Embed Size (px)
Citation preview

Improving the therapeutic potential of
human granzyme B and evaluation of
granzyme M as novel effector molecules in
cytolytic fusion proteins for the treatment of
Serpin B9-positive cancer
Von der Fakultät für Mathematik, Informatik und Naturwissenschaften
der RWTH Aachen University zur Erlangung des akademischen Grades einer
Doktorin der Naturwissenschaften genehmigte Dissertation
vorgelegt von
Diplom-Bioingenieurin
Sonja Schiffer, geb. Hermes
aus Neuss
Berichter: Universitätsprofessor Dr. rer. nat. Rainer Fischer Universitätsprofessor Dr. rer. nat. Dr. rer. medic. Stefan Barth
Tag der mündlichen Prüfung: 29. November 2013
Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar.


Content ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
i
ABBREVIATIONS ............................................................................................................................................... V
1 INTRODUCTION........................................................................................................................................ 1
1.1 CANCER .................................................................................................................................................... 1
1.2 TARGETED CANCER THERAPY ......................................................................................................................... 2
1.2.1 Immunotoxins .................................................................................................................................... 3
1.2.2 Humanization of immunotoxins ........................................................................................................ 4
1.3 GRANZYMES .............................................................................................................................................. 5
1.3.1 Granzyme B ....................................................................................................................................... 5
1.3.2 Granzyme M ...................................................................................................................................... 7
1.4 TUMOR ESCAPE MECHANISMS TO EVADE IMMUNOSURVEILLANCE ......................................................................... 8
1.4.1 The granzyme B inhibitor PI-9 ........................................................................................................... 9
1.5 HODGKIN LYMPHOMA AND THE CD30 RECEPTOR ........................................................................................... 11
1.6 CHRONIC AND ACUTE MYELOMONOCYTIC LEUKEMIA AND THE CD64 RECEPTOR .................................................... 14
1.7 MOLECULAR MODELING ............................................................................................................................ 16
1.8 AIMS AND OBJECTIVES ............................................................................................................................... 17
2 MATERIAL AND METHODS ..................................................................................................................... 19
2.1 CHEMICAL AND BIO-CHEMICAL MATERIAL ...................................................................................................... 19
2.1.1 Equipment ....................................................................................................................................... 19
2.1.2 Chemicals and consumables ............................................................................................................ 20
2.1.3 Kits, enzymes and standards ........................................................................................................... 20
2.1.4 Synthetic oligonucleotides and plasmids ......................................................................................... 21
2.1.5 Antibodies and antibody fragments ................................................................................................ 22
2.1.6 Bacterial strains ............................................................................................................................... 23
2.1.7 Mammalian cell lines....................................................................................................................... 24
2.1.8 Primary cells .................................................................................................................................... 25
2.1.9 Media and buffers ........................................................................................................................... 25
2.2 MOLECULAR-BIOLOGICAL METHODS ............................................................................................................. 27
2.2.1 Polymerase chain reaction .............................................................................................................. 27
2.2.2 Site-directed mutagenesis ............................................................................................................... 28
2.2.3 Agarose gel electrophoresis ............................................................................................................ 29
2.2.4 Zero blunt TOPO PCR cloning .......................................................................................................... 29
2.2.5 Endonuclease restriction of DNA ..................................................................................................... 29
2.2.6 Ligation of DNA fragments .............................................................................................................. 29
2.2.7 Transformation of plasmid DNA into competent E. coli .................................................................. 30
2.2.8 Preparation of plasmid DNA from E. coli ......................................................................................... 30
2.2.9 Sequencing of DNA .......................................................................................................................... 30
2.3 CELL CULTURE .......................................................................................................................................... 30
2.3.1 Cultivation of cell lines and primary cells ........................................................................................ 30

Content ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
ii
2.3.2 Transfection of cell lines .................................................................................................................. 31
2.3.3 Protein delivery into cells via streptolysin O .................................................................................... 31
2.4 PROTEIN-BIOCHEMICAL METHODS ................................................................................................................ 32
2.4.1 SDS-PAGE ......................................................................................................................................... 32
2.4.2 Western blot analysis ...................................................................................................................... 32
2.4.3 Bacterial expression and purification .............................................................................................. 33
2.4.4 Mammalian expression and purification ......................................................................................... 33
2.4.5 Protein quantification ...................................................................................................................... 34
2.4.6 Labeling of SNAP constructs ............................................................................................................ 35
2.4.7 Detection of endogenous PI-9 from cell lines or primary cells......................................................... 35
2.4.8 Serum stability assays ..................................................................................................................... 36
2.4.9 ELISA for detection of CD30 receptor in blood................................................................................. 36
2.5 FLOW CYTOMETRIC ANALYSIS ...................................................................................................................... 36
2.5.1 Binding analysis ............................................................................................................................... 36
2.5.2 Determination of Kd values .............................................................................................................. 37
2.5.3 Determination of specific cell death of primary cells ...................................................................... 37
2.6 CONFOCAL MICROSCOPY ............................................................................................................................ 37
2.7 FUNCTIONAL CHARACTERIZATION OF RECOMBINANT PROTEINS .......................................................................... 37
2.7.1 Protease assays ............................................................................................................................... 37
2.7.2 Apoptosis assays ............................................................................................................................. 39
2.7.3 Viability assay .................................................................................................................................. 39
2.8 IMMUNOHISTOCHEMISTRY ......................................................................................................................... 40
2.9 IN VIVO EXPERIMENTS ................................................................................................................................ 41
2.9.1 Mouse strains, housing and maintenance of animals ..................................................................... 41
2.9.2 Handling of mice and anesthesia .................................................................................................... 41
2.9.3 Establishment of xenograft subcutaneous tumor models ............................................................... 41
2.9.4 In vivo optical imaging by Cri-Maestro system ............................................................................... 42
2.9.5 Biological activity of Gb-Ki4(scFv) and GbR201K-Ki4(scFv) ............................................................. 42
2.10 STATISTICAL ANALYSIS ................................................................................................................................ 42
3 RESULTS ................................................................................................................................................. 43
3.1 PI-9 EXPRESSION IN CANCER CELL LINES ......................................................................................................... 43
3.2 IDENTIFICATION OF MUTATIONAL SITES BY MOLECULAR MODELING ..................................................................... 44
3.3 GENERATION AND PREPARATION OF GRANZYME B CONSTRUCTS AND ITS MUTANTS ................................................ 46
3.4 PREPARATION AND FUNCTIONAL CHARACTERIZATION OF RECOMBINANT PI-9 ....................................................... 48
3.5 ESTABLISHMENT OF A TEST SYSTEM FOR GRANZYME B MUTANTS ........................................................................ 49
3.6 IN VITRO FUNCTIONAL CHARACTERIZATION OF WILD-TYPE AND MUTANT GB-H22(SCFV) AND GB-KI4(SCFV) .............. 52
3.6.1 Proteolytic activity of granzyme B in absence and presence of PI-9 ............................................... 52
3.6.2 Target cell binding ........................................................................................................................... 54

Content ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
iii
3.6.3 Internalization activity ..................................................................................................................... 55
3.6.4 Cytotoxic activity against PI-9-negative cell lines............................................................................ 57
3.6.5 Apoptotic activity on PI-9-negative cell lines ................................................................................... 58
3.7 SELECTION OF THE MOST PROMISING GRANZYME B MUTANT ON PI-9-POSITIVE CELL LINES ..................................... 59
3.7.1 Cytotoxic activity on PI-9-positive cell lines ..................................................................................... 59
3.7.2 Apoptotic activity on PI-9-positive cell lines .................................................................................... 60
3.8 IN VIVO BIOLOGICAL ACTIVITY OF GB-KI4(SCFV) AND GBR201K-KI4(SCFV) ........................................................ 61
3.8.1 Stability of Gb-Ki4(scFv) constructs in mouse serum ....................................................................... 61
3.8.2 Establishment of Hodgkin lymphoma xenograft models ................................................................ 62
3.8.3 In vivo effects of GbR201K-Ki4(scFv) on PI-9-negative L540cy cells ................................................ 65
3.8.4 Comparative in vivo effects of Gb-Ki4(scFv) and GbR201K-Ki4(scFv) on PI-9-positive L428 cells .... 66
3.9 EX VIVO STUDIES WITH CMML AND AMML PATIENT-DERIVED MONOCYTES ........................................................ 68
3.9.1 PI-9 expression in patient-derived PBMCs ....................................................................................... 68
3.9.2 Receptor phenotyping of PBMCs ..................................................................................................... 69
3.9.3 Target cell binding and internalization............................................................................................ 70
3.9.4 Cytotoxic activity on primary cells ................................................................................................... 72
3.9.5 Specificity of target cell death ......................................................................................................... 74
3.9.6 Serum stability of Gb-H22(scFv) fusion proteins in patient-derived serum ..................................... 77
3.10 EVALUATION OF GRANZYME M AS NOVEL EFFECTOR MOLECULE IN CYTOLYTIC FUSION PROTEINS ............................... 77
3.10.1 Generation and preparation of granzyme M fusion proteins ..................................................... 77
3.10.2 Functional characterization of granzyme M fusion proteins ...................................................... 79
3.10.3 Target cell binding ...................................................................................................................... 80
3.10.4 Cytotoxic activity of granzyme M fusion proteins ....................................................................... 81
3.10.5 Ex vivo studies with Gm-H22(scFv) .............................................................................................. 84
4 DISCUSSION ........................................................................................................................................... 86
4.1 THE THERAPEUTIC POTENTIAL OF GRANZYME B-BASED CYTOLYTIC FUSION PROTEINS .............................................. 86
4.2 IMPACT OF ENDOGENOUS PI-9 EXPRESSION IN CANCER CELLS ON THE PRO-APOPTOTIC ACTIVITY OF GRANZYME B ........ 89
4.3 GENERATION AND IDENTIFICATION OF A PI-9 INDEPENDENT GRANZYME B VARIANT .............................................. 94
4.3.1 Evaluation of in silico findings and comparison with in vitro enzymatic activity ............................ 94
4.3.2 Selection of the optimal granzyme B mutant .................................................................................. 95
4.4 EFFICACY OF THE GBR201K MUTANT IN VIVO ................................................................................................ 98
4.5 EX VIVO DETERMINATIONS ON PRIMARY LEUKEMIC CELLS ................................................................................ 101
4.5.1 CMML and AMML primary cells as target for CD64-specific fusion proteins ................................ 101
4.5.2 Specific cytotoxic efficacy of CD64-specific fusion proteins on primary CMML and AMML cells .. 103
4.5.3 The role of PI-9 and the potential of the determined cytolytic fusion proteins for personalized
medicine ..................................................................................................................................................... 105
4.6 APPLICATION OF GRANZYME M AS AN ALTERNATIVE EFFECTOR MOLECULE IN CYTOLYTIC FUSION PROTEINS ............... 106
4.6.1 Efficacy of granzyme M based CFPs .............................................................................................. 107

Content ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
iv
4.6.2 Clinical potential of granzyme M as effector molecule in cytolytic fusion proteins ...................... 109
5 OUTLOOK ............................................................................................................................................. 111
6 SUMMARY ........................................................................................................................................... 113
7 LITERATURE ......................................................................................................................................... 115
8 APPENDIX ................................................................................................................................................. I
8.1 DNA AND AMINO ACID SEQUENCE OF GBR201K ............................................................................................... I
8.2 SYNTHETIC DNA SEQUENCE OF GM-H22(SCFV) IN PMS VECTOR ......................................................................... I
9 INDEX OF FIGURES AND TABLES .............................................................................................................. IV
10 PUBLICATIONS ........................................................................................................................................ VI

Abbreviations ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
v
Abbreviations
AA acrylamide
ADC antibody-drug-conjugate
AML acute myeloid leukemia
AMML acute myelomonocytic leukemia
AP alkaline phosphatase
APC allophycocyanin
BCIP 5-bromo-4-chloro-3-indolyl phosphate
BG O(6)-benzyl guanine
BID BH3-interacting domain death agonist
BSA bovines serum albumin
CFP cytolytic fusion protein
CML chronic myeloid leukemia
CMML chronic myelomonocytic leukemia
CTL cytotoxic T lymphocyte
dd double distilled
DMSO dimethylsulfoxide
DNA deoxyribonucleic acid
dNTPs deoxyribonucleotide triphosphates
DSMZ German Collection for Microorganisms and Cell Cultures
DT diphtheria toxin
DTT dithiothreitol
EBV Epstein-Barr virus
ECS enterokinase cleavage site
E. coli Escherichia coli
EDTA ethylenediamin tetraacetate
EGF epidermal growth factor
EGFP enhanced green fluorescent protein
ETA’ Pseudomonas exotoxin A, mutant with deleted binding domain
et al. et altera
FACS fluorescence-activated cell sorting
FC fragment crystallizable
FCS fetal calf serum
FITC fluorescein isothiocyanate
FPLC fast protein liquid chromatography
FSC forward scatter
g relative centrifuge force
GAM goat anti mouse
Gb granzyme B
GF gel filtration
Gm granzyme M
g gram
h hour(s)
HAMA human anti-mouse antibody
Hb hemoglobin
His-tag polyhistidine motif
HL Hodgkin lymphoma
HRPO horse radish peroxidase
HRS Hodgkin and Reed-Sternberg
IC50 half maximal inhibitory concentration
ICAD inhibitor of caspase-activated DNase
IFN γ interferon gamma
Ig immunglobulin
IL interleukin
IMAC immobilized metal ion affinity chromatography
IPTG isopropyl-β-D-thiogalactoside
IRES internal ribosomal entry site
IT immunotoxin
IVS synthetical intron
Kat2 Katushka2
kDa kilo Dalton
L liter
LB Luria Bertani
M molarity (mol/L)
mA milli ampere
min minute(s)
MFI mean fluorescence intensity
MTD maximum tolerable dose

Abbreviations ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
vi
n nano
NBT nitro blue tetrazolium chloride
n.d. not determined
NFκB nuclear factor 'kappa light chain enhancer' of activated B cells
NK natural killer
nm nanometer
ns not significant
p pico
PAGE polyacrylamide gel electrophoresis
PBMC peripheral blood mononuclear cell
PBS phosphate buffered saline
PBST PBS + 0.05 % (v/v) Tween-20
PCR polymerase chain reaction
PE phycoerythrin
pH negative log of the concentration of hydrogen ions in solution (pondus
hydrogenii)
PI propidium iodide
pI isoelectric point
PLT platelets
RCL reactive center loop
pNA para-nitroanilin
PO peroxidase
RNA ribonucleic acid
rpm rounds per minute
RT room temperature
scFv single chain variable fragment
SD standard deviation
SDS sodiumdodecylsulfate
sec seconds
siRNA small interfering RNA
SSC sideward scatter
TAE Tris-acetate-EDTA
Taq Thermus aquaticus
TB Terrific Broth
TNF tumor necrosis factor
Tris Tris(hydroxymethyl)aminomethane
Tween 20 polyoxyethylene (20) sorbitan monolaurate
U unit
UV ultraviolet
V volts
v/v volume per volume
WBC white blood cells
XIAP X-linked inhibitor of apoptosis protein
w/v weight per volume
XTT sodium3’-[1-(phenylaminocarbonyl)-3,4- tetrazolium]-bis(4-methoxy-6- nitro)benzene sulfonic acid hydrate
One and three letter code of amino acids
A Ala Alanine
C Cys Cysteine
D Asp Aspartic acid
E Glu Glutamic acid
F Phe Phenylalanine
G Gly Glycine
H His Histidine
I Ile Isoleucine
K Lys Lysine
M Met Methionine
N Asn Asparagine
P Pro Proline
A Ala Alanine
Q Gln Glutamine
R Arg Arginine
S Ser Serine
T Thr Threonine
V Val Valine
W Trp Tryptophan
Y Tyr Tyrosine

1 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
1 Introduction
1.1 CANCER
Cancer is one of the most severe health problems in the world. In the United States more than
1.6 million new cancer cases occurred in 2012 and more than half a million people died from this
disease (estimated by the American Cancer Society 2012). During the last five years, overall cancer
incidence rates remained almost stable whereby death rates decreased by 1.8 % per year in men and
by 1.6 % per year in women. Nevertheless, one in four deaths in the United States is still due to
cancer. The four leading types of cancer are lung, bronchus, colorectal and prostate for men or
breast cancer for women. The lifetime probability of developing an invasive cancer is 45 % for men
and 38 % for women. Cancer cells are characterized by genome instability, which generates the
genetic diversity that expedites carcinogenesis and tumor cell proliferation; and, inflammation, which
promotes the multiple hallmark functions of cancer. Tumors exhibit a high level of complexity having
many distinct cell types, which interact in a heterotypic way. The hallmark functions of cancer
comprise continuation of proliferative signaling, resistance to cell death, induction of angiogenesis,
evasion of growth suppressors, enabling of replicative immortality and activation of invasion and
metastasis [1]. During the last few years, further features have been identified, namely
reprogramming of energy metabolism and evasion of immune destruction. Most important for the
survival of tumors is their recruitment of normal cells in order to create a special ‘tumor
microenvironment’ [2].
Typical cancer treatment strategies including surgery, chemotherapy and radiotherapy are limited by
their corresponding serious and often life-threatening side effects, caused by the concurrent killing of
nonmalignant cells. Furthermore, many tumor cells become resistant to drugs, substantially reducing
any positive response to the treatment and resulting in a poor prognosis. In addition, surgical
resection of primary tumors results in high relapse rates due to incomplete removal of tumor cells as
well as the ineffective control of metastatic disease. Even more life-threatening are tumor stem cells
since they are the main initiators of tumor cell proliferation and often survive classical treatment
approaches. Tumor stem cells will often reactivate after therapy leading to a relapse. These facts
clearly illustrate the strong demand for both more potent and yet more selective, targeted treatment
options [3,4].

2 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
1.2 TARGETED CANCER THERAPY
Immunotherapeutic approaches have the potential to provide the needed selective, targeted
treatment options to specifically eliminate malignant cells. Immunotherapy generally involves the
induction of several mechanisms that interfere with both carcinogenesis and with tumor cell
proliferation. Active immunotherapy provides a more specialized approach, in which the immune
system itself is stimulated to remove malignant cells [5,6]. Targeted therapy involves either the direct
activation or inactivation of (soluble) enzymes, growth factors or surface receptors in signaling
pathways that are necessary for tumor maintenance and proliferation (e.g. growth factor receptors
such as EGFR (Epidermal Growth Factor Receptor), Her2, BRAF and Kit) or the indirect targeted
delivery of effector molecules to tumor cells, leading to growth arrest or remission [7,8]. This may
involve drugs that penetrate the tumor cell plasma membrane allowing the targeting of intracellular
tumor antigens, or the use of monoclonal antibodies (mAbs) that target cell surface molecules. Due
to these different mechanisms of action, the potential of targeted therapies is most promising for the
treatment of chemoresistant cells.
Monoclonal antibody therapy (passive immunotherapy) is a rapidly-expanding immunotherapeutic
treatment platform for cancer. At least 12 therapeutic mAbs for oncologic indications have already
been approved by the FDA [9] and several more are in clinical development [10]. However, the
activity of mAbs is limited by their relatively large size, resulting in poor tumor penetration.
Therefore high doses are needed to effectively compete with serum IgGs, which in turn lead to
severe side-effects. Furthermore unfavorable Fc receptor polymorphisms can reduce the impact of
Fc-mediated effector functions. For these reasons, unmodified antibodies are rarely suitable as
therapeutics [11,12].
Several strategies have been developed to enhance the anti-tumor activity of mAbs, including the
humanization of antibodies [13], the optimization of antibody effector functions [14] and the fusion
of antibodies to a potent cytotoxic drug. In the early 1900s, Paul Ehrlich considered the idea of a
‘magic bullet’ forming the basis of the antibody-drug conjugate (ADC); and, immunotoxin (IT)
concepts in which antibodies or antibody fragments specifically targeting tumor antigens deliver
cytotoxic payloads and trigger their internalization into abnormal cells [15].
ADCs usually comprise a full-size antibody chemically linked to a toxin, drug or radionuclide. The
therapeutic potential of ADCs has been demonstrated in numerous preclinical studies [11,16]. The
FDA has approved Brentuximab Vedotin, a CD30-directed ADC consisting of the anti-CD30 mAb
Brentuximab linked to the antimitotic agent monomethyl auristatin E for the treatment of Hodgkin
lymphoma and anaplastic large cell lymphoma [17]. After binding and internalization this agent binds
to tubulin which arrests the cell cycle leading to apoptosis of the targeted cells. In 2013, an ADC

3 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
called Ado-trastuzumab Emtansine (Kadcyla) received FDA approval for treatment of HER2-positive,
metastatic breast cancer in patients who have already undergone unsuccessful treatment with the
mAb Trastuzumab (Herceptin) and a taxane. Ado-trastuzumab Emtasine is a conjugate between the
Trastuzumab portion and a chemotherapeutic molecule called DM1, which attacks the cancer cells
after Her2 targeting [18]. The FDA also approved Gemtuzumab Ozogamicin (Mylotarg®), a humanized
anti-CD33 mAb conjugated to calicheamicin for the treatment of acute myeloid leukemia (AML) [19],
although this has now been withdrawn from the market due to adverse systemic effects.
1.2.1 Immunotoxins
ITs comprise an effector domain, which is a naturally-occurring toxin that inhibits protein synthesis or
induces apoptosis by modifying signal transduction pathways [20,21]. Toxins used in the design of
conventional highly effective recombinant ITs originate from plants, like Ricin A; or, bacteria, like
Pseudomonas exotoxin A (ETA) and Diphtheria Toxin (DT). The toxins inhibit protein biosynthesis by
ADP ribosylation of eukaryotic elongation factor 2, leading to cell death. Cell death-inducing toxins
use different mechanisms of action than chemotherapeutics. Therefore, ITs are promising for the
treatment of chemoresistant tumors or residual diseases to achieve for example a ‘chronic’ status of
the cancer. Furthermore ITs utilize a gentler approach than ADCs which indicates a high potential for
use in post-remission therapies. In addition, the use of ITs for patients who have endured lengthy
chemotherapy can result in improved quality of life and better clinical outcomes.
The first generation of ITs comprised full-size antibodies chemically conjugated to whole-protein
toxins. These ITs, however, have several drawbacks, including lack of specificity, poor stability, and
the tendency to be heterogeneous. Improvements were necessary. Investigation of protein structure
and function showed the presence of discrete cell binding domains within the toxins and led to the
design of the next generations of ITs. In second generation ITs, these cell binding domains were
removed, thereby diminishing the binding to normal cells. Third generation ITs were generated via
recombinant DNA techniques resulting in truncated toxins genetically linked to shorter antibody
fragments (scFv), leaving only the antigen binding variable domains of an antibody, or natural
ligands. These improvements yielded ITs having small size and diminished immunogenic response,
resulting in easier manufacturing, better tumor penetration and increased stability [22].
The selection of proper target molecules specific for tumor cells is one of the challenges in targeted
cancer therapy. But, advancements in phage display technology and protein engineering have
enabled and improved the selection and generation of high-affinity targeting residues [23].
Early generation ITs have shown promising results in in vitro studies and preclinical tests and have
also been tested in a small number of clinical trials [24]. The most prominent example is Denileukin

4 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Diftitox (Ontak®), a recombinant IT comprising DT and interleukin-2 (IL-2), which has been approved
by the FDA for the treatment of T-cell lymphoma [25].
However, the major limitation of clinical deployment of these early generation ITs is that they are
limited by their immunogenicity since they have non-human components. During long-term cancer
treatment requiring repeated doses, these components can induce neutralizing antibodies in patients
with a functional immune system. A murine origin of the binding moiety for example induces a
human anti-mouse antibody (HAMA) response in some patients. HAMA responses not only reduce
treatment efficiency, but also have been shown to cause allergic reactions, in extreme cases leading
to life-threatening complications such as renal failure [26].
These issues reduce the therapeutic potential of these early ITs. The complete humanization of ITs is
therefore an important criterion for the development of fourth-generation molecules called human
cytolytic fusion proteins (CFPs) [4].
1.2.2 Humanization of immunotoxins
There are several strategies that can be used to humanize the tumor-selective ligand. For example,
the effector moiety can be fused to human cytokines or growth factors that target tumor receptors,
e.g., IL-2, IL-4, EGF (epidermal growth factor), TGFα (tumor growth factor alpha) or the CD30 ligand
[24]. Recombinant DNA technology can also be used to engineer antibodies for reduced
immunogenicity. Chimeric antibodies comprising a murine Fab fragment and a human Fc region can
reduce HAMA responses, because the constant region makes the most significant contribution to
immunogenicity. Another strategy is the generation of humanized antibodies by grafting murine
complementarity determining regions (CDR) onto human V-region frameworks, an approach known
as CDR replacement [27]. Complete humanization is also possible by selecting the V-regions from
human antibody libraries or using transgenic knock-out mice or rats in which the gene loci for the
light and heavy antibody chains are replaced by their human counterparts [28]. ScFv antibodies used
for the production of CFPs can be derived from humanized or human monoclonal antibodies or
selected directly from a human scFv library [4].
There are a number of approaches that can be used to decrease the immunogenic responses of ITs
caused by the cell-death inducing domains found in bacterial or plant toxins. For example, the T or B
cell epitopes of ETA can be modified and ITs can be derivated by polyethylene glycol or
immunosuppressive agents can be co-applied [29,30]. A more favorable approach to achieve
complete humanization uses human cytolytic proteins, such as enzymes that induce cell death by a
variety of mechanisms. Pro-apoptotic human proteins that are potentially useful for targeted cancer
therapy include RNases such as angiogenin as well as death receptor ligands such as sTRAIL (tumor

5 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
necrosis factor (TNF) -related apoptosis-inducing ligand) and Fas ligand and pro-apoptotic members
of the Bcl-2 family [31]. In addition, enzymes with tumor suppressive functionality, such as DAPK2
(death-associated protein kinase 2), allow targeted restoration of tumor suppressor activity in cancer
cells [4]. Granzymes (serine proteases released by cytoplasmic granules within cytotoxic T
lymphocytes (CTLs) and natural killer (NK) cells) are another prominent group of human pro-
apoptotic molecules.
1.3 GRANZYMES
Cytotoxic granules contain a variety of granule-associated proteases. The secretion of cytotoxic
granules is the primary defense against virally infected and malignant cells. Among these granule-
associated proteases are granzymes, serine proteases, which play a significant role in this defense.
Five different human granzymes (A, B, H, K and M), each with unique species-dependent expression
patterns and substrate specificities have been discovered [32,33]. Granzymes promote apoptosis;
and regulate B-cell proliferation, the cleavage of extracellular matrix proteins, the induction of
cytokine secretion, the activation of cytokines and the control of viral infections [34]. The main focus
of this study was granzyme B and granzyme M.
1.3.1 Granzyme B
Recombinant granzyme B is the most important and well-studied member of the human granzyme
family. Its inherent pro-apoptotic activity has been demonstrated in vitro in several proof-of-concept
studies including Stahnke et al. (AML), Liu et al. (melanoma), Dalken et al. (breast carcinoma) and
Kanatani et al. (ovarian, breast, endometrial and prostate carcinoma) (Table 9). Granzyme B is also
the most powerful pro-apoptotic member of the serine proteases stored as a macro-molecular
complex with the proteoglycan serglycin and the pore forming protein perforin within CTLs and NK
cells [35]. During NK cell mediated immune surveillance, secretory granules are depleted into the
immunological synapse formed after specific recognition of abnormal cells via an elaborated
repertoire of cell surface receptors [36]. The precise mechanism by which granzyme B is taken up
into cells and released from the endosome into the cytosol is still controversial. Two possible models
for delivery of granzyme B into target cells, which have been reviewed recently, are an endocytosis
dependent mechanism of vesicle disruption or a direct pore-mediated delivery of the apoptosis
inducing cargo. Following the recognition of the target cell, granules release their cytolytic content
into the immunological synapse that directs granzyme B, most likely by the assistance of perforin, to
the target cell’s cytosol [37]. Once released, granzyme B recruits multiple signal transduction
pathways to fulfill its death inducing function.

6 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
The classical apoptosis induction by granzyme B is comparable to the Fas mediated pathway. It
involves the cleavage of several executive and downstream caspases and is mediated by both the
caspase-pathway and the BID (BH3-interacting domain death agonist) -pathway. Direct activation of
the effector caspase 3 is controlled by the inhibitor of apoptosis protein (IAP) that has to be
inactivated by e.g. HtrA2/OMI, a pro-apoptotic molecule released from disrupted mitochondria (see
below). Successful processing of pro-caspase 3 leads to the cleavage of several downstream death
substrates including the inhibitor of the caspase-activated DNase (CAD) ICAD, poly(ADP ribose)
polymerase (PARP), DNA-dependent protein kinase (DNA-PK), the nuclear mitotic apparatus protein
(NuMA) and the nuclear-envelope intermediate-filament protein lamin B. This results in the typical
morphological changes associated with classical apoptosis, i. e. DNA fragmentation, chromatin
condensation, membrane blebbing, cell shrinkage and the formation of apoptotic bodies. The
activation of the BID pathway, either directly by granzyme B or indirectly via the activation of
caspase-8, leads to the cleavage of mitochondrial BID involving BAX (BCL-2-associated X protein) and
BAK (BCL-2 antagonist). The translocation of tBID (truncated BID) to the mitochondria results in the
permeabilization of the outer mitochondrial membrane and thus mitochondrial depolarization and
the subsequent release of cytochrome c (cyt c) and other pro-apoptotic molecules like endonuclease
G and HtrA2/OMI. Cyt c presence in the cytosol is crucial for the apoptosome formation by assembly
to the apoptotic protease activation factor APAF-1 which in turn leads to autocatalytical activation of
pro-caspase-9 and subsequent processing of the effector caspase-3. In addition to the BID-mediated
loss of mitochondrial transmembrane potential, granzyme B can directly induce mitochondrial
depolarization without the assistance of cytosolic mediators, but the exact mechanisms remain
unclear [4,38].
Granzyme B is also known as ‘Asp-ase’ because it cleaves downstream of aspartic or glutamic acid
residues giving it substrate specificity similar to that of caspases. Consequently, granzyme B can
directly target and activate the downstream caspase substrates ICAD, PARP, DNA-PK, NuMA and
lamin B. Taken together, these modes of action show that granzyme B is not only a highly potent
inducer of apoptosis due to its status as the major mediator of cellular immunity, but also because of
its ability to induce apoptosis at multiple levels using distinct pathways. Thus granzyme B has the
potential to circumvent anti-apoptotic mechanisms of the tumor cells, as described below (1.4) [4].
Recombinant expression of granzyme B has been proven successful in different expression systems
of pro- and eukaryotic origin including Escherichia coli (E. coli), Pichia pastoris (P. pastoris), insect
cells and mammalian cells such as HEK293T, HeLa, Jurkat and COS [4,37]. To be active granzyme B
requires a free N-terminus. The native enzyme is expressed as an inactive zymogen containing an N-
terminal prepro-leader sequence. The signal peptide is cleaved by a signal peptidase in the
endoplasmic reticulum to produce an inactive proenzyme with an N-terminal pro-peptide, protecting

7 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
the cell from the induction of apoptosis, thus the pro-peptide must be removed to generate an active
protease [39]. In the Golgi apparatus, a mannose-6-phosphate tag is attached which directs
granzyme B to the cytotoxic granules. Here the pro-peptide is removed by the dipeptidyl peptidase I
(DPPI) (also known as cathepsin C) [40]. Several strategies have been developed to accomplish the
correct in vitro or in vivo processing and activation of recombinant granzyme B, including the here
applied insertion of an enterokinase cleavage site (ECS) upstream of the sequence of the mature
granzyme B polypeptide, allowing activation after purification by in vitro processing. Secretory
expression of the active protein in P. Pastoris can be achieved by the direct N-terminal fusion of the
mature polypeptide to the Saccharomyces cerevisiae (S. cerevisiae) mating factor α prepro-leader
sequence allowing activation by local Kex2-protease. Secretion and functional in vivo activation by a
host cell signal peptidase has been achieved in insect cells and COS cells using the native sequence of
the granzyme B precursor protein with the propeptide deleted, or the genetic fusion of a furin
recognition motif allowing in vivo processing by endogenous P. pastoris furin-like proteases. In
addition, in vitro and in vivo activation has been achieved by either co-expression of rat DPPI in COS
cells or subsequent incubation with bovine spleen DPPI [4].
The crystal structure of granzyme B has been elucidated before by two groups [41,42]. As for all
proteases, the catalytic activity of granzyme B depends on a serine residue at the active site, one of a
triad of residues corresponding to His57, Asp102 and Ser195 in chymotrypsin. The key residue for
contact with the P11 substrate residue is Arg226 [34]. The optimal tetrapeptide recognition motif (P4-
P1) is IEPD, which is similar to that of caspase 8 and caspase 9 and thereby reflects their common
role in the activation of downstream caspases such as caspase-3 [42].
1.3.2 Granzyme M
Granzyme M is classified as ‘Met-ase’ since it cleaves specifically after amino acids with long aliphatic
side chains such as methionine, leucine, or norleucine at the P1 position1 [43]. It is predominantly
expressed within NK cells, NKT cells, γd-T cells and CD8+ effector T cells [44,45]. Recently, it was
shown that granzyme M is involved in TLR4-driven inflammation and endotoxicosis, and as such it
contributes to the inflammatory response to bacterial lipopolysaccharide by inducing serum
cytokines such as interferon (IFN) γ, IL-1α, IL-1β and TNF α [46]. However, the exact mechanism
remains unclear.
Many groups have reported the cytotoxic efficacy of granzyme M towards tumor cells [47,48].
However, the precise molecular mechanism remains controversial in the literature. It has been
1 The active sites in enzymes can be divided in subsides (S) which consist of single amino acids. The corresponding positions (P) of the substrate have the same numbering as the subsides they occupy. The positions are counted from the point of cleavage. P1 is the primary determinant of specificity.

8 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
described that granzyme M activates a caspase-dependent pathway involving cleavage of ICAD,
poly(ADPribose) polymerase (PARP), heat shock protein 75 (HSP 75), survivin and ezrin as well as the
Fas-associated protein with death domain (FADD), but not Bid [49-52]. Thus, as described for
granzyme B, classical mechanisms of apoptosis are triggered such as phosphatidyl serine (PS)
exposure, mitochondrial swelling and loss of mitochondrial transmembrane potential, cyt c release,
and reactive oxygen species (ROS) production. Other groups did not report those effects, but rather a
caspase-independent form of cell death which is not blocked by Bcl-2 expression [53-55]. Other
substrates of granzyme M can be nucleophosmin [54] and α-tubulin leading to a disruption of the
microtubule network [53]. Granzyme M also specifically cleaves the granzyme B inhibitor PI-9 [56],
therefore the application of this enzyme increases the potential to kill even immune-resistant tumor
cells, particularly when applied in combination with granzyme B (see 1.4).
As with granzyme B, granzyme M requires a free N-terminus for enzymatic activity. Recombinant
expression of the active protein has so far only been reported to be successful in the eukaryotic
expression system P. pastoris featuring the S. cerevisiae mating factor α prepro-leader sequence as
described above for granzyme B (1.3.1).
1.4 TUMOR ESCAPE MECHANISMS TO EVADE IMMUNOSURVEILLANCE
There are many examples of the immune system operating as a significant barrier to tumor
formation and progression, especially with non-virus-induced cancer types [57]. This has been proven
by experiments where mice that are deficient in various components of the immune system (e.g.
missing functions of CTLs and NK cells) have increased tumor development [58]. This is further
illustrated in patients with colon and ovarian tumors where prognosis was better if the tumors were
heavily infiltrated with CTLs and NK cells [59].
However, there is also evidence that tumor cells are able to escape the body’s immune surveillance,
thus avoiding this barrier to tumorigenesis and tumor progression. Infiltrating immune cells can bind
to cancer cells and thereby take the place of the tumor’s disconnected epithelial surrounding cells,
allowing them to survive in ectopic microenvironments by suppression of cell death pathway
stimulation [2]. Furthermore, actively immunosuppressive tumors can recruit infiltrating immune
cells, which comprise regulatory T cells and myeloid-derived suppressor cells. Both types are
suppressive towards the activity of CTLs.
Another example of an escape mechanism of tumor cells is the secretion of immunosuppressive
factors such as TGF-β leading to paralysis of CTLs and NK cells [60]. In several studies on breast
cancer it was also shown that tumor-associated macrophages provide survival signals to cancer cells

9 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
such that tissue-protective and therapy-induced mechanisms no longer trigger cell death [61]. Tumor
cells’ strategies to overcome innate and adaptive immune responses also include impaired tumor
antigen presentation by the downregulation of MHC class I molecules, and the expression of factors
that either directly kill or inhibit effector cells or recruit regulatory T-cells and thereby induce
immunotolerance [62]. In addition, tumors can regulate apoptosis by the overexpression of different
classes of anti-apoptotic proteins. For example, FLICE-inhibitory proteins (FLIPs) interfere with signal
transduction by death receptor pathways, anti-apoptotic proteins of the Bcl-2 family regulate
apoptosis at the mitochondrial level and IAP (inhibitors of apoptosis proteins) family members inhibit
caspase activity [4]. Another escape mechanism from immune response, which is discussed in detail
in chapter 1.4.1, is displayed by the expression of serpin B9 (PI-9) in many tumor cells. PI-9 is the
main inhibitor of granzyme B (1.3.1).
Over the last decade the importance of cancer stem cells has also been discussed (1.1), noting that
tumors are maintained by their own stem cells. Central for this concept is the observation that not all
tumors cells are equal. Conventional therapeutic approaches like chemotherapy or radiation may kill
the majority of the tumor cells, but rare cancer stem cells may be able to survive and later multiply.
Additionally, quiescent tumor cells that have drifted to distant sites, will avoid removal by curative
surgical treatment of a primary tumor and this may be responsible for metastasis that can appear
many years after the surgical treatment [63].
The above described new findings, especially the complexity of the tumors, and the presence of
infiltrating immune cells and the maintenance of the tumors by cancer stem cells are connected with
unfavorable clinical outcome [2], greatly influence the development of new treatment approaches
against human cancer. Development of new treatment approaches requires consideration of the
many different aspects and influences within the tumor microenvironment. These aspects and
influences show that combinatorial therapies will be more and more important in eliminating as
many of these multiple characteristic malignant cells as possible, including the ‘heart’ of the tumor -
cancer stem cells. Therapeutics comprising multiple apoptosis induction mechanisms are highly
advantageous to overcome the resistance of tumors and to eliminate residual malignant cells.
1.4.1 The granzyme B inhibitor PI-9
As mentioned above, the expression of the granzyme B inhibitor PI-9 plays an important role in one
tumor cell escape mechanism from immune response (1.4). PI-9 is a member of the clade B serpin
superfamily, a group of structurally-related proteins that regulate proteases in the vertebrate
adaptive and innate immune systems [64,65]. Since granzymes are an integral component of the
human immune system they are tightly controlled at the posttranslational level by serpins. PI-9 is the
best-known granzyme-regulating serpin. This 42 kDa protein accumulates in the cytosol of

10 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
lymphocytes to avoid self-inflicted injury caused by granule leakage and the subsequently
misdirected activity of granzyme B. Cells at immune-privileged sites therefore express PI-9. These
sites include the lens capsula, ovaries, placenta, testes and embryonic stem cells [66,67]. The
inhibition of granzyme B by PI-9 involves a stoichiometric interaction, leading to the irreversible
inactivation of both the enzyme and the inhibitor [4]. Many studies have focused on the expression
profile of PI-9 within different cell types and its induction by different stimuli (see also 4.2). In
addition to effector T cells, PI-9 is expressed by inflammatory cells [68] and in human peripheral
blood mesenchymal stem cells [69]. It is also present in cells infected by cytomegalovirus (CMV),
Epstein-Barr virus (EBV) and the BK polyomavirus [70-72] as well as in endothelial, mesothelial cells
[73] and dendritic cells [66]. PI-9 can be induced in different cell lines by nuclear factor κB (NFκB),
activator protein-1 (AP-1), as well as pro-inflammatory cytokines such as TNFα, and
lipopolysaccharides [68,72,74]. It is also induced by phorbol myristate acetate and IL-2 in CD8+ T cells
[75,76], by viral dsRNA sensors in human renal tubular epithelial cells [70] and by estrogens and
estrogen receptor-α in breast cancer cells [77]. The expression of PI-9 in renal tubular cells is
upregulated following renal allograft rejection, suggesting that its induction could be used to confer
resistance against granzyme B-mediated cytotoxic effects leading to improved graft survival [78].
The widespread expression of PI-9 has to be taken into account when developing therapeutic
strategies based on granzyme B, particularly because malignant cells can also involve multiple
mechanisms to escape the pro-apoptotic signals induced by CTLs and NK cells [79]. Hence, many
studies have reported a direct correlation between the presence of PI-9 and a reduced pro-apoptotic
granzyme B activity [64,80]. Examples for tumor cells that have been found to express PI-9 to
circumvent immune response and therefore might be resistant to granzyme B based
immunotherapeutics are summarized in Table 10 and Table 11. In addition, high expression of PI-9
has been discussed in context of poor clinical prognosis (compare 4.2).
The crystal structure of PI-9 has not been elucidated yet, but serpins share a characteristic
metastable structure and efficient inhibitory activity [65]. The three-dimensional structure comprises
nine α-helices, three β-helices and a variable reactive center loop (RCL), which is the primary site of
interaction with target proteases and is therefore critical for serpin specificity. Members of this
protein family have high sequence homology in a region of about 350 amino acids and are presumed
to have structural similarity [81]. The amino acid residues in the RCL of PI-9 are necessary for its
activity and the residue at the P1 position is a glutamic acid [82,83], explaining its specificity for
granzyme B which favors acidic P1 amino acids (1.3.1). The RCL of serpins acts as a pseudo-substrate,
which is recognized by the protease so the two molecules dock and form a reversible ‘Michaelis
complex’ (initial enzyme-substrate complex) (Figure 1). Rapidly, the protease cleaves at the favored
P1 position resulting in major structural rearrangements within the serpin that involve alternative

Introduction _____________________________________________________________________________________________________________________________________________________________________________________________________________
conformations for the RCL, β-sheet A and the
stoichiometric complex, in which both proteins remain inactive
substrate. The interaction between
(stoichiometry of inhibition) ~ 1
intensively and during their studies they
binds less efficiently to PI-9 [86]. This mutant is used as
Figure 1: X-ray crystal structure of the non
(purple) and S195A trypsin (cyan) (PDB file 1K9O
To visualize the initial complex, mutated proteins were used. the surface of the protease. This complex can be used to superimpose the crystal structure of uncomplexed granzyme B (PDB file 1IAU) and the
1.5 HODGKIN LYMPHOMA
The above mentioned infiltrating immune cells
lymphoma (HL). 150 years ago Thomas Hodgkin already described several cases of a disea
was later associated with Hodgkin’s diseas
disease. Its hallmarks are the B
Reed-Sternberg cells (HRS cells), first published in 1900
1 % of cells in the tumor tissue, although some cases may show more than 10
majority of cells in Hodgkin lymphoma lesions are a mixed infiltrate of various types of cells of the
immune system including T cells, B cells, plasma cells, neutrophils,
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
sheet A and the attached strand 1 of β-sheet resulting in a covalent
stoichiometric complex, in which both proteins remain inactive [84,85]. Thus, PI
substrate. The interaction between granzyme B and PI-9 has a Kass value of 1.7 × 10
~ 1 [4,82]. Sun et al. investigated the importance of the RCL of PI
ensively and during their studies they revealed that a mutant of granzyme B
. This mutant is used as a reference in this study.
ray crystal structure of the non-covalent Michaelis complex of A353K Manduca sexta
(PDB file 1K9O [87]).
mutated proteins were used. The RCL of the serpin interacts This complex can be used to superimpose the crystal structure of uncomplexed
and the active serpin 1K (PDB file 1SEK) for further investigations of interactions
HODGKIN LYMPHOMA AND THE CD30 RECEPTOR
infiltrating immune cells (1.4) play a key role in the development of Hodgkin
Thomas Hodgkin already described several cases of a disea
was later associated with Hodgkin’s disease or better HL since it was realized that it is a lymphoid
B-cell derived mononucleated Hodgkin cells and the multinucleated
), first published in 1900 [89,90]. They usually account for only about
cells in the tumor tissue, although some cases may show more than 10
majority of cells in Hodgkin lymphoma lesions are a mixed infiltrate of various types of cells of the
including T cells, B cells, plasma cells, neutrophils, eosinophils and mast cells
Protease
(S195A rat trypsin)
Serpin
(A353K Manduca sexta
RCL
11 ________________________________________________________________________________________________________
sheet resulting in a covalent
. Thus, PI-9 acts as a suicide
value of 1.7 × 106 M−1s−1; SI
investigated the importance of the RCL of PI-9
B (granzyme B K27A)
Manduca sexta serpin B1
interacts extensively with This complex can be used to superimpose the crystal structure of uncomplexed
for further investigations of interactions.
play a key role in the development of Hodgkin
Thomas Hodgkin already described several cases of a disease [88] that
since it was realized that it is a lymphoid
mononucleated Hodgkin cells and the multinucleated
usually account for only about
cells in the tumor tissue, although some cases may show more than 10 % HRS cells. The
majority of cells in Hodgkin lymphoma lesions are a mixed infiltrate of various types of cells of the
eosinophils and mast cells [91].
Protease
(S195A rat trypsin)
Manduca sexta B1)

12 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Today HL is sub-divided according to differences in morphology and phenotype of the cells and the
composition of the cellular infiltrate into classical HL (cHL, 95 % of all cases) and nodular lymphocyte-
predominant HL (NLPHL, 5 % of all cases) [91]. CHL is further classified into nodular sclerosis, mixed
cellularity, lymphocyte depletion and lymphocyte-rich HL lymphoma. HRS cells of cHL probably
originate from germinal center B cells that have acquired disadvantageous immunoglobulin variable
chain gene mutations which usually results in cell death. Lymphocytic and histiocytic cells of NLPHL
apparently derive from antigen-selected germinal center B cells. Only a few cases of cHL originate
from T cells. The extent to which the cHL cells undergo reprogramming of gene expression is unique
among human lymphomas. They succeed in expression of multiple genes that are actually typical for
other types of cells within the immune system and lose the expression of most B cell-typical genes
[92]. NFkB, a key anti-apoptosis factor in HL, is one of the signaling pathways and transcription
factors that are de-regulated within HRS cells [93]. This transcription factor is activated by various
external stimuli of which many trigger inflammation as well. Its inactive form is present in the
cytoplasm of almost every cell type, bound to an inhibitory protein called IkB, of which IkBα is the
best studied. Upon stimulation of NFkB, IkB kinases are activated which lead to phosphorylation and
ubiquitin-mediated degradation of IkB, so active NFkB is released. NFkB activity comprises its
translocation into the nucleus where it induces the expression of dozens of different genes involved
in cellular processes as diverse as the immune system, in inflammation, cell proliferation, and
inhibition of apoptosis as well as in angiogenesis, invasion and metastasis [94,95]. Recent studies
showed that the constitutive activation of NFkB in chronic inflammation is a central molecular link
between inflammation and cancer development [96]. NFkB was found to be expressed in many HL
cell lines as well [93]. Discussions are ongoing if stimulation of CD30, a receptor highly over-
expressed on HL cells, activates NFkB, leading to increased resistance of the cells towards apoptosis
[97-100].
Other hypotheses on how HRS cells circumvent immune attack by CTLs or NK cells include the
formation of an inflammatory microenvironment resulting from the attraction of many nonmalignant
inflammatory cells into the lymphoma tissue. Thus, unlike most other human malignancies, the HRS
cells are vastly outnumbered by the surrounding nonmalignant inflammatory cells. 99 % of the tumor
mass consists of reactive inflammatory cells (lymphocytes, histiocytes, eosinophils, fibroblasts,
neutrophils, plasma cells) [96]. Thus, the histological appearance is reminiscent of an inflammatory
process as seen for viral infections, which supports the widely accepted finding of a causative role of
EBV infection in HL in epidemiological studies. In 40 % of cHL cases in the western world, HRS cells
contain EBV [101,102].
HL is one of the most curable cancer types. In recent years combinatorial treatment approaches
using chemotherapy and radiation therapy have led to promising cure rates. The estimated 5- and

13 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
10- year relative survival for patients with HL is 85 % and 80 %, respectively [103,104]. However,
patients suffer from high toxicity of current therapeutic regimens and in addition there are still
patients that relapse even after autologous hematopoietic stem-cell transplantation which is
reserved for patients with recurrent or progressive HL after failure of conventional therapy. Those
patients have bad prognosis and limited therapeutic options. Hence, efforts have been made to find
alternative therapies for those resistant cancer subtypes.
The CD30 receptor is an activation-induced antigen that is predominantly expressed on lymphoid
cells. CD30 has been shown to be over-expressed on malignant cells of HL, anaplastic large cell
lymphoma (ALCL) and adult T cell leukemia. Therefore it is a promising target for the treatment of
those HL patients which do not respond to standard therapy [105,106]. CD30 is a glycosylated type I
transmembrane protein and belongs to the TNF receptor superfamily [107]. It can be proteolytically
cleaved in close proximity to the cell membrane by the membrane-anchored metalloproteinase TNF-
α converting enzyme (TACE), leading to the release of the soluble ectodomain of CD30. This is found
at low levels in the serum of healthy donors but is detected at elevated levels in patients suffering
from CD30-positive neoplasms, viral infections, and autoimmune disorders. Hence, in most cases the
level of soluble CD30 provides a prognostic marker [108].
The most recent novel immunotherapeutic agent that targets CD30 is the above mentioned
Brentuximab Vedotin (1.2.1) for the treatment of HL patients that have not responded to prior
chemotherapy or autologous stem cell transplantation. Other immunotherapeutic agents targeting
this receptor have been described in context of in vitro, pre-clinical and clinical studies. Apart from a
CFP consisting of the human RNase angiogenin and the natural ligand of CD30 (CD30L) [109] there
are several other anti-CD30 human IL-2 fusion proteins that have Ber-H2(scFv), among others, as a
binding domain [110]. In addition the construct Ki4(scFv)-ETA’, which as with Ber-H2(scFv), does not
bind soluble CD30 receptor and even prevents shedding [111], has shown promising results in vitro
and in vivo [112]. However, it has the disadvantage of containing a toxic compound of non-human
origin that has the potential to provoke undesirable immune responses. Therefore, fusion to human
effector molecules would be advantageous.

14 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
1.6 CHRONIC AND ACUTE MYELOMONOCYTIC LEUKEMIA AND THE CD64
RECEPTOR
Chronic myelomonocytic leukemia (CMML) is one of the three rare myeloid neoplasms termed
myelodysplastic/ myeloproliferative diseases according to the World Health Organization (WHO).
This clonal hematopoietic stem cell disorder is characterized by persistent monocytosis in peripheral
blood, at least one dysplasia component within the bone marrow and a percentage lower than 20 %
of promonocytes and blasts in peripheral blood and bone marrow [113]. It can be further classified
depending on the amount of blasts into CMML 1 (< 5 % blasts in peripheral blood, < 10 % blasts in
bone marrow) and CMML 2 (< 20 % blasts in peripheral blood, 10 - 19 % blasts in bone marrow)
[114]. CMML mainly occurs in elderly people with median survival of 15 – 20 months and leukemic
transformation rates in 15 – 30 % (both depending on type 1 or 2) [115]. Other diagnostic criteria
include immunophenotyping, whereby CMML and monocytic AML show overlapping antigen
expression patterns. Only the expression of CD56 combined with underexpression of a myeloid
marker like CD15 or CD13 seems to be unique to CMML monocytes [116]. Furthermore an over-
expression of CD33 [117,118] and also of CD14 on marrow monocytes was found in CMML compared
to reactive monocytosis and normal marrow samples [116]. In addition, clonal cytogenetic
abnormalities are found in 20 – 40 % of patients with CMML, but none is specific [119-121]. The close
similarity among the other myelodysplastic/ myeloproliferative diseases such as atypical chronic
myeloid leukemia (CML) and juvenile myelomonocytic leukemia (JMML) means a constant challenge
for diagnosis as well as the appropriate therapy. New molecular markers have been described in
recent years such as mutations in TET2 (Tet Methylcytosine Dioxygenase 2) or CBL (Casitas B-lineage
Lymphoma) [122]. However, these aberrations are non specific for CMML and no targeted therapies
are available for these genetic changes today. Because of this and its rare occurrence, the
establishment of new therapeutic approaches specific to CMML is difficult.
Conventional treatment of CMML is very heterogeneous. It usually starts with the ‘watch and wait
strategy’. The only curative approach for CMML is allogeneic hematopoietic stem cell
transplantation, but this treatment option is often not applicable in patients affected by CMML due
to the age distribution of the disease. Actually, the gold standard treatment is to decrease the
amount of white blood cells in peripheral blood in the beginning with low dose cytarabine,
hydroxycarbamide or etoposide. Another option is supportive care with transfusion. Depending on
the variant of the disease, compounds which had been used to treat myelodysplastic syndrome were
explored for usage in CMML, especially the demethylating agents azacytidin und decitabin,
topoisomerase inhibitors such as valproic acid and farnesyltransferase inhibitors like lonafarnib.
However, information on the significance of these drugs is very limited due to the rare occurrence of

15 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
CMML [123]. In patients with CMML 2, a treatment with DNA methyltransferase-inhibitors has also
been shown to be effective by reducing the rate of transformation into AML and to reduce the
transfusion need. However approaches based on molecular or biological characteristics of the
disease are urgently needed [123]. Those new treatment strategies are unmet medical need for
these patients.
In some patients with a history of CMML, the disease converts into acute myelomonocytic leukemia
(AMML) which is also called AML-M4, the fourth sub-category of AML according to FAB (French-
American-British) classification. This disease is diagnosed in 15-20 % of all AML cases. It occurs in all
age groups, but is more common in older adults, and is initially treated with chemotherapy. AML-M4
cases have a population of < 20 % myeloblasts and monoblasts and < 80 % of total nucleated cells are
monoblasts. As indicated above, the expression patterns of AMML do not distinguish significantly
from those of CMML. In general, myeloblasts express CD13, CD33, CD34 and CD117 whereas
monocytoid cells including monoblasts and promonocytes express CD11b, CD11c, CD13, CD14, CD33
and CD64 [114,118]. Treatment of AMML includes usually intensive multidrug chemotherapy and in
some cases allogeneic hematopoietic stem cell transplantation.
Treatment approaches of other leukemic diseases such as AML comprise targeting of the 72 kDa
glycoprotein CD64, the high affinity receptor for human IgG (Fc γ receptor I, FcγRI), which is
expressed on cells of the myeloid lineage, i. e. monocytes, macrophages and dendritic cells, but is
also over-expressed on the surface of leukemic cells [124,125]. The corresponding cells can be
activated by several cytokines, such as IFNγ, which plays a crucial role in inflammatory responses. It
also leads to a 5- to 10-fold increase of CD64 expression as well as enhanced receptor activity [126],
whereby the amount of CD64 receptors is no prime requisite for sensitivity of the cells, but rather
their activation state. This means that an increased CD64 expression is not necessarily accompanied
by a greater sensitivity to cell-death inducing agents, but rather their corresponding activation via
e.g. IFNγ [127,128]. The advantages of CD64 for targeted delivery of cytotoxic effector molecules
include its highly efficient internalization rate and its limited expression, so normal, non-activated
CD64-expressing cells are not affected by the fusion proteins [127,129,130]. The specific killing of
target cells via CD64 can be accomplished by the fusion of cell-death inducing moieties to H22(scFv),
a humanized single chain antibody fragment [131,132]. The potential of fusions of H22(scFv) to both
Ricin A and ETA' as therapeutic agents against AML or inflammatory cells has been shown previously
[129,133] as well as the efficacy of Gb-H22(scFv) against AML cell lines and primary cells (Table 9).

