely to be treatment related (see below).
Epidemiology Only 1 case to date, a 55 year old male patient who has had a non Hodgkin lymphoma (NHL) with t(14;18)(q32;q21) 3 years before onset of the ANLL.
Evolution Complete remission of the ANLL was achieved, but the NHL relapsed and an advanced gastric carcinoma was found and the patient died shortly afterwards.
Genes involved and Proteins Note: This translocation appears to be closely related to other translocations involving NUP98 and an homeodomain bearing protein, i.e. the t(2;11)(q31;p15), with HOXD13 or with HOXD11 involvement, the t(7;11)(p15;p15), with HOXA9 or with HOXA13 involvement, the t(9;11)(q34;p15), with PRRX2 involvement, and the t(11;12)(p15;q13) with HOXC11 or with HOXC13 involvement.
PRRX1 Location: 1q23
Protein Protein with a homeodomain.
Results of the chromosomal anomaly Hybrid gene Description In frame fusion of NUP98 to PRRX1 exon 2; no reciprocal fusion transcript.
Fusion protein Description The chimeric protein contains the N-term half of NUP98, including the docking site to the homeodomain of PRRX1.
Oncogenesis The PRRX1 homeodomain may be upregulated.
References Nakamura T, Yamazaki Y, Hatano Y, Miura I. NUP98 is fused to PMX1 homeobox gene in human acute myelogenous leukemia with chromosome translocation t(1;11)(q23;p15). Blood 1999;94:741-747.
Hatano Y, Miura I, Kume M, Miura AB. Translocation (1;11)(q23;p15), a novel simple variant of translocation (7;11)(p15;p15), in a patient with AML (M2) accompanied by non-Hodgkin lymphoma and gastric cancer. Cancer Genet Cytogenet 2000;117:-23.
This article should be referenced as such: Huret JL. t(1;11)(q23;p15). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):114.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 115
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
t(2;9)(p11;p13) Jean-Loup Huret
Genetics, Dept Medical Information, UMR 8125 CNRS, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France
Published in Atlas Database: September 2005
Online updated version: http://AtlasGeneticsOncology.org/Anomalies/t0209p11p13ID1404.html DOI: 10.4267/2042/38302
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity
G- banding - Courtesy Melanie Zenger and Claudia Haferlach.
Clinics and pathology Disease Pre B acute lymphocytic leukemia (ALL).
Epidemiology Only one case to date. Cytogenetic Sole anomaly.
Genes PAX5 was rearranged.
References Lu XY, Harris CP, Cooley L, Margolin J, Steuber PC, Sheldon M, Rao PH, Lau CC. The utility of spectral karyotyping in the cytogenetic analysis of newly diagnosed pediatric acute lymphoblastic leukemia. Leukemia 2002;16:2222-2227.
This article should be referenced as such: Huret JL. t(2;9)(p11;p13). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):115.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 116
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
t(3;7)(q27;p12-13) Jean-Loup Huret
Genetics, Dept Medical Information, UMR 8125 CNRS, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France
Published in Atlas Database: September 2005
Online updated version: http://AtlasGeneticsOncology.org/Anomalies/t0307q27p12ID2010.html DOI: 10.4267/2042/38303
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics and pathology Disease Non Hodgkin lymphoma (NHL).
Phenotype / cell stem origin 2 cases of diffuse large B-cell lymphoma (DLBL) with BCL6/ZNFN1A1 involvement, 1 NHL with BCL6 involvement, 1 follicular, predominantly large cell B-NHL and one T-NHL without gene ascertainement.
Epidemiology 5 cases to date, aged 40-70 years, sex ration: 3M:1F.
Prognosis The 2 patients with BCL6/ZNFN1A1 involvement died at 16 and 17 months after diagnosis.
Cytogenetics Additional anomalies del(6) in two cases, del(13q) in two cases, +16 in two cases, +12 in one case.
Genes involved and Proteins BCL6 Location: 3q27
Protein Transcription factor; belongs to the Krüppel family, with a N-term BTB/POZ domain and 6 zinc fingers; transcription repressor.
ZNFN1A1 (Ikaros) Location: 7p12
Protein Transcription regulator; can repress transcription through the recruitment of histone deacetylase complexes; role in conjunction with Aiolos; hemopoietic-specific zinc finger protein regulator of B and T-cell differentiation.
Results of the chromosomal anomaly Hybrid gene Description 5’ Ikaros - 3’ BCL6 fusion transcript; it is supposed that substitution of the promoter of BCL6 may be responsible for BCL6 deregulation.
References Konishi H, Sakurai M, Nakao H, Maseki N, Kaneko Y, Yagiri Y, Notohara K, Frizzera G. Chromosome abnormalities in malignant lymphoma in patients from Kurashiki: histological and immunophenotypic correlations. Cancer Res 1990;50:2698-2703.
Bastard C, Deweindt C, Kerckaert JP, Lenormand B, Rossi A, Pezzella F, Fruchart C, Duval C, Monconduit M, Tilly H. LAZ3 rearrangements in non-Hodgkin's lymphoma: correlation with histology, immunophenotype, karyotype, and clinical outcome in 217 patients. Blood 1994;83:2423-2427.
Brown KE, Guest SS, Smale ST, Hahm K, Merkenschlager M, Fisher AG. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell 1997;91:845-854.
Georgopoulos K, Winandy S, Avitahl N. The role of the Ikaros gene in lymphocyte development and homeostasis. Annu Rev Immunol 1997;15:155- 176.
Nichogiannopoulou A, Trevisan M, Friedrich C, Georgopoulos K. Ikaros in hemopoietic lineage determination and homeostasis. Semin Immunol 1998;10:119-125.

t(3;7)(q27;p12-13) Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 117
Ichinohasama R, Miura I, Funato T, Sato I, Suzuki C, Saito Y, Decoteau JF, Myers JB, Kadin ME, Sawai T, Ooya K. A recurrent nonrandom translocation (3;7)(q27;p12) associated with BCL-6 gene rearrangement in B-cell diffuse large cell lymphoma. Cancer Genet Cytogenet 1998;104:19-27.
Tanaka K, Arif M, Eguchi M, Guo SX, Hayashi Y, Asaoku H, Kyo T, Dohy H, Kamada N. Frequent allelic loss of the RB, D13S319 and D13S25 locus in myeloid malignancies with deletion/translocation at 13q14 of chromosome 13, but not in lymphoid malignancies. Leukemia 1999;13:1367-1373.
Hosokawa Y, Maeda Y, Ichinohasama R, Miura I, Taniwaki M, Seto M. The Ikaros gene, a central regulator of lymphoid differentiation, fuses to the BCL6 gene as a result of t(3;7)(q27;p12) translocation in a patient with diffuse large B-cell lymphoma. Blood 2000;95:2719-2721.
Payne KJ, Huang G, Sahakian E, Zhu JY, Barteneva NS, Barsky LW, Payne MA, Crooks GM. Ikaros isoform x is selectively expressed in myeloid differentiation. J Immunol 2003;170:3091-3098.
Liberg D, Smale ST, Merkenschlager M. Upstream of Ikaros. Trends Immunol 2003;24:567-570.
This article should be referenced as such: Huret JL. t(3;7)(q27;p12-13). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):116-117.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 118
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
t(3;16)(q27;p11) Jean-Loup Huret
Genetics, Dept Medical Information, UMR 8125 CNRS, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France
Published in Atlas Database: September 2005
Online updated version: http://AtlasGeneticsOncology.org/Anomalies/t0316q27p11ID1357.html DOI: 10.4267/2042/38304
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity
G- banding - Courtesy Melanie Zenger and Claudia Haferlach.
Clinics and pathology Disease Diffuse large cell lymphoma.
Epidemiology Only one case to date, a 64 year old male patient.
Evolution Complete remission was obtained.
Cytogenetics Cytogenetics, morphological Sole anomaly within the subclone
Genes involved and Proteins BCL6 Location: 3q27
DNA / RNA Spans on 25 kb; 11 exons.
Protein Sequence-specific DNA binding transcriptional repressor. IL21R (or IL21R-alpha) Location: 16p11
DNA / RNA Spans on 48 kb; 10 exons.
Protein IL21R is, in fact, an heterodimer made of IL21R-alpha, and the common gamma chain (CD132); IL21Ra is the private receptor of IL21; IL21/IL21R, selectively expressed in lymphoid cells, activates the JAK/STAT transcription signaling pathway; IL21/IL21R plays a role in proliferation, differenciation of T cells, B cells, natural killers (NK) cells, and dendritic cells.; promotes and inhibit immune responses.

t(3;16)(q27;p11) Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 119
Results of the chromosomal anomaly Hybrid gene Description The IL21R promoter region replaces the regulatory sequences of BCL6; the chimeric mRNA consists of the 2 non coding exons 1a/1b of IL21R and the coding exons of BCL6.
Fusion protein Oncogenesis Enhanced expression of BCL6.
References Ichinohasama R, Miura I, Shishido T, Matsumoto K, Shimizu Y, Miki T, DeCoteau JF, Kadin ME, Ooya K. Translocation (3;16)(q27;p11) in a patient with diffuse large B-cell lymphoma associated with BCL6 gene rearrangement. Cancer Genet Cytogenet 1998;103:133-139.
Ueda C, Akasaka T, Kurata M, Maesako Y, Nishikori M, Ichinohasama R, Imada K, Uchiyama T, Ohno H. The gene for interleukin-21 receptor is the partner of BCL6 in t(3;16)(q27;p11), which is recurrently observed in diffuse large B-cell lymphoma. Oncogene 2002;21:368-376.
This article should be referenced as such: Huret JL. t(3;16)(q27;p11). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):118-119.

Leukaemia Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 120
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
inv(7)(p15q34), t(7;7)(p15;q34) Barbara Cauwelier, Nicole Dastugue, Anne Hagemeijer, Frank Speleman
Centrum Medische Genetica Gent- CMGG, Medical Research Building- MRB, 2 nd floor, room 120.024, De Pintelaan 185, B-9000 Ghent, Belgium (BC, FS); Center for Human Genetics, University of Leuven, Leuven, Belgium (AH); Génétique des Hémopathies, Laboratoire d'Hématologie, Hôpital Purpan, 31000, Toulouse Cedex, France (ND)
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Anomalies/t0707p15q34ID1384.html DOI: 10.4267/2042/38305
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics and pathology Disease T cell acute lymphoblastic leukemia (T-ALL) and non-Hodgkin lymphoma (T-NHL).
Phenotype / cell stem origin T lineage; occurs at early stage of T cell development (CD2-, CD4+, CD8-).
Epidemiology 3.5 % of T-ALL or T-NHL.
Clinics Hepato and/or splenomegaly, lymphadenopathy, mediastinal mass, moderate WBC count (15 to 100 X 109l).
Cytology FAB L1 or L2.
Cytogenetics
inv(7)(p15q34) G- banding (left) and R- banding (right).
Cytogenetics morphological This rearrangement remains undetected in poor quality metaphases. In less condensed, well banded metaphases, the abnormality may be suspected as del(7)(p15) or clearly as an inv(7)(p15q34).
FISH results of inv(7) (Fig.1a and b) and t(7;7) (Fig.1c and d) case using TRB flanking (Fig.1a and 1c) and HOXA (Fig.1b and 1d) flanking probes.
Cytogenetics molecular inv(7)(p15q34) and t(7;7)(p15;q34) can be detected by FISH using either TRB and HOXA flanking probes which gives a split signal of both probes in these cases. A fusion signal will be detected when combining the proximal TCR/distal HOXA flanking or the distal TCR/proximal HOXA flanking FISH probe.

