Embed Size (px)
Citation preview

Immunocytochemical Characterizationof Quisqualic Acid-
and N-Methyl-D-Aspartate-InducedExcitotoxicity in the Retina of Chicks
ANDY J. FISCHER,* RUTH L. PICKETT SELTNER, JOHNNY POON,
AND WILLIAM K. STELL
Department of Anatomy and Lion’s Sight Center, Faculty of Medicine, University of Calgary,Calgary, Alberta, Canada T2N 4N1
ABSTRACTA single, large dose of N-methyl-D-aspartate (NMDA) or quisqualic acid (QA) injected into
the chick eye has been shown previously to destroy many retinal amacrine cells and to induceexcessive ocular growth accompanied by myopia. The purpose of this study was to identifydistinct populations of retinal cells, particularly those believed to be involved in regulatingocular growth, that are sensitive to NMDA or QA. Two µmol of NMDA or 0.2 µmol of QA wereinjected unilaterally into eyes of 7-day-old chicks, and retinas were prepared for observation 1,3, or 7 days later. Retinal neurons were identified by using immunocytochemistry, and cellscontaining fragmented DNA were identified by 38-nick-end labelling in frozen sections. NMDAand QA destroyed many amacrine cells, including those immunoreactive for vasoactiveintestinal polypeptide, Met-enkephalin, and choline acetyltransferase, but they had littleeffect upon tyrosine hydroxylase-immunoreactive cells. Other cells affected by both QA andNMDA included those immunoreactive for glutamic acid decarboxylase, g-aminobutyric acid,parvalbumin, serotonin, and aminohydroxy methylisoxazole propionic acid (AMPA) receptorsubunits GluR1 and GluR2/3. Cells largely unaffected by QA or NMDA included bipolar cellsimmunoreactive for protein kinase C (a and b isoforms) and amacrine cells immunoreactivefor glucagon. DNA fragmentation was detected maximally in many amacrine cells and in somebipolar cells 1 day after exposure to QA or NMDA. We propose that excitotoxicity caused by QAand NMDA induces apoptosis in specific populations of amacrine cells and that these actionsare responsible for the ocular growth-specific effects of QA and NMDA reported elsewhere.J. Comp. Neurol. 393:1–15, 1998. r 1998 Wiley-Liss, Inc.
Indexing terms: retina; immunocytochemistry; quisqualic acid; N-methyl-D-aspartate;
excitotoxicity; myopia
Ocular growth is regulated precisely by visual experi-ence. Myopia (near-sightedness) resulting from excessiveocular growth is commonly studied in chicks and can beinduced by depriving the eye of patterned images. This iscalled ‘‘form-deprivation myopia’’ (FDM; for review, seeWallman, 1993). Myopia can also be induced by excitotox-ins, including kainate, quisqualic acid (QA), and N-methyl-D-aspartate (NMDA; Wildsoet and Pettigrew, 1988; Ehrlichet al., 1990; Fischer et al., 1997). Both form-deprivationand excitotoxins are thought to elicit their growth-alteringeffects by disrupting the activity of retinal neurons. It iscurrently believed that retinal modulation of ocular growthis mediated by subpopulations of amacrine cells, includingthose that release or respond to acetylcholine, dopamine,vasoactive intestinal polypeptide (VIP), and enkephalin
(ENK; Wallman, 1993; Seltner and Stell, 1995; Seltner etal., 1997). The administration of a large dose of NMDA orQA to the chick retina has been shown to leave photorecep-tor, bipolar, and ganglion cells intact but to destroy manyamacrine cells (Morgan, 1987; Barrington et al., 1989;Ehrlich et al., 1990; Tung et al., 1990; Sheppard et al.,
Grant sponsor: Medical Research Council of Canada; Grant sponsor: RoyAllen Fund; Grant sponsor: Marigold Foundation; Grant sponsor: Edwin L.and John E. Gustus Endowment in Vision Disorders.
*Correspondence to: Andy Fischer, Lion’s Sight Center/Department ofAnatomy, Faculty of Medicine, University of Calgary, 3330 Hospital Dr.NW, Calgary, Alberta, Canada T2N 4N1. E-mail: [email protected]
Received 14 July 1997; Revised 31 October 1997; Accepted 3 November1997
THE JOURNAL OF COMPARATIVE NEUROLOGY 393:1–15 (1998)
r 1998 WILEY-LISS, INC.

1991). The specific subsets of amacrine cells that aresensitive to NMDA or QA have not been identified. It ispossible that excessive ocular growth resulting from reti-nal exposure to excitotoxins is mediated by the differentialdestruction of amacrine cells that contribute to growth-regulating retinal pathways. Although the degree of myo-pia caused by excitotoxins is small (about 22 diopters)compared with that caused by form-deprivation (about210 diopters), excitotoxins have different effects on thegrowth-promoting pathways in the retina that are acti-vated by form-deprivation. Growth-promoting mecha-nisms are disrupted in NMDA-intoxicated retinas, becausemyopia can no longer be enhanced by form-deprivation(Fischer et al., 1997). In contrast, at least some growth-promoting mechanisms are intact in QA-intoxicated reti-nas, because myopia can be enhanced by form-deprivation(Erhlich et al., 1990; Fischer and Stell, unpublished re-sults). Therefore, it is expected that NMDA and QA mightdestroy different subtypes of amacrine cells involved inocular growth regulation, thereby differentially affectinggrowth-enhancing pathways that are activated by visualform-deprivation.
The purpose of this study was to identify subsets ofamacrine cells that might be involved in the retinal controlof ocular growth and the pathogenesis of myopia. To dothis, we characterized the effects of NMDA or QA on cellsbelieved to participate in the regulation of postnatal oculargrowth and FDM by using antisera raised against differ-ent cytological markers (either transmitters, enzymes,transporters, or receptors). We also characterized the timecourse of cell damage in retinal neurons treated withNMDA or QA by probing in situ for DNA fragmentation.We showed that most amacrine cells previously implicatedin growth regulation, including those immunoreactive forENK, choline acetyltransferase (ChAT), and VIP, are de-stroyed by QA as well as by NMDA, thereby making itunlikely that these cells participate in the progression ofFDM. However, dopaminergic amacrine cells survive expo-sure to both NMDA and QA; therefore, these cells, alongwith other unidentified subsets of amacrine cells, remainas candidates for growth-modulators.