16 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
1.7 MOLECULAR MODELING
Molecular modeling can be applied to model or mimic the behavior of molecules and for studying the
interaction between more than one molecule and large molecular systems. The underlying features
of the computational calculations use atoms as the smallest individual unit. Alanine scanning is a fast
computational approach for the prediction of energetically important amino acid residues in protein-
protein interfaces [134,135]. To determine the role of a specific side chain group of a specific residue,
systematic mutations to alanine are performed. Alanine has a small and inert functional group;
therefore it is possible to measure the effect of the deletion of potential crucial side chains. The
output is a list of ‘hot spots’ or amino acid side chains that are predicted to significantly destabilize
the interface when mutated to alanine [136]. Despite the large size of binding interfaces, single
residues can contribute to a large fraction of the binding free energy in an interface [134]. The
disadvantages of alanine scanning are that it does not allow the investigation of the impact of
mutations other than alanine; it does not include hydration at the protein-protein interface in spite
of the fact that water molecules may play a very important role for complex stability; and, it does not
fully take into account the complete flexibility properties of the complex. Hence, alanine scanning
provides only very approximate results. To overcome these limitations, these first predictions are
succeeded by molecular dynamics (MD) simulations in a vacuum (gas-phase simulation), in the
presence of solvent molecules (explicit solvent simulation) or in empirical mathematically expressed
solvents (implicit solvent simulation). Subsequently, those calculations allow predicting which of the
suggested mutation sites (from alanine scanning) destabilize the determined protein-protein
complex the most.
Today, molecular modeling methods are routinely used in pharmaceutical research to investigate
protein folding, enzyme catalysis, protein stability, conformational changes associated with bio-
molecular function, and molecular recognition of proteins, DNA, and membrane complexes to e.g.
design novel drug candidates.

17 Introduction
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
1.8 AIMS AND OBJECTIVES
Conventional cancer treatment usually involves chemotherapy and radiotherapy, which can cause
severe side effects. More targeted approaches are therefore preferred, such as immunotherapy
based on immunotoxins (ITs) comprising an antibody-based ligand that specifically binds to diseased
cells, and a pro-apoptotic protein. However, if the toxin component is derived from bacteria or plants
it can trigger a neutralizing immune response; therefore, human effector molecules are more
suitable. This results in the fourth generation of ITs, human CFPs. The most prominent example of
these fourth generation ITs, that has been successfully tested in cytotoxicity assays against different
cancer cells in vitro and ex vivo, comprises granzyme B as pro-apoptotic candidate. There are three
main reasons for the application of granzyme B as part of CFPs. First, it is a human effector molecule
so the risk of immunogenic responses is limited. Second, the use of granzyme B is not only beneficial
due to its high apoptotic efficiency as the major mediator of cellular immunity, but also because of its
capacity to induce apoptosis at multiple levels so the cancer cells’ lack of sensitivity due to
deregulated apoptotic pathways or overexpression of certain inhibitors may be overcome. Third, it is
relatively flexible concerning heterologous expression. On the downside, during the last decade, the
presence of the endogenous inhibitor PI-9 has been discovered in different types of cancer cells. This
diminishes the universal potential of granzyme B in targeted tumor therapy. Since the expression of
PI-9 has especially been described in the context of treatment-resistant cells and poor clinical
outcome, it becomes evident that alternative therapeutic approaches become necessary to kill these
cells. The aim of this work was the evaluation of human CFPs which induce cell-death independent of
the endogenous expression of PI-9. This should be achieved by highly cytotoxic granzyme B mutants
resistant to PI-9 inhibition.
The following steps will be pursued to achieve this goal (overview shown in Figure 2):
• Elucidation of the most important amino acids responsible for the interaction between the
serine protease and the RCL of its specific inhibitor by molecular homology modeling.
• Identification of suitable amino acids to replace the identified residues with the aim to
prevent an irreversible inactivation of the enzyme by covalent complex formation without
affecting proteolytic activity.
• Verification of the desired effects in silico (cooperation with German Research School for
Simulation Sciences in Juelich, Prof. Dr. rer. nat. Paolo Carloni).
• Generation of different fusion constructs comprising granzyme B and the CD30-targeting
Ki4(scFv) and CD64-targeting H22(scFv), mammalian secretory expression and purification of
the granzyme B mutants.
• Identification of the most promising mutants by in vitro PI-9 inhibition assay.

Introduction _____________________________________________________________________________________________________________________________________________________________________________________________________________
• Set up of a cell-based test system to identify the
• Establishment of xenograft tumor models
towards PI-9-positive and
• Ex vivo verification of the efficacy of the selected mutant on
primary monocytic cells from
Another potent granzyme, granzyme
of human effector molecules for CPFs
effective in killing tumor cells. I
PI-9, so its application increases the potential to kill even
the way for combinatorial treatment with granzyme
The following steps were taken
granzyme-based CFPs:
• Generation of different
Ki4(scFv) and CD64-targeting H22(scFv), mammalian secretory expression and purification
• Determination of the enz
• Verification of the cytotoxic functionality
lines in vitro and CMML and AMML patient
granzyme B-based constructs.
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
based test system to identify the optimal mutant.
Establishment of xenograft tumor models using HL cell lines to confirm anti
positive and -negative tumor cells in vivo.
verification of the efficacy of the selected mutant on PI-9-positive and
primary monocytic cells from CMML and AMML patients.
granzyme M, will also be investigated in this study to extent the portfolio
for CPFs. As with granzyme B, granzyme M has been
In addition, granzyme M specifically cleaves the granzyme
its application increases the potential to kill even immune-resistant tumor cells
the way for combinatorial treatment with granzyme B.
taken to verify the potential of this enzyme in context of
Generation of different fusion constructs comprising granzyme M and
targeting H22(scFv), mammalian secretory expression and purification
nzymatic activity of the novel constructs.
Verification of the cytotoxic functionality of PI-9-positive and PI-9-negative
CMML and AMML patient-derived cells ex vivo in comparison to
based constructs.
Figure 2: Workflow of this study.
18 ________________________________________________________________________________________________________
L cell lines to confirm anti-tumor activity
positive and -negative
to extent the portfolio
M has been proven highly
specifically cleaves the granzyme B inhibitor
resistant tumor cells or to pave
to verify the potential of this enzyme in context of novel human
nd the CD30-targeting
targeting H22(scFv), mammalian secretory expression and purification.
negative HL or AML cell
in comparison to

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
19
2 Material and methods
2.1 CHEMICAL AND BIO-CHEMICAL MATERIAL
2.1.1 Equipment
The equipment and devices used in this work are listed below.
Autoclaves: Varioklav (H+P) 75S (Thermo Electron Corporation)
Balances: Precision and analysis balances, Talent: TE6101, TE64, TE31025, TE12000 (Sartorius)
Cell counter: Casy1 (Schaerfe System)
Centrifuges: Table centrifuge 5415D and 5415R (Eppendorf), Multifuge 3 S-R / 1S (Heraeus Instruments), Avanti J-25I Centrifuge (Beckman Coulter), Rotina 420 R (‘Hettich Zentrifuge’)
Cryo sections: Cryostat CM 3050 (Leica)
DNA sequencing: ABI Prism 3730 Capillary-Sequencer (Applied Biosystems)
End-over-end shaker (CMV): Innova TM 4000 incubator shaker or Innova TM 4430 incubator shaker (New Brunswick Scientific)
Flow cytometry: FACScalibur (Becton Dickinson), FACS-Vantage with DiVa option (Becton Dickinson)
FPLC: Aekta FPLC (Amersham Pharmacia Biotech)
Gelelectrophoresis and Electroblotting:
Mini Protean III gel chamber (BioRad Laboratories, Inc.), Protein Gel-Apparatus and supplies Mini PROTEAN II™ (BioRad), Gel Air Dryer (BioRad), Mini Trans-Blot Cell (BioRad)
Ice machine: Icematic D201 (Castel Mac)
Incubators: Thermomixer compact (Eppendorf AG), Incubator Function Line Type UT12 (Heraeus Instruments); cell culture: CD210 (Binder)
Liquid handling: Pipetman P (Gilson), Multichannel Pipettes (Eppendorf)
Microplate reader: ELISAreader ELx808 (Bio-TEK Instruments) and Epoch (Bio-TEK Instruments)
Microscopes: TCS SP5 confocal laser microscope (Leica), DMIL inverted microscope with mercury fluorescence light source (Leica), DM2000 (Leica)
PCR-Cyclers: Primus 96 Plus (MWG-Biotech), Programmable Thermal Controller PTC-200™ (MJ Research Inc.)
pH-Meter: Basic Meter PB-11 (Sartorius)
Photometer: Biophotometer (Eppendorf),
Software: AIDA image analyzer (Raytest), GraphPad Prism 4.0, WinMDI 2.8
Sonication: Bandelin Sonopuls HD 2070 (Bandelin Electronic) Micro tip MS72/73
Transiluminator: Molecular Imager Gel Doc XR System (BioRad)
Waterbaths: TE31025, TE12000 (Sartorius)

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
20
2.1.2 Chemicals and consumables
If not otherwise indicated, the chemicals used were purchased from the following companies in the
highest available purity: BD Biosciences (Heidelberg), Bio-Rad (Munich), Qualilab (Olivet, France),
Merck (Darmstadt), Roche Diagnostics (Mannheim), Roth (Karlsruhe), Invitrogen (San Diego, USA),
Sigma (Munich), MWG-Biotech (Ebersberg), KMF Laborchemie Handels GmbH (Siegburg), Peprotech
GmbH (Hamburg).
Expendable materials were usually purchased from: BD Biosciences (Heidelberg), Biozym
(Oldendorf), Eppendorf (Hamburg), Greiner (Solingen), Invitrogen (San Diego, USA) , Labomedic
(Bonn), Millipore (Eschborn), Pierce Biotechnology (Rockford, USA), Qiagen (Hilden), Schott (Mainz),
Whatman (Bender & Hobheim, Bruchsal), Zeiss (Oberkochen).
2.1.3 Kits, enzymes and standards
If not otherwise indicated, the restriction enzymes, Quick ligase, T4-DNA-Ligase and Phusion High
Fidelity DNA-polymerase used were purchased from: Invitrogen (San Diego, USA), MWG-Biotech
(Ebersberg) and New Englands Biolab (NEB) (Frankfurt am Main).
The following kits from Macherey-Nagel (Dueren) were used for isolation and purification of DNA or
RNA according to the manufacturer’s instructions:
Plasmid isolation kit (Mini): NucleoSpin® Plasmid
Plasmid isolation kit (Maxi): NucleoBond® AX
DNA-purification kit: NucleoSpin® Extract II
RNA purification kit: NucleoSpin® RNA II
Protein standards:
Pre-stained broad range marker: 175 / 80 / 58 / 46 / 30 / 25 / 17 / 7 kDa; #P7708, NEB (Frankfurt am
Main)
Protein broad range marker: 212 / 158 / 116 / /97.2 / 66.4 / 55.6 / 42.7 / 34.6 / 27.0 / 20.0 / 14.3 /
6.5 kDa, #P7702, NEB (Frankfurt am Main)
Nucleic acid standards:
2-log ladder: 10.0 / 8.0 / 6.0 / 5.0 / 4.0 / 3.0 / 2.0 / 1.5 / 1.2 / 1.0 / 0.9 / 0.8 / 0.7 / 0.6 / 0.5 / 0.4 / 0.3
/ 0.2 / 0.1 kb, #N3200, NEB (Frankfurt am Main)

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
21
2.1.4 Synthetic oligonucleotides and plasmids
Synthetic oligonucleotides were purchased from MWG-Biotech (Ebersberg) in standard quality. The
DNA oligonucleotides used in this work are listed in Table 1.
Table 1: List of oligo nucleotides used for cloning and DNA sequencing.
Oligo name Oligo sequence
Sequencing
T7 promotor 5‘-TAATACGACTCACTATAGGG-3‘
Hinterhis rev 3‘-CCAACAGCTGGCCCTCGCAGACAG-5‘
3’Gb seq 3‘-CTTCACCTGGGCCTTGTTGC-5‘
5‘ pMS 5‘-GGGCGGTAGGCGTGTACGGTGGG-3‘
5‘ PI9 seq 5‘-AGTGGAATGAACCGTTTGAC-3‘
3‘ PI9 seq 3‘-CGAGCTTAAACGTGGCCTCC-5‘
Cloning
5‘ pMS 5‘-GGGCGGTAGGCGTGTACGGTGGG-3‘
3‘ Gb (BlpI) 3‘-CGCGCTCAGCGTAGCGTTTCATGGTTTTC-5‘
5‘ PI9 (XbaI) 5‘-GCGTCTAGAGAAACTCTTTCTAATGCAAGTGGTAC-3‘
5‘ PI9 (NheI) 5‘-GCCGCTAGCCACCATGGAAACTCTTTCTAATGC-3‘
3‘ PI9 (NotI) 3‘-CGGCGCGGCCGCTGGCGATGAGAACCTGCCACAG-5‘
3‘ PI9 (EcoRI) 3‘-CGCGAATTCTCATGGCGATGAGAACCTGCCACAG-5‘
SOE primer
5‘ Gb SOE K27A 5‘-CAGAAGTCTCTGGCGAGGTGCGGTGGCTTCC-3‘
5‘ Gb SOE R28A 5‘-CAGAAGTCTCTGAAGGCGTGCGGTGGCTTCC -3‘
5‘ Gb SOE R28E 5‘-CAGAAGTCTCTGAAGGAGTGCGGTGGCTTCC-3‘
5‘ Gb SOE R28K 5‘-CAGAAGTCTCTGAAGAAGTGCGGTGGCTTCC-3‘
5‘-EGb S195A 5‘-CTTTAAGGGGGACGCTGGAGGCCCTCTTGTGTG-3‘
3‘ Gb SOE K27A 3‘-GGAAGCCACCGCACCTCGCCAGAGACTTCTG-5‘
3‘ Gb SOE R28A 3‘-GGAAGCCACCGCACGCCTTCAGAGACTTCTG-5‘
3‘ Gb SOE R28E 3‘-GGAAGCCACCGCACTCCTTCAGAGACTTCTG-5‘
3‘ Gb SOE R28K 3‘-GGAAGCCACCGCACTTCTTCAGAGACTTCTG-5‘
3‘-EGb S195A 3‘-CACACAAGAGGGCCTCCAGCGTCCCCCTTAAAG-5‘

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
22
The underlying plasmids for all molecular cloning procedures were pMS for mammalian expression
[137] or pMT for bacterial expression [133]. All cloning work was adapted to the characteristics of the
chosen vectors, such as corresponding restriction sites, and performed according to standard
methods (2.2).
For expression of all recombinant granzyme B and granzyme M constructs the already published
plasmids pMS-L-EGb-H22-IV [138] and the earlier generated pMS-L-EGb-Ki4-IV and pMS-L-EGb-IV
were applied as template plasmids. The granzyme M sequence was prepared earlier during a
bachelor thesis by PCR using specific primers based on a full-length cDNA clone (IRATp970D0643D)
from the German Resource Centre for Genome Research (RZPD).
For expression of recombinant human PI-9, cDNA was derived from RNA preparations of target cell
lines by reverse transcription and specific primers (2.1.4). The derived sequence was either cloned
into the pMS vector, deleting the Igκ leader sequence for cytosolic mammalian expression
(pMS-PI9-IV) or into the pMT plasmid to express PI-9 in E. coli (pMT-L-PI9).
2.1.5 Antibodies and antibody fragments
The following antibodies and antibody conjugates were used in this work for western blot (2.4.2) or
flow cytometric (2.5) detection of recombinant or soluble proteins with the indicated dilutions:
- GAM-AP (1:5000) or GAM-PO (1:5000) or GAM-FITC (1:100): Goat-anti-mouse polyclonal
antibody directed against the Fc-part of murine IgGs, conjugated to Alkaline Phosphatase
(AP), horseradish peroxidase (PO) or Fluorescein-Isothiocyanat (FITC) (Sigma, Munich)
- anti-His (1:5000): monoclonal mouse-anti-penta-His antibody directed against C- or
N-terminal Histidin Tags (His-tags) of recombinant proteins (Sigma, Munich)
- anti-His Alexa488 (1:300): monoclonal mouse-anti-penta-His Alexa Fluor®488 antibody
directed against the C- or N-His-tags of recombinant proteins, conjugated to fluorescent dye
Alexa Fluor 488 (Qiagen, Hilden)
- anti-human Gb-PE (1:50): monoclonal mouse-anti-granzyme B-PE antibody directed against
human granzyme B, conjugated to R-Phycoerythrin (PE) (BD Bioscience, Heidelberg)
- anti-human Gm (1:500): monoclonal mouse-anti-granzyme M antibody directed against
human granzyme M, clone 4D11 (Abnova, Heidelberg)
- anti-human Gb (1:1000): monoclonal mouse-anti-granzyme B antibody directed against
human granzyme B, clone 2C5 (Santa Cruz, San Diego)
- anti-human PI-9 (1:300): monoclonal mouse-anti-PI-9 antibody directed against human PI-9,
clone 7D8 (Santa Cruz, San Diego)

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
23
The following antibodies were used for the flow cytometric (2.5) detection of surface antigens on
cancer cell lines or primary leukemic cells according to the manufacturer’s instructions. They are
monoclonal mouse-anti-human antibodies conjugated either to FITC, PE, Allophycocyanin (APC),
Alexa 647 or 488 and detect the antigens as indicated by their name.
- anti-CD64-FITC (AbD Serotec, Duesseldorf)
- anti-CD64-488 (Biolegend, San Diego, USA)
- anti-CD64-Alexa 647 (AbD Serotec, Duesseldorf)
- anti-CD14-APC (eBioscience, Frankfurt)
- anti-CD14-FITC (Dako, Hamburg)
- anti-CD56-APC (eBioscience, Frankfurt)
- anti-CD56-PE (eBioscience, Frankfurt)
- anti-CD33-APC (eBioscience, Frankfurt)
- anti-CD25-PE (eBioscience, Frankfurt)
In addition, the single chain antibody fragments (scFv) listed in Table 2 were used in this study.
Table 2: Origin and specificity of antibody fragments.
Antibody/ligand specificity origin/specification
H22(scFv) human CD64 humanized monoclonal antibody H22
Ki4(scFv) human CD30 mouse monoclonal antibody Ki4
425(scFv) human EGF receptor, extracellular domain III (no mouse cross reactivity)
mouse monoclonal antibody 425
2.1.6 Bacterial strains
The following E. coli strains were used for selection and amplification of plasmid DNA, and for
expression of recombinant protein respectively:
DH5α: F- φ80 lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17(rk-, mk+) phoA supE44thi-1 gyrA96
relA1 tonA (resistant to phage T1) (Invitrogen, Darmstadt)
BL21 StarTM (DE3): F- ompT hsdSB (rB-mB-) gal dcm rne131 (DE3) (Invitrogen, Darmstadt)
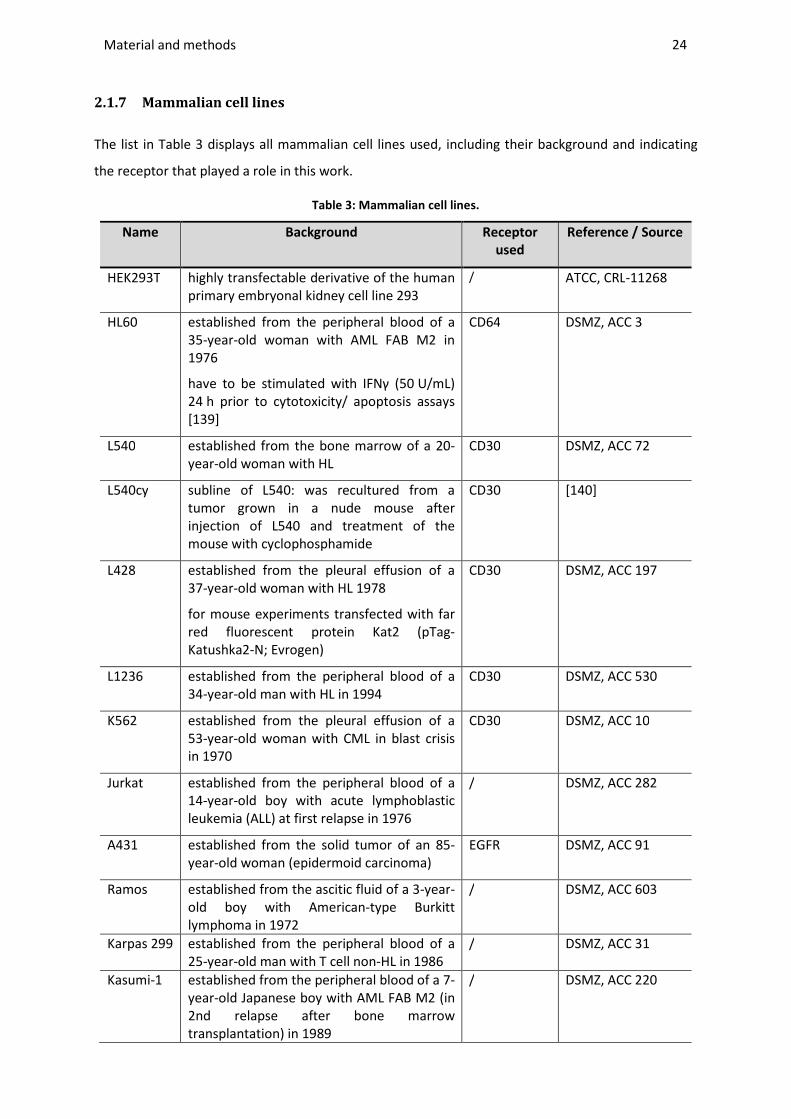
Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
24
2.1.7 Mammalian cell lines
The list in Table 3 displays all mammalian cell lines used, including their background and indicating
the receptor that played a role in this work.
Table 3: Mammalian cell lines.
Name Background Receptor
used
Reference / Source
HEK293T highly transfectable derivative of the human primary embryonal kidney cell line 293
/ ATCC, CRL-11268
HL60 established from the peripheral blood of a 35-year-old woman with AML FAB M2 in 1976
have to be stimulated with IFNγ (50 U/mL) 24 h prior to cytotoxicity/ apoptosis assays [139]
CD64 DSMZ, ACC 3
L540 established from the bone marrow of a 20-year-old woman with HL
CD30 DSMZ, ACC 72
L540cy subline of L540: was recultured from a tumor grown in a nude mouse after injection of L540 and treatment of the mouse with cyclophosphamide
CD30 [140]
L428 established from the pleural effusion of a 37-year-old woman with HL 1978
for mouse experiments transfected with far red fluorescent protein Kat2 (pTag-Katushka2-N; Evrogen)
CD30 DSMZ, ACC 197
L1236 established from the peripheral blood of a 34-year-old man with HL in 1994
CD30 DSMZ, ACC 530
K562 established from the pleural effusion of a 53-year-old woman with CML in blast crisis in 1970
CD30 DSMZ, ACC 10
Jurkat established from the peripheral blood of a 14-year-old boy with acute lymphoblastic leukemia (ALL) at first relapse in 1976
/ DSMZ, ACC 282
A431 established from the solid tumor of an 85-year-old woman (epidermoid carcinoma)
EGFR DSMZ, ACC 91
Ramos established from the ascitic fluid of a 3-year-old boy with American-type Burkitt lymphoma in 1972
/ DSMZ, ACC 603
Karpas 299 established from the peripheral blood of a 25-year-old man with T cell non-HL in 1986
/ DSMZ, ACC 31
Kasumi-1 established from the peripheral blood of a 7-year-old Japanese boy with AML FAB M2 (in 2nd relapse after bone marrow transplantation) in 1989
/ DSMZ, ACC 220
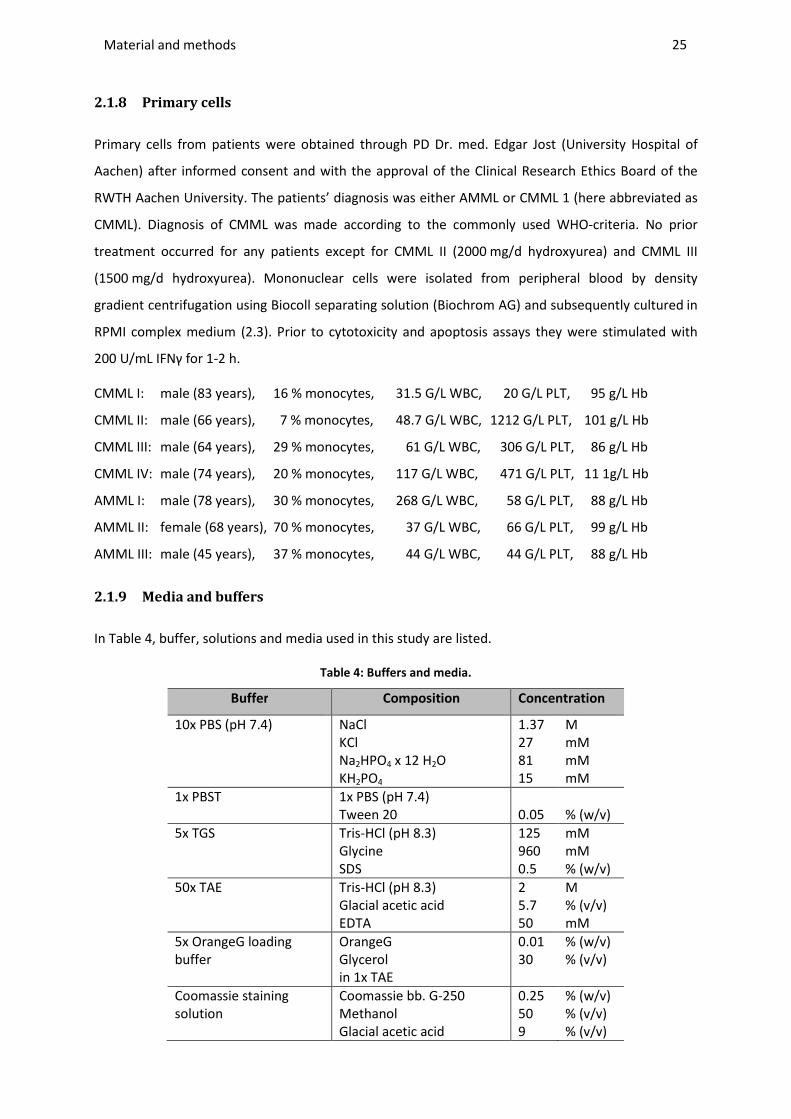
Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
25
2.1.8 Primary cells
Primary cells from patients were obtained through PD Dr. med. Edgar Jost (University Hospital of
Aachen) after informed consent and with the approval of the Clinical Research Ethics Board of the
RWTH Aachen University. The patients’ diagnosis was either AMML or CMML 1 (here abbreviated as
CMML). Diagnosis of CMML was made according to the commonly used WHO-criteria. No prior
treatment occurred for any patients except for CMML II (2000 mg/d hydroxyurea) and CMML III
(1500 mg/d hydroxyurea). Mononuclear cells were isolated from peripheral blood by density
gradient centrifugation using Biocoll separating solution (Biochrom AG) and subsequently cultured in
RPMI complex medium (2.3). Prior to cytotoxicity and apoptosis assays they were stimulated with
200 U/mL IFNγ for 1-2 h.
CMML I: male (83 years), 16 % monocytes, 31.5 G/L WBC, 20 G/L PLT, 95 g/L Hb
CMML II: male (66 years), 7 % monocytes, 48.7 G/L WBC, 1212 G/L PLT, 101 g/L Hb
CMML III: male (64 years), 29 % monocytes, 61 G/L WBC, 306 G/L PLT, 86 g/L Hb
CMML IV: male (74 years), 20 % monocytes, 117 G/L WBC, 471 G/L PLT, 11 1g/L Hb
AMML I: male (78 years), 30 % monocytes, 268 G/L WBC, 58 G/L PLT, 88 g/L Hb
AMML II: female (68 years), 70 % monocytes, 37 G/L WBC, 66 G/L PLT, 99 g/L Hb
AMML III: male (45 years), 37 % monocytes, 44 G/L WBC, 44 G/L PLT, 88 g/L Hb
2.1.9 Media and buffers
In Table 4, buffer, solutions and media used in this study are listed.
Table 4: Buffers and media.
Buffer Composition Concentration
10x PBS (pH 7.4) NaCl KCl Na2HPO4 x 12 H2O KH2PO4
1.37 27 81 15
M mM mM mM
1x PBST 1x PBS (pH 7.4) Tween 20
0.05
% (w/v)
5x TGS Tris-HCl (pH 8.3) Glycine SDS
125 960 0.5
mM mM % (w/v)
50x TAE Tris-HCl (pH 8.3) Glacial acetic acid EDTA
2 5.7 50
M % (v/v) mM
5x OrangeG loading buffer
OrangeG Glycerol in 1x TAE
0.01 30
% (w/v) % (v/v)
Coomassie staining solution
Coomassie bb. G-250 Methanol Glacial acetic acid
0.25 50 9
% (w/v) % (v/v) % (v/v)

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
26
Table 4: Cont.
Buffer Composition Concentration
Coomassie destaining solution
Methanol Glacial acetic acid
10 10
% (v/v) % (v/v)
5x (reducing) protein loading buffer
Tris-HCl (pH 6.8) Glycerol SDS Bromphenolblue (ß-Mercaptoethanol)
62.5 30 4 0.05 10
mM % (v/v) % (w/v) % (w/v) % (v/v)
SDS PAGE gel (separating gel)
Acrylamide/Bisacrylamide (30/1) Tris-HCl (pH 8.8) SDS (10 % (w/v)) TEMED APS (20 % (w/v))
x 375 0.1 0.1 0.1
mL mM % (w/v) % (v/v) % (v/v)
SDS PAGE gel (stacking gel)
Acrylamide/Bisacrylamide (30/1) Tris-HCl (pH 6.8) SDS TEMED APS
5 150 0.1 0.1 0.1
% (w/v) mM % (w/v) % (v/v) % (v/v)
AP buffer Tris-HCl (pH 9.6) NaCl MgCl2
100 100 5
mM mM mM
Transfer buffer for western blotting
Tris-HCl (pH 8.3) Glycine SDS Methanol
25 192 0.05 20
mM mM % (w/v) % (v/v)
Stripping buffer
Tris-HCl (pH 6.8) SDS β-mercaptoethanol
62.5 2 100
mM % (w/v) mM
For FPLC protein purification: 4x binding buffer Tris-HCl (pH 7.4)
NaCl Imidazol
200 1.2 40
mM M mM
Wash buffer Tris-HCl (pH 7.4) NaCl Imidazol
50 300 40
mM mM mM
Elution buffer Tris-HCl (pH 7.4) NaCl Imidazol
50 300 500
mM mM mM
Media
LB (pH 7.5) NaCl Peptone Yeast extract (Agar
1 0.5 1.5 1.6
% (w/v) % (w/v) % (w/v) % (w/v))
SOC Peptone Yeast extract NaCl KCl MgCl2 MgSO4 Glucose
2 % 0,5 % 10 25 10 10 20
(w/v) (w/v) mM mM mM mM mM

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
27
Their application will be described in later chapters. All media were autoclaved for
20 min/121 °C/1 bar. If necessary, buffers were sterile filtrated with a 0.2 µm PES membrane. The
following antibiotics were applied in media for bacterial cultivation, if necessary, with the indicated
concentrations: Ampicillin (100 µg/µL), Kanamycin (50 µg/µL) and Chloramphenicol (25 µg/µL).
2.2 MOLECULAR-BIOLOGICAL METHODS
The described methods followed standard techniques according to Sambrook and Russell [141].
2.2.1 Polymerase chain reaction
The polymerase chain reaction (PCR) for the amplification of DNA segments was performed using
Phusion High-Fidelity DNA Polymerase (NEB), a proofreading polymerase with 3´→5´ exonuclease
activity resulting in higher accuracy during amplification. The standard volume of a PCR procedure
was 50 μL and composed of 1x HF buffer including MgCl2, 10 pmol 5’ primer, 10 pmol 3’ primer,
0.2 mM dNTPs. After the addition of 50 ng of the template and double distilled water (ddH2O),
0.5 % (v/v) polymerase was added.
The following standard protocol with 30 cycles was used:
30 sec 98 °C Loop30 cycles start 10 sec 98 °C
20 sec 55 °C 30 sec 72 °C
Loop30 cycles end 5 min 72 °C
To verify the successful transformation of a specific insert into bacterial cells, a colony PCR reaction
was run using home-made Taq DNA polymerase. Therefore colonies were picked and heated at 95 °C
for 15 min in 20 µl ddH2O in order to destroy the bacterial cells. After spinning off the cell debris
10 µl of the mixture was added to the PCR mixture containing 1x PCR buffer, 1 mM MgCl2, 10 pmol
5’ primer, 10 pmol 3’ primer, 0.2 mM dNTPs, 1 U Taq DNA polymerase in 25 µL total volume.
The following protocol with 30 cycles was applied:
5 min 96 °C Loop30 cycles start 1 min 96 °C
30 sec 55 °C 45 sec 72 °C
Loop30 cycles end 5 min 72 °C

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
28
The purification of the PCR products was done according to the manufacturer’s instructions
(Macherey-Nagel Kits, 2.1.3).
2.2.2 Site-directed mutagenesis
Site-directed mutagenesis was conducted by overlap extension PCR using specific primers during an
SOE (splicing by overlapping extension) PCR. The primer pairs for PCR 1 containing the corresponding
mutations are listed in Table 1. To obtain the two fragments, the primer 5’ pMS was used to generate
the 5’ to 3’ fragment and the primer 3’ Gb (BlpI) was used to generate the 3’ to 5’ fragment. A
construct containing the desired single chain sequence as ligand, including the wild-type sequence of
granzyme B, was used as a template. (2.1.4). The PCR mixtures and programs were as follows.
PCR 1:
1x HF buffer
10 ng template DNA
10 pmol 5’ primer
10 pmol 3’ primer
0.2 mM dNTPs
0.02 U/µL Phusion DNA polymerase
Ad 50 µL ddH2O
Program 1:
30 sec 98 °C
Loop30 cycles start 10 sec 98 °C
20 sec 60 °C
30 sec 72 °C
Loop end
5 min 72 °C
Annealing:
2 µL fragment 1
2 µL fragment 2
1x HF buffer
0.2 mM dNTPs
0.02 U/µL Phusion DNA polymerase
Ad 50 µL ddH2O
Program 2:
30 sec 98 °C
Loop7 cycles start 10 sec 98 °C
1 min 50 °C
30 sec 72 °C
Loop end
5 min 72 °C
Amplification:
10 µL annealing product
1x HF buffer
10 pmol 5’ primer (5’ pMS)
10 pmol 3’ primer (3’ Gb (BlpI))
0.2 mM dNTPs
0.02 U/µL Phusion DNA polymerase
Ad 50 µL ddH2O
Program 3:
30 sec 98 °C
Loop25 cycles start 10 sec 98 °C
20 sec 60 °C
30 sec 72 °C
Loop end
5 min 72 °C

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
29
2.2.3 Agarose gel electrophoresis
To analyze the size and concentration of the PCR products (2.2.1), isolated plasmids (2.2.8) and
restriction fragments (2.2.5), analytical agarose gel electrophoresis was carried out in 1.2 % (w/v)
agarose at field strength of 120 V after addition of 5x OrangeG loading buffer (Table 4).
Electrophoresis was run in 1x TAE buffer with 0.3 μg/mL ethidium bromide. 0.33-0.6 μg 2-Log DNA
Ladder (NEB) was used as the standard for determining the size and concentration of the fragments
(2.1.3). DNA bands were visualized on a UV transilluminator at a wavelength of 302 nm. The bands
obtained were documented with a video documentation system using the software ‘Quantity One
Document Software’ version 4.6 (BioRad, Munich).
2.2.4 Zero blunt TOPO PCR cloning
Since the digestion of PCR products (2.2.1) was often inefficient, the PCR products were cloned into a
TOPO vector according to the manufacturer’s instructions of Zero Blunt® TOPO® PCR Cloning Kit
(Invitrogen, Darmstadt). After growing of the corresponding bacteria, the purified plasmids could be
digested as described below (2.2.5).
2.2.5 Endonuclease restriction of DNA
The double digestion with endonucleases and buffers from NEB (Frankfurt am Main) was performed
according to the manufacturer’s instructions in a total volume of 50 µL with 1 to 2 µg DNA. The
isolation of restricted DNA occurred via preparative agarose gel electrophoresis (2.2.3). After
separation of DNA fragments, desired DNA bands were excised using a clean scalpel on a UV
transilluminator. The isolation of the DNA from the gel was performed according to the
manufacturer's protocol (2.1.3). The concentration of the purified DNA was determined using
analytical agarose gel electrophoresis (2.2.3) or by measuring the absorbance at 260 nm. The
extracted and purified DNA was then used for ligation (2.2.6). For control digestions, 5 times less
than the recommended volume was used and the specific bands were visualized as described above
(2.2.3).
2.2.6 Ligation of DNA fragments
Two purified restriction digested fragments (2.2.5) were ligated with T4 DNA ligase and Quick ligase
respectively, using the buffer system specified by the manufacturer with a total volume of 20 μL. The
ligation mix contained the insert in triplicate molar excess of vector quantity, and the mixture was
incubated overnight at 16 °C and for 5 min at room temperature (RT) respectively. Afterwards, the
whole ligation mixture was transformed into heat shock competent bacteria (2.2.7), as explained

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
30
below.
2.2.7 Transformation of plasmid DNA into competent E. coli
Heat shock competent cells (2.1.6) were prepared with Rhubidium-chloride according to Hanahan
[142]. The aliquoted cells were thawed on ice before the DNA was added (using either 20 µL ligation
mix (2.2.6) or 30 ng purified plasmid DNA (2.2.8) for re-transformation). After incubation for 30 min
on ice, the cells were incubated at 42 °C for 30 sec (heat shock). The transformed bacteria were
stored on ice for 2 min and then mixed with 800 μL of sterile SOC medium. For regeneration, the
bacteria were incubated for 1 h at 37 °C before they were plated on LB agar plates (2.1.9) containing
the corresponding antibiotic (ampicillin for pMS-plasmids, kanamycin for pMT-plasmids). The plates
were incubated overnight at 37 °C and colonies were analyzed using colony PCR (2.2.1) the next day.
Replica plates were prepared for at least three clones.
For the final clones glycerol stocks were prepared in 25 % (v/v) glycerol solution and stored at -80 °C.
2.2.8 Preparation of plasmid DNA from E. coli
For small scale DNA preparation, 3 mL of LB medium supplemented with the appropriate antibiotic
(2.1.9) was inoculated and then the recombinant plasmid DNA (2.2.8) was isolated using a
NucleoSpin plasmid kit according to the manufacturer’s instructions (2.1.3). The concentration was
determined at 260 nm.
2.2.9 Sequencing of DNA
To verify the desired sequence of recombinant clones (2.2.7, 2.2.8), sequencing was carried out at
the Fraunhofer IME sequencing facility by the chain termination method using the ‘Applied
Biosystems 3700 DNA Analyzer’ and the BigDye™ cycle sequencing kit. 160 ng/kb plasmid DNA was
added to ddH2O in a total volume of 28 μL with 20 pmol of sequencing primer (2.1.4). The resulting
sequences were evaluated with the ‘CLC Main Workbench 6.0’ and ‘Clone Manager 9.0’ software.
2.3 CELL CULTURE
2.3.1 Cultivation of cell lines and primary cells
All mammalian cell lines (Table 3) as well as the primary monocytic cells (2.1.8) were cultured in T25
or T75 cell culture flasks (Greiner) in RPMI1640 (Gibco) supplemented with 10 % (v/v) heat-
inactivated fetal calf serum (FCS) (Gibco), 50 μg/mL penicillin and 50 μg/mL streptomycin. The cells

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
31
were maintained at 37 °C in a humidified atmosphere of 5 % CO2. After transfection of HEK293T cells,
100 µg/mL Zeocin was added for selection purposes.
When large amounts of secretory expressed protein were intended, triple flasks (VWR, Darmstadt) or
cell factories (Thermo Fisher Scientific, Waltham, USA) were inoculated, so supernatant volumes
above 1 L could be collected easily for purification.
For viability and apoptosis assays 200 or 50 U/mL Interferon (IFN) γ was added for stimulation of
CD64 expression in the primary cells or HL60 cell line, respectively.
2.3.2 Transfection of cell lines
The physical or chemical transfection of different mammalian cell lines was performed according to
the manufacturer’s instructions (Table 5) and using their protocols for optimization. As
recommended, the cells were kept in serum-free RPMI medium or RPMI containing 10 % (v/v) FCS
without antibiotics. The efficiency of transfection was monitored via flow cytometric analysis.
Table 5: Reagents for physical or chemical transfection of mammalian cell lines.
Name Company
Physical:
Amaxa Nucleofection Kit V Lonza (Cologne)
Neon Transfection systems Life technologies (Darmstadt)
Chemical:
Nanofectin Kit PAA (Pasching, Austria)
Attractene Transfection reagent
Qiagen (Hilden)
GeneJuice Transfection Reagent
Novagen (Darmstadt)
TransIT-Jurkat MirusBio (Madison, USA)
Ingenio Electroporation Kit MIR 50109
MirusBio (Madison, USA)
Roti-Fect Reagent Roth (Karlsruhe)
2.3.3 Protein delivery into cells via streptolysin O
Reversible permeabilization of cells can be achieved via streptolysin O (Sigma, Munich). For protein
delivery 5*106 cells/mL were washed once with HEPES buffer (20 mM HEPES, pH 7.4, 150 mM NaCl)
and re-suspended in 100 µL HEPES buffer containing 0.5 % (w/v) BSA, 5 mM DTT and 10 mM glucose.

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
32
The cells were incubated with 8 µg/mL streptolysin O for 15 min at 37 °C in a 96-well plate.
Afterwards the cell-death inducing protein was added and cells were further incubated for 4 h. The
permeabilization step was conducted in the absence of Ca2+, whereby resealing of the cells was
induced by addition of 1.25 mM Ca2+ 1 h after addition of the cell-death inducing protein. Apoptotic
effects were monitored via Annexin V/ PI assay as described in 2.7.2.1.
2.4 PROTEIN-BIOCHEMICAL METHODS
2.4.1 SDS-PAGE
To determine the amount and identity of proteins (2.4.3, 2.4.4) according to their molecular weight,
analytical protein separation was performed via discontinuous SDS- poly-acryl-amide (SDS-PAA) gel
electrophoresis (SDS-PAGE) based on the method of Laemmli [143]. The denaturing SDS-PAA gel was
prepared with the above mentioned buffers (Table 4). Before the gel was loaded, the samples were
mixed with Laemmli sample buffer and heated for 5 min at 95 °C to destroy the structure of the
proteins. The gel was run with 160 mV in ‘Mini-Protean III’-protein gel electrophoresis chamber
(BioRad) until the samples reached the bottom of the gel. The gel was dyed in a coomassie brilliant
blue solution for 10-30 min and destained with the described buffer (Table 4). With the help of the
loaded standard (2.1.3) the proteins could be identified.
2.4.2 Western blot analysis
Western blot analysis was performed using the ‘Mini Trans-Blot® Electrophoretic Transfer Cell’ (BIO-
RAD) according to the manufacturer's instructions with transfer buffer (Table 4). After the transfer
(250 mA for 90 min or 40 V overnight at 4 °C) of proteins, separated by SDS-PAGE, on a nitrocellulose
membrane (Protran, Whatman), blocking was performed using 2.5 % (w/v) milk powder resolved in
PBST either for 1 h at RT or overnight at 4 °C. After washing three times with PBST, the membrane
was incubated with the primary antibody in PBST for 1 h at RT. The same procedure was followed for
incubation with the secondary antibody. After final washing in PBST and water, the membrane was
developed in the dark depending on the conjugation of the second antibody (2.1.5) if not otherwise
indicated using NBT/BCIP [3.33 % (w/v) NBT, 1.67 % (w/v) BCIP in dimethylformamide] added to the
100x volume of AP buffer (Table 4) for AP-labeled antibodies or with an enhanced
chemiluminescence (ECL) system (Thermo Scientific) and LAS-3000 reader (Fujifilm) for PO-
conjugated ones.

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
33
2.4.3 Bacterial expression and purification
For the expression of recombinant PI-9, the E. coli expression strain BL21 (DE3) was used (2.1.6). To
evaluate successful protein expression, optimal conditions and sufficient purification steps, small
scale experiments were performed in a volume of 300 mL according to the published stress-
expression protocols [144] or the protocols from Qiagen (‘The QiaExpressionistTM’). In order to obtain
enough protein for further determinations, fermentation with 4 L volume (BioFlo 110 fermenter,
400-1000 rpm) was run in synthetic minimal medium (16.6 g/L KH2PO4, 4.6 g/L NH4(H2PO4), 70 mg/L
CaCl2*2H2O, 140 g/L FeSO4*7H2O, 1.5 g/L MgSO4*7H2O, 2.1 g/L citric acid, 0.2 g/L L-methionine,
0.2 g/L L-arginine-hydrochloride, 1 mL/L PTM1-trace metals, 25 mg/L Kanamycin, 0.5 mL/L Antifoam)
supplemented with 20 g/L glucose. Inoculation was prepared with 400 mL bacteria culture grown at
37°C overnight in LB medium. The bacteria were harvested 24 h after induction with 1 mM Isopropyl
Thiogalactoside (IPTG).
After harvesting and centrifugation (4000 g, 15 minutes, 4°C) the bacterial pellet was re-suspended in
lysis buffer (50 mM NaH2PO4, pH 8.0, 500 mM NaCl, 0.5 mM DTT) and sonicated six times for 60 s,
70 %, 9 cycles (2.1.1). Due to its enzymatic activity towards proteases, the addition of protease
inhibitor was disclaimed. Cell debris was removed by centrifugation at 30.000 g, 4 °C for 30 min. The
supernatant was supplemented with 10 mM imidazol and purified via Immobilized Metal-ion Affinity
Chromatography (IMAC) and Fast Performance Liquid Chromatography (FPLC) as described below
(2.4.4.2). After a second IMAC step, the protein was re-buffered into 20 mM Tris, pH 7.4, 1 mM DTT
and further purified via anion exchange chromatography (Q-Sepharose XL, 1 mL, GE Healthcare) with
a salt gradient of 0.05–1 M NaCl. To achieve satisfying purity, gel filtration chromatography followed
using a Superdex 75 (GE Healthcare) column in 20 mM Tris, pH 7.5, 50 mM NaCl and 1 mM DTT.
2.4.4 Mammalian expression and purification
All granzyme B- and granzyme M-based constructs were produced secretory in mammalian HEK293T
cells. In addition, PI-9 was expressed cytosolically within these cells.
2.4.4.1 Expression of recombinant protein in HEK293T cells
HEK293T cells were used as the expression cell line (2.1.6). The cells were transfected with 1 µg DNA
according to the manufacturer’s instructions using RotiFect (Roth) (2.3.2). The pMS plasmid used
(2.1.4) encodes for the bicistronic EGFP reporter, so expression of the target protein could be verified
by the green fluorescence via fluorescence microscopy. To obtain high producing cultures
fluorescence activated cell sorting was performed to select high reporter EGFP producing cells.

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
34
The secretory expression was enabled by the signal peptide sequence of immunoglobulin kappa light
chain (Igκ) encoded by the pMS plasmid (2.1.4). Protein containing supernatants were kept sterile
and were stored at 4 °C until sufficient volumes were collected for purification (2.4.4.2). In case of
cytosolically expressed protein, the cells were harvested, washed with PBS and stored at -20 °C until
further processing (2.4.4.3).
2.4.4.2 Purification of recombinant protein from supernatant
The secreted protein could be purified from the cell culture supernatant via IMAC and FPLC. The
cleared supernatant was supplemented with 10 mM imidazole before loading onto an XK16/20
column (Amersham/ GE Healthcare) containing 8 mL Sepharose 6 Fast Flow resin (GE Healthcare).
The buffers used are listed in Table 4. Afterwards the elution buffer was exchanged with adequate
buffer to enable enterokinase digestion (20 mM Tris-HCl, 200 mM NaCl) via a HiPrep 26/10 desalting
column (GE Healthcare). To obtain higher purity of proteins especially for in vivo experiments, the
protein solution was additionally loaded onto a gel filtration chromatography column (Superdex 75,
GE Healthcare) using the enterokinase buffer.
In order to concentrate the protein solutions, Vivaspin 6 ultrafiltration spin columns (10 kDa MWCO;
Sartorius) were centrifuged at 4000 g and 4 °C. Aliquoted proteins were stored at -80 °C. To activate
granzyme-based constructs enterokinase was added to the protein (0.02 U/µg) with 2 mM CaCl2 for
16 h incubation at 23 °C prior to use. Activated protein was stored after the addition of 1 mM DTT at
-80 °C. The purified proteins or cell lysates were analyzed via SDS-PAGE under reducing conditions
and coomassie staining (2.4.1) or western blots (2.4.2).
2.4.4.3 Purification of recombinant protein from cell lysate
HEK293T cells transfected with pMS-PI9-IV (2.1.4) were harvested and lysed within a native buffer
(50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 % (v/v) Triton X100). Incubation of the cells in buffer took
place for 30 min on ice. Cell debris was then removed by centrifugation at 13.000 g for 15 min at 4 °C
and the supernatant containing the desired protein was purified as described in 2.4.4.2.
2.4.5 Protein quantification
Three different methods were applied for the determination of protein concentration. For purified
protein the concentration was determined after SDS-PAGE and coomassie staining using ‘AIDA Image
Analyzer’ Software (Raytest Isotopenmessgerate). The concentration could be correlated to the
additionally loaded bovine serum albumin (BSA) standard (100 ng, 250 ng, 500 ng, 1000 ng). The
values were confirmed by Bradford assay in a 96-well plate using 1x Roti-Nanoquant (Roth). All
samples were pipetted in triplicates. A stock solution with 1 μg/μL purified BSA (0, 0.5, 1, 2, 4, 6, 8,

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
35
10 μg/well) was used as a calibration standard. In order to determine the total protein concentration
of cell extracts Bradford or BCA assay (Thermo Scientific Pierce) was performed according to each
manufacturer’s instructions. Absorption was measured at 595 nm for BSA and at 562 nm for BCA in a
microplate reader. Data were analyzed with MS Office Excel software, and the total protein
concentration of the samples was determined according to the BSA standard curve.
2.4.6 Labeling of SNAP constructs
The establishment of the SNAP (engineered version of the human DNA-repair enzyme O(6)-
alkylguanine DNA alkyltransferase) -tag technology and the preparation of the corresponding
constructs and their covalent conjugation to substrates containing O(6)-benzylguanine (BG) via a
stable thioether bond in a rapid and highly specific self-labeling reaction has been described before
[145]. Here, in HEK293T cells, secretory expressed and subsequently purified SNAP-tag fusion
proteins (Ki4(scFv)-SNAP and H22(scFv)-SNAP) were conjugated with the BG-modified dyes BG Alexa
Fluor 647 (BG 647), BG Alexa Fluor 747 (BG 747) or BG Vista Green (Covalys Biosciences AG,
Witterswil) by incubation in the dark with 1.5-3-fold molar excess of the dye overnight at 4 °C.
Residual dye was removed using PD 10 columns (GE Healthcare). Stained proteins were visualized
after separation by SDS-PAGE with either a UV transilluminator (BioRad Gel Doc XR) or with a CRi
Maestro imaging system using blue, red and near-infrared filter sets.
2.4.7 Detection of endogenous PI-9 from cell lines or primary cells
For the detection of cytosolic expressed PI-9 within tumor cell lines or primary cells, 106 cells were
lysed within 50 µL lysis buffer (PBS supplemented with 1 % (v/v) Triton X-100) for 30 min on ice. The
cell extract was cleared via centrifugation and the protein concentration was determined with
Bradford reagent (2.4.5). 40 µg total protein amount were loaded on a SDS-gel for western blot
analysis. PI-9 was detected by incubation for 1 h with anti-human PI-9 (2.1.5) in PBST. After washing
with PBST, GAM-PO was added and signals could be detected as described in 2.4.2.
In parallel, detection was performed via flow cytometry (2.5.1) which allows more quantitative
analysis. Therefore 106 cells were washed with PBS and resuspended in 500 µl Cytofix/ Cytoperm (BD
Bioscience, Heidelberg). After incubation for 20 min on ice and washing of the cells, a blocking step
was included with 5 % (w/v) BSA in 200 µl PBS for 20 min on ice. The cells were washed with PBS
containing 0.2 % (v/v) Tween 20 and incubated with anti-human PI-9 (2.1.5) for 30 min on ice. The
second antibody GAM-FITC (2.1.5) was added to the washed cells and incubated as described above.
The final wash step and resuspension of the cells was performed in PBS comprising 0.2 % (v/v)
Tween 20. The measured shift during flow cytometric analysis due to a specific binding of the

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
36
antibody to the fixed and permeabilized cells was quantified as MFI (Median Fluorescence Intensity)
in WinMDI 2.8. MFIs due to low unspecific binding of the secondary antibody were subtracted.
2.4.8 Serum stability assays
70 ng/µL purified protein was incubated with 50 % murine serum (AbD Serotec, Duesseldorf) or
human serum (obtained by centrifugation of 500 to 1000 µL patient-derived blood samples (2.1.8)
for 10 min, 300x g at RT) at 37 °C for different time periods between 0 and 48 h. The amount of
functional remaining protein binding was measured via flow cytometric analysis (2.5.1), here with
400-fold dilution of 7.5 µL of the mixture.
2.4.9 ELISA for detection of CD30 receptor in blood
Mouse serum of L540cy bearing mice was obtained by centrifugation of 10-30 µL mouse-derived
blood samples for 10 min and 300x g at RT and stored at -20 °C until use. The concentration of
soluble CD30 receptor within blood serum was evaluated by an ELISA set-up as previously published
[146]. High binding ELISA plates were coated with 50 µl Ki2 monoclonal antibody (240 µg/mL) in PBS
overnight at 4 °C. After washing with PBST and blocking in PBS containing 5 % (w/v) milk powder for
1 h at RT, 50 µl CD30-containing mouse serum (1:5 dilution in PBS) was added and incubated for 2 h
at RT. Plates were washed three times and 50 µl Biotin coupled Ki3 monoclonal antibody (270 ng/µL,
1:100 dilution in PBS) was added for a 1 h incubation at RT. For detection, plates were first incubated
with 50 µl Streptavidin-PO (1:200 in PBS, Abcam, Cambridge, UK) for 1 h; then washed in PBST and
then incubated for 1 h with 50 µl ABTS solution (Roche Diagnostics, Mannheim). Plates were read out
at 405 nm in a microplate reader.
2.5 FLOW CYTOMETRIC ANALYSIS
2.5.1 Binding analysis
The binding of fusion proteins to the target cells (Table 3) or the endogenous protein thereof (2.4.7)
was evaluated via flow cytometric analysis (CellQuest Version 3.3 (Becton Dickinson, Heidelberg))
and WinMDI 2.8. For standard binding analysis 4*105 cells were washed with PBS and incubated with
50 nM purified protein in 100-300 µL PBS for 30 min on ice. After 2 wash cycles the cells were
incubated with anti-His Alexa 488 (2.1.5) for 30 min on ice, in the dark. Negative controls with cell
lines not expressing the target receptor were run in parallel. Monocytic cells of primary PBMCs were
gated in forward and sideward scatter (FSC/ SSC) dotplot prior to evaluation.

