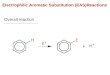
Embed Size (px)
DESCRIPTION
Authors: P.S. Skell and W. L. Hall.Year: 1963.Journal: Journal of American Chemical Society.Volume:85.Number: 18.Pages: 2851-2852.
Citation preview

Sept. 20, 1963 COMMUNICATIONS TO THE EDITOR 2851
nearly total racemization in refluxing CC14-NBS to the effect of temperature observed in reactions of (+)-1- bromo-2-methylbutane with molecular bromine . 7
Earlier implied that Br. rather than succini- midyl radical abstracted a hydrogen atom from the substrate. The present study indicates that the alkyl radical intermediate is not brominated by NBS, but presumably by molecular Br2 present in steady low concentration.
Br. + R H --+ HBr + R . R . + Brz + RBr + Br.
S B S + HBr ---f Br2 + succinimide
Acknowledgment.-This research was supported by the Directorate of Chemical Sciences, Air Force Office of Scientific Research.
DEPARTMENT OF CHEMISTRY THE PENSSYLVANIA STATE UNIVERSITY UNIVERSITY PARK, PENSSYLVASIA
(8) E thy l Corporation Fellow, 1962-1963
P. S. SKELL D. L. TULEEN P. D. READIO*
RECEIVED JUNE 24, 1963
Importance of “Gegenion” in Electrophilic Processes. cis and trans E l Reaction Conditions
Sir : Studies of the solvolyses of alkyl halides, tosylates,
and sulfonium ion salts in aqueous ethanol led to the concept of electrophilic processes in which solvent-aided ionizations yield carbonium ions which are partitioned among the available reaction routes, such as ejection of a proton to produce olefin (El ) , or addition of a nucleophile (SN1) . The choice between these paths was considered to be independent of the method of generating the carbonium ion. Re-examination of the data employed to support this position shows deviations from this generalization. Again and again instances have been reported which indicate that the composition of the products in these processes are strongly de- pendent upon the nature of the leaving group and sol- ~ e n t . ~ , ~ Recently, attention has been sharply focused on this subject by Cram and S a h y ~ n , ~ and Winstein and C o ~ i v e r a . ~ Both groups suggest that the “gegen- ions” play an important role in determining the prod- ucts from these processes.
A study of solvolytic elimination reactions in the 3-deuterio-2-butyl tosylate system has helped to elucidate the role of both solvent and “gegenion.” This system is convenient for studying the stereo- chemistry, since trans-elimination yields, in the case of erythro-3-deuterio-2-butyl tosylate, deuterated cis- 2-butene and undeuterated trans-%butene. The stereo- chemical purity of the tosylates was demonstrated to be better than 95yo by examination of the butenes obtained by reaction with potassium ethoxide in ethanol. The extent of cis- or trans-elimination is
known by separating the butene product mixture into its components and assaying their deuterium content by mass spectrometry, correcting for the small amounts of
(1) K . A Cooper , E. D . Hughes, C . K. Ingold, and B. J. MacXul ty ,
(2) M . S. Silver, J . A m . Chem. SOC., 85, 3482 (1961). (3) W . B . Smith and W . H . Watson , Jr., i b i d . , 84, 3174 (1962). ( 4 ) D. J. C r a m and M. R. V . Sahyun , i b i d . , 86, 1257 (1963) ( 5 ) S. Winstein and M. Cocivera, i b i d . , 86, 1702 (1963).
J . Chem. SOL., 2038 (1948).
