Embed Size (px)
DESCRIPTION
immunohistochemistry
Citation preview

UNIT 21.4ImmunohistochemistryFlorence M. Hofman1 and Clive R. Taylor1
1University of Southern California, Los Angeles, California
ABSTRACT
This unit describes several methods for localizing specific antigens in various tissue andcell preparations using immunohistochemistry (IHC). Protocols describe preparation ofsuitable material for IHC including fresh, unfixed, frozen tissue specimens; unfixed cells,either freshly isolated or derived from suspension or adherent cultures; or fixed, paraffin-embedded tissue sections. By careful selection of reagents, it is possible to detect twoor even three antigens simultaneously. For antigens that are sensitive to fixative, it maybe necessary to unmask the antigen by the antigen-retrieval technique. If there is cross-reactivity between the secondary antibody and antigens present in the target cells ortissue, the secondary antibody can be preabsorbed. Several new, sensitive amplificationtechniques are currently available. The different IHC protocols are represented schemat-ically and summarized in a table that also lists advantages and disadvantages of eachapproach. Causes of background staining and ways to eliminate it are also discussed.Curr. Protoc. Immunol. 103:21.4.1-21.4.26. C©2013 by John Wiley & Sons, Inc.
Keywords: immunohistochemistry � antigen retrieval � double staining � staining fixedtissues � staining frozen tissues � staining cell cultures � immunochemistry
INTRODUCTION
This unit describes several methods for localizing specific antigens in various tissue andcell preparations using immunohistochemistry (IHC). Suitable material for IHC includesfresh, unfixed, frozen tissue specimens (see Basic Protocol); unfixed cells, either freshlyisolated or derived from suspension or adherent cultures (see Alternate Protocol 1); orfixed, paraffin-embedded tissue sections (see Alternate Protocol 2).
Support Protocol 1 describes “antigen retrieval,” a technique that is especially useful forantigens that have been “masked” by fixation. If there is cross-reactivity between thesecondary antibody and antigens present in the target cells or tissue, the secondary anti-body can be preabsorbed; Support Protocol 2 provides a method that enables polyclonalsecondary antibodies to be absorbed with mouse serum to eliminate cross-reactivity.Cross-reactivity of the secondary antibody is usually a problem only in staining rat ormouse tissue, because these animals are the most common animal sources of MAbs. Itis particularly useful when monoclonal antibodies (MAbs) generated in rats are usedon mouse tissue or MAbs generated in mice are used on rat tissue. Support Protocol 3provides a strategy to help reduce background immunofluorescence staining, a problemthat is relevant for both frozen and formalin fixed sections.
By careful selection of reagents, it is possible to detect two or more antigens simultane-ously (see Alternate Protocol 3).
Success of immunostaining depends on a number of factors: the source, specificity,and quality of the primary antibody, and whether it is monoclonal or polyclonal; thecondition of the specimen; the fixation procedure; and the antigen-detection strategy.These components are highly interdependent (see Critical Parameters). The different IHCprotocols are represented schematically in Figure 21.4.1 and summarized in Table 21.4.1,
Current Protocols in Immunology 21.4.21-21.4.26, November 2013Published online November 2013 in Wiley Online Library (wileyonlinelibrary.com).DOI: 10.1002/0471142735.im2104s103Copyright C© 2013 John Wiley & Sons, Inc.
Microscopy
21.4.1
Supplement 103

Key
primary Ab labeled with enzyme
secondary Ab labeled with enzyme
treatment with biotinylated primary Abfollowed by the addition of avidin-enzymecomplex
treatment with biotinylated secondaryAb followed by addition of biotin-avidincomplex with the enzyme bound throughthe biotin
enzyme-substrate complex
avidin
biotin-avidin complex
biotin
avidin-enzyme complex
primary Ab secondary Ab
antigenic determinantson specimen
A
B
Figure 21.4.1 Diagrammatic illustration of (A) direct, (B) indirect, and (C) three-step immuno-histochemistry.
which also lists advantages and disadvantages of each approach. With each additionalstep there is an increase in sensitivity, but unfortunately this is offset by a possibleincrease in nonspecific background staining. Causes of background staining and ways toeliminate it are discussed in the Commentary (see Critical Parameters).
BASICPROTOCOL
IMMUNOHISTOCHEMISTRY FOR FROZEN TISSUE SECTIONS
The following is a typical immunohistochemistry (IHC) protocol for frozen sections oftissues from human and rodent sources; any frozen solid tissues can be used, including,spleen, thymus, and lymph nodes. Cells or tissues from other species may be studiedusing appropriate immunological reagents. Essentially, staining is detected by sequen-tial applications of a specific primary antibody and one or more reagents that amplify
Immuno-histochemistry
21.4.2
Supplement 103 Current Protocols in Immunology

Table 21.4.1 Summary of Basic Immunohistochemistry Protocols
Protocol Uses Disadvantages
Direct
Enzyme-conjugated primaryantibody
When primary antibody isproduced in the same speciesas tissue to be stained; fordouble staining; whensecondary antibody produceshigh background
Requires significantquantities of primaryantibody to directly label withthe enzyme. Gives weakstaining because of minimalamplification.
Two-step
Primary antibody +enzyme-conjugatedsecondary antibody orPrimary antibody +Polymer-conjugated withsecondary antibody and HRP
When quantity of primaryantibody is limited; when thedirect protocol does notproduce sufficient signal andfurther amplification isrequired.
May demonstrate increasedbackground due to secondaryantibody
Three-step
Primary antibody +biotinylated secondaryantibody + avidin-conjugatedbiotinylated enzyme complex
For increased signalamplification
May produce increasedbackground due toendogenous biotin
the signal and are ultimately linked to an enzyme. In the presence of the appropriatesubstrate, the enzyme causes precipitation of a colored reaction product at the site ofthe antigen. This protocol is written for a horseradish peroxidase (HRP)–based detectionsystem; however other enzymes and colored substrate combinations may be used (seeTable 21.4.2).
Materials
Tissue, freshly isolatedPBS, pH 7.4 (APPENDIX 2A)OCT embedding compound (Miles Labs)Liquid nitrogenPoly-L-lysine-coated slides (see recipe; also commercially available)Acetone (reagent grade) or 4% (w/v) paraformaldehyde/PBS0.3% (v/v) H2O2/PBSBlocking solution: 5% (v/v) goat serum or 1% (w/v) BSA in PBSPrimary antibody diluted in either PBS, 1% (v/v) goat serum/PBS, or 0.1% (v/v)
saponin/PBSAmplification system:
Biotin-avidin system, used for over 30 years because of its sensitivity,convenience, and economic value. As such the biotin-avidin protocol hasbecome the yardstick for measuring reactivity in the research setting. Touse the protocol below with the biotin-avidin system, a biotinylatedsecondary antibody directed against the species from which the primaryantibody was derived is required (Hsu et al., 1981).
Polymer-based detection system (available from multiple vendors), currentlypreferred over earlier biotin-based methods, especially for automatedsystems (Taylor and Shi, 2013; Fig. 21.4.2). The polymer-basedtechnology is useful because of lack of reactivity with endogenous biotin,and also greater stability and sensitivity. Polymer-based systems areavailable both for HRP and alkaline phosphatase (AP), as a second Microscopy
21.4.3
Current Protocols in Immunology Supplement 103

Table 21.4.2 Commonly Used Immunocytochemistry Enzymes and Substrates
Enzyme Substrate; color Advantages Disadvantages
Horseradishperoxidase
3,3′-Diaminobenzidine(DAB); brown
Intense color;permanent
Endogenous peroxidaseactivity
3-Amino-9-ethylcarbazole (AEC); red
Intense color;contrasts well withblue for doublestaining
Endogenous peroxidaseactivity
True Blue; blue Greatly increasedsensitivity
Most effective for fixedtissues only
4-Chloro-1-naphthol(4-CN); blue-black
Color contrast Soluble in alcohol
Vina green; green Color contrast Soluble in alcohol
Alkalinephosphatase
Nitroblue tetrazolium(NBT) and 5-bromo-4-chloro-3- indoylphosphate (BCIP); blue
Intense color Endogenous alkalinephosphatase activity
Blue Substrate; blue Less intense color;but better for doublestaining
Endogenous alkalinephosphatase activity
Fast red TR Red; red Color contrast fordouble staining
Endogenous alkalinephosphates activity
Glucose oxidase Nitroblue tetrazolium(NBT); blue
No endogenousenzyme activity
Low staining intensity(high concentration ofprimary and secondaryantibodies required foreffectiveness)
step 1primary antibody
step 2polymer containingsecondary antibody and enzyme
antigen
key:
enzyme
primary antibody secondary antibody
polymer, antibody, enzyme complex
Figure 21.4.2 Diagrammatic illustration of the two-step polymer staining technique.
Immuno-histochemistry
21.4.4
Supplement 103 Current Protocols in Immunology