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
37
2.5.2 Determination of Kd values
For the determination of the affinity constant (Kd) different concentrations of the fusion proteins
(0-16.7 nM, 1:2 dilution) were incubated with the cells and evaluated by flow cytometric analysis
(2.5.1) and non-linear-regression determinations in GraphPad Prism 4.0.
2.5.3 Determination of specific cell death of primary cells
The specific reduction of the target cell population was determined by staining primary cells with
anti-CD64 Alexa647 (AbD Serotec, Duesseldorf) to localize the population within the FSC/SSC dotplot
during flow cytometry. These cells were gated with WinMDI 2.8 so their reduction after treatment
could be evaluated accordingly and compared to untreated control.
To further specify reduction of the target cell population specific antibodies displayed in 2.1.5 were
incubated with the treated cells and reduction of the stained target cell population could be
compared to the untreated control by flow cytometric analysis.
2.6 CONFOCAL MICROSCOPY
All images were taken with a LEICA confocal microscope (TCS SP5) equipped with the following
lasers: 458 nm, 488 nm, 561 nm and 633 nm. A 40x optical magnification was used. Cells were
stained with SNAP-based constructs (2.4.6) as indicated and prepared as described above for flow
cytometry (2.5), resuspended in 50 µL PBS and pipetted onto a microscope slide.
2.7 FUNCTIONAL CHARACTERIZATION OF RECOMBINANT PROTEINS
2.7.1 Protease assays
2.7.1.1 Colorimetric substrate assays
The activity of 100 nM granzyme B-based constructs after enterokinase digestion was detected by
cleavage of 200 µM of the synthetic substrate Ac-IETD-pNA (Calbiochem/Merck, Darmstadt) which
mimics the cleavage site of pro-caspase 3. The reaction was monitored in 96-well plates in a
microplate reader at 405 nm and 37 °C at 1 or 2 min intervals for 1 h. Afterwards, time was plotted
against the absorbance so differences in activity could be evaluated. After complex formation
(2.7.1.4) and measurement of the remaining activity the velocity of the reaction was calculated from
the linear slope of the reaction within the first 10-12 min and converted to pmol/min with the help of

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
38
the corresponding conversion factor (µM/A405nm) according to the manufacturer’s protocol of the
Caspase-8 Assay kit (Calbiochem).
Granzyme M activity, after enterokinase digestion, was evaluated using the colorimetric substrate
Suc-Ala-Ala-Pro-Leu-pNA (Bachem, Bubendorf, Switzerland) according to a published protocol [56].
The change in absorbance at 405 nm was measured as above with the difference that here only the
absolute change was recorded, not the kinetics.
2.7.1.2 Determination of Michaelis-Menten Kinetics
To analyze the activity of the wild-type protein and its most promising mutant, Km values were
evaluated according to the manufacturer’s protocol of the Caspase-8 Assay kit (Calbiochem). The
enzymatic reaction was prepared and measured as described above (2.7.1.1). For evaluation, change
in absorbance at 405 nm during the first 12 min of the reaction was determined. Km values were
derived by non-linear curve fitting to the Michaelis–Menten equation using GraphPad Prism 4.0.
2.7.1.3 Cleavage of PI-9 by granzyme M
The activity of the granzyme M-based constructs was verified by their ability to cleave recombinant
PI-9 (1.3.2). The fusion protein was incubated at a 1:3 molar ratio with PI-9 at 25 °C in 100 mM HEPES
(pH 7.4), 200 mM NaCl, 0.01 % (v/v) Tween 20 and 1 mM DTT. After 24 h the reaction mixture was
analyzed by SDS-PAGE and western blotting using anti-human PI-9 and GAM-PO as described in 2.4.2.
2.7.1.4 Formation of the granzyme B and PI-9 Complex
The complex of recombinant PI-9 and Gb-H22(scFv) and its mutants was formed using a 5:1 molar
ratio (PI-9:Gb-H22(scFv)) under reducing conditions in 20 mM Tris-HCl (pH 7.4), 50 mM NaCl and
1 mM DTT. 600 ng of wild-type or mutant granzyme B construct was incubated with or without PI-9
for 1 h at 37 °C and the retained enzymatic activity was evaluated as described above (2.7.1.1). For
western blot analysis (2.4.2) adequate amounts containing at least 100 ng of granzyme B-based
protein was prepared for SDS-PAGE in the same manner. For detection of specific bands, anti-human
PI-9, anti-human Gb or anti-His were used followed by the second antibody GAM-PO (2.1.5).
Endogenous complex formation was determined after incubation using Gb-Ki4(scFv) as described for
apoptosis assay (2.7.2.1). Analysis was done as described above using anti-human Gb and GAM-PO.

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
39
2.7.2 Apoptosis assays
Apoptosis was documented via Annexin V/ propidium iodide (PI) staining (2.7.2.1) or via
determination of caspase 3/7 activation (2.7.2.2) within the target cells.
2.7.2.1 Annexin V/ PI Assay
Annexin V binds to phosphatidylserine which is exposed on the surface of apoptotic cells. Thus, dead
cells can be visualized using labeled Annexin V protein. In addition, PI enters through the permeable
membrane of dead cells, so necrotic and apoptotic cells can be distinguished. 2*105 cells/mL were
incubated at 37 °C, 5 % CO2, with different concentrations of fusion protein (between 11 and
100 nM), depending on target cells and construct, for 48 h in 12-well plates containing 1 mL
supplemented RPMI medium (2.3.1). Afterwards, the cells were washed in PBS and the pellet was re-
suspended either in 100 µL 1x Annexin V binding buffer (10 mM HEPES (pH 7.5), 140 mM NaCl,
0.5 mM CaCl2) supplemented with 2.5 µL Annexin V-FITC (eBioscience, Frankfurt) or in 450 µL cell-
free culture supernatant from HEK293T cells expressing Annexin V-labeled green fluorescent protein
(EGFP, [147]) supplemented with 10x Annexin V binding buffer and 5 µg/mL PI. The incubation with
labeled Annexin V protein took place in the dark, either at RT for 15 min or on ice for 20 min
respectively. Results were analyzed via flow cytometry (2.5).
2.7.2.2 Caspase 3/7 assay
To determine the caspase 3/7 activity within apoptotic, granzyme B-treated cells (2.7.2.1), a pre-
luminescent caspase 3/7-DEVD-aminoluciferine substrate was applied (Caspase-Glo™ 3/7 Assay,
Promega). Caspase 3/7 is a direct substrate of granzyme B. If it is cleaved after granzyme B delivery
into the target cells and thereby activated, it can cleave the pre-luminescent caspase 3/7-DEVD-
aminoluciferine substrate so free unbound aminoluciferine is released. This is subsequently used by
luciferase whereby a luminescence signal is produced. For the evaluation, cells were transferred into
96-well plates and the read out was done in a microplate reader. The amount of luminescence was
directly proportional to the activity of caspase 3/7.
2.7.3 Viability assay
The cytotoxic effect of the granzyme B-based fusion proteins was monitored using the ability of
metabolic active cells to reduce the tetrazolium salt XTT ((sodium 2,3,-bis(2-methoxy-4-nitro-5-
sulfophenyl)-5-[(phenylamino)-carbonyl]-2H-tetrazolium) inner salt, Life Technologies (Invitrogen)) to
orange colored compounds of formazan. The dye intensity was measured by a microplate reader and
is directly proportional to the number of living cells.

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
40
For Gb-Ki4(scFv) and its mutants 2*105 cells were plated in 1 mL in 12-well plates with 1:3 dilutions
and incubated for 48 or 72 h at 37 °C and 5 % CO2. For Gb-H22(scFv) and its variants and
Gm-H22(scFv), cells were incubated in 96-well plates with 5.000 IFNγ stimulated HL60 cells (or
control cell lines) or 20.000 primary cells per 100 µL with 1:3 or 1:5 dilutions of protein for 72 h at
37 °C. In a different approach, XTT was used for read out of viable cells after incubation with single
doses of fusion protein (concentrations as indicated, depending on target cells and construct) in 12-
well plates and evaluated after transfer of 100 µL of each approach into 96-well plates as described
above. 50 µL XTT was added to a 100 µL cell suspension and extinction was measured after a 4 h
incubation at 450 nm at a reference wavelength of 630 nm.
Viability curves and IC50 values were evaluated with GraphPad Prism4.0. Therefore dilution effects by
the protein volume used were normed using respective buffer controls. Zeocin was used as the
negative control.
Competition assays were carried out with H22(scFv) by adding 10 nM Gb-H22(scFv) or Gm-H22(scFv)
to IFNγ stimulated HL60 cells and incubating them for 48 h in the presence or absence of 10 or
100 nM H22(scFv). The buffer control was assigned a viability of 100 % and the converted XTT signal
was read out as above.
2.8 IMMUNOHISTOCHEMISTRY
Resected cryo-conserved tumors were cut into 8 µm sections on a freezing microtome (Cryostat CM
3050 (Leica) and mounted on coated slides. After drying for 48-72 h, sections were fixed for 10 min
with dry acetone and air-dried. In order to detect endothelial cells, slides were incubated with
naphthol AS-BI phosphate (sodium salt, 50 mg/100 mL; Sigma, Munich) as substrate and new fuchsin
(10 mg/100 mL; Merck, Darmstadt) as chromogen dissolved in 0.1 M Tris-HCl (pH 8.5) resulting in
pink/red staining. By omitting the addition of levamisole to the reaction mixture, endothelial cells
became visible. Slides were counterstained with hematoxylin.
In order to visualize remaining CD30 receptor expression in resected tumors, 12.5 nM
Ki4(scFv)-SNAP-BG-Vista-Green diluted in PBS was added to the slides and incubated for 30 min in
the dark at RT. After washing, images were collected via a Leica confocal microscope (2.6).

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
41
2.9 IN VIVO EXPERIMENTS
The experiments were officially approved by the local Animal Care and Use Review Committee (AZ:
87-51.04.2010.A278). All animals received human care in accordance with the requirements of the
‘German Tierschutzgesetz’, §8 Abs. 1 and in accordance with the Guide for the Care and Use of
Laboratory Animals published by the National Institute of Health.
2.9.1 Mouse strains, housing and maintenance of animals
For animal experiments 6 to 8 week-old female BALBc nu/nu mice (Charles River GmbH) were used.
Mice were housed in the ‘Institut fuer Versuchstierkunde’ of the university hospital, Aachen, during
tumor take rate studies, and in the animal facility of the Fraunhofer IME during the treatment
experiments.
2.9.2 Handling of mice and anesthesia
Anesthesia of the mice was performed either by intramuscular administration of 75 mg/kg Ketamin –
10 mg/kg Xylazin for periods up to 30 min or by isoflurane for shorter times up to 2 min.
Proteins were administered intravenously (i. v.) with volumes up to 100 μL. Samples were brought up
to RT before injection. Before and after image acquisition, anesthetized mice were kept warm on a
hot-water bag. As in previous imaging experiments, the mice injected with far red fluorescent protein
Kat2 (pTag-Katushka2-N; Evrogen) transfected cells were fed a purified, chlorophyll-free diet
(AIN93G, SSNIFF GmbH) 7 days before the imaging experiments were started whereas mice injected
with L540cy were fed a normal diet (SSNIFF GmbH).
Blood samples (10-30 µL) were obtained through the tail vein of the mice.
2.9.3 Establishment of xenograft subcutaneous tumor models
Before treatment of the mice with fusion proteins could be started, the tumor take rates of the
potential target cell lines had to be determined. For injection of tumor cells, L428 cells transfected
with Kat2 or L540cy cells (not transfected) were used after washing with PBS and resuspension in
50 % BD Matrigel™ Basement Membrane Matrix High Concentration, Growth Factor Reduced (BD
Bioscience, Heidelberg). 5*106 cells in a volume of 30 µL were injected subcutaneously into the right
hind limb of 5 mice for L428 and 5 mice for L540cy. Tumor growth was monitored by molecular
imaging with the CRi-Maestro Imaging System (2.9.4) in the case of Kat2 transfected L428; for L540cy
triplet caliper measurements were executed. Based on the evaluated tumor take rates and tumor
sizes, adequate treatment schedules could be set up.

Material and methods ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
42
2.9.4 In vivo optical imaging by Cri-Maestro system
In vivo optical imaging was performed with the CRi-Maestro Imaging System (Cri Inc., Woburn, MA,
USA) and analyzed with the Maestro Spectral Imaging Software. The Maestro imager has a Xenon
light source and allows measurements through the complete visible and near infrared light spectrum
by applying different filter sets. Whole mouse images were taken with constant stage height and
illumination settings. The Kat2 signal of the transfected L428 cells was visualized with the yellow filter
set (630–850 nm). To identify and discriminate the individual spectra within the images, spectral
libraries were defined. Therefore the different dye spectra were unmixed from the cube images. The
component images, which display the targeted signal, were used to define the tumor to background
ratios (TBRs) by drawing a region of interest (ROI) around the tumor and a larger area on the back
representing average background signal. The thereby calculated values for the tumor size in mm² for
the region of interest were used to calculate the tumor regression.
2.9.5 Biological activity of Gb-Ki4(scFv) and GbR201K-Ki4(scFv)
To determine the in vivo biological activity of Gb-Ki4(scFv) and GbR201K-Ki4(scFv), the cell lines L428
(Kat2 transfected) and L540cy were injected into the mice respectively as described in 2.9.3. The
mice were randomized equally into groups and received 50 µg GbR201K-H22(scFv) (as non-binding
control), Gb-Ki4(scFv) or GbR201K-Ki4(scFv) i. v. per treatment day. Tumor size was measured as
described in 2.9.3 and 2.9.4. Relative tumor size was calculated by setting the tumor size on first day
of treatment to 100 % and adapting the following values accordingly.
In vivo targeting of CD30-positive L540cy cells was accomplished by injection of 0.75 nmol
Ki4(scFv)-SNAP-BG-747 into L540cy bearing mice. 6 h later, the mice were imaged with the CRi-
Maestro system using the deep red filter set (730-950 nm) (2.9.4).
2.10 STATISTICAL ANALYSIS
Data were analyzed using a two-tailed t-test in GraphPad Prism 4.0, with p < 0.05 considered as
statistically significant.

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
43
3 Results
3.1 PI-9 EXPRESSION IN CANCER CELL LINES
The high potential of granzyme B-based CFPs has already been reported by several groups in vitro
and in vivo; however, one of the major challenges for its therapeutic application is the expression of
the specific granzyme B inhibitor PI-9 in many tumor cells (see 1.4.1). Based on these findings and the
observed limitations of cytotoxic effects induced by different granzyme B-based CFPs within this
group, the PI-9 expression profiles of the most commonly used cancer cell lines were evaluated.
Therefore total soluble protein of native cell extracts (2.4.7) of different cancer cell lines (2.1.6) was
separated by SDS-PAGE and transferred to a nitrocellulose membrane. The expression of
endogenous PI-9 could be confirmed by specific anti-human PI-9 staining of a 42 kDa protein band
correlating to the calculated molecular weight. An overview of the results for all tested cell lines is
displayed in Table 6. Representative results are shown for CD30-positive cell lines in Figure 3A.
Table 6: Overview of the PI-9 expression profile of different cancer cell lines.
(++: strong, +: moderate, (+): low, -: no expression)
Cell line Cell type Target receptor PI-9 expression
(protein level)
L428 HL CD30 ++ L1236 HL CD30 ++
L540/ L540cy HL CD30 - K562 Chronic myeloid leukemia in blast crisis CD30 +
Karpas 299 T cell lymphoma CD30 - L3.6pl Pancreatic carcinoma EGFR ++ PT45 Pancreatic adenocarcinoma EGFR - A431 Epidermoid carcinoma EGFR - HL60 AML CD64 - U937 Histiocytic lymphoma CD64 - Jurkat T cell leukemia / -
Kasumi 1 AML / (+)
PI-9-positive as well as PI-9–negative cell lines expressing the tumor marker CD30 were identified.
Corresponding qualitative results from western blotting were confirmed by flow cytometry providing
more quantitative data. For this purpose triplicate flow cytometric analysis of fixed and
permeabilized cells (2.4.7) was performed (Figure 3B).
Jurkat cells were PI-9-negative according to Godal et al. [148] so they could be used as a negative
control. The CML cell line K562, also previously published to express PI-9 at moderate level [148],
showed the lowest expression level compared to the Hodgkin lymphoma (HL) cell lines L428 and
L1236 which express similar amounts of PI-9. L428 retained their PI-9 expression after transfection
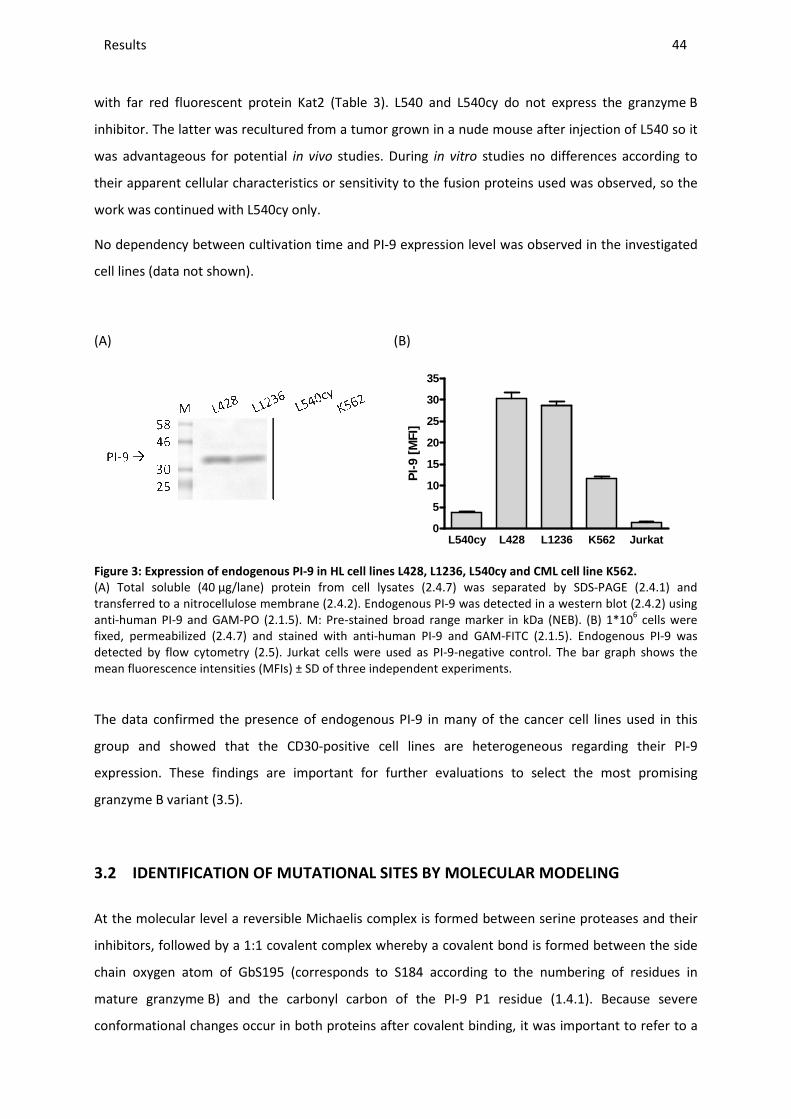
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
with far red fluorescent protein Kat2 (
inhibitor. The latter was recultured from a tumor grown in a nude mouse after injection of L540 so it
was advantageous for potential
their apparent cellular characteristics or sensi
work was continued with L540cy only.
No dependency between cultivation time and
cell lines (data not shown).
(A)
Figure 3: Expression of endogenous
(A) Total soluble (40 µg/lane) protein from cell lysates (transferred to a nitrocellulose membraneanti-human PI-9 and GAM-PO (2.1.5fixed, permeabilized (2.4.7) and stained with antidetected by flow cytometry (2.5). mean fluorescence intensities (MFIs) ± SD
The data confirmed the presence of endogenous PI
group and showed that the
expression. These findings are
granzyme B variant (3.5).
3.2 IDENTIFICATION OF MU
At the molecular level a reversible Michaelis complex is formed
inhibitors, followed by a 1:1 covalent complex whereby a covalent bond is
chain oxygen atom of GbS195
mature granzyme B) and the carbonyl carbon of the PI
conformational changes occur in both proteins after covalent binding, it was important to refer to a
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
with far red fluorescent protein Kat2 (Table 3). L540 and L540cy do not express the granzyme
The latter was recultured from a tumor grown in a nude mouse after injection of L540 so it
for potential in vivo studies. During in vitro studies no differences according to
their apparent cellular characteristics or sensitivity to the fusion proteins used was observed, so the
work was continued with L540cy only.
cultivation time and PI-9 expression level was observed in the investigated
(B)
L540cy L428 L12360
5
10
15
20
25
30
35
PI-
9 [M
FI]
endogenous PI-9 in HL cell lines L428, L1236, L540cy and CML cell line K562.
protein from cell lysates (2.4.7) was separated by transferred to a nitrocellulose membrane (2.4.2). Endogenous PI-9 was detected in a western b
2.1.5). M: Pre-stained broad range marker in kDa (NEB). and stained with anti-human PI-9 and GAM-FITC (2.1.5).
Jurkat cells were used as PI-9-negative control. The bar graph shows the fluorescence intensities (MFIs) ± SD of three independent experiments.
The data confirmed the presence of endogenous PI-9 in many of the cancer cell lines used in this
showed that the CD30-positive cell lines are heterogeneous regarding their PI
important for further evaluations to select the most promising
IDENTIFICATION OF MUTATIONAL SITES BY MOLECULAR MODELING
reversible Michaelis complex is formed between serine proteases and their
, followed by a 1:1 covalent complex whereby a covalent bond is formed
(corresponds to S184 according to the numbering of residues in
and the carbonyl carbon of the PI-9 P1 residue (1.4.1
conformational changes occur in both proteins after covalent binding, it was important to refer to a
_______________________________________________________________________________________________
44
L540cy do not express the granzyme B
The latter was recultured from a tumor grown in a nude mouse after injection of L540 so it
studies no differences according to
was observed, so the
ion level was observed in the investigated
L1236 K562 Jurkat
9 in HL cell lines L428, L1236, L540cy and CML cell line K562.
SDS-PAGE (2.4.1) and 9 was detected in a western blot (2.4.2) using
stained broad range marker in kDa (NEB). (B) 1*106 cells were . Endogenous PI-9 was
The bar graph shows the
9 in many of the cancer cell lines used in this
heterogeneous regarding their PI-9
important for further evaluations to select the most promising
LECULAR MODELING
between serine proteases and their
formed between the side
(corresponds to S184 according to the numbering of residues in
1.4.1). Because severe
conformational changes occur in both proteins after covalent binding, it was important to refer to a

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
45
structure describing the initial Michaelis complex, in which the protease was inactivated by mutating
S195 to alanine; allowing the interacting amino acids responsible for complex formation to be
identified. The published rat trypsin - M. sexta serpin B1 complex [87] was found to be suitable for
fulfilling the prerequisites. In addition, the crystal structure of human granzyme B bound to a five-
residue peptide, solved at 2 Å resolution [42], is known. Human granzyme B and rat trypsin have a
sequence identity of 36 % according to results from homology modeling (MODELLERv7 software). In
addition, it is known that these proteins have similar length and are closely functionally and
structurally related. Thus, the backbone of human granzyme B structure could be fitted with that of
rat trypsin. Because the crystal structure of human PI-9 has not yet been solved, its structure had to
be built using molecular homology modeling based on the X-ray structure of the rat trypsin - M. sexta
serpin B1 complex. The sequence identity between M. sexta serpin B1 and human PI-9 was 27 %.
In cooperation with the German Research School for Simulation Sciences in Juelich (Prof. Dr. rer. nat.
Paolo Carloni), the modeled complex was inserted into a water box and key interactions at the
interface of the two proteins were screened by Baker’s alanine scanning procedure using ROBETTA
server (AMBER ff99SB, TIP3P and Aaqvist force fields were used for the protein, water, and counter
ions respectively, evaluation of long-range electrostatic interactions via particle mesh Ewald
method). The residues R28 and R201, according to the numbering of residues in mature granzyme B,
were found to have the largest destabilization in terms of loss of contacts. According to the
respective charge of the amino acids (neutral, opposite and same charge as the original amino acid,
respectively) arginine was substituted with alanine, glutamic acid and lysine by site-directed
mutagenesis (2.2.2). To investigate a potential supportive effect of both sites at once, the double
mutant R28A/R201A was considered as well, so in total, seven putative PI-9 resistant variants were
generated in silico: R28A, R28K, R28E, R201A, R201K, R201E and R28A-R201A. Their influence on
complex formation was analyzed during 50 ns molecular dynamic simulations in aqueous solution
(NAMD 2.7 package). In all of these mutated complexes the structure of the active sites was
maintained according to molecular modeling results. The three mutants R28K, R201A and R201K
were predicted to be the most destabilized, whereas the others were almost as stable as those of the
wild-type complex. Further details about the molecular simulation studies are published in Losasso et
al. [149].

Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
3.3 GENERATION AND PREPARAT
MUTANTS
The effect of the amino acid exchanges described above on the
sensitivity towards PI-9 was evaluate
both generated prior to this work
malignancies has been demonstrated before
cytotoxic effects on the HL target cell line L540.
cytotoxicity and apoptosis could be shown on L540 as well as L540cy
the protein granzyme B lacking a cell
structures are displayed in Figure
(A)
(B) I
Figure 4: Overview of initial granzyme B
and after Enterokinase cleavage (B)
(A) Schematic structure of the binary eukaryotic expression vectorH22(scFv) and EGb. Igκ: signal peptide sequencecleavage site, Gb: granzyme B encoding sequence, scFv: single chain variable fragment, tag, IVS/IRES: synthetic intron and internal ribosome entry site, Mutated granzyme B sequences could be inserted via expressed in HEK293T cells and purified from supernatant via chromatography (2.4.4). Activation of the protein was (2.4.4) resulting in higher running band in presenceseparated by SDS-PAGE and stained with coomassie(NEB); II: (E)Gb-Ki4(scFv), M: Pre-stained broad range marker in kDa (NEBmarker in kDa (NEB).
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
PREPARATION OF GRANZYME B CONSTRUCTS AND
amino acid exchanges described above on their functionality as well as their
9 was evaluated using the initial constructs Gb-H22(scFv) and Gb
both generated prior to this work. The cytotoxic potential of Gb-H22(scFv) against CD64
malignancies has been demonstrated before [138]. Gb-Ki4(scFv) was cloned earlier
cytotoxic effects on the HL target cell line L540. In this study, the protocol was optimized
apoptosis could be shown on L540 as well as L540cy cells (Figure
B lacking a cell-binding ligand was prepared (Gb). The corresponding
Figure 4A.
II III
granzyme B constructs (A) and SDS-PAGE of the purified fusion proteins before
(B).
Schematic structure of the binary eukaryotic expression vector pMS encoding for EGbIgκ: signal peptide sequence (L) of immunoglobulin kappa light chain, ECS: enterokinase
B encoding sequence, scFv: single chain variable fragment, ron and internal ribosome entry site, EGFP: enhanced green fluorescent protein.
B sequences could be inserted via XbaI/ BlpI cloning site. (B) expressed in HEK293T cells and purified from supernatant via IMAC and subsequent gel filtration
. Activation of the protein was achieved via incubation with 0.02resulting in higher running band in presence (a) and lower bands in absence (b)
PAGE and stained with coomassie. I: (E)Gb-H22(scFv), M: Protein broad range marker in kDa stained broad range marker in kDa (NEB), III: (E)Gb, M: Protein broad range
_______________________________________________________________________________________________
46
CONSTRUCTS AND ITS
functionality as well as their
H22(scFv) and Gb-Ki4(scFv),
H22(scFv) against CD64-positive
earlier but did not show
the protocol was optimized (2.7.3) so
Figure 12A). In addition
corresponding schematic
PAGE of the purified fusion proteins before
pMS encoding for EGb-Ki4(scFv), EGb-of immunoglobulin kappa light chain, ECS: enterokinase
B encoding sequence, scFv: single chain variable fragment, H: 6x histidine (His6)-GFP: enhanced green fluorescent protein.
Fusion proteins were and subsequent gel filtration
0.02 U/µg enterokinase of ECS (E). Protein was
in broad range marker in kDa ), III: (E)Gb, M: Protein broad range

Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
The secretory expression, enabled by the Igκ leader (
expressed in an inactive form by insertion of an enterokinase cleavage site at its N
prevent killing of the expressing cells, was performed
fusion proteins into the supernatant should allow correct post
granzyme B has two glycosylation sites. The protein was purified by affinity chromatography
selecting for the His6-tag and gel filtration (
PAGE (2.4.5). The identity of the protein was confirmed by western blot analysis using an anti
Gb antibody (2.4.2). To obtain a free N
granzyme B, the protein was incubated with enterokinase (0.02
Gb-H22(scFv) and Gb-Ki4(scFv) are shown in
whereby successful cleavage was verified by
the proteins was quantified by AIDA analysis (
The putative PI-9 resistant mutants described in
splice-overlap extension (SOE)-PCR
cloned into the corresponding pMS vector encoding for the wild
addition, the enzymatically inactive variant S184A was generated as control protein
granzyme B mutant K27A which has been described
overview of all constructs generat
Figure 5: List of generated point-mutated variants (
modeling (3.2).
All of the constructs needed,
determinations, have successfully been generated and produced in HEK293T cells, followed by
cleavage by enterokinase.
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
The secretory expression, enabled by the Igκ leader (2.4.4), of the granzyme
expressed in an inactive form by insertion of an enterokinase cleavage site at its N
prevent killing of the expressing cells, was performed in HEK293T cells (2.4.4.1). The secretion of the
fusion proteins into the supernatant should allow correct post-translational modification because
wo glycosylation sites. The protein was purified by affinity chromatography
tag and gel filtration (2.4.4.2), with a yield of 1-2 mg/L, as de
). The identity of the protein was confirmed by western blot analysis using an anti
). To obtain a free N-terminus, which is important for the proteolytic activity of
B, the protein was incubated with enterokinase (0.02 U/µg). The purified fusion proteins
are shown in Figure 4B before and after digestion with enterokinase,
whereby successful cleavage was verified by the reduction of molecular weight. The concentration of
the proteins was quantified by AIDA analysis (2.4.5).
mutants described in 3.2 were generated via site-directed mutagenesis
PCR (2.2.2) with specific primers (2.1.4), and the new sequences were
cloned into the corresponding pMS vector encoding for the wild-type-based con
the enzymatically inactive variant S184A was generated as control protein
which has been described before to be resistant to PI
generated is summarized in Figure 5.
mutated variants (2.2.2) arisen from literature (#: [87]
needed, including the respective mutations suggested from
have successfully been generated and produced in HEK293T cells, followed by
_______________________________________________________________________________________________
47
B proteins, which is
expressed in an inactive form by insertion of an enterokinase cleavage site at its N-terminus to
). The secretion of the
translational modification because
wo glycosylation sites. The protein was purified by affinity chromatography
mg/L, as determined by SDS-
). The identity of the protein was confirmed by western blot analysis using an anti-human
terminus, which is important for the proteolytic activity of
U/µg). The purified fusion proteins
B before and after digestion with enterokinase,
the reduction of molecular weight. The concentration of
directed mutagenesis by
and the new sequences were
based constructs (2.1.4). In
the enzymatically inactive variant S184A was generated as control protein and a
to be resistant to PI-9 inhibition [86]. An
];
##: [86]) or molecular
including the respective mutations suggested from in silico
have successfully been generated and produced in HEK293T cells, followed by

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
48
3.4 PREPARATION AND FUNCTIONAL CHARACTERIZATION OF RECOMBINANT PI-9
Recombinant PI-9 had to be produced in order to evaluate the enzymatic activity of the new
granzyme B mutants in the presence of the inhibitor in vitro during a substrate assay to pre-select the
most promising mutants. As during the initially planned mammalian expression in HEK293T cells, only
low amounts of PI-9 could be produced (2.1.4, data not shown); therefore another alternative for the
production of PI-9 was pursued, namely expression in E. coli. The PI-9-encoding sequence was cloned
into the E. coli expression vector pMT (2.1.4) by HindIII/NotI restriction sites (Figure 6A). The
periplasmic expression under osmotic stress (2.4.3) has been proven to be successful in small scale
by SDS-PAGE and western blotting (data not shown); so a 4 L batch fermentation was carried out. As
shown in Figure 6B, the four subsequent purification steps included two IMAC purifications, one
purification step via anion exchange chromatography and a final run using a gel filtration column
(2.4.3). The purified protein was sterile filtered and stored at -80 °C in the presence of 1 mM DTT.
Functionality of the produced PI-9 was confirmed by its ability to form covalent, SDS-stable
complexes with recombinant wild-type granzyme B-based proteins. Therefore PI-9 and wild-type
Gb-H22(scFv) were incubated in a 5:1 molar ratio for 1 h at 37 °C under reducing conditions in
presence of 1 mM DTT (2.7.1.4). Successful complex formation and thereby functionality of the
produced PI-9 was verified via western blot analysis (Figure 6C).
Complexes were formed between PI-9 and free Gb which was present as a degradation product
within the Gb-H22(scFv) sample running at approximately 77 kDa and between full length
Gb-H22(scFv) and PI-9 resulting in a complex above 100 kDa (correlates with 175 kDa band of marker
which is inaccurate at high molecular sizes). The complex could not be verified using anti-His, maybe
due to sterical hindrance of the antigen binding site; therefore the complex was verified using anti-
human Gb and anti-human PI-9. During analysis with anti-His it turned out that the band running
below the more intense band was not functional, since it remained within the sample after
incubation with granzyme B in contrast to the higher running band (Figure 6C, III, lane 2).

Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
(A)
(C)
I
II
Figure 6: Preparation of recombinant PI
type Gb-H22(scFv).
(A) Schematic structure of the prokaryotic expression vector pMT encoding for PIsequence for periplasmic localizatioNotI cloning site. (B) Recombinant PIsubsequently via anion exchange chromatography (MonoQ column) and finally via gel filtrationchromatography (GF) (2.4.4). the band of active PIfurther experiments. The band below the 42assays (C,III). (C) To demonstrate the wild-type Gb-H22(scFv) for 1 h at 37°C transferred to a nitrocellulose membrane (western blot using anti-human Gb ((2.1.5); 1: Gb-H22(scFv), 2: Gb-H22(scFv)complex are indicated in grey, full-length protein and complex (red rectangle) thereof in blackbroad range marker in kDa (NEB).
Functional PI-9 could be produced
proteolytic activity of the new granzyme
3.5 ESTABLISHMENT OF A T
For selection of the most promising
suitable in vitro test system based on PI
Within this thesis different strategies were pu
cell lines expressing cytosolic PI-
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
(B)
II
III
Preparation of recombinant PI-9 and functionality determination via complex formation with
Schematic structure of the prokaryotic expression vector pMT encoding for PI-9. pelBlocalization, H: 6x histidine tag, PI-9: PI-9 encoding sequence, inserted via
Recombinant PI-9 was expressed in E. coli (2.4.3) and purified subsequently via anion exchange chromatography (MonoQ column) and finally via gel filtration
the band of active PI-9 protein is indicated by an arrow. Fraction 6 was used for and below the 42 kDa band was inactive protein, as prove
the functionality of the produced recombinant PI-9 at 37°C (2.7.1.4) before the protein mixture was separated by SDS
transferred to a nitrocellulose membrane (2.4.2). Complexed and uncomplexed protein was detected in a human Gb (I), anti-human PI-9 (II) or anti-His monoclonal antibodies
(scFv) and PI-9, 3: PI-9. Gb cleaved during purification and the corresponding length protein and complex (red rectangle) thereof in black
9 could be produced by bacterial expression and is essential to determine
proteolytic activity of the new granzyme B mutants in presence of PI-9 (3.6.1).
ESTABLISHMENT OF A TEST SYSTEM FOR GRANZYME B MUTANTS
promising mutant, able to induce apoptosis within PI
based on PI-9-positive and –negative cell lines had to be
sis different strategies were pursued: First, the generation of transiently transfected
-9 or the delivery of recombinant granzyme B protein
_______________________________________________________________________________________________
49
omplex formation with wild-
9. pelB: signal peptide 9 encoding sequence, inserted via HindIII/
and purified twice via IMAC, subsequently via anion exchange chromatography (MonoQ column) and finally via gel filtration
Fraction 6 was used for proven during functionality
it was incubated with protein mixture was separated by SDS-PAGE and
and uncomplexed protein was detected in a monoclonal antibodies (III) and GAM-PO
purification and the corresponding length protein and complex (red rectangle) thereof in black. M: Pre-stained
essential to determine the retained
RANZYME B MUTANTS
o induce apoptosis within PI-9-expressing cells, a
had to be established.
the generation of transiently transfected
protein into inherently

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
50
PI-9-positive cells. Second, the identification of PI-9-positive as well as -negative cell lines
overexpressing identical target receptors. The different approaches which were evaluated in parallel
are described in detail in the following.
1) The direct delivery of the protein into potential target cells is independent of receptor
expression and specific binders, and thereby parameters based on affinity, internalization and
endosomal release. This approach was followed by generating a granzyme B construct lacking
the binding moiety (Figure 4A). Protein delivery into the cytosol of PI-9-positive L428 and L1236
cells as well as PI-9-negative Jurkat cells, among others, (3.1) was analyzed with the pore-
forming reagent streptolysin O (2.3.3), an oxygen-labile streptococcal hemolytic exotoxin. The
delivery of granzyme B variants was only successful for Jurkat cells as shown by western blot
analysis of total soluble protein of cell lysates and resulting cytotoxic effects. The efficacy was
determined via apoptosis assays done 4 h after Gb delivery into the cells (2.7.2.1). In contrast,
translocation efficiencies for L428 and L1236 cells were not sufficient to compare the effect of
different granzyme B variants to inherently PI-9-positive or -negative cells. Since Jurkat cells
could not be transfected with PI-9 even though all common transfection reagents and methods
were tested (Table 5), this approach was not further pursued.
2) Since the functionality of Gb-H22(scFv) was established before with AML cell line U937 and
primary CD64-positive leukemic cells, this construct could be used to test the new granzyme B
mutants targeting the CD64-positive AML cell line HL60 (U937 lost its sensitivity to Gb-H22(scFv)
for unknown reasons and was therefore no longer applicable for this purpose). HL60 cells were
tested to be PI-9-negative (Table 6), so PI-9 transfection of this cell line would allow comparable
assays to confirm resistance of granzyme B mutants against PI-9. For this purpose different
transfection methods were evaluated using varying conditions as listed in Table 5. However, as
for Jurkat cells (3.5, 1)) all transfection efficiencies were too low to establish a stable transfected
cell line.
3) The other available granzyme B-based construct was Gb-Ki4(scFv) whose cytotoxic potential
could be evaluated in this study using the CD30-positive cell lines L540 and L540cy (2.1.6). Both
target cell lines were tested to be PI-9-negative (Figure 3). As was seen with HL60 cells,
transfection with PI-9 was not successful for L540cy cells (3.5, 2). Alternatively, the in vitro anti-
tumor efficacy in cells expressing both endogenous PI-9 and the CD30 receptor was tested.
Here, depending on the ability of the target cell lines to express PI-9 or not, no induction of
apoptosis was determined by an Annexin V/ PI assay after incubation with wild-type
Gb-Ki4(scFv) for 48 h. Results are shown in Figure 7A. Apoptotic cells were only observed in the
PI-9-negative cell line L540cy. No cell death was induced in PI-9-positive L428 cells. To verify that
complex formation with the inhibitor was the cause for the cytotoxic limitation, the cells were
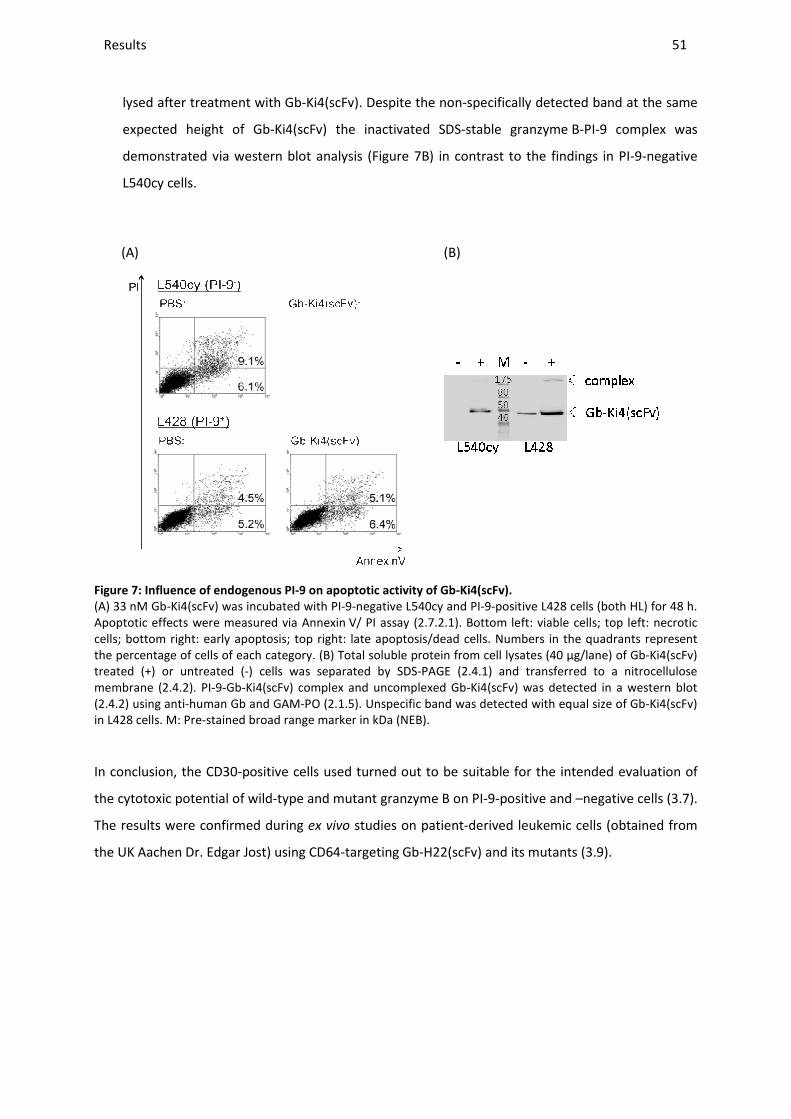
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
lysed after treatment with Gb
expected height of Gb-Ki4(scFv)
demonstrated via western blot analysis
L540cy cells.
(A)
Figure 7: Influence of endogenous PI
(A) 33 nM Gb-Ki4(scFv) was incubated with Apoptotic effects were measured via Annexincells; bottom right: early apoptosis; top right: late apoptosis/dead cells. Numbers in the quadrants represthe percentage of cells of each category.treated (+) or untreated (-) cells was separated by SDSmembrane (2.4.2). PI-9-Gb-Ki4(scFv) complex and uncomplexed Gb(2.4.2) using anti-human Gb and GAMin L428 cells. M: Pre-stained broad range marker in kDa (NEB).
In conclusion, the CD30-positive cells
the cytotoxic potential of wild-type and mutant granzyme
The results were confirmed during
the UK Aachen Dr. Edgar Jost) using CD64
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
lysed after treatment with Gb-Ki4(scFv). Despite the non-specifically detected band at the same
Ki4(scFv) the inactivated SDS-stable granzyme B
rn blot analysis (Figure 7B) in contrast to the findings in PI
(B)
PI-9 on apoptotic activity of Gb-Ki4(scFv).
was incubated with PI-9-negative L540cy and PI-9-positive L428 cellsApoptotic effects were measured via Annexin V/ PI assay (2.7.2.1). Bottom left: viable cells; top left: necrotic cells; bottom right: early apoptosis; top right: late apoptosis/dead cells. Numbers in the quadrants represthe percentage of cells of each category. (B) Total soluble protein from cell lysates (40 µg/lane) of
) cells was separated by SDS-PAGE (2.4.1) and transferred to a nitrocellulose Ki4(scFv) complex and uncomplexed Gb-Ki4(scFv) was detected in a western blot
human Gb and GAM-PO (2.1.5). Unspecific band was detected with equal size stained broad range marker in kDa (NEB).
positive cells used turned out to be suitable for the intended
type and mutant granzyme B on PI-9-positive and
The results were confirmed during ex vivo studies on patient-derived leukemic cells (obtained from
the UK Aachen Dr. Edgar Jost) using CD64-targeting Gb-H22(scFv) and its mutants
_______________________________________________________________________________________________
51
specifically detected band at the same
B-PI-9 complex was
in contrast to the findings in PI-9-negative
positive L428 cells (both HL) for 48 h. Bottom left: viable cells; top left: necrotic
cells; bottom right: early apoptosis; top right: late apoptosis/dead cells. Numbers in the quadrants represent (B) Total soluble protein from cell lysates (40 µg/lane) of Gb-Ki4(scFv)
) and transferred to a nitrocellulose Ki4(scFv) was detected in a western blot
equal size of Gb-Ki4(scFv)
for the intended evaluation of
positive and –negative cells (3.7).
derived leukemic cells (obtained from
and its mutants (3.9).

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
52
3.6 IN VITRO FUNCTIONAL CHARACTERIZATION OF WILD-TYPE AND MUTANT
GB-H22(SCFV) AND GB-KI4(SCFV)
The main challenge during generation of granzyme B mutants was to avoid a negative effect of the
mutations on the active site of the enzyme and its tertiary structure. To exclude hampering of
functionality, the wild-type and mutated granzyme B-based fusion proteins were tested in
comparative assays regarding their enzymatic activity, their binding affinity, and their specific cell
cytotoxicity.
3.6.1 Proteolytic activity of granzyme B in absence and presence of PI-9
The influence of the single point mutations on the granzyme B proteolytic activity was determined
via a colorimetric substrate assay based on the synthetic granzyme B substrate Ac-IETD-pNA, which
mimics the cleavage and activation site of human procaspase-3 (2.7.1.1). Results for Gb-Ki4(scFv) and
GbR201K-Ki4(scFv) are shown in Figure 8.
0 10 20 30 40 50 600.0
0.1
0.2
0.3
0.4
0.5
0.6
GbR201K-Ki4Gb-Ki4
Time [min]
E40
5 n
m
Figure 8: Enzymatic activity of Gb-Ki4(scFv) and GbR201K-Ki4(scFv).
The proteolytic activity of activated protein was measured via a colorimetric assay based on the synthetic granzyme B substrate Ac-IETD-pNA (2.7.1.1). Reaction was documented for 1 h with a 2 min interval at 37 °C and 405 nm in an Elisa plate reader. Graphs were plotted using GraphPad Prism 4.0.
No differences were observed regarding the measured enzymatic kinetics so it could be concluded
that the active site was not affected by these mutations.
Enzymatic activity of Gb-H22(scFv) and GbR201K-H22(scFv) was additionally quantified via Michaelis-
Menten kinetics. KM values were comparable for both constructs with 104 ± 32 µM for the wild-type
and 105 ± 13 µM for the mutant.
A comparative overview of the activity of all generated mutants, including GbK27A, a mutant
published before and supposed to bind less efficiently to PI-9 (1.4.1), is depicted as grey bars in

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
53
Figure 9. Except for the double mutant R28A/R201A and the single mutant R201E, no significant
differences were measured in comparison to the wild-type protein.
Gb-H22
GbK27
A-H22
GbR28
A-H22
GbR28
E-H22
GbR28
K-H22
GbR28
1A/R
201A-H
22
GbR20
1A-H
22
GbR201E
-H22
GbR20
1K-H
22
0.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035without PI-9with PI-9
E40
5nm
/min
Figure 9: Enzymatic activity of Gb-H22(scFv) wild-type and its mutants in the presence and absence of PI-9.
Gb-H22(scFv) was incubated either with our without recombinant PI-9 according to 2.7.1.4. The proteolytic activity was measured via a colorimetric assay based on the synthetic granzyme B substrate Ac-IETD-pNA (2.7.1.1, 2.7.1.4). The graph shows means ± SD of 3 to 6 independent experiments [149].
To confirm the earlier obtained in silico findings (3.23.2) in vitro, the proteolytic activity of wild-type
and mutant granzyme B was analyzed after challenge with recombinant PI-9 in a comparative
enzymatic assay. In the presence of PI-9, GbR28K retained 76 % of its original activity, and GbR201A
retained 46 %. The enzymatic activity of GbR201K was almost the same in the presence or absence of
PI-9 (6 % reduction after PI-9 challenge), so this variant was the least inhibited variant according to
this assay. The catalytic activity of the other mutated granzyme B variants decreased relative to wild-
type granzyme B, or was even lower in the case of the double mutant. Their remaining activity after
complexing ranged from 0.5 % for the double mutant (R28A-R201A) to 25 % for R28E. An exception
was R28A with a remaining activity of 54 % as opposed to the computed predictions [149]. Even
though the result of the latter was promising, this variant was not further tested since it did not show
good results during preliminary cytotoxicity studies on PI-9—positive and –negative cells (data not
shown). The PI-9 insensitivity of the earlier described granzyme B variant K27A (see above) could not
be reproduced either during the in silico or during the in vitro experiments.
The most promising constructs with retained proteolytic activity after incubation with recombinant
PI-9, GbR28K, GbR201A and GbR201K, were further tested, always in comparison to GbK27A and Gb
wild-type.

Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
3.6.2 Target cell binding
To exclude an influence of the point
activity of the granzyme B variants, flow
Gb-Ki4(scFv) or Gb-H22(scFv) variants
human Gb-PE or anti-His Alexa 488
Representative results for Gb-Ki4(scFv) and GbR201K
(A)
(B)
Figure 10: Binding analysis of granzyme B
50 nM Gb-based constructs were incubated with binding was detected with anti-His Alexa488 (GbR201K-Ki4(scFv) on CD30-positive L428 and CD30GbR201K-H22(scFv) on CD64-positive HL60 and
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
To exclude an influence of the point-mutations on the tertiary structure and possibly the binding
variants, flow cytometry-based binding assays were performed with all
variants. The specific detection of cellular binding was done by anti
His Alexa 488 (2.1.5).
Ki4(scFv) and GbR201K-Ki4(scFv) are shown in Figure
granzyme B-based CFPs.
based constructs were incubated with 4*105 target and control cell lines His Alexa488 (2.1.5) and flow cytometry (2.5.1). (A) Binding of Gb
ve L428 and CD30-negative HL60 cell line. (B) Binding of Gbpositive HL60 and CD64-negative Ramos cell line.
_______________________________________________________________________________________________
54
mutations on the tertiary structure and possibly the binding
were performed with all
of cellular binding was done by anti-
Figure 10A.
target and control cell lines for 30 min on ice and (A) Binding of Gb-Ki4(scFv) and
. (B) Binding of Gb-H22(scFv) and

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
55
No difference in binding activity was found for any of the new constructs compared to the wild-type,
on CD30-positive HL cell lines L428, L1236 and L540cy, or on the CML cell line K562. No unspecific
binding to the CD30-negative AML cell line HL60 was detected.
The corresponding Kd values for Gb-Ki4(scFv) and GbR201K-Ki4(scFv) on all of the cell lines used are
summarized in Table 7 and are all in the same nM range, independent of both the mutation and the
level of the CD30 receptor expression. The latter was evaluated since significant differences were
observed during previous flow cytometric analysis. CD30 expression level was set to 100 % for L540cy
since for this cell line the highest shift in fluorescence was measured. K562 was determined to have
the lowest CD30 expression.
Table 7: Affinity constants and relative receptor expression level of CD30-positive cell lines.
(n. d.: not determined)
Cell line KD of Gb-Ki4(scFv)
[nM]
KD of GbR201K-Ki4(scFv)
[nM]
CD30 expression
[% MFI]
L540cy 3.6 5.7 100
L428 1.5 1.9 32
L1236 n. d. n. d. 35
K562 5.2 4.8 7
Specific binding of Gb-H22(scFv) and its mutants was determined using CD64-positive HL60 cells and
CD64-negative Burkitt lymphoma cell line Ramos as a control. Data are shown representatively for
Gb-H22(scFv) and GbR201K-H22(scFv) in Figure 10B. The affinity constant (Kd) for each of these
constructs was measured to be 2.1 nM; which shows specific and high affinity to the receptor.
The described data indicated that the point-mutations within granzyme B did not affect the high
affinity binding of the determined CFPs to their respective target cells.
3.6.3 Internalization activity
Since marked dissimilarities in CD30 receptor expression were observed (Table 7) and to assure that
cytotoxic differences were not due to subsequent diminished internalization, target cell
internalization was analyzed by confocal microscopy (2.6). For that purpose cells were incubated
with Ki4(scFv)-SNAP-BG-647, a red fluorescent protein based on the SNAP-tag technology (2.4.6), at
37 °C or 4 °C. Incubation times were adapted to CD30 expression levels so K562 was incubated for 4 h
with the fluorescent fusion protein whereas the other cell lines were investigated after 2 h. This
longer incubation time for K562 was chosen based on the low CD30 expression to allow for additional
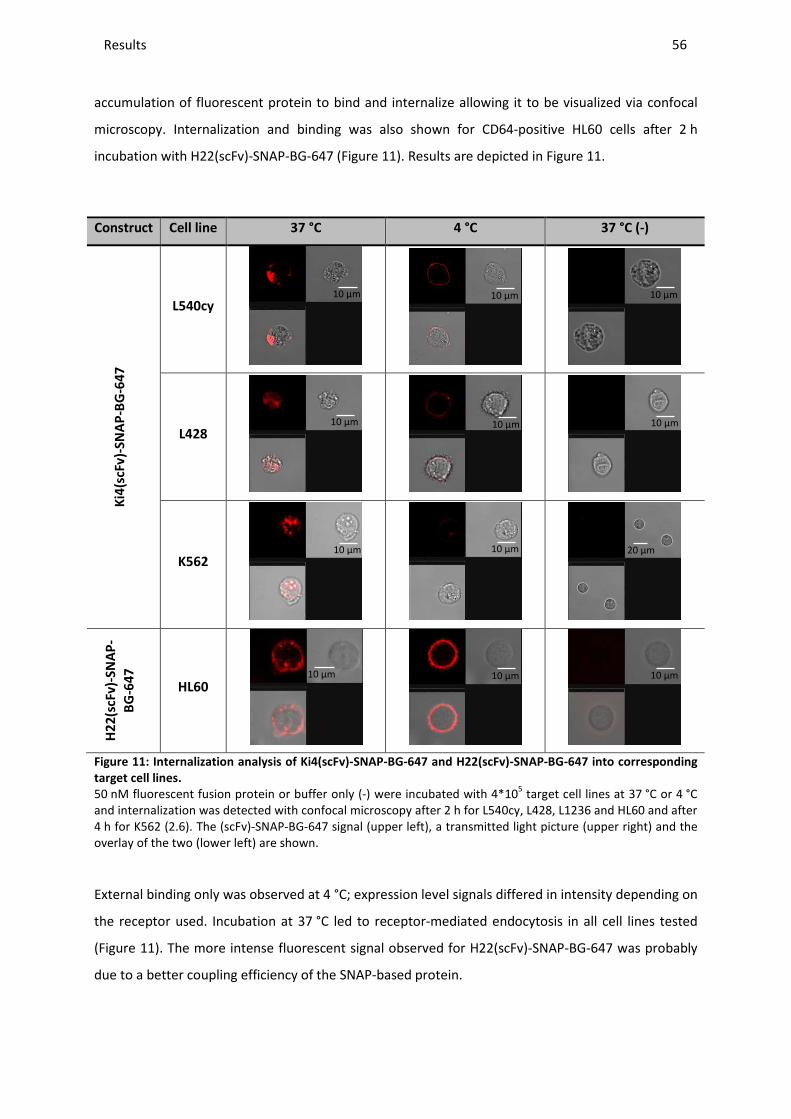
Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
56
accumulation of fluorescent protein to bind and internalize allowing it to be visualized via confocal
microscopy. Internalization and binding was also shown for CD64-positive HL60 cells after 2 h
incubation with H22(scFv)-SNAP-BG-647 (Figure 11). Results are depicted in Figure 11.
Construct Cell line 37 °C 4 °C 37 °C (-)
Ki4
(scF
v)-
SN
AP
-BG
-64
7
L540cy
L428
K562
H2
2(s
cFv
)-S
NA
P-
BG
-64
7
HL60
Figure 11: Internalization analysis of Ki4(scFv)-SNAP-BG-647 and H22(scFv)-SNAP-BG-647 into corresponding
target cell lines.
50 nM fluorescent fusion protein or buffer only (-) were incubated with 4*105 target cell lines at 37 °C or 4 °C and internalization was detected with confocal microscopy after 2 h for L540cy, L428, L1236 and HL60 and after 4 h for K562 (2.6). The (scFv)-SNAP-BG-647 signal (upper left), a transmitted light picture (upper right) and the overlay of the two (lower left) are shown.
External binding only was observed at 4 °C; expression level signals differed in intensity depending on
the receptor used. Incubation at 37 °C led to receptor-mediated endocytosis in all cell lines tested
(Figure 11). The more intense fluorescent signal observed for H22(scFv)-SNAP-BG-647 was probably
due to a better coupling efficiency of the SNAP-based protein.
10 µm 10 µm 10 µm
10 µm 10 µm 10 µm
10 µm 10 µm 20 µm
10 µm 10 µm 10 µm
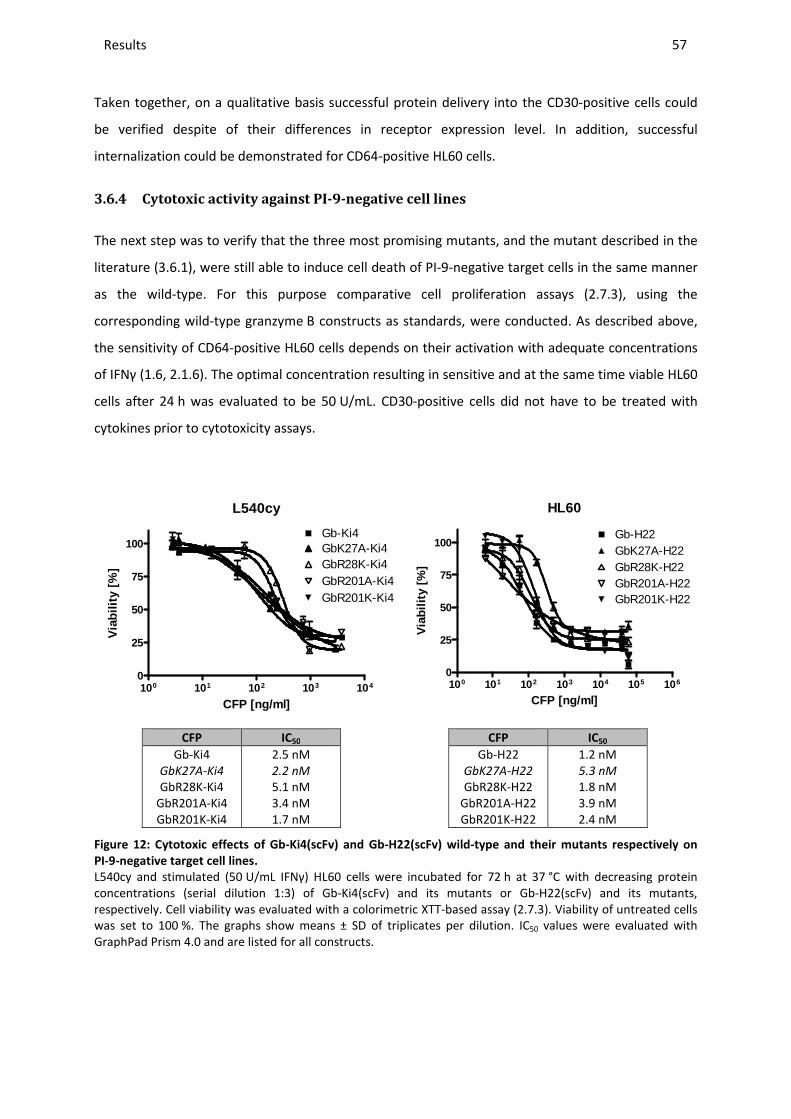
Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
57
Taken together, on a qualitative basis successful protein delivery into the CD30-positive cells could
be verified despite of their differences in receptor expression level. In addition, successful
internalization could be demonstrated for CD64-positive HL60 cells.
3.6.4 Cytotoxic activity against PI-9-negative cell lines
The next step was to verify that the three most promising mutants, and the mutant described in the
literature (3.6.1), were still able to induce cell death of PI-9-negative target cells in the same manner
as the wild-type. For this purpose comparative cell proliferation assays (2.7.3), using the
corresponding wild-type granzyme B constructs as standards, were conducted. As described above,
the sensitivity of CD64-positive HL60 cells depends on their activation with adequate concentrations
of IFNγ (1.6, 2.1.6). The optimal concentration resulting in sensitive and at the same time viable HL60
cells after 24 h was evaluated to be 50 U/mL. CD30-positive cells did not have to be treated with
cytokines prior to cytotoxicity assays.
L540cy
100 101 102 103 1040
25
50
75
100
GbR28K-Ki4GbR201A-Ki4GbR201K-Ki4
GbK27A-Ki4Gb-Ki4
CFP [ng/ml]
Via
bilit
y [%
]
HL60
100 101 102 103 104 105 1060
25
50
75
100Gb-H22GbK27A-H22GbR28K-H22GbR201A-H22GbR201K-H22
CFP [ng/ml]
Via
bilit
y [%
]
CFP IC50
Gb-Ki4 GbK27A-Ki4
GbR28K-Ki4 GbR201A-Ki4 GbR201K-Ki4
2.5 nM 2.2 nM 5.1 nM 3.4 nM 1.7 nM
CFP IC50
Gb-H22 GbK27A-H22
GbR28K-H22 GbR201A-H22 GbR201K-H22
1.2 nM 5.3 nM 1.8 nM 3.9 nM 2.4 nM
Figure 12: Cytotoxic effects of Gb-Ki4(scFv) and Gb-H22(scFv) wild-type and their mutants respectively on
PI-9-negative target cell lines.
L540cy and stimulated (50 U/mL IFNγ) HL60 cells were incubated for 72 h at 37 °C with decreasing protein concentrations (serial dilution 1:3) of Gb-Ki4(scFv) and its mutants or Gb-H22(scFv) and its mutants, respectively. Cell viability was evaluated with a colorimetric XTT-based assay (2.7.3). Viability of untreated cells was set to 100 %. The graphs show means ± SD of triplicates per dilution. IC50 values were evaluated with GraphPad Prism 4.0 and are listed for all constructs.

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
58
Due to the absence of PI-9 in L540cy and HL60 cells, no differences of functionality should occur
during the performed assays. This could be confirmed by the dose-depending curves whose values
were determined after incubation of the target cells with the fusion proteins Gb-Ki4(scFv) and
Gb-H22(scFv) and their mutants. The IC50 values (between 1.7 and 5.1 nM or 1.2 and 5.3 nM,
respectively, summarized in Figure 12) were all in the same range not differing significantly from the
wild-type. No cytotoxic effects were determined on target receptor negative cell lines HL60 for
Gb-Ki4(scFv) constructs and Ramos for Gb-H22(scFv) constructs (data not shown).
The specificity of cell death induced by GbR201K-H22(scFv) was additionally confirmed by a
competitive assay (2.7.3). Therefore cells were incubated with 10 nM of the fusion protein in the
absence and presence of 10 or 100 nM H22(scFv). 48 h incubation of HL60 cells with
GbR201K-H22(scFv) alone resulted in a remaining cell viability of 44 % compared to 100 % of
untreated cells determined via XTT assay. Cytotoxic effects were significantly reduced by 14 % or
25 % in presence of 10 nM H22(scFv) or 100 nM H22(scFv), respectively. Incubation with H22(scFv)
alone did not reduce cell viability significantly (data not shown).
3.6.5 Apoptotic activity on PI-9-negative cell lines
To verify apoptosis induction as underlying mechanism of the observed cell death, an Annexin V/ PI
assay was performed as described in 2.7.2.1. Results are depicted in Figure 13.
(A)
Buffer
Gb-Ki4
GbK27
A-Ki4
GbR28
K-Ki4
GbR20
1A-K
i4
GbR201K
-Ki4
0
25
50
75
100 L540cy
Via
bilit
y [%
]
(B)
Buffer
Gb-H22
GbR201K
-H22
0
25
50
75
100 HL60K562
Via
bilit
y [%
]
Figure 13: Apoptotic effects of Gb-Ki4(scFv) or Gb-H22(scFv) wild-type and their mutants respectively on PI-9-
negative target cell lines.
21 nM CFPs were incubated with target cell lines for 48 h at 37°C. Apoptotic effects were measured via Annexin V/ PI assay (2.7.2.1), normalized to buffer control and converted to viability. The bar graphs show means ± SD of three independent experiments. (A) Result after incubation of Gb-Ki4(scFv) and its mutants with CD30+ L540cy cell line. (B) Result after incubation of Gb-H22(scFv) and its mutants with stimulated (50 U/mL IFNγ) CD64+ HL60 cell line. CD64 K562 cell line was used as negative control.

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
59
Here, up to 60 % of the targeted L540cy cells were apoptotic after incubation with Gb-Ki4(scFv) or its
mutants (Figure 13A). Receptor-negative HL60 cells were not affected by the fusion proteins (data
not shown) confirming specificity of apoptosis induction.
In case of Gb-H22(scFv) and its most promising mutant GbR201K-H22(scFv), apoptosis induction after
incubation with these CFPs was observed in more than 50 % of the stimulated HL60 cells, whereas no
unspecific effect could be detected on the CD64-negative CML cell line K562 (Figure 13B).
Apoptosis induction was additionally confirmed via a colorimetric caspase 3/7 assay (2.7.2.2) which is
a good indicator for apoptosis after treatment with granzyme B since caspase 3 is a direct substrate
of the enzyme. After incubation of HL60 cells with 33 nM Gb-H22(scFv) or GbR201K-H22(scFv) for
48 h the specific signal for caspase activity was in both cases increased 2.5-fold compared to the
buffer control. The same increase was determined for the positive control treated with Zeocin (data
not shown).
Since the above data showed no differences in cytotoxic potential for the tested granzyme B variants
on PI-9-negative cell lines, they were all further tested on PI-9-positive cells to select the most
promising one.
3.7 SELECTION OF THE MOST PROMISING GRANZYME B MUTANT ON PI-9-
POSITIVE CELL LINES
As described in 3.5, the in vitro test system used for this purpose was based on CD30-positive cell
lines which can be targeted with the construct Gb-Ki4(scFv). Decreased apoptotic effects were
observed in PI-9-expressing cells after incubation with Gb-Ki4(scFv) in contrast to PI-9-negative cells
(Figure 7A). Based on the assumption that the observed resistance to cell death is related to the
expression of the endogenous granzyme B inhibitor (Figure 7B) and subsequently depending on the
remaining sensitivity of the novel constructs towards the inhibitor, differences in their cytotoxic
potential were expected here.
3.7.1 Cytotoxic activity on PI-9-positive cell lines
To select the optimal granzyme B variant, the in vitro anti-tumor efficacy of the granzyme B mutants
GbR28K-Ki4(scFv), GbR201A-Ki4(scFv) and GbR201K-Ki4(scFv) was tested on the PI-9-positive HL cell
lines L428 and L1236 as well as the PI-9-positive CML cell line K562 and compared with the effects
triggered by the wild-type Gb-Ki4(scFv) or GbK27A-Ki4(scFv), respectively (2.7.3).
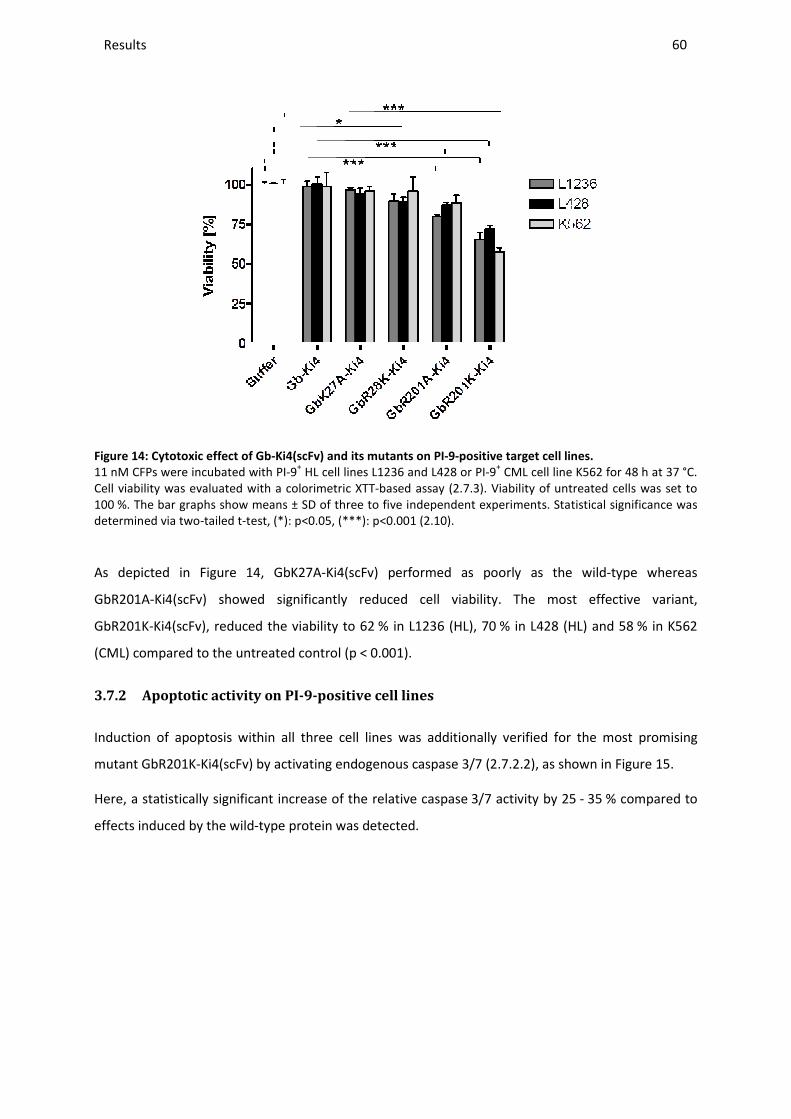
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
Figure 14: Cytotoxic effect of Gb-Ki4(scFv) and its
11 nM CFPs were incubated with PICell viability was evaluated with a colorimetric XTT100 %. The bar graphs show means ± SD of three to five determined via two-tailed t-test, (*)
As depicted in Figure 14, GbK27A
GbR201A-Ki4(scFv) showed significantly
GbR201K-Ki4(scFv), reduced the viability to 62
(CML) compared to the untreated control (p
3.7.2 Apoptotic activity on PI
Induction of apoptosis within all three cell lines was
mutant GbR201K-Ki4(scFv) by activating
Here, a statistically significant increase of the relative caspase
effects induced by the wild-type protein was detected.
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Ki4(scFv) and its mutants on PI-9-positive target cell lines.
PI-9+ HL cell lines L1236 and L428 or PI-9+ CML cell lineiability was evaluated with a colorimetric XTT-based assay (2.7.3). Viability of untreate
s ± SD of three to five independent experiments. Statistic(*): p<0.05, (***): p<0.001 (2.10).
, GbK27A-Ki4(scFv) performed as poorly as the wild
showed significantly reduced cell viability. The most effective
Ki4(scFv), reduced the viability to 62 % in L1236 (HL), 70 % in L428 (HL) and 58
(CML) compared to the untreated control (p < 0.001).
PI-9-positive cell lines
Induction of apoptosis within all three cell lines was additionally verified for
activating endogenous caspase 3/7 (2.7.2.2), as shown in
Here, a statistically significant increase of the relative caspase 3/7 activity by 25
type protein was detected.
_______________________________________________________________________________________________
60
positive target cell lines.
K562 for 48 h at 37 °C. Viability of untreated cells was set to
tatistical significance was
as the wild-type whereas
reduced cell viability. The most effective variant,
% in L428 (HL) and 58 % in K562
y verified for the most promising
, as shown in Figure 15.
3/7 activity by 25 - 35 % compared to
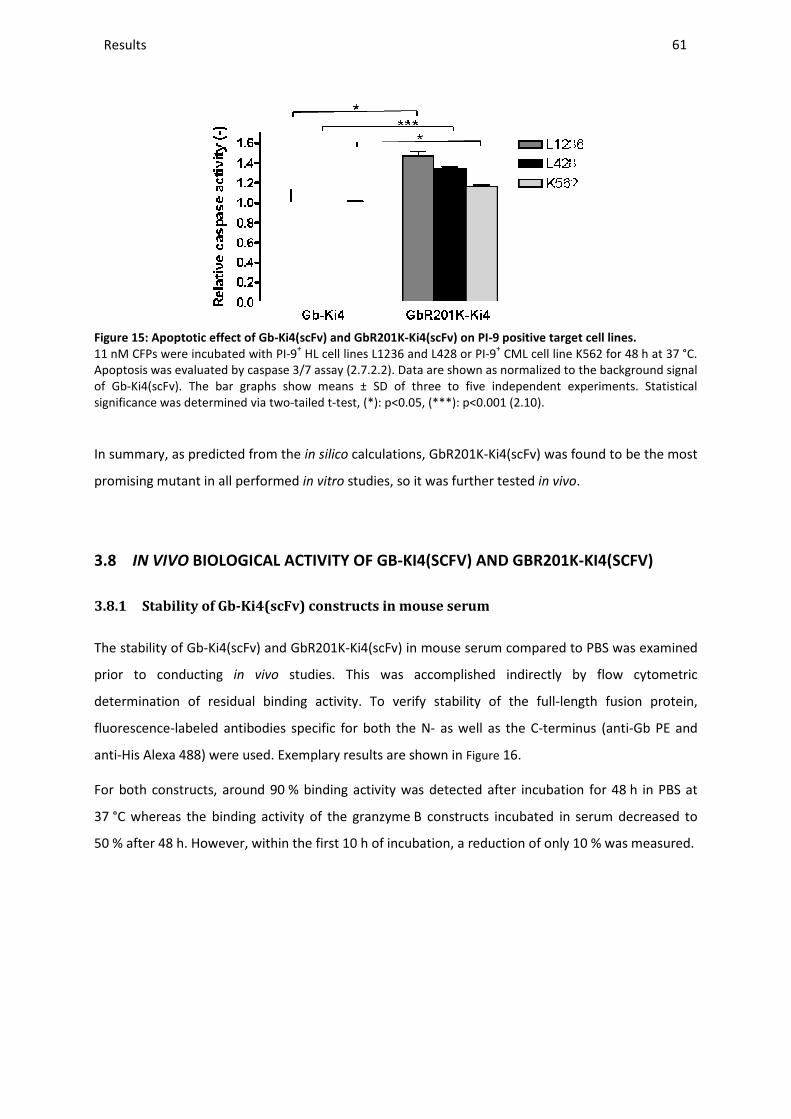
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
Figure 15: Apoptotic effect of Gb-Ki4(scFv) and
11 nM CFPs were incubated with PIApoptosis was evaluated by caspaseof Gb-Ki4(scFv). The bar graphs show means ± SD of three to five independent experiments. significance was determined via two
In summary, as predicted from the
promising mutant in all performed
3.8 IN VIVO BIOLOGICAL ACTIVITY O
3.8.1 Stability of Gb-Ki4(scFv) constructs in mouse serum
The stability of Gb-Ki4(scFv) and Gb
prior to conducting in vivo
determination of residual binding activity.
fluorescence-labeled antibodies specific for both the N
anti-His Alexa 488) were used. Exemplary results are shown in
For both constructs, around 90
37 °C whereas the binding activity of the granzyme
50 % after 48 h. However, within the first 10
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Ki4(scFv) and GbR201K-Ki4(scFv) on PI-9 positive target cell lines.
PI-9+ HL cell lines L1236 and L428 or PI-9+ CML cell line K562 for 48Apoptosis was evaluated by caspase 3/7 assay (2.7.2.2). Data are shown as normalized to
The bar graphs show means ± SD of three to five independent experiments. significance was determined via two-tailed t-test, (*): p<0.05, (***): p<0.001 (2.10).
as predicted from the in silico calculations, GbR201K-Ki4(scFv) was found to be the most
promising mutant in all performed in vitro studies, so it was further tested in vivo
IOLOGICAL ACTIVITY OF GB-KI4(SCFV) AND GBR201K
Ki4(scFv) constructs in mouse serum
Ki4(scFv) and GbR201K-Ki4(scFv) in mouse serum compared to PBS
studies. This was accomplished indirectly by
determination of residual binding activity. To verify stability of the full-length fusion protein
labeled antibodies specific for both the N- as well as the C-terminus (anti
Exemplary results are shown in Figure 16.
% binding activity was detected after incubation for 48
°C whereas the binding activity of the granzyme B constructs incubated in serum decreased to
h. However, within the first 10 h of incubation, a reduction of only 10
_______________________________________________________________________________________________
61
9 positive target cell lines.
CML cell line K562 for 48 h at 37 °C. Data are shown as normalized to the background signal
The bar graphs show means ± SD of three to five independent experiments. Statistical
Ki4(scFv) was found to be the most
in vivo.
K-KI4(SCFV)
compared to PBS was examined
indirectly by flow cytometric
length fusion protein,
terminus (anti-Gb PE and
% binding activity was detected after incubation for 48 h in PBS at
B constructs incubated in serum decreased to
a reduction of only 10 % was measured.

Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
(A)
(C)
Serum stability [%]
Time [h] Gb-Ki4(scFv)0
0.5 2 4
10 24
32.5 48
100 100 96 99 90 65 63 49
Figure 16: Serum stability of Gb-Ki4(scFv) and GbR201K
70 ng/µL of each fusion protein wasafter different time points (2.4.8). Ranti-His Alexa 488 (2.5.1). Data exemplary of percentage serum and PBS stability
Serum stability was comparable for both the wild
point mutation.
3.8.2 Establishment of Hodgkin lymphoma
Before testing Gb-Ki4(scFv) and GbR201K
based on PI-9-negative cell line L540cy
To do this, L540cy cells were resuspended in
injected into 5 mice. Each mouse was injected in
(1*107 cells and 5*106 cells in 30
concentrations. Tumors, 2-5 mm in diameter,
visibility and accessibility, the tu
sizes were above 10 mm, so the
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
(B)
Serum stability [%] PBS stability [%]
(scFv) GbR201K-Ki4(scFv) Gb-Ki4(scFv) GbR201K100 107 97 99 90 69 62 53
100 - -
99 -
91 -
84
Ki4(scFv) and GbR201K-Ki4(scFv).
as incubated in 50 % mouse serum (A) or PBS (B) and samples were taken ). Residual binding activity to L540cy was measured via flow
exemplary shown for GbR201K-Ki4(scFv) and selected time pointsstability based on MFIs and related to 0 h respectively.
Serum stability was comparable for both the wild-type and the mutant indicating no influence of the
Hodgkin lymphoma xenograft models
Ki4(scFv) and GbR201K-Ki4(scFv) in vivo, subcutaneous xenograft
negative cell line L540cy, and on the PI-9-positive cell line L428, had to be established.
cells were resuspended in 50 % Matrigel™ to support tumor growth
. Each mouse was injected in both hind legs each with a different cell densit
cells in 30 µL) (2.9.3). A 100 % tumor take rate was obtained
mm in diameter, became clearly visible after 12 days.
tumors could be measured with a caliper. After three weeks
so the mice were sacrificed.
_______________________________________________________________________________________________
62
PBS stability [%]
GbR201K-Ki4(scFv) 100
- -
99 -
91 -
89
% mouse serum (A) or PBS (B) and samples were taken measured via flow cytometry using
and selected time points. (C) Overview
type and the mutant indicating no influence of the
subcutaneous xenograft tumor models
had to be established.
% Matrigel™ to support tumor growth and were then
different cell density
% tumor take rate was obtained from both cell
became clearly visible after 12 days. Due to their clear
After three weeks tumor

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
63
As the tumors seemed to be encapsulated, the resected tumors were also examined for the presence
of blood vessels. Tumor-derived tissue sections were stained with alkaline phosphatase without
levamisole to visualize any endothelial cells (2.8). Low vascularization was determined as
representatively shown in Figure 17A, therefore it had to be determined if intravenously applied
constructs could be delivered to the tumors. For that purpose, the reported CD30 ectodomain
shedding (1.5) was turned to account, since its level in mouse blood serum could provide information
on a connection of tumors with the bloodstream. The CD30 concentration in blood serum was
quantified with an earlier established ELISA set-up (2.4.9). It was found that it clearly correlated to
tumor size, such that the higher the CD30 level in blood serum the bigger the tumors (Figure 17B). In
addition, specific in vivo targeting of tumors was performed using Ki4(scFv)-SNAP-BG-747 and
molecular imaging (2.9.4). After injection into L540cy tumor-bearing mice localization of the signal in
the tumor was observed (Figure 17C) confirming continuous in vivo CD30 receptor expression and
successful delivery of the Ki4(scFv)-derived construct.
No obvious tumor growth was detected with L428 cells after tumor cell injection as described above,
so caliper measurements were impossible. Therefore, based on earlier obtained findings in this group
indicating the successful correlation between caliper determinations and the signals obtained by
optical imaging [150], Kat2 transfected L428 cells (2.1.6) were used that enable the visualization of
even slow and transient tumor growth via molecular imaging (Figure 17D). 5*106 cells were injected
into the right hind limb of 5 mice. Tumor growth was monitored by optical imaging using a Cri-
Maestro system with the yellow filter set (630-850 nm) and an exposure time of 500 ms every other
day. Tumor sizes remained constant for at least one week. Afterwards spontaneous tumor remission
was observed. A tumor take rate of 90 % was determined.
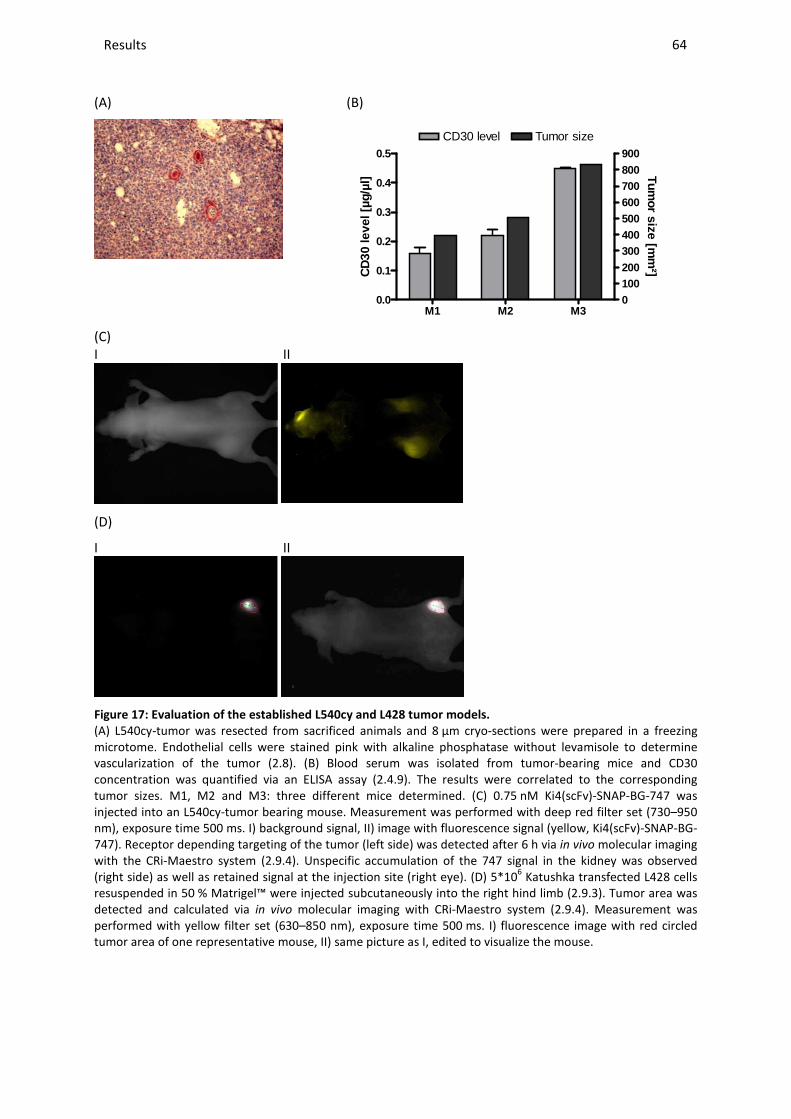
Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
64
(A)
(B)
M1 M2 M30.0
0.1
0.2
0.3
0.4
0.5
0100200300400500600700800900
CD30 level Tumor size
CD
30 le
vel [
µg/µ
l]
Tumor size [m
m²]
(C) I II
(D)
I II
Figure 17: Evaluation of the established L540cy and L428 tumor models.
(A) L540cy-tumor was resected from sacrificed animals and 8 µm cryo-sections were prepared in a freezing microtome. Endothelial cells were stained pink with alkaline phosphatase without levamisole to determine vascularization of the tumor (2.8). (B) Blood serum was isolated from tumor-bearing mice and CD30 concentration was quantified via an ELISA assay (2.4.9). The results were correlated to the corresponding tumor sizes. M1, M2 and M3: three different mice determined. (C) 0.75 nM Ki4(scFv)-SNAP-BG-747 was injected into an L540cy-tumor bearing mouse. Measurement was performed with deep red filter set (730–950 nm), exposure time 500 ms. I) background signal, II) image with fluorescence signal (yellow, Ki4(scFv)-SNAP-BG-747). Receptor depending targeting of the tumor (left side) was detected after 6 h via in vivo molecular imaging with the CRi-Maestro system (2.9.4). Unspecific accumulation of the 747 signal in the kidney was observed (right side) as well as retained signal at the injection site (right eye). (D) 5*106 Katushka transfected L428 cells resuspended in 50 % Matrigel™ were injected subcutaneously into the right hind limb (2.9.3). Tumor area was detected and calculated via in vivo molecular imaging with CRi-Maestro system (2.9.4). Measurement was performed with yellow filter set (630–850 nm), exposure time 500 ms. I) fluorescence image with red circled tumor area of one representative mouse, II) same picture as I, edited to visualize the mouse.

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
65
Based on all described pre-testing, the L540cy tumor model can be regarded as well established so a
treatment schedule starting approximately after two weeks when tumor size was 2 to 5 mm was set-
up. Also, an appropriate L428-based tumor model could successfully be established with the help of
in vivo optical imaging technique so a treatment schedule starting one day after cell injection could
be implemented.
3.8.3 In vivo effects of GbR201K-Ki4(scFv) on PI-9-negative L540cy cells
No difference in cytotoxic activity between Gb-Ki4(scFv) and its mutant GbR201K-Ki4(scFv) was
detected on PI-9-negative cells in vitro, so only the mutant was used to confirm its effectiveness on
PI-9-negative cells in vivo. As a control the non-binding fusion protein GbR201K-H22(scFv) was used.
The treatment schedule followed is shown in Figure 18A. Mice were injected with cells as described
above (3.8.2), and then randomized in two groups (N = 3 for control group, N = 6 for treatment
group) as soon as the tumor size was 2 – 5 mm according to caliper measurements (after 16 days).
The mice were treated with 50 µg protein for 5 consecutive days. Treatment was then continued
every other day until day 13. CFPs were administered i. v. by retro-orbital injection. The size of the
tumors on day 1 of the treatment was set to 100 %. Results are shown in Figure 18B.
In addition, L540cy tumor derived tissue sections were stained after excision by
Ki4(scFv)-SNAP-BG-Vista-Green to confirm continued expression of CD30 after treatment with the
Ki4(scFv)-based construct (Figure 18C).
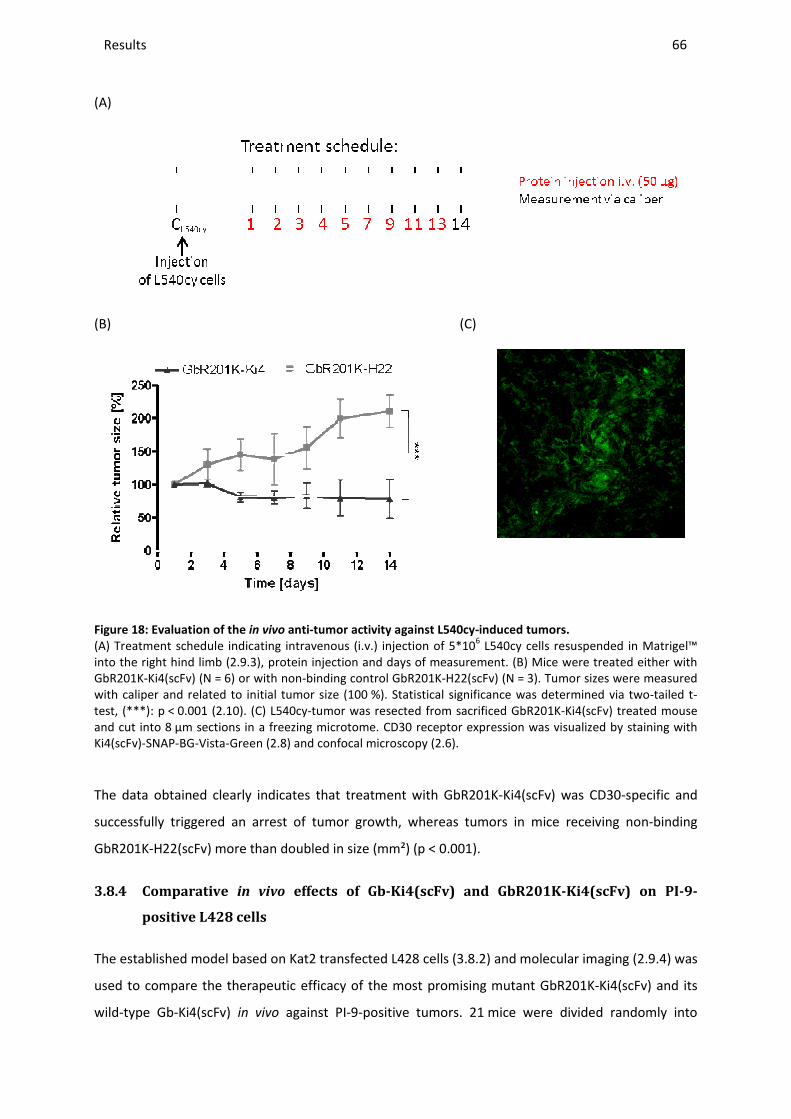
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
(A)
(B)
Figure 18: Evaluation of the in vivo
(A) Treatment schedule indicating intravenous (i.v.) injection of into the right hind limb (2.9.3), protein injection and days of measurement. (B) GbR201K-Ki4(scFv) (N = 6) or with nonwith caliper and related to initial tumor test, (***): p < 0.001 (2.10). (C) L540cand cut into 8 µm sections in a freezing microtome. CD30 receptor Ki4(scFv)-SNAP-BG-Vista-Green (2.8
The data obtained clearly indicate
successfully triggered an arrest of tumor growth, whereas tumors in mice receiving non
GbR201K-H22(scFv) more than doubled in size
3.8.4 Comparative in vivo
positive L428 cells
The established model based on Kat2 transfected L428 cells (
used to compare the therapeutic efficacy
wild-type Gb-Ki4(scFv) in vivo against PI
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
(C)
anti-tumor activity against L540cy-induced tumors.
(A) Treatment schedule indicating intravenous (i.v.) injection of 5*106 L540cy cells resuspended in Matrigel, protein injection and days of measurement. (B) Mice were treated either with
6) or with non-binding control GbR201K-H22(scFv) (N = 3). Tumor sizecaliper and related to initial tumor size (100 %). Statistical significance was determined via two
L540cy-tumor was resected from sacrificed GbR201K-Ki4(scFv) a freezing microtome. CD30 receptor expression was visualized by
2.8) and confocal microscopy (2.6).
clearly indicates that treatment with GbR201K-Ki4(scFv) was CD30
triggered an arrest of tumor growth, whereas tumors in mice receiving non
doubled in size (mm²) (p < 0.001).
effects of Gb-Ki4(scFv) and GbR201K-Ki4(scFv)
he established model based on Kat2 transfected L428 cells (3.8.2) and molecular imaging (
o compare the therapeutic efficacy of the most promising mutant GbR201K
against PI-9-positive tumors. 21 mice were divided
_______________________________________________________________________________________________
66
induced tumors.
resuspended in Matrigel™ Mice were treated either with
umor sizes were measured Statistical significance was determined via two-tailed t-
Ki4(scFv) treated mouse visualized by staining with
was CD30-specific and
triggered an arrest of tumor growth, whereas tumors in mice receiving non-binding
Ki4(scFv) on PI-9-
) and molecular imaging (2.9.4) was
of the most promising mutant GbR201K-Ki4(scFv) and its
divided randomly into
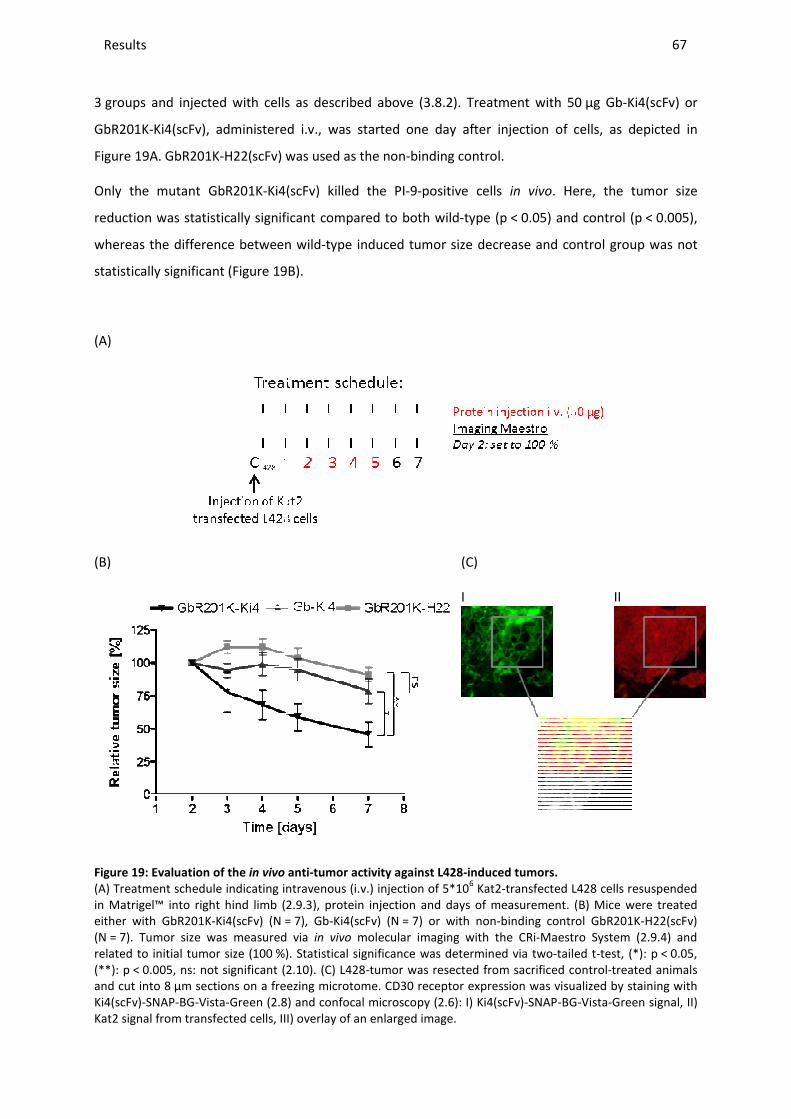
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
3 groups and injected with cells as described above (
GbR201K-Ki4(scFv), administered
Figure 19A. GbR201K-H22(scFv) was used as
Only the mutant GbR201K-Ki4(scFv)
reduction was statistically significant compared to both wild
whereas the difference between wild
statistically significant (Figure 19
(A)
(B)
Figure 19: Evaluation of the in vivo
(A) Treatment schedule indicating intravenous (i.v.) injection of in Matrigel™ into right hind limb (2.9.3either with GbR201K-Ki4(scFv) (N =(N = 7). Tumor size was measured viarelated to initial tumor size (100 %). (**): p < 0.005, ns: not significant (2.10and cut into 8 µm sections on a freezing microtome. Ki4(scFv)-SNAP-BG-Vista-Green (2.8Kat2 signal from transfected cells, III)
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
injected with cells as described above (3.8.2). Treatment with 50
administered i.v., was started one day after injection of cells
H22(scFv) was used as the non-binding control.
Ki4(scFv) killed the PI-9-positive cells in vivo. Here, the tumor size
reduction was statistically significant compared to both wild-type (p < 0.05) and control (p
whereas the difference between wild-type induced tumor size decrease and control group was not
19B).
(C)
I
anti-tumor activity against L428-induced tumors.
(A) Treatment schedule indicating intravenous (i.v.) injection of 5*106 Kat2-transfected L428 cells2.9.3), protein injection and days of measurement. (B) = 7), Gb-Ki4(scFv) (N = 7) or with non-binding control GbR201K
size was measured via in vivo molecular imaging with the CRi-Maestro System (%). Statistical significance was determined via two-tailed t
2.10). (C) L428-tumor was resected from sacrificed controlµm sections on a freezing microtome. CD30 receptor expression was visualized by staining with
2.8) and confocal microscopy (2.6): I) Ki4(scFv)-SNAP-BGKat2 signal from transfected cells, III) overlay of an enlarged image.
_______________________________________________________________________________________________
67
. Treatment with 50 µg Gb-Ki4(scFv) or
jection of cells, as depicted in
Here, the tumor size
0.05) and control (p < 0.005),
type induced tumor size decrease and control group was not
II
L428 cells resuspended , protein injection and days of measurement. (B) Mice were treated
binding control GbR201K-H22(scFv) Maestro System (2.9.4) and
tailed t-test, (*): p < 0.05, tumor was resected from sacrificed control-treated animals
CD30 receptor expression was visualized by staining with BG-Vista-Green signal, II)

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
68
The continuous presence of CD30 receptor after treatment with Ki4(scFv)-specific CFPs in L428-
derived tumors was verified by immunohistochemical analysis of tissue sections (2.8). Successful
receptor staining with Ki4(scFv)-SNAP-BG-Vista-Green as well as the sustained fluorescence of the
cytosolically expressed Kat2 protein is shown in Figure 19C.
During the animal experiments, all mice involved were active and did not show any symptoms of
disease caused by treatment.
These results confirmed the earlier in vitro data. GbR201K-Ki4(scFv) was able to kill PI-9-positive as
well as PI-9-negative cells in contrast to the wild-type which only killed the latter. To further verify
these findings, ex vivo studies were performed.
3.9 EX VIVO STUDIES WITH CMML AND AMML PATIENT-DERIVED MONOCYTES
Effective apoptosis induction of Gb-H22(scFv) in AML patient-derived primary cells which are known
to overexpress CD64 has been shown before [138]. Therefore they are a valuable target for this CFP
(1.6). In this study the rare diseases CMML as well as AMML were put into focus to determine if
these cells are also suitable for an immunotherapeutic granzyme B-based approach. In addition,
during these ex vivo studies, the potential of the newly generated granzyme B variants could be
further evaluated.
Peripheral blood samples of leukemic patients, mostly prior to any clinical treatment, were obtained
from the university hospital Aachen (PD Dr. med. Edgar Jost) after pre-testing for over-expression of
CD64. PBMCs were isolated via Ficoll gradient, cultivated in supplemented RPMI medium (2.1.8) and
used for the following investigations.
3.9.1 PI-9 expression in patient-derived PBMCs
As described for the investigation of cancer cell lines previously (3.1), primary cells were tested for
their ability to express PI-9 via western blot analysis (2.4.7). To evaluate if the cells up-regulate
endogenous PI-9 during cultivation, or as a result of external stimuli, primary cells were harvested
and lysed immediately after isolation from peripheral blood and again after different time points
either in the presence or absence of IFNγ. The addition of IFNγ prior to cytotoxicity assays was shown
to be essential in this study to retain the activation status of the isolated cells and thereby sensitivity
to the used CFPs (1.6).
The results, representatively displayed in Figure 20A for CMML IV, indicated that PI-9 expression was
independent of the addition of IFNγ, but dependent on cultivation time. Except for CMML II, all
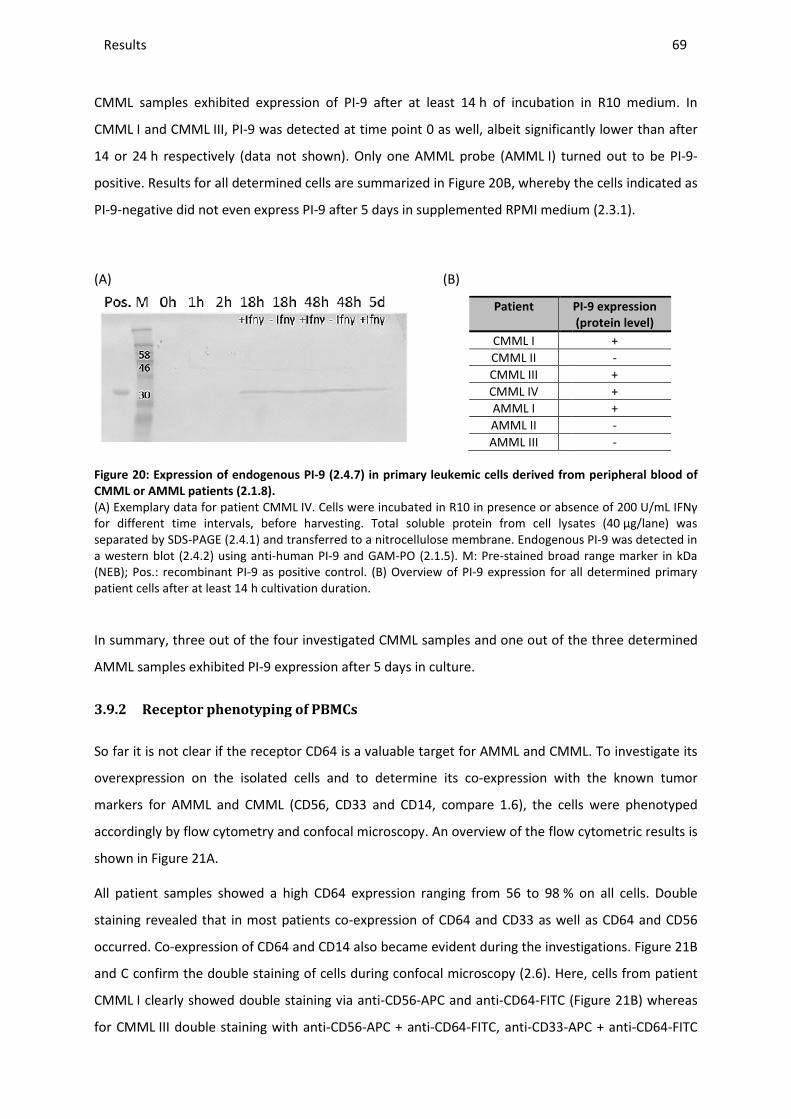
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
CMML samples exhibited expression of PI
CMML I and CMML III, PI-9 was detected at time point 0 as well, albeit significantly lower than after
14 or 24 h respectively (data not show
positive. Results for all determined cells are summarized in
PI-9-negative did not even express PI
(A)
Figure 20: Expression of endogenous PI
CMML or AMML patients (2.1.8).
(A) Exemplary data for patient CMMLfor different time intervals, before harvesting. separated by SDS-PAGE (2.4.1) and transferred to a nitrocellulose membrane. Endogenousa western blot (2.4.2) using anti-human PI(NEB); Pos.: recombinant PI-9 as positive control. patient cells after at least 14 h cultivation duration.
In summary, three out of the four investigated CMML samples and one out of the three determined
AMML samples exhibited PI-9 expression after 5 days in culture.
3.9.2 Receptor phenotyping of PBMCs
So far it is not clear if the receptor CD64 is
overexpression on the isolated cells and to determine its co
markers for AMML and CMML (CD56, CD33 and CD14, compare
accordingly by flow cytometry and confocal microscopy
shown in Figure 21A.
All patient samples showed a high CD64 expression ranging from 56 to 98
staining revealed that in most patients co
occurred. Co-expression of CD64 and CD14 also became evident during the investigations.
and C confirm the double staining of cells during confocal micro
CMML I clearly showed double staining via anti
for CMML III double staining with anti
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
CMML samples exhibited expression of PI-9 after at least 14 h of incubation in R10 medium. In
9 was detected at time point 0 as well, albeit significantly lower than after
h respectively (data not shown). Only one AMML probe (AMML I) turned out to be PI
ll determined cells are summarized in Figure 20B, whereby the cells indicated as
negative did not even express PI-9 after 5 days in supplemented RPMI medium
(B)
Patient
CMML I CMML II CMML III CMML IV AMML I AMML II AMML III
Expression of endogenous PI-9 (2.4.7) in primary leukemic cells derived from peripheral blood of
) Exemplary data for patient CMML IV. Cells were incubated in R10 in presence or absence of 200before harvesting. Total soluble protein from cell lysates
) and transferred to a nitrocellulose membrane. Endogenoushuman PI-9 and GAM-PO (2.1.5). M: Pre-stained broad range marker in kDa
9 as positive control. (B) Overview of PI-9 expression for all determined primary ivation duration.
In summary, three out of the four investigated CMML samples and one out of the three determined
9 expression after 5 days in culture.
henotyping of PBMCs
if the receptor CD64 is a valuable target for AMML and CMML. To investigate its
expression on the isolated cells and to determine its co-expression with the known tumor
markers for AMML and CMML (CD56, CD33 and CD14, compare 1.6), the cells were phenotyped
by flow cytometry and confocal microscopy. An overview of the flow cytometric r
All patient samples showed a high CD64 expression ranging from 56 to 98 % on all cells. Double
staining revealed that in most patients co-expression of CD64 and CD33 as well as CD64 and CD
expression of CD64 and CD14 also became evident during the investigations.
and C confirm the double staining of cells during confocal microscopy (2.6). Here, cells from patient
I clearly showed double staining via anti-CD56-APC and anti-CD64-FITC (
III double staining with anti-CD56-APC + anti-CD64-FITC, anti-CD33-APC + anti
_______________________________________________________________________________________________
69
h of incubation in R10 medium. In
9 was detected at time point 0 as well, albeit significantly lower than after
I) turned out to be PI-9-
whereby the cells indicated as
9 after 5 days in supplemented RPMI medium (2.3.1).
PI-9 expression
(protein level)
+ - + + + - -
leukemic cells derived from peripheral blood of
in presence or absence of 200 U/mL IFNγ Total soluble protein from cell lysates (40 µg/lane) was
) and transferred to a nitrocellulose membrane. Endogenous PI-9 was detected in stained broad range marker in kDa
9 expression for all determined primary
In summary, three out of the four investigated CMML samples and one out of the three determined
d CMML. To investigate its
expression with the known tumor
), the cells were phenotyped
An overview of the flow cytometric results is
% on all cells. Double
expression of CD64 and CD33 as well as CD64 and CD56
expression of CD64 and CD14 also became evident during the investigations. Figure 21B
). Here, cells from patient
FITC (Figure 21B) whereas
APC + anti-CD64-FITC

