Embed Size (px)
Citation preview

10/1/2014
1
Hemolytic Anemia: Evolving Pathogenesis and new Treatment Paradigms
Matt GrahamUT‐Erlanger Oncology and Hematology
October 2, 2014
Disclosures
• I have no conflicts of interest or financial disclosures for this presentation
• Off‐label rituximab usage described in this presentation
Hemolysis
• Shortened RBC Survival– RBC survival is less than 120 days– RBCs die daily and there is age independent random hemolysis on the order of 0.05 to 0.5 % per day
• Compensatory increase in EPO production– Increased RBC production– Increased reticulocyte % and absolute reticulocyte count
Typical Clinical Manifestations
• Dyspnea• Fatigue• Hyperdynamic state• Palpitations• Lethargy • Confusion• Pallor• Jaundice• Splenomegaly
Laboratory Findings
• Spectrum of Anemia• Reticulocytosis• Increased MCV• Low haptoglobin• Elevated LDH• Unconjugated hyperbilirubinemia• + DAT
AE Lichtin. The Merck Manual. 2013

10/1/2014
2
DAT Pearls
• Weakly + test in 1 in 10K healthy donors• + in 5‐10% of hospitalized patients without any evidence of hemolysis; complement‐mediated
• Low titers of autoantibody may appear negative – Cannot detect fewer than 100‐500 Ab molecules– Cannot detect fewer than 400‐1100 C’
• Commonly used reagents, Erlanger’s included, cannot identify IgA or IgM autoantibodies
AE Lichtin. The Merck Manual. 2013/Up To Date AE Lichtin. The Merck Manual. 2013
Classification of Hemolytic Anemias
• Hereditary Hemolytic Disorders– RBC enzyme defects– RBC membrane defects– Hemoglobinopathies– Thalassemias
• Acquired Hemolytic Disorders– Immune‐mediated hemolytic anemias– Microangiopathic hemolytic anemias (MAHAs)– Paroxysmal nocturnal hemoglobinuria (PNH)– Spur cell anemia– Direct toxins
Immune‐mediated Hemolytic Anemias
• Paroxysmal Cold Hemoglobinuria• Warm Autoantibody Hemolytic Anemia• Cold Agglutinin Disease
Paroxysmal Cold Hemoglobinuria (PCH)
• One of the 1st hematologic syndromes recognized clinically
• Red to brown urine after cold exposure described in 1872
• Donath and Landsteiner’s work identified the causative antibody in 1904
MA Lichtman. In: Hematology. 2000
PCH
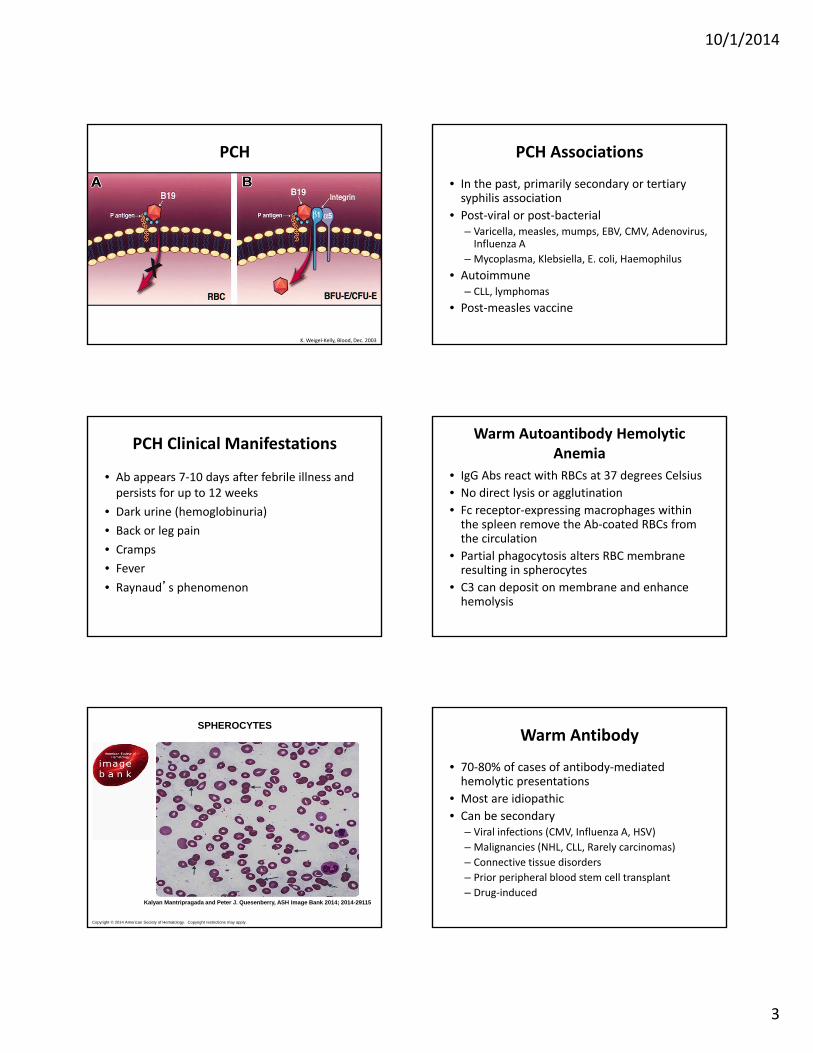
• A polyclonal, cold‐reacting IgG• Fixes complement directly• Thermal amplitude varies greatly• Blood in the periphery cools and Ab and 1st 2 components of complement are fixed to the RBC surface
• Lysis occurs when the RBC are subsequently warmed (complement cascade completes)
10/1/2014
3
PCH
K. Weigel‐Kelly, Blood, Dec. 2003
PCH Associations
• In the past, primarily secondary or tertiary syphilis association
• Post‐viral or post‐bacterial– Varicella, measles, mumps, EBV, CMV, Adenovirus, Influenza A
– Mycoplasma, Klebsiella, E. coli, Haemophilus• Autoimmune
– CLL, lymphomas• Post‐measles vaccine
PCH Clinical Manifestations
• Ab appears 7‐10 days after febrile illness and persists for up to 12 weeks
• Dark urine (hemoglobinuria)• Back or leg pain• Cramps• Fever• Raynaud’s phenomenon
Warm Autoantibody Hemolytic Anemia
• IgG Abs react with RBCs at 37 degrees Celsius• No direct lysis or agglutination • Fc receptor‐expressing macrophages within the spleen remove the Ab‐coated RBCs from the circulation
• Partial phagocytosis alters RBC membrane resulting in spherocytes
• C3 can deposit on membrane and enhance hemolysis
Copyright © 2014 American Society of Hematology. Copyright restrictions may apply.
Kalyan Mantripragada and Peter J. Quesenberry, ASH Image Bank 2014; 2014-29115
SPHEROCYTESWarm Antibody
• 70‐80% of cases of antibody‐mediated hemolytic presentations
• Most are idiopathic• Can be secondary
– Viral infections (CMV, Influenza A, HSV)– Malignancies (NHL, CLL, Rarely carcinomas)– Connective tissue disorders – Prior peripheral blood stem cell transplant– Drug‐induced

10/1/2014
4
Drug‐Induced
• Alpha‐methyldopa• Fludarabine• Procainamide• Diclofenac• Quinidine, Quinine• Sulfa drugs
• Isoniazid• Amphotericin• Penicillins• Cephalosporins• Tetracycline• Ribavirin
AE Lichtin. The Merck Manual. 2013/Up To Date
Warm‐Reactive Treatment
• Treat any known underlying condition• Stop any implicated drug• Folic Acid 5 mg daily• Prednisone 1 mg/kg/day• Splenectomy• Cytotoxic Drugs
– Cyclophosphamide• Immunosuppresants
– Cyclosporine, mycophenolate mofetil, azathioprine• IVIG
Rituximab
• Chimeric, monoclonal anti‐CD20 antibody• Multiple case studies and retrospective reports
• Off label usage
www.rituxan.comby Genentech
• 64 patients with newly‐dx WAIHA• Prednisolone + rituximab vs prednisolone alone
• After 12 months, 75% on P‐R vs 36% P alone with satisfactory response
• After 36 months, 70% vs 45% remission• No significant difference in adverse events
Transfusion Concerns
• Pan‐agglutination with usual cross‐matching with all normal donor cells
• Allo‐reactive Abs may be present in up to 1/3 of patients with AIHA
• Undetected ALLO‐ Abs could cause massive hemolysis after transfusion