MATERIALS AND METHODS
Animals
One-day-old male leghorn chickens (Gallus gallus domes-ticus) were obtained from Lillydale Hatchery (Linden,Alberta, Canada) and kept under fluorescent lights on acycle of 12 hours light, 12 hours dark (lights on at 7:00a.m.). Prior to experimentation, chicks were held for 1week in a stainless-steel brooder at about 25°C. Thereaf-ter, chicks were kept in clear Nalgene cages at an irradi-ance level of about 0.8 cd/m2, varying somewhat withdirection of gaze. Chicks received water and Purina chickstarter ad libitum.
Intraocular injections
Chicks were anaesthetized with 1.5% halothane in 50%N20 and 50% O2 prior to injection. Injections were madeinto the vitreous chamber by using a 25-µl Hamiltonsyringe with a 26-gauge needle. Penetration of the needlewas made consistently into the dorsonasal quadrant of theeye. The left eye (control) was injected with 20 µl of sterilesaline, and the right eye (treated) was injected with 20 µl
of 0.1 M (2,000 nmol) NMDA or 20 µl of 0.01 M (200 nmol)QA dissolved in sterile saline. Doses of NMDA and QAwere similar to those used in prior studies in chick eyes(Sattayasai and Ehrlich, 1987; Ehrlich et al., 1990; Tung etal., 1990; Sheppard et al., 1991). Assuming that thevolume of liquid vitreous within an eye was 150 µl, theinitial maximum concentration of NMDA presented to theretina was about 11.7 mM, and that of QA was about 1.17mM. All drugs were obtained from Research BiochemicalsInternational (Natick, MA).
Tissue dissection, fixation, and sectioning
Chicks were killed by chloroform inhalation. Then, eyeswere removed from the orbit, and most of the attachedconnective tissues and muscles were trimmed away. Eyeswere hemisected equatorially, and the gel vitreous wasremoved from the posterior eye cup. Eye cups were fixedfor 30 minutes at 20°C in 4% paraformaldehyde plus 2%sucrose in 0.1 M phosphate buffer, pH 7.4, except forsamples to be labelled with antisera to serotonin, g-aminobutyric acid (GABA), or glutamic acid decarboxylase(GAD), which were fixed for 24 hours at 4°C. Fixedsamples were washed three times in phosphate-bufferedsaline (PBS; 0.05 M phosphate buffer, 195 mM NaCl, and 3mM NaN3, pH 7.4), cryoprotected in PBS plus 30% sucrose,soaked in embedding medium (O.C.T. compound; Tissue-Tek, Elkhart, IN) for 10 minutes, and freeze-mounted ontosectioning blocks. Vertical sections 12–13 µm thick werecut consistently from the posterior pole of the eye in thenasotemporal plane near the dorsal edge of the pecten andwere thaw-mounted onto gelatin-coated glass slides. Sec-tions from control and treated eyes from the same indi-vidual were placed together in pairs on each slide to ensureequal exposure to reagents. Sections were air dried, ringedwith rubber cement to form a well for antibody solutions,and stored at 220°C until use.
Immunocytochemistry
Sections were washed three times in PBS, covered with150 µl of 5% normal goat serum (NGS) diluted in PBS plus0.3% Triton X-100, and incubated for 1 hour at roomtemperature in a humidified chamber. After incubation,the NGS solution was aspirated, and sections were coveredwith primary antibody solution (150 µl of antibody dilutedin PBS plus 0.3% Triton X-100 and 0.01% NaN3) andincubated for about 24 hours at 20°C in a humidifiedchamber. The sources and working dilutions of antibodiesare listed in Table 1. The slides were washed three times inPBS, covered with the secondary antibody solution (150 µlof 1:1,500 Cy3-conjugated goat-anti-rabbit or goat-anti-mouse immunoglobulin [IgG]; Biological Detection Sys-tems, Pittsburgh, PA; or 1:100 fluorescein isothiocyanate[FITC]-conjugated goat-anti-rat IgG; Sigma, Oakville, On-tario), and incubated for at least 2 hours at room tempera-ture in a humidified chamber. Finally, samples werewashed three times in PBS, rubber cement was removedfrom the slides, and sections were mounted under cover-slips in 4:1 (v/v) glycerol to water for observation under anepifluorescence microscope with FITC or rhodamine filtercombinations. Photographs were taken on T-Max 400 film(Kodak, Rochester, NY), and negatives were developed inT-Max Developer (Kodak) according to the manufacturer’sinstructions.
2 A.J. FISCHER ET AL.

Histology
Slides were warmed to 20°C, washed three times in PBSand incubated under 200 µl 0.1% (w/v) toluidine blue forabout 2 minutes. The stain was drained away, and slideswere washed three times in PBS and mounted, as de-scribed above, for microscopy in transmitted white light.
Labelling of fragmented DNA
Retinal sections were obtained as described above fromchicks 1, 3, and 7 days after NMDA or QA treatment.Slides were warmed to 20°C and washed once in PBS,followed by one wash in PBS plus 0.3% Triton X-100, andtwo more washes in normal PBS. Sections were thencovered with 100 µl of incubation medium (0.5 nmolCy3-conjugated dCTP, 20 units of 38-terminal deoxynucleo-tidyl transferase [Amersham, Little Chalfont, United King-dom], 100 mM sodium cacodylate, 2 mM CoCl2, and 0.25mM b-mercaptoethanol in sterile saline, pH 7.2) andincubated for 1 hour in a humidified chamber at 37°C. Thesections were then washed three times in PBS, mounted in4:1 (v/v) glycerol to water, and coverslips were added forobservation by epifluorescence with a rhodamine filtercombination.
Measurements, cell counts, and statisticalanalyses
Errors were calculated as the standard deviation of eachsample, which was comprised of at least six individuals pergroup. To compare data from treated and control eyes,statistical significance was assessed by using a pairedtwo-tailed Student’s t-test. Percentage inner plexiformlayer (IPL) depth was calculated as the distance from the
border of the IPL and the inner nuclear layer (INL) dividedby the total IPL thickness, multiplied by 100. All thicknessmeasurements were made from photomicrographs of cen-tral retina, whereas all cell counts were made from centralretina under the microscope of at least 100 cells perindividual on at least four different sections.