inv(7)(p15q34), t(7;7)(p15;q34) Cauwelier B et al.
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 121
Probes TRB flanking probes: RP11-1220K2 and RP11-556I13 HOXA flanking probes: RP1-167F23 and RP5-1103I5
Additional anomalies Most patients show no additional karyotypic abnormalities.
Genes involved and Proteins Note: Chromosomal disruption of the HOXA gene cluster (7p15) following chromosomal rearrangement, leads to upregulation of HOXA gene expression, which are normally weakly expressed in T-ALL. Further studies are needed to determine the various patterns of HOXA gene upregulation but from present data, HOXA10 seems most consistently involved in keeping with the breakpoint position near HOXA9. The upregulation of HOXA10 expression is thought to result from enhancers embedded within the TRB locus, which is translocated upstream from these genes. Upregulation of HOXA genes has also been described for other subgroups of T-ALL i.e. the CALM-AF10 and MLL rearranged T-ALLs indicating a more general role for HOXA genes in T-ALL development. HOXA, together with HOXB, HOXC and HOXD, belongs to the class I homeobox genes and compromises 11 HOXA cluster genes: HOXA1, HOXA2, HOXA3, HOXA4, HOXA5, HOXA6, HOXA7, HOXA9, HOXA10, HOXA11, HOXA13. Given the breakpoint position 5’ to HOXA10 and its consistent overexpression in all TRB-HOXA rearranged cases, we currently assume that this gene exerts a specific oncogenic effect in this subgroup of T-ALLs.
HOXA10 (alias: PL, HOX1.8, HOX1H, HOX1)
Location: 7p15
Note: DNA-binding transcription factor which regulates gene expression, morphogenesis, and differentiation. More specifically, it may function in fertility, embryo viability, and regulation of hematopoietic lineage commitment. Two transcript variants encoding different isoforms have been found for this gene. HOXA10 expression is normally present
in hematopoietic stem cells and developing T-cells with decreasing expression as T-cells mature.
DNA / RNA 2 transcripts: - transcript variant 1 (isoform a): 2 exons, transcript 2618 bp, protein 393 amino-acids. - transcript variant 2 (isoform b): 2 exons, transcript 2241 bp, protein 94 amino-acids.
Protein DNA binding, transcription factor activity.
TRB Location: 7q34
Note: The human TRB locus at 7q34 spans 620 kb and consists of 82-85 genes. Enhancer sequences have been characterized 5.5kb 3' from TRBC2.
Protein Proteins encoded by the TRB locus are the T-cell receptor beta chains.
Results of the chromosomal anomaly Fusion protein Description No fusion protein, but ectopic expression of HOXA10.
Oncogenesis Little is known about the target genes for HOXA10. Cyclin-dependent kinase inhibitor p21 (alias CDKN1A, CIP1, WAF1) was shown to be a transcriptional target of HOXA10 in differentiating myelomonocytic cells. However, a potential role of p21 in HOXA10 driven oncogenesis has not been proved so far. In vitro transfection experiments with HOXA9 and HOXA10 showed upregulation of several genes of the Wnt pathway (Wnt10b, Frizzled1, Frizzled5) which are essential in hematopoietic stem cell renewal.
To be noted Translocations involving the TRB genes frequently result from errors of the recombinase enzyme complexe (RAG1, RAG2, etc.), responsable of the Immunoglobulin and T cell receptor V-J and V-D-J rearrangements.
References Bromleigh VC, Freedman LP. p21 is a transcriptional target of HOXA10 in differentiating myelomonocytic cells. Genes Dev 2000 Oct 15;14(20):2581-2586.
Taghon T, Thys K, De Smedt M, Weerkamp F, Staal FJ, Plum J, Leclercq G. Homeobox gene expression profile in human hematopoietic multipotent stem cells and T-cell progenitors: implications for human T-cell development. Leukemia 2003 Jun;17(6):1157-1163.

inv(7)(p15q34), t(7;7)(p15;q34) Cauwelier B et al.
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 122
Ferrell CM, Dorsam ST, Ohta H, Humphries RK, Derynck MK, Haqq C, Largman C, Lawrence HJ. Activation of stem-cell specific genes by HOXA9 and HOXA10 homeodomain proteins in CD34+ human cord blood cells. Stem Cells 2005 May;23(5):644-655.
Soulier J, Clappier E, Cayuela JM, Regnault A, García-Peydró M, Dombret H, Baruchel A, Toribio ML, Sigaux F. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood 2005 Jul 1;106(1):274-286.
Speleman F, Cauwelier B, Dastugue N, Cools J, Verhasselt B, Poppe B, Van Roy N, Vandesompele J, Graux C, Uyttebroeck
A, Boogaerts M, De Moerloose B, Benoit Y, Selleslag D, Billiet J, Robert A, Huguet F, Vandenberghe P, De Paepe A, Marynen P, Hagemeijer A. A new recurrent inversion, inv(7)(p15q34) leads to transcriptional activation of HOXA10 en HOXA11 in a subset of T-cell acute lymphoblastic leukemias. Leukemia 2005;19 (3):358-366.
This article should be referenced as such: Cauwelier B, Dastugue N, Hagemeijer A, Speleman F. inv(7)(p15q34), t(7;7)(p15;q34). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):120-122.

Leukaemia Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 123
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
t(9;22)(p24;q11.2) Stefan K Bohlander
Department of Medicine III, University Hospital Grosshadern, Marchioninistr. 15, D-81377 Munich, Germany or/ bGSF, Clinical Cooperative Group Leukemia, Marchioninistr. 25, D-81377 Munich, Germany
Published in Atlas Database: November 2005
Online updated version: http://AtlasGeneticsOncology.org/Anomalies/t0922p24q11ID1331.html DOI: 10.4267/2042/38306
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Note: Only one case with this translocation has been reported yet.
G-banded chromosomes showing t(9;22)(p24;q11.2).
Clinics and pathology Disease Typical chronic myeloid leukemia (CML).
Phenotype / cell stem origin Hematopoietic stem cell?
Epidemiology Only one case described so far.
Treatment No response to Imatinib!
Prognosis Blast crisis developed 20 months after initial diagnosis. The patient died 24 months after initial.
Cytogenetics Cytogenetics molecular FISH with a BCR/ABL probe (dual color dual fusion) will show a split of the BCR signal but no fusion signals and two normal ABL signals.
Additional anomalies 7q deletion and trisomy 19 was found at blast crisis.
Genes involved and Proteins BCR1 Location: 22q11.2
JAK2 Location: 9p24
Protein Janus activated kinase 2, protein tyrosine kinase.
Results of the chromosomal anomaly Hybrid gene Note: Only the BCR-JAK2 fusion transcript was detected. The reciprocal JAK2-BCR fusion transcript could not be amplified.
Detection protocole The fusion transcript can be detected by RT-PCR using the 5' BCR sense primer: 5'-cagaactcgcaacagtccttc-3' (bp 1602-1622) and the 3' JAK2 antisense primer: 5'tcataccggcacatctccacac-3' (bp 3100-3081). A PCR product of 300 bp should be expected. Please note that since only one case is known, the breakpoints may vary slightly in future cases. This might necessitate the design of different primers.
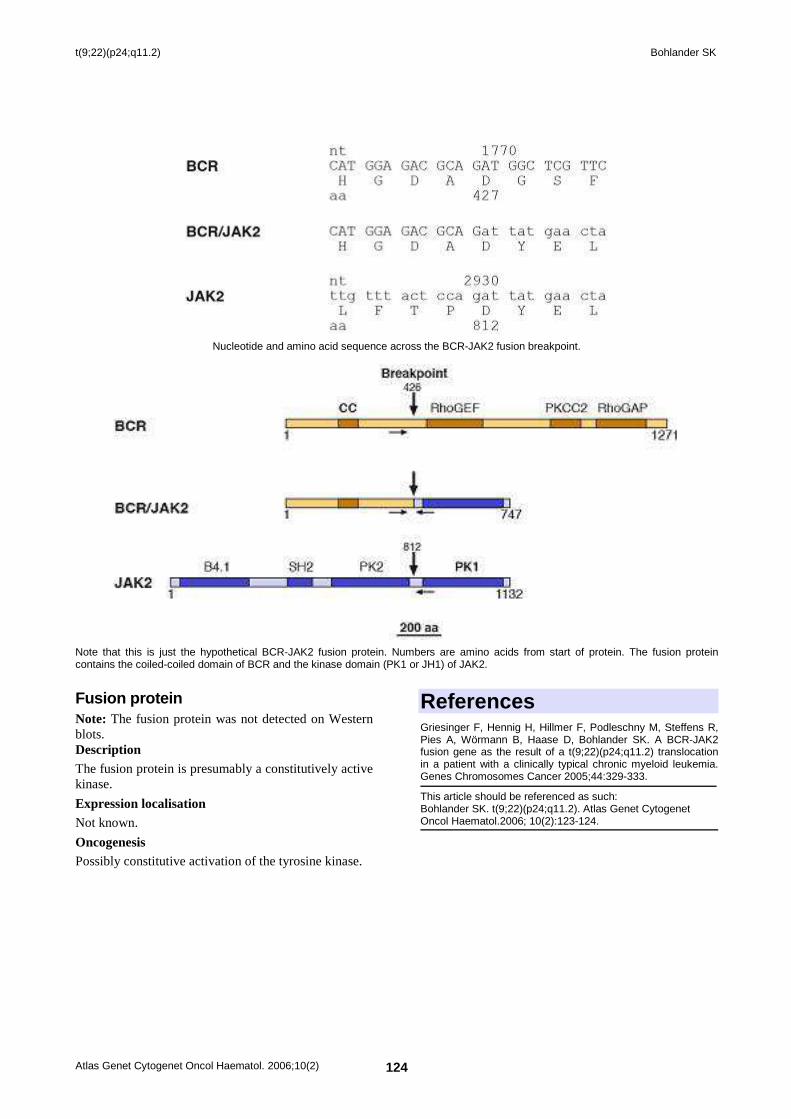
t(9;22)(p24;q11.2) Bohlander SK
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 124
Nucleotide and amino acid sequence across the BCR-JAK2 fusion breakpoint.
Note that this is just the hypothetical BCR-JAK2 fusion protein. Numbers are amino acids from start of protein. The fusion protein contains the coiled-coiled domain of BCR and the kinase domain (PK1 or JH1) of JAK2.
Fusion protein Note: The fusion protein was not detected on Western blots. Description The fusion protein is presumably a constitutively active kinase.
Expression localisation Not known.
Oncogenesis Possibly constitutive activation of the tyrosine kinase.
References Griesinger F, Hennig H, Hillmer F, Podleschny M, Steffens R, Pies A, Wörmann B, Haase D, Bohlander SK. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer 2005;44:329-333.
This article should be referenced as such: Bohlander SK. t(9;22)(p24;q11.2). Atlas Genet Cytogenet Oncol Haematol.2006; 10(2):123-124.
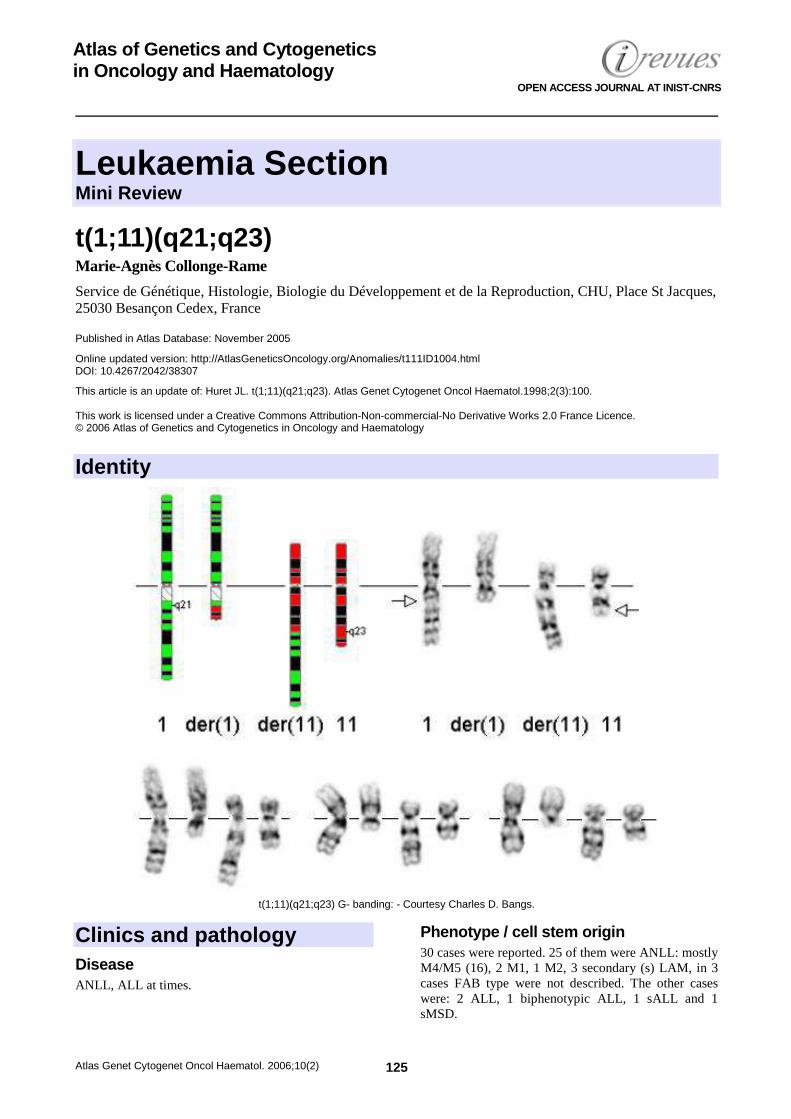
Leukaemia Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 125
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
t(1;11)(q21;q23) Marie-Agnès Collonge-Rame
Service de Génétique, Histologie, Biologie du Développement et de la Reproduction, CHU, Place St Jacques, 25030 Besançon Cedex, France
Published in Atlas Database: November 2005
Online updated version: http://AtlasGeneticsOncology.org/Anomalies/t111ID1004.html DOI: 10.4267/2042/38307
This article is an update of: Huret JL. t(1;11)(q21;q23). Atlas Genet Cytogenet Oncol Haematol.1998;2(3):100. This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity
t(1;11)(q21;q23) G- banding: - Courtesy Charles D. Bangs.
Clinics and pathology Disease ANLL, ALL at times.
Phenotype / cell stem origin 30 cases were reported. 25 of them were ANLL: mostly M4/M5 (16), 2 M1, 1 M2, 3 secondary (s) LAM, in 3 cases FAB type were not described. The other cases were: 2 ALL, 1 biphenotypic ALL, 1 sALL and 1 sMSD.