TABLE I PERCENT OLEFIN FORMED BY CZT-ELIMINATION (MECHANISM A )
trans- CIS-
Solvent Tosylate 2-Butene, 2-Butene,
Solvent PK. isomer % 70 Nitrobenzene - 11 3 erythro- 98 + 2 95 f 2 Nitrobenzene -11 3 threo- 8 9 f 2 9 9 f 2 Glacial acetic -6 1 erythro- 82 f 2 65 f 2
TABLE I1 PER CENT OLEFIN FORMED BY ~ T ~ ~ S - E L I M I N A T I O N (MECHANISM B)
acid
trans- CIS-
Solvent P K ~ isomer 7c 70 Solvent Tosylate 2-Butene, 2-Butene,
-0Et in eth- erythro- 100 100 anol
lcetamide 0 0 threo- 9 2 f 2 6 9 f 2 .ketamide 0 0 erythro- 81 f 2 91 f 2 80YG aqueous --2 erythro- 66 f 2 84 f 2
ethanol
intercontamination The results of these experiments are summarized in Tables I and 11.
Ionization of the tosylate group and movement of a hydrogen atom through a 60’ clockwise arc would pro- duce the same carbonium ion from both threo- and erythro-tosylates However, in solvolytic eliminations
OTs
cH3$rH3 H D
erythro-
1
OTs
cH3@1H3 H H
threo-
1
I H
(80yc aqueous ethanol, anhydrous acetamide, glacial acetic acid, 90yc formic acid, and nitrobenzene) these isomeric tosylates yield olefins with widely differing extents of deuteration. These large differences suggest that these tosylates do not react through a common intermediate. This leaves as the only alternative the conclusion that the product precursor has the tosylate group intimately associated with the same face of the a-carbon atom to which it was attached in the starting ester.
In nitrobenzene olefin is produced by a nearly stereo- specific cis-elimination, whereas in acetamide or aqueous ethanol the reaction follows a predominantly trans path. This alteration of mechanism from one extreme to another can be correlated with the basicities of the solvents,6 the less basic solvents favoring cis- and the more basic solvents trans-eliminations,
The cis-elimination is rationalized by removal of the P-proton by the parting tosylate group which must re- main on the same face of the carbonium ion until it has removed a proton from an adjacent carbon atom.
By contrast, trans-eliminations, which occur in the more basic solvents, require attack by the solvent from
(6) E. M. Arne t t , ”Progress in Physical Organic Chemistry,” Vol. 1, S. Cohen, A. Streitwieser, Jr . , and R. W: T a f t , E d . , Interscience Publishers, S e w York, Ii. Y . , in press.

2852 COMMUNICATIONS TO THE EDITOR Vol. 85
the antiperiplanar position, essentially as in the concerted E2 elimination. Thus, the difference between E2 and E l in basic solvents may only be a matter of degree
Hac /C H3 B
/ -pTs //,yHs H., - /c=c
'Solvent D \ H H CHZ
of polarization of the bond between the leaving group and the a-carbon.
A41though it is not possible completely to rule out gegcnion free carbonium ions in each of the solvents, i t is clear that they do not play a major role in these solvolytic eliminations. Streitwieser and Walsh' have demonstrated that the solvolytic displacement of tosylate by acetate group in the 2-octyl system occurs with 100% inversion in glacial acetic acid. .-I11 of these resiilts are best accommodated by rejjection of the idea that in these soholytic processes the fate of the carbonium ion is independent of i f s origin.
Acknowledgment.-This work was supported by the U. S. Xrmy Research Office (Durham).
DEPARTMENT OF CHEMISTRY P. S. SKELL THE PENSSYLVASIA STATE LXIVERSITY M' L. HALL USIYERSITY PARK, PENNSYLVANIA
RECEIVED JULY 3, 1963
17) A Streitwieser, J r . . a n d T 11. Wal ih , T e l r a h e d t o n L e t t e r s , 27 (1963).
The Cyclononatetraenyl Anion Sir :
The cyclononatetraenyl anion I should have a closed shell configuration of 10 T electrons and should be highly resonance stabilized. I That i t should be aromatic is, however, questionable for two reasons: (1) The energy required to distort the angles of its bonds might be excessively large? and (2) the nine-membered ring might valence tautomerize3 spontaneously to form a single bond a t the expense of a double Nevertheless, the cyclononatetraenyl anion does appear to be aromatic. Its synthesis is the subject of this report.