10/1/2014
5
L. Dean, Blood Groups and Red Cell Antigens, 2005.
Transfusion Concerns
• If patient never pregnant or no history of transfusion, low probability of an alloantibody
• Adequate testing for alloantibodies may take up to 6 hours– Testing diluted serum against RBC panel taking advantage of differences in reaction strength
– Autoadsorption technique• Remove the autoantibody and use these ‘clean’ cells to keep removing autoantibody, leaving allo‐Ab behind for ID
Petz LD. British Journal of Haematology, 124, 712‐716
Transfusion Concerns
• Severe anemia may preclude obtaining a large enough volume of RBCs for autoadsorptionprocedure and not useful in those transfused within 3 months– Allogeneic adsorption may be needed
• Use several samples of allogeneic red cells of varying phenotypes responsible for clinically important hemolytic reactions to remove the autoAb
Petz LD. British Journal of Haematology, 124, 712‐716
Transfusion Concerns
• The clinician must balance risk and benefit of transfusion
• No patient should die from lack of transfusion, give most compatible blood
• Transfuse the minimal amount of blood required to alleviate symptoms and maintain a tolerable hgb/hct
• Transfuse slowly and with close monitoring
Cold‐Reactive/Cold Agglutinin Dz
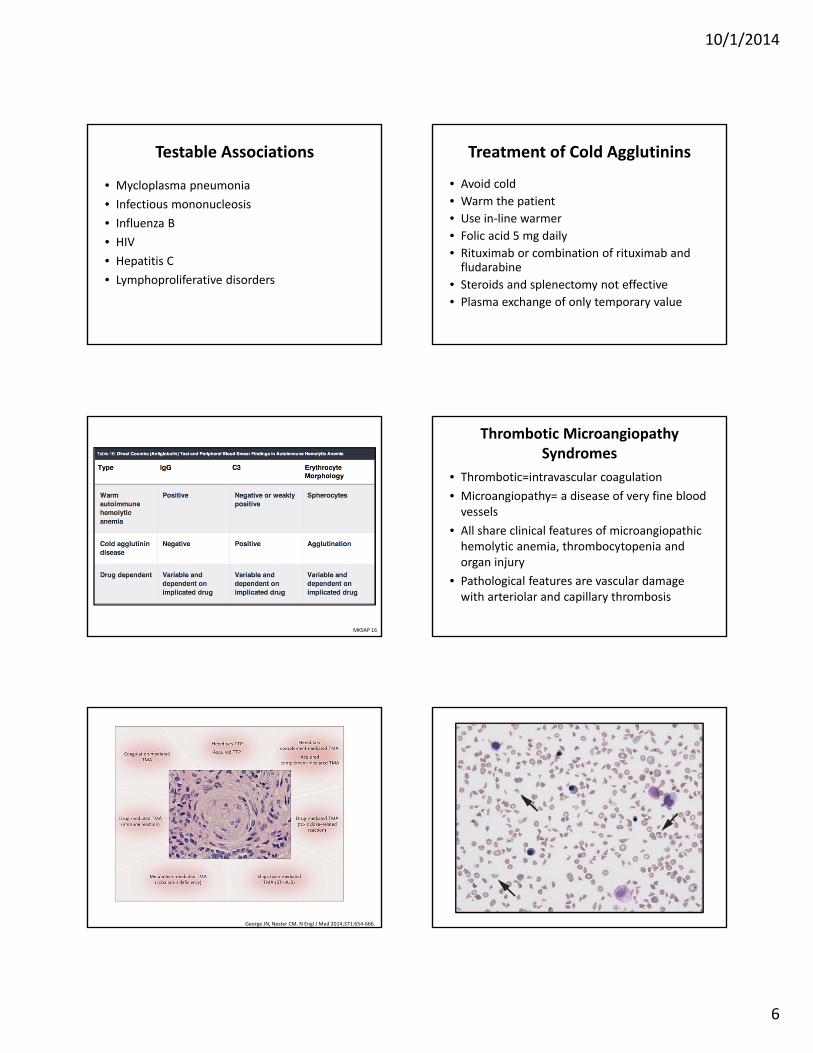
• IgM complement‐fixing antibodies• Anti‐I and anti‐i most common• Ab binds to RBCs and causes agglutination at low temperatures (4 degrees Celsius)
• Intravascular hemolysis• DAT positive for complement only since IgM is released at 37 degrees Celsius
Copyright © 2011 American Society of Hematology. Copyright restrictions may apply.
John Lazarchick, ASH Image Bank 2011; 2011-1053
COLD AGGLUTININ DISEASE
10/1/2014
6
Testable Associations
• Mycloplasma pneumonia• Infectious mononucleosis• Influenza B• HIV• Hepatitis C• Lymphoproliferative disorders
Treatment of Cold Agglutinins
• Avoid cold• Warm the patient• Use in‐line warmer• Folic acid 5 mg daily• Rituximab or combination of rituximab and fludarabine
• Steroids and splenectomy not effective• Plasma exchange of only temporary value
MKSAP 16
Thrombotic MicroangiopathySyndromes
• Thrombotic=intravascular coagulation• Microangiopathy= a disease of very fine blood vessels
• All share clinical features of microangiopathichemolytic anemia, thrombocytopenia and organ injury
• Pathological features are vascular damage with arteriolar and capillary thrombosis
Pathological Features of the Nine Primary Thrombotic Microangiopathy (TMA) Syndromes.
George JN, Nester CM. N Engl J Med 2014;371:654‐666.

10/1/2014
7
Thrombotic Thrombocytopenic Purpura (TTP)
• Definition: Coombs’ negative microangiopathic hemolytic anemia and thrombocytopenia in the absence of an alternative explanation
• Minimum criteria: Thrombocytopenia
Rock. Br J Haematol 2000 Theodore Warkentin GWBR 2011
Theodore Warkentin GWBR 2011 Theodore Warkentin GWBR 2011
Theodore Warkentin GWBR 2011Theodore Warkentin GWBR 2011

10/1/2014
8
Complement‐Mediated TMA (CM‐TMA)
• 1st recognized as a familial disorder in 1975• Two brothers with TMA were found to have a deficiency of complement factor H
• Associated mutations in the gene encoding complement factor H (CFH) characterized 1998
• Loss‐of‐function mutations in regulatory genes or gain‐of‐function effector gene mutations
• Functional deficiencies in complement factors
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Diagnostic Criteria
• Serum creatinine above the normal range• MAHA• Thrombocytopenia• ADAMTS13 activity of 5% or more• Negative stool tests for Shiga toxin‐producing infection
CM‐TMA Diagnosis
• Plasma levels of C3, C4, CF‐H, ‐B and ‐I are not helpful
• Need access to cost effective, rapid and commercially available genetic studies
• Noris et al. published an in vitro assay to measure serum‐induced endothelial C5b‐9 deposition to monitor complement activation and treatment effectiveness
Noris et al. Blood. 2014;124(11):1715‐1726.
Eculizumab is now considered the standard in CM‐TMA. This is a monoclonal antibody that is a complement protein C5 inhibitor that can terminate complement‐mediated hemolysis and thrombotic microangiopathy.
Theodore Warkentin GWBR 2011 Neal Young GWBR 2010
10/1/2014
9
Highlights
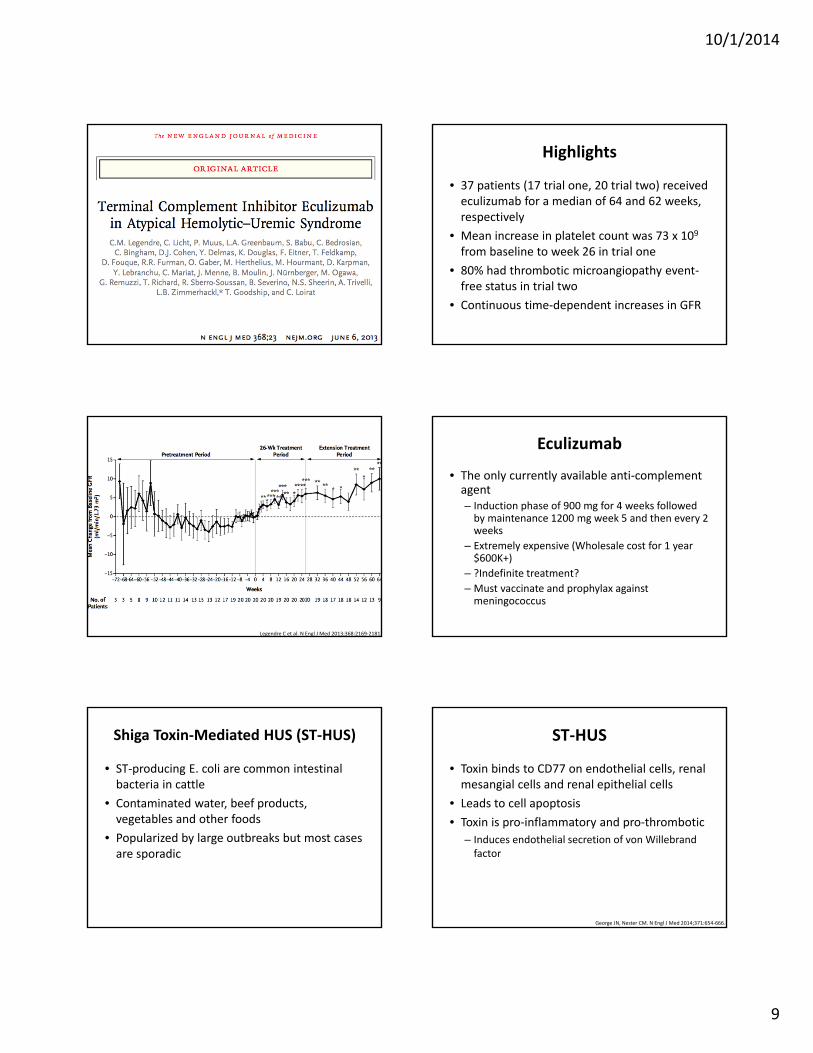
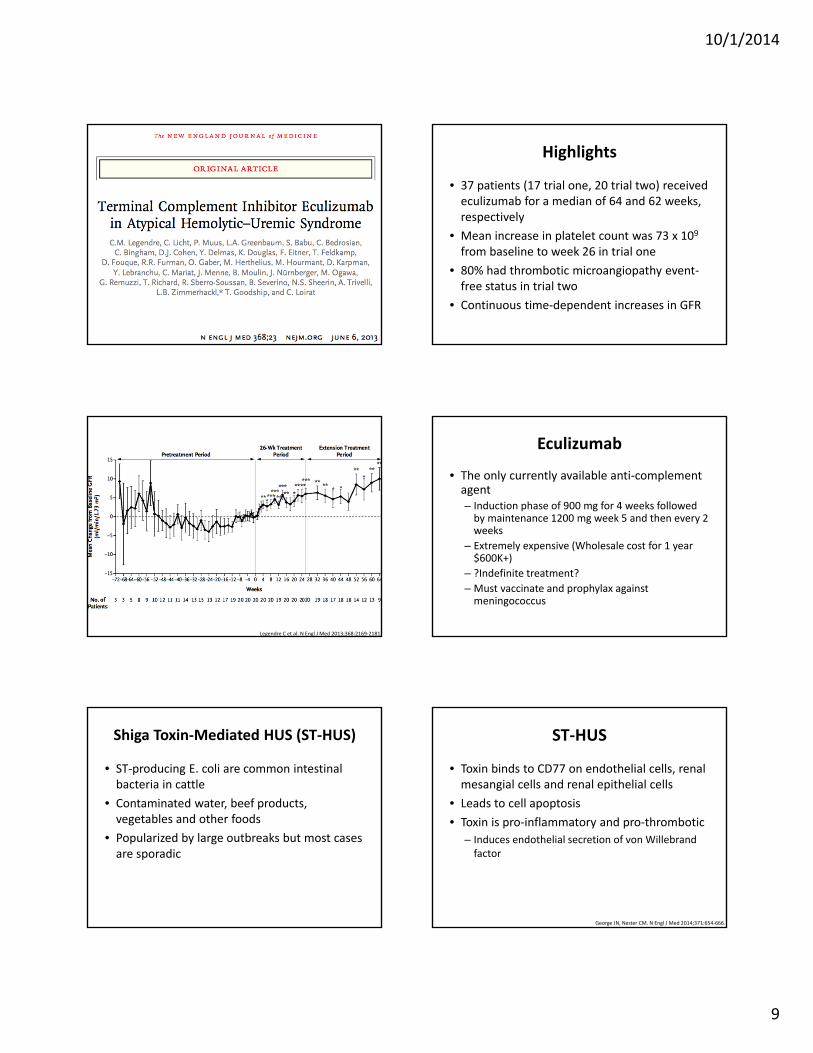
• 37 patients (17 trial one, 20 trial two) received eculizumab for a median of 64 and 62 weeks, respectively
• Mean increase in platelet count was 73 x 109 from baseline to week 26 in trial one
• 80% had thrombotic microangiopathy event‐free status in trial two
• Continuous time‐dependent increases in GFR
End Points.
Legendre C et al. N Engl J Med 2013;368:2169‐2181.
Eculizumab
• The only currently available anti‐complement agent– Induction phase of 900 mg for 4 weeks followed by maintenance 1200 mg week 5 and then every 2 weeks
– Extremely expensive (Wholesale cost for 1 year $600K+)
– ?Indefinite treatment?– Must vaccinate and prophylax against meningococcus
Shiga Toxin‐Mediated HUS (ST‐HUS)
• ST‐producing E. coli are common intestinal bacteria in cattle
• Contaminated water, beef products, vegetables and other foods
• Popularized by large outbreaks but most cases are sporadic
ST‐HUS
• Toxin binds to CD77 on endothelial cells, renal mesangial cells and renal epithelial cells
• Leads to cell apoptosis• Toxin is pro‐inflammatory and pro‐thrombotic
– Induces endothelial secretion of von Willebrandfactor
George JN, Nester CM. N Engl J Med 2014;371:654‐666.