RESULTS
Histology
The appearance and thickness of retinal layers did notchange significantly in control eyes over the course of thestudy (Table 2). In contrast, the appearance and thicknessof retinal layers treated with QA or NMDA varied dramati-cally. One day after treatment with NMDA, there was alarge increase in the thickness of the IPL, whereas thethickness of other retinal layers was unchanged (Table 2,Fig. 1b). Three days after NMDA treatment, there wereareas of reduced thickness in the IPL accompanied byfolding and detachment of the retina, between which theretina remained attached and the thickness of the IPL wasnot significantly different from that in the control (Table 2,Fig. 1c). In contrast, in areas of retinal folding, thethickness of the INL was not significantly affected, but,between folds, the thickness of the INL was reduced (Table2, Fig. 1c). Seven days after NMDA treatment, the thick-nesses of both the IPL and the INL were severely reduced,whereas that of the optic fiber layer (OFL) was increased(Table 2, Fig. 1d).
One day after retinal exposure to QA, the thickness ofthe IPL was significantly increased, whereas that of theINL was reduced (Table 2, Fig. 1b). Three days after
TABLE 1. Antisera
Antiserum Antigen Species/type SourceWorkingdilution
1465 Choline acetyltransferase (ChAT) Rabbit polyclonal Dr. M. Epstein (U. of Wisconsin) 1:1,000a-GABA g-Aminobutyric acid (GABA) Rabbit polyclonal Chemicon (Temecula, CA) 1:100R24 GABA transporter (GAT-1) Rabbit polyclonal Drs. R. Jahn and L. Edelman (Yale) 1:1,0008305034 Glucagon Rabbit polyclonal Dr. J. Walsh (UCLA) 1:1,000634 Glutamic acid decarboxylase (GAD-65) Rabbit polyclonal Dr. C. Brandon (Chicago School of Medicine) 1:1,0009T Glutamate receptor 1 (GluR1) Rabbit polyclonal Chemicon 1:5025-7 Glutamate receptor 2/3 (GluR2/3) Rabbit polyclonal Chemicon 1:50LEP 100 Lysosomal membrane glycoprotein Mouse monoclonal Hybridoma Bank (U. Of Iowa) 1:501473 Met-enkephalin (ENK) Rabbit polyclonal Dr. J. Walsh 1:1,0003A10 Neurofilament-associated antigen (NAA) Mouse monoclonal Hybridoma Bank 1:50aPA Parvalbumin Mouse monoclonal Sigma (Mississauga, Ontario) 1:1,600RPN536 Protein kinase C (PKC) a and b isoforms Mouse monoclonal Amersham (Oakville, Ontario) 1:505-HT serotonin (5-HT) Rabbit polyclonal Inc Star (Stillwater, MN) 1:800S-10 Somatostatin Rat monoclonal Dr. A. Buchan (U. of British Columbia) 1:40016 Tyrosine hydroxylase (TH) Rabbit polyclonal Dr. W. Tank (U. of Rochester) 1:1,000VP31 Vasoactive intestinal polypeptide (VIP) Rat monoclonal Dr. A. Buchan 1:80
TABLE 2. Percent Thickness of the IPL, INL, and OFL After Treatment With NMDA or QA1
TreatmentRetinallayer
Time after treatment (days)
1 3 3 (Retinal fold) 7
NMDA IPL 174.5 6 26.8**** 105.3 6 6.3 66.0 6 11.2**** 37.4 6 7.0****NMDA INL 94.9 6 4.0* 68.6 6 12.0**** 94.8 6 12.6 74.2 6 8.4****NMDA OFL 94.5 6 19.3 102.4 6 32.9 207.7 6 33.2****QA IPL 169.1 6 8.0**** 123.4 6 9.5*** 55.8 6 4.8****QA INL 81.8 6 5.6**** 84.8 6 8.4** 82.7 6 3.2****QA OFL 124.5 6 21.8* 109.9 6 21.0 194.9 6 30.5****
1The percent thickness was derived by dividing the treated by the control measurements and multiplying by 100. Each sample is comprised of measurements from six individuals.Values are x 6 S.D. Significance was assessed by using a two-tailed Student t-test to compare differences between treated and control conditions at each time interval and for eachtreatment. IPL, inner plexiform layer; INL, inner nuclear layer; OFL, optic fiber layer; NMDA, N-methyl-D-apartate; QA, quisqualic acid.*P , 0.05.**P , 0.005.***P , 0.0005.****P , 0.0001.
QA- AND NMDA-INDUCED RETINAL EXCITOTOXICITY 3
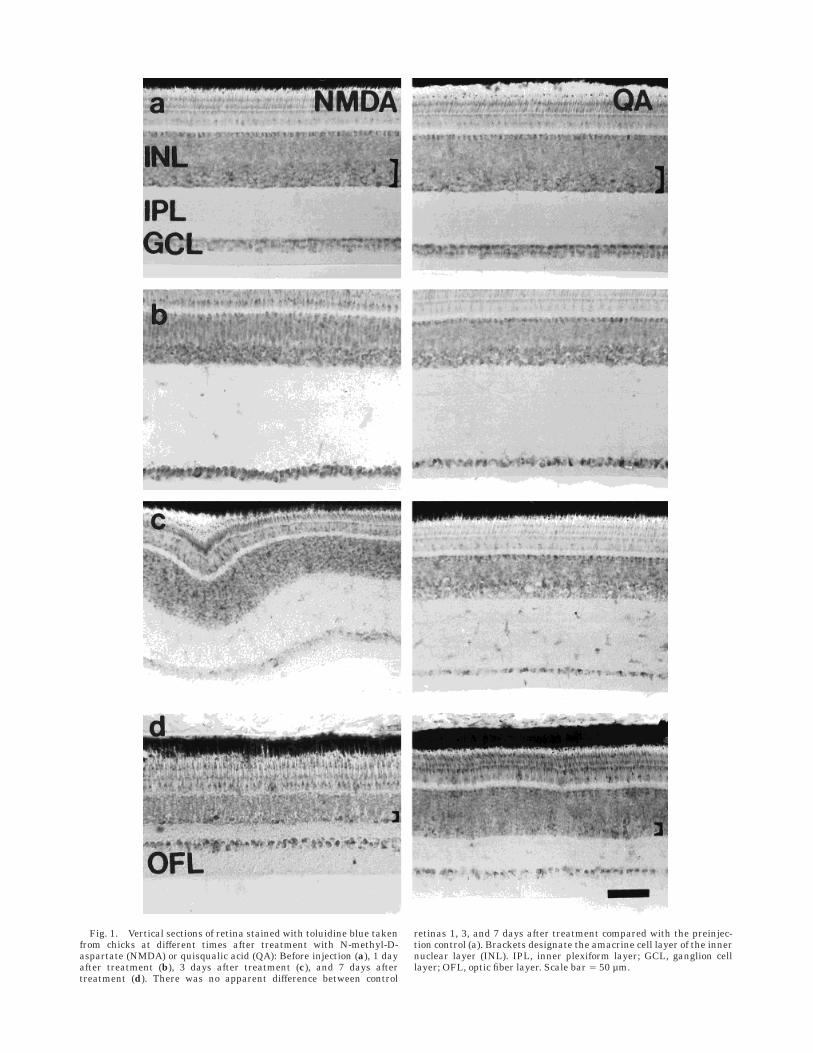
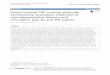
Fig. 1. Vertical sections of retina stained with toluidine blue takenfrom chicks at different times after treatment with N-methyl-D-aspartate (NMDA) or quisqualic acid (QA): Before injection (a), 1 dayafter treatment (b), 3 days after treatment (c), and 7 days aftertreatment (d). There was no apparent difference between control
retinas 1, 3, and 7 days after treatment compared with the preinjec-tion control (a). Brackets designate the amacrine cell layer of the innernuclear layer (INL). IPL, inner plexiform layer; GCL, ganglion celllayer; OFL, optic fiber layer. Scale bar 5 50 µm.