enzyme for differential color staining, including double stains. To use thisprotocol with a polymer-based detection system, a polymer-conjugatedsecondary antibody/HRP complex is required.
HRP-conjugated avidin-biotin complex (ABC): e.g., ABC Elite (for use withhuman tissue) or ABC Standard (for use with mouse tissue; both from VectorLabs
Substrate (Table 21.4.1):Diaminobenzidine (DAB) is prepared according to manufacturer’s
instructions (several variants exist including proprietary formats)A number of additional substrates yielding differently colored reaction
products are available, including 3-amino 9-ethyl carbazole (AEC) (red),4-CN blue-black, Vina green for HRP, and Fast Red for alkalinephosphatase (AP)
Hematoxylin counterstain (Mayer’s; Sigma)Aquamount (Thermo Scientific Shandon)
Petri dishesAluminum foil, cut into �3.5 × 3.5–cm pieces and labeled on the outside using a
waterproof markerCryostatCoplin jar or staining dish with slide rackPap pen (Research Products International or Thermo Scientific Shandon)Staining chamber (Fig. 21.4.3A)Glass coverslipsClear nail polish (optional)
Freeze the tissue sample
1. Place the freshly isolated tissue in a petri dish containing PBS and cut into thedesired number of approximately 0.5-cm3 pieces.
2. Place one tissue piece into the center of a labeled, precut piece of aluminumfoil.
3. Apply OCT embedding compound to the fragment in sufficient quantity to com-pletely cover the tissue. Fold the foil to create a secure envelope.
OCT embedding compound is used to prevent dehydration at the edges of the sampleand to provide greater surface area for manipulation of the frozen tissue.
4. Drop the foil envelope directly into liquid nitrogen and allow the specimen toremain there for 5 to 10 min. Subsequently, remove the aluminum envelope withforceps and store at −70° or −120°C (for long-term storage).
Cryosection the tissue
5. To prepare the tissue for cryosectioning, bring the aluminum foil envelope contain-ing the tissue to the cryostat on dry ice (preferable to liquid nitrogen), and open theenvelope inside the cryostat.
The cryostat should always be at approximately −20°C and, like a refrigerator, shouldnot be turned off except for cleaning. The tissue should be left in the cryostat for about10 to 15 min to bring the tissue to −20°C, for easier cutting.
6. Cover the cryostat chuck with a thin layer of OCT embedding compound.Place the tissue on the OCT-coated chuck in the appropriate orientation. Allowthe tissue/OCT/chuck complex to freeze solid (�10 min at −20°C) inside thecryostat.
Microscopy
21.4.5
Current Protocols in Immunology Supplement 103

45 cm2 cm
30 cm
6 cm
tissue sections
frosted wax pen outline
A
B
glass slide
Figure 21.4.3 (A) Staining chamber. (B) Arrangement of sections on slide.
Covering the cryostat chuck is particularly important for such tissues as eye or brainwhere orientation is critical. The temperature should be between −18° and −20°C; anycolder or warmer makes cutting the tissue difficult.
7. Set the cryostat to cut 5- to 8-µm-thick sections.
If the tissue contains experimental cells that are particularly large (e.g., brain), 8- to10-µm-thick sections should be cut.
8. As the ribbon of tissue comes off the frozen block, place a poly-L-lysine-coatedslide under the sections and collect a maximum of three sections clustered togetheron each slide (Fig. 21.4.3B).
Due to the temperature differential, as the section touches the slide, the section willflatten and stick to the slide.
9. Allow the glass slide with the cryostat sections to air dry, preferably overnight atroom temperature.
Dried slides are ready for fixation.
Fix sections
10. Immerse the slides for 5 min in a Coplin jar or staining dish containing reagent-grade acetone. Allow the slides to air dry for 10 min at room temperature.
Immuno-histochemistry
21.4.6
Supplement 103 Current Protocols in Immunology

As a fixation procedure, acetone should always be tried first, because acetone pro-vides optimal retention of antigenic determinants and thus maximal staining. However,acetone fixation often distorts morphology, depending on the specific tissue used. Iftissue morphology is critical, paraformaldehyde fixation can be used. Fixation for 10min in 4% paraformaldehyde/PBS is an alternative, more gentle procedure that hasbeen shown to preserve some degree of antigenicity, although not as well as acetone.Paraformaldehyde, however, produces superior morphology (see Critical Parameters).
Coplin jars can be used for fixing and washing up to five slides; a staining dish witha slide rack can be used for up to 20 slides. These containers are useful because theyexpose the slides to a large volume of solution that completely covers the slides.
11. Using a Pap pen, outline the tissue sections on the glass slide (Fig. 21.4.3B).
Encircling the sections using a Pap pen (water-repellant wax) creates a boundary thatprevents the reagent from spreading over the entire surface of the slide. When thesections are outlined in this manner, small quantities of reagent (50 µl) can be used withminimal risk of drying.
Some antigens are not stable when stored, so it is recommended that slides be stainedas soon as possible after preparation and fixation to obtain optimal staining. If slidesmust be stored, storage at −70°C is usually adequate. However, it may be necessary totest slides over time to determine if and when a particular epitope loses its ability tobind antibody.
Stain the tissue
12. Place the slide in a staining chamber (Fig. 21.4.3A) and add PBS to cover the slide.Incubate 5 min at room temperature.
IMPORTANT NOTE: Take special precautions to prevent slides from drying duringthe staining procedure; it is best to add reagents to no more than three to four slidesat a time. When in doubt about drying, cover the slides with more reagent. Also, linethe staining chamber with paper towels soaked in sterile water to maintain a moistenvironment.
Use a squeeze bottle or pipet to add the PBS, but do not apply directly to the tissue,because the force of the liquid could lift up or destroy the tissue.
Perform all incubation and washing procedures in the staining chamber. If it is necessaryto interrupt the protocol, the slides may remain in PBS at any wash step for up to 2 hrwithout deleterious effects.
13. Remove the PBS by tipping the slide so liquid drains into the staining chamber.
14. Treat the slides with 0.3% H2O2/PBS for 5 min or until bubbling stops.
If bubbling has not stopped after 5 min, reapply H2O2/PBS for another 5 min and, ifnecessary, repeat until bubbling stops.
This incubation is used to eliminate endogenous peroxidase activity; if the enzyme to beused in amplification is not peroxidase, pretreatment with H2O2 is not necessary. Simi-larly, pretreatment with H2O2 is not needed if the tissue has no endogenous peroxidaseactivity. This can be determined by performing steps 22 and 23 of this protocol on thetest slide (this should take �20 min). If there is red precipitate on the slide (substratewithout antibody), then peroxide pretreatment is necessary.
15. Add blocking solution to the slide and incubate 15 min. Tilt the slide to remove thesolution. Before going to the next step (applying the antibody), remove as much ofthe liquid as possible without touching the tissue or letting the section dry.
Remove excess liquid by blotting the edge of the slide on a paper towel so the antibodywill not be unnecessarily diluted when it is added to the slide.
16. Apply diluted primary antibody in sufficient quantity to cover the tissue within thearea marked by the wax pen. Incubate 60 min at room temperature.
Microscopy
21.4.7
Current Protocols in Immunology Supplement 103

Depending on the specificity of the primary antibody, it may be necessary to optimizeworking dilution (see Critical Parameters). Optimization may be performed using cellsuspensions (see Alternate Protocol 1).
For convenience, it is possible to incubate the slides with primary antibody overnight(approximately 18 hr), at room temperature or at 4°C. For overnight staining, the work-ing dilution needs to be re-evaluated and usually modified by increasing the dilution.
Using mouse monoclonal primary antibody on mouse tissues presents a major problem(see Alternate Protocol 3). Several commercially available blocking kits have beendeveloped.
17. Wash the slide twice in PBS, each time for 10 min.
Amplify signal
18a. For biotin-avidin system: Apply diluted biotinylated secondary antibody to thetissue within the wax outline. Incubate 30 min at room temperature.
The use of the biotin-avidin system is has been the gold standard—however, due todecreased sensitivity, background in specific organs such as kidney and liver, and fewerincubation steps, the popularity of this system has decreased . However, this method isuseful for a wide variety of primary antibodies. Furthermore, biotinylated secondaryantibody can be diluted to the optimal concentration, making it more economical.Biotinylated antibodies that recognize mouse, rabbit, goat, and rat primary antibodiesare available.
The secondary antibody should recognize immunoglobulin (Ig) from the species that isthe source of primary antibody (e.g., biotinylated goat anti-mouse is used to recognizea mouse monoclonal primary antibody). Secondary rodent-specific polyclonal anti-sera may need to be preabsorbed against normal rodent serum to remove backgroundreactivity with rodent Ig antigens (see Support Protocol 2).
18b. For the polymer system: Apply diluted polymer-conjugated secondary antibody/HRP complex to the tissue section for 30 min at room temperature.
The polymer-based amplification technique is currently used in many research and mostclinical laboratories. This method is more sensitive than the biotin-avidin system dueto the increased numbers of peroxidase enzymes and secondary antibody moleculesbound to this structure. Background is reduced because no biotin is involved. Thepolymer system is a two-step technique since the secondary antibody and HRP are bothbound together. However the polymer reagent comes pre-diluted, and is therefore lesseconomical.
The polymer system is limited to use with rabbit and mouse primary antibodies.
19. Wash the slide twice in PBS, each time for 10 min.
Detect bound antibodies
20. For the avidin-biotin technique: Incubate the slides with avidin-biotin complex(ABC) conjugated to HRP for 20 min.
21. Wash the slide twice in PBS, each time for 10 min.
22. Add DAB or AEC substrate solution to the slide and incubate 5 to 10 min. Checkthe slide after 5 min for color development.
Always prepare the working substrate solution immediately before adding to the slide,and add H2O2 as the final ingredient. If this solution is not used within 5 min ofpreparation, the staining intensity will be significantly diminished.
To evaluate the slide, place a white paper towel underneath the slide and look for brownor red color. If the slide looks dark, stop the reaction immediately by washing the slidein tap water. To examine the slide more closely, tilt it to remove most of the liquid, then
Immuno-histochemistry
21.4.8
Supplement 103 Current Protocols in Immunology