10/1/2014
10
ST‐HUS Presentation, Diagnosis & Treatment
• Severe abdominal pain and bloody diarrhea begin within several days of consumption
• Renal failure and thrombocytopenia begin as GI symptoms resolve
• Stool analyses diagnostic during acute colitis phase
• Supportive treatment– Aggressive hydration– Plasma exchange and anti‐complement therapy inconclusive
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Drug‐induced MAHA
• Ticlopidine, clopidogrel• Quinine • Mitomycin‐C, cisplatin and bleomycin• Cyclosporine, tacrolimus and sirolimus• Penicillamine, penicillins
Metabolism‐Mediated TMA
• Occurs in infants, only reported in one adult• Mutations in gene encoding the methylmalonic aciduria and homocystinuriatype C protein (MMACHC)
• Hyperhomocysteinemia, low methionine and methlymalonic aciduria with normal folateand B12 levels
• Parenteral hydroxycobalamin treatment of choice
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Common Disorders Associated with Microangiopathic Hemolytic Anemia and Thrombocytopenia.
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Neal Young GWBR 2010 Neal Young GWBR 2010
10/1/2014
11
TREATMENT OF PNH
• Anticoagulation in the face of thrombosis– Prophylaxis may be considered if more than 50% of cells are CD55‐ or CD59‐deficient.
• If marked bone marrow failure, treatment consists of immunosuppressive therapy or allogeneic bone marrow transplantation– Transplant preferred with young patient with a donor with bone marrow failure
• Eculizumab– Decreases transfusions and thromboses and improves quality of life but does not reverse bone marrow failure
Spur Cell Hemolytic Anemia
• Secondary to severe impaired liver function or cirrhosis
• Unable to esterify cholesterol causing free cholesterol to bind to the red cell membrane, increasing its surface area
Allen DW Blood 1994
January 1, 2003; Blood: 101 (1)
Direct Toxins
• Arsenic hydride• Lead• Copper• Chlorates• Insect, Spider and Snake Venoms• Heat• Radiation
Williams Hematology. 2001. Chapter 53.
Thank you for your time and attention! References
The Merck Manual 2013Up To DateLichtman MA, Spivak JL, eds. Ueber paroxysmale Haemoglo‐binurie [Concerning paroxysmal hemoglobinuria]. In: Hematology: Landmark Papers of the Twentieth Century. San Diego, Calif: Academic Press; 2000:21.Weigel‐Kelley KA et al. α5β1 integrin as a cellular coreceptor for human parvovirus B19: requirement of functional activation of β1 integrin for viral entry. Blood. December 2003, Vol. 102:12.The American Society of Hematology Image BankBirgens H et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013 Nov;163(3):393‐9. Dean L. Blood Groups and Red Cell Antigens [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2005. Available from: http://www.ncbi.nlm.nih.gov/books/NBK2261/Petz LD. Br J Haematol. 2004 Mar;124(6):712‐6. Review.Medical Knowledge Self‐Assessment Program 16George JN et al. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014 Aug 14;371(7):654‐66. Rock GA. Management of thrombotic thrombocytopenic purpura. Br J Haematol. 2000 Jun;109(3):496‐507.The George Washington Board Review Series 2010 and 2011.Noris M et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. September 2014, Vol. 124:11.Legendre CM et al. Terminal complement inhibitor eculizumab in atypical hemolytic‐uremic syndrome. N Engl J Med. 2013; 368:2169‐2181Allen DW et al. Abnormal phospholipid metabolism in spur cell anemia: decreased fatty acid incorporation intophosphatidylethanolamine and increased incorporation into acylcarnitine in spur cell anemia erythrocytes. Blood 1994;84:1283‐1287. Beutler E. In: Williams Hematology, Chapter 53. 2001.

10/1/2014
1
Hemolytic Anemia: Evolving Pathogenesis and new Treatment Paradigms
Matt GrahamUT‐Erlanger Oncology and Hematology
October 2, 2014
Disclosures
• I have no conflicts of interest or financial disclosures for this presentation
• Off‐label rituximab usage described in this presentation
Hemolysis
• Shortened RBC Survival– RBC survival is less than 120 days– RBCs die daily and there is age independent random hemolysis on the order of 0.05 to 0.5 % per day
• Compensatory increase in EPO production– Increased RBC production– Increased reticulocyte % and absolute reticulocyte count
Typical Clinical Manifestations
• Dyspnea• Fatigue• Hyperdynamic state• Palpitations• Lethargy • Confusion• Pallor• Jaundice• Splenomegaly
Laboratory Findings
• Spectrum of Anemia• Reticulocytosis• Increased MCV• Low haptoglobin• Elevated LDH• Unconjugated hyperbilirubinemia• + DAT
AE Lichtin. The Merck Manual. 2013

10/1/2014
2
DAT Pearls
• Weakly + test in 1 in 10K healthy donors• + in 5‐10% of hospitalized patients without any evidence of hemolysis; complement‐mediated
• Low titers of autoantibody may appear negative – Cannot detect fewer than 100‐500 Ab molecules– Cannot detect fewer than 400‐1100 C’
• Commonly used reagents, Erlanger’s included, cannot identify IgA or IgM autoantibodies
AE Lichtin. The Merck Manual. 2013/Up To Date AE Lichtin. The Merck Manual. 2013
Classification of Hemolytic Anemias
• Hereditary Hemolytic Disorders– RBC enzyme defects– RBC membrane defects– Hemoglobinopathies– Thalassemias
• Acquired Hemolytic Disorders– Immune‐mediated hemolytic anemias– Microangiopathic hemolytic anemias (MAHAs)– Paroxysmal nocturnal hemoglobinuria (PNH)– Spur cell anemia– Direct toxins
Immune‐mediated Hemolytic Anemias
• Paroxysmal Cold Hemoglobinuria• Warm Autoantibody Hemolytic Anemia• Cold Agglutinin Disease
Paroxysmal Cold Hemoglobinuria (PCH)
• One of the 1st hematologic syndromes recognized clinically
• Red to brown urine after cold exposure described in 1872
• Donath and Landsteiner’s work identified the causative antibody in 1904
MA Lichtman. In: Hematology. 2000
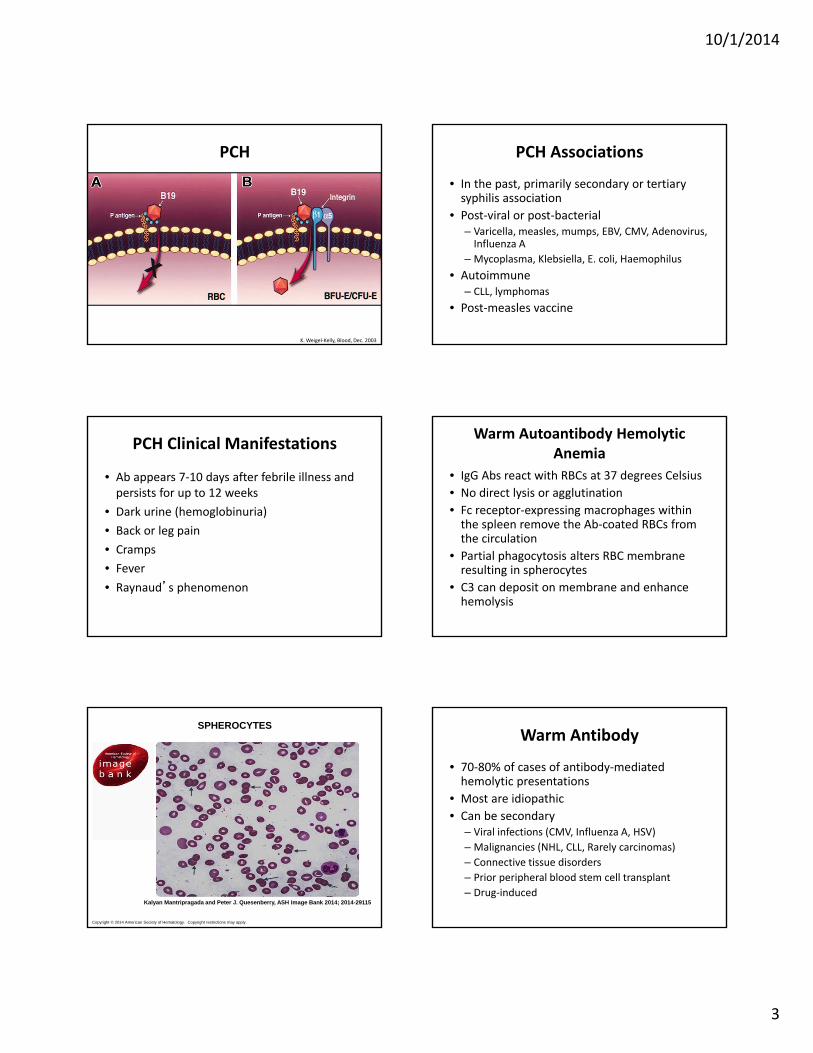
PCH
• A polyclonal, cold‐reacting IgG• Fixes complement directly• Thermal amplitude varies greatly• Blood in the periphery cools and Ab and 1st 2 components of complement are fixed to the RBC surface
• Lysis occurs when the RBC are subsequently warmed (complement cascade completes)
10/1/2014
3
PCH
K. Weigel‐Kelly, Blood, Dec. 2003
PCH Associations
• In the past, primarily secondary or tertiary syphilis association
• Post‐viral or post‐bacterial– Varicella, measles, mumps, EBV, CMV, Adenovirus, Influenza A
– Mycoplasma, Klebsiella, E. coli, Haemophilus• Autoimmune
– CLL, lymphomas• Post‐measles vaccine
PCH Clinical Manifestations
• Ab appears 7‐10 days after febrile illness and persists for up to 12 weeks
• Dark urine (hemoglobinuria)• Back or leg pain• Cramps• Fever• Raynaud’s phenomenon
Warm Autoantibody Hemolytic Anemia
• IgG Abs react with RBCs at 37 degrees Celsius• No direct lysis or agglutination • Fc receptor‐expressing macrophages within the spleen remove the Ab‐coated RBCs from the circulation
• Partial phagocytosis alters RBC membrane resulting in spherocytes
• C3 can deposit on membrane and enhance hemolysis
Copyright © 2014 American Society of Hematology. Copyright restrictions may apply.
Kalyan Mantripragada and Peter J. Quesenberry, ASH Image Bank 2014; 2014-29115
SPHEROCYTESWarm Antibody
• 70‐80% of cases of antibody‐mediated hemolytic presentations
• Most are idiopathic• Can be secondary
– Viral infections (CMV, Influenza A, HSV)– Malignancies (NHL, CLL, Rarely carcinomas)– Connective tissue disorders – Prior peripheral blood stem cell transplant– Drug‐induced