exposure to QA, the thickness of the IPL remained slightlyincreased, and the thickness of the INL remained reduced(Table 2, Fig. 1c). Seven days after QA treatment, thethicknesses of both the IPL and the INL were substantiallyreduced (Table 2, Fig. 1d), whereas the thickness of theOFL was nearly double that of the control (Table 2, Fig.1d). Twenty-one days after exposure to NMDA or QA, thereductions in thickness of the IPL and INL remainedunchanged from 7 days after treatment, whereas thethickness of the OFL approached that of control retinas(results not shown).
In all cases, QA- or NMDA-induced thinning of the INLappeared to result from reduced numbers of amacrine cellsin the proximal INL. Amacrine cells were distinguishedfrom other cells in the INL by their relatively intensestaining with toluidine blue and the known location ofamacrine cells in the proximal half of the INL. In addition,the number and density of cell bodies in the ganglion celllayer (GCL) appeared to be reduced by both QA and NMDAtreatments. The loss of amacrine and ganglion cell bodieswas not uniform across the retina. The loss of amacrinecells, in particular, was greatest in the nasal half of theretina near the site of the injection. In addition, 1 and 3days after QA or NMDA treatment, significant numbers ofirregularly shaped cell bodies were scattered throughoutthe IPL, which was practically acellular in control retinas(Fig. 1).
NMDA- and QA-induced changes in ENK,ChAT, VIP, and TH immunoreactivity
Immunohistochemical changes in retinas were testedonly 7 days after exposure to either NMDA or QA, becauseexcitotoxin-induced lesions have been shown to becomestabilized within 1 week after treatment (Dvorak andMorgan, 1983).
ENK. Antiserum to ENK labelled many cells that hadsomata near the middle of the INL and dendrites inseveral distinct sublaminae of the IPL (Fig. 2a). These cellsare the ENK-neurotensin-somatostatin-like-immunoreac-tive (ENSLI-IR) amacrine cells that have been describedpreviously (Brecha et al., 1979; Watt et al., 1985; Morganet al., 1994; Watt and Florack, 1994). After NMDA treat-ment, ENSLI cells disappeared almost entirely, with only13.2 6 7.4% of their cell bodies remaining detectable.ENK-IR neurites in the IPL were almost entirely disinte-grated, with remnants no longer clearly stratified andforming only short, narrow arbors extending into theproximal IPL (Fig. 2a). After retinal exposure to QA, fewerthan 1% of these ENK-IR amacrine cells remained (Fig.2a). In all cases of QA treatment (n 5 6), a small patch ofENK-IR amacrine cells (five to ten cells per section)remained near the central retina, medial to the optic nervehead. The dendritic arbors of these residual cells werenarrow but retained their stratification in the IPL (Fig.2a).
ChAT. Antiserum directed against ChAT revealed nu-merous cells in the INL and GCL as well as in two robustlylabelled strata in the IPL (Fig. 2b), exactly as describedpreviously. Millar et al. (1987) identified three subtypes ofChAT-IR amacrine cells in the chick retina, which includedtype I cholinergic amacrine cells, with cell bodies at theIPL/INL border and processes in stratum 2 (15–25%depth) of the IPL; type II cholinergic amacrine cells, withcell bodies in the GCL and processes in stratum 4 (65–75%depth) of the IPL; and type III cholinergic amacrine cells,
with cell bodies near the middle of the INL and processesdiffusely distributed in strata 1 (0–10% depth) and 3–5(45–85% depth) of the IPL. In the present study, we foundthat nearly half (46.2 6 9.1%; 937 cells counted from sevenindividuals) of the type III ChAT-IR amacrine cells werealso immunoreactive for somatostatin, i.e., ENSLI cells(Fig. 3), and that all ENSLI cells (n 5 433) were alsoChAT-IR. ENSLI and non-ENSLI type III cholinergicamacrine cells could be distinguished further by structuraland immunoreactive criteria. The type III non-ENSLI(type IIINE) cholinergic amacrine cells (ChAT immunoreac-tivity only) were weakly immunoreactive for ChAT andhad smaller, irregularly shaped cell bodies, with a thinlayer of cytoplasm surrounding the nucleus and a slenderprimary neurite. In contrast, the type III ENSLI (type IIIE)cholinergic amacrine cells were strongly immunoreactivefor ChAT and had larger, rounder cell bodies, a largercytoplasmic volume, and a thick primary neurite. Thesefindings indicate that ENSLI amacrine cells as well as afourth, unidentified type of amacrine cell may also utilizeacetylcholine as a neurotransmitter.