add a coverslip and examine the slide using a light microscope. If the positive controlslide is understained, reapply a freshly prepared substrate solution.
23. Transfer the slide to a Coplin jar or staining dish and wash 10 min in runningtap water. Direct the stream of water at a corner of the container to provide goodcirculation and to avoid contact with the tissue on the slide.
Counterstain tissue
24. Immerse slide in hematoxylin counterstain for 30 sec to 2 min.
Hematoxylin is reusable and should be kept in a covered staining dish on the benchtop.However, carryover of water from the slides into the hematoxylin will eventually dilutethe stain. Thus, after five to ten uses, the incubation time required will steadily increase.When nuclear staining is no longer crisp and clear, the stain should be replaced withfresh hematoxylin.
25. Wash the slide 10 min in tap water.
26. Place 1 to 2 drops of Aquamount on the tissue.
27. Place a coverslip over the tissue and, if desired, seal with clear nail polish to preventdrying. Allow the nail polish to dry thoroughly before examining the slide under alight microscope.
Sealed slides can be stored indefinitely.
ALTERNATEPROTOCOL 1
IMMUNOHISTOCHEMICAL STAINING OF CULTURED CELLS
Immunohistochemistry (IHC) of cells in culture is an excellent system to complementstudies on tissue sections. Tissues provide a static picture of a process, and cultured cellscan provide insights into active processes. Single-cell preparations from cultured cellscan be used to evaluate a variety of immunologic reagents, including those that recog-nize cytokines and activation antigens as well as adhesion molecules. It is also easier toidentify the subcellular localization of particular antigens (e.g., nuclear, Golgi, or surface-associated antigens) in single-cell preparations. In addition, IHC of cell preparations, aswith tissue sections, can be combined with in situ hybridization, in situ apoptosis, ordouble immunostaining (see Alternate Protocol 3) to examine several parameters simul-taneously. Use of cultured cells versus tissue sections often facilitates visual interpretationof staining results when the multiple staining protocol (see Alternate Protocol 3) is beingperformed; cell preparations should be used to optimize conditions for different proce-dures, and may be used as a preliminary protocol for tissue staining. Cultured cells canalso be used as a positive control for determining the optimal concentration of antibodyfor a transiently expressed antigen such as adhesion molecules (e.g., E-selectin). Cells inculture are ideal for the study of transient antigens because these cells can be activatedand fixed at the appropriate time for optimal antigen expression. After the cells are ina single-cell suspension, a cytospin centrifuge is used to prepare slides. Fixation andstaining follow the procedure described in the Basic Protocol. Adherent cells can also bestained in situ.
Additional Materials (also see Basic Protocol)
Cultured cells in appropriate mediumCell suspension medium: any isotonic balanced salt solution (e.g., RPMI) with 1%
to 2% (v/v) FBS or 1% (w/v) BSAComplete medium with 10% (v/v) FBS (e.g., RPMI-10; APPENDIX 2A)0.5 mM EDTA (APPENDIX 2A) or 0.5% (w/v) trypsin/0.5 mM EDTA
Microscopy
21.4.9
Current Protocols in Immunology Supplement 103

Sorvall RT 6000 centrifuge and H-1000B rotor (or equivalent)Cytospin centrifuge (Shandon/Lipshaw), including chambers and filters
Additional reagents for counting viable cells by trypan blue exclusion (APPENDIX 3B)
1. Culture cells in the appropriate medium under conditions appropriate for the exper-iment.
Either suspension cells or adherent cells are suitable for IHC. Cells can be cultured inany of a variety of tissue culture vessels—Petri dishes or flasks. Adherent cells can alsobe cultured on sterile coverslips in dishes or in glass chambered slides.
2a. For adherent cells to be stained in situ: At the time appropriate for staining, washthe cells twice with PBS, each time for 10 min. Air dry the slides overnight at roomtemperature. Proceed with staining, beginning at step 7.
For cells grown in suspension cultures
2b. Harvest the cultures, count viable cells (APPENDIX 3B), and resuspend cells in cellsuspension medium at 0.5 × 106 viable cells/ml.
Cells should always be counted and percent viability determined using trypan blueexclusion as described in APPENDIX 3B, because dead cells usually exhibit nonspecificpositive staining.
To harvest adherent cells, wash the cells twice in cold PBS. Incubate the cells briefly in0.5% trypsin/0.5 mM EDTA or 0.5 mM EDTA (EDTA alone is preferred for studies ofcell-surface antigens because trypsin destroys many surface antigens). When the cellshave detached, add 2 to 3 vol complete medium that contains 10% FBS. Centrifuge thecells 10 min at 200 × g (1000 rpm in H-1000B rotor), room temperature. Resuspendthe cells in a small (1- to 5-ml) amount of cell suspension medium and determine theviable cell count. Adjust the volume of the cell suspension to give 0.5 × 106 viablecells/ml.
To harvest suspension cells, transfer cells and medium to a centrifuge tube and centrifuge10 min at 200 × g. Resuspend the cell pellet in a small amount of cell suspension medium(usually 1 to 2 ml) and determine the viable cell count. Adjust the volume of the cellsuspension to give 0.5 × 106 viable cells/ml.
3b. Assemble the cytospin apparatus, consisting of slide, filter, and sample chamber.
4b. Carefully resuspend the cells and add 150 to 200 µl (105 cells) to the cytospinchamber.
This cell number is appropriate for leukocytes; for larger cells such as endothelial cells,fewer cells (�5 × 104 cells/slide) should be used.
Cells should be added to the chambers as quickly as possible. If it takes >5 min tofill all the chambers, the cells will settle and cluster in the funnels and deposit on theslide as a densely packed crescent, rather than a single thin layer of cells in a circularpattern. It may be difficult to interpret the results when cells are densely packed on theslide.
5b. Centrifuge the cytospin assembly 5 min at �70 × g (800 rpm), room temperature.
6b. Remove the cytospin assembly, separate the slides from the filters, and allow theslides to air dry overnight at room temperature.
Dried filters can be reused 2 or 3 times.
7. Fix and stain the slides (see Basic Protocol, steps 10 to 27).
Immuno-histochemistry
21.4.10
Supplement 103 Current Protocols in Immunology

ALTERNATEPROTOCOL 2
IMMUNOHISTOCHEMISTRY OF FIXED PARAFFIN-EMBEDDEDSECTIONS
Formalin- or paraformaldehyde-fixed tissue retains excellent morphology; however, theantigen of interest may have been denatured or altered during the fixation process suchthat recognition by primary antibody is diminished or completely obliterated. This is truefor a wide range of antigens. This problem is addressed by the use of a combination ofrecently developed reagents specific for fixed antigens, as well as the “antigen retrieval”protocol for unmasking antigens (Table 21.4.3).
Table 21.4.3 Partial List of Antibodies that React withFormalin-Fixed Human Cell Determinants
Antigens Sourcea
T cell
CD3 D, N
CD4 N
CD6 B
CD43 B, D, N, S
CD45 RO D, S
B cell
CD20 B, D, N, S
CD21 D, N
CD45 RA B, D, N
CD23 N
Plasma cell B, D
Other leukocytes
CD68 (macrophages) B, D, S
CD15 (granulocytes) D, N, S
CD57 (natural killer) B, D, N
CD45 (common leukocyte) B, D, S
Miscellaneous
CD31 (endothelial cells) B, D, S
CD34 (endothelial cells) B, N
CD35 (dendritic reticulum) D
HLA Dr B, D, S
ICAM-1 B
Myelin basic protein B, D
bcl2 B, D, S
p53 B, D, S
Ki-67 (proliferation) B, D, S
PCNA (proliferation) B, D, S
Cytomegalovirus D, N, S
Epstein-Barr Virus D, N
Human immunodeficiency virus D
aCommercial suppliers: B = BioGenex; D = Dako; N = Novocastra Lab; andS = Signet Lab. See APPENDIX 5. Microscopy
21.4.11
Current Protocols in Immunology Supplement 103

Additional Materials (also see Basic Protocol)
Tissue: fixed, ideally in 10% (v/v) neutral buffered formalin or 4% (w/v)paraformaldehyde in PBS, pH 7.4 (APPENDIX 2A), paraffin-embedded, andsectioned using a microtome (5 µm thickness).
Staining dishes containing 100%, 95%, and 80% ethanolXylenesHistoclear0.3% (v/v) H2O2/methanol
1. Immerse the tissue fragment in 10% neutral buffered formalin or 4% paraformalde-hyde in PBS for 8 to 24 hr, depending on the size of tissue.
The optimal size for the tissue fragment is 5 × 5 × 3 mm. Tissue should not be left infixative for more than 48 hr. If tissue cannot be processed by 48 hr, then the tissue shouldbe transferred to 70% ethanol; the tissue can remain in this solution for several weeks,
If tissue is already embedded in paraffin, begin with step 4.
2. Process the tissue: Dehydrate 15 min in 80% ethanol; 15 and then 20 min in 95%ethanol; three times, 20 min each, in 100% ethanol; and finally, three times, 20 mineach, in xylenes. Embed tissue in paraffin and cut sections 5- to 8-µm thick. Place onpoly-L-lysine-coated or SuperPlus slides (as described above).
Tissue processing and sectioning are standard procedures and usually performed in adedicated histology laboratory on a fee-for-service basis.
3. After the tissue sections are applied onto the slide, place slides in the oven for 1 hr at60°C.
This drying procedure removes water from beneath the tissue section and makes the sectionadhere better to the slide.
4. Deparaffinize the slides by placing them for two 5-min changes each in Histoclear,100% ethanol, and 95% ethanol. Between changes, quickly blot the edge of the slideon a paper towel to remove excess liquid, but do not allow the slide to dry.
Histoclear is a substitute for potentially toxic xylenes.
Immersion steps are performed using either Coplin jars or staining dishes with slide racks,depending on the number of slides that are being processed. The containers are filled sothat the slides are completely immersed when they are inside the container. The solutionsare reusable; however, they must be replaced once or twice a week depending on thenumber of slides processed and the amount of liquid carried over into the containers withthe slides. Usually, the solutions are changed after every 50 slides. However, because thesereagents absorb water from the atmosphere, they should be changed at least every 2 weeksno matter how many slides have been treated.
5. To block endogenous peroxidase, incubate the slide 10 min in 0.3% H2O2/methanol.
This step should always be performed when peroxidase is the enzyme used for amplifica-tion; it should be omitted when other enzymes (Table 21.4.2) are used for detection.
6. Wash slides twice in PBS, 10 min each at room temperature.
7. Proceed with immunohistochemistry (see Basic Protocol, steps 15 to 27).
ALTERNATEPROTOCOL 3
IMMUNOHISTOCHEMISTRY TO DETECT TWO ANTIGENSSIMULTANEOUSLY
There are several features of immunohistochemistry that make it possible to use thismethodology to detect two antigens simultaneously. This method is often referred toas the double-staining method. Because this procedure uses the light microscope to
Immuno-histochemistry
21.4.12
Supplement 103 Current Protocols in Immunology