t(1;11)(q21;q23) Collonge-Rame MA
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 126
Epidemiology Most cases were infants (10/23) and children (7/23), range is 4 months - 62 years, balanced sex ratio (14F/12M on 26 cases). Prognosis Yet unknown.
Cytogenetics Cytogenetics morphological Presents as der(11)t(1;11)(q21;q23) in 9 of the 30 cases. Unbalanced form is identified in the 4 ALL and in all of the secondary cases.
Additional anomalies Balanced translocation is present as sole anomaly in 16/21 cases, and as part of simple karyotype in 5/21 cases; +19, +22 were recurrent. Additional abnormalities were observed in 8 of the 9 cases showing the der(11); karyotype of 5 cases were highly complex.
Variants Two three-way translocations were identified: t(1;11;3)(q21;q23;q21) and t(1;11;4)(q21;q23;p16).
Genes involved and Proteins AF1q Location: 1q21
DNA / RNA 1.8 kb mRNA.
Protein 9 kDa.
MLL Location: 11q23
DNA / RNA 21 exons, spanning over 100 kb; 13-15 kb mRNA.
Protein 431 kDa; contains two DNA binding motifs (a AT
hook, and Zinc fingers), a DNA methyl transferase motif, a bromodomain; transcriptional regulatory factor; nuclear localisation transcriptional regulatory factor; nuclear localisation.
Results of the chromosomal anomaly Hybrid gene Description 5' MLL - 3' AF1q; breakpoints: between exons 6 and 7 in MLL and within the 5' untranslated region in AF1q.
Fusion protein Description N-term -- AT hook (DNA binding) and DNA methyltransferase motif from MLL fused to the entire AF1q on the der(11); the reciprocal on der(1) is out of frame.
References Meloni-Balliet AM, Morgan R, Piatt J, Sandberg AA. Translocation t(1;11)(q21;q23), a new subgroup within M4 acute nonlymphocytic leukemia. Cancer Genet Cytogenet 1989 Feb;37(2):269-71.
Tse W, Zhu W, Chen HS, Cohen A. A novel gene, AF1q, fused to MLL in t(1;11) (q21;q23), is specifically expressed in leukemic and immature hematopoietic cells. Blood 1995 Feb 1;85(3):650-6.
Harrison CJ, Cuneo A, Clark R, Johansson B, Lafage-Pochitaloff M, Mugneret F, Moorman AV, Secker-Walker LM. Ten novel 11q23 chromosomal partner sites. Leukemia 1998 May;12(5):811-822.
Busson-Le Coniat M, Salomon-Nguyen F, Hillion J, Bernard OA, Berger R. MLL-AF1q fusion resulting from t(1;11) in acute leukemia. Leukemia 1999;13:302-306.
So CW, Ma SK, Wan SK, Chan GCF, Ha SY, Chan LC. Analysis of MLL-derived transcripts in infant acute monocytic leukaemia with a complex translocation (1;11;4)(q21;q23;p16). Cancer Genet Cytogenet 2000 Feb;117(1):24-27.
This article should be referenced as such: Collonge-Rame MA. t(1;11)(q21;q23). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):125-126.

Solid Tumour Section Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 127
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
Angiomatoid fibrous histiocytoma (AFH) Carolina Vicente-Dueñas, Isidro Sánchez-García
Laboratorio 13, Instituto de Biologia Molecular y Celular del Cancer (IBMCC), Centro de Investigacion del Cancer, Campus Unamuno, 37.007-Salamanca, Spain
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Tumors/AngiomFibHistiocytID5204.html DOI: 10.4267/2042/38308
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: Angiomatoid malignant fibrous histiocytoma (AMFH)
Classification Note: Angiomatoid fibrous histiocytoma is a rare soft tissue tumor of low-grade malignancy that usually occurs in children and young adults. Eighty-eight percent of patients are 30 years of age or younger. Enzinger in 1979 first designated the tumor as angiomatoid malignant fibrous histiocytoma. The tumour was later renamed angiomatoid fibrous histiocytoma because of its slow growth and rare metastasis. This tumor forms solid, lobulated sheets of plump to spindled cells having histiocytic features adjacent to areas of haemorrhage.
Clinics and pathology Note: This tumour typically affects children and young adults, presenting as a painless, slowly growing subcutaneous soft tissue mass that is usually located in the extremities and less commonly in the trunk, head, and neck. Only 18% of reported cases involved deep structures, such as skeletal muscle or periosteum.
Disease Symptoms of anemia, weight loss, and fever are observed in a minority of cases; local symptoms, such as pain or tenderness, are extremely rare.
Embryonic origin The cell of origin of AFH remains unclear. It is probably that AFH arises from a pluripotent
mesenchymal cell due to ultrastructural morphology supports histiocytic, vascular, smooth, and striated muscle differentiation. Clinics Clinically, the tumor is often mistaken for hematoma or hemangioma. The diagnosis of angiomatoid fibrous histiocytoma is made on the basis of histopathology and immunohistochemical studies. Three microscopic findings are characteristic of AFH: (1) solid arrays or nests of histiocyte-like cells, (2) hemorrhagic cyst-like spaces, and (3) aggregates of chronic inflammatory cells. Multifocal recent and old hemorrhages are a striking feature in this tumor. These spaces resemble vascular spaces, but they are not lined by endothelium. Inflammatory cells present include lymphocytes and plasma cells. A thick pseudocapsule and occasional germinal centers give this tumor a resemblance to a lymph node. Immunohistochemical studies are helpful in differential diagnosis of AFH. It was reported that the histiocytic marker CD68 was positive in 9 of 19 (47%) cases of angiomatoid fibrous histiocytoma. Immunopositivity for myoid or myofibroblastic markers in more than 50% of cases has also been reported.
Treatment Local recurrence has been reported in 11% of patients and distant metastasis in 1%; wide excision is recommended as the treatment of angiomatoid fibrous histiocytoma. Local recurrence is attributed to the infiltrative margin and deep location of the tumour. Angiomatoid fibrous histiocytoma in the head and neck also can frequently recur, which may be a result of the difficulty of performing a wide local excision. If the tumor is unresectable or has metastasized, adjuvant chemotherapy may be helpful.

Angiomatoid fibrous histiocytoma (AFH) Vicente-Dueñas, Sánchez-García I
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 128
t(12;16)(q13;p11) G- banding - Courtesy G. Reza Hafez, Eric B. Johnson, and Sara Morrison-Delap.
Cytogenetics Note: This disease is characterised by the translocations: t(12;16)(q13;p11) and t(12;22)(q13;q12).
Cytogenetics molecular FUS-ATF1 fusion gene in the t(12;16)(q13; p11). EWSR1-ATF1 fusion gene in the t(12;22)(q13;q12).
Genes involved and Proteins FUS (TLS) Location: 16p11
DNA/RNA FUS gene consists of 15 exons located within 11 kb of genomic DNA.
Protein FUS protein, provisionally designated TLS (translocated in liposarcoma), and then called FUS, contains an RNA-recognition motif and is a component of nuclear riboprotein complexes. Lack of FUS in mice causes lethality into neonatal period, it influences lymphocyte development in a non-cell-intrinsic manner, it has an intrinsic role in the proliferative responses of B cells to specific mitogenic stimuli, and it is required for the maintenance of genomic stability. The involvement of a nuclear riboprotein in these processes in vivo indicates that FUS is important in genome maintenance.
Somatic mutations: FUS has been also shown a partner of gene fusions linked in other malignancies: fused to ERG in acute myeloid leukaemia with t(16;21)(p11;q22), fused to CREB3L2 in low-grade fibromyxoid sarcoma (LGFMS) by a translocation between chromosome bands 7q33-q34 (CREB3L2) and 16p11 (FUS), fused to ATF1 in histiocytoma or fused to CHOP gene in Myxoid Liposarcoma with t(13;16)(q13;p11).
ATF1 Location: 12q13
DNA/RNA 816 bp mRNA
Protein ATF1 gene encodes a member of the CREB-ATF basic leucine-zipper (bZIP) family of transcription factors. This protein of 271 amino acids has a nuclear localization. Function: DNA binding protein, binds the consensus sequence: 5'GTGACGT (A/C) (A/G)-3'; cAMP-inducible transcription factor (cAMP-responsive enhancer-binding protein (CRE), like CREB. Is a member of the CREB protein family.
Somatic mutations: t(12;22)(q13;q12) in Angiomatoid Fibrous histiocytoma ATF1-EWSR1. It is also rearranged in clear cell sarcoma (CCS) with t(12;22) (q13;q12), creating an EWSR1-ATF1 fusion gene.
EWSR1 Location: 22q12
DNA/RNA DNA spans over 40 kb; open reading frame: 2.0 kb, 17 exons. Transcription 2.4 kb mRNA; centromere to telomere direction; differential splicing.
Protein 656 amino acids; serine-tyrosine tandem repeats. It has a wide expression and functions as a RNA binding.
Somatic mutations: Ewing tumours with: t(11;22)(q24;q12) → FLI1 - EWSR1. Ewing tumours: including Ewing's Sarcoma and periphral primitive neuroectodermal tumour. Ewing tumours with t(21;22)(q21;q12) → ERG - EWSR1. Ewing tumours (Ewing's Sarcoma and peripheral primitive neuroectodermal tumour). Ewing tumours with t(7;22)(p22;q12) → ETV1 - EWSR1. Ewing tumours (Ewing's Sarcoma and peripheral primitive neuroectodermal tumour). Ewing tumours with t(17;22)(q12;q12) → E1AF - EWSR1. Ewing tumours (Ewing's Sarcoma and peripheral primitive neuroectodermal tumour). t(11;22)(p13;q12) / Intra abdominal desmoplastic small round cell sarcoma (IADSRCT) → WT1 - EWSR1. t(12;22)(q13;q12) / Angiomatoid Fibrous Histiocytoma → ATF1 - EWSR1. t(9;22)(q22;q12) / Myxoid Chondrosarcoma → TEC - EWSR1.