After NMDA treatment, there was a significant reduc-tion in retinal ChAT immunoreactivity. In all cases (n 5 7)type II, type IIINE, and type IIIE cholinergic amacrine cellbodies and neurites were almost entirely destroyed. Incontrast, type I cholinergic amacrine cells appeared to beonly moderately affected (Table 3, Fig. 2b). ChAT-IR neu-rites in the proximal IPL were nearly nonexistent, whereasthose arising from type I cells in the distal IPL appearedonly moderately less abundant compared with those incontrol retinas (Fig. 2b).
Following exposure to QA, nearly all type I cholinergicamacrine cells were destroyed or were entirely depleted ofChAT immunoreactivity (Table 3, Fig. 2b). Similarly, alltype IIINE cholinergic amacrine cells and most of the typeIIIE cells were no longer detectable (Table 3, Fig. 2b). In allcases (n 5 7), most type II cholinergic amacrine cells werelost; the residual type II cells were scattered across theretina (Table 3, Fig. 2b), except in the temporal retina(region farthest from the site of injection) where they weremore abundant and their processes in the IPL appearednearly normal (Table 3, Fig. 2b).
TH. Antiserum to TH labelled sparsely distributed cellbodies with neurites in three different strata in the IPL, at0–10%, 35%, and 75% depths (Fig. 2c), exactly as describedpreviously (Su and Watt, 1987). In all cases (n 5 8),treatment with NMDA caused little or no loss of TH-IRamacrine cells and some depletion of TH-IR neurites in thedistal IPL, whereas neurites in the more proximal layerswere less abundant and were compressed into the remain-ing IPL (Fig. 2c). Following exposure to QA, there wassome loss of TH-IR neurites and cells, mostly from thecentral retina (Fig. 2c).
VIP. Antiserum to VIP weakly labelled many cellbodies near the border of the INL and IPL in addition tosparsely distributed neurites at 0–10%, 35%, and 70% IPLdepths, as described previously (Fig. 2d; Brecha, 1983;Seltner and Stell, 1995). After NMDA treatment, morethan 90% of the VIP-IR amacrine cells were no longerdetectable, and the residual neurites in the IPL werealmost entirely disintegrated (Fig. 2d). Similarly, VIP-IRamacrine cells in the INL and neurites in the IPL were nolonger detectable after retinal exposure to QA (Fig. 2d).Depletion of ChAT, ENK, and VIP immunoreactivities was
QA- AND NMDA-INDUCED RETINAL EXCITOTOXICITY 5
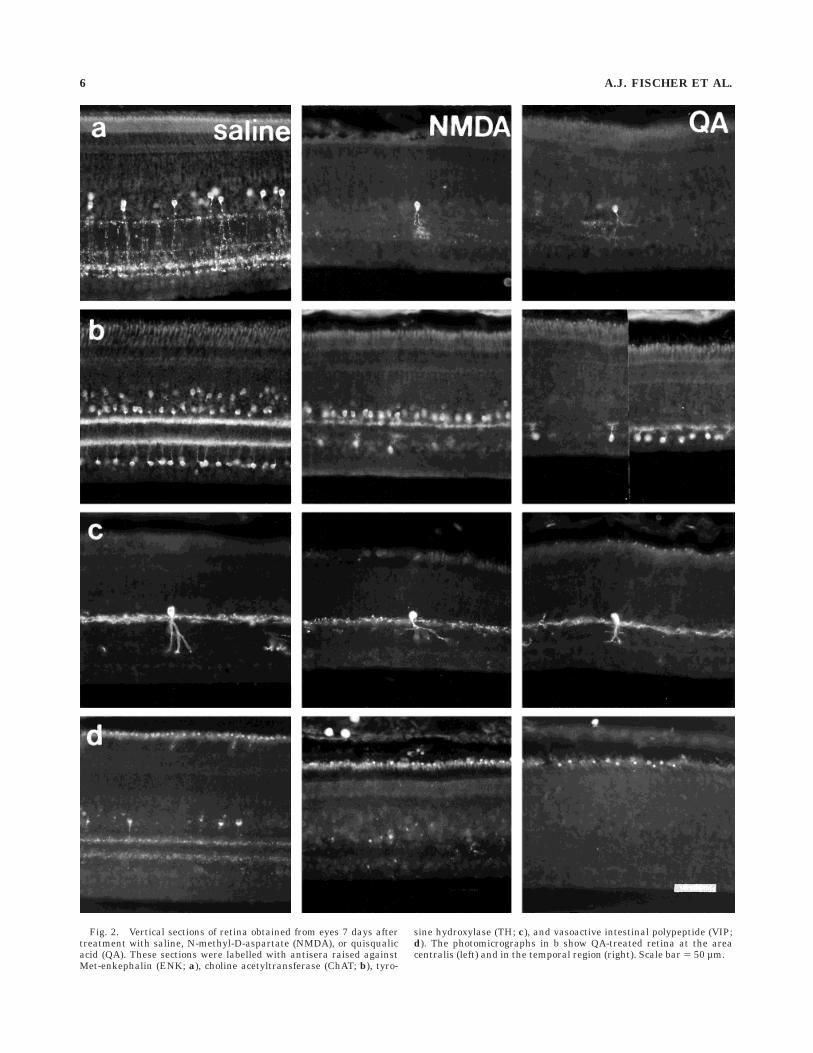
Fig. 2. Vertical sections of retina obtained from eyes 7 days aftertreatment with saline, N-methyl-D-aspartate (NMDA), or quisqualicacid (QA). These sections were labelled with antisera raised againstMet-enkephalin (ENK; a), choline acetyltransferase (ChAT; b), tyro-
sine hydroxylase (TH; c), and vasoactive intestinal polypeptide (VIP;d). The photomicrographs in b show QA-treated retina at the areacentralis (left) and in the temporal region (right). Scale bar 5 50 µm.
6 A.J. FISCHER ET AL.
evident as soon as 24 hours after treatment with NMDA orQA (results not shown).