10/1/2014
4
Drug‐Induced
• Alpha‐methyldopa• Fludarabine• Procainamide• Diclofenac• Quinidine, Quinine• Sulfa drugs
• Isoniazid• Amphotericin• Penicillins• Cephalosporins• Tetracycline• Ribavirin
AE Lichtin. The Merck Manual. 2013/Up To Date
Warm‐Reactive Treatment
• Treat any known underlying condition• Stop any implicated drug• Folic Acid 5 mg daily• Prednisone 1 mg/kg/day• Splenectomy• Cytotoxic Drugs
– Cyclophosphamide• Immunosuppresants
– Cyclosporine, mycophenolate mofetil, azathioprine• IVIG
Rituximab
• Chimeric, monoclonal anti‐CD20 antibody• Multiple case studies and retrospective reports
• Off label usage
www.rituxan.comby Genentech
• 64 patients with newly‐dx WAIHA• Prednisolone + rituximab vs prednisolone alone
• After 12 months, 75% on P‐R vs 36% P alone with satisfactory response
• After 36 months, 70% vs 45% remission• No significant difference in adverse events
Transfusion Concerns
• Pan‐agglutination with usual cross‐matching with all normal donor cells
• Allo‐reactive Abs may be present in up to 1/3 of patients with AIHA
• Undetected ALLO‐ Abs could cause massive hemolysis after transfusion

10/1/2014
5
L. Dean, Blood Groups and Red Cell Antigens, 2005.
Transfusion Concerns
• If patient never pregnant or no history of transfusion, low probability of an alloantibody
• Adequate testing for alloantibodies may take up to 6 hours– Testing diluted serum against RBC panel taking advantage of differences in reaction strength
– Autoadsorption technique• Remove the autoantibody and use these ‘clean’ cells to keep removing autoantibody, leaving allo‐Ab behind for ID
Petz LD. British Journal of Haematology, 124, 712‐716
Transfusion Concerns
• Severe anemia may preclude obtaining a large enough volume of RBCs for autoadsorptionprocedure and not useful in those transfused within 3 months– Allogeneic adsorption may be needed
• Use several samples of allogeneic red cells of varying phenotypes responsible for clinically important hemolytic reactions to remove the autoAb
Petz LD. British Journal of Haematology, 124, 712‐716
Transfusion Concerns
• The clinician must balance risk and benefit of transfusion
• No patient should die from lack of transfusion, give most compatible blood
• Transfuse the minimal amount of blood required to alleviate symptoms and maintain a tolerable hgb/hct
• Transfuse slowly and with close monitoring
Cold‐Reactive/Cold Agglutinin Dz
• IgM complement‐fixing antibodies• Anti‐I and anti‐i most common• Ab binds to RBCs and causes agglutination at low temperatures (4 degrees Celsius)
• Intravascular hemolysis• DAT positive for complement only since IgM is released at 37 degrees Celsius
Copyright © 2011 American Society of Hematology. Copyright restrictions may apply.
John Lazarchick, ASH Image Bank 2011; 2011-1053
COLD AGGLUTININ DISEASE
10/1/2014
6
Testable Associations
• Mycloplasma pneumonia• Infectious mononucleosis• Influenza B• HIV• Hepatitis C• Lymphoproliferative disorders
Treatment of Cold Agglutinins
• Avoid cold• Warm the patient• Use in‐line warmer• Folic acid 5 mg daily• Rituximab or combination of rituximab and fludarabine
• Steroids and splenectomy not effective• Plasma exchange of only temporary value
MKSAP 16
Thrombotic MicroangiopathySyndromes
• Thrombotic=intravascular coagulation• Microangiopathy= a disease of very fine blood vessels
• All share clinical features of microangiopathichemolytic anemia, thrombocytopenia and organ injury
• Pathological features are vascular damage with arteriolar and capillary thrombosis
Pathological Features of the Nine Primary Thrombotic Microangiopathy (TMA) Syndromes.
George JN, Nester CM. N Engl J Med 2014;371:654‐666.

10/1/2014
7
Thrombotic Thrombocytopenic Purpura (TTP)
• Definition: Coombs’ negative microangiopathic hemolytic anemia and thrombocytopenia in the absence of an alternative explanation
• Minimum criteria: Thrombocytopenia
Rock. Br J Haematol 2000 Theodore Warkentin GWBR 2011
Theodore Warkentin GWBR 2011 Theodore Warkentin GWBR 2011
Theodore Warkentin GWBR 2011Theodore Warkentin GWBR 2011

10/1/2014
8
Complement‐Mediated TMA (CM‐TMA)
• 1st recognized as a familial disorder in 1975• Two brothers with TMA were found to have a deficiency of complement factor H
• Associated mutations in the gene encoding complement factor H (CFH) characterized 1998
• Loss‐of‐function mutations in regulatory genes or gain‐of‐function effector gene mutations
• Functional deficiencies in complement factors
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Diagnostic Criteria
• Serum creatinine above the normal range• MAHA• Thrombocytopenia• ADAMTS13 activity of 5% or more• Negative stool tests for Shiga toxin‐producing infection
CM‐TMA Diagnosis
• Plasma levels of C3, C4, CF‐H, ‐B and ‐I are not helpful
• Need access to cost effective, rapid and commercially available genetic studies
• Noris et al. published an in vitro assay to measure serum‐induced endothelial C5b‐9 deposition to monitor complement activation and treatment effectiveness
Noris et al. Blood. 2014;124(11):1715‐1726.
Eculizumab is now considered the standard in CM‐TMA. This is a monoclonal antibody that is a complement protein C5 inhibitor that can terminate complement‐mediated hemolysis and thrombotic microangiopathy.
Theodore Warkentin GWBR 2011 Neal Young GWBR 2010
10/1/2014
9
Highlights
• 37 patients (17 trial one, 20 trial two) received eculizumab for a median of 64 and 62 weeks, respectively
• Mean increase in platelet count was 73 x 109 from baseline to week 26 in trial one
• 80% had thrombotic microangiopathy event‐free status in trial two
• Continuous time‐dependent increases in GFR
End Points.
Legendre C et al. N Engl J Med 2013;368:2169‐2181.
Eculizumab
• The only currently available anti‐complement agent– Induction phase of 900 mg for 4 weeks followed by maintenance 1200 mg week 5 and then every 2 weeks
– Extremely expensive (Wholesale cost for 1 year $600K+)
– ?Indefinite treatment?– Must vaccinate and prophylax against meningococcus
Shiga Toxin‐Mediated HUS (ST‐HUS)
• ST‐producing E. coli are common intestinal bacteria in cattle
• Contaminated water, beef products, vegetables and other foods
• Popularized by large outbreaks but most cases are sporadic
ST‐HUS
• Toxin binds to CD77 on endothelial cells, renal mesangial cells and renal epithelial cells
• Leads to cell apoptosis• Toxin is pro‐inflammatory and pro‐thrombotic
– Induces endothelial secretion of von Willebrandfactor
George JN, Nester CM. N Engl J Med 2014;371:654‐666.

10/1/2014
10
ST‐HUS Presentation, Diagnosis & Treatment
• Severe abdominal pain and bloody diarrhea begin within several days of consumption
• Renal failure and thrombocytopenia begin as GI symptoms resolve
• Stool analyses diagnostic during acute colitis phase
• Supportive treatment– Aggressive hydration– Plasma exchange and anti‐complement therapy inconclusive
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Drug‐induced MAHA
• Ticlopidine, clopidogrel• Quinine • Mitomycin‐C, cisplatin and bleomycin• Cyclosporine, tacrolimus and sirolimus• Penicillamine, penicillins
Metabolism‐Mediated TMA
• Occurs in infants, only reported in one adult• Mutations in gene encoding the methylmalonic aciduria and homocystinuriatype C protein (MMACHC)
• Hyperhomocysteinemia, low methionine and methlymalonic aciduria with normal folateand B12 levels
• Parenteral hydroxycobalamin treatment of choice
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Common Disorders Associated with Microangiopathic Hemolytic Anemia and Thrombocytopenia.
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Neal Young GWBR 2010 Neal Young GWBR 2010
10/1/2014
11
TREATMENT OF PNH
• Anticoagulation in the face of thrombosis– Prophylaxis may be considered if more than 50% of cells are CD55‐ or CD59‐deficient.
• If marked bone marrow failure, treatment consists of immunosuppressive therapy or allogeneic bone marrow transplantation– Transplant preferred with young patient with a donor with bone marrow failure
• Eculizumab– Decreases transfusions and thromboses and improves quality of life but does not reverse bone marrow failure
Spur Cell Hemolytic Anemia
• Secondary to severe impaired liver function or cirrhosis
• Unable to esterify cholesterol causing free cholesterol to bind to the red cell membrane, increasing its surface area
Allen DW Blood 1994
January 1, 2003; Blood: 101 (1)
Direct Toxins
• Arsenic hydride• Lead• Copper• Chlorates• Insect, Spider and Snake Venoms• Heat• Radiation
Williams Hematology. 2001. Chapter 53.
Thank you for your time and attention! References
The Merck Manual 2013Up To DateLichtman MA, Spivak JL, eds. Ueber paroxysmale Haemoglo‐binurie [Concerning paroxysmal hemoglobinuria]. In: Hematology: Landmark Papers of the Twentieth Century. San Diego, Calif: Academic Press; 2000:21.Weigel‐Kelley KA et al. α5β1 integrin as a cellular coreceptor for human parvovirus B19: requirement of functional activation of β1 integrin for viral entry. Blood. December 2003, Vol. 102:12.The American Society of Hematology Image BankBirgens H et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013 Nov;163(3):393‐9. Dean L. Blood Groups and Red Cell Antigens [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2005. Available from: http://www.ncbi.nlm.nih.gov/books/NBK2261/Petz LD. Br J Haematol. 2004 Mar;124(6):712‐6. Review.Medical Knowledge Self‐Assessment Program 16George JN et al. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014 Aug 14;371(7):654‐66. Rock GA. Management of thrombotic thrombocytopenic purpura. Br J Haematol. 2000 Jun;109(3):496‐507.The George Washington Board Review Series 2010 and 2011.Noris M et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. September 2014, Vol. 124:11.Legendre CM et al. Terminal complement inhibitor eculizumab in atypical hemolytic‐uremic syndrome. N Engl J Med. 2013; 368:2169‐2181Allen DW et al. Abnormal phospholipid metabolism in spur cell anemia: decreased fatty acid incorporation intophosphatidylethanolamine and increased incorporation into acylcarnitine in spur cell anemia erythrocytes. Blood 1994;84:1283‐1287. Beutler E. In: Williams Hematology, Chapter 53. 2001.