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
70
and anti-CD14-APC + anti-CD64-FITC showed heterogeneity of the cell population, as indicated by
flow cytometry before (Figure 21C).
(A)
Tumor marker Receptor expression [%]
CMML I CMML II CMML III CMML IV AMML I AMML II AMML III
CD64 79 59 56 98 (7.4) 82 59 (18) 82 (31) CD56 68 3 57 65 n.d. 7 30
CD64+CD56 60 2 36 64 n.d. n.d. 28 CD14 50 39 59 97 n.d. 31 30
CD64+CD14 45 n.d. 44 97 n.d. n.d. 30 CD33 n.d. n.d. 75 98 n.d. 63 31
CD64+CD33 n.d. n.d. 50 97 n.d. 59 30 CD56+CD14 46 3 32 97 n.d. 2 n.d. CD33+CD14 n.d. n.d. 46 51 n.d. 23 20
(B) CD56-APC/
CD64-FITC
(C) CD56-APC/ CD33-APC/ CD14-APC/
CD64-FITC CD64-FITC CD64-FITC
Figure 21: Phenotyping of primary leukemic cells.
PBMCs were isolated from peripheral blood of leukemic patients (CMML or AMML, 2.1.8) and incubated with specific antibodies (2.1.5) according to manufacturer’s instructions (2.5.3). Binding was detected by flow cytometry (2.5.3) or confocal microscopy (2.6) (A) Overview of expression profile of patient-derived primary leukemic cells determined via flow cytometry; %: portion of receptor positive cells within the whole isolated cell population; n.d.: not determined; numbers in brackets: strong CD64 expression detected. (B-C) Confocal microscopy of cells from patient CMML I (B) and CMML III (C) stained with antibodies (2.1.5) as indicated.
As a result of the described observations, targeting AMML and CMML patient-derived cells and
potentially killing them with the CD64-specific H22(scFv)-based CFPs was pursued.
3.9.3 Target cell binding and internalization
After detection of CD64 expression, specific binding of the H22(scFv)-derived fusion proteins was
likewise determined. Exemplary results are shown in Figure 22A for cells from patient CMML IV after
incubation with Gb-H22(scFv), GbR201K-H22(scFv) or H22(scFv)-ETA’, respectively. All constructs
bound specifically to the cells.
10 µm 30 µM 10 µM 30 µM

Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
Specific CD64-based internalization was demonstrate
H22(scFv)-SNAP-BG-647 or H22(scFv)
CMML III and CMML IV in Figure
indicating that no unspecific binding occurred to CD64
(A)
I
(B) I
Figure 22: Binding and internalization a
(A) 50 nM H22(scFv)-fused constructs were incubated with peripheral blood of patient CMML IV for 30(2.1.5) and flow cytometry (2.5.1). incubated with 4*105 CD64-positive primary cells from patient 10 min at 37 °C and internalization was detected with c
Since specific binding and internalization
performed to evaluate the cell death inducing
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
based internalization was demonstrated after incubation of primary cells with
or H22(scFv)-SNAP-BG-Vista-Green as representatively depicted for patient
Figure 22B. Some cells were not stained by the CD64
indicating that no unspecific binding occurred to CD64-negative cells.
II
II
inding and internalization analysis with primary leukemic cells (2.1.8).
fused constructs were incubated with 4*105 CD64-positive primary cells CMML IV for 30 min on ice. Specific binding was detected with anti
). (B) 50 nM H22(scFv)-SNAP-BG-647 or H22(scFv)-SNAPpositive primary cells from patient CMML III (I) or CMML IV (II) respectively
and internalization was detected with confocal microscopy (2.6).
specific binding and internalization to the primary cells was hereby verified
cell death inducing efficacy of the corresponding CFPs.
10 µM
_______________________________________________________________________________________________
71
d after incubation of primary cells with
y depicted for patient
B. Some cells were not stained by the CD64-specific construct,
positive primary cells derived from ected with anti-His Alexa488
SNAP-BG-Vista-Green was (I) or CMML IV (II) respectively for
verified, studies could be
corresponding CFPs.
10 µM
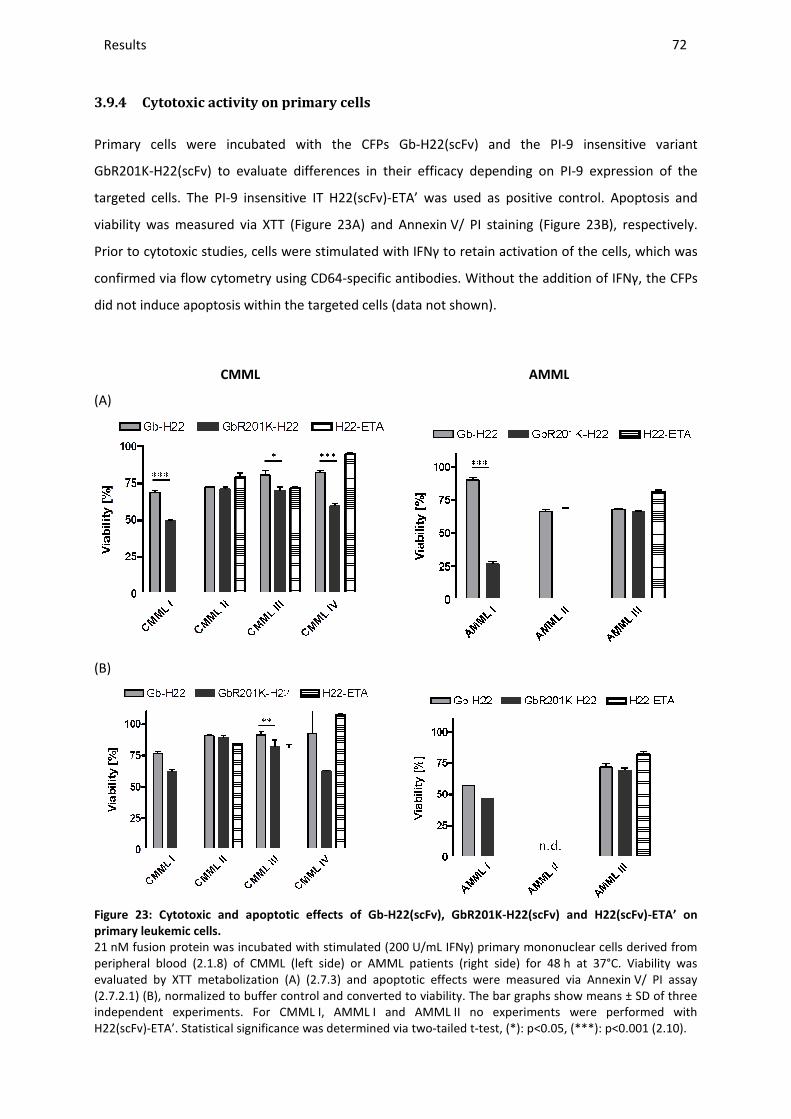
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
3.9.4 Cytotoxic activity on primary cells
Primary cells were incubated with the
GbR201K-H22(scFv) to evaluate differences in their efficacy depending on
targeted cells. The PI-9 insensitive
viability was measured via XTT
Prior to cytotoxic studies, cells were stimulated with IFNγ to retain activation of the cells, which was
confirmed via flow cytometry using CD64
did not induce apoptosis within the targeted cells (data not shown).
CMML
(A)
(B)
Figure 23: Cytotoxic and apoptotic
primary leukemic cells.
21 nM fusion protein was incubated with stimulated (200peripheral blood (2.1.8) of CMML (leftevaluated by XTT metabolization (A) ((2.7.2.1) (B), normalized to buffer control and converted to viabilityindependent experiments. For CMMLH22(scFv)-ETA’. Statistical significance was determined via two
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Cytotoxic activity on primary cells
Primary cells were incubated with the CFPs Gb-H22(scFv) and the PI-9
H22(scFv) to evaluate differences in their efficacy depending on PI-
sensitive IT H22(scFv)-ETA’ was used as positive control
XTT (Figure 23A) and Annexin V/ PI staining (Figure
cells were stimulated with IFNγ to retain activation of the cells, which was
confirmed via flow cytometry using CD64-specific antibodies. Without the addition of IFNγ, the CFPs
did not induce apoptosis within the targeted cells (data not shown).
AMML
and apoptotic effects of Gb-H22(scFv), GbR201K-H22(scFv) and H22(scFv)
nM fusion protein was incubated with stimulated (200 U/mL IFNγ) primary mononuclear cells derived from ) of CMML (left side) or AMML patients (right side) for 48 h at 37°C. Viability was
by XTT metabolization (A) (2.7.3) and apoptotic effects were measured via Annexin, normalized to buffer control and converted to viability. The bar graphs show means ± SD of three
For CMML I, AMML I and AMML II no experiments were performed with Statistical significance was determined via two-tailed t-test, (*): p<0.05, (***)
_______________________________________________________________________________________________
72
9 insensitive variant
-9 expression of the
positive control. Apoptosis and
Figure 23B), respectively.
cells were stimulated with IFNγ to retain activation of the cells, which was
specific antibodies. Without the addition of IFNγ, the CFPs
H22(scFv) and H22(scFv)-ETA’ on
IFNγ) primary mononuclear cells derived from h at 37°C. Viability was
) and apoptotic effects were measured via Annexin V/ PI assay The bar graphs show means ± SD of three
II no experiments were performed with p<0.05, (***): p<0.001 (2.10).

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
73
A statistically significant increased reduction in cell viability was observed after XTT evaluation for
GbR201K-H22(scFv) compared to the wild-type protein for all PI-9-positive cells CMML I, CMML III
and CMML IV as well as AMML I. Hence, more than 50 % of cells died in the presence of
GbR201K-H22(scFv) in sample CMML I whereas only 30 % died after incubation with the wild-type
protein. For CMML II almost 30 % of the cells were eliminated. The results for PI-9-positive CMML III
after incubation with GbR201K-H22(scFv) were in the same range, whereby the effect of the wild-
type fusion was 10 % lower. In CMML IV the difference between the mutant and Gb-H22(scFv) is
even more evident. Here more than 40 % of the monocytes died in the presence of
GbR201K-H22(scFv) but only 18 % were killed by the unmodified version. The largest difference and
thereby the most significant evidence for the superior performance of the mutant compared to the
wild-type was observed in PI-9-positive cells from patient AMML I. Here 75 % of the cells could be
killed by GbR201K-H22(scFv), but only 15 % by Gb-H22(scFv). In contrast the cytotoxic efficacy of
both constructs was the same on PI-9-negative CMML II, AMML II and AMML III cells (Figure 23A).
The cytotoxic efficacy H22(scFv)-ETA’ was as high as for the mutant granzyme B construct in case of
patient CMML III. In contrast, for all other determined cells, the performance of H22(scFv)-ETA’ was
significantly lower than for Gb-H22(scFv) or GbR201K-H22(scFv).
To confirm that cell death occurred through apoptosis, Annexin V/ PI assays were additionally
performed. The viability detected here were in general higher than the ones measured via XTT;
however the same tendencies were seen (Figure 23B).
Incubation of the primary cells with the inactive variant GbS184A-H22(scFv) or the non-binding
GbR201K-Ki4(scFv) did not reduce cell viability (data not shown).
In addition, the concentration-dependent cytotoxic effect of the granzyme B-based constructs was
compared using XTT. As indicated by the data above, the most evident results for the better
performance of the mutant were obtained for AMML I and are shown in Figure 24.
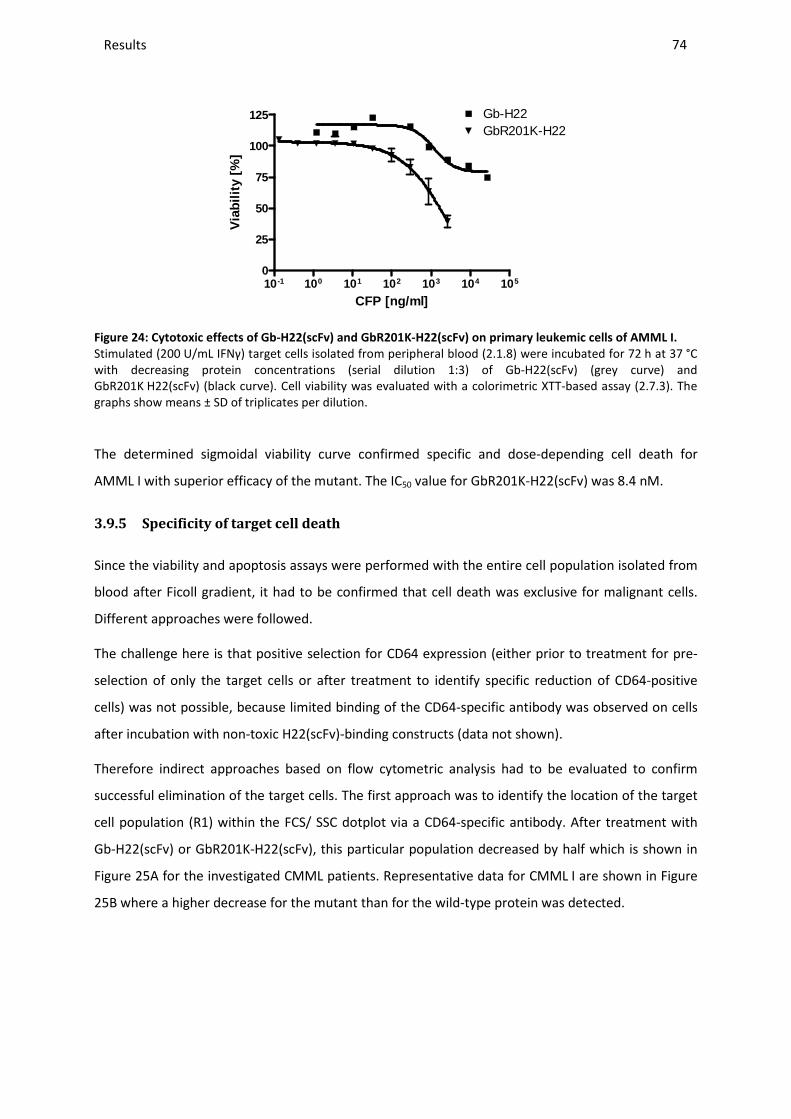
Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
74
10 -1 100 101 102 103 104 1050
25
50
75
100
125 Gb-H22GbR201K-H22
CFP [ng/ml]
Via
bilit
y [%
]
Figure 24: Cytotoxic effects of Gb-H22(scFv) and GbR201K-H22(scFv) on primary leukemic cells of AMML I.
Stimulated (200 U/mL IFNγ) target cells isolated from peripheral blood (2.1.8) were incubated for 72 h at 37 °C with decreasing protein concentrations (serial dilution 1:3) of Gb-H22(scFv) (grey curve) and GbR201K H22(scFv) (black curve). Cell viability was evaluated with a colorimetric XTT-based assay (2.7.3). The graphs show means ± SD of triplicates per dilution.
The determined sigmoidal viability curve confirmed specific and dose-depending cell death for
AMML I with superior efficacy of the mutant. The IC50 value for GbR201K-H22(scFv) was 8.4 nM.
3.9.5 Specificity of target cell death
Since the viability and apoptosis assays were performed with the entire cell population isolated from
blood after Ficoll gradient, it had to be confirmed that cell death was exclusive for malignant cells.
Different approaches were followed.
The challenge here is that positive selection for CD64 expression (either prior to treatment for pre-
selection of only the target cells or after treatment to identify specific reduction of CD64-positive
cells) was not possible, because limited binding of the CD64-specific antibody was observed on cells
after incubation with non-toxic H22(scFv)-binding constructs (data not shown).
Therefore indirect approaches based on flow cytometric analysis had to be evaluated to confirm
successful elimination of the target cells. The first approach was to identify the location of the target
cell population (R1) within the FCS/ SSC dotplot via a CD64-specific antibody. After treatment with
Gb-H22(scFv) or GbR201K-H22(scFv), this particular population decreased by half which is shown in
Figure 25A for the investigated CMML patients. Representative data for CMML I are shown in Figure
25B where a higher decrease for the mutant than for the wild-type protein was detected.
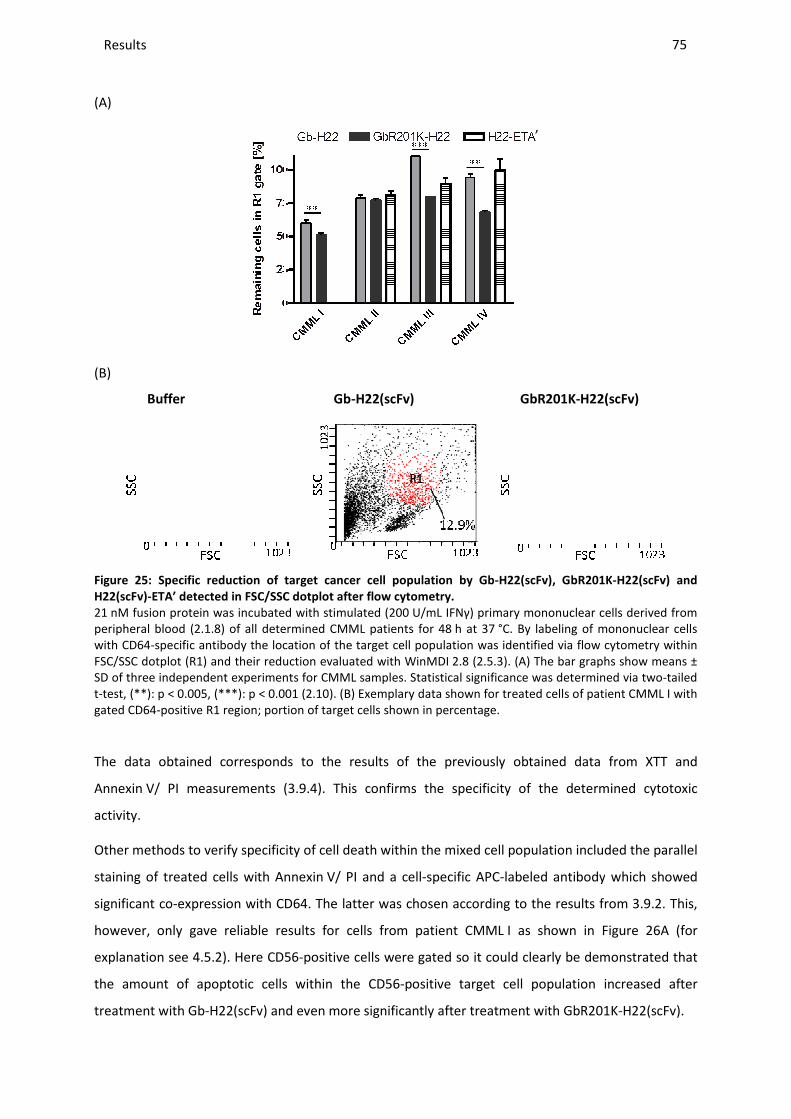
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
(A)
(B)
Buffer Gb
Figure 25: Specific reduction of target
H22(scFv)-ETA’ detected in FSC/SSC dotplot after flow cytometry
21 nM fusion protein was incubated with stimulated (200peripheral blood (2.1.8) of all determined with CD64-specific antibody the location ofFSC/SSC dotplot (R1) and their reduction evaluated with WinMDISD of three independent experiments for CMML samples. t-test, (**): p < 0.005, (***): p < 0.001 (gated CD64-positive R1 region; portion
The data obtained correspond
Annexin V/ PI measurements (
activity.
Other methods to verify specificity of cell death within the mixed cell population
staining of treated cells with Annexin
significant co-expression with CD64. The latter was chosen accord
however, only gave reliable results for cells from patient CMML
explanation see 4.5.2). Here CD56
the amount of apoptotic cells within the CD56
treatment with Gb-H22(scFv) and even
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Buffer Gb-H22(scFv) GbR201K
target cancer cell population by Gb-H22(scFv), GbR201K
ETA’ detected in FSC/SSC dotplot after flow cytometry.
nM fusion protein was incubated with stimulated (200 U/mL IFNγ) primary mononuclear cells derived from all determined CMML patients for 48 h at 37 °C. By labeling of mononuclear cells
ic antibody the location of the target cell population was identified via flow cytometry and their reduction evaluated with WinMDI 2.8 (2.5.3). (A) The bar graphs show means ±
SD of three independent experiments for CMML samples. Statistical significance was determined via two0.001 (2.10). (B) Exemplary data shown for treated cells of patient
portion of target cells shown in percentage.
corresponds to the results of the previously obtained
V/ PI measurements (3.9.4). This confirms the specificity of the determined cytotoxic
to verify specificity of cell death within the mixed cell population
Annexin V/ PI and a cell-specific APC-labeled antibody
expression with CD64. The latter was chosen according to the results from
however, only gave reliable results for cells from patient CMML I as shown in
CD56-positive cells were gated so it could clearly be
the amount of apoptotic cells within the CD56-positive target cell population increased after
H22(scFv) and even more significantly after treatment with GbR201K
‚
_______________________________________________________________________________________________
75
GbR201K-H22(scFv)
GbR201K-H22(scFv) and
primary mononuclear cells derived from By labeling of mononuclear cells
via flow cytometry within (A) The bar graphs show means ±
Statistical significance was determined via two-tailed treated cells of patient CMML I with
obtained data from XTT and
specificity of the determined cytotoxic
to verify specificity of cell death within the mixed cell population included the parallel
labeled antibody which showed
ing to the results from 3.9.2. This,
I as shown in Figure 26A (for
clearly be demonstrated that
positive target cell population increased after
GbR201K-H22(scFv).
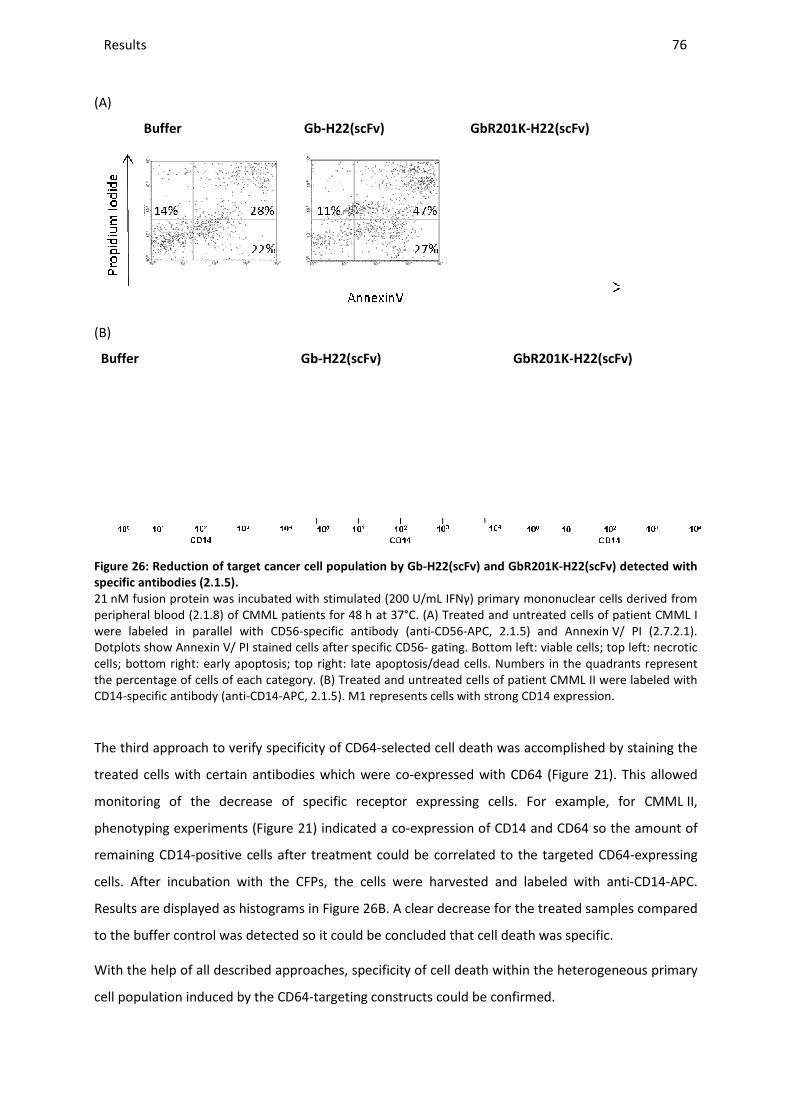
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
(A)
Buffer
(B)
Buffer
Figure 26: Reduction of target cancer cell population
specific antibodies (2.1.5).
21 nM fusion protein was incubated with stimulated (200peripheral blood (2.1.8) of CMML patients for 48were labeled in parallel with CD56Dotplots show Annexin V/ PI stained cells after specific CD56cells; bottom right: early apoptosis; top right: late apoptosis/dead cells. Numbers in the quadrants represent the percentage of cells of each category.CD14-specific antibody (anti-CD14-APC,
The third approach to verify specificity
treated cells with certain antibodies
monitoring of the decrease of
phenotyping experiments (Figure
remaining CD14-positive cells after treatment
cells. After incubation with the
Results are displayed as histograms in
to the buffer control was detected
With the help of all described approaches, specificity of cell death within the heterogeneous primary
cell population induced by the CD64
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Gb-H22(scFv) GbR201K-H22(scFv)
Gb-H22(scFv) GbR201K-
eduction of target cancer cell population by Gb-H22(scFv) and GbR201K-H22(scFv)
nM fusion protein was incubated with stimulated (200 U/mL IFNγ) primary mononuclear cells derived from ) of CMML patients for 48 h at 37°C. (A) Treated and untreated cells
were labeled in parallel with CD56-specific antibody (anti-CD56-APC, 2.1.5) and AnnexinV/ PI stained cells after specific CD56- gating. Bottom left: viable cells; top left: necrotic
cells; bottom right: early apoptosis; top right: late apoptosis/dead cells. Numbers in the quadrants represent the percentage of cells of each category. (B) Treated and untreated cells of patient CMML II were
APC, 2.1.5). M1 represents cells with strong CD14 expression.
to verify specificity of CD64-selected cell death was accomplished by staining the
certain antibodies which were co-expressed with CD64 (Figure
the decrease of specific receptor expressing cells. For example, for
Figure 21) indicated a co-expression of CD14 and CD64 so
after treatment could be correlated to the targeted
fter incubation with the CFPs, the cells were harvested and labeled with anti
Results are displayed as histograms in Figure 26B. A clear decrease for the treated samples compared
to the buffer control was detected so it could be concluded that cell death was specific.
approaches, specificity of cell death within the heterogeneous primary
cell population induced by the CD64-targeting constructs could be confirmed.
_______________________________________________________________________________________________
76
(scFv)
-H22(scFv)
H22(scFv) detected with
IFNγ) primary mononuclear cells derived from Treated and untreated cells of patient CMML I
) and Annexin V/ PI (2.7.2.1). Bottom left: viable cells; top left: necrotic
cells; bottom right: early apoptosis; top right: late apoptosis/dead cells. Numbers in the quadrants represent Treated and untreated cells of patient CMML II were labeled with
M1 represents cells with strong CD14 expression.
was accomplished by staining the
Figure 21). This allowed
example, for CMML II,
CD64 so the amount of
the targeted CD64-expressing
the cells were harvested and labeled with anti-CD14-APC.
B. A clear decrease for the treated samples compared
so it could be concluded that cell death was specific.
approaches, specificity of cell death within the heterogeneous primary

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
77
3.9.6 Serum stability of Gb-H22(scFv) fusion proteins in patient-derived serum
Serum stability is an important pre-requisite for later systemic, intravenous application of the
immunotherapeutic. Hence, Gb-H22(scFv) and GbR201K-H22(scFv) were incubated in either 50 %
human patient serum derived from patient CMML III or PBS at 37 °C for 1, 6 or 24 h (2.4.8). Stability
was determined by measuring residual binding activity via flow cytometry as described for the
Ki4(scFv)-based constructs (3.8.1). Results are summarized in Table 8.
Table 8: Serum stability of Gb-H22(scFv) and GbR201K-H22(scFv) in human patient serum (CMML III) or PBS.
Stability in serum [%] Stability in PBS [%]
Time [h] Gb-H22(scFv) GbR201K-H22(scFv) Gb-H22(scFv) GbR201K-H22(scFv) 0 100 100 100 100 1 98 96 96 96 6 80 74 92 93
24 66 62 92 92
Serum stability was comparable for both variants and could be confirmed for at least 6 h. After 24 h
serum stability was clearly reduced whereas retained binding activity after incubation in PBS was
significantly higher (reduction of 8 %). Hence, the serum- and time-dependent stability for both
constructs was determined.
3.10 EVALUATION OF GRANZYME M AS NOVEL EFFECTOR MOLECULE IN CYTOLYTIC
FUSION PROTEINS
Granzyme M has been described to possess anti-tumor activity (1.3.2), so in this study CFPs
containing this serine protease as an alternative to granzyme B were generated. To confirm the
potential of granzyme M, it was fused to different tumor-specific single chains and functionality
assays were performed. To evaluate the results properly, experiments were prepared in comparison
to the corresponding granzyme B-based CFP.
3.10.1 Generation and preparation of granzyme M fusion proteins
The fusion constructs comprised of granzyme M and the CD64-specific H22(scFv) or CD30-specific
Ki4(scFv) (2.1.5) are shown in Figure 27A. They were generated as described in 2.1.4 by replacing the
granzyme B sequence with that of granzyme M within the corresponding constructs.

Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
(A)
(B)
Figure 27: Overview of granzyme
Gm-H22(scFv) (B).
(A) Schematic structure of the binary eukaryotic expression vector pMS EGm-Ki4(scFv). Ig-κ: signal peptide sequencecleavage site, H: 6x histidine tag, IVS/IRES: synthetic intron and internal ribosome entry site, EGFP: enhanced green fluorescent protein. Enterokinase cleavage site indicated with in HEK293T cells and purified from supernatant via 0.02 U/µg Enterokinase (2.4.4). Protein w(2.4.1) or transferred to a nitrocellulose membrane (2.4.2). Specific protein bands were detected at 61.5kDa (NEB).
As in the case of the granzyme
N-terminus to facilitate expression in eukaryotic cells.
supernatant should allow correct post
glycosylation sites. The protein was purified by affinity chromatography selecting for the His
with a yield of 1 mg/L, as determined by SDS
The identity of the protein was confirmed by western blot analysis using an anti
(2.4.2). Under denaturing conditions, SDS
150 kDa, a band with the expected molecular weight of 61.5
Hence, the intended granzyme
expressed in HEK293T cells.
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
I II
Overview of granzyme M-based constructs (A) and SDS-PAGE and western blot analysis
of the binary eukaryotic expression vector pMS encoding forκ: signal peptide sequence (L) of immunoglobulin kappa light chain, ECS: enterokinase
cleavage site, H: 6x histidine tag, IVS/IRES: synthetic intron and internal ribosome entry site, EGFP: enhanced Enterokinase cleavage site indicated with arrow. (B) Fusion proteins were expressed
in HEK293T cells and purified from supernatant via IMAC (2.4.4). Activation of the protein was done with Protein was separated by SDS-PAGE and either stained with coomassie (I)
) or transferred to a nitrocellulose membrane for western blotting with anti-human Specific protein bands were detected at 61.5 kDa and ~150 kDa. M: Pre-stained broad range marker in
granzyme B-based CFPs, an enterokinase site precedes the granzyme
terminus to facilitate expression in eukaryotic cells. The secretion of the fusion proteins into the
allow correct post-translational modification because granzyme
ation sites. The protein was purified by affinity chromatography selecting for the His
mg/L, as determined by SDS-PAGE (2.4.5) and activated with enterokinase (
The identity of the protein was confirmed by western blot analysis using an anti-
). Under denaturing conditions, SDS-PAGE revealed, apart from a band at approximately
a band with the expected molecular weight of 61.5 kDa (Figure 27B).
Hence, the intended granzyme M constructs could successfully be generated and
_______________________________________________________________________________________________
78
and western blot analysis of
encoding for EGm-H22(scFv) and of immunoglobulin kappa light chain, ECS: enterokinase
cleavage site, H: 6x histidine tag, IVS/IRES: synthetic intron and internal ribosome entry site, EGFP: enhanced Fusion proteins were expressed
). Activation of the protein was done with stained with coomassie (I)
human Gm and GAM-AP stained broad range marker in
an enterokinase site precedes the granzyme M
secretion of the fusion proteins into the
translational modification because granzyme M has three
ation sites. The protein was purified by affinity chromatography selecting for the His6-tag,
d with enterokinase (2.4.4.2).
-human Gm antibody
a band at approximately
generated and secretory

Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
3.10.2 Functional characterization of granzyme M
Successful activation after enterokinase cleavage
ability of granzyme M to cleave PI
synthetic granzyme M substrate, Suc
Western blot analysis with anti-human PI
Gm-H22(scFv) in a 3:1 molar ratio. Recombinant PI
unknown reasons (3.4) but cleavage by granzyme
lower band (Figure 28A). The same results were obtained for Gm
The enzymatic functionality of the fusion protein was also verified by the applied colorimetric assay.
No activity was detected for the uncleaved variant EGm
dependent increase in extinction was observed for
(A)
Figure 28: Enzymatic activity of Gm
(A) Recombinant PI-9 (3.4) and Gm-was separated by SDS-PAGE and transferred to a nitrocellulose membrane (was detected in a western blot using antiin kDa (NEB). (B) The proteolytic activity of activated protein was measured via a colorimetric assay based on the synthetic granzyme M substrate Suc405 nm for different concentrations as indicated and compared to uncleaved variant EGmgraphs show means ± SD of three independent experiments.
These results confirm that enzymatically
expression in HEK293T cells and subsequent enterokinase digestion
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Functional characterization of granzyme M fusion proteins
after enterokinase cleavage was tested by two methods, one based on the
to cleave PI-9 (2.7.1.3), and the other one using a colorimetric assay with a
, Suc-Ala-Ala-Pro-Leu-pNA (2.7.1.1).
human PI-9 confirmed the cleavage of PI-9 after 24
H22(scFv) in a 3:1 molar ratio. Recombinant PI-9 was partially degraded during purification for
eavage by granzyme M was nevertheless clearly visible, intensifying the
ame results were obtained for Gm-Ki4(scFv) (data not shown).
The enzymatic functionality of the fusion protein was also verified by the applied colorimetric assay.
tected for the uncleaved variant EGm-H22(scFv), whereas a concentration
dependent increase in extinction was observed for active Gm-H22(scFv) (Figure 28
(B)
Buffer
EGm-H
22 1µ
M
Gm-H22
200
nM
Gm-H22
1 µM
Gm-H
22 2
µM0.00
0.25
0.50
0.75
1.00
Abs
orba
nce
405n
m
Gm-H22(scFv) tested by cleavage of PI-9 or a colorimetric substrate
-H22(scFv) were incubated for 24 h at 37 °C (2.7.1.3) before protein mixture PAGE and transferred to a nitrocellulose membrane (2.4.2). Active and inactivated PI
was detected in a western blot using anti-human PI-9 and GAM-PO (2.1.5) M: Pre-stained broad range marker oteolytic activity of activated protein was measured via a colorimetric assay based on
substrate Suc-Ala-Ala-Pro-Leu-pNA (2.7.1.1). Reaction was documentednm for different concentrations as indicated and compared to uncleaved variant EGm
graphs show means ± SD of three independent experiments.
enzymatically active granzyme M fusion proteins could be produced via
K293T cells and subsequent enterokinase digestion.
_______________________________________________________________________________________________
79
ed by two methods, one based on the
ng a colorimetric assay with a
9 after 24 h incubation with
9 was partially degraded during purification for
M was nevertheless clearly visible, intensifying the
Ki4(scFv) (data not shown).
The enzymatic functionality of the fusion protein was also verified by the applied colorimetric assay.
H22(scFv), whereas a concentration-
28B).
Gm-H22
1 µM
Gm-H
22 2
µM
9 or a colorimetric substrate.
) before protein mixture ). Active and inactivated PI-9 stained broad range marker
oteolytic activity of activated protein was measured via a colorimetric assay based on ). Reaction was documented at 37 °C and
nm for different concentrations as indicated and compared to uncleaved variant EGm-H22(scFv). The bar
could be produced via

Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
3.10.3 Target cell binding
The antigen-specific binding of all
corresponding target cell lines.
CD64-positive AML cell line HL60 (
(Figure 29B). CD64-negative L428
(A) (B
(C)
Figure 29: Binding analysis of Gm-H22(scFv
50 nM Gm-H22(scFv) was incubated with 4*10blood of patient CMML IV (B) for 30 min on icecytometry (2.5.1). CD64- L428 cell line was
An obvious fluorescence shift was detected on CD64
attested for the granzyme M-based CFP. No binding was observed on the control cell line L428
confirming that the binding activity was s
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
specific binding of all granzyme M-based CFPs was tested by flow cytometry using the
target cell lines. Exemplary results are shown for Gm-H22(scFv) targeting human
positive AML cell line HL60 (Figure 29A) and CD64-positive primary cells from a CMML patient
negative L428 cells were used as the control cell line (Figure 29
(A) (B)
H22(scFv).
H22(scFv) was incubated with 4*105 HL60 cells (A) or primary leukemic cells derived from peripheral for 30 min on ice. Binding was detected with anti-His Alexa488 (
L428 cell line was used as negative control.
fluorescence shift was detected on CD64-positive cells, so specific binding could be
based CFP. No binding was observed on the control cell line L428
confirming that the binding activity was selective for the targeted receptor.
_______________________________________________________________________________________________
80
ed by flow cytometry using the
H22(scFv) targeting human
positive primary cells from a CMML patient
29C).
(A) or primary leukemic cells derived from peripheral His Alexa488 (2.1.5) and flow
so specific binding could be
based CFP. No binding was observed on the control cell line L428
The same exclusive

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
81
target-specific binding was confirmed for Gm-Ki4(scFv) using CD30-positive cell lines (data not
shown).
3.10.4 Cytotoxic activity of granzyme M fusion proteins
In accordance with the viability and apoptosis assays performed and described for Gb-H22(scFv)
(3.6.4 and 3.6.5), the CD64-positive AML cell line HL60 was used for the evaluation of Gm-H22(scFv).
The concentration-dependent cytotoxicity of Gm-H22(scFv) was evaluated using an XTT-based
colorimetric cell proliferation assay (2.7.3) with IFNγ stimulated HL60 cells. Unstimulated HL60 cells
and CD64-negative L428 cells were used as negative control.
As shown in Figure 30A, the viability of the stimulated HL60 cells declined significantly, whereas CD64
receptor negative L428 cells and unstimulated HL60 cells remained unaffected. The IC50 values were
between 1.2 and 6.4 nM which is in the same range as shown before in Figure 12B for Gb-H22(scFv).
A non-specific granzyme M fusion protein showed no evidence of cytotoxicity (data not shown).
The antigen-dependency of Gm-H22(scFv)-induced cytotoxicity was confirmed by carrying out a
competitive assay which showed a statistically significant dose-dependent reduction in cytotoxicity in
the presence of free H22(scFv) (Figure 30B). There was no significant difference between the viability
of cells incubated with free H22(scFv) alone or with the buffer alone.
The mechanism of Gm-H22(scFv) toxicity in HL60 cells was further characterized by carrying out an
Annexin V/ PI apoptosis assay (Figure 30C). When stimulated HL60 cells were incubated with 66 nM
Gm-H22(scFv) for 48 h, the number of apoptotic cells stained with Annexin V/ PI was 65 % greater
than in control experiments lacking the protein. This was in accordance with the results obtained
after treatment with Gb-H22(scFv) and demonstrated efficient programmed cell death. No difference
between experimental cultures and controls was detected for the CD64-negative CML cell line K562.
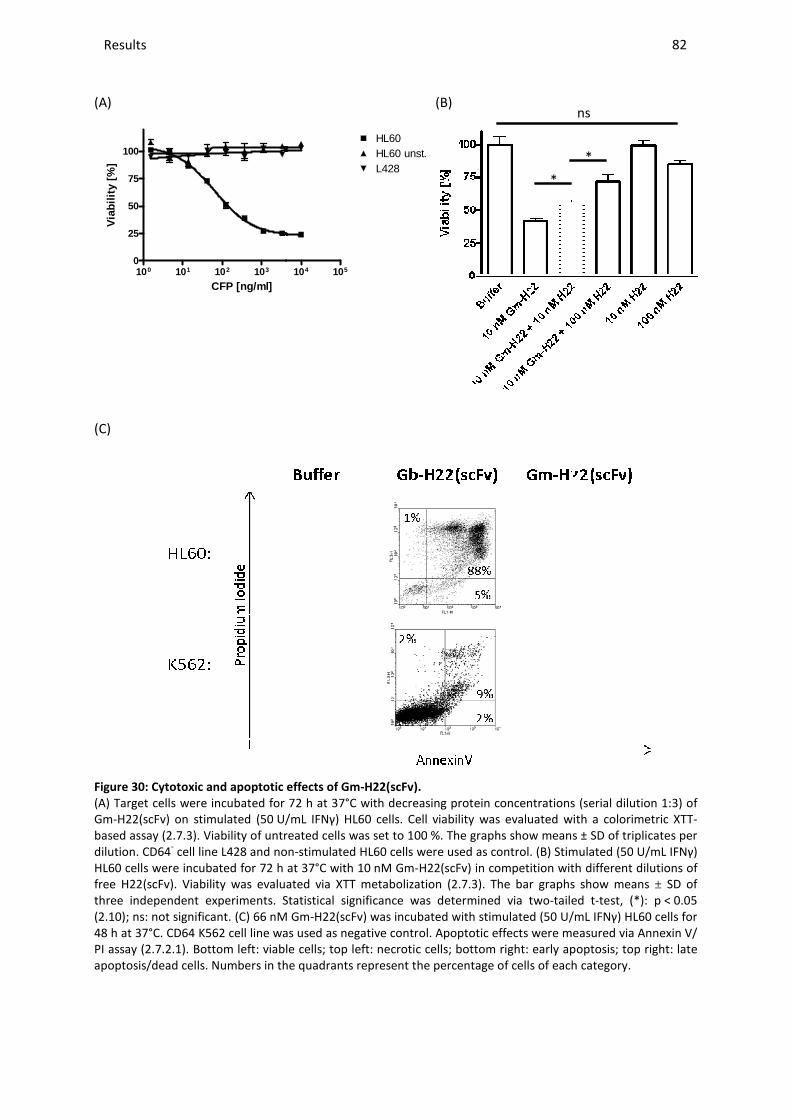
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
(A)
100 101 102 1030
25
50
75
100
CFP [ng/ml]
Via
bilit
y [%
]
(C)
Figure 30: Cytotoxic and apoptotic effects of Gm
(A) Target cells were incubated for 72 Gm-H22(scFv) on stimulated (50 U/mbased assay (2.7.3). Viability of untreated cells was set to 100dilution. CD64- cell line L428 and nonHL60 cells were incubated for 72 h free H22(scFv). Viability was evaluated viathree independent experiments. Statistical significance was determined via two(2.10); ns: not significant. (C) 66 nM G48 h at 37°C. CD64 K562 cell line was used as PI assay (2.7.2.1). Bottom left: viable cells; top left: necrotic cells; bottom right: early apoptosis; top right: late apoptosis/dead cells. Numbers in the quadrants represent the percentage of cells of each category.
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
104 105
L428
HL60HL60 unst.
(B)
totic effects of Gm-H22(scFv).
Target cells were incubated for 72 h at 37°C with decreasing protein concentrations (U/mL IFNγ) HL60 cells. Cell viability was evaluated with a colorimetric XTT
Viability of untreated cells was set to 100 %. The graphs show means ± SD of triplicates per non-stimulated HL60 cells were used as control. (B) Stimulated (50
at 37°C with 10 nM Gm-H22(scFv) in competition with different dilutions of Fv). Viability was evaluated via XTT metabolization (2.7.3). The bar graphs show means
Statistical significance was determined via two-tailed tnM Gm-H22(scFv) was incubated with stimulated (50 U/m
K562 cell line was used as negative control. Apoptotic effects were measured via AnnexinBottom left: viable cells; top left: necrotic cells; bottom right: early apoptosis; top right: late
cells. Numbers in the quadrants represent the percentage of cells of each category.
*
_______________________________________________________________________________________________
82
protein concentrations (serial dilution 1:3) of iability was evaluated with a colorimetric XTT-
%. The graphs show means ± SD of triplicates per Stimulated (50 U/mL IFNγ)
H22(scFv) in competition with different dilutions of The bar graphs show means ± SD of
tailed t-test, (*): p < 0.05 U/mL IFNγ) HL60 cells for
were measured via Annexin V/ Bottom left: viable cells; top left: necrotic cells; bottom right: early apoptosis; top right: late
cells. Numbers in the quadrants represent the percentage of cells of each category.
*
ns
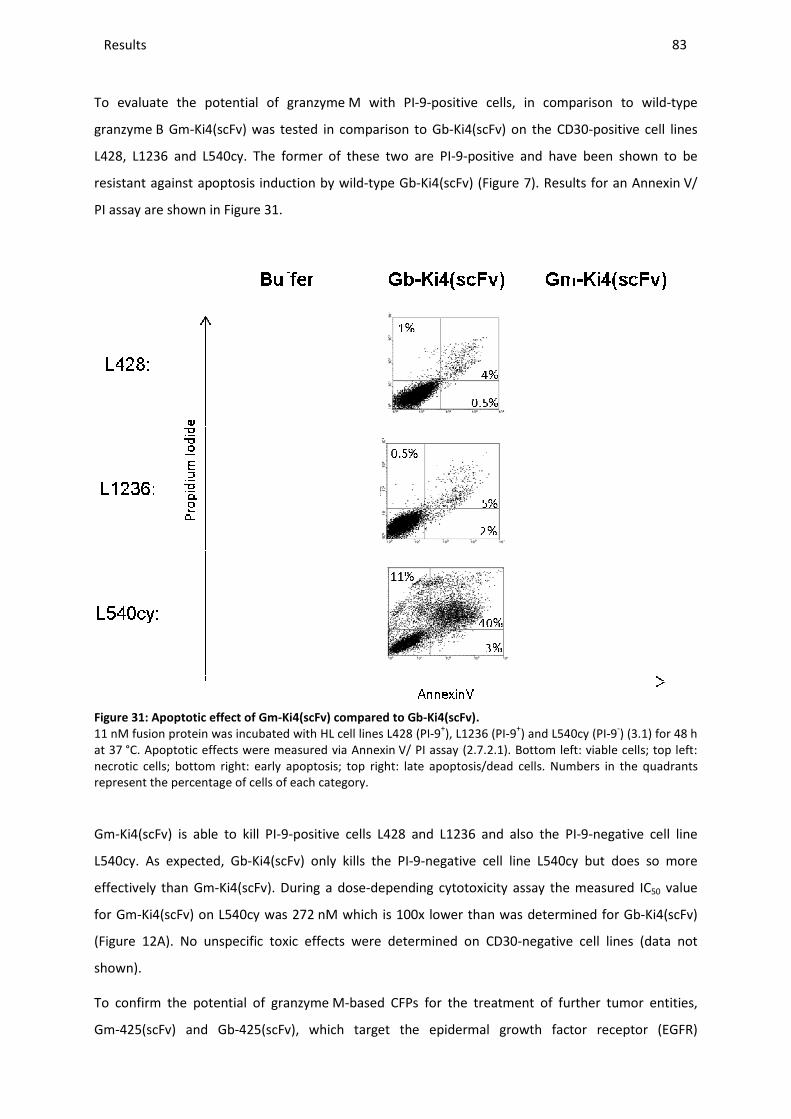
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
To evaluate the potential of granzyme
granzyme B Gm-Ki4(scFv) was tested in comparison to Gb
L428, L1236 and L540cy. The former
resistant against apoptosis induction by wild
PI assay are shown in Figure 31.
Figure 31: Apoptotic effect of Gm-Ki4(scFv) compared to
11 nM fusion protein was incubated with HL cell lines L428 (PIat 37 °C. Apoptotic effects were measured via Annexinnecrotic cells; bottom right: early apoptosis; top right: late apoptosis/dead cells. Numbers in the quadrants represent the percentage of cells of each category
Gm-Ki4(scFv) is able to kill PI-9
L540cy. As expected, Gb-Ki4(scFv) only kills
effectively than Gm-Ki4(scFv). During a dose
for Gm-Ki4(scFv) on L540cy was 272
(Figure 12A). No unspecific toxic effects were determined on CD3
shown).
To confirm the potential of granzyme
Gm-425(scFv) and Gb-425(scFv)
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
To evaluate the potential of granzyme M with PI-9-positive cells, in comparison to wild
Ki4(scFv) was tested in comparison to Gb-Ki4(scFv) on the CD30
he former of these two are PI-9-positive and have been shown to be
apoptosis induction by wild-type Gb-Ki4(scFv) (Figure 7). Results for an Annexin
Ki4(scFv) compared to Gb-Ki4(scFv).
nM fusion protein was incubated with HL cell lines L428 (PI-9+), L1236 (PI-9+) and L540cy (PI°C. Apoptotic effects were measured via Annexin V/ PI assay (2.7.2.1). Bottom left
necrotic cells; bottom right: early apoptosis; top right: late apoptosis/dead cells. Numbers in the quadrants represent the percentage of cells of each category.
9-positive cells L428 and L1236 and also the PI
Ki4(scFv) only kills the PI-9-negative cell line L540cy but
Ki4(scFv). During a dose-depending cytotoxicity assay the measured IC
Ki4(scFv) on L540cy was 272 nM which is 100x lower than was determined for Gb
A). No unspecific toxic effects were determined on CD30-negative cell lines (data not
To confirm the potential of granzyme M-based CFPs for the treatment of further tumor entities
425(scFv), which target the epidermal growth factor receptor (EGFR)
_______________________________________________________________________________________________
83
in comparison to wild-type
CD30-positive cell lines
and have been shown to be
Results for an Annexin V/
) and L540cy (PI-9-) (3.1) for 48 h Bottom left: viable cells; top left:
necrotic cells; bottom right: early apoptosis; top right: late apoptosis/dead cells. Numbers in the quadrants
PI-9-negative cell line
0cy but does so more
depending cytotoxicity assay the measured IC50 value
determined for Gb-Ki4(scFv)
negative cell lines (data not
based CFPs for the treatment of further tumor entities,
epidermal growth factor receptor (EGFR)

Results ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
84
overexpressed in a variety of tumor cells [151], were analyzed in comparative assays. Both granzyme
constructs showed a similar specific cytotoxic as well as apoptotic effects against the EGFR-positive
epidermoid cancer cell line A431, but did not affect the EGFR-negative cell line L540cy (data not
shown).
Concluding, granzyme M was fused to three different cell-binding moieties and showed specific anti-
tumor activity towards the corresponding target cells while leaving receptor negative cells
unaffected. It is also able to kill cells in presence of the granzyme B inhibitor PI-9 in contrast to the
wild-type granzyme B construct.
3.10.5 Ex vivo studies with Gm-H22(scFv)
Apart from in vivo studies, ex vivo experiments with primary cells are important assays to determine
the efficacy of novel immunotherapeutics. The characteristics of these patient-derived cells predict
the later effects in humans much better than artificial cell lines (see also 4.5). Hence, the potential of
the novel human effector molecule granzyme M fused to H22(scFv) was tested comparative to
Gb-H22(scFv) using the same primary cells as used in the experiments described in 3.9. In addition,
by parallel experiments with GbR201K-H22(scFv) the prospective of granzyme M on PI-9-expressing
cells in comparison to this newly established PI-9 insensitive variant could also be assessed.
Cytotoxic effects were monitored using the XTT assay (Figure 32A) or by Annexin V/ PI staining
(Figure 32B) as described in 3.9.4. For Gm-H22(scFv) both assays indicated that cell viability was
reduced by about 40 % in all samples. The same was observed for GbR201K-H22(scFv). Significant
differences of these both constructs compared to the wild-type Gb-H22(scFv) were evident for cells
from patient CMML IV, tested PI-9-positive before (Figure 20A). Here only 15 % of the cells could be
killed by the wild-type granzyme B-based construct.
The specificity of the cytotoxic effect and thereby the selective reduction of the CD64-positive target
cell population was demonstrated here in the same manner as described earlier in 3.9.5 (data not
shown).
To further evaluate the cytotoxic effect of Gm-H22(scFv) on primary cells, dose-dependent cell
viability assays were prepared for cells from patient AMML II (Figure 32C) and AMML III (Figure 32D).
The sigmoidal curves evidently demonstrated the dose-dependency of cell death triggered by
Gm-H22(scFv) ex vivo, even though general cell viability remained rather high.
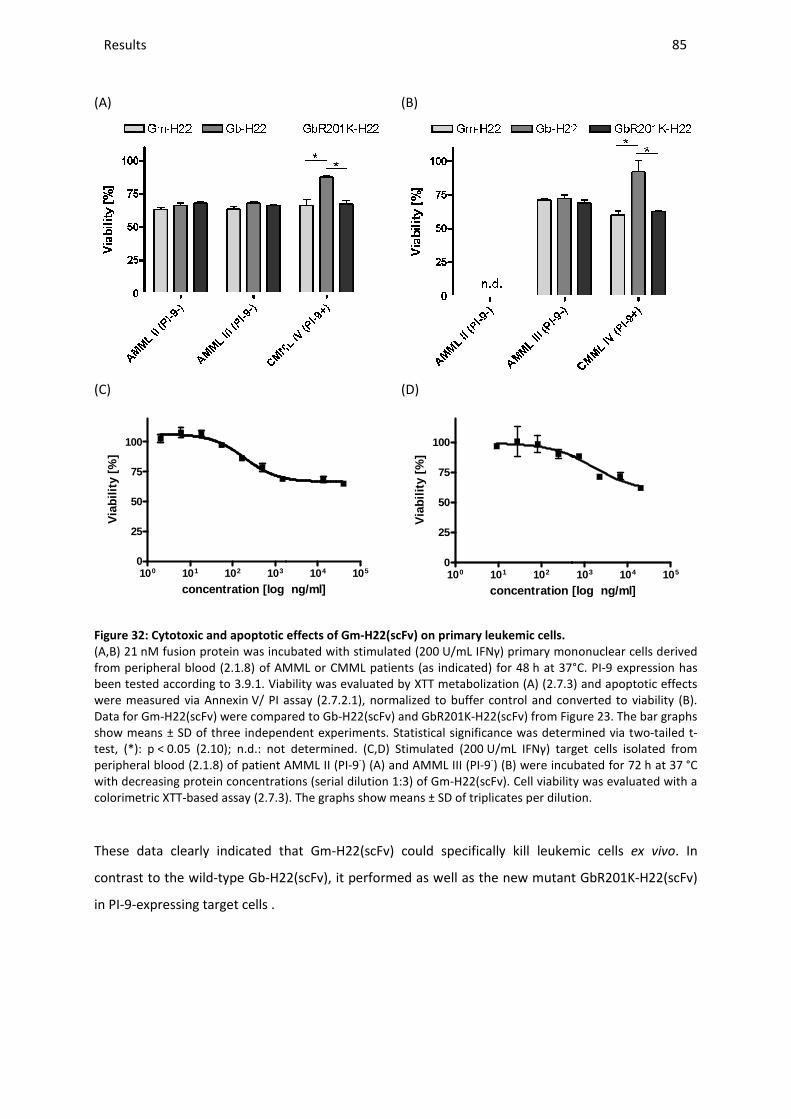
Results _____________________________________________________________________________________________________________________________________________________________________________________________________________
(A)
(C)
100 101 102 1030
25
50
75
100
concentration [log ng/ml]
Via
bilit
y [%
]
Figure 32: Cytotoxic and apoptotic effects of Gm
(A,B) 21 nM fusion protein was incubated with stimulated (200from peripheral blood (2.1.8) of AMML or CMML patients (as indicated) for 48been tested according to 3.9.1. Viability was evaluated by XTT metabolization (A) (were measured via Annexin V/ PI assay (Data for Gm-H22(scFv) were compared to Gbshow means ± SD of three independent experiments.test, (*): p < 0.05 (2.10); n.d.: not determined.peripheral blood (2.1.8) of patient AMML II (PIwith decreasing protein concentrations (serial colorimetric XTT-based assay (2.7.3).
These data clearly indicated that Gm
contrast to the wild-type Gb-H22(scFv),
in PI-9-expressing target cells .
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
(B)
104 105
concentration [log ng/ml]
(D)
100 101 1020
25
50
75
100
concentration [log ng/ml]
Via
bilit
y [%
]
Cytotoxic and apoptotic effects of Gm-H22(scFv) on primary leukemic cells.
nM fusion protein was incubated with stimulated (200 U/mL IFNγ) primary mononuclear cells derived ) of AMML or CMML patients (as indicated) for 48 h at 37°C. PI
. Viability was evaluated by XTT metabolization (A) (2.7.3PI assay (2.7.2.1), normalized to buffer control and converted to viability (B).
H22(scFv) were compared to Gb-H22(scFv) and GbR201K-H22(scFv) from Figure show means ± SD of three independent experiments. Statistical significance was determined via two
); n.d.: not determined. (C,D) Stimulated (200 U/mL IFNγ) target cells ) of patient AMML II (PI-9-) (A) and AMML III (PI-9-) (B) were incubated for 72
with decreasing protein concentrations (serial dilution 1:3) of Gm-H22(scFv). Cell viability was evaluated with a ). The graphs show means ± SD of triplicates per dilution.
These data clearly indicated that Gm-H22(scFv) could specifically kill leukemic cells
H22(scFv), it performed as well as the new mutant GbR201K
_______________________________________________________________________________________________
85
103 104 105
concentration [log ng/ml]
IFNγ) primary mononuclear cells derived at 37°C. PI-9 expression has 2.7.3) and apoptotic effects
), normalized to buffer control and converted to viability (B). Figure 23. The bar graphs
Statistical significance was determined via two-tailed t-U/mL IFNγ) target cells isolated from
were incubated for 72 h at 37 °C iability was evaluated with a
The graphs show means ± SD of triplicates per dilution.
leukemic cells ex vivo. In
as well as the new mutant GbR201K-H22(scFv)

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
86
4 Discussion
The use of immunotoxins (ITs) highly increases the efficacy of cancer therapy leaving healthy cells
mostly unaffected. However, the clinical deployment of early generation ITs was limited due to their
immunogenicity, resulting in the generation of neutralizing antibodies that exclude repeated
applications. Therefore complete humanization of ITs is an important prerequisite for the successful
development of more effective ITs [24,31], called cytolytic fusion proteins (CFPs). This concept is based
on the use of endogenous human enzymes that induce cell death, fused to humanized or even fully
human antibody fragments. Candidate human enzymes include proteases, RNases and kinases
[4,31,109,152,153]. In this thesis, the focus was granzymes, which are human immunoproteases
involved in the granule-mediated apoptosis of virus-infected or transformed tumor cells. The
efficiency of granzyme B was elucidated in relation to the expression of its specific inhibitor PI-9.
Since limited effects were observed for the wild-type protein on PI-9-positive cells, the intention was
to generate novel variants via molecular modeling and site-directed mutagenesis, which are
enzymatically active even in presence of PI-9. When fused to the CD30-targeting Ki4(scFv) or CD64-
targeting H22(scFv), the most promising variant was eventually selected and its efficacy was proven
in vitro and in vivo on CD30-positive Hodgkin lymphoma and CML cell lines, as well as ex vivo on
CD64-positive primary leukemic cells. In addition, another member of the granzyme family,
granzyme M, was introduced in this study bearing the potential to become a novel effector molecule
in context of CFPs. Combined with different cancer cell-specific ligands, its cytotoxic efficacy could
successfully be shown in vitro as well as ex vivo, with the advantage to be active despite the presence
of the inhibitor PI-9. In the following, the hallmarks of granzyme B and its PI-9 insensitive mutants as
well as of granzyme M will be discussed in detail focusing on their therapeutic potential and
applicability for later clinical use as immunotherapeutics.
4.1 THE THERAPEUTIC POTENTIAL OF GRANZYME B-BASED CYTOLYTIC FUSION
PROTEINS
Granzyme B has been evaluated as a CFP targeting CD64-positive cells during a previous PhD thesis in
this work-group and was published before by Stahnke et al. [138]. In this study Gb-H22(scFv) was
successfully used to selectively kill AML-derived HL60 cells and primary leukemic cells (Figure 12 and
Figure 23). In addition the in vitro protocols for another granzyme B-based CFP, Gb-Ki4(scFv), have
been optimized to allow IC50 value determination. The IC50 values were comparable with the ones
determined for Gb-H22(scFv).