Angiomatoid fibrous histiocytoma (AFH) Vicente-Dueñas, Sánchez-García I
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 129
Result of the chromosomal anomaly Hybride Gene Description FUS-ATF1 fusion gene t(12;16)(q13;p11). EWSR1-ATF1 fusion gene t(12;22)(q13;q12).
Fusion protein FUS/ATF-1
Description The fusion gene contains the N-terminus of FUS and the DNA binding domain of ATF-1 with a glycine to valine transition at the junction. This is similar to the EWS/ATF1 chimeric protein found in CCS (clear cell sarcoma). The normal ATF1 gene is transcribed from centromere to telomere, while the transcription of FUS seems to proceed in the opposite direction. Hence, the formation of FUS/ATF-1 is possible only if another genomic aberration, such as an inversion, occurs in addition to the chromosomal translocation. Such an event would be analogous to the formation of EWS/ERG in Ewing sarcoma and EWS/CHOP in myxoid liposarcoma.
EWSR1-ATF1
Description See the diagram below. EWSR1 is interrupted at codon 265 (exon 7) and fused to codon 110 (exon 5) of ATF-1, resulting in an in-frame junction. The chimaeric protein is composed of the N-terminal domain of EWS linked to the bZIP domain of ATF-1. The fusion gene EWSR1-ATF1 can be associated with different tumor types (Clear cell sarcoma (CCS) and Angiomatoid fibrous histiocytoma (AFH)). Activation of the EWSR1-ATF1 oncogene is probably an early step in the transformation process, but the overall gene expression patterns are likely to vary considerably between AFH and CCS, in keeping with their clinopathologic differences. EWS/ATF1 functions as a potent constitutive activator of several cAMP-inducible promoters when assayed by transfection in cells lacking EWS/ATF1. EWSR1 like FUS is an RNA-binding protein. Both are involved as the N-terminal part of fusion proteins in a number of sarcomas in combination with various transcription factor partners suggested to be tumor-specific. It has been previously shown that the N-terminal parts of EWSR1 and FUS are fused to certain transcription factors, resulting in a common or very similar oncogenic potential having the same tumor phenotype.
This fusion was generated by a translocation between chromosomal bands 16p11 and 12q 13, harbouring the FUS and ATF1 genes, respectively. FUS is interrupted at codon 175 (exon5) and fused to condon 110 (exon 5) of ATF1, resulting in an in-frame junction with a glycine to valina (GGT to GTT) transition.

Angiomatoid fibrous histiocytoma (AFH) Vicente-Dueñas, Sánchez-García I
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 130
References Enzinger FM. Angiomatoid malignant fibrous histiocytoma: a distinct fibrohistiocytic tumor of children and young adults simulating a vascular neoplasm. Cancer 1979;44:2147-2157.
Costa MJ and Weiss SW. Angiomatoid malignant fibrous histiocytoma. A follow-up study of 108 cases with evaluation of possible histologic predictors of outcome. Am J Surg Pathol 1990;14:1126-1132.
Smith ME, Costa MJ and Weiss SW. Evaluation of CD68 and other histiocytic antigens in angiomatoid malignant fibrous histiocytoma. Am J Surg Pathol 1991;15:757-763.
Zucman J, Delattre O, Desmaze C, Epstein AL, Stenman G, Speleman F, Fletchers CD, Aurias A and Thomas G. EWS and ATF-1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat Genet 1993;4:341-345.
Zucman J, Melot T, Desmaze C, Ghysdael J, Plougastel B, Peter M, Zucker JM, Triche TJ, Sheer D, Turc-Carel C and et al. Combinatorial generation of variable fusion proteins in the Ewing family of tumours. Embo J 1993;12:4481-4487.
Giovannini M, Biegel JA, Serra M, Wang JY, Wei YH, Nycum L, Emanuel BS and Evans GA. EWS-erg and EWS-Fli1 fusion transcripts in Ewing's sarcoma and primitive neuroectodermal tumors with variant translocations. J Clin Invest 1994;94:489-496.
Ichikawa H, Shimizu K, Hayashi Y and Ohki M. An RNA-binding protein gene, TLS/FUS, is fused to ERG in human myeloid leukemia with t(16;21) chromosomal translocation. Cancer Res 1994;54:2865-2868.
Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ and Denny CT. A second Ewing's sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 1994;6:146-151.
Panagopoulos I, Hoglund M, Mertens F, Mandahl N, Mitelman F and Aman P. Fusion of the EWS and CHOP genes in myxoid liposarcoma. Oncogene 1996;12:489-494.
Dal Cin P, Sciot R, Panagopoulos I, Aman P, Samson I, Mandahl N, Mitelman F, Van den Berghe H and Fletcher CD. Additional evidence of a variant translocation t(12;22) with EWS/CHOP fusion in myxoid liposarcoma: clinicopathological features. J Pathol 1997;182:437-441.
Fanburg-Smith JC and Miettinen M. Angiomatoid 'malignant' fibrous histiocytoma: a clinicopathologic study of 158 cases and further exploration of the myoid phenotype. Hum Pathol 1999;30:1336-1343.
Kim J and Pelletier J. Molecular genetics of chromosome translocations involving EWS and related family members. Physiol Genomics 1999;1:127-138.
Jacobs IA and Chevinsky A. Angiomatoid fibrous histiocytoma: a case report and review of the literature. Dermatol Surg 2000;26:491-492.
Wang H, Jafri J, Recant W and Montag AG. Pathologic quiz case: a large cystic thigh mass in a 10-year-old boy. Arch Pathol Lab Med 2000;124:783-784.
Waters BL, Panagopoulos I and Allen EF. Genetic characterization of angiomatoid fibrous histiocytoma identifies fusion of the FUS and ATF-1 genes induced by a chromosomal translocation involving bands 12q13 and 16p11. Cancer Genet Cytogenet 2000;121:109-116.
Asakura S, Tezuka N, Inoue S, Kihara N and Fujino S. Angiomatoid fibrous histiocytoma in mediastinum. Ann Thorac Surg 2001;72:283-285.
Raddaoui E, Donner LR and Panagopoulos I. Fusion of the FUS and ATF1 genes in a large, deep-seated angiomatoid fibrous histiocytoma. Diagn Mol Pathol 2002;11:157-162.
Shing DC, McMullan DJ, Roberts P, Smith K, Chin SF, Nicholson J, Tillman RM, Ramani P, Cullinane C and Coleman N. FUS/ERG gene fusions in Ewing's tumors. Cancer Res 2003;63:4568-4576.
Li CS, Chan WP, Chen WT, Chang CP, Shih LS, Chen RC and Tu HY. MRI of angiomatoid fibrous histiocytoma. Skeletal Radiol 2004;33:604-608.
Hallor KH, Mertens F, Jin Y, Meis-Kindblom JM, Kindblom LG, Behrendtz M, Kalén A, Mandahl N and Panagopoulos I. Fusion of the EWSR1 and ATF1 genes without expression of the MITF-M transcript in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 2005;44:97-102.
Lemos MM, Karlen J and Tani E. Fine-needle aspiration cytology of angiomatoid malignant fibrous histiocytoma. Diagn Cytopathol 2005;33:116-121.
Pérez-Mancera PA and Sánchez-García I. Understanding mesenchymal cancer: the liposarcoma-associated FUS-DDIT3 fusion gene as a model. Seminars in Cancer Biology 2005;15:206-214.
This article should be referenced as such: Vicente-Dueñas, Sánchez-García I. Angiomatoid fibrous histiocytoma (AFH). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):127-130.