NMDA- and QA-induced changes in otheramacrine cell markers
GABA. Numerous GABA-IR cell bodies were presentin the amacrine and horizontal cell layers of the INL and inthe GCL (Fig. 4a). Immunoreactive GABA was also de-tected in neurites throughout the IPL, in sparsely distrib-uted processes in the outer plexiform layer (OPL), and innerve fibers extending vertically through the INL to the
TABLE 3. Percentage of ChAT-IR Amacrine Cells Remaining 7 DaysAfter Retinal Exposure to NMDA or QA1
Type of cholinergiccell NMDA-treated QA-treated
Type I 78.8 6 13.9* 3.8 6 5.9**Type II 27.4 6 26.3** C: 12.4 6 7.8**
P: 94.2 6 4.8**Type III (a 1 b) 8.4 6 5.9** 3.2 6 4.1**
1C, central retina; P, peripheral retina; ChAT-IR, choline acetyltransferase-immunoreac-tive.*Significance of difference from control retinas: P , 0.01.**P , 0.0005.
Fig. 3. Vertical section of retina from an untreated eye that wasdouble-labelled with antisera raised against choline acetyltransferase(ChAT; a) and somatostatin (b). The same section was photographedthrough fluorescein isothiocyanate (FITC; a) and rhodamine (b) filter
sets. Arrowheads indicate cells that contain both ChAT and somatosta-tin immunoreactivity. INL, inner nuclear layer; IPL, inner plexiformlayer; GCL, ganglion cell layer. Scale bar 5 50 µm.
QA- AND NMDA-INDUCED RETINAL EXCITOTOXICITY 7
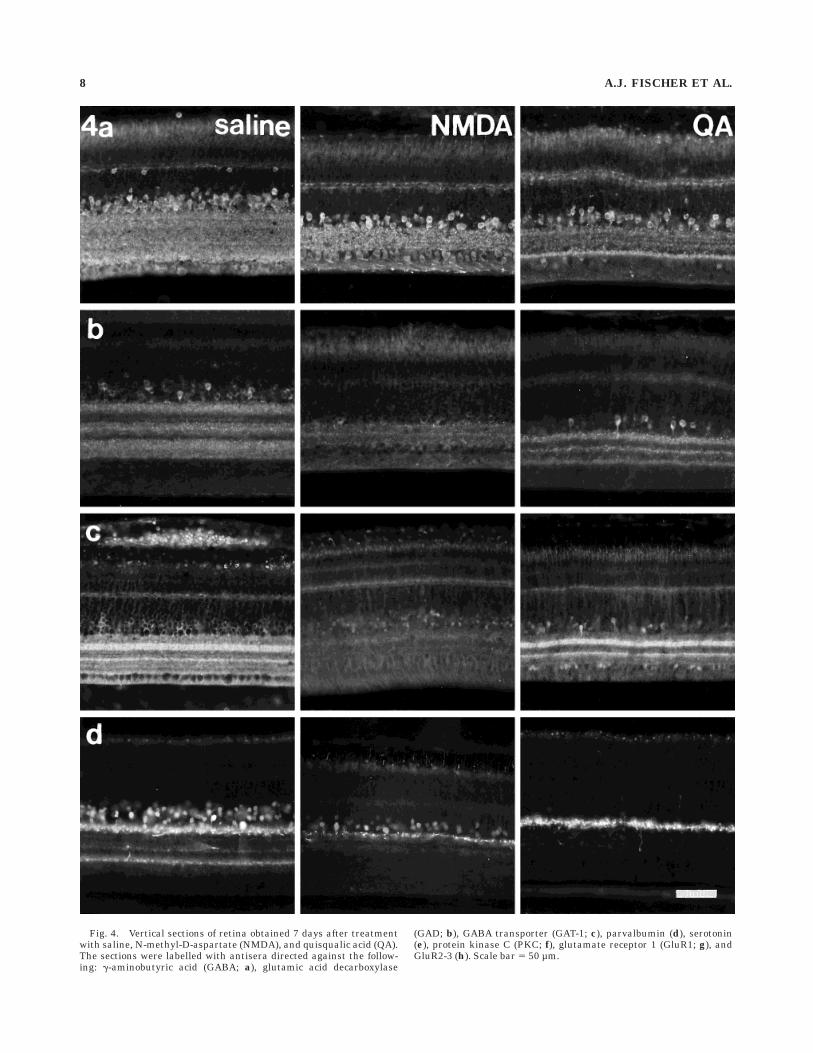
Fig. 4. Vertical sections of retina obtained 7 days after treatmentwith saline, N-methyl-D-aspartate (NMDA), and quisqualic acid (QA).The sections were labelled with antisera directed against the follow-ing: g-aminobutyric acid (GABA; a), glutamic acid decarboxylase
(GAD; b), GABA transporter (GAT-1; c), parvalbumin (d), serotonin(e), protein kinase C (PKC; f), glutamate receptor 1 (GluR1; g), andGluR2-3 (h). Scale bar 5 50 µm.