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
87
Other groups have also shown the great anti-tumor potential of granzyme B-based CFPs as
summarized in Table 9. Apart from its low immunogenicity, granzyme B has several inherent
advantages as an immunotherapeutic component like high cytotoxic efficacy reflecting its status as
an effector molecule of the cellular immune system, broad range of apoptosis inducing mechanisms
with the potential to circumvent anti-apoptotic proteins (1.3.1). Furthermore, compared to other cell
death inducing proteins like microtubule-associated proteins which exclusively kill proliferating cells
[153], it is independent of cell proliferation, which is advantageous for non- or low-proliferating
malignant diseases like CMML. Also, the production of granzyme B (and CFPs derived therefrom) in
various heterologous expression systems (E. coli, Pichia pastoris, insect cells and mammalian cells
such as HEK293T, HeLa, Jurkat and COS [4]) is relatively straightforward, which reflects a significant
prerequisite before biopharmaceuticals can enter clinical studies.
The involvement of granzyme B in natural cellular defense, however, is a mixed blessing in targeted
cancer therapy since the efficacy of ITs/ CFPs depends on the sensitivity of tumor cells to pro-
apoptotic signals. One of the major challenges in both targeted cancer therapy and standard
chemotherapy approaches, that induce programmed cell death, is the tendency for tumor cells to
develop either escape mechanisms or resistance to pro-apoptotic signals allowing host
immunosurveillance. This limits the clinical application of granzyme B insofar as that it is specifically
inhibited by PI-9, which usually protects cells from misdirected granzyme B (1.4.1), but is also
upregulated in certain tumors (4.2). Other limiting factors are, that granzyme B has a high isoelectric
point (pI ~ 10), resulting in a positive surface charge contributing to non-specific binding to negatively
charged cell surface proteoglycans via electrostatic interactions. In addition, it is, as all CFPs in
general, released slowly after uptake by the receptor-mediated endocytosis which reduces its
cytotoxic impact [4]. In this study, the PI-9-related challenge is focused and in detail discussed in the
following sections, whereas solutions to the latter factors will be introduced in chapter 5.
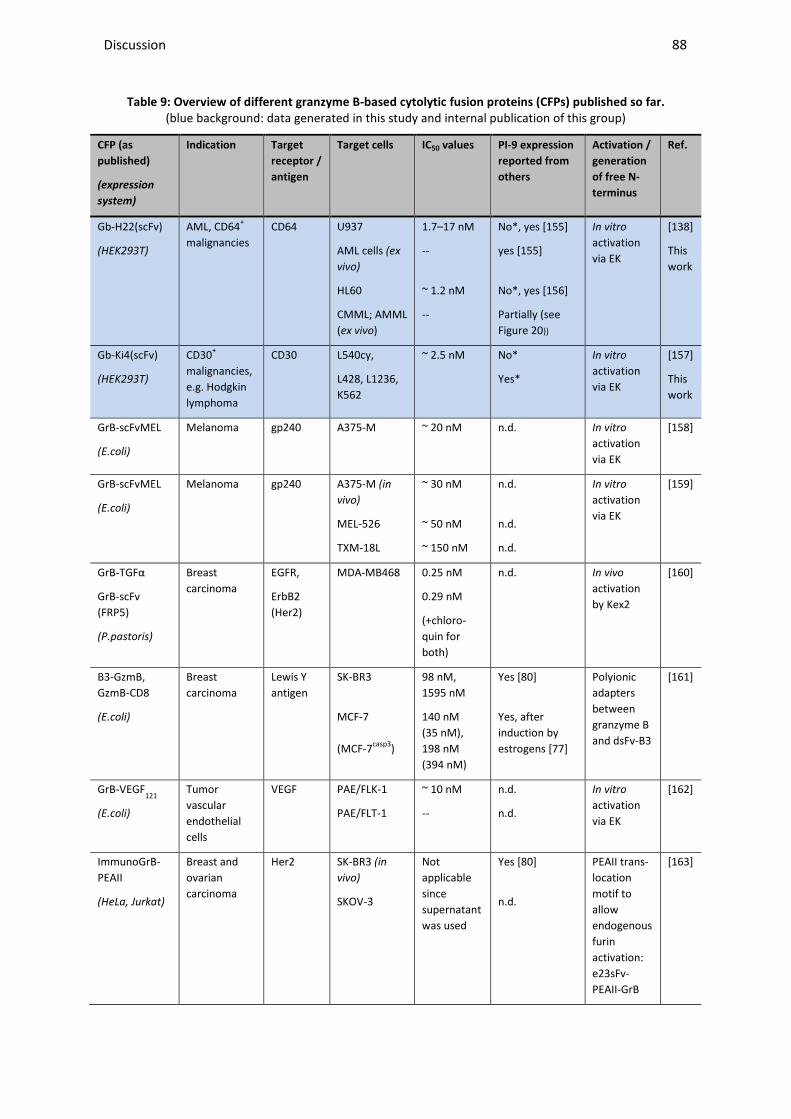
Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
88
Table 9: Overview of different granzyme B-based cytolytic fusion proteins (CFPs) published so far.
(blue background: data generated in this study and internal publication of this group)
CFP (as
published)
(expression
system)
Indication Target
receptor /
antigen
Target cells IC50 values PI-9 expression
reported from
others
Activation /
generation
of free N-
terminus
Ref.
Gb-H22(scFv)
(HEK293T)
AML, CD64+
malignancies
CD64 U937
AML cells (ex
vivo)
HL60
CMML; AMML
(ex vivo)
1.7–17 nM
--
~ 1.2 nM
--
No*, yes [155]
yes [155]
No*, yes [156]
Partially (see
Figure 20))
In vitro
activation
via EK
[138]
This
work
Gb-Ki4(scFv)
(HEK293T)
CD30+
malignancies,
e.g. Hodgkin
lymphoma
CD30 L540cy,
L428, L1236,
K562
~ 2.5 nM No*
Yes*
In vitro
activation
via EK
[157]
This
work
GrB-scFvMEL
(E.coli)
Melanoma gp240 A375-M
~ 20 nM
n.d. In vitro
activation
via EK
[158]
GrB-scFvMEL
(E.coli)
Melanoma gp240 A375-M (in
vivo)
MEL-526
TXM-18L
~ 30 nM
~ 50 nM
~ 150 nM
n.d.
n.d.
n.d.
In vitro
activation
via EK
[159]
GrB-TGFα
GrB-scFv
(FRP5)
(P.pastoris)
Breast
carcinoma
EGFR,
ErbB2
(Her2)
MDA-MB468 0.25 nM
0.29 nM
(+chloro-
quin for
both)
n.d. In vivo
activation
by Kex2
[160]
B3-GzmB,
GzmB-CD8
(E.coli)
Breast
carcinoma
Lewis Y
antigen
SK-BR3
MCF-7
(MCF-7casp3)
98 nM,
1595 nM
140 nM
(35 nM),
198 nM
(394 nM)
Yes [80]
Yes, after
induction by
estrogens [77]
Polyionic
adapters
between
granzyme B
and dsFv-B3
[161]
GrB-VEGF121
(E.coli)
Tumor
vascular
endothelial
cells
VEGF PAE/FLK-1
PAE/FLT-1
~ 10 nM
--
n.d.
n.d.
In vitro
activation
via EK
[162]
ImmunoGrB-
PEAII
(HeLa, Jurkat)
Breast and
ovarian
carcinoma
Her2 SK-BR3 (in
vivo)
SKOV-3
Not
applicable
since
supernatant
was used
Yes [80]
n.d.
PEAII trans-
location
motif to
allow
endogenous
furin
activation:
e23sFv-
PEAII-GrB
[163]
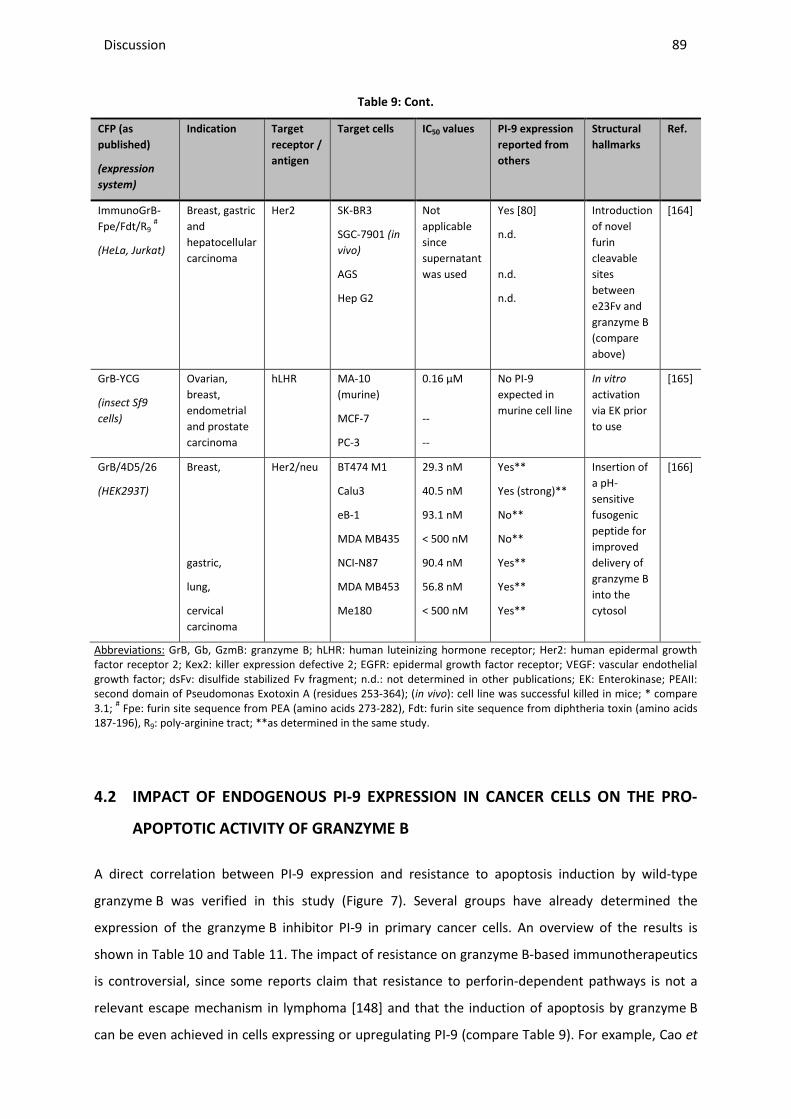
Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
89
Table 9: Cont.
CFP (as
published)
(expression
system)
Indication Target
receptor /
antigen
Target cells IC50 values PI-9 expression
reported from
others
Structural
hallmarks
Ref.
ImmunoGrB-
Fpe/Fdt/R9 #
(HeLa, Jurkat)
Breast, gastric
and
hepatocellular
carcinoma
Her2 SK-BR3
SGC-7901 (in
vivo)
AGS
Hep G2
Not
applicable
since
supernatant
was used
Yes [80]
n.d.
n.d.
n.d.
Introduction
of novel
furin
cleavable
sites
between
e23Fv and
granzyme B
(compare
above)
[164]
GrB-YCG
(insect Sf9
cells)
Ovarian,
breast,
endometrial
and prostate
carcinoma
hLHR MA-10
(murine)
MCF-7
PC-3
0.16 µM
--
--
No PI-9
expected in
murine cell line
In vitro
activation
via EK prior
to use
[165]
GrB/4D5/26
(HEK293T)
Breast,
gastric,
lung,
cervical
carcinoma
Her2/neu BT474 M1
Calu3
eB-1
MDA MB435
NCI-N87
MDA MB453
Me180
29.3 nM
40.5 nM
93.1 nM
< 500 nM
90.4 nM
56.8 nM
< 500 nM
Yes**
Yes (strong)**
No**
No**
Yes**
Yes**
Yes**
Insertion of
a pH-
sensitive
fusogenic
peptide for
improved
delivery of
granzyme B
into the
cytosol
[166]
Abbreviations: GrB, Gb, GzmB: granzyme B; hLHR: human luteinizing hormone receptor; Her2: human epidermal growth factor receptor 2; Kex2: killer expression defective 2; EGFR: epidermal growth factor receptor; VEGF: vascular endothelial growth factor; dsFv: disulfide stabilized Fv fragment; n.d.: not determined in other publications; EK: Enterokinase; PEAII: second domain of Pseudomonas Exotoxin A (residues 253-364); (in vivo): cell line was successful killed in mice; * compare 3.1; # Fpe: furin site sequence from PEA (amino acids 273-282), Fdt: furin site sequence from diphtheria toxin (amino acids 187-196), R9: poly-arginine tract; **as determined in the same study.
4.2 IMPACT OF ENDOGENOUS PI-9 EXPRESSION IN CANCER CELLS ON THE PRO-
APOPTOTIC ACTIVITY OF GRANZYME B
A direct correlation between PI-9 expression and resistance to apoptosis induction by wild-type
granzyme B was verified in this study (Figure 7). Several groups have already determined the
expression of the granzyme B inhibitor PI-9 in primary cancer cells. An overview of the results is
shown in Table 10 and Table 11. The impact of resistance on granzyme B-based immunotherapeutics
is controversial, since some reports claim that resistance to perforin-dependent pathways is not a
relevant escape mechanism in lymphoma [148] and that the induction of apoptosis by granzyme B
can be even achieved in cells expressing or upregulating PI-9 (compare Table 9). For example, Cao et

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
90
al. have not observed any relationship between PI-9 levels and cell sensitivity in their granzyme B
fusion protein targeting Her2/neu-positive cell lines [166]. Another example is a CFP comprising
granzyme B and dsFv-B3, targeting the Lewis Y carbohydrate antigen overexpressed e. g. on
squamous cell lung carcinoma or lung adenocarcinoma, which has been shown to kill the human
breast cancer lines SK-BR3 and MCF-7 in an antigen-dependent manner [161]. The reported IC50
values are rather low compared to the ETA-based reference, which might reflect the expression of
PI-9 in SK-BR3 cells and the ability of MCF-7 cells to upregulate PI-9 in the presence of estrogen [77].
However, it is difficult to confirm in retrospect whether these cells actually expressed PI-9, because
cell lines can differ depending on their source, cultivation conditions, and cell cycle phases [167].
Indeed, several studies have revealed a direct correlation between PI-9 expression and the loss of
granzyme B pro-apoptotic activity in vitro and in xenograft mouse models [64,80,168-170].
As a consequence of these published data, and since a limited effect induced by some granzyme B-
based CFPs has been observed in our group, a closer look at the PI-9 protein expression profiles in
potential target cell lines was carried out in this work (3.1). Hereby, the influence of cultivation time
on the PI-9 level could be excluded via investigations with a Hoechst sorting reagent indicating that
all cells in one cultivation flask were in the same maturation status. Jurkat cells were found to be
PI-9-negative as suggested by published data [148], so they could be used as negative control. The
PI-9 expression shown within K562 (CML cell line) is consistent with earlier findings whereas the
absence of PI-9 in the AML cell lines HL-60 and U937 is controversial [82,148,155,156]. The latter
might be related to differences in detection methods, cultivation conditions and the fact that PI-9 can
be triggered by external stimuli (compare Table 10 and Table 14).
It was found that the PI-9 expression level differed among the EGFR-positive and CD30-positive target
cell lines (Table 6). The latter were chosen for further analysis (3.5) to compare cell lines which were
initially derived from patients with the same cancer type (compare details in 4.3.2). Flow cytometric
analysis indicated a heterogeneous expression of PI-9 among these cells (Figure 3B). This is in
accordance to an earlier study (compare Table 11) showing that 6 out of 57 tested HL patients
expressed the inhibitor [171]. In addition, PI-9 expression was also upregulated upon EBV infection,
which is a main cause of B-type HL [72] and was recently even directly linked to EBV-positive cases of
HL [172]. Other data, shown in Table 10 and Table 11, also indicate that PI-9 is expressed in a
heterogeneous manner within the tumor cell population, leading to the survival of a small number of
malignant cells surrounded by innate immune cells. PI-9 was expressed primarily in tumors
containing many granzyme B-positive infiltrating CTLs and the high incidence of PI-9-positive tumor
cells often predicted an unfavorable clinical outcome [173]. This suggest that PI-9-positive tumor
cells are positively selected as they evade immunosurveillance, which is clinically relevant for the
treatment of hematological disorders or solid tumors in which PI-9 expression is upregulated during
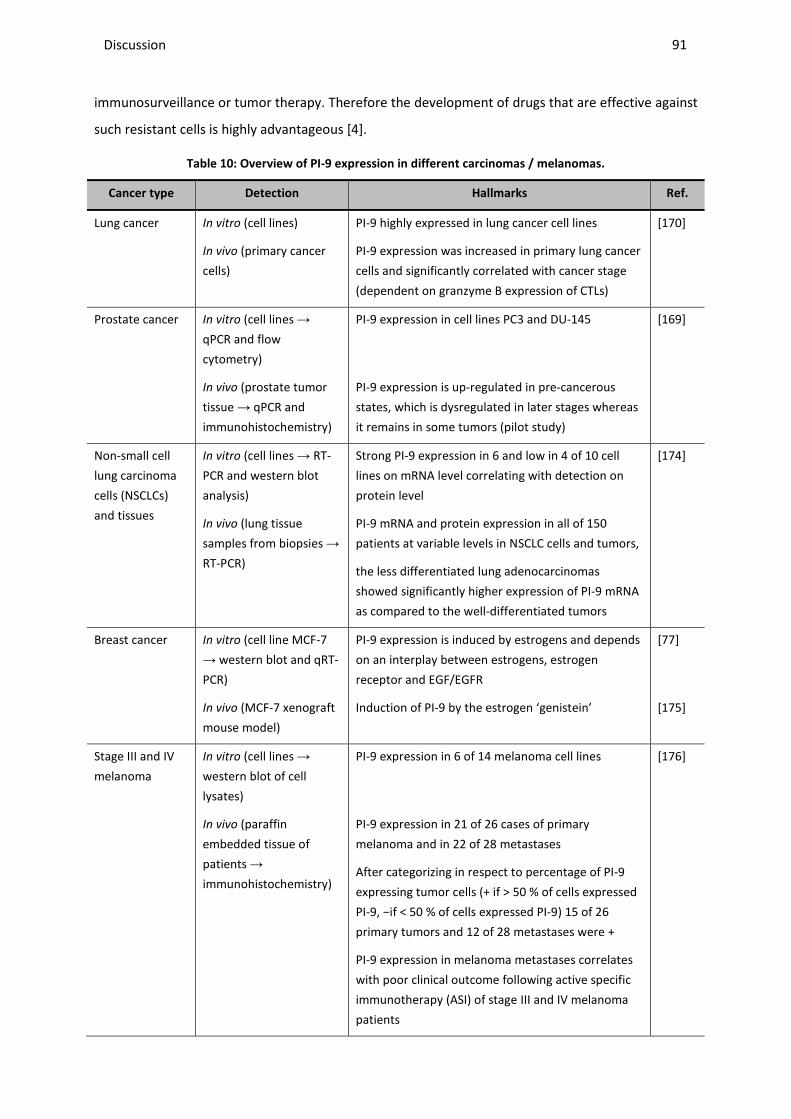
Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
91
immunosurveillance or tumor therapy. Therefore the development of drugs that are effective against
such resistant cells is highly advantageous [4].
Table 10: Overview of PI-9 expression in different carcinomas / melanomas.
Cancer type Detection Hallmarks Ref.
Lung cancer In vitro (cell lines)
In vivo (primary cancer
cells)
PI-9 highly expressed in lung cancer cell lines
PI-9 expression was increased in primary lung cancer
cells and significantly correlated with cancer stage
(dependent on granzyme B expression of CTLs)
[170]
Prostate cancer In vitro (cell lines →
qPCR and flow
cytometry)
In vivo (prostate tumor
tissue → qPCR and
immunohistochemistry)
PI-9 expression in cell lines PC3 and DU-145
PI-9 expression is up-regulated in pre-cancerous
states, which is dysregulated in later stages whereas
it remains in some tumors (pilot study)
[169]
Non-small cell
lung carcinoma
cells (NSCLCs)
and tissues
In vitro (cell lines → RT-
PCR and western blot
analysis)
In vivo (lung tissue
samples from biopsies →
RT-PCR)
Strong PI-9 expression in 6 and low in 4 of 10 cell
lines on mRNA level correlating with detection on
protein level
PI-9 mRNA and protein expression in all of 150
patients at variable levels in NSCLC cells and tumors,
the less differentiated lung adenocarcinomas
showed significantly higher expression of PI-9 mRNA
as compared to the well-differentiated tumors
[174]
Breast cancer In vitro (cell line MCF-7
→ western blot and qRT-
PCR)
In vivo (MCF-7 xenograft
mouse model)
PI-9 expression is induced by estrogens and depends
on an interplay between estrogens, estrogen
receptor and EGF/EGFR
Induction of PI-9 by the estrogen ‘genistein’
[77]
[175]
Stage III and IV
melanoma
In vitro (cell lines →
western blot of cell
lysates)
In vivo (paraffin
embedded tissue of
patients →
immunohistochemistry)
PI-9 expression in 6 of 14 melanoma cell lines
PI-9 expression in 21 of 26 cases of primary
melanoma and in 22 of 28 metastases
After categorizing in respect to percentage of PI-9
expressing tumor cells (+ if > 50 % of cells expressed
PI-9, −if < 50 % of cells expressed PI-9) 15 of 26
primary tumors and 12 of 28 metastases were +
PI-9 expression in melanoma metastases correlates
with poor clinical outcome following active specific
immunotherapy (ASI) of stage III and IV melanoma
patients
[176]

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
92
Table 10: Cont.
Cancer type Detection Hallmarks Ref.
Nasopharyngeal
carcinoma
In vivo (formalin-fixed,
paraffin-embedded
tumor biopsies →
immunohistochemistry)
PI-9 expression in 3 of 43 cases
Presence of many tumor infiltrating activate CTLs
within patient biopsies is related to bad clinical
outcome
[177]
Melanoma,
breast,
cervical and
colon carcinoma
In vitro (cell lines → PCR)
In vivo (primary colon
carcinoma cells → PCR)
PI-9 expression in a subset of determined tumor
lines (e.g. MCF-7, SK-BR3)
PI-9 expression in 2 of 4 primary surgical specimens
[80]
Strategies to optimize the efficacy of granzyme B-based CFPs on primarily PI-9-positive cells include
either the downregulation of PI-9 expression/ activity in target cells or the generation of granzyme B
variants that are insensitive to PI-9 (3.5). The former may be achieved by RNA interference (RNAi) or
antisense technology to knock down PI-9 gene expression. The successful use of siRNA to inhibit the
expression of PI-9 has been described in stem cells [69] and in breast cancer [77]. However, interest
in development of siRNA-based therapies has declined following the Phase III failure of Bevasiranib,
which targeted the expression of vascular endothelial growth factor (VEGF) for the treatment of age-
related wet macular degeneration [178]. In alternative approaches, the NFκB inhibitor pyrrolidin
dithiocarbamate (PTDC) has been used to suppress PI-9 gene expression in PBMCs cultivated in vitro
[72], whereas the serine protease granzyme M (found in the cytotoxic granules of NK cells) can
inhibit PI-9 at the protein level [56]. Hence, the genetic fusion of granzyme M to a specific binding
domain facilitating cellular uptake could lead to the inactivation of endogenous PI-9 so the co-
administration of granzymes M and B could potentially clear the way for granzyme B-based therapy
(further discussed in 4.6.2) [4].
Those strategies, however, are indirect approaches which require the administration of granzyme B
in combination with a separate therapeutic modality. In contrast, as primarily intended in this study,
granzyme B can be modified in such a way that it retains its enzymatic activity but becomes
insensitive to PI-9. In an analogous approach, a human tissue-type plasminogen activator was
mutated at a few critical contact residues to achieve resistance against a complex mixture of
endogenous serpins [37,179].
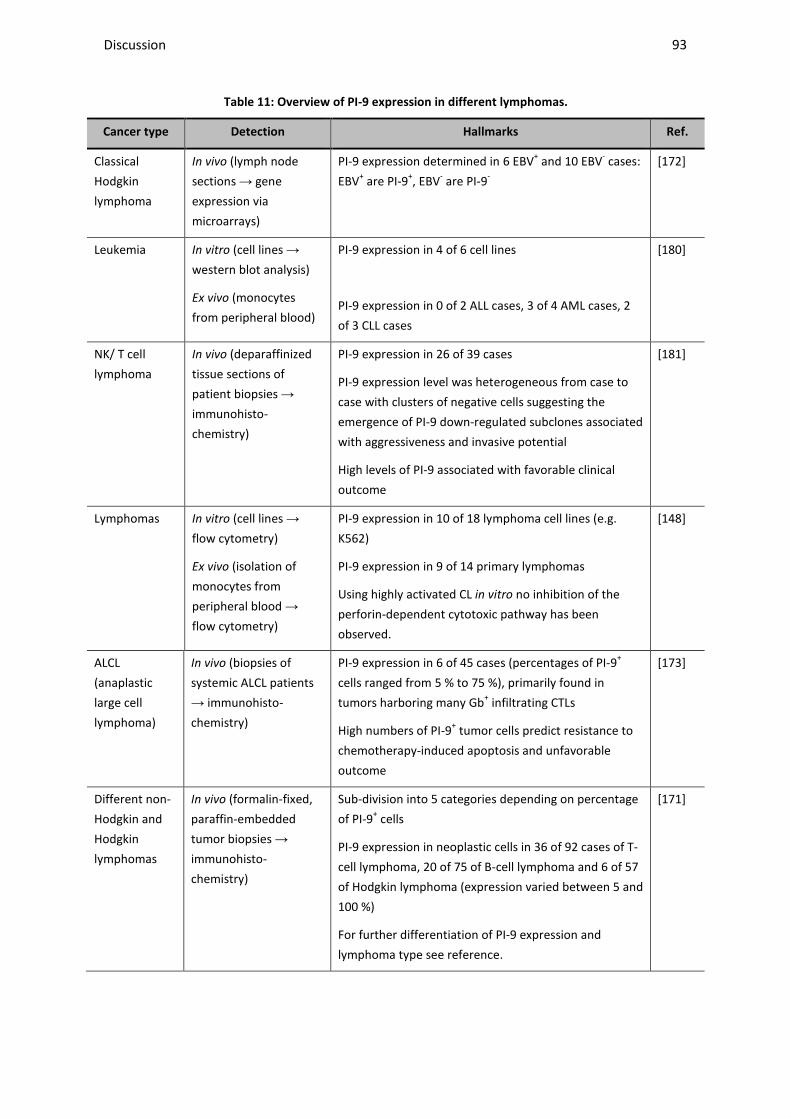
Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
93
Table 11: Overview of PI-9 expression in different lymphomas.
Cancer type Detection Hallmarks Ref.
Classical
Hodgkin
lymphoma
In vivo (lymph node
sections → gene
expression via
microarrays)
PI-9 expression determined in 6 EBV+ and 10 EBV- cases:
EBV+ are PI-9+, EBV- are PI-9-
[172]
Leukemia In vitro (cell lines →
western blot analysis)
Ex vivo (monocytes
from peripheral blood)
PI-9 expression in 4 of 6 cell lines
PI-9 expression in 0 of 2 ALL cases, 3 of 4 AML cases, 2
of 3 CLL cases
[180]
NK/ T cell
lymphoma
In vivo (deparaffinized
tissue sections of
patient biopsies →
immunohisto-
chemistry)
PI-9 expression in 26 of 39 cases
PI-9 expression level was heterogeneous from case to
case with clusters of negative cells suggesting the
emergence of PI-9 down-regulated subclones associated
with aggressiveness and invasive potential
High levels of PI-9 associated with favorable clinical
outcome
[181]
Lymphomas In vitro (cell lines →
flow cytometry)
Ex vivo (isolation of
monocytes from
peripheral blood →
flow cytometry)
PI-9 expression in 10 of 18 lymphoma cell lines (e.g.
K562)
PI-9 expression in 9 of 14 primary lymphomas
Using highly activated CL in vitro no inhibition of the
perforin-dependent cytotoxic pathway has been
observed.
[148]
ALCL
(anaplastic
large cell
lymphoma)
In vivo (biopsies of
systemic ALCL patients
→ immunohisto-
chemistry)
PI-9 expression in 6 of 45 cases (percentages of PI-9+
cells ranged from 5 % to 75 %), primarily found in
tumors harboring many Gb+ infiltrating CTLs
High numbers of PI-9+ tumor cells predict resistance to
chemotherapy-induced apoptosis and unfavorable
outcome
[173]
Different non-
Hodgkin and
Hodgkin
lymphomas
In vivo (formalin-fixed,
paraffin-embedded
tumor biopsies →
immunohisto-
chemistry)
Sub-division into 5 categories depending on percentage
of PI-9+ cells
PI-9 expression in neoplastic cells in 36 of 92 cases of T-
cell lymphoma, 20 of 75 of B-cell lymphoma and 6 of 57
of Hodgkin lymphoma (expression varied between 5 and
100 %)
For further differentiation of PI-9 expression and
lymphoma type see reference.
[171]

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
94
4.3 GENERATION AND IDENTIFICATION OF A PI-9 INDEPENDENT GRANZYME B
VARIANT
4.3.1 Evaluation of in silico findings and comparison with in vitro enzymatic activity
As described in the previous chapter, a granzyme B variant that can kill both PI-9-positive and PI-9-
negative tumor cells will provide a significant therapeutic advantage, particularly for the treatment of
relapsing tumors. Since the underlying reaction is typical for the interaction between proteases and
serpins the necessary structural hallmarks could be predicted in silico using a structurally related
Michaelis complex formed between a rat trypsin and M. sexta serpin B1 (3.2).
Using state-of-art molecular modeling techniques key residues at the interface of the two proteins
which might decrease their affinity could be identified (3.2). Among wild-type granzyme B residues
involved in complexing, only residues not located in its active site were considered so the catalytic
activity was not expected to be affected. First results were generated using computational alanine
scanning mutagenesis, a convenient and fast strategy to investigate the determinants of protein
complex interactions [182]. However, hydration at the protein/ protein interface is not included in
these calculations even though water molecules may play a very important role for complex stability
[183]. Another disadvantage is that only alanine mutations can be simulated and the flexibility
properties of the complex are omitted. Therefore, molecular dynamics simulations of wild-type and
mutated complexes were performed in water solution which on the one hand demands a greater
computational cost but on the other hand allows the qualitative assessment of the effect of all
proposed mutations. Those mutations were decided considering the charge of the original amino
acid, arginine, so in the end, including one double mutant, seven new granzyme B mutants were
investigated (R28A, R28E, R28K, R28A/R201A, R201A, R201E, R201K) [149].
In vitro the corresponding mutations were introduced into the wild-type sequence of granzyme B via
site-directed mutagenesis. The protein was produced via the established secretory expression in
HEK293T cells [137,138,184] allowing natural glycosylation patterns within granzyme B which
harbors two N-glycosylation sites [185]. These sites were not affected by the mutations. No
differences in production rates were observed compared to the wild-type protein and all variants
could successfully be cleaved by enterokinase (3.3).
To test the remaining in vitro proteolytic activity after site-directed mutagenesis and challenge with
PI-9, the inhibitor had to be recombinantly produced since pure protein was not commercially
available. Bacterial expression turned out to be the most efficient method to obtain sufficient
amounts of protein (3.4). The identity of the protein was confirmed by mass spectrometric analysis
(data not shown). The successful complex formation of the produced protein could be verified after

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
95
incubation with recombinant granzyme B-based CFPs by detection of a SDS-stable complex on
western blots by different antibodies (Figure 6). Because only the upper band disappeared after
incubation with granzyme B (3.4), it could be concluded that the lower band might be the earlier
described inactive variant of PI-9 lacking the RCL which resulted in the small decrease in molecular
weight compared with intact PI-9 [56]. The same samples were used in a colorimetric granzyme B
substrate assay and the inhibition of enzymatic activity by complex formation between wild-type
granzyme B and the produced recombinant PI-9 could be directly confirmed. This assay was
consequently used as the read-out system to identify the most promising PI-9-insensitive granzyme B
mutants. Here, it was found that the in silico calculations were highly consistent with the in vitro
results: GbR201A-, GbR201K- and GbR28K-based fusion proteins (to H22(scFv)) in the absence of PI-9
feature catalytic activity similar to that of wild-type granzyme B indicating that the active site was not
affected. In the presence of PI-9, GbR201K-H22(scFv) retains almost its entire catalytic activity
whereas GbR28K-H22(scFv) and GbR201A-H22(scFv) kept three quarter or half of their enzymatic
potential, respectively. The measured Km values (~ 104 µM) for GbR201K-H22(scFv) was almost
identical to the one determined for the wild-type fusion protein (3.6.1) and consistent with published
data for recombinant granzyme B [83,86].
The PI-9 insensitivity of the recently described granzyme B variant K27A by Sun et al. [86] could not
be reproduced neither during in silico nor in vitro experiments. The reason might be that this earlier
structural in silico prediction was based on a complex with a modeled truncated PI-9 version
consisting of only 12 residues (334–345) of the RCL. The latter is a solvent exposed stretch of amino
acids representing the primary site of interaction with human granzyme B. Subsequently, different H-
bonds and salt bridges have been considered by Sun et al. resulting in different predictions.
Inconsistencies seen in the in vitro assays could be explained by a different expression system for
human granzyme B and PI-9 (P. pastoris with Sun et al.) leading to differences in glycosylation
patterns [186], which can influence complex formation. Differences in protein quality used in this
thesis and in the previous study could also have an effect.
With the combination of the applied in silico and in vitro techniques it was possible to pre-select
three out of eight generated granzyme B mutants, which retained their enzymatic activity even after
incubation with recombinant PI-9.
4.3.2 Selection of the optimal granzyme B mutant
The above described results give a first insight into the functionality of the newly generated mutants.
The three most promising variants (R28K, R201A, R201K) were further tested in vitro according to
their binding activity and affinity to target cells, stability and cytotoxic as well as apoptotic efficacy.
K27A was used as a comparative control. A sufficient evaluation of their PI-9 sensitivity required PI-9-

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
96
negative as well as PI-9-positive cells. CD30-positive cell lines were used based on reasons described
in detail earlier (4.2). To target those cells Ki4(scFv)-based fusion proteins were generated whose
potential had been confirmed previously in combination with ETA’ whereby IC50 values of 43 - 85 pM
were determined [112]. CD30-targeting murine Ki4(scFv) is an outstanding anti-CD30 single chain as
it does not bind soluble CD30 receptor [187]. This is important since it has been reported that CD30
ectodomain shedding occurs at high incidences [108], which could lead to an off-target capturing of
the CFP. The H22(scFv)-based fusion proteins which were used during the above discussed in vitro
characterization (4.3.1) were also employed during the evaluation of the mutants on PI-9-negative
cells as a reference in comparative assays.
It was found that both wild-type and mutants specifically bound to the target cells and did not exhibit
unspecific binding to CD30- or CD64-negative control cell lines, respectively (Figure 10). Kd for the
wild-type and the R201K mutated variant were also in the same nM range on the respective cell lines
(3.6.2) and correlated to the affinity of 3*10-9 M published for Ki4(scFv) alone [187] and 10-8 to
10-10 M reported for H22(scFv) [188]. This excluded a steric hindrance of antibody fragment binding
by the mutated granzyme B fusions. Also a negative influence of the mutations on apoptosis inducing
potential (Figure 13) as well as the cytotoxic activity could be excluded, since the observed IC50 values
were comparable to the respective values determined with the wild-type protein (Figure 12).
Compared to other published granzyme B-based CFPs (Table 9), the determined IC50 values exceed
the average.
The next step was to compare the ability of the generated mutants to kill PI-9 expressing cells and
eventually select the most promising one. For this goal, different strategies were considered: either
direct delivery of granzyme B and its variants into the cells or indirect delivery based on specific
targeting of tumor antigens by genetic fusion to tumor specific ligands. Since the former approach
was not successful (compare (3.5, 1)), strategies involving indirect, receptor-mediated delivery of the
protein into the cells were pursued instead. Here, the modification of originally PI-9-negative cell
lines to express PI-9 was first intended to exclude any cell-based differences (other than the
expression of PI-9) which occur when different types of naturally PI-9-positive or -negative cell lines
are used. The resistance of the cell lines used towards transfection with pMS-PI9-IV, however, limited
this approach. Since HEK293T cells successfully produced cytosolic PI-9 after transfection with
pMS-PI9-IV, it could be excluded that the reasons for the transfection difficulties were based on the
chosen plasmid DNA, but rather were due to the cells themselves. It has been reported that certain
cell lines are intrinsically easier to transfect than others, although the exact reason for these
differences is currently unknown. Additionally, even clonal variability in DNA uptake has been
reported in certain mouse cells [189].

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
97
An alternative solution was to use cell lines equal in antigen-expression, but different in PI-9
expression. As described in 4.2, CD30-positive cell lines belong to this category which also showed
differences in sensitivity towards Gb-Ki4(scFv), whereby a limited apoptotic effect was observed on
PI-9-positive L428 cell line whereas successful cell death induction was seen on PI-9-negative L540cy
cells (Figure 7B). Limited internalization due to differences in receptor expression (Table 7) could be
excluded as a reason for variations in cytotoxicity since complete internalization could be verified
with the help of Ki4(scFv)-SNAP-BG-647 (Figure 11) and similar Kd values were measured for all cell
lines used (Table 7).
Based on these findings, an adequate cell-based test system could be set up for the further
evaluation of the generated mutants and selection of the most promising variant. As suggested by
the previous substrate assay (4.3.1), the wild-type and the already published mutant GbK27A were
only effective in PI-9-negative cells (Figure 14). GbR201K-Ki4(scFv) was the most effective as it was
able to significantly reduce the viability of both PI-9-positive (L428, L1236, K562) and PI-9-negative
(L540cy) cell lines. These results were verified in an assay based on caspase 3/7 (Figure 15), which is a
strong indicator for apoptosis induction, especially in case of granzyme B induced cell death (1.3.1).
The cytotoxic effects triggered by GbR201K-Ki4(scFv) in L428 and L1236 were rather low compared to
those in L540cy. The addition of endosomolytic agents like chloroquine led to improved effects in
previous studies [160], but did not increase cytotoxicity here; neither did the increase in protein
concentrations. However, the observed differences can be explained by the published, lower
sensitivity of L428 and L1236 to apoptosis induction [97]. As described in 1.5, one of the signaling
pathways characteristic for HL involves NFκB. In these previous studies L428 and L1236 were much
more resistant to apoptosis induction by the anti-CD30 antibody 5F11 than L540 due to the
expression of this anti-apoptotic protein leading to induced resistance to CD30-mediated cell death
in HL cells. These effects can be explained by the defective NFκB inhibitor IκBα in L428 or the instable
inhibitor in L1236 cells causing constitutively active NFκB, and thereby apoptosis resistance [190-
192]. In one study it was shown that NFκB could be down-regulated through the proteasome
inhibitor bortezomib after CD30-mediated NFκB activation with the human CD30 antibody 5F11 [97].
The same protocol was adapted in this thesis as well. However, here, the addition of bortezomib did
not improve cytotoxicity. Nevertheless, even the currently observed low cytotoxic effects can have
high medical impact, especially if they lead to the elimination of treatment resistant cells responsible
for the minimal residual disease which are potentially related to PI-9 expression (4.2). On the other
hand the CD30-based system applied here has to be regarded as a model system to select the most
promising granzyme B mutant and this was successfully accomplished.

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
98
In summary, based on a computational model of the complex between granzyme B and its inhibitor
PI-9, seven new granzyme B variants were generated and their in silico predicted potential was
successfully confirmed during comparative in vitro studies with the wild-type protein and an earlier
suggested mutant. Finally, the variant GbR201K was selected as the most promising and was used for
further evaluation as fusion to CD30-targeting Ki4(scFv) in vivo (4.4) as well as CD64-targeting
H22(scFv) ex vivo on patient derived leukemic cells (4.5).
4.4 EFFICACY OF THE GBR201K MUTANT IN VIVO
To confirm the differences between the in vitro performance of wild-type and mutant granzyme B
R201K, comparative studies on the biological activity were done employing the above used CD30-
positive cell lines L540cy (PI-9-negative) and L428 (PI-9-positive).
First, the serum stability of GbR201K-based CFPs was compared to that of the wild-type, and no
influence of the point-mutations on stability was observed (Figure 16 and Table 8). Murine serum
itself was found to already be very cytotoxic towards the target cells. Therefore a proper
discrimination between cell death induced by the fusion proteins or the serum was unfeasible. The
isolation of the protein from the serum via IMAC was not done as then the protein concentration
would be depending on His6-tag binding efficacy and not just serum stability. Alternatively, residual
binding activity of the constructs to the target cells was used as measure for serum stability, whereby
the detection of full length CFP was ensured by the employment of both His6-tag and granzyme B-
specific antibodies. Via this method, stability for at least 10 h was observed for both constructs. The
half life of biopharmaceuticals strongly depends on their composition and the intended application
and varies highly ranging from minutes to days [193]. The stability measured here is lower than the
one published for the ETA’-based construct Ki4(scFv)-ETA’ which retained 85% to 90% of initial
binding activity for at least 24 h [112]. However, according to Kampmeier et al. a 10 h stability should
suffice for our application. In their study the tumor-specific SNAP-based signal was strongest after
this time point in a subcutaneous mouse tumor model [194].
Two different xenograft mouse models were established in this study, one based on the PI-9-positive
L428 cells and one on PI-9-negative L540cy cells. So far in vivo studies for HL have been strongly
restricted to a single cell line, L540cy, that achieves a sufficient tumor take rate in subcutaneous and
disseminated mouse models [140]. However, tumor take rate related problems for L540cy have been
experienced before in this group (Dr. rer. nat. Theo Thepen, personal communication). Therefore, it
was decided to focus on a subcutaneous tumor model that is easier to monitor and faster to evaluate
than a disseminated one. Via resuspension of the cells in Matrigel™, which leads to a fixed

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
99
accumulation of the cells and has been described to be advantageous to establish difficult tumor
models [195], a high tumor take rate could be achieved (3.8.2). Because of the observed
encapsulation and limited vascularization (Figure 17B) the connection of the tumor to the
bloodstream was tested by quantitative analysis of soluble tumor-derived CD30 receptor in blood
samples of tumor-bearing mice (3.8.2). The direct correlation observed between tumor size and
CD30 concentration in blood suggested that intravenous application of the CFP could be successful.
In addition, the localization of the fluorescent signal after injection of Ki4(scFv)-SNAP-BG-747 into
L540cy tumor-bearing mice (Figure 17A) verified the specific delivery and the continuous CD30
receptor expression on the tumors themselves in vivo. The latter was also confirmed by staining of
tissue sections prepared from both L540cy and L428 tumors after treatment with the Ki4(scFv)-
derived constructs (Figure 18C and Figure 19C). Following, the usability of the established tumor
model was completely demonstrated. The translocation of the construct to the low vascularized
tumors was possibly supported by extravascular transport playing an important role within tumor
tissue [196]. Several studies in the past have changed the understanding of the interstitial-lymphatic
continuum of tumors from a passive to a more active one, so the delivery of tumor therapeutics is
possible [197]. To optimize transport conditions in future animal experiments, the intravascular
transport could be improved by the addition of dopamine, which supports the distribution and
normalization of dysfunctional blood vessels in tumors and has significantly increased the
concentration of anticancer drugs in tumor tissues in previous studies [198].
As mentioned, no tumor mouse model was available for PI-9-positive L428 cells. Such a model was,
however, necessary for the evaluation of the novel granzyme B mutant. Via Kat2 expression by these
slowly and transiently growing cells in vivo the monitoring of tumor growth became possible via
optical imaging (Figure 17C). The direct correlation between caliper measurements and tumor size
determined by the imaging technique was proven recently confirming the reliability of the imaging
measurements [150]. Hence, a novel HL tumor model based on the cell line L428 was established in
this work.
Since no difference in cytotoxic activity between Gb-Ki4(scFv) and its mutant GbR201K-Ki4(scFv) was
detected on PI-9-negative cells in vitro, only the mutant was used to confirm its effectiveness on PI-9-
negative cells in vivo. Up-regulation of PI-9 in L540cy as an immune escape mechanism could be
excluded by cell lysis and subsequent western blots of L540cy tumor-derived cells using PI-9-specific
antibodies (data not shown). To treat L428 tumors both constructs were used, to confirm differential
influence of endogenous PI-9 on the biological activity of wild-type and mutant granzyme B. To
exclude unspecific effects by granzyme B itself, instead of using a buffer control, the non-binding
control GbR201K-H22(scFv) was chosen. The obtained results clearly demonstrated that only the
mutant was able to kill the PI-9-negative tumor cells as well as the PI-9-positive ones in vivo so this

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
100
variant has the potential to replace the wild-type in the future, since this only kills PI-9-negative cells
(Figure 18B and Figure 19B). With this, the results of the previously obtained in vitro analyses could
be confirmed after the establishment of appropriate tumor mouse models.
In this study 50 µg of CFP per treatment day and injection was applied which was well tolerated by
the mice. In a previous study by Gehrmann et al. much higher injected concentrations of 20 µg
human granzyme B per gram body weight have been reported without inducing adverse side-effects,
meaning that after injection of active granzyme B the investigated organs exhibited no
pathophysiological changes and individual organs exhibited normal tissue architecture, with no
evidence of infarcts or internal bleeding [199]. Therefore it seems that active human granzyme B at
doses to efficiently reduce tumor size, does not induce detectable histological or morphological
changes in normal organs. However, those conclusions have to be regarded with care since in our
case immune suppressed mice were used and also species-dependent substrate specificities have to
be considered which can distort the results. Those species-dependent substrate specificities leading
to lacking cell death induction of granzyme B in foreign species have been described by others in
vitro [200]. In addition the same could be shown and published in the time course of this work as
well ex vivo (data not shown) when the cytotoxic efficacy of CD64-specific human CFPs and ETA’-
based IT was tested on murine and human derived macrophages [201]. In that study, Gb-H22(scFv)
only killed human derived macrophages, leaving the murine derived cells unaffected. These data
suggest that human granzyme B is not able to cause any side-effects in mice since it cannot even kill
target cells after specific delivery. Therefore, the estimation of maximum tolerated doses (MTDs) of
human granzyme B in humans becomes a challenge because the results from other species might be
misleading. Possible side-effects in humans could be expected from unspecific binding to cells or
tissue, due to the high positive surface charge of granzyme B [202], which can mediate electrostatic
interactions with the negatively-charged heparin sulfate proteoglycans found on the surface of
almost all cells. Also its potentially excessive extracellular activity, which is important for tissue
remodeling during an immune response, can cause pathophysiological tissue destruction [203]. It
seems that the only choice for a predictive pre-clinical model system will be primates due to their
close relationship to humans. Corresponding studies have not yet been reported and neither has the
efficacy of human granzyme B been tested in any primate cells. Another important fact regarding the
in vivo experiments described here is that we use subcutaneous tumor models. These models are
artificial because it does not reflect the biology of HL in humans (1.5). However, the focus here was
on the differences in cell death induction by the wild-type versus the mutant and not on the disease
itself. Therefore this model was sufficient for our studies. In summary, the exclusive biological
activity of the novel granzyme B mutant R201K on PI-9-positive as well as PI-9-negative cells in