Cancer Prone Disease Section Mini Review
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 131
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
Alagille syndrome (AGS) Michèle Meunier-Rotival, Michelle Hadchouel
INSERM E0020, 80 rue du Général Leclerc, F-94276 Le Kremlin-Bicêtre Cedex, France
Published in Atlas Database: October 2005
Online updated version: http://AtlasGeneticsOncology.org/Kprones/AlagilleID10090.html DOI: 10.4267/2042/38309
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: Alagille-Watson syndrome (AWS); arteriohepatic dysplasia (AHD); cholestasis with peripheral pulmonary stenosis; hepatic ductular paucity, syndromatic Note: syndrome associating 5 major features (complete syndrome): paucity of interlobular bile ducts, pulmonary artery stenosis, butterfly-like vertebrae, posterior embryotoxon and a peculiar face. Only the 2 first ones are symptomatic. Incomplete syndrome is very frequent. Other features have been described involving kidney, cardiac and vascular anomalies including tetralogy of Fallot, ear, pancreas, intestine etc. Inheritance: autosomal dominant with a highly variable expressivity and nearly complete penetrance; frequency is about 1/70,000-100,000 live newborns; 60-70% are sporadic cases.
Clinics Phenotype and clinics - liver: jaundice, pruritus, xanthomas, bile duct paucity, biochemical cholestasis and hypercholesterolemia. Liver transplantation is performed in about 25% cases. - cardiovascular system: peripheral pulmonary stenosis, coarctation of aorta, tetralogy of Fallot, ventricular or atrial septal defects, patent ductus arteriosus, truncus arteriosus, right ventricule hypoplasia. - systemic vascular system: coarctation of aorta, middle aortic syndrome, arterial hypoplasia (hepatic, renal, carotid, celiac), moyamoya disease, hypoplastic portal vein branch, intracranial bleeding. - vertebrae and skeleton: butterfly-like vertebrae, spina bifida, abnormal progression of interpedicular distances, shortening of distal phalanges and metacarpal bones, clinodactily.
- eye: posterior embryotoxon, retinal pigmentation, iris strands, cataract, myopia, strabismus, glaucoma, optic disc drusen, fundus hypopigmentation, blindness. - kidney: mesangiolipidosis, tubular dysfunction, tubulointerstitial nephritis, renal hypoplasia, renal agenesis, horseshoe kidney, cysts. - ear: temporal bone abnormalities, chronic otitis media, deafness. - larynx: high pitched voice. - pancreas: diabetes, exocrine pancreatic insufficiency. - gut: small bowell atresia or stenosis. - lung: tracheal and bronchial stenosis. - face: prominent forehead, deep-set eyes, mild hypertelorism, straight nose, small pointed chin. - growth retardation. - mental retardation (?).
Neoplastic risk Very rare hepatocellular carcinoma.
Treatment No specific treatment.
Prognosis Major contributors to morbidity arise from bile duct paucity or cholestatic liver disease (including liver transplantation), cardiac disease, and renal disease.
Cytogenetics 3-7% of patients with Alagille syndrome have deletions of part or totality of the JAG1 gene in 20p and, in rare instances translocations : del(20p), del(20)(p11.2), del(20)(p12.3-p11.23), del(20)(p13-p12.2), ins(7;20), t(2;20).
Other findings Note: There is no phenotype-genotype correlation.
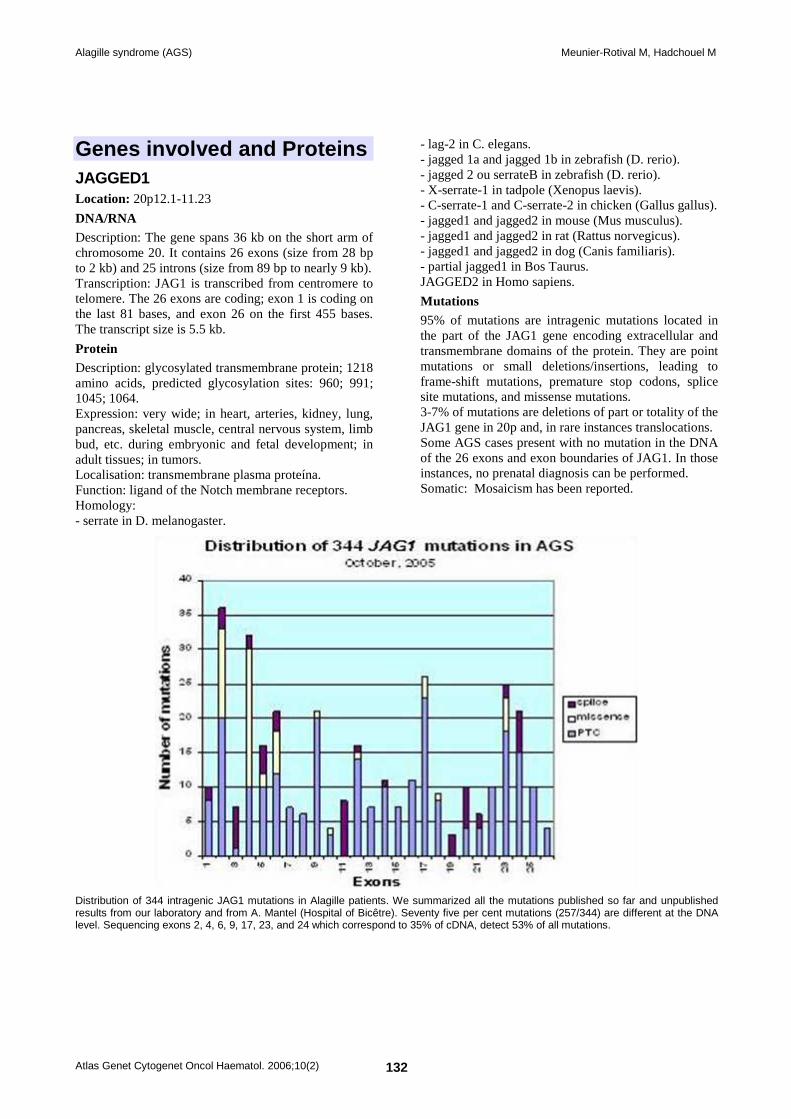
Alagille syndrome (AGS) Meunier-Rotival M, Hadchouel M
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 132
Genes involved and Proteins JAGGED1 Location: 20p12.1-11.23
DNA/RNA Description: The gene spans 36 kb on the short arm of chromosome 20. It contains 26 exons (size from 28 bp to 2 kb) and 25 introns (size from 89 bp to nearly 9 kb). Transcription: JAG1 is transcribed from centromere to telomere. The 26 exons are coding; exon 1 is coding on the last 81 bases, and exon 26 on the first 455 bases. The transcript size is 5.5 kb.
Protein Description: glycosylated transmembrane protein; 1218 amino acids, predicted glycosylation sites: 960; 991; 1045; 1064. Expression: very wide; in heart, arteries, kidney, lung, pancreas, skeletal muscle, central nervous system, limb bud, etc. during embryonic and fetal development; in adult tissues; in tumors. Localisation: transmembrane plasma proteína. Function: ligand of the Notch membrane receptors. Homology: - serrate in D. melanogaster.
- lag-2 in C. elegans. - jagged 1a and jagged 1b in zebrafish (D. rerio). - jagged 2 ou serrateB in zebrafish (D. rerio). - X-serrate-1 in tadpole (Xenopus laevis). - C-serrate-1 and C-serrate-2 in chicken (Gallus gallus). - jagged1 and jagged2 in mouse (Mus musculus). - jagged1 and jagged2 in rat (Rattus norvegicus). - jagged1 and jagged2 in dog (Canis familiaris). - partial jagged1 in Bos Taurus. JAGGED2 in Homo sapiens.
Mutations 95% of mutations are intragenic mutations located in the part of the JAG1 gene encoding extracellular and transmembrane domains of the protein. They are point mutations or small deletions/insertions, leading to frame-shift mutations, premature stop codons, splice site mutations, and missense mutations. 3-7% of mutations are deletions of part or totality of the JAG1 gene in 20p and, in rare instances translocations. Some AGS cases present with no mutation in the DNA of the 26 exons and exon boundaries of JAG1. In those instances, no prenatal diagnosis can be performed. Somatic: Mosaicism has been reported.
Distribution of 344 intragenic JAG1 mutations in Alagille patients. We summarized all the mutations published so far and unpublished results from our laboratory and from A. Mantel (Hospital of Bicêtre). Seventy five per cent mutations (257/344) are different at the DNA level. Sequencing exons 2, 4, 6, 9, 17, 23, and 24 which correspond to 35% of cDNA, detect 53% of all mutations.

Alagille syndrome (AGS) Meunier-Rotival M, Hadchouel M
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 133
References Watson GH, Miller V. Arteriohepatic dysplasia: familial pulmonary arterial stenosis with neonatal liver disease. Arch Dis Child 1973;48:459-466.
Alagille D, Odievre M, Gautier M, Dommergues JPJ. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental and sexual development, and cardiac murmur. Pediatrics 1975;86:63-71.
Alagille D, Estrada A, Hadchouel M, Gautier M, Odievre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr 1987;110:195-200.
Anad F, Burn J, Matthews D, Cross I, Davison BC, Mueller R, Sands M, Lillington DM, Eastham E. Alagille syndrome and deletion of 20p. J Med Genet 1990;27:729-737. (Review).
Dhorne-Pollet S, Deleuze JF, Hadchouel M, Bonaiti-Pellie C. Segregation analysis of Alagille syndrome. J Med Genet 1994;31:453-457.
Spinner NB, Rand EB, Fortina P, Genin A, Taub R, Semeraro A, Piccoli DA. Cytologically balanced t(2;20) in a two-generation family with Alagille syndrome: cytogenetic and molecular studies. Am J Hum Genet 1994;55:238-243.
Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 1997;16:243-251.
Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet 1997;16:235-242.
Krantz ID, Colliton RP, Genin A, Rand EB, Li L, Piccoli DA, Spinner NB. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet 1998;62:1361-1369.
Crosnier C, Driancourt C, Raynaud N, Dhorne-Pollet S, Pollet N, Bernard O, Hadchouel M, Meunier-Rotival M. Mutations in JAGGED1 gene are predominantly sporadic in Alagille syndrome. Gastroenterology 1999;116:1141-1148.
Gray GE, Mann RS, Mitsiadis E, Henrique D, Carcangiu ML, Banks A, Leiman J, Ward D, Ish-Horowitz D, Artavanis-Tsakonas S. Human ligands of the Notch receptor. Am J Pathol 1999;154:785-794.
Krantz ID, Smith R, Colliton RP, Tinkel H, Zackai EH, Piccoli DA, Goldmuntz E, Spinner NB. Jagged1 mutations in patients ascertained with isolated congenital heart defects. Am J Med Genet 1999;84:56-60.
Crosnier C, Lykavieris P, Meunier-Rotival M, Hadchouel M. Alagille syndrome. The widening spectrum of arteriohepatic dysplasia. Clin Liver Dis 2000;4:765-778. (Review).
Crosnier C, Attie-Bitach T, Encha-Razavi F, Audollent S, Soudy F, Hadchouel M, Meunier-Rotival M, Vekemans M. JAGGED1 gene expression during human embryogenesis elucidates the wide phenotypic spectrum of Alagille syndrome. Hepatology 2000;32:574-581.
Jones EA, Clement-Jones M, Wilson DI. JAGGED1 expression in human embryos: correlation with the Alagille syndrome phenotype. J Med Genet 2000;37:658-662.
Crosnier C, Driancourt C, Raynaud N, Hadchouel M, Meunier-Rotival M. Fifteen novel mutations in the JAGGED1 gene of patients with Alagille syndrome. Hum Mutat 2001;17:72-73.
Eldadah ZA, Hamosh A, Biery NJ, Montgomery RA, Duke M, Elkins R, Dietz HC. Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Hum Mol Genet 2001;10:163-169.
Giannakudis J, Ropke A, Kujat A, Krajewska-Walasek M, Hughes H, Fryns JP, Bankier A, Amor D, Schlicker M, Hansmann I. Parental mosaicism of JAG1 mutations in families with Alagille syndrome. Eur J Hum Genet 2001;9:209-216.
Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut 2001;49:431-435. (Review).
Morrissette JD, Colliton RP, Spinner NB. Defective intracellular transport and processing of JAG1 missense mutations in Alagille syndrome. Hum Mol Genet 2001;10:405-413.
Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in Alagille syndrome. Hum Mutat 2001;17:18-33. (Review).
Yuan ZR, Okaniwa M, Nagata I, Tazawa Y, Ito M, Kawarazaki H, Inomata Y, Okano S, Yoshida T, Kobayashi N, Kohsaka T. The DSL domain in mutant JAG1 ligand is essential for the severity of the liver defect in Alagille syndrome. Clin Genet 2001;59:330-337.
Le Caignec C, Lefevre M, Schott JJ, Chaventre A, Gayet M, Calais C, Moisan JP. Familial deafness, congenital heart defects, and posterior embryotoxon caused by cysteine substitution in the first epidermal-growth-factor-like domain of jagged 1. Am J Hum Genet 2002;71:180-186.
Lu F, Morrissette JJ, Spinner NB. Conditional JAG1 mutation shows the developing heart is more sensitive than developing liver to JAG1 dosage. Am J Hum Genet 2003;72(4):1065-1070.
Lykavieris P, Crosnier C, Trichet C, Meunier-Rotival M, Hadchouel M. Bleeding tendency in children with Alagille syndrome. Pediatrics 2003;111:167-170.
Ropke A, Kujat A, Graber M, Giannakudis J, Hansmann I. Identification of 36 novel Jagged1 (JAG1) mutations in patients with Alagille syndrome. Hum Mutat 2003;21:100.
Kamath BM, Spinner NB, Emerick KM, Chudley AE, Booth C, Piccoli DA, Krantz ID. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation 2004;109:1354-1358. (Review).
Boyer J, Crosnier C, Driancourt C, Raynaud N, Gonzales M, Hadchouel M, Meunier-Rotival M. Expression of mutant JAGGED1 alleles in patients with Alagille syndrome. Hum Genet 2005;116:445-453.
Jurkiewicz D, Popowska E, Glaser C, Hansmann I, Krajewska-Walasek M. Twelve novel JAG1 gene mutations in polish Alagille syndrome patients. Hum Mutat 2005;25:321.
Kim B, Park SH, Yang HR, Seo JK, Kim WS, Chi JG. Hepatocellular carcinoma occurring in Alagille syndrome. Pathol Res Pract 2005;201:55-60.
This article should be referenced as such: Meunier-Rotival M, Hadchouel M. Alagille syndrome (AGS). Atlas Genet Cytogenet Oncol Haematol.2006;10(2):131-133.