10/1/2014
1
Hemolytic Anemia: Evolving Pathogenesis and new Treatment Paradigms
Matt GrahamUT‐Erlanger Oncology and Hematology
October 2, 2014
Disclosures
• I have no conflicts of interest or financial disclosures for this presentation
• Off‐label rituximab usage described in this presentation
Hemolysis
• Shortened RBC Survival– RBC survival is less than 120 days– RBCs die daily and there is age independent random hemolysis on the order of 0.05 to 0.5 % per day
• Compensatory increase in EPO production– Increased RBC production– Increased reticulocyte % and absolute reticulocyte count
Typical Clinical Manifestations
• Dyspnea• Fatigue• Hyperdynamic state• Palpitations• Lethargy • Confusion• Pallor• Jaundice• Splenomegaly
Laboratory Findings
• Spectrum of Anemia• Reticulocytosis• Increased MCV• Low haptoglobin• Elevated LDH• Unconjugated hyperbilirubinemia• + DAT
AE Lichtin. The Merck Manual. 2013

10/1/2014
2
DAT Pearls
• Weakly + test in 1 in 10K healthy donors• + in 5‐10% of hospitalized patients without any evidence of hemolysis; complement‐mediated
• Low titers of autoantibody may appear negative – Cannot detect fewer than 100‐500 Ab molecules– Cannot detect fewer than 400‐1100 C’
• Commonly used reagents, Erlanger’s included, cannot identify IgA or IgM autoantibodies
AE Lichtin. The Merck Manual. 2013/Up To Date AE Lichtin. The Merck Manual. 2013
Classification of Hemolytic Anemias
• Hereditary Hemolytic Disorders– RBC enzyme defects– RBC membrane defects– Hemoglobinopathies– Thalassemias
• Acquired Hemolytic Disorders– Immune‐mediated hemolytic anemias– Microangiopathic hemolytic anemias (MAHAs)– Paroxysmal nocturnal hemoglobinuria (PNH)– Spur cell anemia– Direct toxins
Immune‐mediated Hemolytic Anemias
• Paroxysmal Cold Hemoglobinuria• Warm Autoantibody Hemolytic Anemia• Cold Agglutinin Disease
Paroxysmal Cold Hemoglobinuria (PCH)
• One of the 1st hematologic syndromes recognized clinically
• Red to brown urine after cold exposure described in 1872
• Donath and Landsteiner’s work identified the causative antibody in 1904
MA Lichtman. In: Hematology. 2000
PCH
• A polyclonal, cold‐reacting IgG• Fixes complement directly• Thermal amplitude varies greatly• Blood in the periphery cools and Ab and 1st 2 components of complement are fixed to the RBC surface
• Lysis occurs when the RBC are subsequently warmed (complement cascade completes)
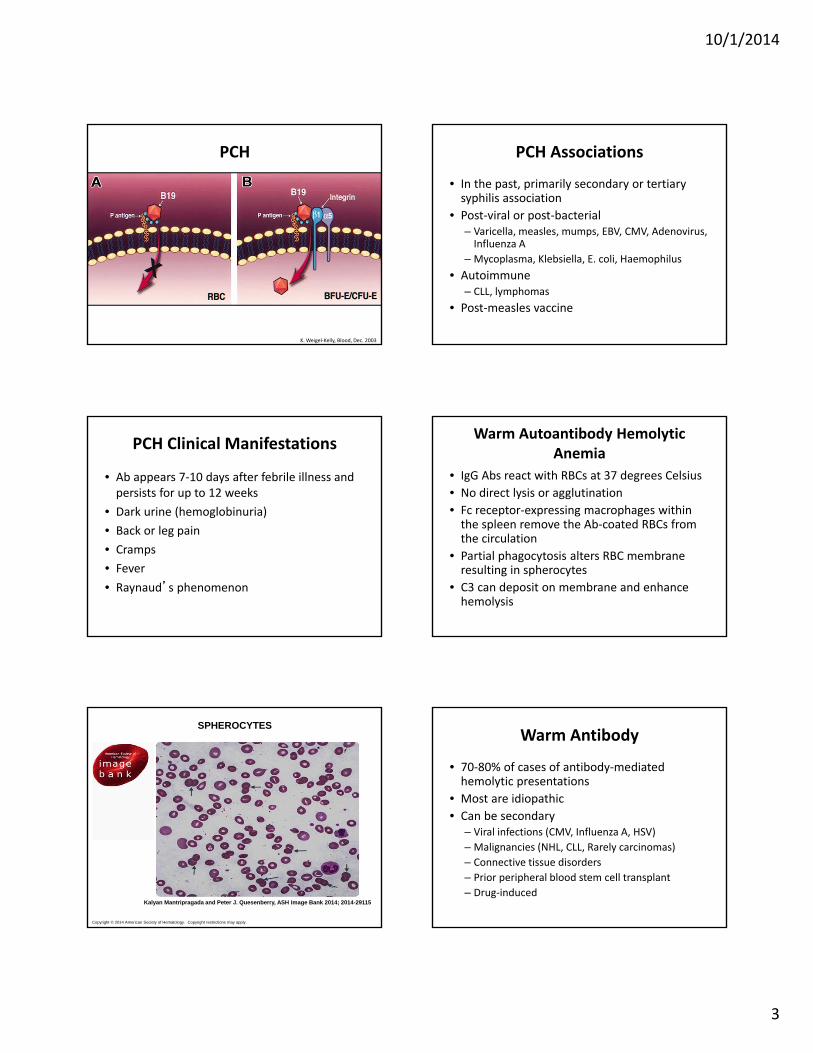
10/1/2014
3
PCH
K. Weigel‐Kelly, Blood, Dec. 2003
PCH Associations
• In the past, primarily secondary or tertiary syphilis association
• Post‐viral or post‐bacterial– Varicella, measles, mumps, EBV, CMV, Adenovirus, Influenza A
– Mycoplasma, Klebsiella, E. coli, Haemophilus• Autoimmune
– CLL, lymphomas• Post‐measles vaccine
PCH Clinical Manifestations
• Ab appears 7‐10 days after febrile illness and persists for up to 12 weeks
• Dark urine (hemoglobinuria)• Back or leg pain• Cramps• Fever• Raynaud’s phenomenon
Warm Autoantibody Hemolytic Anemia
• IgG Abs react with RBCs at 37 degrees Celsius• No direct lysis or agglutination • Fc receptor‐expressing macrophages within the spleen remove the Ab‐coated RBCs from the circulation
• Partial phagocytosis alters RBC membrane resulting in spherocytes
• C3 can deposit on membrane and enhance hemolysis
Copyright © 2014 American Society of Hematology. Copyright restrictions may apply.
Kalyan Mantripragada and Peter J. Quesenberry, ASH Image Bank 2014; 2014-29115
SPHEROCYTESWarm Antibody
• 70‐80% of cases of antibody‐mediated hemolytic presentations
• Most are idiopathic• Can be secondary
– Viral infections (CMV, Influenza A, HSV)– Malignancies (NHL, CLL, Rarely carcinomas)– Connective tissue disorders – Prior peripheral blood stem cell transplant– Drug‐induced

10/1/2014
4
Drug‐Induced
• Alpha‐methyldopa• Fludarabine• Procainamide• Diclofenac• Quinidine, Quinine• Sulfa drugs
• Isoniazid• Amphotericin• Penicillins• Cephalosporins• Tetracycline• Ribavirin
AE Lichtin. The Merck Manual. 2013/Up To Date
Warm‐Reactive Treatment
• Treat any known underlying condition• Stop any implicated drug• Folic Acid 5 mg daily• Prednisone 1 mg/kg/day• Splenectomy• Cytotoxic Drugs
– Cyclophosphamide• Immunosuppresants
– Cyclosporine, mycophenolate mofetil, azathioprine• IVIG
Rituximab
• Chimeric, monoclonal anti‐CD20 antibody• Multiple case studies and retrospective reports
• Off label usage
www.rituxan.comby Genentech
• 64 patients with newly‐dx WAIHA• Prednisolone + rituximab vs prednisolone alone
• After 12 months, 75% on P‐R vs 36% P alone with satisfactory response
• After 36 months, 70% vs 45% remission• No significant difference in adverse events
Transfusion Concerns
• Pan‐agglutination with usual cross‐matching with all normal donor cells
• Allo‐reactive Abs may be present in up to 1/3 of patients with AIHA
• Undetected ALLO‐ Abs could cause massive hemolysis after transfusion