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
101
contrast to the wild-type protein could successfully be demonstrated in vivo based on the newly
established xenograft tumor models.
4.5 EX VIVO DETERMINATIONS ON PRIMARY LEUKEMIC CELLS
Before biopharmaceuticals are approved for clinical studies, their in vitro and in vivo efficacy has to
be tested extensively. These experimental data are based either on animal models or on cell-based
assays. While animal models are commonly used for toxicity and metabolism studies, their
extrapolation to humans is restricted. Furthermore, they are both expensive and have low-
throughput which usually limits their use to the late stages of preclinical testing. Cell-based assays
are continuously being improved to help reverse the increasing trend of costly late-stage drug
failures. These improvements are of interest to the pharmaceutical industry. Use of standard well
established cell lines has the benefit of phenotypic stability and homogeneity, unlimited life span and
ease of culture. However, they cannot mimic the complexity and heterogeneity of e. g. tumor cells
and for some diseases appropriate cell lines are not even available. As a consequence, ex vivo
models, comprising primary cells, are coming more and more into focus because they are thought to
simulate more closely the actual cells. With the help of ex vivo models, the molecular mechanisms
underlying tumor behavior can be elucidated and the potential of new anticancer treatment
candidates evaluated. In addition to the great value for the characterization of new drugs in general,
another advantage of ex vivo determinations is that individual patients can be studied enabling
predictions on effectiveness of new drug candidates prior to clinical application [204]. These aspects
are especially valuable in context of personalized medicine. However, primary cells are variable and
often can only be cultured for a few passages before they reach senescence or undergo undesirable
phenotypic changes. In addition, the availability of patient material is often very limited and the
heterogeneity due to differences in patients’ treatment history restricts the comparison between
different experiments. As a consequence, established cell lines have their value in providing first
indication into the functionality of new drug candidates yielding reproducible data with standardized
methods. Those results, however, should always be confirmed using appropriate primary cell
material to approach the complex clinical situation and the real diverse hallmarks of the disease.
4.5.1 CMML and AMML primary cells as target for CD64-specific fusion proteins
Based on the above rationale, the potential of wild-type granzyme B and its most promising mutant
was evaluated ex vivo in addition to in vitro and in vivo studies. For this purpose the CD64-specific
constructs Gb-H22(scFv) and its mutant GbR201K-H22(scFv), whose in vitro efficacy has extensively

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
102
been tested during the time-course of this work (3.6.4 and 3.6.5), were applied and their efficacy was
compared to the established IT H22(scFv)-ETA’. As target, the leukemic rare diseases CMML as well
as AMML (1.6) have been chosen, since specific therapeutics against CMML and AMML represent
unmet medical need for the patients. Research of corresponding targeted therapeutics is restricted
by the lack of frequent and specific genetic aberrations known until now in this disease and the lack
of mouse and in vitro models of the chronic disease. Thus, studies with primary cells, as performed
within this thesis, are the only available system to test new immunotherapeutics.
The hematological malignancy leukemia is especially suitable for immunotherapeutic treatment with
CFPs because the circulating cancer cells are easily accessible and incomplete tumor penetration due
to the absence of vascularization is not a limiting factor [205]. Numerous studies have been
described in literature dealing with immunotherapeutics against primary leukemic cells, e.g. via IL-3
[206], IL-4 [207] or CD33 [208,209]. Examples have been introduced in 1.2.1 and 1.6.
Before cytotoxic assays on those primary cells were performed it was first evaluated, if CD64 is a
valuable target to treat CMML and AMML patients, since this tumor marker has not been described
for these diseases so far (1.6). Primary monocytes were isolated from fresh peripheral blood of
largely newly diagnosed patients (university hospital, Aachen) (2.1.8). The high affinity receptor CD64
is generally a promising candidate target in leukemia therapy. It is superior to most of the other
investigated targets due to its characteristics of efficiently internalizing fused ligands as well as its
restricted expression pattern, leaving normal, non-activated CD64-expressing cells unaffected (see
below and 1.6). The potential of CD64-targeting H22(scFv)-ETA’ as well as the ability of Gb-H22(scFv)
to specifically kill primary AML cells has been shown before [129,138]. The advantage of the latter is
that its cytotoxic efficacy is based on a human effector molecule preventing expected immunogenic
responses provoked by the non-human cell-death inducing agents and thereby allowing multiple
treatment cycles.
During phenotyping of the primary cells for CD64, CD56, CD14 and CD33 receptor expression (3.9.2),
a clear over-expression of CD64 cells was found on the CMML cells, enabling specific binding of
H22(scFv)-derived constructs (Figure 22A). The expression of CD64 has been discussed before in
context of CMML, where a decrease of this marker has been described [115,210]. However,
published data are based on immunohistochemical staining of bone marrow which is different to our
results based on flow cytometric analyses of monocytic cells. The detected overexpression of CD64
on the primary AMML cells was in accordance to published data [114]. Through double staining of
patient derived cells, significant co-expression of CD64 and CD56 as well as co-expression of the
malignant cell markers CD33 and CD14 with CD64, respectively, was observed via flow cytometry and
confocal microscopy (Figure 21). The finding that the same cells express CD64 in parallel to the

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
103
already known tumor markers confirmed the assumption that CD64 is a promising target to eliminate
malignant cells in CMML and AMML patients. In addition, this knowledge is valuable for the
verification of the specific cell death described in the following section.
To evaluate if Gb-H22(scFv) and GbR201K-H22(scFv) are actually stable in CMML and AMML patient-
derived serum, which is important for their potential clinical application, their serum stability was
determined (Table 8). The stability of the IT H22(scFv)-ETA’ has been evaluated before, whereby after
24 h 77 % of the original binding ability was retained after incubation in mouse serum [211]. This is
10 % more than determined for the CFPs after 24 h in patient serum and corresponds to the
determined stability of the CFPs after 6 h. Since H22(scFv) internalizes very fast and its affinity is very
high (Kd = 2.1 nM (3.6.2)) [132], this was sufficient (see also 1.6 and 4.4).
4.5.2 Specific cytotoxic efficacy of CD64-specific fusion proteins on primary CMML and
AMML cells
As indicated above, the characterization of CFPs using primary cells has some drawbacks (4.5). Firstly,
patients have a different clinical history and samples of patients whose treatment has not started yet
are obtainable just once. Secondly, in the present case it has to be taken into account that CMML
and AMML are rare diseases limiting the number of available patients and that the cell number
within their blood samples is irregular. These facts excluded a reproduction of the obtained results
using the cells from the individual patient or direct comparison of the investigations. For these
reasons, as many evaluation techniques as possible were used to obtain as much information as
possible and to increase reliability and elucidate specificity.
The standard XTT and Annexin V measurements after treatment with the H22(scFv)-based constructs
confirmed cell death of up to 50 % even though the results differ slightly; with the XTT-based data
showing higher toxic effects than observed during flow cytometry (Figure 23). This difference might
be due to the fact that classical cell proliferation assays do not discriminate between apoptotic and
necrotic cells as is the case for apoptosis assays. Hence, necrotic cells and very small cell fragments
could skew flow cytometric measurements. Additionally, dose-depending viability assays showed a
typical sigmoidal course despite the fact that still high overall viability remained (around 50 %). The
measured cytotoxic impact and especially the high overall remaining viability after incubation of the
primary cells with the CFPs were in contrast to the results obtained using homogeneous cell lines.
This is probably due to the heterogeneous nature of the primary cell population since it contains a
variable proportion of CD64-positive target cells together with many others; and the applied
measurement techniques are influenced by those non-target cells. This phenomenon was also
described previously in cytotoxicity studies conducted on primary AML cells [208,209,212]. The

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
104
enrichment of those cells, e. g. by magnetic cell sorting using CD64-specific antibodies, would have
occupied the receptors and interfered with the binding and internalization of the fusion protein.
It was observed that the percentage of dead cells did not correlate with the initially detected
proportion of CD64 cells within the cell population. This discrepancy is due to the fact that the
amount of the CD64 receptors is not related to the sensitivity of the cells. It is rather their activation
state, influenced by mediators like IFNγ as used in the current tests (1.6). Thus, if the CD64-
expressing cells are not activated, they are not affected by the immunotherapeutic. This is also
important for later clinical studies with CD64-specific CFPs since thereby it can be avoided that
healthy CD64-positive cells are also killed [213]. Controls using the antibody alone and also the
inactive granzyme B variant GbS184A-H22(scFv) [214] confirmed that cell death was induced by the
effector domain and not just by receptor binding and internalization. Therefore, the proof-of-concept
of the immunotherapeutics tested here could successfully be demonstrated.
Determinations regarding the specificity of primary cell death were performed by four different
approaches. First, using a competitive viability assay the antigen-specificity of cell death could be
confirmed (3.6.4). Hence, it could be concluded that even in a mixture of different cells only the
target cells would be specifically targeted. Second, efficient CD64-depending internalization into
primary cells was shown by confocal microscopy (Figure 22B). Third, non-specific effects by
granzyme B were excluded by incubation of the cells with the non-binding control Gb-Ki4(scFv).
Fourth, based on co-staining experiments of the heterogeneous cell population (4.5.1), the selective
reduction of the target cells was determined via flow cytometry using specific antibodies. The parallel
staining with Annexin V and a target cell-specific antibody to specifically determine apoptosis of the
targeted cells was only possible in one case (Figure 26A), likely due to the loss of receptor expression
by apoptotic cells. Thus, many apoptotic cells would have been omitted during evaluation of the
data. The same was seen during preliminary in vitro assays as well, where co-staining with Annexin V
and CD56 of the CD56/CD64-positive cells and subsequent gating of CD56-positive cells prior to
Annexin V-based evaluation significantly differed from the results obtained without prior gating (data
not shown). As an alternative the position of the CD64-positive target cell population in FSC/ SSC
dotplot has been identified so that the decrease of this population could be estimated after
treatment of the cells (3.9.5). Because the obtained results correlated reproducibly with the results
from viability and apoptosis assays (Figure 23 and Figure 25), this method was verified to be a
reliable measure for specific cell death as was proposed earlier [212]. Another measure for specific
cell death was the reduction of the target population identified and stained with an adequate
antibody (shown for CD14-staining in Figure 26B) which has been chosen according to phenotyping
(Figure 21). Since the incidence of blocking by the H22(scFv)-based constructs was observed on HL60
in pre-tests, the use of CD64-specific antibodies was denied. In summary, the results obtained here

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
105
provide clear evidence that CMML and AMML cells can both be selectively killed by granzyme B-
based CFPs.
4.5.3 The role of PI-9 and the potential of the determined cytolytic fusion proteins for
personalized medicine
To correlate the cytotoxic effects of the new PI-9 resistant granzyme B variant GbR201K-H22(scFv) on
primary cells with the expression of the inhibitor, endogenous PI-9 levels were investigated
immediately after isolation from peripheral blood and also after cultivation for different time
intervals by western blotting. The reason for the observed upregulation of PI-9 in cell culture after
day one is not known (Figure 20), but has been observed before in the AML cell line U937 [180].
Since the expression of PI-9 in lymphocytes and monocytes of PBMCs derived from normal blood has
been described before [72] and was determined here as well (data not shown), it could not be
explicitly concluded if the detected PI-9 expression is an escape mechanism in CMML and AMML to
protect against CTL attack, or the usual PI-9 level in PBMCs. The previously found mild up-regulation
of PI-9 by IFNγ in primary monocytes after in vitro cultivation could not be corroborated here (Figure
20B), however this might be due to the applied different detection method based on flow cytometry
[72]. Nevertheless, the detection of PI-9 within the target cells potentially diminishes the efficacy of
wild-type granzyme B based immunotherapy. Therefore these cells also represented a potential ex
vivo test system to evaluate the new granzyme B mutant R201K, especially in comparison with the
granzyme M-based construct Gm-H22(scFv) which will further be elucidated in 4.6.1. Indeed a
correlation between PI-9 expression and cytotoxic efficacy of the mutant was observed. In
comparison to wild-type Gb-H22(scFv) a significantly higher cytotoxic activity of GbR201K-H22(scFv)
was observed against cells with a distinct PI-9 expression. As observed during the in vitro studies on
cell lines, no difference between the effect of the mutant and its wild-type occurred in cells not
expressing the granzyme B inhibitor. However, to state that PI-9 expression is a hallmark of CMML or
AMML in vivo, further investigations are required involving a higher number of patients and a more
sensitive and in particular more quantitative detection method such as qPCR. The PI-9 expression
level might then be an important marker for patient stratification in terms of personalized therapies
(see below).
Unexpectedly and in contrast to results obtained on cell lines, lower cytotoxic effects by
H22(scFv)-ETA’ were observed, which acts independently of PI-9, compared to Gb-H22(scFv) and
GbR201K-H22(scFv) ex vivo, even though the bacterial toxin-based IT has been proven to be highly
effective before in preclinical in vitro and in vivo studies [211]. The reason might be that, as described
previously for the similarly operating DT [215], cells were rather resistant to ETA’ (1.2.1) due to the
cells’ lack in proliferation ex vivo. This is especially characteristic for CMML, where even in vivo cells

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
106
exhibit low cell cycle activity. In addition, this finding and also the fact that compared to wild-type
Gb-H22(scFv) the other two CFPs GbR201K-H22(scFv) and Gm-H22(scFv) were partly more effective,
might indicate that ex vivo studies can be used as a predictive screening method to determine
optimal sensitivity of malignant cells from specific patients. This is one of the long-term aims in
personalized medicine: providing the correct drug to the right patient with the appropriate dose.
Over the past decade, the proof of this concept has been reported for imatinib, a competitive
tyrosine-kinase inhibitor, which has dramatically improved the prognosis of CML patients with BCR–
ABL translocation (‘breakpoint cluster region’-‘Abelson’, chromosomal defect in Philadelphia
chromosome) [216].
In summary, three major conclusions can be drawn from the here presented data: First, due to the
co-expression with several CMML specific markers like CD56, CD33 and CD14, CD64 is a potential
specific target for the treatment of CMML. Second, the applied CD64-targeting CFPs kill the target
cells in a selective manner whereby the PI-9 independent granzyme B mutant R201K is as active as its
wild-type on PI-9-negative cells but is superior if the target cells have up-regulated the inhibitor.
Third, the applied CFPs are superior in cell death-induction compared to the bacterial based IT
H22(scFv)-ETA’.
4.6 APPLICATION OF GRANZYME M AS AN ALTERNATIVE EFFECTOR MOLECULE IN
CYTOLYTIC FUSION PROTEINS
As described earlier (1.2.2), research on targeted immunotherapy focuses more and more on human
effector molecules fulfilling the cell-death inducing function instead of bacterial or plant toxins.
Proteins of the granzyme family are most promising candidates for immunotherapeutic approaches
and the successful application of granzyme B fusion proteins in immunotherapeutic approaches has
been discussed and investigated extensively in this study. Depending on treatment history and
developed, specific anti-apoptotic responses (1.4), it will be advantageous for individualized medicine
to extend the portfolio of highly efficient human effector molecules for immunotherapeutic
approaches. Hence, an alternative serine proteinase, granzyme M, was evaluated in this study for its
use as effector molecule in human CFPs. The cytotoxic potential of granzyme M has already been
confirmed independently by several groups (1.3.2) [49-55]. Its ability to kill tumor cells has previously
been described in vivo, whereby it was demonstrated that adoptively transferred NK cells from
granzyme M deficient mice were unable to inhibit tumor growth compared to wild-type NK cells [48].
In addition, the prospective of granzyme M is especially supported by the fact that it induces

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
107
apoptosis via distinct pathways compared to granzyme B and that it is not inhibited by PI-9, but
instead is able to inactivate the inhibitor (1.3.2).
Here, granzyme M was fused to two different well-established cancer cell-targeting antibody
fragments, H22(scFv) and Ki4(scFv). All constructs were successfully expressed secretory in
mammalian HEK293T cells to ensure correct post-translational modification, in contrast to other
groups which used yeast as an expression system. After purification higher band at 150 kDa was
observed during SDS-PAGE and western blot analysis (Figure 27B). Such a band was detected for the
granzyme B constructs as well, but since it was much weaker, it was not further investigated. In this
case, mass spectrometric analysis (performed in context of a master thesis) identified an additional
protein apart from granzyme M within this band, called α-2-macroglobulin precursor [217]. This is a
plasma protein, which is synthesized mainly in the liver, but also locally by macrophages, fibroblasts,
and adrenocortical cells; it is also a component of the medium supplement FCS and of human serum.
After secretory expression in HEK293T cells cultivated in synthetic, complete serum-free medium
(Panserin293S, PAN Biotech GmbH) this band disappeared in the purified sample (data not shown),
which confirmed the assumption that granzyme M complexes with α-2-macroglobulin from FCS. The
major function of α-2-macroglobulin is the inhibition of proteinases. It does not inactivate the
proteinase, but hinders the access of large substrates to its active site. This is accomplished via
cleavage of a so called ‘bait’ region in the structure of α-2-macroglobulin, located near the middle of
the polypeptide chain, which triggers a conformational change and consequent entrapment of the
proteinase [218]. An inhibition, however, was not observed in this study, as the constructs were still
highly enzymatically active as well as cytotoxic towards the target cells even if, according to the SDS-
PAGE, the complete fusion protein was trapped (only higher band visible and only weak band at
expected height). It could be the fact that the complex, whose half life has been described to be only
a few minutes [219], disintegrates before granzyme M is internalized into the targeted cells. This
suggests an advantage for later use in humans, as by complexing the granzyme M-based
immunotherapeutic with the human serum component α-2-macroglobulin, its serum stability could
potentially be increased as the plasma protein acts as a carrier after i. v. injection of the
immunotherapeutic. This, however, has to be further investigated to exclude unspecific binding,
internalization, and killing of non-target cells as the α-2-macroglobulin receptor is expressed in many
tissues and cells, especially in fibroblasts and hepatocytes [219].
4.6.1 Efficacy of granzyme M based CFPs
All generated and purified constructs were enzymatically active after purification as proved by
cleavage of recombinant PI-9 and a synthetic substrate (Figure 28) and bound specifically to their
respective target cells (Figure 29). Subsequently a cell-specific cytotoxic activity could be attested for

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
108
all constructs, whereas control cell lines remained unaffected (Figure 30 and Figure 31). To evaluate
the potential of granzyme M-based CFPs their cytotoxic effects were compared with the equivalent
constructs based on granzyme B. Thereby, it could be demonstrated that Gm-H22(scFv) triggers
comparable cytotoxic and apoptotic effects as Gb-H22(scFv) (Figure 12 and Figure 30A,C) in a specific
manner (Figure 30B). The same was true when granzyme M was fused to the epidermal growth
factor receptor (EGFR)-specific antibody 425(scFv), which has previously been fused to ETA' and the
efficacy of this construct has been demonstrated in vitro and in vivo [150,220]. Again Gm-425(scFv)
was as cytotoxic active as Gb-425(scFv) during viability as well as in apoptosis assays using the EGFR-
positive, PI-9-negative cell line A431 (data not shown). These results indicated that granzyme M has
indeed high potential as a novel effector molecule in CFPs.
Differences were observed for CD30-specific constructs. Here, 100x lower cytotoxic efficacy, was
observed for Gm-Ki4(scFv), which was in correlation with repeatedly observed lower apoptotic
effects during Annexin V/ PI assays (3.10.4). During later studies in context of a Master thesis,
however, the performance of both constructs was comparable, so the differences detected here
might be due to the protein preparation used. The observations on L540cy were independent of PI-9.
In presence of PI-9, Gm-Ki4(scFv) was advantageous regarding cell death induction compared to wild-
type Gb-Ki4(scFv) (Figure 31). Comparisons with GbR201K-Ki4(scFv) were not performed here due to
time restrictions. Even though the detected effects were low (compare explanations in 4.3.2), it still
suggested that granzyme M could be active despite of the ability of the target cells to express PI-9.
To further evaluate these findings, the cytotoxic potential of the CD64-targeting CFPs based on
granzyme M, granzyme B as well as granzyme B R201K, was comparatively tested on PI-9-positive
primary leukemic cells ex vivo (4.5.2). Hereby, due to a limited availability of patient material and the
delayed production of Gm-H22(scFv), only the last three patients could be considered, of which only
one was PI-9-positive. The obstacles which have to be overcome when working with primary cells
have already been discussed earlier (4.5). As shown for granzyme B constructs, Gm-H22(scFv) was
also able to bind CD64-positive AMML and CMML primary cells and selectively kill them (3.10.5).
When comparing the effects induced by Gm-H22(scFv), Gb-H22(scFv) and GbR201K-H22(scFv) in the
presence of PI-9, it became obvious that the two new constructs are both advantageous compared to
the wild-type Gb-H22(scFv) (Figure 32). Hence, as indicated on CD30-positive cell lines, the
insensitivity of granzyme M-based CFPs towards PI-9 could be confirmed.
In summary, apart from the verified ability to specifically kill cancerous cells in vitro and ex vivo,
granzyme M is obviously able to induce apoptosis in the presence of PI-9 to the same extent as the
new granzyme B mutant, whereas no difference in cytotoxicity between the granzyme B-based and
granzyme M-based constructs was seen in cells lacking PI-9. Consequently, granzyme M has the

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
109
potential to serve either as an alternative to granzyme B or to improve the therapeutic efficiency of
wild-type granzyme B by combinatorial immunotherapeutic treatment of PI-9 positive tumor entities.
These aspects are further discussed in the following section.
4.6.2 Clinical potential of granzyme M as effector molecule in cytolytic fusion proteins
Based on the above described findings, granzyme M can extent the palette of available human pro-
apoptotic compounds for CFPs, which can be of great importance in the context of immunotherapy
and especially for personalized medicine. Hereby, most effective cytolytic moieties and also best
binding ligands need to be selected during pre-tests to achieve optimal therapeutic outcome for the
patient. Preliminary indications for this have already been reported here during ex vivo studies by the
application of three different CFPs on primary patient material (Figure 32A,B). In addition,
granzyme M cannot be just valuable as such but also in combination with other immunotherapeutics,
especially granzyme B-based constructs. For the latter there are two rationales:
First, combination therapy with both granzymes has significant potential as these enzymes trigger
different apoptotic pathways. Details on granzyme B and granzyme M substrates have been
introduced in 1.3. The differences reflect the sequence specificity of each enzyme, i.e. methionine or
leucine for granzyme M [43] and aspartate or glutamate for granzyme B [34]. Hereby it is evident
that granzyme M is unique among the granzyme family, due to its specific expression by NK cells,
which may have phylogenetically evolved early to serve a specialized function in innate immunity
[45]. Furthermore it has been suggested that there are still unidentified substrates, which, in the
presence of the protease, transiently induce a functional active-site conformation and leads to even
more pathways for granzyme M to induce cell death [221]. Taken together, a combination of
granzyme B and granzyme M may increase the cytotoxicity by synergistic effects and reduce the
likelihood that cells can evolve resistance, e. g. by overexpression of particular inhibitors. Hereby, it
would be more effective to use different target receptors for each enzyme, to prevent competition
or steric hindrance of cell surface binding sites. Such combinations have been tested during a Master
thesis related to this work in vitro on L540cy cells, which express both HL-specific receptors CD30 and
CD25. These can be targeted by the already evaluated ligands Ki4(scFv) and RFT5(scFv) [222]
respectively. Results after combination treatment with Gm-Ki4(scFv) and GbR201K-RFT5n(scFv) or
GbR201K-Ki4(scFv) and GbR201K-RFT5n(scFv) indeed suggest synergistic effects of granzyme B and
granzyme M.
Second, the described ability of granzyme M to cleave PI-9 [56] holds potential for a combination
approach, since thereby it could prevent inhibition of granzyme B. The ability of the granzyme M
constructs to directly cleave PI-9 was only demonstrated in vitro using recombinant PI-9 during this
work (Figure 28A). Corresponding determinations should be done in the future by either incubating

Discussion ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
110
native cell lysates of PI-9-positive cells with a granzyme M based CFP or by cell lysis and subsequent
western blot analysis of those cells after treatment with a corresponding construct. In fact, the high
potential of this approach is supported by the finding, that the interaction between PI-9 and
granzyme M has a high stoichiometry of inhibition (SI) value of 63, which means that PI-9 is a much
better substrate than inhibitor of granzyme B [56]. By cleaving PI-9, granzyme M could therefore
clear the way for apoptosis induced by granzyme B. Hence, granzyme M could be useful, not only in a
therapeutic context, especially against treatment-resistant cells on its own, but also in combination
with granzyme B.
In contrast to granzyme B, granzyme M has not been investigated as extensively, so in the future
both promising, as well as adverse effects may be detected on its prospective role as
immunotherapeutic. Known so far is e.g. that on the down side, as described for granzyme B, a
specific endogenous inhibitor, called serpin B4, has recently been suggested to form a typical SDS-
stable serpin–protease complex with granzyme M in vitro and thereby inhibited NK cell-mediated cell
death in Hela cells [223]. However, the affinity between the two proteins is of a second-order rate
constant of 1.3 x 104 M-1 s-1 which is two orders of magnitude below that of granzyme B and PI-9 and,
in general, below the range of biologically-relevant interactions. This rather weak inhibition might be
overcome by CFP-related excessive delivery of the effector molecule to the cells. Furthermore, the in
vivo expression of this inhibitor has not been reported and any potential escape mechanisms in
tumors remain to be investigated. Other serpins present in plasma, like α1-antichymotrypsin and α1-
proteinase inhibitor, could also become a limiting factor for granzyme M activity in blood as
suggested earlier [56]. Their inhibitory relevance has to be evaluated in the future in serum-stability
assays, and respective modifications, as shown in this study for granzyme B, should be considered.
Other modifications, also in accordance to granzyme B, might involve the cationic sites within
granzyme M that may lead to off-target effects [221]. Hence, current literature on this topic should
be monitored carefully, as in vivo studies should be pursued to further evaluate the potential of
granzyme M in immunotherapeutic approaches.
Concluding, with the novel granzyme B mutant, identified during in silico homology modeling, it was
demonstrated on in vitro, in vivo as well as ex vivo level that residual therapy-resistant cancer cells
can be killed, provided the resistance is due to PI-9 expression as an escape mechanism. Additionally,
the cytotoxic potential and the PI-9 insensitivity of granzyme M-based fusion proteins was verified in
vitro and ex vivo clearing the way for a potential combination treatment with both granzymes.

Outlook ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
111
5 Outlook
The presented data emphasize the great efficacy of granzyme B-based cytolytic fusion proteins (CFPs)
in vitro, in vivo as well as ex vivo and especially verify the necessity and potential of directed
modifications, here to pave the way for inhibitor-insensitive treatment. On this basis further
investigations and optimizations are now ongoing in our group concerning the structure of the
corresponding CFPs as well as their extended application.
Structural modifications include the introduction of an additional modification to reduce the high
positive surface charge of granzyme B (pI ~ 10) which is mainly caused by two major heparan-binding
motifs and might result in off-target effects by intrinsic binding to heparin sulfate proteoglycans. In a
previous study, the basic amino acid residues involved in heparan sulfate binding were substituted
for alanine which did not interfere with ligand binding or enzyme activity and reduced the negative
impact of extracellular matrix effects [224]. Hence, specific cytotoxicity of granzyme B could be
increased by using this granzyme B variant. Furthermore, the improvement of endosomal release
after receptor mediated endocytosis within the target cells by certain adapter sequences is now
being investigated in-house (confidential) which suggest that the engineering of granzyme B-based
CFPs by the insertion of functional adapter elements will improve their therapeutic potential. As an
alternative, a pH-sensitive fusogenic peptide has been introduced recently which increased specific
cytotoxicity compared with the construct lacking the peptide, and demonstrated enhanced cellular
uptake and improved delivery of granzyme B to the cytosol of target cells [166]. In addition, another
promising mutational site is presently evaluated in our group which was also suggested from in silico
determinations to be promising to render granzyme B resistant to PI-9 inhibition. The potential of
structure related modifications of the human molecules, however, introduce novel sequences with
potential antigenicity in vivo which have to be evaluated carefully.
Further structural related improvements which are required for translation into clinical application to
reduce potential immunogenicity include either removal of the His6-tag after IMAC purification or
applying alternative purification strategies like ion exchange chromatography independent of any
tags. In addition, the applied murine Ki4(scFv) should either be humanized or replaced by the human
CD30L, which has been shown promising results in combination with human RNase angiogenin
before [187] to prevent HAMA response.
Regarding the extended application of the CFPs, the economic and up-scaled production of active
granzyme B and granzyme M has to be considered. Therefore the yeast Hansenula polymorpha is
now under investigation as a potential alternative expression system. It harbors the additional
advantage to become independent of expensive enterokinase processing of the granzymes by
insertion of the mating factor α prepro-leader sequence allowing straightforward purification of

Outlook ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
112
active granzyme from the culture supernatant based on local Kex2-protease activity in the trans-
Golgi compartment. Furthermore, expression of E. coli is presently explored to increase protein yield
with lower expenses.
The described ex vivo findings need to be verified on additional patient material and evaluated in
terms of personalized medicine. Those investigations will be most valuable in combination with
information on the previous treatment and anamnesis. To further evaluate CD64 as valuable
potential therapeutic target also for CMML and AMML in the clinical it will now be investigated if
CD64 is indeed over-expressed on bone marrow samples of AMML and CMML patients. A
corresponding set-up for immunohistochemical determinations with H22 antibody fragments is
already established.
In addition, further in vivo experiments are planned with the described constructs which include the
establishment of a disseminated tumor model using the AML cell line HL60. The same might be
indicated for Hodgkin lymphoma since for this disease a disseminated model is less artificial than the
subcutaneous tumor model applied in this study. Furthermore, the novel variant GbR201K is now
used in combination with several other binders to kill target cells despite the presence of
endogenous PI-9.
The CFP-based treatment of tumor cells, even in presence of PI-9, was shown to be feasible using the
new effector molecule granzyme M. Further combination experiments are planned to test the
potential of granzyme M to clear the way for granzyme B activity by inactivation of PI-9 within the
target cells. These studies will be performed as soon as an adequate PI-9-positive cell line with both
receptors co-expressed becomes available. In addition, the potential of granzyme M-based CFPs has
to be shown in vivo as well using the xenograft tumor models already established for the
granzyme B-based constructs. For both constructs the species-dependent substrate-specificity has to
be taken into account when these enzymes undergo further studies using other than xenograft
tumor models or the in our groups well established mouse inflammation model [213], especially
when proceeding towards clinical application. These specificities can significantly obscure e.g.
immunogenic or unspecific effects, so special care has to be taken when planning pre-clinical studies.
In accordance to granzyme B, novel variants for granzyme M independent of the plasma serpins α1-
antichymotrypsin, α1-proteinase inhibitor and the endogenous inhibitor serpin B4, if detected in vivo
as well, can also be established. Additionally, a granzyme M variant, which does not bind to α-2-
macroglobulin anymore, is planned to prevent its entrapment in human serum.
The here developed novel constructs may eventually be used in the clinic in combination with
conventional treatment approaches helping to reduce the necessary dose of non-specific drugs,
avoiding adverse effects and promoting long-term remission.

Summary ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
113
6 Summary
Current cancer treatments often lack specificity and therefore also kill healthy cells, resulting in
severe side effects. To improve the specificity of cancer treatment, recombinant chimeric proteins
such as immunotoxins have been developed, consisting of a tumor-selective binding domain and a
cytotoxic protein primarily of bacterial or plant origin. To circumvent immunogenic responses, recent
studies have focused on the development of fully human cytolytic fusion proteins (CFPs), containing
pro-apoptotic components of human origin. Most promising are enzymes from granule-associated
immune defense. The best studied is the serine protease granzyme B. However, its anti-tumor
efficacy is limited by irreversible inhibition by serpin B9 (PI-9) which is naturally co-expressed in the
cytosol of cytotoxic immune cells to avoid auto-toxicity by misdirected granzyme B. Several tumors
have been described to overexpress PI-9 as a mechanism for immune escape. As a very high number
of PI-9-positive tumor cells predicts unfavorable clinical outcome, the specific killing of these often
treatment-resistant cells may strongly increase therapeutic success and life span.
In this study different cell lines were found to express PI-9, including CD30-positive HL cell lines or
primary leukemic cells, which decreased the cytotoxic efficacy of granzyme B-based CFPs during
former studies. The aim of this work was therefore to improve the therapeutic potential of
granzyme B by the generation of PI-9 resistant mutants. Therefore the interacting amino acids
between granzyme B and PI-9 were elucidated by in silico simulations based on the known crystal
structure of granzyme B as well as a complex between rat trypsin and Manduca sexta serpin B1. Two
corresponding amino acids, R28 and R201, were identified and exchanged to Ala, Lys or Glu
respectively, by site-directed mutagenesis. The resulting eight mutants, including a double mutant
(R28A-R201A) and a published mutant (K27A), were further tested in vitro with regard to an
increased resistance against PI-9 inhibition. The new mutants were fused to H22(scFv) and Ki4(scFv)
which bind CD64 (high-affinity receptor for IgG, over-expressed e. g. on AML cells) and CD30 (cell
membrane protein of the tumor necrosis factor receptor family, over-expressed e. g. on HL cells)
respectively. Proteins were expressed in HEK293T cells and purified via affinity chromatography from
the supernatant. Three mutants were identified by enzymatic activity assays to be as active as the
wild-type in absence of PI-9 and especially retained most of their proteolytic activity after challenge
with recombinant PI-9. These mutants were comparable to the corresponding wild-type constructs in
respect to binding affinity (between 1.5 and 5.7 nM), cytotoxic and apoptotic activity (IC50 between
1.2 and 5.3 nM) as well as serum stability (6 - 10 h).
The mutant R201K demonstrated the best cytotoxic activity against both CD30-expressing PI-9-
positive and -negative tumor cells in vitro. To evaluate the influence of the mutation on the biological
activity appropriate xenograft tumor models were set up. For HL, only PI-9-negative L540cy cells are

Summary ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
114
currently established. In addition, a novel tumor model with the PI-9-expressing HL cell line L428 was
established here based on optical in vivo imaging. In these proof-of-principle models, it could be
verified in vivo that only the mutant is able to kill cells both in the presence and absence of the
inhibitor, this in contrast to the wild-type which only killed PI-9-negative L540cy tumor cells in vivo.
This is analogous to ex vivo studies with primary cells from patients with the rare diseases CMML and
AMML. These cells were targeted via the CD64 receptor. The data additionally demonstrated that the
new constructs perform even better than H22(scFv)-ETA’. The differences in apoptotic efficacy
between the applied constructs suggest ex vivo studies to be a predictive screening method for
personalized medicine to enable a patient-specific treatment selection.
In addition, a novel human granzyme-based CFP with a pro-apoptotic moiety based on granzyme M is
firstly described here. This serine protease is known to be able to specifically cleave and inactivate
PI-9. It may be a valuable addition to the palette of human effector molecules, in particular for the
development of personalized medicine in which the optimal cytolytic protein and targeting molecule
combination can be selected on a per-patient basis. Different fusion constructs, comprising
granzyme M targeting CD64- and CD30-positive cancer cell lines were generated, expressed and
purified analogous to the granzyme B-based constructs. During in vitro and ex vivo studies
granzyme M was proved to be a potent human anti-tumor agent (IC50 between 1.2 and 6.4 nM for
Gm-H22(scFv)), even in presence of PI-9. CFPs combining granzyme B and granzyme M offer potential
for improved therapeutic approaches by simultaneously triggering different apoptotic pathways to
circumvent possible overexpression of inhibitors within the target cell or by potentiating granzyme B
activity by inactivating PI-9.
In summary, during this thesis two new effector molecules, granzyme M and a PI-9 resistant variant
of granzyme B, granzyme B R201K, have successfully been generated, produced and evaluated in
vitro, ex vivo and in parts also in vivo in regard to their cytotoxic potential and for use in
immunotherapy to treat leukemic diseases or HL. These effector molecules, either individually or in
combination, also bear clinical relevance for the treatment of solid tumors or other hematological
disorders that potentially express PI-9 or selected PI-9-positive cells under immune surveillance or
tumor therapy.

Literature ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
115
7 Literature
1. HANAHAN, D. and WEINBERG, R.A., The hallmarks of cancer. Cell, 2000. 100(1): p. 57-70. 2. HANAHAN, D. and COUSSENS, L.M., Accessories to the crime: functions of cells recruited to the tumor
microenvironment. Cancer Cell, 2012. 21(3): p. 309-22. 3. HILLEN, T., ENDERLING, H., and HAHNFELDT, P., The tumor growth paradox and immune system-mediated
selection for cancer stem cells. Bull Math Biol, 2013. 75(1): p. 161-84. 4. HEHMANN-TITT, G., SCHIFFER, S., BERGES, N., MELMER, G., and BARTH, S., Improving the Therapeutic Potential of
Human Granzyme B for Targeted Cancer Therapy. Antibodies, 2013. 2(1): p. 19-49. 5. KLEBANOFF, C.A., ACQUAVELLA, N., YU, Z.Y., and RESTIFO, N.P., Therapeutic cancer vaccines: are we there yet?
Immunological Reviews, 2011. 239: p. 27-44. 6. KREITMAN, R.J., Immunotoxins for targeted cancer therapy. AAPS J, 2006. 8(3): p. 532-51. 7. VANNEMAN, M. and DRANOFF, G., Combining immunotherapy and targeted therapies in cancer treatment. Nat
Rev Cancer, 2012. 12(4): p. 237-51. 8. WALDMANN, T.A., Immunotherapy: past, present and future. Nat Med, 2003. 9(3): p. 269-77. 9. SCOTT, A.M., ALLISON, J.P., and WOLCHOK, J.D., Monoclonal antibodies in cancer therapy. Cancer Immun, 2012.
12: p. 14. 10. REICHERT, J.M., Antibody-based therapeutics to watch in 2011. MAbs, 2011. 3(1): p. 76-99. 11. SCHRAMA, D., REISFELD, R.A., and BECKER, J.C., Antibody targeted drugs as cancer therapeutics. Nat Rev Drug
Discov, 2006. 5(2): p. 147-59. 12. CARTER, P., Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer, 2001. 1(2): p. 118-29. 13. RYBAK, S.M., HOOGENBOOM, H.R., MEADE, H.M., RAUS, J.C., SCHWARTZ, D., and YOULE, R.J., Humanization of
immunotoxins. Proc Natl Acad Sci U S A, 1992. 89(8): p. 3165-9. 14. NATSUME, A., NIWA, R., and SATOH, M., Improving effector functions of antibodies for cancer treatment:
Enhancing ADCC and CDC. Drug Des Devel Ther, 2009. 3: p. 7-16. 15. CARTER, P.J. and SENTER, P.D., Antibody-drug conjugates for cancer therapy. Cancer Journal, 2008. 14(3): p. 154-
69. 16. LAMBERT, J.M., Drug-conjugated monoclonal antibodies for the treatment of cancer. Curr Opin Pharmacol, 2005.
5(5): p. 543-9. 17. YOUNES, A., YASOTHAN, U., and KIRKPATRICK, P., Brentuximab vedotin. Nat Rev Drug Discov, 2012. 11(1): p. 19-
20. 18. TRAYNOR, K., Ado-trastuzumab emtansine approved for advanced breast cancer. Am J Health Syst Pharm, 2013.
70(7): p. 562. 19. BROSS, P.F., BEITZ, J., CHEN, G., CHEN, X.H., DUFFY, E., KIEFFER, L., et al., Approval summary: gemtuzumab
ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res, 2001. 7(6): p. 1490-6. 20. KREITMAN, R.J., Recombinant toxins for the treatment of cancer. Curr Opin Mol Ther, 2003. 5(1): p. 44-51. 21. ALLEN, T.M., Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer, 2002. 2(10): p. 750-63. 22. PASTAN, I., HASSAN, R., FITZGERALD, D.J., and KREITMAN, R.J., Immunotoxin treatment of cancer. Annu Rev Med,
2007. 58: p. 221-37. 23. WINTER, G., GRIFFITHS, A.D., HAWKINS, R.E., and HOOGENBOOM, H.R., Making antibodies by phage display
technology. Annu Rev Immunol, 1994. 12: p. 433-55. 24. MADHUMATHI, J. and VERMA, R.S., Therapeutic targets and recent advances in protein immunotoxins. Curr Opin
Microbiol, 2012. 15(3): p. 300-9. 25. DANG, N.H., PRO, B., HAGEMEISTER, F.B., SAMANIEGO, F., JONES, D., SAMUELS, B.I., et al., Phase II trial of
denileukin diftitox for relapsed/refractory T-cell non-Hodgkin lymphoma. Br J Haematol, 2007. 136(3): p. 439-47. 26. HWANG, W.Y. and FOOTE, J., Immunogenicity of engineered antibodies. Methods, 2005. 36(1): p. 3-10. 27. JONES, P.T., DEAR, P.H., FOOTE, J., NEUBERGER, M.S., and WINTER, G., Replacing the complementarity-
determining regions in a human antibody with those from a mouse. Nature, 1986. 321(6069): p. 522-5. 28. MENORET, S., ISCACHE, A.L., TESSON, L., REMY, S., USAL, C., OSBORN, M.J., et al., Characterization of
immunoglobulin heavy chain knockout rats. Eur J Immunol, 2010. 40(10): p. 2932-41. 29. SIEGALL, C.B., HAGGERTY, H.G., WARNER, G.L., CHACE, D., MIXAN, B., LINSLEY, P.S., et al., Prevention of
immunotoxin-induced immunogenicity by coadministration with CTLA4Ig enhances antitumor efficacy. J Immunol, 1997. 159(10): p. 5168-73.
30. TSUTSUMI, Y., ONDA, M., NAGATA, S., LEE, B., KREITMAN, R.J., and PASTAN, I., Site-specific chemical modification
with polyethylene glycol of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and
reduces animal toxicity and immunogenicity. Proc Natl Acad Sci U S A, 2000. 97(15): p. 8548-53. 31. MATHEW, M. and VERMA, R.S., Humanized immunotoxins: a new generation of immunotoxins for targeted cancer
therapy. Cancer Sci, 2009. 100(8): p. 1359-65. 32. BOTS, M. and MEDEMA, J.P., Granzymes at a glance. J Cell Sci, 2006. 119(Pt 24): p. 5011-4. 33. DE POOT, S.A.H., WESTGEEST, M., HOSTETTER, D.R., VAN DAMME, P., PLASMAN, K., DEMEYER, K., et al., Human
and mouse granzyme M display divergent and species-specific substrate specificities. Biochem J 2011. 437: p. 431-42.

Literature ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
116
34. TRAPANI, J.A., Granzymes: a family of lymphocyte granule serine proteases. Genome Biol, 2001. 2(12): p. REVIEWS3014.
35. TRAPANI, J.A. and SUTTON, V.R., Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr Opin Immunol, 2003. 15(5): p. 533-43.
36. LANIER, L.L., NK cell recognition. Annu Rev Immunol, 2005. 23: p. 225-74. 37. KURSCHUS, F.C. and JENNE, D.E., Delivery and therapeutic potential of human granzyme B. Immunol Rev, 2010.
235(1): p. 159-71. 38. CHOWDHURY, D. and LIEBERMAN, J., Death by a thousand cuts: granzyme pathways of programmed cell death.
Annu Rev Immunol, 2008. 26: p. 389-420. 39. KAM, C.M., HUDIG, D., and POWERS, J.C., Granzymes (lymphocyte serine proteases): characterization with natural
and synthetic substrates and inhibitors. Biochim Biophys Acta, 2000. 1477(1-2): p. 307-23. 40. EDWARDS, K.M., DAVIS, J.E., BROWNE, K.A., SUTTON, V.R., and TRAPANI, J.A., Anti-viral strategies of cytotoxic T
lymphocytes are manifested through a variety of granule-bound pathways of apoptosis induction. Immunol Cell Biol, 1999. 77(1): p. 76-89.
41. ESTEBANEZ-PERPINA, E., FUENTES-PRIOR, P., BELORGEY, D., BRAUN, M., KIEFERSAUER, R., MASKOS, K., et al., Crystal structure of the caspase activator human granzyme B, a proteinase highly specific for an Asp-P1 residue. Biol Chem, 2000. 381(12): p. 1203-14.
42. ROTONDA, J., GARCIA-CALVO, M., BULL, H.G., GEISSLER, W.M., MCKEEVER, B.M., WILLOUGHBY, C.A., et al., The
three-dimensional structure of human granzyme B compared to caspase-3, key mediators of cell death with
cleavage specificity for aspartic acid in P1. Chem Biol, 2001. 8(4): p. 357-68. 43. SMYTH, M.J., O'CONNOR, M.D., TRAPANI, J.A., KERSHAW, M.H., and BRINKWORTH, R.I., A novel substrate-binding
pocket interaction restricts the specificity of the human NK cell-specific serine protease, Met-ase-1. J Immunol, 1996. 156(11): p. 4174-81.
44. BADE, B., BOETTCHER, H.E., LOHRMANN, J., HINK-SCHAUER, C., BRATKE, K., JENNE, D.E., et al., Differential
expression of the granzymes A, K and M and perforin in human peripheral blood lymphocytes. International Immunology, 2005. 17(11): p. 1419-28.
45. SAYERS, T.J., BROOKS, A.D., WARD, J.M., HOSHINO, T., BERE, W.E., WIEGAND, G.W., et al., The restricted
expression of granzyme M in human lymphocytes. J Immunol, 2001. 166(2): p. 765-71. 46. ANTHONY, D.A., ANDREWS, D.M., CHOW, M., WATT, S.V., HOUSE, C., AKIRA, S., et al., A role for granzyme M in
TLR4-driven inflammation and endotoxicosis. J Immunol, 2010. 185(3): p. 1794-803. 47. SUSANTO, O., TRAPANI, J.A., and BRASACCHIO, D., Controversies in granzyme biology. Tissue Antigens, 2012.
80(6): p. 477-87. 48. PEGRAM, H.J., HAYNES, N.M., SMYTH, M.J., KERSHAW, M.H., and DARCY, P.K., Characterizing the anti-tumor
function of adoptively transferred NK cells in vivo. Cancer Immunol Immunother, 2010. 59(8): p. 1235-46. 49. HU, D., LIU, S., SHI, L., LI, C., WU, L., and FAN, Z., Cleavage of survivin by Granzyme M triggers degradation of the
survivin-X-linked inhibitor of apoptosis protein (XIAP) complex to free caspase activity leading to cytolysis of target
tumor cells. J Biol Chem, 2010. 285(24): p. 18326-35. 50. HUA, G., ZHANG, Q., and FAN, Z., Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation
and protects cells from granzyme M-mediated apoptosis. J Biol Chem, 2007. 282(28): p. 20553-60. 51. WANG, S., XIA, P., SHI, L., and FAN, Z., FADD cleavage by NK cell granzyme M enhances its self-association to
facilitate procaspase-8 recruitment for auto-processing leading to caspase cascade. Cell Death Differ, 2012. 19(4): p. 605-15.
52. LU, H., HOU, Q., ZHAO, T., ZHANG, H., ZHANG, Q., WU, L., et al., Granzyme M directly cleaves inhibitor of caspase-
activated DNase (CAD) to unleash CAD leading to DNA fragmentation. J Immunol, 2006. 177(2): p. 1171-8. 53. BOVENSCHEN, N., DE KONING, P.J., QUADIR, R., BROEKHUIZEN, R., DAMEN, J.M., FROELICH, C.J., et al., NK cell
protease granzyme M targets alpha-tubulin and disorganizes the microtubule network. J Immunol, 2008. 180(12): p. 8184-91.
54. CULLEN, S.P., AFONINA, I.S., DONADINI, R., LUTHI, A.U., MEDEMA, J.P., BIRD, P.I., et al., Nucleophosmin is cleaved
and inactivated by the cytotoxic granule protease granzyme M during natural killer cell-mediated killing. J Biol Chem, 2009. 284(8): p. 5137-47.
55. KELLY, J.M., WATERHOUSE, N.J., CRETNEY, E., BROWNE, K.A., ELLIS, S., TRAPANI, J.A., et al., Granzyme M mediates
a novel form of perforin-dependent cell death. J Biol Chem, 2004. 279(21): p. 22236-42. 56. MAHRUS, S., KISIEL, W., and CRAIK, C.S., Granzyme M is a regulatory protease that inactivates proteinase inhibitor
9, an endogenous inhibitor of granzyme B. J Biol Chem, 2004. 279(52): p. 54275-82. 57. TENG, M.W., SWANN, J.B., KOEBEL, C.M., SCHREIBER, R.D., and SMYTH, M.J., Immune-mediated dormancy: an
equilibrium with cancer. J Leukoc Biol, 2008. 84(4): p. 988-93. 58. KIM, R., EMI, M., and TANABE, K., Cancer immunoediting from immune surveillance to immune escape.
Immunology, 2007. 121(1): p. 1-14. 59. NELSON, B.H., The impact of T-cell immunity on ovarian cancer outcomes. Immunol Rev, 2008. 222: p. 101-16. 60. YANG, L., PANG, Y., and MOSES, H.L., TGF-beta and immune cells: an important regulatory axis in the tumor
microenvironment and progression. Trends Immunol, 2010. 31(6): p. 220-7. 61. SHREE, T., OLSON, O.C., ELIE, B.T., KESTER, J.C., GARFALL, A.L., SIMPSON, K., et al., Macrophages and cathepsin
proteases blunt chemotherapeutic response in breast cancer. Genes Dev, 2011. 25(23): p. 2465-79. 62. VESELY, M.D., KERSHAW, M.H., SCHREIBER, R.D., and SMYTH, M.J., Natural innate and adaptive immunity to
cancer. Annu Rev Immunol, 2011. 29: p. 235-71.

Literature ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
117
63. CLEVERS, H., The cancer stem cell: premises, promises and challenges. Nat Med, 2011. 17(3): p. 313-9. 64. BOTS, M., L, V.A.N.B., RADEMAKER, M.T., OFFRINGA, R., and MEDEMA, J.P., Serpins prevent granzyme-induced
death in a species-specific manner. Immunol Cell Biol, 2006. 84(1): p. 79-86. 65. HUNTINGTON, J.A., READ, R.J., and CARRELL, R.W., Structure of a serpin-protease complex shows inhibition by
deformation. Nature, 2000. 407(6806): p. 923-6. 66. BLADERGROEN, B.A., STRIK, M.C., BOVENSCHEN, N., VAN BERKUM, O., SCHEFFER, G.L., MEIJER, C.J., et al., The
granzyme B inhibitor, protease inhibitor 9, is mainly expressed by dendritic cells and at immune-privileged sites. J Immunol, 2001. 166(5): p. 3218-25.
67. BUZZA, M.S., HOSKING, P., and BIRD, P.I., The granzyme B inhibitor, PI-9, is differentially expressed during
placental development and up-regulated in hydatidiform moles. Placenta, 2006. 27(1): p. 62-9. 68. KANNAN-THULASIRAMAN, P. and SHAPIRO, D.J., Modulators of inflammation use nuclear factor-kappa B and
activator protein-1 sites to induce the caspase-1 and granzyme B inhibitor, proteinase inhibitor 9. J Biol Chem, 2002. 277(43): p. 41230-9.
69. EL HADDAD, N., MOORE, R., HEATHCOTE, D., MOUNAYAR, M., AZZI, J., MFARREJ, B., et al., The novel role of
SERPINB9 in cytotoxic protection of human mesenchymal stem cells. J Immunol, 2011. 187(5): p. 2252-60. 70. HEUTINCK, K.M., KASSIES, J., FLORQUIN, S., TEN BERGE, I.J., HAMANN, J., and ROWSHANI, A.T., SerpinB9
expression in human renal tubular epithelial cells is induced by triggering of the viral dsRNA sensors TLR3, MDA5
and RIG-I. Nephrol Dial Transplant, 2012. 27(7): p. 2746-54. 71. ROWSHANI, A.T., STRIK, M.C., MOLENAAR, R., YONG, S.L., WOLBINK, A.M., BEMELMAN, F.J., et al., The granzyme
B inhibitor SERPINB9 (protease inhibitor 9) circulates in blood and increases on primary cytomegalovirus infection
after renal transplantation. J Infect Dis, 2005. 192(11): p. 1908-11. 72. CLASSEN, C.F., BIRD, P.I., and DEBATIN, K.M., Modulation of the granzyme B inhibitor proteinase inhibitor 9 (PI-9)
by activation of lymphocytes and monocytes in vitro and by Epstein-Barr virus and bacterial infection. Clin Exp Immunol, 2006. 143(3): p. 534-42.
73. BUZZA, M.S., HIRST, C.E., BIRD, C.H., HOSKING, P., MCKENDRICK, J., and BIRD, P.I., The granzyme B inhibitor, PI-9,
is present in endothelial and mesothelial cells, suggesting that it protects bystander cells during immune
responses. Cell Immunol, 2001. 210(1): p. 21-9. 74. BARRIE, M.B., STOUT, H.W., ABOUGERGI, M.S., MILLER, B.C., and THIELE, D.L., Antiviral cytokines induce hepatic
expression of the granzyme B inhibitors, proteinase inhibitor 9 and serine proteinase inhibitor 6. J Immunol, 2004. 172(10): p. 6453-9.
75. HORIE, O., SAIGO, K., MURAYAMA, T., and RYO, R., Differential expression of proteinase inhibitor-9 and granzyme
B mRNAs in activated immunocompetent cells. Tohoku J Exp Med, 2005. 205(2): p. 103-13. 76. HIRST, C.E., BUZZA, M.S., BIRD, C.H., WARREN, H.S., CAMERON, P.U., ZHANG, M., et al., The intracellular granzyme
B inhibitor, proteinase inhibitor 9, is up-regulated during accessory cell maturation and effector cell degranulation,
and its overexpression enhances CTL potency. J Immunol, 2003. 170(2): p. 805-15. 77. JIANG, X., ELLISON, S.J., ALARID, E.T., and SHAPIRO, D.J., Interplay between the levels of estrogen and estrogen
receptor controls the level of the granzyme inhibitor, proteinase inhibitor 9 and susceptibility to immune
surveillance by natural killer cells. Oncogene, 2007. 26(28): p. 4106-14. 78. ROWSHANI, A.T., FLORQUIN, S., BEMELMAN, F., KUMMER, J.A., HACK, C.E., and TEN BERGE, I.J., Hyperexpression
of the granzyme B inhibitor PI-9 in human renal allografts: a potential mechanism for stable renal function in
patients with subclinical rejection. Kidney Int, 2004. 66(4): p. 1417-22. 79. WALKER, P.R., SAAS, P., and DIETRICH, P.Y., Role of Fas ligand (CD95L) in immune escape: the tumor cell strikes
back. J Immunol, 1997. 158(10): p. 4521-4. 80. MEDEMA, J.P., DE JONG, J., PELTENBURG, L.T., VERDEGAAL, E.M., GORTER, A., BRES, S.A., et al., Blockade of the
granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a
mechanism for immune escape by tumors. Proc Natl Acad Sci U S A, 2001. 98(20): p. 11515-20. 81. GETTINS, P., PATSTON, P.A., and SCHAPIRA, M., The role of conformational change in serpin structure and
function. Bioessays, 1993. 15(7): p. 461-7. 82. SUN, J., BIRD, C.H., SUTTON, V., MCDONALD, L., COUGHLIN, P.B., DE JONG, T.A., et al., A cytosolic granzyme B
inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J Biol Chem, 1996. 271(44): p. 27802-9.
83. POE, M., BLAKE, J.T., BOULTON, D.A., GAMMON, M., SIGAL, N.H., WU, J.K., et al., Human cytotoxic lymphocyte
granzyme B. Its purification from granules and the characterization of substrate and inhibitor specificity. J Biol Chem, 1991. 266(1): p. 98-103.
84. HEDSTROM, L., Serine protease mechanism and specificity. Chem Rev, 2002. 102(12): p. 4501-24. 85. MARSZAL, E. and SHRAKE, A., Serpin crystal structure and serpin polymer structure. Arch Biochem Biophys, 2006.
453(1): p. 123-9. 86. SUN, J., WHISSTOCK, J.C., HARRIOTT, P., WALKER, B., NOVAK, A., THOMPSON, P.E., et al., Importance of the P4'
residue in human granzyme B inhibitors and substrates revealed by scanning mutagenesis of the proteinase
inhibitor 9 reactive center loop. J Biol Chem, 2001. 276(18): p. 15177-84. 87. YE, S., CECH, A.L., BELMARES, R., BERGSTROM, R.C., TONG, Y., COREY, D.R., et al., The structure of a Michaelis
serpin-protease complex. Nat Struct Biol, 2001. 8(11): p. 979-83. 88. HODGKIN, T., On some Morbid Appearances of the Absorbent Glands and Spleen. Med Chir Trans, 1832. 17: p. 68-
114.