Ia, h l T = Li+ IIa, S = C1 I I Ia , S = Cl
c , S = H c , X = H b , h l t = R C b, X = OCHa b, S = OCHs
Cold chloroform reacts with dipotassium cyclo- octatetraenide4 in tetrahydrofuran (THF) to yield, after aqueous work-up, 9-chlorobicyclo [6.1 .O]nonatriene (IIa) , b.p. 28-32' (0.2 mm.), in 327, yield; A:::" 218 mp (log E 3 . 5 7 ) . *lnal. Calcd. for CsH9C1: C.
; H. j . 94 ; C1, 23.23 Found: C, i l . 0 6 ; H, C1, 2,'3.42. Similarly, dichloromethyl methyl
ether reacts to yield 9-methoxybicyclo [6.1 .O]nonatriene ( I I b ) in 267' yield; b.p. 54-56 (1.1 mm. ) ; Xfl;:" 233 mp (log E 3.69). .Inal. Calcd. for CIoH110: C,Sl .O9; H,S.11. Found: C,80.80; H,8 .18 . SIeth- ylene chloride yields bicyclo [6.1 .0]nonatriene5 (IIc) in 4ijT; yield.
IIa was identified by its ultraviolet spectrum and by its n.in.r. spectrum. The latter exhibited a multiplet
11 I t - Huckel mulecular orbital delocalization energy is i3 5 3 (21 'The ccimpressiona! harrier i huwever, overcome by its next lower
homolog. Lhr cyclr , ,xtatetraenyI d nion IT. J K a t z , W H R e i n m u t h , and D i th . J . A m ( 'hem Soc. 84, 02 i 19l i2) , a n d references there in]
J . Katz . J A m ( h e m . .Sof , 83, :3781 (1900) Yogel. A u ~ e m . C'hem , 73, A - I R ( I O i i l ) , and private c< ,mmunica t im
a t 4.1 (rel. int. 6 ) and a coupled ( J = 4 c.P.s.) triplet a t 7.54 (rel. int. 1) and doublet a't 8.14 T (rel. int. 2) . I Ib exhibited a similar n.m.r. spectrum: a multiplet a t 4.2 (rel. int. Ci) , a singlet a t 6.70 (rel. int. 3 ) , and a coupled ( J = 4 c.P.s.) triplet a t 7.40 (rel. int. 1) and doublet a t 8.40 T (rel. int. 2). Only the anti isomers appear to be found, in striking contrast to the forma- tion of mixtures of comparable amounts of syn and anti isomers, the former predominating, when chloro-6 and methoxycarbene' are added to other olefins?
Heating I Ia to 70' yields l-chloro-X,9-dihydroindene ( I I Ia ; z'lnal. Found: C, 71.03; H , 6.01; C1, 23.24), identified by its ultraviolet spectrum [AX:,:::'' 2(j2 ( 3 . 5 2 ) , 270 mp (log E 3.14)] and its n.m.r. spectrum (4.2, 5.27, (5.6 T. rel. int. 6 : 1 : 2 , all multiplets). Lithium aluminum hydride reduces I I Ia to S.9-dihydroindeney (IIIc) [XXr);::" 262 (3 .37) , 271 mp (log E X S I ) , maleic anhydride adduct, m.p. 141,5"J, which was identical with that obtained by heating IIc."