10/1/2014
5
L. Dean, Blood Groups and Red Cell Antigens, 2005.
Transfusion Concerns
• If patient never pregnant or no history of transfusion, low probability of an alloantibody
• Adequate testing for alloantibodies may take up to 6 hours– Testing diluted serum against RBC panel taking advantage of differences in reaction strength
– Autoadsorption technique• Remove the autoantibody and use these ‘clean’ cells to keep removing autoantibody, leaving allo‐Ab behind for ID
Petz LD. British Journal of Haematology, 124, 712‐716
Transfusion Concerns
• Severe anemia may preclude obtaining a large enough volume of RBCs for autoadsorptionprocedure and not useful in those transfused within 3 months– Allogeneic adsorption may be needed
• Use several samples of allogeneic red cells of varying phenotypes responsible for clinically important hemolytic reactions to remove the autoAb
Petz LD. British Journal of Haematology, 124, 712‐716
Transfusion Concerns
• The clinician must balance risk and benefit of transfusion
• No patient should die from lack of transfusion, give most compatible blood
• Transfuse the minimal amount of blood required to alleviate symptoms and maintain a tolerable hgb/hct
• Transfuse slowly and with close monitoring
Cold‐Reactive/Cold Agglutinin Dz
• IgM complement‐fixing antibodies• Anti‐I and anti‐i most common• Ab binds to RBCs and causes agglutination at low temperatures (4 degrees Celsius)
• Intravascular hemolysis• DAT positive for complement only since IgM is released at 37 degrees Celsius
Copyright © 2011 American Society of Hematology. Copyright restrictions may apply.
John Lazarchick, ASH Image Bank 2011; 2011-1053
COLD AGGLUTININ DISEASE
10/1/2014
6
Testable Associations
• Mycloplasma pneumonia• Infectious mononucleosis• Influenza B• HIV• Hepatitis C• Lymphoproliferative disorders
Treatment of Cold Agglutinins
• Avoid cold• Warm the patient• Use in‐line warmer• Folic acid 5 mg daily• Rituximab or combination of rituximab and fludarabine
• Steroids and splenectomy not effective• Plasma exchange of only temporary value
MKSAP 16
Thrombotic MicroangiopathySyndromes
• Thrombotic=intravascular coagulation• Microangiopathy= a disease of very fine blood vessels
• All share clinical features of microangiopathichemolytic anemia, thrombocytopenia and organ injury
• Pathological features are vascular damage with arteriolar and capillary thrombosis
Pathological Features of the Nine Primary Thrombotic Microangiopathy (TMA) Syndromes.
George JN, Nester CM. N Engl J Med 2014;371:654‐666.

10/1/2014
7
Thrombotic Thrombocytopenic Purpura (TTP)
• Definition: Coombs’ negative microangiopathic hemolytic anemia and thrombocytopenia in the absence of an alternative explanation
• Minimum criteria: Thrombocytopenia
Rock. Br J Haematol 2000 Theodore Warkentin GWBR 2011
Theodore Warkentin GWBR 2011 Theodore Warkentin GWBR 2011
Theodore Warkentin GWBR 2011Theodore Warkentin GWBR 2011

10/1/2014
8
Complement‐Mediated TMA (CM‐TMA)
• 1st recognized as a familial disorder in 1975• Two brothers with TMA were found to have a deficiency of complement factor H
• Associated mutations in the gene encoding complement factor H (CFH) characterized 1998
• Loss‐of‐function mutations in regulatory genes or gain‐of‐function effector gene mutations
• Functional deficiencies in complement factors
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Diagnostic Criteria
• Serum creatinine above the normal range• MAHA• Thrombocytopenia• ADAMTS13 activity of 5% or more• Negative stool tests for Shiga toxin‐producing infection
CM‐TMA Diagnosis
• Plasma levels of C3, C4, CF‐H, ‐B and ‐I are not helpful
• Need access to cost effective, rapid and commercially available genetic studies
• Noris et al. published an in vitro assay to measure serum‐induced endothelial C5b‐9 deposition to monitor complement activation and treatment effectiveness
Noris et al. Blood. 2014;124(11):1715‐1726.
Eculizumab is now considered the standard in CM‐TMA. This is a monoclonal antibody that is a complement protein C5 inhibitor that can terminate complement‐mediated hemolysis and thrombotic microangiopathy.
Theodore Warkentin GWBR 2011 Neal Young GWBR 2010
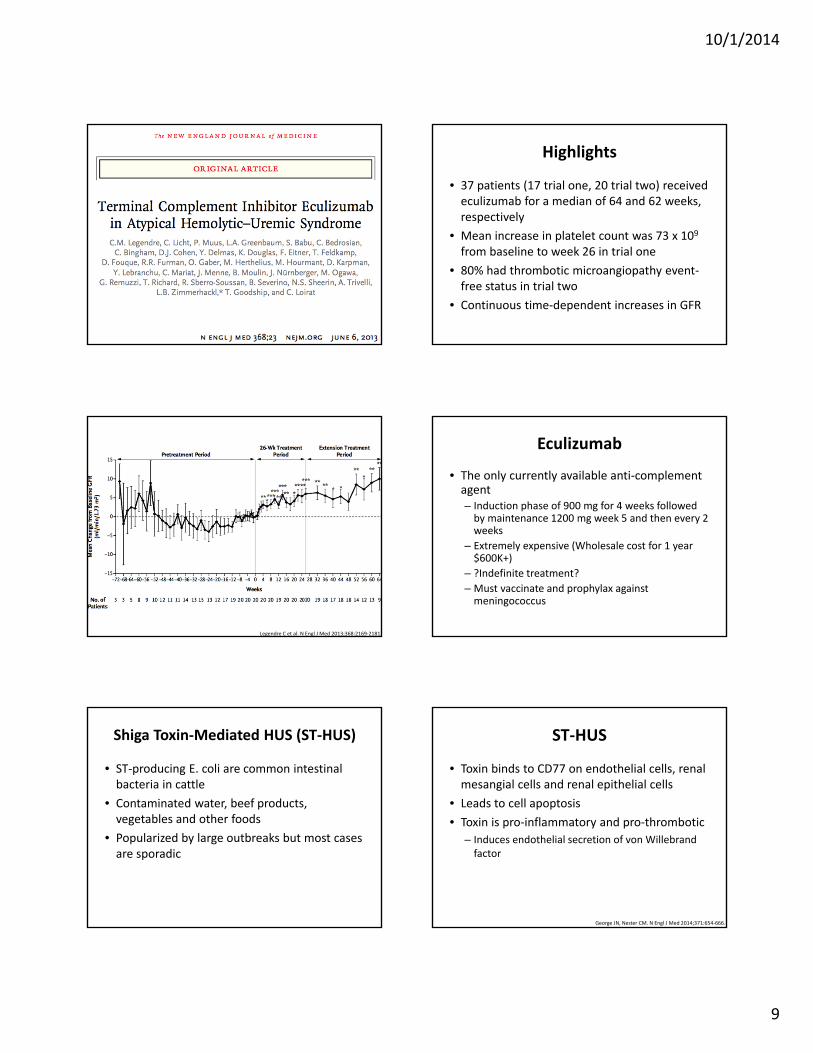
10/1/2014
9
Highlights
• 37 patients (17 trial one, 20 trial two) received eculizumab for a median of 64 and 62 weeks, respectively
• Mean increase in platelet count was 73 x 109 from baseline to week 26 in trial one
• 80% had thrombotic microangiopathy event‐free status in trial two
• Continuous time‐dependent increases in GFR
End Points.
Legendre C et al. N Engl J Med 2013;368:2169‐2181.
Eculizumab
• The only currently available anti‐complement agent– Induction phase of 900 mg for 4 weeks followed by maintenance 1200 mg week 5 and then every 2 weeks
– Extremely expensive (Wholesale cost for 1 year $600K+)
– ?Indefinite treatment?– Must vaccinate and prophylax against meningococcus
Shiga Toxin‐Mediated HUS (ST‐HUS)
• ST‐producing E. coli are common intestinal bacteria in cattle
• Contaminated water, beef products, vegetables and other foods
• Popularized by large outbreaks but most cases are sporadic
ST‐HUS
• Toxin binds to CD77 on endothelial cells, renal mesangial cells and renal epithelial cells
• Leads to cell apoptosis• Toxin is pro‐inflammatory and pro‐thrombotic
– Induces endothelial secretion of von Willebrandfactor
George JN, Nester CM. N Engl J Med 2014;371:654‐666.

10/1/2014
10
ST‐HUS Presentation, Diagnosis & Treatment
• Severe abdominal pain and bloody diarrhea begin within several days of consumption
• Renal failure and thrombocytopenia begin as GI symptoms resolve
• Stool analyses diagnostic during acute colitis phase
• Supportive treatment– Aggressive hydration– Plasma exchange and anti‐complement therapy inconclusive
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Drug‐induced MAHA
• Ticlopidine, clopidogrel• Quinine • Mitomycin‐C, cisplatin and bleomycin• Cyclosporine, tacrolimus and sirolimus• Penicillamine, penicillins
Metabolism‐Mediated TMA
• Occurs in infants, only reported in one adult• Mutations in gene encoding the methylmalonic aciduria and homocystinuriatype C protein (MMACHC)
• Hyperhomocysteinemia, low methionine and methlymalonic aciduria with normal folateand B12 levels
• Parenteral hydroxycobalamin treatment of choice
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Common Disorders Associated with Microangiopathic Hemolytic Anemia and Thrombocytopenia.
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Neal Young GWBR 2010 Neal Young GWBR 2010
10/1/2014
11
TREATMENT OF PNH
• Anticoagulation in the face of thrombosis– Prophylaxis may be considered if more than 50% of cells are CD55‐ or CD59‐deficient.
• If marked bone marrow failure, treatment consists of immunosuppressive therapy or allogeneic bone marrow transplantation– Transplant preferred with young patient with a donor with bone marrow failure
• Eculizumab– Decreases transfusions and thromboses and improves quality of life but does not reverse bone marrow failure
Spur Cell Hemolytic Anemia
• Secondary to severe impaired liver function or cirrhosis
• Unable to esterify cholesterol causing free cholesterol to bind to the red cell membrane, increasing its surface area
Allen DW Blood 1994
January 1, 2003; Blood: 101 (1)
Direct Toxins
• Arsenic hydride• Lead• Copper• Chlorates• Insect, Spider and Snake Venoms• Heat• Radiation
Williams Hematology. 2001. Chapter 53.
Thank you for your time and attention! References
The Merck Manual 2013Up To DateLichtman MA, Spivak JL, eds. Ueber paroxysmale Haemoglo‐binurie [Concerning paroxysmal hemoglobinuria]. In: Hematology: Landmark Papers of the Twentieth Century. San Diego, Calif: Academic Press; 2000:21.Weigel‐Kelley KA et al. α5β1 integrin as a cellular coreceptor for human parvovirus B19: requirement of functional activation of β1 integrin for viral entry. Blood. December 2003, Vol. 102:12.The American Society of Hematology Image BankBirgens H et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013 Nov;163(3):393‐9. Dean L. Blood Groups and Red Cell Antigens [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2005. Available from: http://www.ncbi.nlm.nih.gov/books/NBK2261/Petz LD. Br J Haematol. 2004 Mar;124(6):712‐6. Review.Medical Knowledge Self‐Assessment Program 16George JN et al. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014 Aug 14;371(7):654‐66. Rock GA. Management of thrombotic thrombocytopenic purpura. Br J Haematol. 2000 Jun;109(3):496‐507.The George Washington Board Review Series 2010 and 2011.Noris M et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. September 2014, Vol. 124:11.Legendre CM et al. Terminal complement inhibitor eculizumab in atypical hemolytic‐uremic syndrome. N Engl J Med. 2013; 368:2169‐2181Allen DW et al. Abnormal phospholipid metabolism in spur cell anemia: decreased fatty acid incorporation intophosphatidylethanolamine and increased incorporation into acylcarnitine in spur cell anemia erythrocytes. Blood 1994;84:1283‐1287. Beutler E. In: Williams Hematology, Chapter 53. 2001.