first primary antibody
Key
second primaryantibody
firstprimary antibody
firstsecondary antibody
tissue with two distinctantigens
biotinylated firstsecondary antibody
avidin-biotin-peroxidasecomplex + substrate
alkaline phosphatase- conjugatedsecond secondary antibody + substrate
alkaline phosphatase+ substrate
avidin/biotinperoxidase +substrate
avidin/biotin
avidinbiotin
secondprimary antibody
secondsecondary antibody
Figure 21.4.4 Double-staining (see Alternate Protocol 3).
visualize the reaction product, it is possible to identify reaction products in the contextof tissue morphology and topography based on their different colors. The staining resultsare relatively permanent. It is often possible to localize the antigens on a cellular orsubcellular level. There are a number of possible combinations of enzymes and substrates(see Table 21.4.2).
Several factors should be considered in designing a double-staining experiment. Therelative cellular locations of the antigens to be stained are important. In sequentialstaining, to reduce steric interference, intracytoplasmic or nuclear antigens should bestained before surface antigens are labeled. The primary antibodies for the two differentantigens must be from different species. Ideally, one primary antibody could be a mousemonoclonal antibody (MAb) and the second a rabbit polyclonal antibody. With thisarrangement, the two secondary antibodies would be specific for mouse Ig and rabbit Ig.The final consideration is whether the substrate used in the first staining process alters ordiminishes the antigenicity of the second antigen.
The double-staining procedure is essentially two single-staining procedures performedconsecutively (Fig. 21.4.4). To obtain excellent color contrast, the double-stainingprotocol described here uses alkaline phosphatase–conjugated secondary antibody and Microscopy
21.4.13
Current Protocols in Immunology Supplement 103

alkaline phosphatase blue substrate (Vector Labs), followed by horseradish peroxidase(HRP)–conjugated secondary antibody with AEC substrate (red; see Basic Protocol,steps 18 to 23). Other enzyme-substrate combinations are possible (see Table 21.4.2).Alternatively, two ‘stains’ may be performed together if a mouse primary antibody isused for one target antigen and a rabbit monoclonal antibody for the second. The de-tection system then employs both mouse and rabbit linker systems simultaneously, eachwith a separate chromogen. Rabbit monoclonal antibodies are widely available against agrowing range of antigens, making this the preferred method.
With automated staining platforms, many manufactures now offer ‘double staining’kits, designed to run on their specific platforms. Using these kits greatly simplifies theprocedure, and increases the chances of a good outcome. However, if these kits are used,the manufacturer’s instructions should be followed, although incubation times may beadjusted for different research applications.
Additional Materials (also see Basic Protocol)
Slides with cryostat sections (see Basic Protocol, steps 1 to 9), tissue culture cells(see Alternate Protocol 1, steps 1 to 6), or deparaffinized paraffin-embeddedsections (see Alternate Protocol 2, steps 1 to 6)
Primary antibodyAlkaline phosphatase–conjugated secondary antibody (Vector Labs)0.125 M levamisole/H2OAlkaline phosphatase Blue Substrate (Vector Labs or Kirkegaard & Perry)Staining chamber (Fig. 21.4.3) wrapped in foil
Stain with first antibody
1. Incubate slide of cryostat sections, cultured cells, or deparaffinized paraffin-embedded sections 30 to 60 min in the primary antibody for the first antigen.Include slides for appropriate controls (see Table 21.4.4). Wash slide twice in PBS,each time for 10 min.
The primary antibody used here should be prepared in a different animal than that usedto prepare the antibody detected using the HRP detection system.
2. Wash slide twice in PBS, each time for 10 min.
3. Incubate slide 30 min in alkaline phosphatase–conjugated secondary antibody.
The alkaline phosphatase–conjugated secondary antibody should recognize Ig from thespecies used to prepare the first primary antibody.
4. Wash slide twice in PBS, each time for 10 min.
5. Dilute 0.125 M levamisole 1:1 (v/v) in PBS. Incubate the slide 30 min in thissolution.
Levamisole blocks human alkaline phosphatase, which is present in a wide range of celltypes, but it does not inhibit bovine alkaline phosphatase, which is the enzyme conjugatedto the secondary antibody.
6. Prepare the alkaline phosphatase Blue Substrate according to the manufacturers’instructions.
The substrate should not be allowed to stand >3 min before it is applied to the slide.This substrate provides the best contrast to the red product produced by the horseradishperoxidase reaction.
7. Remove the slide from the levamisole solution and add the substrate solution imme-diately to the slide. Incubate 45 to 60 min in a foil-wrapped slide chamber at roomtemperature, examining the slide every 15 min for color development.
Immuno-histochemistry
21.4.14
Supplement 103 Current Protocols in Immunology
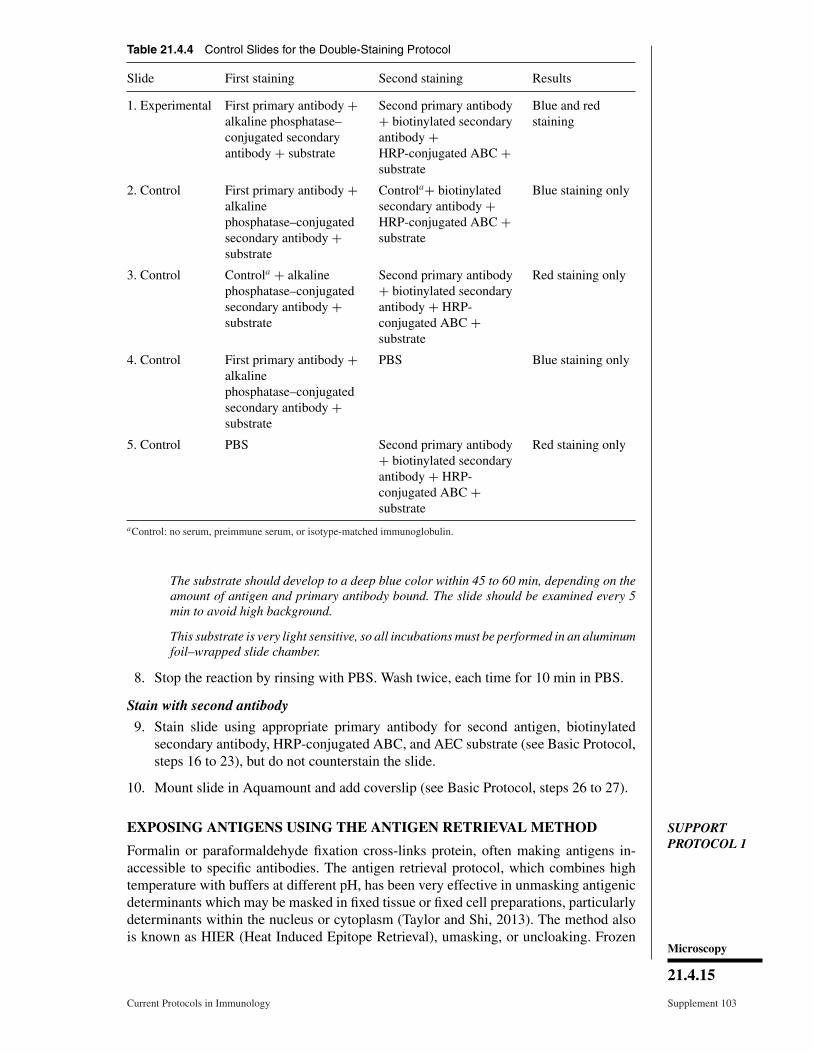
Table 21.4.4 Control Slides for the Double-Staining Protocol
Slide First staining Second staining Results
1. Experimental First primary antibody +alkaline phosphatase–conjugated secondaryantibody + substrate
Second primary antibody+ biotinylated secondaryantibody +HRP-conjugated ABC +substrate
Blue and redstaining
2. Control First primary antibody +alkalinephosphatase–conjugatedsecondary antibody +substrate
Controla+ biotinylatedsecondary antibody +HRP-conjugated ABC +substrate
Blue staining only
3. Control Controla + alkalinephosphatase–conjugatedsecondary antibody +substrate
Second primary antibody+ biotinylated secondaryantibody + HRP-conjugated ABC +substrate
Red staining only
4. Control First primary antibody +alkalinephosphatase–conjugatedsecondary antibody +substrate
PBS Blue staining only
5. Control PBS Second primary antibody+ biotinylated secondaryantibody + HRP-conjugated ABC +substrate
Red staining only
aControl: no serum, preimmune serum, or isotype-matched immunoglobulin.
The substrate should develop to a deep blue color within 45 to 60 min, depending on theamount of antigen and primary antibody bound. The slide should be examined every 5min to avoid high background.
This substrate is very light sensitive, so all incubations must be performed in an aluminumfoil–wrapped slide chamber.
8. Stop the reaction by rinsing with PBS. Wash twice, each time for 10 min in PBS.
Stain with second antibody
9. Stain slide using appropriate primary antibody for second antigen, biotinylatedsecondary antibody, HRP-conjugated ABC, and AEC substrate (see Basic Protocol,steps 16 to 23), but do not counterstain the slide.
10. Mount slide in Aquamount and add coverslip (see Basic Protocol, steps 26 to 27).
SUPPORTPROTOCOL 1
EXPOSING ANTIGENS USING THE ANTIGEN RETRIEVAL METHOD
Formalin or paraformaldehyde fixation cross-links protein, often making antigens in-accessible to specific antibodies. The antigen retrieval protocol, which combines hightemperature with buffers at different pH, has been very effective in unmasking antigenicdeterminants which may be masked in fixed tissue or fixed cell preparations, particularlydeterminants within the nucleus or cytoplasm (Taylor and Shi, 2013). The method alsois known as HIER (Heat Induced Epitope Retrieval), umasking, or uncloaking. Frozen
Microscopy
21.4.15
Current Protocols in Immunology Supplement 103