IIa reacts with lithium in THF to give lithium cyclo- nonatetraenide ( I a ) . On quenching with H20, I I Ic is formed, and on quenching with DzO, i t is formed (24y6 .yield) with the incorporation of 1 atom of deuterium.'" Similarly, I Ib reacts with potassium to give potassium cyclononatetraenide ( I Ib), which, on quenching with DQ yields monodeuterated IIIc. I 1
The n.m.r. spectra of potassium and of lithium cyclo- nonatetraenide in completely deuterated T H F consist of only one single very sharp peak at 2.96 and 33.15 7, respectively.12 These spectra show that the cyclo- nonatetraenyl anion is aromatic.'3 If the cyclo- nonatetraenyl anion and the cyclooctatetraenyl dianion are compared, their ring currentsL4 should be sirnilar, but the protons in the former should be less shielded that in the latter because the negative charge density associated with each carbon atom is rather than z , g , That is, the cyclononatetraenyl anion's proton n.m.r. should'6,Li appear (I '4 - y ) X 10 = l . 1p .p .m. to lower fields than the resonance (4.3 T) of the cyclo- octatetraenyl d i a n i ~ n . ~ The agreement with the ob- served chemical shift is excellent.
The ultraviolet spectra of l a [XzT 252 mp (5,0), :318 mp (3,9), 325 mp (log t .'3.9)] and Ib are similar.18
I t is remarkable that the cyclononatetraenyl anion has the all-cis stereochemistry.
(13) (a) G I > Clriss a n d L E Closs. J . A m . ( ' hew . Soc. . 83, .-,72:3 i l900) ; (b ) G. 1,. Closs, R A Sloss, a n d J . J . Coyle, ibiif , 84, 1985 ( I S H ? ) .
( 7 ) V , Schollkopf, A Lercn, and W. Pitteroff, Tetrahedron L e t l e r s , 241 (19ii2); L!. Y Schhllkopf and G J I.ehman, i b i d . , !&> ( 1 9 i i P ) ; V. Schiillkopf and H. Kuppers. t b i , I , , 102 ( I Q l i 3 ) .
( 8 ) T h e stereochemical assignment is based on the assumption t h a t t h e small size of t h e spin-spin coupling cons tan t implies t h e nizti stereochemistry [ a f t e r Closs, ref lib) T h e presence of a trace of t h e o ther is<,mer in samples of I l b i c indicated h y a small second methoxyl peak 0.0: p p . m abuve t h e main peak
(9) K Alder a n d F. H. Flock, Chem Ber , 87, 1910 (1954) f l 0 ) F o u n d . 9 .40 , 9,liu; 9.38; a t o m 5% excess 11. (Fall ing drop analyses
b y Josef S e m e t h . Urbana , 111.1 (11) !I I f i a t o m 'T excess I) . ( 1 2 ) hleaqured using t h e trace protonated solvent peaks a s internal
s tandards (13) I, 11 Jackrnan, F Sondheimer, Y . Amiel, I ) A. Ben-Efra im, Y .
Garini, R . X'olovsky, and A A Bothner -By. J A m ( 'hem. SOL , 84, 4307 ( I R ( i 2 )
(14) J A Pople. TI;. G . Schneider, and H J Bcrnstein, "High~Iiesll lution Suclear Magnetic Resonance, " h1cGraw-Hill Book Co , Inc , S e w Yiirk,
( l i ) 'The point dipole approximation of Pople:l leads t o the iireriiction t h a t t h e protons in cyclononatetraenide should be more shielded t h a n those in cyclooctatetraenide b y 0 02 p.p rn
Fraenkel, R . E. Car te r , A. SlcLachlan, and J . H . Richards, J A m C h e m Soc , 82, 5840 ;19601; ( b ) T Schaefer and \V. G Schneider, CUM J Chem . 41, B( i l i (19f;3!.
( 1 7 ) I f t h e propurtionali ty cons tan t i- 10 i I " h t h e shift shrnlld be 1 .i 1 3 . 1 ) m . (18) T h e spectra are in agreement with t h a t repurted for tetraethyl-
amrncmiurn cgclononatetraenide. synthesized independently b y E A I,al,ancette a n d R . E. Benson. J . A m ('hein So i . , 8k, 2853 (1Ul i3) We thank theqe au thors for communicating this result t o us prior t o publication
N. Y , !Q.i8, p 180 ff
( I f ; ) (a, G