8 A.J. FISCHER ET AL.
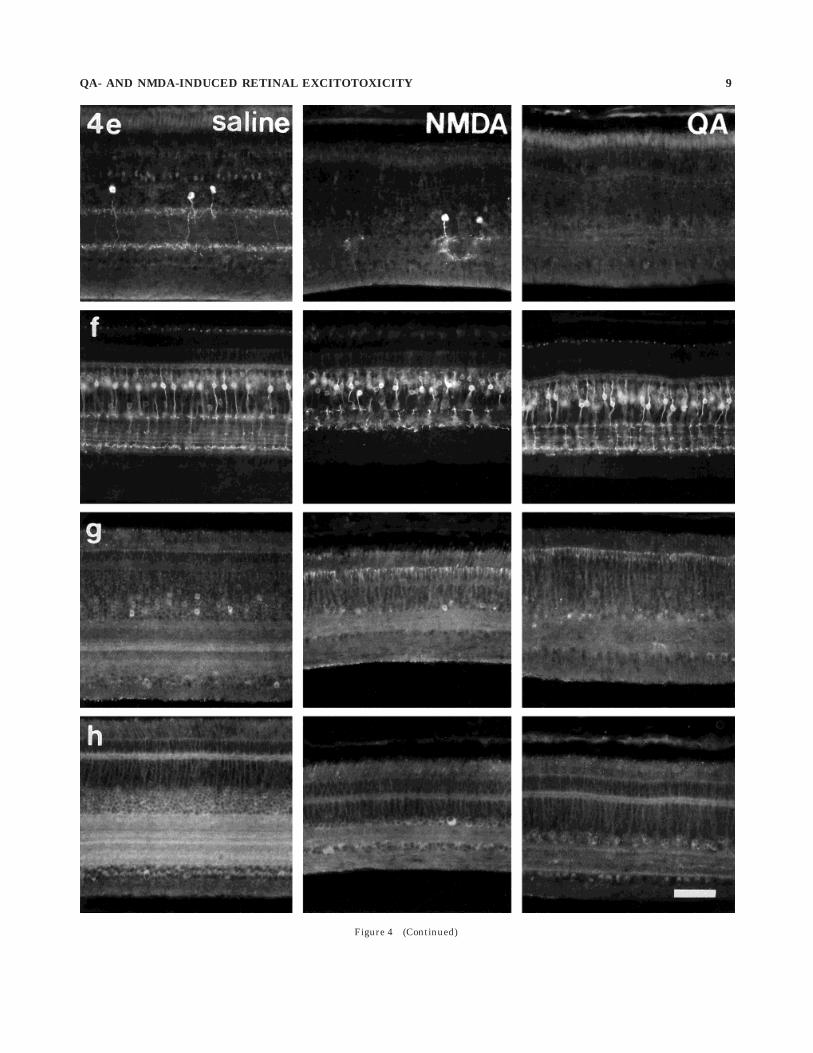
Figure 4 (Continued)
QA- AND NMDA-INDUCED RETINAL EXCITOTOXICITY 9

OPL (Fig. 4a). NMDA treatment destroyed many GABA-ergic amacrine cells in the INL, whereas GABA immunore-activity in the OPL, horizontal cells, and cells in the GCLappeared unaffected (Fig. 4a). Although the labellingintensity of GABA-IR dendrites in the IPL was unchangedby exposure to NMDA, stratification was no longer evident(Fig. 4a). Similarly, after retinal exposure to QA there wasa substantial loss of GABA-IR amacrine cells and cells inthe GCL, no apparent loss of GABA-IR horizontal cells,and a significant depletion of GABA immunoreactivity inthe IPL from 0% to 85% depth (Fig. 4a).
GAD. Some amacrine cells, many cells in the GCL, andstrata at 0–20%, 35–60%, and 70–100% IPL depths wereGAD-IR (Fig. 4b). NMDA treatment greatly reduced num-bers of GAD-IR amacrine cell bodies and abolished mostGAD immunoreactivity in the IPL (Fig. 4b). In contrast,there was little loss of GAD-IR amacrine cells in QA-treated retinas and only partial loss of GAD-IR neurites,mostly in the two proximal strata of the IPL (Fig. 4b)
GABA transporter 1. Immunoreactivity for GABAtransporter 1 (GAT-1) was seen in many amacrine cells, insome cells in the GCL, and in two broad strata at 20–35%and 50–55% IPL depths (Fig. 4c). Following treatmentwith NMDA, little GAT-1 immunoreactivity remained inthe IPL, and most GAT-1-IR amacrine cells were no longerdetectable (Fig. 4c). After treatment with QA, GAT-1-IRstrata remained in the IPL, whereas many GAT-1-IRamacrine cells were lost, and those remaining appeared tobe hyperimmunoreactive (Fig. 4c).
Parvalbumin. Parvalbumin immunoreactivity waspresent in numerous amacrine cells, in densely innervatedstrata at 0–10% and 70% IPL depths, and in a weaklyinnervated stratum at about 40% IPL depth (Fig. 4d).NMDA treatment resulted in the destruction of many(43.0 6 14.7%; n 5 6) parvalbumin-IR amacrine cells andof most neurites in the proximal IPL but left processes inthe distal IPL intact (Fig. 4d). After QA treatment, parval-bumin was detected in only a few (,1%) weakly immunore-active amacrine cells and in neurites at the IPL/INLborder (Fig. 4d). These neurites were also immunoreactivefor neurofilament-associated antigen (results not shown),and therefore might originate at least in part from dis-placed ganglion cells.
Serotonin. Serotonin immunoreactivity was presentin sparsely distributed amacrine cells, with cell bodieslocated near the middle of the INL and neurites ramifyingin strata at 0–15% and 65–85% IPL depth, as well as someweakly immunoreactive bipolar cells (Fig. 4e), as describedpreviously (Millar et al., 1988). After exposure to NMDA,most serotonin-IR amacrine cells (73.6 6 25.0%; n 5 6)were undetectable, and the density of serotonin-IR pro-cesses within the IPL was severely reduced (Fig. 4e). AfterQA, serotonin-IR amacrine cells in the INL and theirneurites in the IPL were entirely absent (Fig. 4e). Both QAand NMDA had little or no effect on serotonin-IR bipolarcells.
Protein kinase C a and b isoforms. Antiserum toprotein kinase C (PKC) labelled two different types ofbipolar cells. The first, weakly immunoreactive bipolar celltype had small somata in the distal INL, whereas thesecond, strongly immunoreactive bipolar cell type hadlarger somata near the middle of the INL and sent axonterminals mainly into the distal (0–10% depth) and proxi-mal (80–90% depth) levels of the IPL (Fig. 4f). PKCimmunoreactivity was also present in three weakly immu-
noreactive strata at 45%, 65%, and 75% IPL depths(Fig. 4f). In addition, some amacrine cells were also weaklyimmunoreactive for PKC (Fig. 4f). This distribution closelymatches that described previously (Negishi et al., 1988).NMDA and QA treatments did not significantly affect theabundance or distribution of either type of PKC-IR bipolarcell, except as a consequence of the reduction in INL andIPL thickness (Fig. 4f). However, following exposure toNMDA or QA, PKC-IR amacrine cells were less abundant.
AMPA-type glutamate receptors. Antiserum to gluta-mate receptor 1 (GluR1) labelled some ganglion andamacrine cells and weakly labelled strata at 0–15%,40–60%, and 70–100% IPL depths (Fig. 4g). After treat-ment with NMDA, immunoreactivity remained in the IPL,but lamination was no longer evident, and GluR1-IRamacrine and ganglion cells both appeared unchanged(Fig. 4g). In contrast, QA treatment abolished most GluR1immunoreactivity in the retina (Fig. 4g).