tissues or fresh cell preparations normally have well preserved and exposed antigens withminimal protein cross-linking, and they cannot withstand the high temperature requiredfor antigen retrieval.
With the growing use of automated staining systems many leading manufacturers nowsell ‘antigen retrieval’ solutions that have been formulated and proven for use with theirrespective systems. The use of these reagents should be considered in these circumstances;they also perform effectively in manual protocols.
Materials
Slides prepared from fixed tissue or cultured cells (see Alternate Protocol 1, steps 1to 5)
Sodium citrate buffer: 0.1 M citric acid/0.1 M sodium citrate, pH 6.0PBS (APPENDIX 2A)
Microwave oven, standard model operating at a frequency of 2450 MHz withhighest power setting 600 W
Plastic Coplin jars with vented screw caps
1. Wash slide prepared from fixed tissue or cultured cells in distilled water twice, 5 mineach.
2. Dilute sodium citrate buffer to 1 mM and fill plastic Coplin jars three-quarters full.
3. Add slide to Coplin jar of sodium citrate buffer and loosely cover the jar with a ventedscrew cap. Heat 5 min in a microwave oven set at the highest power setting.
4. Check the jar for fluid level and add distilled water if necessary to bring the level tothree-quarters full.
5. Heat the slide again in the microwave oven for 5 min at the highest power setting.
6. Remove the jar from the microwave oven and cool 15 min.
7. Wash the slides twice, 10 min each, in PBS.
The slide is now ready for immunohistochemistry (see Basic Protocol, steps 15 to 27).
SUPPORTPROTOCOL 2
ABSORPTION OF SECONDARY POLYCLONAL ANTISERA TOELIMINATE BACKGROUND
This absorption protocol is particularly useful when monoclonal antibodies (MAbs)generated in rats are used on mouse tissue or MAbs generated in mice are used on rattissue. Nonspecific background is due to the cross-reactivity of secondary antibodies withIg epitopes in the tissue specimen. For example, a polyclonal anti-rat reagent used as asecondary antibody may cross-react with naturally occurring mouse Ig epitopes in mousetissue, particularly in mouse hematopoietic organs. Therefore, the polyclonal secondaryantibody should be absorbed with mouse serum (Fig. 21.4.5). This cross-reactivity ofthe secondary reagent is usually a problem only in staining rat or mouse tissue, becausethese animals are the most common animal sources of MAbs.
Materials
Polyclonal secondary antibody: e.g., biotinylated goat anti–rat Ig or biotinylatedgoat anti–mouse Ig
Normal serum from the same species as the tissueSorvall RT 6000 centrifuge and H-1000B rotor (or equivalent)
1. Add undiluted polyclonal secondary antibody to an equal volume of appropriatenormal serum in a small microcentrifuge tube. Incubate 18 to 24 hr at 4°C.
Immuno-histochemistry
21.4.16
Supplement 103 Current Protocols in Immunology

unabsorbed biotinylatedpolyclonal goat anti-ratantibody with a mixtureof specificities
unabsorbed goat anti-ratantibody cross-reacts withmouse Ig epitopes (and ratprimary antbody)
mouse tissue
absorbed goat anti-ratantibody binds specificallyto rat MAb
mouse tissue
absorption removes goatanti-rat antibody thatcross-reacts with mouseIg epitopes
Keybiotinylated goatanti-rat antibodythat cross-reactswith mouse Igepitopes
biotinylated goatanti-rat antibody
primary monoclonalrat anti-mouseantibody
mouse tissue mouse Ig epitopes
mouseserum
Figure 21.4.5 Absorption of cross-reactive antibodies to epitopes in mouse tissue from sec-ondary reagents (see Support Protocol 2).
2. Centrifuge the mixture 10 min at 800 × g (2000 rpm in H-1000B rotor), roomtemperature.
3. Remove the supernatant and use at the appropriate dilution for the secondary reagent.
SUPPORTPROTOCOL 3
METHOD FOR REDUCING BACKGROUND STAINING
The ability to stain mouse tissues is important to the researcher. As mentioned previously,many primary antibodies are prepared as mouse monoclonal reagents. Thus, using anyanti-mouse secondary antibody on mouse tissue will result in unacceptable levels ofbackground. This is particularly relevant when staining immune organs, which expresshigh levels of Ig. This problem is important for both frozen and formalin-fixed sections,but to a lesser extent for cell preparations because cells can be washed to remove exoge-nous Ig. There are several approaches to reducing background Ig staining. The methodsuggested below uses a proprietary blocking reagent (M.O.M. Mouse Ig Blocking; Microscopy
21.4.17
Current Protocols in Immunology Supplement 103

Vector Laboratories) and biotinylated anti-mouse Ig as the secondary antibody. Thereare several approaches to reducing background Ig staining. The method suggested belowuses biotinylation of the primary antibody before using the reagent. This technique hasbeen somewhat successful if the manufacturer’s instructions are faithfully followed. Thestreptavidin-HRP reagent is used as the detection mechanism.
Additional Materials (also see Basic Protocol)
Mouse monoclonal primary antibody (e.g., Dako)M.O.M. mouse immunodetection kit (Vector Laboratories, cat. no. MKB-2213)Horseradish peroxidase–conjugated streptavidin (streptavidin-HRP; Life
Technologies, cat. no. S0911)
1. Follow steps 1 to 15 of the Basic Protocol.
2. Add mouse blocking reagent to the slide according to the manufacturer’s instructions.
3. Add mouse monoclonal primary antibody at the appropriate concentration and incu-bate for 30 min at room temperature.
4. Wash twice with PBS, each time for 10 min.
5. Add the secondary biotinylated anti-mouse Ig (from M.O.M. kit; follow manufac-turer’s instructions), and incubate for 30 min.
6. Wash twice with PBS, each time for 10 min.
7. Add enough streptavidin-HRP to the cover the specimen on the slide and incubate for30 min at room temperature.
8. Wash twice with PBS, each time for 10 min.
9. Continue with Basic Protocol, steps 21 to 27
REAGENTS AND SOLUTIONSUse deionized, distilled water in all recipes and protocol steps. For common stock solutions, seeAPPENDIX 2A; for suppliers, see APPENDIX 5.
Poly-L-lysine-coated slides
Clean glass slides in 70% ethanol and air dry. Dip the slides individually into poly-L-lysine solution (Sigma) diluted 1:10 in water for 5 min. Dry the slides >30 min ina 55°C oven. Store at 4°C and use within 1 week.
COMMENTARY
Background InformationIt has been almost 40 years since enzyme-
labeled antibodies were first introduced andused to identify specific antigens in rou-tinely processed tissues (Taylor, 1974). Thistechnique, immunohistochemistry (IHC), ismethodologically identical to immunofluo-rescence (IF), which has been in use since1941 (Coons et al., 1941). The increasingpopularity of IHC over IF is due to four mainfactors. First, IF requires special equipment,a fluorescence microscope, and a designateddarkened area. IHC, on the other hand, can beperformed using a standard light microscope.Second, IF requires a dark field, so it is not
possible to examine fluorescence patterns andcell and tissue morphology simultaneously.IHC utilizes a light microscope for visual-ization, so antigen localization and cell andtissue morphology can be observed at thesame time. Third, fluorochrome-based signalslast only 3 to 10 days, depending on the signalintensity and storage conditions. The coloredprecipitate in IHC staining, particularly with3,3′-diaminobenzidine (DAB; Table 21.4.2),remains vibrant for years and providesan excellent permanent record. Fourth, IFfluorochromes are light sensitive, so specialcare must be taken throughout the stainingprocedure to minimize loss of signal. The
Immuno-histochemistry
21.4.18
Supplement 103 Current Protocols in Immunology