Deep Insight Section
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 134
Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
Three-dimensional organization of the mammalian nucleus in normal and tumor cells Sabine Mai
Manitoba Institute of Cell Biology, CancerCare Manitoba, The University of Manitoba, 675 McDermot Avenue, Winnipeg MB R3E 0V9, Canada
Published in Atlas Database: November 2005
Online updated version: http://AtlasGeneticsOncology.org/Deep/3DOrganizNuclID20053.html DOI: 10.4267/2042/38310
This work is licensed under a Creative Commons Attribution-Non-commercial-No Derivative Works 2.0 France Licence. © 2006 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Nuclear organization and genome stability The three-dimensional organization of the genome and nucleus play pivotal roles in tumor development. As Theodor Boveri (1862-1915) postulated a century ago, aberrant chromosome numbers are associated with aberrant cell division and linked to tumor formation (Boveri, 1902; Boveri, 1914). Studying cell division in Ascaris and in sea urchin eggs under normal conditions and following double fertilization, he observed normal as well as aberrant cell divisions. From the latter with aberrant chromosome constitutions he inferred similar changes could occur in cancer cells (Boveri, 1914). A concept of chromosome and centrosome cycles emerged from his work and Boveri's seminal observations are as valid today as they were a century ago. They are often considered the basis for the first genetic model of cancer development (Moritz and Sauer, 1996; Wunderlich, 2002). Today’s researchers in the field of nuclear structure and genome organization are still gathering many details of this organizational puzzle, which is important for determining normal or aberrant nuclear organization and cellular fate.
Imaging of nuclear structures Advances in imaging, and specifically in fluorescent imaging, have contributed to our understanding of the three-dimensional organization of the nucleus. The transition from two-dimensional (2D) to three-dimensional (3D) imaging has allowed us to better understand how the nucleus is spatially organized. In
addition to 3D approaches, live cell imaging has added a new dimension to our ability of developing clear concepts about the dynamics of nuclear organization (Liu and Chang, 2003; Garini et al., 2005). Live cell imaging with 3D resolution is often called four-dimensional (4D) imaging, where the fourth dimension of time is added to the imaging in the x,y, and z planes, that constitute a 3D image. Such 4D studies involve light microscopy and fluorescent imaging approaches, and use fluorescent proteins or fluorescent labeled nucleic acids (Haraguchi et al., 1999; Stephens and Allen, 2003; Gatlin et al., 2003; Solovei et al., 2002; Molenaar et al., 2003; Bystricky et al., 2004, 2005). The images shown below indicate how 2D and 3D images differ with respect to the spatial information given. The same object, a lymphocyte nucleus (blue) with telomeres (red), is shown in 2D (Fig. 1, top) and in 3D (Fig. 1, bottom). Using illustrations such as the one shown in Fig. 2, one can imagine how 3D information is collected. However, 3D movies as published online (Louis et al., 2005) best reflect the spatial order of objects in the nucleus. Since the depth and spatial relationships are lost when imaging a 3D object in one focal plane, i.e. in two dimensions (Fig. 1, top), 3D imaging methods that account for the overall structure of the object are an obvious and necessary choice for any spatially relevant study. 3D imaging whether through confocal or deconvolution approaches give a representation of the true spatial organization of the nucleus (Fig. 1, bottom) and will ultimately allow us to link structure and function. Finally, the dynamic nature of the nucleus becomes evident through time lapse and live cell studies (i.e. Molenaar et al., 2003).

Three-dimensional organization of the mammalian nucleus in normal and tumor cells Mai s
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 135
Figure 1. Two-dimensional (2D) and three-dimensional image of a mouse lymphocyte nucleus and its telomeres. Top: 2D image. The nucleus (blue) is stained with 4’,6-diamidino-2-phenylindole (DAPI). The telomeres (red) are labeled with a Cy3-conjugated peptide-nucleic acid (PNA) probe (DAKO). 2D and 3D images were acquired using a Zeiss Axioplan 2 with cooled AxioCam HR CCD, AxioVision, a PlanApo 63x1.4 oil immersion objective and DAPI and Cy3 filters. The pixel distance in the lateral plane was Dx = Dy = 106 nm; the axial sampling was Dz = 200 nm. Bottom: Three-dimensional (3D image) of the mouse lymphocyte nucleus and its telomeres shown in 2D image (Top). The blue counterstain has been ‘removed’ (i.e. the blue color channel is switched off) to only visualize the telomeres (red).
The normal mammalian nucleus Chromosomal organization
While some laboratories reported that chromosomes are organized randomly in the interphase nucleus (Cerda et al., 1999; Holley et al., 2002; Cornforth et al., 2002), most research groups find a consistent distribution of chromosomes within the mammalian nucleus and observe that the normal mammalian nucleus has a cell-
type–specific shape and structure in which chromosomes are observed in probable non-random territories (Cremer et al., 2001; Parada et al., 2004; Misteli, 2002; Essers et al., 2005). As recently shown for all chromosomes in primary human cells, the order of chromosomes is consistent from cell to cell within the identical primary cell population (Bolzer et al., 2005). In this recent study, the authors examined the organization of all chromosomes in primary human fibroblasts, amniotic fluid cells, and in prometaphase rosettes. Small chromosomes were found in the centre of these nuclei irrespective of their gene density, and larger chromosomes were observed in closer proximity to the nuclear periphery or rosette rim. The cells analyzed by this group and in the above study exhibit flat-ellipsoid nuclei, and their organization may differ from the one in spherical lymphocytes. This conclusion is supported by others who report that the positions of chromosome territories are cell-type-dependent (Parada et al., 2002; Parada and Misteli, 2002; Parada et al., 2004). It appears that different patterns of nuclear chromosome territories are not only dependent on nuclear shape and space, but also on gene-rich and gene-poor chromatin domains (Bolzer et al., 2005) and on the differentiation status of the cell: Nuclei of embryonic stem cells appear to have their very specific 3D chromosomal organization that differs from the position the same chromosomes have in differentiated cells (Wiblin et al., 2005). It was also shown that the 3D nuclear order of chromosomes within the same cell type is inherited during mitosis: daughter cells will show an organizational pattern that resembles their parental cell (Cremer et al., 2001; Gerlich et al., 2003; Essers et al., 2005). Moreover, the chromosomal order within the 3D nuclear space is evolutionarily conserved (Tanabe et al., 2002) strongly suggesting that the structural organization of the nucleus that is relevant to its stability and overall function has been established during evolution.
Chromosome movement or static order in the interphase nucleus?
Are chromosomes at the same position all the time? This is an area of intense research and no consensus has been reached. This is often due to the use of different cell lines, imaging and analysis conditions used. In addition, cell cycle and developmental stages of nuclei examined may have affected the results. The chromosome movements that were reported are of three types; 1) repositioning of sub-chromosomal regions within stable chromosome territories, 2) selective movement of single territories, and 3) small scale refolding events within sub-chromosomal regions (Zink and Cremer, 1998). Positional changes of chromosomes during the cell cycle have been observed by several groups (Walter et al., 2003; Vourc’h et al., 1993; Ferguson and Ward, 1992; Bridger et al., 2000; Chubb
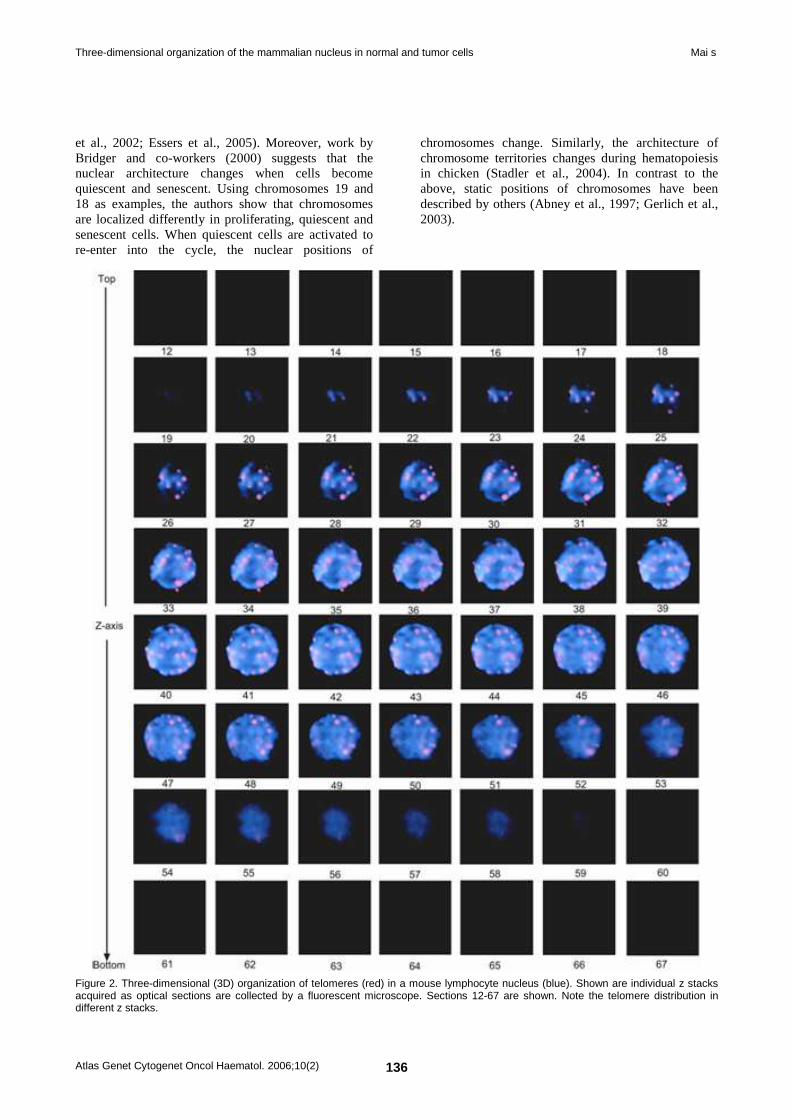
Three-dimensional organization of the mammalian nucleus in normal and tumor cells Mai s
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 136
et al., 2002; Essers et al., 2005). Moreover, work by Bridger and co-workers (2000) suggests that the nuclear architecture changes when cells become quiescent and senescent. Using chromosomes 19 and 18 as examples, the authors show that chromosomes are localized differently in proliferating, quiescent and senescent cells. When quiescent cells are activated to re-enter into the cycle, the nuclear positions of
chromosomes change. Similarly, the architecture of chromosome territories changes during hematopoiesis in chicken (Stadler et al., 2004). In contrast to the above, static positions of chromosomes have been described by others (Abney et al., 1997; Gerlich et al., 2003).
Figure 2. Three-dimensional (3D) organization of telomeres (red) in a mouse lymphocyte nucleus (blue). Shown are individual z stacks acquired as optical sections are collected by a fluorescent microscope. Sections 12-67 are shown. Note the telomere distribution in different z stacks.

Three-dimensional organization of the mammalian nucleus in normal and tumor cells Mai s
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 137
Telomeric organization in normal cells
As shown by live cell imaging, telomere positions in the 3D nucleus are not static. Telomeres can move in the interphase nucleus (Molenaar et al., 2003). The distances they move vary (ibid). Telomeres are organized in a very typical way within the 3D space of the nucleus. Normal lymphocytes of mouse or human origin show a cell cycle-dependent organization of telomeres in their interphase nuclei. In an unperturbed nucleus and under optimal growth conditions, telomeres are widely distributed throughout the nucleus in normal G0/G1 cells (Weierich et al., 2003; Chuang et al., 2004). S-phase cells display a similar pattern of telomere organization and, in addition, show replicative structures of telomeres (Chuang et al., 2004). In G2, telomeres assemble into a telomeric disk, first observed by us (Chuang et al., 2004). Human keratinocyte cell lines with flatter nuclei perform less reorganization of telomeres but also exhibit a dynamic cell-cycle-specific organization (Ermler et al., 2004). Thus, telomeres reorganize in the 3D space of the nucleus during a
normal cell cycle (Fig. 3). At no time during a normal cell cycle do telomeres of normal cells come into such close association that they form clusters or aggregates (Chuang et al., 2004). In fact, telomeres of normal cells do not overlap (Chuang et al., 2004).
Telomeric organization in tumor cells
Telomeres of tumor cell nuclei show an altered 3D nuclear organization. In contrast to normal cells, telomeres of tumor cells form aggregates. One or more telomeric aggregate can be observed per interphase nucleus, and these telomere clusters can be of various sizes (Chuang et al., 2004). Some telomeric aggregates represent telomeric fusions and thus generate dicentric chromosomes. This spatial alteration in the 3D organization of telomeres is causal to the initiation of breakage-bridge-fusion cycles and directly results in genomic instability (Louis et al., 2005). Further studies will elucidate the molecular mechanisms of telomeric aggregate formation.
Figure 3. Overview of the telomeric positions during the cell cycle of normal lymphocytes. The examples shown here are from primary mouse lymphocytes. For details, see Chuang et al., 2004.