10/1/2014
1
Hemolytic Anemia: Evolving Pathogenesis and new Treatment Paradigms
Matt GrahamUT‐Erlanger Oncology and Hematology
October 2, 2014
Disclosures
• I have no conflicts of interest or financial disclosures for this presentation
• Off‐label rituximab usage described in this presentation
Hemolysis
• Shortened RBC Survival– RBC survival is less than 120 days– RBCs die daily and there is age independent random hemolysis on the order of 0.05 to 0.5 % per day
• Compensatory increase in EPO production– Increased RBC production– Increased reticulocyte % and absolute reticulocyte count
Typical Clinical Manifestations
• Dyspnea• Fatigue• Hyperdynamic state• Palpitations• Lethargy • Confusion• Pallor• Jaundice• Splenomegaly
Laboratory Findings
• Spectrum of Anemia• Reticulocytosis• Increased MCV• Low haptoglobin• Elevated LDH• Unconjugated hyperbilirubinemia• + DAT
AE Lichtin. The Merck Manual. 2013

10/1/2014
2
DAT Pearls
• Weakly + test in 1 in 10K healthy donors• + in 5‐10% of hospitalized patients without any evidence of hemolysis; complement‐mediated
• Low titers of autoantibody may appear negative – Cannot detect fewer than 100‐500 Ab molecules– Cannot detect fewer than 400‐1100 C’
• Commonly used reagents, Erlanger’s included, cannot identify IgA or IgM autoantibodies
AE Lichtin. The Merck Manual. 2013/Up To Date AE Lichtin. The Merck Manual. 2013
Classification of Hemolytic Anemias
• Hereditary Hemolytic Disorders– RBC enzyme defects– RBC membrane defects– Hemoglobinopathies– Thalassemias
• Acquired Hemolytic Disorders– Immune‐mediated hemolytic anemias– Microangiopathic hemolytic anemias (MAHAs)– Paroxysmal nocturnal hemoglobinuria (PNH)– Spur cell anemia– Direct toxins
Immune‐mediated Hemolytic Anemias
• Paroxysmal Cold Hemoglobinuria• Warm Autoantibody Hemolytic Anemia• Cold Agglutinin Disease
Paroxysmal Cold Hemoglobinuria (PCH)
• One of the 1st hematologic syndromes recognized clinically
• Red to brown urine after cold exposure described in 1872
• Donath and Landsteiner’s work identified the causative antibody in 1904
MA Lichtman. In: Hematology. 2000
PCH
• A polyclonal, cold‐reacting IgG• Fixes complement directly• Thermal amplitude varies greatly• Blood in the periphery cools and Ab and 1st 2 components of complement are fixed to the RBC surface
• Lysis occurs when the RBC are subsequently warmed (complement cascade completes)
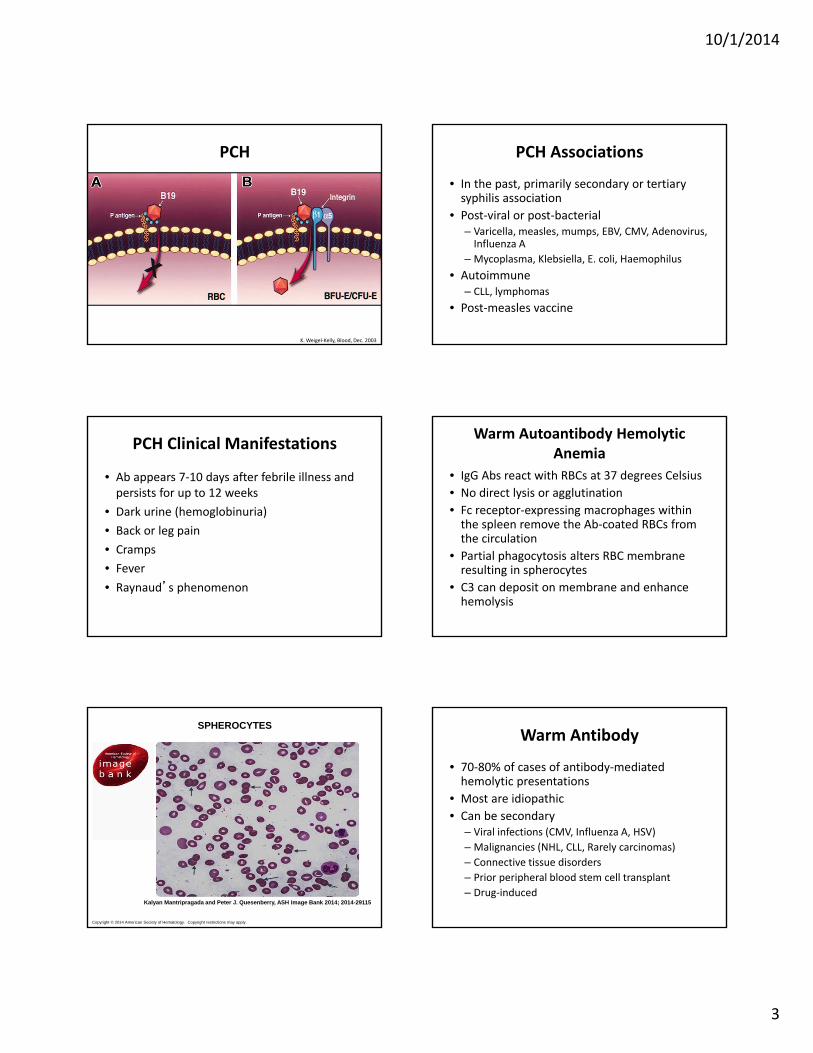
10/1/2014
3
PCH
K. Weigel‐Kelly, Blood, Dec. 2003
PCH Associations
• In the past, primarily secondary or tertiary syphilis association
• Post‐viral or post‐bacterial– Varicella, measles, mumps, EBV, CMV, Adenovirus, Influenza A
– Mycoplasma, Klebsiella, E. coli, Haemophilus• Autoimmune
– CLL, lymphomas• Post‐measles vaccine
PCH Clinical Manifestations
• Ab appears 7‐10 days after febrile illness and persists for up to 12 weeks
• Dark urine (hemoglobinuria)• Back or leg pain• Cramps• Fever• Raynaud’s phenomenon
Warm Autoantibody Hemolytic Anemia
• IgG Abs react with RBCs at 37 degrees Celsius• No direct lysis or agglutination • Fc receptor‐expressing macrophages within the spleen remove the Ab‐coated RBCs from the circulation
• Partial phagocytosis alters RBC membrane resulting in spherocytes
• C3 can deposit on membrane and enhance hemolysis
Copyright © 2014 American Society of Hematology. Copyright restrictions may apply.
Kalyan Mantripragada and Peter J. Quesenberry, ASH Image Bank 2014; 2014-29115
SPHEROCYTESWarm Antibody
• 70‐80% of cases of antibody‐mediated hemolytic presentations
• Most are idiopathic• Can be secondary
– Viral infections (CMV, Influenza A, HSV)– Malignancies (NHL, CLL, Rarely carcinomas)– Connective tissue disorders – Prior peripheral blood stem cell transplant– Drug‐induced

10/1/2014
4
Drug‐Induced
• Alpha‐methyldopa• Fludarabine• Procainamide• Diclofenac• Quinidine, Quinine• Sulfa drugs
• Isoniazid• Amphotericin• Penicillins• Cephalosporins• Tetracycline• Ribavirin
AE Lichtin. The Merck Manual. 2013/Up To Date
Warm‐Reactive Treatment
• Treat any known underlying condition• Stop any implicated drug• Folic Acid 5 mg daily• Prednisone 1 mg/kg/day• Splenectomy• Cytotoxic Drugs
– Cyclophosphamide• Immunosuppresants
– Cyclosporine, mycophenolate mofetil, azathioprine• IVIG
Rituximab
• Chimeric, monoclonal anti‐CD20 antibody• Multiple case studies and retrospective reports
• Off label usage
www.rituxan.comby Genentech
• 64 patients with newly‐dx WAIHA• Prednisolone + rituximab vs prednisolone alone
• After 12 months, 75% on P‐R vs 36% P alone with satisfactory response
• After 36 months, 70% vs 45% remission• No significant difference in adverse events
Transfusion Concerns
• Pan‐agglutination with usual cross‐matching with all normal donor cells
• Allo‐reactive Abs may be present in up to 1/3 of patients with AIHA
• Undetected ALLO‐ Abs could cause massive hemolysis after transfusion

10/1/2014
5
L. Dean, Blood Groups and Red Cell Antigens, 2005.
Transfusion Concerns
• If patient never pregnant or no history of transfusion, low probability of an alloantibody
• Adequate testing for alloantibodies may take up to 6 hours– Testing diluted serum against RBC panel taking advantage of differences in reaction strength
– Autoadsorption technique• Remove the autoantibody and use these ‘clean’ cells to keep removing autoantibody, leaving allo‐Ab behind for ID
Petz LD. British Journal of Haematology, 124, 712‐716
Transfusion Concerns
• Severe anemia may preclude obtaining a large enough volume of RBCs for autoadsorptionprocedure and not useful in those transfused within 3 months– Allogeneic adsorption may be needed
• Use several samples of allogeneic red cells of varying phenotypes responsible for clinically important hemolytic reactions to remove the autoAb
Petz LD. British Journal of Haematology, 124, 712‐716
Transfusion Concerns
• The clinician must balance risk and benefit of transfusion
• No patient should die from lack of transfusion, give most compatible blood
• Transfuse the minimal amount of blood required to alleviate symptoms and maintain a tolerable hgb/hct
• Transfuse slowly and with close monitoring
Cold‐Reactive/Cold Agglutinin Dz
• IgM complement‐fixing antibodies• Anti‐I and anti‐i most common• Ab binds to RBCs and causes agglutination at low temperatures (4 degrees Celsius)
• Intravascular hemolysis• DAT positive for complement only since IgM is released at 37 degrees Celsius
Copyright © 2011 American Society of Hematology. Copyright restrictions may apply.
John Lazarchick, ASH Image Bank 2011; 2011-1053
COLD AGGLUTININ DISEASE
10/1/2014
6
Testable Associations
• Mycloplasma pneumonia• Infectious mononucleosis• Influenza B• HIV• Hepatitis C• Lymphoproliferative disorders
Treatment of Cold Agglutinins
• Avoid cold• Warm the patient• Use in‐line warmer• Folic acid 5 mg daily• Rituximab or combination of rituximab and fludarabine
• Steroids and splenectomy not effective• Plasma exchange of only temporary value
MKSAP 16
Thrombotic MicroangiopathySyndromes
• Thrombotic=intravascular coagulation• Microangiopathy= a disease of very fine blood vessels
• All share clinical features of microangiopathichemolytic anemia, thrombocytopenia and organ injury
• Pathological features are vascular damage with arteriolar and capillary thrombosis
Pathological Features of the Nine Primary Thrombotic Microangiopathy (TMA) Syndromes.
George JN, Nester CM. N Engl J Med 2014;371:654‐666.