enzymes and most substrates used in IHC, onthe other hand, are relatively light insensitive,and therefore the staining procedure can easilybe performed on the open benchtop withoutany unusual precautions. The disadvantage ofsome IHC chromogens is their known carcino-genicity, but using gloves when handling thesereagents is an adequate safety precaution. Theavailability of a variety of enzyme-conjugatedantibodies and colored substrates (see Ta-ble 21.4.2) has made IHC more convenientand accessible to many laboratories.
IHC procedures are continually beingmodified to accommodate new experimentalsituations. In the past, most specimensprocessed for IHC were fixed with formalin,and the antibodies used for detecting antigenswere polyclonal reagents made primarily inrabbits. These polyclonal antibodies identifieda wide range of epitopes and were specificallyselected for binding to epitopes that remainedresistant to the fixation procedures. Althoughthese polyclonal antibodies had a broad rangeof specificities, they frequently gave highlevels of background staining. The advent ofMAbs brought with it a vast improvementin the specificity of staining along with arelative elimination of background staining.This made it possible to detect such diverseelements as membrane receptors, secreted ex-tracellular products, and signal-transductionenzymes. Previously, the disadvantage of us-ing MAbs was that these reagents proved to beuseful only in analyzing fresh, acetone-fixedspecimens, and were not particularly effectivewith formalin-fixed specimens because manyof the epitopes detected by MAbs are formalinsensitive. This problem is being addressedby new methods that include enhanceddetection of relatively low levels of antigenand selective development of MAbs thatrecognize formalin-resistant epitopes. Thesetechniques have greatly expanded the useful-ness of IHC in a wide range of experimentalsituations.
Critical Parameters andTroubleshooting
Immunohistochemistry (IHC), like otherantibody-based techniques, requires carefulselection and titration of antibody reagentsand use of appropriate controls. Linscott’sDirectory of Immunological and BiologicalReagents provides a complete, regularly up-dated listing of commercially available anti-bodies, reagents, and kits. In addition, fixationand the method chosen for signal amplificationcan affect the results.
Primary antibodyThe critical issue in IHC is always the
specificity of the primary antibody. Therefore,whenever possible, MAbs should be used.Often, however, due to the lack of availabil-ity or the necessity for double staining, thisis not always possible. The recent availabilityof a growing range of rabbit monoclonal anti-bodies has greatly improved the situation, in-cluding the ability to perform double stains, asnoted previously. Staining human tissue withrabbit polyclonal antibodies often yields sub-stantial background, because rabbit Ig appearsto bind more readily to human Fc receptorsthan do sera from other species, but this is notan issue for rabbit monoclonals. If an effectivemouse or rabbit monoclonal antibody is notavailable, one option is to use a polyclonal an-tibody prepared in a species other than rabbit;goat and horse sera have been shown empir-ically to give the lowest background levels.However, if only rabbit primary antibodies areavailable, then blocking with 5% (v/v) normalgoat serum is useful in reducing background.If nonspecific staining persists, further dilu-tion of the rabbit primary antibody combinedwith an increase in the concentration of sec-ondary reagent is often effective. To determinewhether the staining observed with the primaryantibody is significant, normal serum from theanimal source of the primary polyclonal an-tibody, or an isotype-matched Ig for MAbs,should be used at a concentration equivalent tothat used in the experimental staining. The nor-mal serum or isotype-matched control slidesshould produce no staining.
The most common primary antibodies cur-rently in use are MAbs generated from mousehybridoma or rabbit cells. Mouse monoclonalantibodies work well on non-murine tissue,but are not usually useful for staining mousespecimens because the secondary antibodymust necessarily be an anti–mouse Ig, and thisresults in high background staining in mousetissue, particularly in hematopoietic tissue andin areas adjacent to blood vessels. Therefore,monoclonal or polyclonal antibodies from rab-bit are recommended when staining mouse tis-sue, even with tissues that are relatively im-munologically sequestered, such as brain. Analternative approach to staining mouse tissuewith a mouse primary MAb is to conjugateit directly with an enzyme (e.g., horseradishperoxidase, HRP) or linking reagent (e.g., bi-otin; see Table 21.4.1). Mouse MAb may beused on certain mouse cell lines (e.g., fibrob-lasts and T cell lines) because these cellsdo not normally have Ig on their surface. Microscopy
21.4.19
Current Protocols in Immunology Supplement 103

Nevertheless, when using mouse MAb onmouse cells, it is always necessary to in-clude a control slide that has been exposed tomouse serum or isotype-matched Ig in place ofthe primary antibody and subsequently treatedidentically. Such a control slide determinesthe extent of nonspecific background bind-ing of the anti-mouse secondary antibody andsubsequent reagents. Mouse tissue specimenscan, however, be successfully stained with ratMAbs, which are becoming more common.The most common secondary antibody for ratMAbs is goat anti–rat Ig. In some tissues,particularly mouse hematopoietic organs, thissecondary reagent can cause background stain-ing because of the cross-reactivity betweenrat Ig and mouse Ig. To identify nonspecificbackground in these experiments, use a con-trol slide in which PBS is substituted for theMAb during primary antibody incubation. Ifa diffuse positive reaction is observed, thesecondary antibody should be absorbed withmouse Ig to remove cross-reactive components(see Support Protocol 2). When mouse MAbsare used to stain rat tissue, the secondary goatanti-mouse reagent should be absorbed withrat serum. With the proper precautions andcontrols, MAbs can be used on rodent tissuesto obtain consistent, clear, and reliable results.
Antibody titrationThe optimal concentration of primary an-
tibody should be determined using a standardcheckerboard titration experiment, taking intoconsideration the manufacturers’ recommen-dations and the source of the reagent. ForMAbs derived from hybridoma supernatantsusing undiluted supernatant, 3- and 10-folddilutions are good starting points. For MAbsderived from ascites fluid, dilutions of 1:10,1:100, and 1:300 are more appropriate. Un-less instructed otherwise by the manufacturer,dilutions of �1:200 for biotinylated horseanti–mouse Ig and 1:400 for biotinylated goatanti–rabbit Ig are optimal. For polyclonal anti-bodies, dilutions ranging from 1:100 to 1:1000should be tested. Titration experiments shouldalways include appropriate controls—e.g., anirrelevant antibody or preimmune serum inplace of a primary polyclonal antibody or anisotype-matched Ig in place of primary MAb.Ideally, these controls should give no back-ground staining at the optimal concentration.The final working concentration for primaryantibodies used on cytocentrifuge preparationsis usually 2- to 3-fold higher than for the sameprimary antibody on tissue sections.
ControlsProper controls must always be included
with each staining procedure. Either preim-mune serum or isotype-matched Ig should beused as a negative control to ensure that thestaining observed is not due to background;a positive control slide should always be in-cluded in each staining experiment to verifythe activity of all the reagents.
In staining tissue, the specificity of the pri-mary antibody is critical and should be eval-uated on both positive and negative controltissues and cell preparations. To test reagentsthat supposedly detect antigenic markers ap-pearing only on activated cells, known posi-tively responding cells should be stimulatedand harvested at a time point that is ap-propriate in terms of response kinetics. Un-stimulated or nonresponding cells should becultured and harvested in parallel. Then, cyto-centrifuge preparations of the cells should bestained with the antibody in question. For ex-ample, to test the primary antibody for the ad-hesion molecule E-selectin, endothelial cellsshould be treated for 3 to 4 hr with an appropri-ate reagent (e.g., TNF or IL-1) to up-regulateexpression of the antigen. Both stimulatedand unstimulated cell preparations should bestained. Unstimulated cells should be rela-tively negative for E-selectin (<2% positive),and stimulated cells should be >50% posi-tive, suggesting that the primary antibody isspecific.
In general, activated human cell lines andperipheral blood leukocytes provide goodsources of positive controls for the titrationof many primary reagents. Activated mousespleen cells are usually a good source of pos-itive control murine cells. Untreated controland stimulated cell preparations should befixed with acetone and stored at −70°C forfuture use in reagent evaluation and titration.
It is important to control for endogenousperoxidase activity when HRP is used asthe detection enzyme, especially in tissuesdisplaying high numbers of macrophages orgranulocytes, as in inflammation. A simplepreliminary test for assessing the extent ofendogenous peroxidase is to hydrate a fixedslide with PBS, apply only the substrate (e.g.,AEC), and incubate for the appropriate reac-tion time (e.g., 10 min). The slides are thenwashed in water and examined for evidence ofstaining; there should be none. If there is reac-tivity, H2O2 pretreatment should be performedto eliminate endogenous peroxidase activity,and then the optimized pretreatment should beperformed on all experimental slides.
Immuno-histochemistry
21.4.20
Supplement 103 Current Protocols in Immunology