Three-dimensional organization of the mammalian nucleus in normal and tumor cells Mai s
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 138
Measurement of nuclear structures Chromosomes
In order to speak of chromosomal positions, one needs to be able to measure them. Tools to achieve this have been developed (Walter et al., 2003; Solovei et al., 2002). For specific questions related to chromosomal positions and overlaps, additional programs are also in use (Louis et al., 2005). What is required for future investigations are additional analytical tools and comparative evaluations of existing tools.
Telomeres
To measure the 3D organization of telomeres, we have developed TeloViewTM (Chuang et al., 2004; Vermolen et al., 2005). Briefly, the program measures telomere positions that can vary during the cell cycle. The nuclear area that contains the telomeres is best characterized as an oblate spheroid. In this spheroid, the two main axes, a and b, are of equal length, while the third axis, c, is shorter. It is this axis that varies during the cell cycle. The 3D organization during the cell cycle is thus reflected by the a/c ratio, and it is 1.4+0.1, 1.5+0.2, and 14+2 respectively for normal lymphocytes in G0/G1, S and G2 (Vermolen et al., 2005). In addition, the program quantitates the 3D fluorescent intensity of each telomere that is found in the nucleus. The relative fluorescent intensity measured is proportional to the telomere size (Poon et al., 1999).
The tumor cell nucleus and nuclear remodeling During tumorigenesis, the nucleus is remodeled (Pienta et al., 1989). We have studied oncogenic remodeling of the mammalian nucleus, using the impact of c-Myc deregulation as example (Louis et al., 2005). Induction of c-Myc leads to the formation of telomeric aggregates that are commonly found in tumor cells and not present in normal cells. Some of the c-Myc-induced telomeric aggregates represent end-to-end chromosomal fusions. Dicentric chromosomes that are generated during c-Myc induction are chromosomal end-to-end fusions that initiate breakage-bridge-fusion (BBF) cycles in the subsequent anaphases. Propagation of such BBF cycles leads to ongoing chromosomal rearrangements and typically generates unbalanced chromosomal translocations and terminal deletions. New BBF cycles will continue until no more telomere free ends are available for fusions (Louis et al., 2005). Thus remodeling of the 3D telomeric organization induces genomic instability that is a hallmark of tumors (Mitelman et al., 2005; Hanahan and Weinberg, 2000; Vogelstein and Kinzler, 2004; Gollin et al., 2005; Weaver and Cleveland, 2005). Nuclear remodeling as a result of c-Myc deregulation does not stop at the telomeric ends. During the process of c-Myc-mediated nuclear remodeling, chromosomes
also alter their normal positions (Louis et al., 2005). When studying mouse chromosomes 5 and 13, 7 and 10, 7 and 17, 15 and 11 in PreB lymphocytes, we found that all of them change their positions as a result of c-Myc deregulation. The generated overlap between the above mentioned chromosome pairs is commonly associated with their involvement in translocations, as our spectral karyotyping (SKY) data suggest. The analysis of chromosomal overlaps, that were measured in interphase nuclei following chromosome painting, and the occurrence of recurrent translocations, found in metaphases after SKY, matched for mouse chromosomes 5 and 13, 7 and 17, 7 and 10 in Myc-activated PreB cells (Louis et al., 2005). However, this relationship did not hold true for chromosomes 11 and 15 (reviewed in Mai and Garini, 2005). Thus, the close vicinity of chromosomes represents most of the time a favorable condition for chromosomal translocations. It is evident that additional factors are required to facilitate the occurrence of translocations. These may include double strand breakage, sequence homologies, and recombination. Future studies will be needed to elucidate these processes further. Data by others support the notion that close spatial proximity of chromosomes or specific chromosomal neighborhoods contributes to translocation frequencies. For example, mouse chromosomes 12 and 15 that are frequently involved in translocations in mouse plasmacytoma are found in closer vicinity to each other in mouse B cells than in mouse hepatocytes (Parada et al., 2004). In human chronic myeloid leukemia, chromosomes 9 and 22 are found in close proximity and this fact has been linked to the predisposition for translocations between the two chromosomes (Neves et al., 1999). A comprehensive study that examined 11,010 human constitutional translocations concluded that the frequency of constitutional translocations depended on three main factors, and these included the chromosome positions, chromosome sizes and specific DNA sequences (Bickmore and Teague, 2002). Other events also contribute to nuclear remodeling. Examples include activated HaRas that leads to chromatin coarsening and loss of heterochromatin aggregates and correlates with metastatic potential (Fischer et al., 1998; Zink et al., 2004), viral infections (Igakura et al., 1998; De Noronha et al., 2001), and DNA damaging agents (Abdel-Halim et al., 2004; Bickmore and Teague, 2002; Gazave et al., 2005). Thus, the impact of a single remodeling-promoting event can be with long lasting consequences to the cell. The generation of an aberrant cell that is able to multiply, evade growth control and apoptosis and form a tumor is one outcome. Telomeric reorganization may also contribute to mental retardation since subtelomeric rearrangements are a frequent cause for these diseases (Kok et al., 2005; Hwang et al., 2005). On the other hand, additional pathways affect nuclear organization. For example, an altered nuclear matrix organization

Three-dimensional organization of the mammalian nucleus in normal and tumor cells Mai s
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 139
contributes to tumor development as shown for HMGY (I) (Takaha et al., 2002; Leman et al., 2003). Structural changes in the nuclear envelope contribute to the Hutchinson-Gilford progeria syndrome as demonstrated for mutations in Lmna that encodes lamins A and C that are structural components of the nuclear envelope (Pollex and Hegele, 2004). We conclude that nuclear organization of the genome, cellular function, genome stability and disease are tightly linked. Irrespective of the type of genetic and/or structural changes present in tumors or in other genetic diseases, the 3D nuclear organization is different from that found in normal cells. It is therefore feasible, in the future, to benefit from the exact knowledge about the 3D structure of the nucleus for diagnosis, monitoring of disease, and therapeutic intervention.
Acknowledgement I would like to thank Drs. Michael Mowat and Francis Wiener for critical reading. I express my sincere thanks to all my lab members and colleagues who were involved in studies reviewed here. My work was funded through CancerCare Manitoba Foundation, the Canadian Institutes of Health Research, the National Cancer Institute of Canada and the Canada Foundation for Innovation.
References Abdel-Halim HI, Imam SA, Badr FM, Natarajan AT, Mullenders LH, Boei JJ. Ionizing radiation-induced instant pairing of heterochromatin of homologous chromosomes in human cells. Cytogenetic and genome research. 2004 ; 104 (1-4) : 193-199.
Abney JR, Cutler B, Fillbach ML, Axelrod D, Scalettar BA. Chromatin dynamics in interphase nuclei and its implications for nuclear structure. The Journal of cell biology. 1997 ; 137 (7) : 1459-1468.
Bickmore WA, Teague P. Influences of chromosome size, gene density and nuclear position on the frequency of constitutional translocations in the human population. Chromosome research . 2002 ; 10 (8) : 707-715.
Bystricky K, Heun P, Gehlen L, Langowski J, Gasser SM. Long-range compaction and flexibility of interphase chromatin in budding yeast analyzed by high-resolution imaging techniques. PNAS. 2004 ; 101 (47) : 16495-16500.
Bystricky K, Laroche T, van Houwe G, Blaszczyk M, Gasser SM. Chromosome looping in yeast: telomere pairing and coordinated movement reflect anchoring efficiency and territorial organization. The Journal of cell biology. 2005 ; 168 (3) : 375-387.
Bolzer A, Kreth G, Solovei I, Koehler D, Saracoglu K, Fauth C, Müller S, Eils R, Cremer C, Speicher MR, Cremer T. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS biology. 2005 ; 3 (5) : page e157.
Boveri T. Ueber mehrpolige Mitosen als Mittel zur Analyse des Zellkerns. Verh Phys Med Gesellschaft Würzburg. 1902 ; 35 : 67-90.
Boveri T. Zur Frage der Entstehung maligner Tumoren. Fischer, Jena. 1914.
Bridger JM, Boyle S, Kill IR, Bickmore WA. Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Current biology : CB. 2000 ; 10 (3) : 149-152.
Chuang TC, Moshir S, Garini Y, Chuang AY, Young IT, Vermolen B, van den Doel R, Mougey V, Perrin M, Braun M, Kerr PD, Fest T, Boukamp P, Mai S. The three-dimensional organization of telomeres in the nucleus of mammalian cells. BMC biology. 2004 ; 2 : page 12.
Chubb JR, Boyle S, Perry P, Bickmore WA. Chromatin motion is constrained by association with nuclear compartments in human cells. Current biology : CB. 2002 ; 12 (6) : 439-445.
Cerda MC, Berríos S, Fernández-Donoso R, Garagna S, Redi C. Organisation of complex nuclear domains in somatic mouse cells. Biology of the cell. 1999 ; 91 (1) : 55-65.
Cremer M, von Hase J, Volm T, Brero A, Kreth G, Walter J, Fischer C, Solovei I, Cremer C, Cremer T. Non-random radial higher-order chromatin arrangements in nuclei of diploid human cells. Chromosome research . 2001 ; 9 (7) : 541-567.
Cornforth MN, Greulich-Bode KM, Loucas BD, Arsuaga J, Vázquez M, Sachs RK, Brückner M, Molls M, Hahnfeldt P, Hlatky L, Brenner DJ. Chromosomes are predominantly located randomly with respect to each other in interphase human cells. The Journal of cell biology. 2002 ; 159 (2) : 237-244.
Ermler S, Krunic D, Knoch TA, Moshir S, Mai S, Greulich-Bode KM, Boukamp P. Cell cycle-dependent 3D distribution of telomeres and telomere repeat-binding factor 2 (TRF2) in HaCaT and HaCaT-myc cells. European journal of cell biology. 2004 ; 83 (11-12) : 681-690.
Essers J, van Cappellen WA, Theil AF, van Drunen E, Jaspers NG, Hoeijmakers JH, Wyman C, Vermeulen W, Kanaar R. Dynamics of relative chromosome position during the cell cycle. Molecular biology of the cell. 2005 ; 16 (2) : 769-775.
Ferguson M, Ward DC. Cell cycle dependent chromosomal movement in pre-mitotic human T-lymphocyte nuclei. Chromosoma. 1992 ; 101 (9) : 557-565.
Fischer AH, Chadee DN, Wright JA, Gansler TS, Davie JR. Ras-associated nuclear structural change appears functionally significant and independent of the mitotic signaling pathway. Journal of cellular biochemistry. 1998 ; 70 (1) : 130-140.
Garini Y, Vermolen BJ, Young IT. From micro to nano: recent advances in high-resolution microscopy. Current opinion in biotechnology. 2005 ; 16 (1) : 3-12.
Gatlin J, Melkus MW, Padgett A, Petroll WM, Cavanagh HD, Garcia JV, Jester JV. In vivo fluorescent labeling of corneal wound healing fibroblasts. Experimental eye research. 2003 ; 76 (3) : 361-371.
Gazave E, Gautier P, Gilchrist S, Bickmore WA. Does radial nuclear organisation influence DNA damage? Chromosome research . 2005 ; 13 (4) : 377-388.
Gerlich D, Beaudouin J, Kalbfuss B, Daigle N, Eils R, Ellenberg J. Global chromosome positions are transmitted through mitosis in mammalian cells. Cell. 2003 ; 112 (6) : 751-764.
Gollin SM. Mechanisms leading to chromosomal instability. Seminars in cancer biology. 2005 ; 15 (1) : 33-42.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 ; 100 (1) : 57-70.
Haraguchi T, Ding DQ, Yamamoto A, Kaneda T, Koujin T, Hiraoka Y. Multiple-color fluorescence imaging of chromosomes and microtubules in living cells. Cell structure and function. 1999 ; 24 (5) : 291-298.
Holley WR, Mian IS, Park SJ, Rydberg B, Chatterjee A. A model for interphase chromosomes and evaluation of radiation-induced aberrations. Radiation research. 2002 ; 158 (5) : 568-580.