Literature ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
118
89. REED, W., Recent Researches concerning the Etiology, Propagation, and Prevention of Yellow Fever, by the United
States Army Commission. J Hyg (Lond), 1902. 2(2): p. 101-19. 90. STERNBERG, C., Über eine eigenartige unter dem Bilde der Pseudoleukämie verlaufende Tuberkolose des
lymphatischen Apparates. Z. Heilkunde, 1898. 19: p. 21-90. 91. KUPPERS, R., The biology of Hodgkin's lymphoma. Nat Rev Cancer, 2009. 9(1): p. 15-27. 92. KUPPERS, R., Molecular biology of Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program, 2009: p. 491-
6. 93. IZBAN, K.F., ERGIN, M., HUANG, Q., QIN, J.Z., MARTINEZ, R.L., SCHNITZER, B., et al., Characterization of NF-kappaB
expression in Hodgkin's disease: inhibition of constitutively expressed NF-kappaB results in spontaneous caspase-
independent apoptosis in Hodgkin and Reed-Sternberg cells. Mod Pathol, 2001. 14(4): p. 297-310. 94. AGGARWAL, B.B., Nuclear factor-kappaB: the enemy within. Cancer Cell, 2004. 6(3): p. 203-8. 95. PAHL, H.L., Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene, 1999. 18(49): p. 6853-
66. 96. KHAN, G., Epstein-Barr virus, cytokines, and inflammation: a cocktail for the pathogenesis of Hodgkin's
lymphoma? Exp Hematol, 2006. 34(4): p. 399-406. 97. BOLL, B., HANSEN, H., HEUCK, F., REINERS, K., BORCHMANN, P., ROTHE, A., et al., The fully human anti-CD30
antibody 5F11 activates NF-{kappa}B and sensitizes lymphoma cells to bortezomib-induced apoptosis. Blood, 2005. 106(5): p. 1839-42.
98. HIRSCH, B., HUMMEL, M., BENTINK, S., FOULADI, F., SPANG, R., ZOLLINGER, R., et al., CD30-induced signaling is
absent in Hodgkin's cells but present in anaplastic large cell lymphoma cells. Am J Pathol, 2008. 172(2): p. 510-20. 99. HORIE, R., HIGASHIHARA, M., and WATANABE, T., Hodgkin's lymphoma and CD30 signal transduction. Int J
Hematol, 2003. 77(1): p. 37-47. 100. HORIE, R., WATANABE, T., MORISHITA, Y., ITO, K., ISHIDA, T., KANEGAE, Y., et al., Ligand-independent signaling by
overexpressed CD30 drives NF-kappaB activation in Hodgkin-Reed-Sternberg cells. Oncogene, 2002. 21(16): p. 2493-503.
101. ANAGNOSTOPOULOS, I., HANSMANN, M.L., FRANSSILA, K., HARRIS, M., HARRIS, N.L., JAFFE, E.S., et al., European
Task Force on Lymphoma project on lymphocyte predominance Hodgkin disease: histologic and immunohistologic
analysis of submitted cases reveals 2 types of Hodgkin disease with a nodular growth pattern and abundant
lymphocytes. Blood, 2000. 96(5): p. 1889-99. 102. CHANG, K.C., KHEN, N.T., JONES, D., and SU, I.J., Epstein-Barr virus is associated with all histological subtypes of
Hodgkin lymphoma in Vietnamese children with special emphasis on the entity of lymphocyte predominance
subtype. Hum Pathol, 2005. 36(7): p. 747-55. 103. ANSELL, S.M., Brentuximab vedotin: delivering an antimitotic drug to activated lymphoma cells. Expert Opin
Investig Drugs, 2011. 20(1): p. 99-105. 104. BRENNER, H., GONDOS, A., and ARNDT, V., Recent major progress in long-term cancer patient survival disclosed by
modeled period analysis. J Clin Oncol, 2007. 25(22): p. 3274-80. 105. GRUSS, H.J., PINTO, A., GLOGHINI, A., WEHNES, E., WRIGHT, B., BOIANI, N., et al., CD30 ligand expression in
nonmalignant and Hodgkin's disease-involved lymphoid tissues. Am J Pathol, 1996. 149(2): p. 469-81. 106. SCHWAB, U., STEIN, H., GERDES, J., LEMKE, H., KIRCHNER, H., SCHAADT, M., et al., Production of a monoclonal
antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin's disease and a subset of normal lymphoid cells. Nature, 1982. 299(5878): p. 65-7.
107. LOCKSLEY, R.M., KILLEEN, N., and LENARDO, M.J., The TNF and TNF receptor superfamilies: integrating
mammalian biology. Cell, 2001. 104(4): p. 487-501. 108. HANSEN, H.P., RECKE, A., REINEKE, U., VON TRESCKOW, B., BORCHMANN, P., VON STRANDMANN, E.P., et al., The
ectodomain shedding of CD30 is specifically regulated by peptide motifs in its cysteine-rich domains 2 and 5. FASEB J, 2004. 18(7): p. 893-5.
109. HUHN, M., SASSE, S., TUR, M.K., MATTHEY, B., SCHINKOTHE, T., RYBAK, S.M., et al., Human angiogenin fused to
human CD30 ligand (Ang-CD30L) exhibits specific cytotoxicity against CD30-positive lymphoma. Cancer Res, 2001. 61(24): p. 8737-42.
110. HIRSCH, B., BRAUER, J., FISCHDICK, M., LODDENKEMPER, C., BULFONE-PAUS, S., STEIN, H., et al., Anti-CD30
human IL-2 fusion proteins display strong and specific cytotoxicity in vivo. Curr Drug Targets, 2009. 10(2): p. 110-7. 111. HORN-LOHRENS, O., TIEMANN, M., LANGE, H., KOBARG, J., HAFNER, M., HANSEN, H., et al., Shedding of the
soluble form of CD30 from the Hodgkin-analogous cell line L540 is strongly inhibited by a new CD30-specific
antibody (Ki-4). Int J Cancer, 1995. 60(4): p. 539-44. 112. BARTH, S., HUHN, M., MATTHEY, B., TAWADROS, S., SCHNELL, R., SCHINKOTHE, T., et al., Ki-4(scFv)-ETA', a new
recombinant anti-CD30 immunotoxin with highly specific cytotoxic activity against disseminated Hodgkin tumors
in SCID mice. Blood, 2000. 95(12): p. 3909-14. 113. VARDIMAN, J.W., IMBERT, M., PIERRE, R., BRUNNING, R.D., BAIN, B., and FLANDRIN, G., Chronic myelomonocytic
leukaemia. In: Jaffe ES, Harris NL, Sten H, Vardiman JW (eds). World Health Organization Classification of Tumors: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues., 2001: p. IARC Press: Lyon, pp 49-52.
114. VARDIMAN, J.W., HARRIS, N.L., and BRUNNING, R.D., The World Health Organization (WHO) classification of the
myeloid neoplasms. Blood, 2002. 100(7): p. 2292-302. 115. ORAZI, A. and GERMING, U., The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with
dysplastic features. Leukemia, 2008. 22(7): p. 1308-19.

Literature ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
119
116. XU, Y., MCKENNA, R.W., KARANDIKAR, N.J., PILDAIN, A.J., and KROFT, S.H., Flow cytometric analysis of monocytes
as a tool for distinguishing chronic myelomonocytic leukemia from reactive monocytosis. Am J Clin Pathol, 2005. 124(5): p. 799-806.
117. MESAROSOVA, A., HRIVNAKOVA, A., KLOBUSICKA, M., and BABUSIKOVA, O., Chronic myeloid leukemia:
correlation between purine metabolism enzyme activities and membrane immunophenotype. Neoplasma, 1995. 42(1): p. 9-14.
118. ROLLINS-RAVAL, M.A. and ROTH, C.G., The value of immunohistochemistry for CD14, CD123, CD33,
myeloperoxidase and CD68R in the diagnosis of acute and chronic myelomonocytic leukaemias. Histopathology, 2012. 60(6): p. 933-42.
119. ORAZI, A., CHIU, R., O'MALLEY, D.P., CZADER, M., ALLEN, S.L., AN, C., et al., Chronic myelomonocytic leukemia: The
role of bone marrow biopsy immunohistology. Mod Pathol, 2006. 19(12): p. 1536-45. 120. FENAUX, P., BEUSCART, R., LAI, J.L., JOUET, J.P., and BAUTERS, F., Prognostic factors in adult chronic
myelomonocytic leukemia: an analysis of 107 cases. J Clin Oncol, 1988. 6(9): p. 1417-24. 121. ONIDA, F., BALL, G., KANTARJIAN, H.M., SMITH, T.L., GLASSMAN, A., ALBITAR, M., et al., Characteristics and
outcome of patients with Philadelphia chromosome negative, bcr/abl negative chronic myelogenous leukemia. Cancer, 2002. 95(8): p. 1673-84.
122. GERMING, U. and NEUKIRCHEN, J., How to treat patients with CMML? Leuk Res, 2013. 37(6): p. 605-6. 123. BACHER, U., HAFERLACH, T., SCHNITTGER, S., KREIPE, H., and KROGER, N., Recent advances in diagnosis,
molecular pathology and therapy of chronic myelomonocytic leukaemia. Br J Haematol, 2011. 153(2): p. 149-67. 124. DUNPHY, C.H. and TANG, W., The value of CD64 expression in distinguishing acute myeloid leukemia with
monocytic differentiation from other subtypes of acute myeloid leukemia: a flow cytometric analysis of 64 cases. Arch Pathol Lab Med, 2007. 131(5): p. 748-54.
125. THEPEN, T., HUHN, M., MELMER, G., TUR, M.K., and BARTH, S., Fcgamma receptor 1 (CD64), a target beyond
cancer. Curr Pharm Des, 2009. 15(23): p. 2712-8. 126. FANGER, M.W., SHEN, L., GRAZIANO, R.F., and GUYRE, P.M., Cytotoxicity mediated by human Fc receptors for IgG.
Immunol Today, 1989. 10(3): p. 92-9. 127. THEPEN, T., VAN VUUREN, A.J.H., KIEKENS, R.C.M., DAMEN, C.A., VOOIJS, W.C., and VAN DE WINKEL, J.G.J.,
Resolution of cutaneous inflammation after local elimination of macrophages. Nature Biotechnology, 2000. 18(1): p. 48-51.
128. HRISTODOROV, D., MLADENOV, R., HUHN, M., BARTH, S., and THEPEN, T., Macrophage-Targeted Therapy: CD64-
Based Immunotoxins for Treatment of Chronic Inflammatory Diseases. Toxins (Basel), 2012. 4(9): p. 676-94. 129. ZHONG, R.K., VAN DE WINKEL, J.G., THEPEN, T., SCHULTZ, L.D., and BALL, E.D., Cytotoxicity of anti-CD64-ricin a
chain immunotoxin against human acute myeloid leukemia cells in vitro and in SCID mice. J Hematother Stem Cell Res, 2001. 10(1): p. 95-105.
130. VAN ROON, J.A., VAN VUUREN, A.J., WIJNGAARDEN, S., JACOBS, K.M., BIJLSMA, J.W., LAFEBER, F.P., et al., Selective elimination of synovial inflammatory macrophages in rheumatoid arthritis by an Fcgamma receptor I-
directed immunotoxin. Arthritis Rheum, 2003. 48(5): p. 1229-38. 131. DE KRUIF, J., TIJMENSEN, M., GOLDSEIN, J., and LOGTENBERG, T., Recombinant lipid-tagged antibody fragments
as functional cell-surface receptors. Nat Med, 2000. 6(2): p. 223-7. 132. GRAZIANO, R.F., TEMPEST, P.R., WHITE, P., KELER, T., DEO, Y., GHEBREMARIAM, H., et al., Construction and
characterization of a humanized anti-gamma-Ig receptor type I (Fc gamma RI) monoclonal antibody. J Immunol, 1995. 155(10): p. 4996-5002.
133. TUR, M.K., HUHN, M., THEPEN, T., STOCKER, M., KROHN, R., VOGEL, S., et al., Recombinant CD64-specific single
chain immunotoxin exhibits specific cytotoxicity against acute myeloid leukemia cells. Cancer Res, 2003. 63(23): p. 8414-9.
134. BOGAN, A.A. and THORN, K.S., Anatomy of hot spots in protein interfaces. J Mol Biol, 1998. 280(1): p. 1-9. 135. CLACKSON, T. and WELLS, J.A., A hot spot of binding energy in a hormone-receptor interface. Science, 1995.
267(5196): p. 383-6. 136. KORTEMME, T. and BAKER, D., A simple physical model for binding energy hot spots in protein-protein complexes.
Proceedings of the National Academy of Sciences of the United States of America, 2002. 99(22): p. 14116-21. 137. STOCKER, M., TUR, M.K., SASSE, S., KRUSSMANN, A., BARTH, S., and ENGERT, A., Secretion of functional anti-
CD30-angiogenin immunotoxins into the supernatant of transfected 293T-cells. Protein Expr Purif, 2003. 28(2): p. 211-9.
138. STAHNKE, B., THEPEN, T., STOCKER, M., ROSINKE, R., JOST, E., FISCHER, R., et al., Granzyme B-H22(scFv), a human
immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Molecular Cancer Therapeutics, 2008. 7(9): p. 2924-32.
139. BALL, E.D., MCDERMOTT, J., GRIFFIN, J.D., DAVEY, F.R., DAVIS, R., and BLOOMFIELD, C.D., Expression of the three
myeloid cell-associated immunoglobulin G Fc receptors defined by murine monoclonal antibodies on normal bone
marrow and acute leukemia cells. Blood, 1989. 73(7): p. 1951-6. 140. VON KALLE, C., WOLF, J., BECKER, A., SCKAER, A., MUNCK, M., ENGERT, A., et al., Growth of Hodgkin cell lines in
severely combined immunodeficient mice. Int J Cancer, 1992. 52(6): p. 887-91. 141. SAMBROOK, J. and RUSSELL, D.W., Molecular cloning: a laboratory manual. . 3rd ed. 2001, Cold Spring Harbor
(NY): Cold Spring Harbor Laboratory. 142. HANAHAN, D., Studies on transformation of Escherichia coli with plasmids. J Mol Biol, 1983. 166(4): p. 557-80.

Literature ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
120
143. LAEMMLI, U.K., Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 1970. 227(5259): p. 680-5.
144. BARTH, S., HUHN, M., MATTHEY, B., KLIMKA, A., GALINSKI, E.A., and ENGERT, A., Compatible-solute-supported
periplasmic expression of functional recombinant proteins under stress conditions. Appl Environ Microbiol, 2000. 66(4): p. 1572-9.
145. KAMPMEIER, F., RIBBERT, M., NACHREINER, T., DEMBSKI, S., BEAUFILS, F., BRECHT, A., et al., Site-specific, covalent
labeling of recombinant antibody fragments via fusion to an engineered version of 6-O-alkylguanine DNA
alkyltransferase. Bioconjug Chem, 2009. 20(5): p. 1010-5. 146. VON TRESCKOW, B., KALLEN, K.J., VON STRANDMANN, E.P., BORCHMANN, P., LANGE, H., ENGERT, A., et al.,
Depletion of cellular cholesterol and lipid rafts increases shedding of CD30. J Immunol, 2004. 172(7): p. 4324-31. 147. STOCKER, M., PARDO, A., HETZEL, C., REUTELINGSPERGER, C., FISCHER, R., and BARTH, S., Eukaryotic expression
and secretion of EGFP-labeled annexin A5. Protein Expr Purif, 2008. 58(2): p. 325-31. 148. GODAL, R., KEILHOLZ, U., UHAREK, L., LETSCH, A., ASEMISSEN, A.M., BUSSE, A., et al., Lymphomas are sensitive to
perforin-dependent cytotoxic pathways despite expression of PI-9 and overexpression of bcl-2. Blood, 2006. 107(8): p. 3205-11.
149. LOSASSO, V., SCHIFFER, S., BARTH, S., and CARLONI, P., Design of human granzyme B variants resistant to serpin
B9. Proteins, 2012. 80(11): p. 2514-22. 150. PARDO, A., STOCKER, M., KAMPMEIER, F., MELMER, G., FISCHER, R., THEPEN, T., et al., In vivo imaging of
immunotoxin treatment using Katushka-transfected A-431 cells in a murine xenograft tumour model. Cancer Immunol Immunother, 2012. 61(10): p. 1617-26.
151. SALOMON, D.S., BRANDT, R., CIARDIELLO, F., and NORMANNO, N., Epidermal growth factor-related peptides and
their receptors in human malignancies. Crit Rev Oncol Hematol, 1995. 19(3): p. 183-232. 152. TUR, M.K., NEEF, I., JAGER, G., TEUBNER, A., STOCKER, M., MELMER, G., et al., Immunokinases, a novel class of
immunotherapeutics for targeted cancer therapy. Curr Pharm Des, 2009. 15(23): p. 2693-9. 153. ROSENBLUM, M.G. and BARTH, S., Development of novel, highly cytotoxic fusion constructs containing granzyme
B: unique mechanisms and functions. Curr Pharm Des, 2009. 15(23): p. 2676-92. 154. SENNVIK, K., BOEKHOORN, K., LASRADO, R., TERWEL, D., VERHAEGHE, S., KORR, H., et al., Tau-4R suppresses
proliferation and promotes neuronal differentiation in the hippocampus of tau knockin/knockout mice. FASEB J, 2007. 21(9): p. 2149-61.
155. GRULLICH, C., FRITSCH, K., and FINKE, J., Constitutive Expression of the Granzyme B Inhibitor PI-9 Protects
Leukemic Cells from Granzyme B Induced Apoptosis. Blood (ASH Annual Meeting Abstracts), 2005. 106(Abstract 3030).
156. CLASSEN, C.F., USHMOROV, A., BIRD, P., and DEBATIN, K.M., The granzyme B inhibitor PI-9 is differentially
expressed in all main subtypes of pediatric acute lymphoblastic leukemias. Haematologica, 2004. 89(11): p. 1314-21.
157. SCHIFFER, S., HANSEN, H., HEHMANN-TITT, G., HUHN, M., FISCHER, R., BARTH, S., et al., Efficacy of an adapted
Granzyme B-based anti-CD30 cytolytic fusion protein against PI-9-positive classical Hodgkin lymphoma cells in a
murine model. Blood Cancer Journal, 2013. 3: p. e106. 158. LIU, Y., CHEUNG, L.H., HITTELMAN, W.N., and ROSENBLUM, M.G., Targeted delivery of human pro-apoptotic
enzymes to tumor cells: In vitro studies describing a novel class of recombinant highly cytotoxic agents. Mol Cancer Ther, 2003. 2(12): p. 1341-50.
159. LIU, Y., ZHANG, W., NIU, T., CHEUNG, L.H., MUNSHI, A., MEYN, R.E., JR., et al., Targeted apoptosis activation with
GrB/scFvMEL modulates melanoma growth, metastatic spread, chemosensitivity, and radiosensitivity. Neoplasia, 2006. 8(2): p. 125-35.
160. DALKEN, B., GIESUBEL, U., KNAUER, S.K., and WELS, W.S., Targeted induction of apoptosis by chimeric granzyme B
fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ, 2006. 13(4): p. 576-85.
161. KURSCHUS, F.C., KLEINSCHMIDT, M., FELLOWS, E., DORNMAIR, K., RUDOLPH, R., LILIE, H., et al., Killing of target
cells by redirected granzyme B in the absence of perforin. FEBS Lett, 2004. 562(1-3): p. 87-92. 162. LIU, Y., CHEUNG, L.H., THORPE, P., and ROSENBLUM, M.G., Mechanistic studies of a novel human fusion toxin
composed of vascular endothelial growth factor (VEGF)121 and the serine protease granzyme B: directed
apoptotic events in vascular endothelial cells. Mol Cancer Ther, 2003. 2(10): p. 949-59. 163. ZHANG, L., ZHAO, J., WANG, T., YU, C.J., JIA, L.T., DUAN, Y.Y., et al., HER2-targeting recombinant protein with
truncated pseudomonas exotoxin A translocation domain efficiently kills breast cancer cells. Cancer Biol Ther, 2008. 7(8): p. 1226-31.
164. WANG, T., ZHAO, J., REN, J.L., ZHANG, L., WEN, W.H., ZHANG, R., et al., Recombinant immunoproapoptotic
proteins with furin site can translocate and kill HER2-positive cancer cells. Cancer Res, 2007. 67(24): p. 11830-9. 165. KANATANI, I., LIN, X., YUAN, X., MANOREK, G., SHANG, X., CHEUNG, L.H., et al., Targeting granzyme B to tumor
cells using a yoked human chorionic gonadotropin. Cancer Chemother Pharmacol, 2011. 68(4): p. 979-90. 166. CAO, Y., MOHAMEDALI, K.A., MARKS, J.W., CHEUNG, L.H., HITTELMAN, W.N., and ROSENBLUM, M.G.,
Construction and Characterization of Novel, Completely Human Serine Protease Therapeutics Targeting Her2/neu. Mol Cancer Ther, 2013.
167. BUEHRING, G.C., EBY, E.A., and EBY, M.J., Cell line cross-contamination: how aware are Mammalian cell culturists
of the problem and how to monitor it? In Vitro Cell Dev Biol Anim, 2004. 40(7): p. 211-5.

Literature ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
121
168. BIRD, C.H., SUTTON, V.R., SUN, J., HIRST, C.E., NOVAK, A., KUMAR, S., et al., Selective regulation of apoptosis: the
cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without
perturbing the Fas cell death pathway. Mol Cell Biol, 1998. 18(11): p. 6387-98. 169. RAY, M., HOSTETTER, D.R., LOEB, C.R., SIMKO, J., and CRAIK, C.S., Inhibition of Granzyme B by PI-9 protects
prostate cancer cells from apoptosis. Prostate, 2012. 72(8): p. 846-55. 170. SORIANO, C., MUKARO, V., HODGE, G., AHERN, J., HOLMES, M., JERSMANN, H., et al., Increased proteinase
inhibitor-9 (PI-9) and reduced granzyme B in lung cancer: mechanism for immune evasion? Lung Cancer, 2012. 77(1): p. 38-45.
171. BLADERGROEN, B.A., MEIJER, C.J., TEN BERGE, R.L., HACK, C.E., MURIS, J.J., DUKERS, D.F., et al., Expression of the
granzyme B inhibitor, protease inhibitor 9, by tumor cells in patients with non-Hodgkin and Hodgkin lymphoma: a
novel protective mechanism for tumor cells to circumvent the immune system? Blood, 2002. 99(1): p. 232-7. 172. TIACCI, E., DORING, C., BRUNE, V., VAN NOESEL, C.J., KLAPPER, W., MECHTERSHEIMER, G., et al., Analyzing
primary Hodgkin and Reed-Sternberg cells to capture the molecular and cellular pathogenesis of classical Hodgkin
lymphoma. Blood, 2012. 120(23): p. 4609-20. 173. TEN BERGE, R.L., MEIJER, C.J., DUKERS, D.F., KUMMER, J.A., BLADERGROEN, B.A., VOS, W., et al., Expression levels
of apoptosis-related proteins predict clinical outcome in anaplastic large cell lymphoma. Blood, 2002. 99(12): p. 4540-6.
174. ROUSALOVA, I., KREPELA, E., PROCHAZKA, J., CERMAK, J., and BENKOVA, K., Expression of proteinase inhibitor-
9/serpinB9 in non-small cell lung carcinoma cells and tissues. Int J Oncol, 2010. 36(1): p. 275-83. 175. JIANG, X.G., PATTERSON, N.M., LING, Y., XIE, J.W., HELFERICH, W.G., and SHAPIRO, D.J., Low Concentrations of the
Soy Phytoestrogen Genistein Induce Proteinase Inhibitor 9 and Block Killing of Breast Cancer Cells by Immune Cells. Endocrinology, 2008. 149(11): p. 5366-73.
176. VAN HOUDT, I.S., OUDEJANS, J.J., VAN DEN EERTWEGH, A.J., BAARS, A., VOS, W., BLADERGROEN, B.A., et al., Expression of the apoptosis inhibitor protease inhibitor 9 predicts clinical outcome in vaccinated patients with
stage III and IV melanoma. Clin Cancer Res, 2005. 11(17): p. 6400-7. 177. OUDEJANS, J.J., HARIJADI, H., KUMMER, J.A., TAN, I.B., BLOEMENA, E., MIDDELDORP, J.M., et al., High numbers of
granzyme B/CD8-positive tumour-infiltrating lymphocytes in nasopharyngeal carcinoma biopsies predict rapid
fatal outcome in patients treated with curative intent. J Pathol, 2002. 198(4): p. 468-75. 178. CHAPPELOW, A.V. and KAISER, P.K., Neovascular age-related macular degeneration: potential therapies. Drugs,
2008. 68(8): p. 1029-36. 179. MADISON, E.L., GOLDSMITH, E.J., GERARD, R.D., GETHING, M.J., and SAMBROOK, J.F., Serpin-resistant mutants of
human tissue-type plasminogen activator. Nature, 1989. 339(6227): p. 721-4. 180. FRITSCH, K., PhD Thesis: Die Bedeutung des Proteinase Inhibitor 9 für die Apoptoseresistenz in leukämischen
Zelllinien, in Medicinal faculty 2008, Albert-Ludwigs-University, Freiburg im Breisgau. 181. BOSSARD, C., BELHADJ, K., REYES, F., MARTIN-GARCIA, N., BERGER, F., KUMMER, J.A., et al., Expression of the
granzyme B inhibitor PI9 predicts outcome in nasal NK/T-cell lymphoma: results of a Western series of 48 patients
treated with first-line polychemotherapy within the Groupe d'Etude des Lymphomes de l'Adulte (GELA) trials. Blood, 2007. 109(5): p. 2183-9.
182. KORTEMME, T., KIM, D.E., and BAKER, D., Computational alanine scanning of protein-protein interfaces. Sci STKE, 2004. 2004(219): p. pl2.
183. ANDRUSIER, N., MASHIACH, E., NUSSINOV, R., and WOLFSON, H.J., Principles of flexible protein-protein docking. Proteins, 2008. 73(2): p. 271-89.
184. GEHRMANN, M., DOSS, B.T., WAGNER, M., ZETTLITZ, K.A., KONTERMANN, R.E., FOULDS, G., et al., A novel
expression and purification system for the production of enzymatic and biologically active human granzyme B. J Immunol Methods, 2011. 371(1-2): p. 8-17.
185. GRIFFITHS, G.M. and ISAAZ, S., Granzymes A and B are targeted to the lytic granules of lymphocytes by the
mannose-6-phosphate receptor. J Cell Biol, 1993. 120(4): p. 885-96. 186. BRETTHAUER, R.K. and CASTELLINO, F.J., Glycosylation of Pichia pastoris-derived proteins. Biotechnol Appl
Biochem, 1999. 30 ( Pt 3): p. 193-200. 187. KLIMKA, A., MATTHEY, B., ROOVERS, R.C., BARTH, S., ARENDS, J.W., ENGERT, A., et al., Human anti-CD30
recombinant antibodies by guided phage antibody selection using cell panning. Br J Cancer, 2000. 83(2): p. 252-60. 188. HULETT, M.D. and HOGARTH, P.M., The second and third extracellular domains of FcgammaRI (CD64) confer the
unique high affinity binding of IgG2a. Mol Immunol, 1998. 35(14-15): p. 989-96. 189. NIKCEVIC, G., KOVACEVIC-GRUJICIC, N., and STEVANOVIC, M., Improved transfection efficiency of cultured human
cells. Cell Biol Int, 2003. 27(9): p. 735-7. 190. WOOD, K.M., ROFF, M., and HAY, R.T., Defective IkappaBalpha in Hodgkin cell lines with constitutively active NF-
kappaB. Oncogene, 1998. 16(16): p. 2131-9. 191. GRUSS, H.J., BOIANI, N., WILLIAMS, D.E., ARMITAGE, R.J., SMITH, C.A., and GOODWIN, R.G., Pleiotropic effects of
the CD30 ligand on CD30-expressing cells and lymphoma cell lines. Blood, 1994. 83(8): p. 2045-56. 192. KRAPPMANN, D., EMMERICH, F., KORDES, U., SCHARSCHMIDT, E., DORKEN, B., and SCHEIDEREIT, C., Molecular
mechanisms of constitutive NF-kappaB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene, 1999. 18(4): p. 943-53.
193. PAVLINKOVA, G., BERESFORD, G.W., BOOTH, B.J., BATRA, S.K., and COLCHER, D., Pharmacokinetics and
biodistribution of engineered single-chain antibody constructs of MAb CC49 in colon carcinoma xenografts. J Nucl Med, 1999. 40(9): p. 1536-46.

Literature ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
122
194. KAMPMEIER, F., NIESEN, J., KOERS, A., RIBBERT, M., BRECHT, A., FISCHER, R., et al., Rapid optical imaging of EGF
receptor expression with a single-chain antibody SNAP-tag fusion protein. Eur J Nucl Med Mol Imaging, 2010. 37(10): p. 1926-34.
195. BECK, R., PEDROSA, R.C., DEJEANS, N., GLORIEUX, C., LEVEQUE, P., GALLEZ, B., et al., Ascorbate/menadione-
induced oxidative stress kills cancer cells that express normal or mutated forms of the oncogenic protein Bcr-Abl.
An in vitro and in vivo mechanistic study. Invest New Drugs, 2011. 29(5): p. 891-900. 196. PATHAK, A.P., ARTEMOV, D., WARD, B.D., JACKSON, D.G., NEEMAN, M., and BHUJWALLA, Z.M., Characterizing
extravascular fluid transport of macromolecules in the tumor interstitium by magnetic resonance imaging. Cancer Res, 2005. 65(4): p. 1425-32.
197. WIIG, H., RUBIN, K., and REED, R.K., New and active role of the interstitium in control of interstitial fluid pressure:
potential therapeutic consequences. Acta Anaesthesiol Scand, 2003. 47(2): p. 111-21. 198. CHAKROBORTY, D., SARKAR, C., YU, H., WANG, J., LIU, Z., DASGUPTA, P.S., et al., Dopamine stabilizes tumor blood
vessels by up-regulating angiopoietin 1 expression in pericytes and Kruppel-like factor-2 expression in tumor
endothelial cells. Proc Natl Acad Sci U S A, 2011. 108(51): p. 20730-5. 199. GEHRMANN, M., STANGL, S., KIRSCHNER, A., FOULDS, G.A., SIEVERT, W., DOSS, B.T., et al., Immunotherapeutic
targeting of membrane Hsp70-expressing tumors using recombinant human granzyme B. PLoS One, 2012. 7(7): p. e41341.
200. CULLEN, S.P., ADRAIN, C., LUTHI, A.U., DURIEZ, P.J., and MARTIN, S.J., Human and murine granzyme B exhibit
divergent substrate preferences. J Cell Biol, 2007. 176(4): p. 435-44. 201. SCHIFFER, S., HRISTODOROV, D., MLADENOV, R., ASLANIAN, E., HUHN, M., R., F., et al., Species-Dependent
Functionality of the Human Cytolytic Fusion Proteins Granzyme B-H22(scFv) and H22(scFv)-Angiogenin in
Macrophages. Antibodies, 2013. 2(1): p. 9-18. 202. BIRD, C.H., SUN, J., UNG, K., KARAMBALIS, D., WHISSTOCK, J.C., TRAPANI, J.A., et al., Cationic sites on granzyme B
contribute to cytotoxicity by promoting its uptake into target cells. Mol Cell Biol, 2005. 25(17): p. 7854-67. 203. BUZZA, M.S., ZAMURS, L., SUN, J., BIRD, C.H., SMITH, A.I., TRAPANI, J.A., et al., Extracellular matrix remodeling by
human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem, 2005. 280(25): p. 23549-58. 204. LANG, D.S., DROEMANN, D., SCHULTZ, H., BRANSCHEID, D., MARTIN, C., RESSMEYER, A.R., et al., A novel human ex
vivo model for the analysis of molecular events during lung cancer chemotherapy. Respir Res, 2007. 8: p. 43. 205. CHRISTIANSEN, J. and RAJASEKARAN, A.K., Biological impediments to monoclonal antibody-based cancer
immunotherapy. Mol Cancer Ther, 2004. 3(11): p. 1493-501. 206. FEURING-BUSKE, M., FRANKEL, A.E., ALEXANDER, R.L., GERHARD, B., and HOGGE, D.E., A diphtheria toxin-
interleukin 3 fusion protein is cytotoxic to primitive acute myeloid leukemia progenitors but spares normal
progenitors. Cancer Res, 2002. 62(6): p. 1730-6. 207. VALLERA, D.A., JIN, N., BALDRICA, J.M., PANOSKALTSIS-MORTARI, A., CHEN, S.Y., and BLAZAR, B.R., Retroviral
immunotoxin gene therapy of acute myelogenous leukemia in mice using cytotoxic T cells transduced with an
interleukin 4/diphtheria toxin gene. Cancer Res, 2000. 60(4): p. 976-84. 208. TEN CATE, B., BREMER, E., DE BRUYN, M., BIJMA, T., SAMPLONIUS, D., SCHWEMMLEIN, M., et al., A novel AML-
selective TRAIL fusion protein that is superior to Gemtuzumab Ozogamicin in terms of in vitro selectivity, activity
and stability. Leukemia, 2009. 23(8): p. 1389-97. 209. SCHWEMMLEIN, M., PEIPP, M., BARBIN, K., SAUL, D., STOCKMEYER, B., REPP, R., et al., A CD33-specific single-
chain immunotoxin mediates potent apoptosis of cultured human myeloid leukaemia cells. Br J Haematol, 2006. 133(2): p. 141-51.
210. SANTOS, I.M., FRANZON, C.M., and KOGA, A.H., Laboratory diagnosis of chronic myelomonocytic leukemia and
progression to acute leukemia in association with chronic lymphocytic leukemia: morphological features and
immunophenotypic profile. Rev Bras Hematol Hemoter, 2012. 34(3): p. 242-4. 211. TUR, M.K., HUHN, M., JOST, E., THEPEN, T., BRUMMENDORF, T.H., and BARTH, S., In vivo efficacy of the
recombinant anti-CD64 immunotoxin H22(scFv)-ETA' in a human acute myeloid leukemia xenograft tumor model. Int J Cancer, 2011. 129(5): p. 1277-82.
212. NORDIGARDEN, A., ZETTERBLAD, J., TRINKS, C., GREEN, H., ELIASSON, P., DRUID, P., et al., Irreversible pan-ERBB
inhibitor canertinib elicits anti-leukaemic effects and induces the regression of FLT3-ITD transformed cells in mice. Br J Haematol, 2011. 155(2): p. 198-208.
213. RIBBERT, T., THEPEN, T., TUR, M.K., FISCHER, R., HUHN, M., and BARTH, S., Recombinant, ETA'-based CD64
immunotoxins: improved efficacy by increased valency, both in vitro and in vivo in a chronic cutaneous
inflammation model in human CD64 transgenic mice. Br J Dermatol, 2010. 163(2): p. 279-86. 214. SILVERMAN, G.A., BIRD, P.I., CARRELL, R.W., CHURCH, F.C., COUGHLIN, P.B., GETTINS, P.G., et al., The serpins are
an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of
inhibition, novel functions, and a revised nomenclature. J Biol Chem, 2001. 276(36): p. 33293-6. 215. JEDEMA, I., BARGE, R.M., FRANKEL, A.E., WILLEMZE, R., and FALKENBURG, J.H., Acute myeloid leukemia cells in G0
phase of the cell cycle that are unresponsive to conventional chemotherapy are sensitive to treatment with
granulocyte-macrophage colony-stimulating factor/diphtheria toxin fusion proteins. Exp Hematol, 2004. 32(2): p. 188-94.
216. DRUKER, B.J., GUILHOT, F., O'BRIEN, S.G., GATHMANN, I., KANTARJIAN, H., GATTERMANN, N., et al., Five-year
follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med, 2006. 355(23): p. 2408-17. 217. ARMSTRONG, P.B. and QUIGLEY, J.P., Alpha2-macroglobulin: an evolutionarily conserved arm of the innate
immune system. Dev Comp Immunol, 1999. 23(4-5): p. 375-90.

Literature ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
123
218. ROBERTS, R.C., Protease inhibitors of human plasma. Alpha-2-macroglobulin. J Med, 1985. 16(1-3): p. 129-224. 219. REHMAN, A.A., AHSAN, H., and KHAN, F.H., Alpha-2-macroglobulin: A physiological guardian. J Cell Physiol, 2012. 220. BRUELL, D., BRUNS, C.J., YEZHELYEV, M., HUHN, M., MULLER, J., ISCHENKO, I., et al., Recombinant anti-EGFR
immunotoxin 425(scFv)-ETA' demonstrates anti-tumor activity against disseminated human pancreatic cancer in
nude mice. Int J Mol Med, 2005. 15(2): p. 305-13. 221. KHURSHID, R., SALEEM, M., AKHTAR, M.S., and SALIM, A., Granzyme M: characterization with sites of post-
translational modification and specific sites of interaction with substrates and inhibitors. Mol Biol Rep, 2011. 38(5): p. 2953-60.
222. BARTH, S., HUHN, M., MATTHEY, B., SCHNELL, R., TAWADROS, S., SCHINKOTHE, T., et al., Recombinant anti-CD25
immunotoxin RFT5(SCFV)-ETA' demonstrates successful elimination of disseminated human Hodgkin lymphoma in
SCID mice. Int J Cancer, 2000. 86(5): p. 718-24. 223. DE KONING, P.J., KUMMER, J.A., DE POOT, S.A., QUADIR, R., BROEKHUIZEN, R., MCGETTRICK, A.F., et al.,
Intracellular serine protease inhibitor SERPINB4 inhibits granzyme M-induced cell death. PLoS One, 2011. 6(8): p. e22645.
224. JABULOWSKY, R.A., OBEROI, P., BAHR-MAHMUD, H., DALKEN, B., and WELS, W.S., Surface Charge-Modification
Prevents Sequestration and Enhances Tumor-Cell Specificity of a Recombinant Granzyme B-TGF alpha Fusion
Protein. Bioconjugate Chemistry, 2012. 23(8): p. 1567-76.

Appendix ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
I
8 Appendix
8.1 DNA AND AMINO ACID SEQUENCE OF GBR201K
The wild-type DNA sequence triplet cga was exchanged by aag in the modified GbR201K (position
601-603).
atcatcgggg gacatgaggc caagccccac tcccgcccct acatggcttt tcttatgatc 60 tgggatcaga agtctctgaa gaggtgcggt ggcttcctga tacgagacga cttcgtgctg 120 acagctgctc actgttgggg aagctccata aatgtcacct tgggggccca caatatcaag 180 gaacaggagc cgacccagca gtttatccct gtgaaaagag ccatccccca tccagcctat 240 aatcctaaga acttctccaa tgacatcatg ctactgcagc tggagagaaa ggccaagcgg 300 accagagctg tgcagcccct caggctacct agcaacaagg cccaggtgaa gccagggcag 360 acatgcagtg tggccggctg ggggcagacg gcccccctgg gaaaacactc acacacacta 420 caagaggtga agatgacagt gcaggaagat cgaaagtgcg aatctgactt acgccattat 480 tacgacagta ccattgagtt gtgcgtgggg gacccagaga ttaaaaagac ttcctttaag 540 ggggactctg gaggccctct tgtgtgtaac aaggtggccc agggcattgt ctcctatgga 600 aagaacaatg gcatgcctcc acgagcctgc accaaagtct caagctttgt acactggata 660 aagaaaacca tgaaacgcta c 681
In accordance to that, the modified GbR201K amino acid sequence comprises a lysine (K) at position
201 instead of wild-type arginine (R).
IIGGHVAKPH SRPYMAYLMI WDQKSLKRCG GFLIRDDFVL TAAHCWGSSI NVTLGAHNIK 60 EQEPTQQFIP VKRAIPHPAY NPKNFSNDIM LLQLERKAKR TRAVQPLRLP SNKAQVKPGQ 120 TCSVAGWGQT APLGKHSHTL QEVKMTVQED RKCESDLRHY YDSTIELCVG DPEIKKTSFK 180 GDSGGPLVCN KVAQGIVSYG KNNGMPPRAC TKVSSFVHWI KKTMKRY 227
8.2 SYNTHETIC DNA SEQUENCE OF GM-H22(SCFV) IN PMS VECTOR
The sequence of Gm-H22(scFv) is shown exemplary for all granzyme M-based constructs generated
in this study. The single chain sequences were exchanged accordingly via appropriate restriction
sites.

Appendix ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
II

Appendix ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
III

Index of Figures and Tables ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
IV
9 Index of Figures and Tables
TABLES
Table 1: List of oligo nucleotides used for cloning and DNA sequencing. ............................................. 21
Table 2: Origin and specificity of antibody fragments. ......................................................................... 23
Table 3: Mammalian cell lines. .............................................................................................................. 24
Table 4: Buffers and media.................................................................................................................... 25
Table 5: Reagents for physical or chemical transfection of mammalian cell lines. .............................. 31
Table 6: Overview of the PI-9 expression profile of different cancer cell lines. ................................... 43
Table 7: Affinity constants and relative receptor expression level of CD30-positive cell lines............. 55
Table 8: Serum stability of Gb-H22(scFv) and GbR201K-H22(scFv) in human patient serum (CMML III)
or PBS. ................................................................................................................................................... 77
Table 9: Overview of different granzyme B-based cytolytic fusion proteins (CFPs) published so far. . 88
Table 10: Overview of PI-9 expression in different carcinomas / melanomas. .................................... 91
Table 11: Overview of PI-9 expression in different lymphomas. .......................................................... 93
FIGURES
Figure 1: X-ray crystal structure of the non-covalent Michaelis complex of A353K Manduca sexta
serpin B1 (purple) and S195A trypsin (cyan) (PDB file 1K9O [87]). ....................................................... 11
Figure 2: Workflow of this study. .......................................................................................................... 18
Figure 3: Expression of endogenous PI-9 in HL cell lines L428, L1236, L540cy and CML cell line K562.44
Figure 4: Overview of initial granzyme B constructs (A) and SDS-PAGE of the purified fusion proteins
before and after Enterokinase cleavage (B). ......................................................................................... 46
Figure 5: List of generated point-mutated variants (2.2.2) arised from literature (#: [87]; ##: [86]) or
molecular modeling (3.2). ..................................................................................................................... 47
Figure 6: Preparation of recombinant PI-9 and functionality determination via complex formation
with wild-type Gb-H22(scFv). ................................................................................................................ 49
Figure 7: Influence of endogenous PI-9 on apoptotic activity of Gb-Ki4(scFv). .................................... 51
Figure 8: Enzymatic activity of Gb-Ki4(scFv) and GbR201K-Ki4(scFv). .................................................. 52
Figure 9: Enzymatic activity of Gb-H322(scFv) wild-type and its mutants in the presence and absence
of PI-9. ................................................................................................................................................... 53
Figure 10: Binding analysis of granzyme B-based CFPs. ........................................................................ 54
Figure 11: Internalization analysis of Ki4(scFv)-SNAP-BG-647 and H22(scFv)-SNAP-BG-647 into
corresponding target cell lines. ............................................................................................................. 56

Index of Figures and Tables ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
V
Figure 12: Cytotoxic effects of Gb-Ki4(scFv) and Gb-H22(scFv) wild-type and their mutants
respectively on PI-9-negative target cell lines. ..................................................................................... 57
Figure 13: Apoptotic effects of Gb-Ki4(scFv) or Gb-H22(scFv) wild-type and their mutants respectively
on PI-9-negative target cell lines. .......................................................................................................... 58
Figure 14: Cytotoxic effect of Gb-Ki4(scFv) and its mutants on PI-9-positive target cell lines. ............ 60
Figure 15: Apoptotic effect of Gb-Ki4(scFv) and GbR201K-Ki4(scFv) on PI-9 positive target cell lines. 61
Figure 16: Serum stability of Gb-Ki4(scFv) and GbR201K-Ki4(scFv). ..................................................... 62
Figure 17: Evaluation of the established L540cy and L428 tumor models. .......................................... 64
Figure 18: Evaluation of the in vivo anti-tumor activity against L540cy-induced tumors. .................... 66
Figure 19: Evaluation of the in vivo anti-tumor activity against L428-induced tumors. ....................... 67
Figure 20: Expression of endogenous PI-9 (2.4.7) in primary leukemic cells derived from peripheral
blood of CMML or AMML patients (2.1.8). ........................................................................................... 69
Figure 21: Phenotyping of primary leukemic cells. ............................................................................... 70
Figure 22: Binding and internalization analysis with primary leukemic cells (2.1.8). ........................... 71
Figure 23: Cytotoxic and apoptotic effects of Gb-H22(scFv), GbR201K-H22(scFv) and H22(scFv)-ETA’
on primary leukemic cells. ..................................................................................................................... 72
Figure 24: Cytotoxic effects of Gb-H22(scFv) and GbR201K-H22(scFv) on primary leukemic cells of
AMML I. ................................................................................................................................................. 74
Figure 25: Specific reduction of target cancer cell population by Gb-H22(scFv), GbR201K-H22(scFv)
and H22(scFv)-ETA’ detected in FSC/SSC dotplot after flow cytometry. .............................................. 75
Figure 26: Reduction of target cancer cell population by Gb-H22(scFv) and GbR201K-H22(scFv)
detected with specific antibodies (2.1.5). ............................................................................................. 76
Figure 27: Overview of granzyme M-based constructs (A) and SDS-PAGE and western blot analysis of
Gm-H22(scFv) (B). .................................................................................................................................. 78
Figure 28: Enzymatic activity of Gm-H22(scFv) tested by cleavage of PI-9 or a colorimetric substrate.
............................................................................................................................................................... 79
Figure 29: Binding analysis of Gm-H22(scFv). ....................................................................................... 80
Figure 30: Cytotoxic and apoptotic effects of Gm-H22(scFv)................................................................ 82
Figure 31: Apoptotic effect of Gm-Ki4(scFv) compared to Gb-Ki4(scFv)............................................... 83
Figure 32: Cytotoxic and apoptotic effects of Gm-H22(scFv) on primary leukemic cells. .................... 85

Publications ________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
VI
10 Publications
Publications (*,# equal contribution)
Schiffer S, Rosinke R, Jost E, Huhn M, Fischer R, Thepen T*, Barth S*. Targeted ex vivo reduction of
CD64-positive monocytes in CMML and AMML using human granzyme B-based cytolytic fusion
proteins. [in revision]
Schiffer S, Letzian S, Jost E, Mladenov R, Hristodorov D, Huhn M, Fischer R, Barth S*. Thepen T*.
Granzyme M as a novel effector molecule for human cytolytic fusion proteins: CD64-specific
cytotoxicity of Gm-H22(scFv) against leukemic cells. Cancer Letters 2013; 341, 178-185.
Schiffer S, Hansen H, Hehmann-Titt G, Huhn M, Fischer R, Barth S*, Thepen T*. Efficacy of an adapted
granzyme B-based anti-CD30 cytolytic fusion protein against PI-9-positive classical Hodgkin
lymphoma cells in a murine model. Blood Cancer Journal 2013; 3: e106.
Hehmann-Titt G*, Schiffer S*, Berges N*, Melmer G, Barth S. Improving the Therapeutic Potential of
Human Granzyme B for Targeted Cancer Therapy. Antibodies 2013; 2(1): 19-49.
Schiffer S*, Hristodorov D*, Mladenov R, Huhn M, Fischer R, Barth S*, Thepen T*. Species-dependent
functionality of the human cytolytic fusion proteins Granzyme B-H22(scFv) and H22(scFv)-
Angiogenin. Antibodies 2013; 2(1): 9-18.
Losasso V*, Schiffer S*, Barth S#, Carloni P#. Design of Human Granzyme B Variants resistant to Serpin
B9. Proteins 2012; 80(11): 2514-22.
Patent applications
Barth S, Schiffer S (2012), International patent application, Novel immunoproteases – EP12191711.6-
2401
Barth S, Schiffer S, Hehmann-Titt G (2011), International patent application, Novel serine protease
variants - EP11007764.1-2401, US61/538,638,PCT/EP2012/068607
Poster presentations
Schiffer S, Hehmann-Titt G, Fischer R, Barth S. Novel approach to circumvent the inhibitory potential
of Serpin B9 in cancer therapy. Biomedica Liège, Belgium (2012)
Schiffer S, Hehmann-Titt G, Thepen T, Huhn M, Fischer R, Barth S. Optimized human Granzyme B
based immunotoxins to circumvent the inhibitory potential of Serpin B9 in cancer therapy. CIMT
Mainz, Germany (2012)

Danksagung
An dieser Stelle möchte ich allen danken, die mich während meiner Dissertation fachlich oder
persönlich unterstützt haben. Mein besonderer Dank gilt:
Prof. Dr. Rainer Fischer für die Möglichkeit diese Arbeit im Fraunhofer Institut für Molekularbiologie
und angewandte Ökologie anzufertigen, sowie die Korrektur dieser Arbeit. Außerdem gilt ihm mein
Dank für die Übernahme des Referats.
Prof. Dr. Dr. Stefan Barth für die Möglichkeit diese Arbeit in seiner Abteilung durchzuführen, seine
wissenschaftliche Betreuung, sein Vertrauen, sowie die gewährte Freiheit bei der Gestaltung meiner
Projekte.
Dr. Grit Hehmann-Titt für die Betreuung dieser Arbeit, alle wertvollen Tipps und Anregungen und die
Korrektur dieser Arbeit. Besonders gilt ihr mein Dank für ihre Aufmunterungen in schwierigeren
Phasen sowie für wertvolle Diskussionen.
Dr. Michael Huhn für seine unermüdliche Unterstützung während dieser Arbeit, die nie abreißende
Flut an neuen Ideen und dass er immer ein offenes Ohr für mich hatte.
Dr. Theo Thepen für die Korrekturen meiner Veröffentlichungen und dieser Arbeit, seine
Unterstützung bei den in vivo Experimenten und seine wertvollen Tipps und Anregungen.
Dr. Edgar Jost für die Bereitstellung des primären Probenmaterials.
Dr. Hinrich Hansen, der mich maßgeblich bei der Verfassung einer meiner Publikationen unterstützt
hat und mir viele motivierende Worte aus Köln geschickt hat.
Prof. Dr. Paolo Carloni, Dr. Valeria Losasso und Dr. Heinrich Delbrück für ihre Hilfe bei der
Modellierung der Mutanten und die gute Zusammenarbeit.
Der ganzen PPD und Pharmedartis Gruppe für die tolle Atmosphäre in der Arbeitsgruppe, durch die
das Lachen nie zu kurz kam. Besonders danke ich hier Nora Frohnecke und Vivian Dietrich für ihre
Freundschaft sowie ihre fachliche und persönliche Unterstützung; Sarah Kirchhoff für ihre Loyalität
und große Unterstützung im Labor; Judith Niesen und Lea Hein für ihr treues Mitleiden und
Mitfreuen; Dmitrij Hristodorov und Radoslav Mladenov für ihr großes Engagement; Jens Bührmann,
Torsten Sieben und Anh-Tuan Pham für ihre sehr verlässliche Hilfe im Labor; besonders Reinhard
Rosinke für seine unermüdliche Unterstützung, seine immer guten Ideen, sowie seine exzellente Hilfe
bei der Proteinaufreinigung, sowie bei der Arbeit mit Primärmaterial.
Meinen Eltern, die mich immer unterstützt haben, mich zu der Person gemacht haben, die ich bin,
und ohne die ich niemals so weit gekommen wäre.
Meinem Mann, der auch in schwierigen Zeiten immer hinter mir stand und meine Höhen und Tiefen
während dieser Arbeit geduldig ertragen hat.

Lebenslauf
Persönliche Daten
Sonja Schiffer
geboren 1984 in Neuss
Ausbildung
2003 Erwerb der Allgemeinen Hochschulreife, Pascal-Gymnasium, Grevenbroich
2003 - 2004 Auslandsaufenthalt in Chicago, USA
2004 – 2009
2009
Studium des Bioingenieurwesens, TU Dortmund; Schwerpunkt Biotechnologie
(Abschluss: Dipl. Ing.)
2007: Auslandssemester an der TH Lund, Schweden
2008: Auslandspraktikum bei „Sumitomo Chemical“ in Osaka, Japan
Diplomarbeit am Max-Planck-Institut für molekulare Physiologie, Dortmund,
Abteilung Strukturbiologie
Betreuung: Prof. Dr. Alfred Wittinghofer (MPI) /
Prof. Dr. Andreas Schmid (TU Dortmund)
Titel: “Screening for functional ‘GAPettes’ directed against constitutive active
Ras proteins”
2009-2013
Promotion am Fraunhofer Institut für Molekularbiologie und angewandte
Ökologie IME, Abteilung Pharmazeutische Produktentwicklung / RWTH Aachen
Betreuung: Prof. Dr. Rainer Fischer /
Prof. Dr. Dr. Stefan Barth
Titel: “Improving the therapeutic potential of human granzyme B and
evaluation of granzyme M as novel effector molecules in cytolytic fusion
proteins for the treatment of Serpin B9-positive cancer“





