The double-staining technique requiresseveral controls to ensure accurate interpreta-tion of results. The control slides are describedin Table 21.4.4. Slide 1 is for the experimentalprocedure, and should always be performedin duplicate, with at least two specimens perslide. Slides 2 through 5 are control slides forvarious steps in the procedure. Slide 2 shouldshow staining equivalent to that of slide 4; thisdemonstrates that the second staining proce-dure does not give background (nonspecificred color) and that no antigen is lost during thelong staining procedure with the use of dif-ferent reagents. Slide 3 should be equivalentto slide 5; this shows that there is no non-specific blue staining from the first protocoland that the first staining procedure did notdestroy, alter, or diminish the antigenicity ofthe determinant recognized by the second pri-mary antibody. Use of alkaline phosphatasewith a blue substrate followed by peroxidasewith AEC has provided the best results.
In double staining, the order of antigensto be labeled should also be carefully consid-ered. Ideally, cytoplasmic antigens should bestained first and surface antigens stained sec-ond. The reverse approach may cause stericinterference. Optimal conditions for each anti-gen should first be determined using a single-staining protocol. If the antigens in questionare both cell-surface determinants, there maybe some degree of interference in double-staining experiments. This can be assessed byexamining control slides for single staining; asmentioned previously; there should be approx-imately the same number of positive cells onslide 4 as on slide 2 and on slide 5 as on slide3. The visible color is also an indicator of po-tential overlap in double staining: if the stainsappear bright red or blue, there is no overlapin staining, but if the color is purple, there issignificant overlap. If the results of a double-staining experiment demonstrate dual-stainingcells (e.g., staining with anti-CD4 and anti-IFN-γ), another set of slides where the knownoutcome is single-staining cells (e.g., stainingwith anti-macrophage and anti-IFN-γ) shouldbe double stained. By comparing the results ofthe two experiments, the individual patterns inthe dual-staining cells should be obvious.
For best results, when double-staining isused for IHC and in situ hybridization or insitu apoptosis, it is advisable to fix the tis-sue, continue with staining, and then begin thesecond procedure. If results with AEC (red)are too faint or undetectable, substitute DAB(brown) for the substrate. In situ hybridiza-tion or apoptosis use either peroxidase or alka-
line phosphatase as the precipitating enzyme.If DAB is used for IHC, alkaline phosphatasesubstrate (Vector Labs) is recommended as thesecond chromogen. For these experiments, thesame controls as described for double IHC (Ta-ble 21.4.4) are recommended to ensure properevaluation of results.
FixationOnce an antibody that recognizes a par-
ticular antigenic determinant based on othertechniques (e.g., immunoblot analysis) is iden-tified, the next step is to take a known pos-itive control cell preparation and fix it us-ing acetone. Acetone is a gentle fixative thatcauses minimal antigenic denaturation, mem-brane permeabilization that is limited but suf-ficient to allow antibodies to enter the cell,and fixation of the tissue that is sufficient forsome degree of morphological identification.Acetone also appears to be effective for local-ization of specific cell surface as well as cyto-plasmic antigens. In the author’s experience,acetone is the fixative of choice for determin-ing optimal antibody concentrations of the vastmajority of both monoclonal and polyclonalprimary antibodies.
Occasionally, acetone-fixed cells known tobe positive for a given antigen fail to yield apositive result. If acetone-fixed positive con-trols show no staining when, for example,these control cells are positive based on othercriteria—e.g., mRNA analysis, immunoblotanalysis, or ELISA—the time interval betweenpreparing the slides and staining may havebeen too long. For example, IFN–γ is a partic-ularly sensitive antigen and must be stainedwithin 24 hr after slide preparation. Othercytokines (e.g., IL-6) are more stable and canbe detected by IHC after slides have beenstored 1 or 2 weeks at room temperature.
Another possible cause for negative resultsusing a known positive control is that theantigen in question (e.g., intracellular cy-tokine) may have leaked out of the cell duringwashing steps because acetone fixation didnot adequately cross-link the cell membrane.In such cases, it is worthwhile to use 4%(w/v) paraformaldehyde/PBS, pH 7.4, anothergentle fixative that has been shown to preservesome antigenic determinants (e.g., IL-6,but not CD4 or CD8). Paraformaldehydefixation cross-links proteins, often sufficientlyto protect cytoplasmic antigens from beingwashed out of the cell. Electron microscope(EM)–grade paraformaldehyde does notappear to have a significant advantage overthe non-EM grade. If positive controls fixed
Microscopy
21.4.21
Current Protocols in Immunology Supplement 103

with paraformaldehyde continue to showno staining, the antibody solution may besupplemented with 0.1% (w/v) saponin or0.1% (v/v) Triton X-100 (final concentration).Because these detergents dissolve lipids, usingthem enhances membrane permeabilizationand removes lipids that may also obscureintracellular antigenic determinants. Saponinor Triton X-100 should also be present in thePBS washes between applications of primaryand secondary antibodies, as this eliminatesor decreases nonspecific background. Saponinshould be prepared as a 10% (w/v) stocksolution, and diluted in PBS on the day ofstaining.
Proteinase K treatment after fixation hasalso been used to increase access to antigens,especially nuclear antigens. The enzyme isusually added to the slides before applicationof the primary antibody. However, this pro-cedure is rather harsh, and requires carefulcalibration of treatment duration versus pro-teinase K concentration. First, try incubatingthe slide 10 min in 10 µg/ml proteinase K (di-luted in PBS) at room temperature. If the tissuecomes off the slide or background is high, firstdecrease the duration of the incubation, thendecrease the proteinase K concentration.
A problem that often occurs in formalin-fixed tissues is extensive denaturation of pro-teins. To determine whether the antibody of in-terest can be used following formalin fixation,one fresh positive sample (cells or cryostat sec-tions) is fixed with acetone and another is fixedwith formalin for �10 min. Both slides arewashed twice in PBS and subjected to IHC (seeBasic Protocol, steps 13 to 27). The stainingintensities of both slides are compared to de-termine the effect of formalin fixation, if any.Because formalin fixation affects many anti-gens and because formalin-fixed tissues arewidely used, many companies have made aneffort to identify and manufacture antibodiesto specific antigenic epitopes that are resistantto formalin fixation, and these antibodies canbe used specifically for formalin-fixed tissue(see Table 21.4.3).
In addition to these new reagents, theantigen retrieval method has become widelyused to “unmask” denatured or coveredantigens resulting from fixation. This methoddescribed in Support Protocol 1 uses a com-bination of high temperature and buffers ofdifferent pH to retrieve or unmask the antigenin formalin- or paraformaldehyde-fixed tissue(Shi et al., 1991). High temperature is usedto reverse cross-linking of proteins. Thebuffer most commonly used in this process is
1 mM sodium citrate buffer, pH 6.0, but 1 mMTris·Cl and 1 mM EDTA at pH 8.0 have alsobeen used. In general, higher-pH solutions aremore effective for intensifying staining, butcare should be used because they may causethe tissue to disintegrate and fall off the slideor occasionally produce nonspecific stainingof the nucleus. Studies have shown that evenusing distilled water (pH 6.0) on slides at hightemperatures enhances staining. The criticalhigh-temperature step is most often performedin a microwave oven at the highest power set-ting. High temperature can also be producedby autoclaving 10 min at 120°C, heating 2 minin a domestic pressure cooker at an operatingpressure of 103 kPa/15 lb/in.2, or heating 20to 40 min in a 100°C water bath. Each antigenmay require different conditions, so heatingconditions and buffer pH may need to beoptimized. Antigen retrieval is most effectivefor cytoplasmic determinants; however, someenhancement of surface receptors has been ob-served (Shi et al., 1995). Additionally, antigenretrieval has provided limited but substantialaccess to antigens in formalin-fixed archivaltissue.
Signal amplificationOnce the primary antibody titer has been
established and the proper fixation procedureidentified, optimal conditions for maximizingthe signal must be identified. There are severalways to intensify staining without increasingthe concentration of primary antibody. Theduration of the primary antibody incubationcan be increased from 30 to 60 min, oreven overnight at room temperature. For anovernight incubation, it is necessary to takeadditional steps to prevent slides from dryingout. Placing a paper towel saturated with ster-ile water in the bottom of the staining chamberis usually sufficient. If the slide has dried atany point during the procedure, it will exhibitdifferent kinds of nonspecific staining; e.g.,a pink to dark red haze over the entire tissue(with AEC substrate), a granular deposit overthe entire tissue, intense staining of nuclei,or staining around the edges of the tissue.Staining intensity may also be increased byincubating the slides 30 min at 37°C in anincubator or commercially available slidewarmer.
Another possible source of backgroundis the peroxidase enzyme. Peroxidase is anaturally occurring enzyme that is foundin macrophages and granulocytes. Endoge-nous peroxidase activity can be eliminatedsimply by a brief incubation in H2O2,
Immuno-histochemistry
21.4.22
Supplement 103 Current Protocols in Immunology

Table 21.4.5 Troubleshooting Guide for Immunohistochemistry
Problem Possible reason Solutions
Tissue has “lace-like”network appearance
Tissue was frozen improperly(e.g., too slowly), not coatedwell with OCT, or frozen andthawed
Exercise greater care infreezing (see Basic Protocol,steps 2-4)
Focusing on a single celllayer is impossible and/orhematoxylin is too dark
Tissue section is too thick Recut sections thinner(�10 µm); check thethickness designation oncryostat, it may be inaccurate
Positive cells are detectedafter incubation with only thesubstrate (negative control)
Endogenous peroxidaseactivity is present
Try longer or multiple H2O2
treatments; if endogenousperoxidase backgroundpersists (e.g., in bonemarrow), use another enzyme(e.g., alkaline phosphatase)
Tissue sections come off theslide during treatment
Slides were not properlycoated with poly-L-lysine
Retreat the same slides or usecommercially preparedprecoated microscope slides(e.g., Superfrost/Plus Slides,Fisher)
In cytocentrifuge preparation,the cells have accumulated ina crescent formation, all toone side
Cell suspension remained inthe cytocentrifuge chambertoo long
Prepare fewer slides at onetime
Entire tissue is brightlystaining
Slide has dried at some pointduring the procedure
Discard slides; avoidallowing the slide to dry
Edge of tissue is stainingintensely
Several reagents haveaccumulated under the tissue
Increase washing time aftersecondary antibody and ABCto 30 min
Staining with AEC isbrownish-yellow instead ofred
Too much substrate hasprecipitated at the antibodysite
Reduce primary antibodyconcentration �50%, untilAEC staining appears red
For a series of slides cut atthe same time, slides stainedimmediately are intenselypositive; slides stained within2 or 3 days are weak ornegative
This antigenic determinant isunstable and may have beenaltered because the sectionswere left at room temperaturefor too long
For consistently satisfactoryresults, perform staining onsections or cytospinpreparations within 24 hr offixation; if storage isnecessary, −70°C is usuallyadequate
Staining for a particularantigen appears weaker thanusual on the positive controlslide
Primary antibody wasimproperly handled—e.g., thereagent was left out on thebench for a long time orinadvertently frozen andthawed repeatedly
Primary antibody should bere-titrated
There is little or no stainingfor the antigen in question,and there appears to beprecipitate on the slide
Primary antibody may becontaminated
Use only sterile tips inhandling all reagents and/oradd 0.1% sodium azide toantibodies, but do not use incell culture studies; discardantibody if contamination isconfirmed
Continued
Microscopy
21.4.23
Current Protocols in Immunology Supplement 103