Three-dimensional organization of the mammalian nucleus in normal and tumor cells Mai s
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 140
Hwang KS, Pearson MA, Stankiewicz P, Lennon PA, Cooper ML, Wu J, Ou Z, Cai WW, Patel A, Cheung SW. Cryptic unbalanced translocation t(17;18)(p13.2;q22.3) identified by subtelomeric FISH and defined by array-based comparative genomic hybridization in a patient with mental retardation and dysmorphic features. American journal of medical genetics. Part A. 2005 ; 137 (1) : 88-93.
Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science (New York, N.Y.). 2003 ; 299 (5613) : 1713-1716.
Kok K, Dijkhuizen T, Swart YE, Zorgdrager H, van der Vlies P, Fehrmann R, te Meerman GJ, Gerssen-Schoorl KB, van Essen T, Sikkema-Raddatz B, Buys CH. Application of a comprehensive subtelomere array in clinical diagnosis of mental retardation. European journal of medical genetics. 2005 ; 48 (3) : 250-262.
Leman ES, Madigan MC, Brünagel G, Takaha N, Coffey DS, Getzenberg RH. Nuclear matrix localization of high mobility group protein I(Y) in a transgenic mouse model for prostate cancer. Journal of cellular biochemistry. 2003 ; 88 (3) : 599-608.
Liu YC, Chiang AS. High-resolution confocal imaging and three-dimensional rendering. Methods (San Diego, Calif.). 2003 ; 30 (1) : 86-93.
Louis SF, Vermolen BJ, Garini Y, Young IT, Guffei A, Lichtensztejn Z, Kuttler F, Chuang TC, Moshir S, Mougey V, Chuang AY, Kerr PD, Fest T, Boukamp P, Mai S. c-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. PNAS. 2005 ; 102 (27) : 9613-9618.
Mai S, Garini Y. Oncogenic remodeling of the three-dimensional organization of the interphase nucleus: c-Myc induces telomeric aggregates whose formation precedes chromosomal rearrangements. Cell cycle (Georgetown, Tex.). 2005 ; 4 (10) : 1327-1331.
Misteli T. Spatial positioning; a new dimension in genome function. Cell. 2004 ; 119 (2) : 153-156.
Mitelman F, Johansson B, Mertens F. Mitelman Database of Chromosome Aberrations in Cancer. http://cgap.nci.nih.gov/Chromosomes/Mitelman. 2005.
Molenaar C, Wiesmeijer K, Verwoerd NP, Khazen S, Eils R, Tanke HJ, Dirks RW. Visualizing telomere dynamics in living mammalian cells using PNA probes. The EMBO journal. 2003 ; 22 (24) : 6631-6641.
Moritz KB, Sauer HW. Boveri's contributions to developmental biology--a challenge for today. The International journal of developmental biology. 1996 ; 40 (1) : 27-47.
Neves H, Ramos C, da Silva MG, Parreira A, Parreira L. The nuclear topography of ABL, BCR, PML, and RARalpha genes: evidence for gene proximity in specific phases of the cell cycle and stages of hematopoietic differentiation. Blood. 1999 ; 93 (4) : 1197-1207.
de Noronha CM, Sherman MP, Lin HW, Cavrois MV, Moir RD, Goldman RD, Greene WC. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science (New York, N.Y.). 2001 ; 294 (5544) : 1105-1108.
Parada LA, McQueen PG, Munson PJ, Misteli T. Conservation of relative chromosome positioning in normal and cancer cells. Current biology : CB. 2002 ; 12 (19) : 1692-1697.
Parada L, Misteli T. Chromosome positioning in the interphase nucleus. Trends in cell biology. 2002 ; 12 (9) : 425-432.
Parada LA, McQueen PG, Misteli T. Tissue-specific spatial organization of genomes. Genome biology. 2004 ; 5 (7) : page R44.
Pienta KJ, Partin AW, Coffey DS. Cancer as a disease of DNA organization and dynamic cell structure. Cancer research. 1989 ; 49 (10) : 2525-2532.
Pollex RL, Hegele RA. Hutchinson-Gilford progeria syndrome. Clinical genetics. 2004 ; 66 (5) : 375-381.
Poon SS, Martens UM, Ward RK, Lansdorp PM. Telomere length measurements using digital fluorescence microscopy. Cytometry. 1999 ; 36 (4) : 267-278.
Stadler S, Schnapp V, Mayer R, Stein S, Cremer C, Bonifer C, Cremer T, Dietzel S. The architecture of chicken chromosome territories changes during differentiation. BMC cell biology. 2004 ; 5 (1) : page 44.
Stephens DJ, Allan VJ. Light microscopy techniques for live cell imaging. Science (New York, N.Y.). 2003 ; 300 (5616) : 82-86.
Solovei I, Cavallo A, Schermelleh L, Jaunin F, Scasselati C, Cmarko D, Cremer C, Fakan S, Cremer T. Spatial preservation of nuclear chromatin architecture during three-dimensional fluorescence in situ hybridization (3D-FISH). Experimental cell research. 2002 ; 276 (1) : 10-23.
Tanabe H, Müller S, Neusser M, von Hase J, Calcagno E, Cremer M, Solovei I, Cremer C, Cremer T. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. PNAS. 2002 ; 99 (7) : 4424-4429.
Takaha N, Hawkins AL, Griffin CA, Isaacs WB, Coffey DS. High mobility group protein I(Y): a candidate architectural protein for chromosomal rearrangements in prostate cancer cells. Cancer research. 2002 ; 62 (3) : 647-651.
Vermolen BJ, Garini Y, Mai S, Mougey V, Fest T, Chuang TC, Chuang AY, Wark L, Young IT. Characterizing the three-dimensional organization of telomeres. Cytometry. Part A : the journal of the International Society for Analytical Cytology. 2005 ; 67 (2) : 144-150.
Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature medicine. 2004 ; 10 (8) : 789-799.
Vourc'h C, Taruscio D, Boyle AL, Ward DC. Cell cycle-dependent distribution of telomeres, centromeres, and chromosome-specific subsatellite domains in the interphase nucleus of mouse lymphocytes. Experimental cell research. 1993 ; 205 (1) : 142-151.
Walter J, Schermelleh L, Cremer M, Tashiro S, Cremer T. Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. The Journal of cell biology. 2003 ; 160 (5) : 685-697.
Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer cell. 2005 ; 8 (1) : 7-12.
Weierich C, Brero A, Stein S, von Hase J, Cremer C, Cremer T, Solovei I. Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosome research . 2003 ; 11 (5) : 485-502.
Wiblin AE, Cui W, Clark AJ, Bickmore WA. Distinctive nuclear organisation of centromeres and regions involved in pluripotency in human embryonic stem cells. Journal of cell science. 2005 ; 118 (Pt 17) : 3861-3868.
Wunderlich V. JMM---past and present. Chromosomes and cancer: Theodor Boveri's predictions 100 years later. Journal of molecular medicine (Berlin, Germany). 2002 ; 80 (9) : 545-548.
Zink D, Cremer T. Cell nucleus: chromosome dynamics in nuclei of living cells. Current biology : CB. 1998 ; 8 (9) : R321-R324.
Zink D, Fischer AH, Nickerson JA. Nuclear structure in cancer cells. Nature reviews. Cancer. 2004 ; 4 (9) : 677-687.

Three-dimensional organization of the mammalian nucleus in normal and tumor cells Mai s
Atlas Genet Cytogenet Oncol Haematol. 2006;10(2) 141
This article should be referenced as such:
Mai S. Three-dimensional organization of the mammalian nucleus in normal and tumor cells. Atlas Genet Cytogenet Oncol Haematol.2006;10(2):134-141.

Atlas of Genetics and Cytogenetics in Oncology and Haematology
OPEN ACCESS JOURNAL AT INIST-CNRS
Instructions to Authors Manuscripts submitted to the Atlas must be submitted solely to the Atlas. Iconography is most welcome: there is no space restriction. The Atlas publishes "cards", "deep insights", "case reports", and "educational items". Cards are structured review articles. Detailed instructions for these structured reviews can be found at: http://AtlasGeneticsOncology.org/Forms/Gene_Form.html for reviews on genes, http://AtlasGeneticsOncology.org/Forms/Leukaemia_Form.html for reviews on leukaemias, http://AtlasGeneticsOncology.org/Forms/SolidTumour_Form.html for reviews on solid tumours, http://AtlasGeneticsOncology.org/Forms/CancerProne_Form.html for reviews on cancer-prone diseases. According to the length of the paper, cards are divided, into "reviews" (texts exceeding 2000 words), "mini reviews" (between), and "short communications" (texts below 400 words). The latter category may not be accepted for indexing by bibliographic databases. Deep Insights are written as traditional papers, made of paragraphs with headings, at the author's convenience. No length restriction. Case Reports in haematological malignancies are dedicated to recurrent -but rare- chromosomes abnormalities in leukaemias/lymphomas. Cases of interest shall be: 1- recurrent (i.e. the chromosome anomaly has already been described in at least 1 case), 2- rare (previously described in less than 20 cases), 3- with well documented clinics and laboratory findings, and 4- with iconography of chromosomes. It is mandatory to use the specific "Submission form for Case reports": see http://AtlasGeneticsOncology.org/Reports/Case_Report_Submission.html. Educational Items must be didactic, give full information and be accompanied with iconography. Translations into French, German, Italian, and Spanish are welcome.
Subscription: The Atlas is FREE!
Corporate patronage, sponsorship and advertising Enquiries should be addressed to [email protected].
Rules, Copyright Notice and Disclaimer Conflicts of Interest: Authors must state explicitly whether potential conflicts do or do not exist. Reviewers must disclose to editors any conflicts of interest that could bias their opinions of the manuscript. The editor and the editorial board members must disclose any potential conflict. Privacy and Confidentiality – Iconography: Patients have a right to privacy. Identifying details should be omitted. If complete anonymity is difficult to achieve, informed consent should be obtained. Property: As "cards" are to evolve with further improvements and updates from various contributors, the property of the cards belongs to the editor, and modifications will be made without authorization from the previous contributor (who may, nonetheless, be asked for refereeing); contributors are listed in an edit history manner. Authors keep the rights to use further the content of their papers published in the Atlas, provided that the source is cited. Copyright: The information in the Atlas of Genetics and Cytogenetics in Oncology and Haematology is issued for general distribution. All rights are reserved. The information presented is protected under international conventions and under national laws on copyright and neighbouring rights. Commercial use is totally forbidden. Information extracted from the Atlas may be reviewed, reproduced or translated for research or private study but not for sale or for use in conjunction with commercial purposes. Any use of information from the Atlas should be accompanied by an acknowledgment of the Atlas as the source, citing the uniform resource locator (URL) of the article and/or the article reference, according to the Vancouver convention. Reference to any specific commercial products, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favouring. The views and opinions of contributors and authors expressed herein do not necessarily state or reflect those of the Atlas editorial staff or of the web site holder, and shall not be used for advertising or product endorsement purposes. The Atlas does not make any warranty, express or implied, including the warranties of merchantability and fitness for a particular purpose, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, and shall not be liable whatsoever for any damages incurred as a result of its use. In particular, information presented in the Atlas is only for research purpose, and shall not be used for diagnosis or treatment purposes. No responsibility is assumed for any injury and/or damage to persons or property for any use or operation of any methods products, instructions or ideas contained in the material herein. See also: "Uniform Requirements for Manuscripts Submitted to Biomedical Journals: Writing and Editing for Biomedical Publication - Updated October 2004": http://www.icmje.org.
http://AtlasGeneticsOncology.org
© ATLAS - ISSN 1768-3262










![Hegel Vol. 10 N 3 DOI :10.4267/2042/70734 - Revues et Congrèsdocuments.irevues.inist.fr/bitstream/handle/2042/... · Pronk, et al. (2005) [28] sur les thérapies alternatives montre](https://img.pdfslide.us/doc/110x75/60fc7b53e2f7471be1365a1a/hegel-vol-10-n-3-doi-104267204270734-revues-et-congr-pronk-et-al-2005.jpg)



