10/1/2014
7
Thrombotic Thrombocytopenic Purpura (TTP)
• Definition: Coombs’ negative microangiopathic hemolytic anemia and thrombocytopenia in the absence of an alternative explanation
• Minimum criteria: Thrombocytopenia
Rock. Br J Haematol 2000 Theodore Warkentin GWBR 2011
Theodore Warkentin GWBR 2011 Theodore Warkentin GWBR 2011
Theodore Warkentin GWBR 2011Theodore Warkentin GWBR 2011

10/1/2014
8
Complement‐Mediated TMA (CM‐TMA)
• 1st recognized as a familial disorder in 1975• Two brothers with TMA were found to have a deficiency of complement factor H
• Associated mutations in the gene encoding complement factor H (CFH) characterized 1998
• Loss‐of‐function mutations in regulatory genes or gain‐of‐function effector gene mutations
• Functional deficiencies in complement factors
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Diagnostic Criteria
• Serum creatinine above the normal range• MAHA• Thrombocytopenia• ADAMTS13 activity of 5% or more• Negative stool tests for Shiga toxin‐producing infection
CM‐TMA Diagnosis
• Plasma levels of C3, C4, CF‐H, ‐B and ‐I are not helpful
• Need access to cost effective, rapid and commercially available genetic studies
• Noris et al. published an in vitro assay to measure serum‐induced endothelial C5b‐9 deposition to monitor complement activation and treatment effectiveness
Noris et al. Blood. 2014;124(11):1715‐1726.
Eculizumab is now considered the standard in CM‐TMA. This is a monoclonal antibody that is a complement protein C5 inhibitor that can terminate complement‐mediated hemolysis and thrombotic microangiopathy.
Theodore Warkentin GWBR 2011 Neal Young GWBR 2010
10/1/2014
9
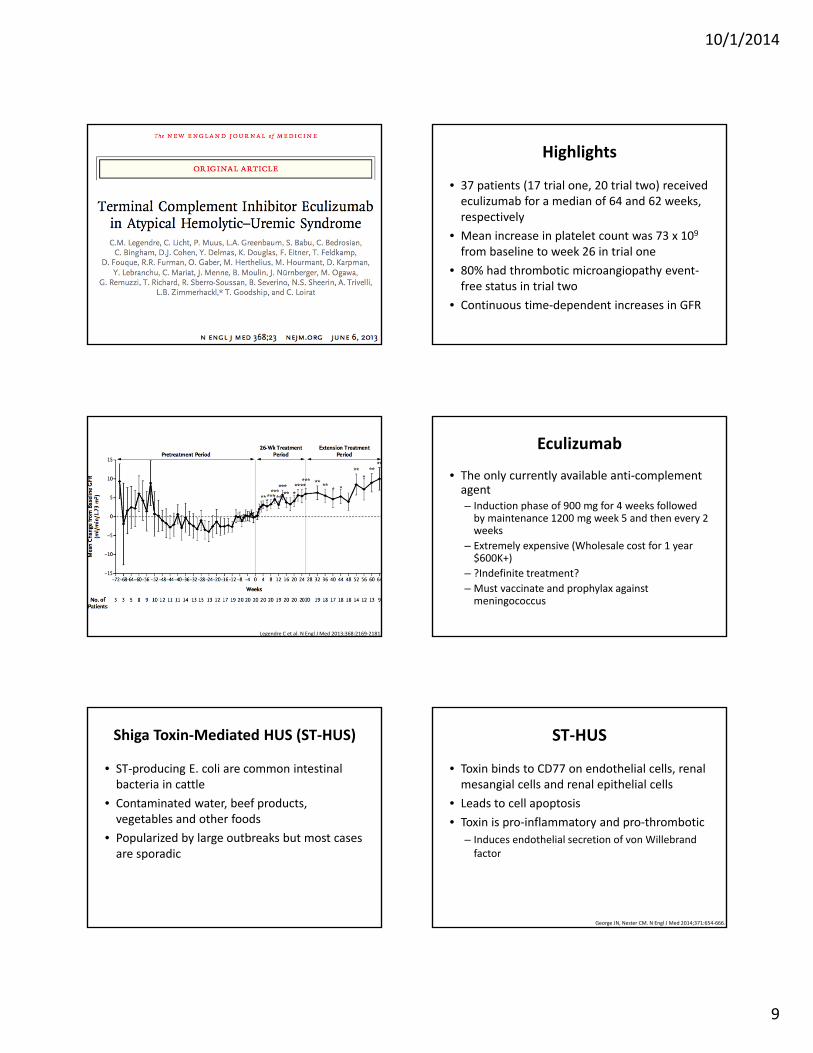
Highlights
• 37 patients (17 trial one, 20 trial two) received eculizumab for a median of 64 and 62 weeks, respectively
• Mean increase in platelet count was 73 x 109 from baseline to week 26 in trial one
• 80% had thrombotic microangiopathy event‐free status in trial two
• Continuous time‐dependent increases in GFR
End Points.
Legendre C et al. N Engl J Med 2013;368:2169‐2181.
Eculizumab
• The only currently available anti‐complement agent– Induction phase of 900 mg for 4 weeks followed by maintenance 1200 mg week 5 and then every 2 weeks
– Extremely expensive (Wholesale cost for 1 year $600K+)
– ?Indefinite treatment?– Must vaccinate and prophylax against meningococcus
Shiga Toxin‐Mediated HUS (ST‐HUS)
• ST‐producing E. coli are common intestinal bacteria in cattle
• Contaminated water, beef products, vegetables and other foods
• Popularized by large outbreaks but most cases are sporadic
ST‐HUS
• Toxin binds to CD77 on endothelial cells, renal mesangial cells and renal epithelial cells
• Leads to cell apoptosis• Toxin is pro‐inflammatory and pro‐thrombotic
– Induces endothelial secretion of von Willebrandfactor
George JN, Nester CM. N Engl J Med 2014;371:654‐666.

10/1/2014
10
ST‐HUS Presentation, Diagnosis & Treatment
• Severe abdominal pain and bloody diarrhea begin within several days of consumption
• Renal failure and thrombocytopenia begin as GI symptoms resolve
• Stool analyses diagnostic during acute colitis phase
• Supportive treatment– Aggressive hydration– Plasma exchange and anti‐complement therapy inconclusive
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Drug‐induced MAHA
• Ticlopidine, clopidogrel• Quinine • Mitomycin‐C, cisplatin and bleomycin• Cyclosporine, tacrolimus and sirolimus• Penicillamine, penicillins
Metabolism‐Mediated TMA
• Occurs in infants, only reported in one adult• Mutations in gene encoding the methylmalonic aciduria and homocystinuriatype C protein (MMACHC)
• Hyperhomocysteinemia, low methionine and methlymalonic aciduria with normal folateand B12 levels
• Parenteral hydroxycobalamin treatment of choice
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Common Disorders Associated with Microangiopathic Hemolytic Anemia and Thrombocytopenia.
George JN, Nester CM. N Engl J Med 2014;371:654‐666.
Neal Young GWBR 2010 Neal Young GWBR 2010
10/1/2014
11
TREATMENT OF PNH
• Anticoagulation in the face of thrombosis– Prophylaxis may be considered if more than 50% of cells are CD55‐ or CD59‐deficient.
• If marked bone marrow failure, treatment consists of immunosuppressive therapy or allogeneic bone marrow transplantation– Transplant preferred with young patient with a donor with bone marrow failure
• Eculizumab– Decreases transfusions and thromboses and improves quality of life but does not reverse bone marrow failure
Spur Cell Hemolytic Anemia
• Secondary to severe impaired liver function or cirrhosis
• Unable to esterify cholesterol causing free cholesterol to bind to the red cell membrane, increasing its surface area
Allen DW Blood 1994
January 1, 2003; Blood: 101 (1)
Direct Toxins
• Arsenic hydride• Lead• Copper• Chlorates• Insect, Spider and Snake Venoms• Heat• Radiation
Williams Hematology. 2001. Chapter 53.
Thank you for your time and attention! References
The Merck Manual 2013Up To DateLichtman MA, Spivak JL, eds. Ueber paroxysmale Haemoglo‐binurie [Concerning paroxysmal hemoglobinuria]. In: Hematology: Landmark Papers of the Twentieth Century. San Diego, Calif: Academic Press; 2000:21.Weigel‐Kelley KA et al. α5β1 integrin as a cellular coreceptor for human parvovirus B19: requirement of functional activation of β1 integrin for viral entry. Blood. December 2003, Vol. 102:12.The American Society of Hematology Image BankBirgens H et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013 Nov;163(3):393‐9. Dean L. Blood Groups and Red Cell Antigens [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2005. Available from: http://www.ncbi.nlm.nih.gov/books/NBK2261/Petz LD. Br J Haematol. 2004 Mar;124(6):712‐6. Review.Medical Knowledge Self‐Assessment Program 16George JN et al. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014 Aug 14;371(7):654‐66. Rock GA. Management of thrombotic thrombocytopenic purpura. Br J Haematol. 2000 Jun;109(3):496‐507.The George Washington Board Review Series 2010 and 2011.Noris M et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. September 2014, Vol. 124:11.Legendre CM et al. Terminal complement inhibitor eculizumab in atypical hemolytic‐uremic syndrome. N Engl J Med. 2013; 368:2169‐2181Allen DW et al. Abnormal phospholipid metabolism in spur cell anemia: decreased fatty acid incorporation intophosphatidylethanolamine and increased incorporation into acylcarnitine in spur cell anemia erythrocytes. Blood 1994;84:1283‐1287. Beutler E. In: Williams Hematology, Chapter 53. 2001.





