Table 21.4.5 Troubleshooting Guide for Immunohistochemistry, continued
Problem Possible reason Solutions
Faint background is seen allover slide
Blocking was insufficient Block with 5% goat serumbefore primary antibody isapplied, and dilute primaryantibody in 1% goat serum
Background staining is seenaround all blood vessels inmouse tissue
Absorption of secondaryantibody was insufficient
Absorb secondary antibodywith mouse serum overnightat 4°C (Support Protocol 2)
Staining is seen in theabsence of primary antibody
Secondary antibody isproducing background
Use 0.1% Triton X-100 inPBS during the washes afteraddition of secondaryantibody
Red particles are seen ontissue
AEC is precipitating in stocksolution
Prepare new AEC stocksolution
Red (AEC) color intensity isfainter than usual on positivecontrol
There is too little H2O2 in theAEC; too much liquid on theslide when AEC was added
Prepare new working AECsolution and treat again
Slides fade immediately afteradding coverslip
Mounting medium containsalcohol, which solubilizesAEC
Change to alcohol-freemounting medium or useDAB as substrate
Bubbles are seen on the slide Coverslip was not placed onthe slide slowly
Remove coverslip by placingslide in 37°C water until thecoverslip slides off, thencarefully add a fresh coverslip
(Note: do not removecoverslip manually as tissueswill come off)
leaving insignificantly low levels of back-ground reactivity.
The substrates for peroxidase are com-monly 3-amino-9-ethyl carbazole (AEC),which gives a bright red precipitate, and 3,3′-diaminobenzidine (DAB), which results in anintense brown precipitate. The DAB signal canbe intensified further with a nickel sulfate solu-tion. A new commercially available substratefor peroxidase, TrueBlue, from KPL increasesthe sensitivity of staining by 20- to 50-foldand provides a bright blue reaction product.However, the pH of the substrate solution israther acidic (pH 6.0), so it works best onparaffin-embedded, formalin-fixed tissue; it isnot suitable for frozen sections or cell prepara-tions. The peroxidase reaction develops into anintensely colored precipitate with a very lowrate of diffusion, thereby producing crisp stain-ing. The signal is long lasting, often vivid for6 months to several years. Alkaline phos-phatase or glucose oxidase may also be usedas the detection enzyme (see Table 21.4.2).
Although both alkaline phosphatase and glu-cose oxidase produce less intense signals thanperoxidase, these enzyme labels are used fordouble-staining experiments (Alternate Proto-col 3). A wide range of substrates that produceprecipitates of different colors (e.g., black,blue, red, purple) are available (Vector Labs,Dako) to produce specific contrasting signals.
Colloidal gold–labeled secondary antibod-ies, which are used for immunogold staining,are very sensitive and produce minimal back-ground. With the availability of smaller (1-and 5-nm) particles, these conjugates have be-come more useful (De Waele et al., 1991).However, silver enhancement is generally nec-essary to maximize contrast. The one advan-tage of the immunogold method is that eachblack dot theoretically represents a single anti-genic determinant; therefore, this techniqueis potentially more quantitative than enzy-matic methods, which depend on the concen-tration and duration of enzyme activity. Thegranularity of the staining product makes it
Immuno-histochemistry
21.4.24
Supplement 103 Current Protocols in Immunology

Figure 21.4.6 Immunohistochemistry of virus-infected mouse liver tissue. Tissue was stainedwith rabbit anti–lymphocytic choriomeningitis virus (LCMV) antibody using alkaline phosphatasesubstrate (blue) and mouse monoclonal anti-CD4 using peroxidase substrate (red) without coun-terstaining. Arrows indicate single positive cells with no overlap in staining. Magnification is 100×.For the color version of this figure, go to http://www/currentprotocols.com/protocol/im2104.
difficult to interpret the results, and methodsfor signal quantitation are still crude. For elec-tron microscopy, immunogold is an excellenttechnique.
For guidance concerning the problems mostoften encountered in IHC experiments andmethods for troubleshooting and resolvingthem, see Table 21.4.5.
Anticipated ResultsThe goal of an immunohistochemistry
(IHC) experiment is to obtain clearly definedstaining in the context of tissue morphologythat will be reproducible and remain readablefor years. There have been several attemptsto address the sensitivity of IHC. The variousstaining systems have been compared (Eliaset al., 1989), and the results indicate that thesensitivity of the primary antibody is the pri-mary critical parameter. With this in mind, itis not advisable to use IHC as a method forquantifying the amount of antigen at the cellu-lar level, but rather, it should be used as a tech-nique for demonstrating the absence or pres-ence of an antigen and its distribution. Percentpositive cells in cell preparations can be de-
termined by counting the number of positiveand negative cells and in tissue preparations bycounting the number of positive cells per unitarea. However, IHC is not suitable for deter-mining the actual number of molecules provid-ing the specific signal observed in either tissueor cell preparations. With the constant devel-opment of new reagents and protocols, the ver-satility and usefulness of this procedure shouldincrease. Figures 21.4.6 and 21.4.7 show re-sults of double-staining experiments.
Time ConsiderationsBoth cryostat sections and cytocentrifuge
cell preparations require an overnight dryingstep before fixation. The optimal three-stepprotocol (Basic Protocol) requires anywherefrom 4 hr to 24 hr, depending on the length oftime needed for primary antibody incubation.Double staining usually takes between a mini-mum of 8 hr and a maximum of 24 hr becauseit is not advisable to incubate overnight withboth primary antibodies. Thus, starting fromfresh tissue or cells, the entire procedure canbe completed within 48 hr.
Microscopy
21.4.25
Current Protocols in Immunology Supplement 103

Figure 21.4.7 Immunohistochemistry of brain tissue derived from animals with experimentalallergic encephalomyelitis. Tissue was stained with rabbit anti-nitrotyrosine using alkaline phos-phatase substrate (blue) and mouse monoclonal anti-CD11 using peroxidase substrate (red)without counterstaining. Arrows indicate positive double staining for a nitrotyrosine subpopula-tion of CD11-positive cells. Magnification is 400×. For the color version of this figure, go tohttp://www/currentprotocols.com/protocol/im2104.
Literature CitedCoons, A.H., Creech, H.J., and Jones, R.N. 1941.
Immunological properties of an antibody con-taining a fluorescent group. Proc. Soc. Exp. Biol.Med. 47:200-202.
De Waele, M., Renmans, W., Segers, E., Jochmans,K., Salmon, I., Depardieu, C., Dehou, M.-F.,and Van Camp, B. 1991. Leukemia and lym-phoma immunophenotyping in cell smears withimmunogold-silver staining. Am. J. Clin. Pathol.96:351-359.
Elias, J.M., Margiotta, M., and Gabore, D. 1989.A comparison of the peroxidase-anti-peroxidase(PAP), avidin-biotin complex (ABC), and la-beled avidin-biotin complex (LAB) methods fordetection of glucagon in paraffin-embedded hu-man pancreas. Am. J. Clin. Pathol. 92:62-67.
Hsu, S.M., Raine, L., and Fanger, H. 1981. Theuse of avidin-biotin peroxidase complex (ABC)in immunoperoxidase technique: A comparisonbetween ABC and unlabeled antibody (PAP)procedures. J. Histochem. Cytochem. 29:577-580.
Shi, S.R., Key, M.E., and Kalra, K.L. 1991. Antigenretrieval in formalin-fixed, paraffin-embeddedtissues: An enhancement method for immuno-histochemical staining based on microwaveoven heating tissue sections. J. Histochem. Cy-tochem. 39:741-748.
Shi, S.R., Imam, A., Young, L., Cote, R.J.,and Taylor, C.R. 1995. Antigen retrieval im-munohistochemistry under the influence of pHusing monoclonal antibodies. J. Histochem.Cytochem. 43:193-201.
Taylor, C.R. 1974. The nature of Reed-Sternbergcells and other malignant cells. Lancet 2:802-807.
Taylor, C.R. and Shi, S-R. 2013. Techniquesof Immunohistochemistry: Principles, pitfallsand standardization. In Comprehensive Di-agnostic Immunohistochemistry, Fourth edi-tion (D.J. Dabbs, ed.) pp. 1-40 Elsevier,New York.
Key ReferencesTaylor and Shi, 2013. See above.An in-depth historical background analysis andcritical overviews of theoretical and practical as-pects of immunohistochemistry techniques, with ap-pendix listings of commercial sources for antibod-ies, reagents, and equipment.This is an excellent chapter discussing the latestadvances in immunohistohemistry techniques, forthe laboratory investigator and clinical laboratory.The development of the protocols and troubleshoot-ing analyses are very useful for those planning toestablish these methodologies.Immuno-
histochemistry
21.4.26
Supplement 103 Current Protocols in Immunology





