Antiserum to GluR2/3 labelled most if not all amacrineand ganglion cells and diffusely labelled the IPL withimmunoreactivity concentrated at 0–10%, 40%, 55%, and75% IPL depths (Fig. 4h). Anti-GluR2/3 also weakly la-belled the OPL and produced punctate labelling in theOFL (Fig. 4h). After NMDA treatment, stratification ofGluR2/3 immunoreactivity in the IPL was lost, and manyGluR2/3-IR amacrine cells also disappeared from the INL(Fig. 4h), whereas GluR2/3 immunoreactivity remained inthe GCL, OPL, and in putative displaced ganglion cells inthe INL (Fig. 4h). QA treatment resulted in the loss ofmany cells immunoreactive for GluR2/3 and the loss ofimmunoreactive strata from the IPL (Fig. 4h).
Glucagon. Antiserum to glucagon robustly labelledsparsely distributed amacrine cells with large somata atthe IPL/INL border, one densely innervated stratum at0–10% IPL depth, and one sparsely innervated stratum at35% IPL depth (results not shown), as described previ-ously (Kiyama et al., 1985). After exposure to NMDA orQA, the abundance and labelling intensity of glucagon-IRsomata and processes at the 0–10% level of the IPL wereunaffected, whereas there was a slight reduction in theabundance of glucagon-IR neurites at 35% IPL depth(results not shown).
Cells responding to QA or NMDAby fragmentation of nuclear DNA
No nuclei were labelled for DNA fragmentation inretinas from control eyes at any time following treatment(Fig. 5a). Four hours after injection of NMDA, manyscattered nuclei in the INL, mostly in the amacrine celllayer, contained fragmented DNA (results not shown). Oneday after exposure to NMDA, DNA fragmentation wasdetected in increased numbers of nuclei in the amacrinecell layer of the INL as well as in a few nuclei in both theGCL and the bipolar cell layer (Fig. 5b). About 45% of allamacrine cells were nick-end-labelled 1 day after NMDAtreatment, with the greatest density near the site ofinjection (nasal retina) and lower densities farther fromthe injection site (toward temporal retina). Three daysafter NMDA treatment, DNA fragmentation was stilldetectable, mostly in amacrine cells. Less than 10% of theremaining amacrine cells as well as a few cells (,1%) inthe bipolar cell layer of the INL contained fragmentedDNA (Fig. 5c). Seven days after NMDA treatment, frag-mented DNA was present in less than 1% of the residual
10 A.J. FISCHER ET AL.
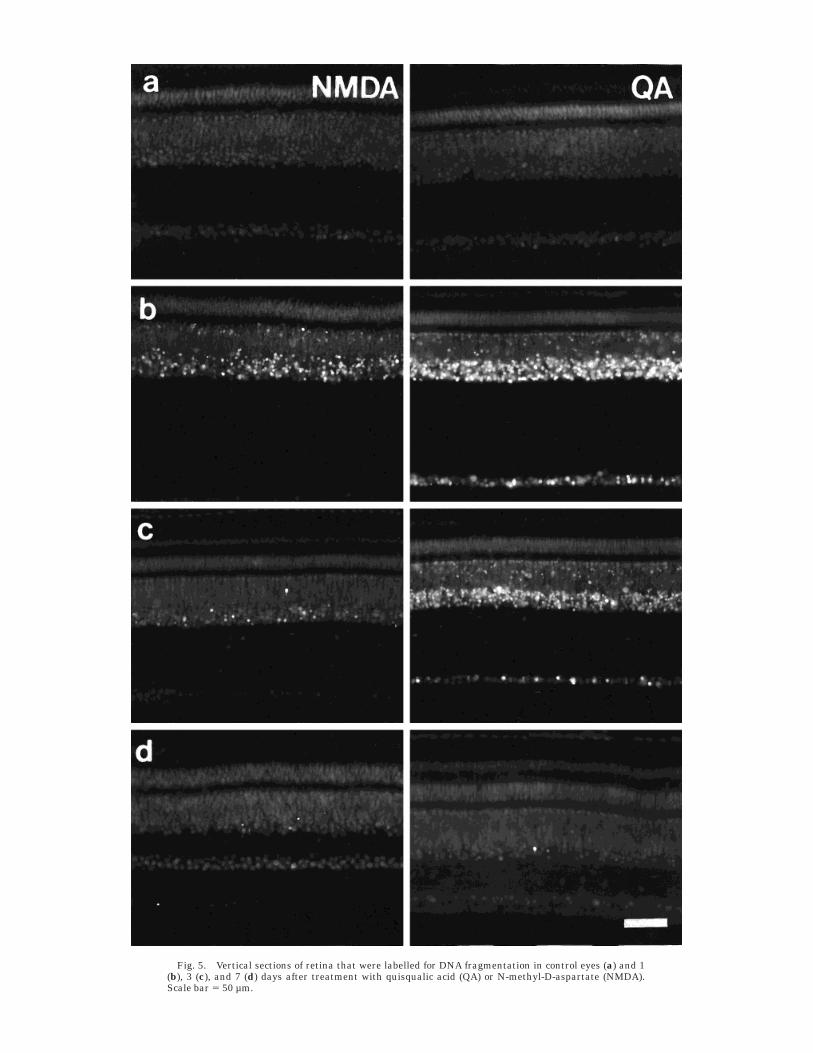
Fig. 5. Vertical sections of retina that were labelled for DNA fragmentation in control eyes (a) and 1(b), 3 (c), and 7 (d) days after treatment with quisqualic acid (QA) or N-methyl-D-aspartate (NMDA).Scale bar 5 50 µm.
amacrine cells, rarely in bipolar cells, and never in gan-glion cells (Fig. 5d).
Four hours after exposure to QA, no retinal cells werelabelled for DNA fragmentation (results not shown). How-ever, 24 hours after QA treatment, most nuclei in theamacrine cell layer of the INL and a few nuclei in the GCLwere intensely labelled (Fig. 5b). Three days after QAtreatment, the nuclei of most amacrine cells were weaklylabelled, although a few were robustly labelled (Fig. 5c).There were also some weakly labelled nuclei in the GCLand bipolar cell layer of the INL (Fig. 5c). Very few of theresidual amacrine cell nuclei were weakly labelled 7 daysafter treatment (Fig. 5d). Nuclei in the outer nuclear layerwere never labelled at any time following retinal exposureto NMDA or QA.
Accumulation of cells immunoreactivefor lysosomal membrane in NMDA-
and QA-treated retinas
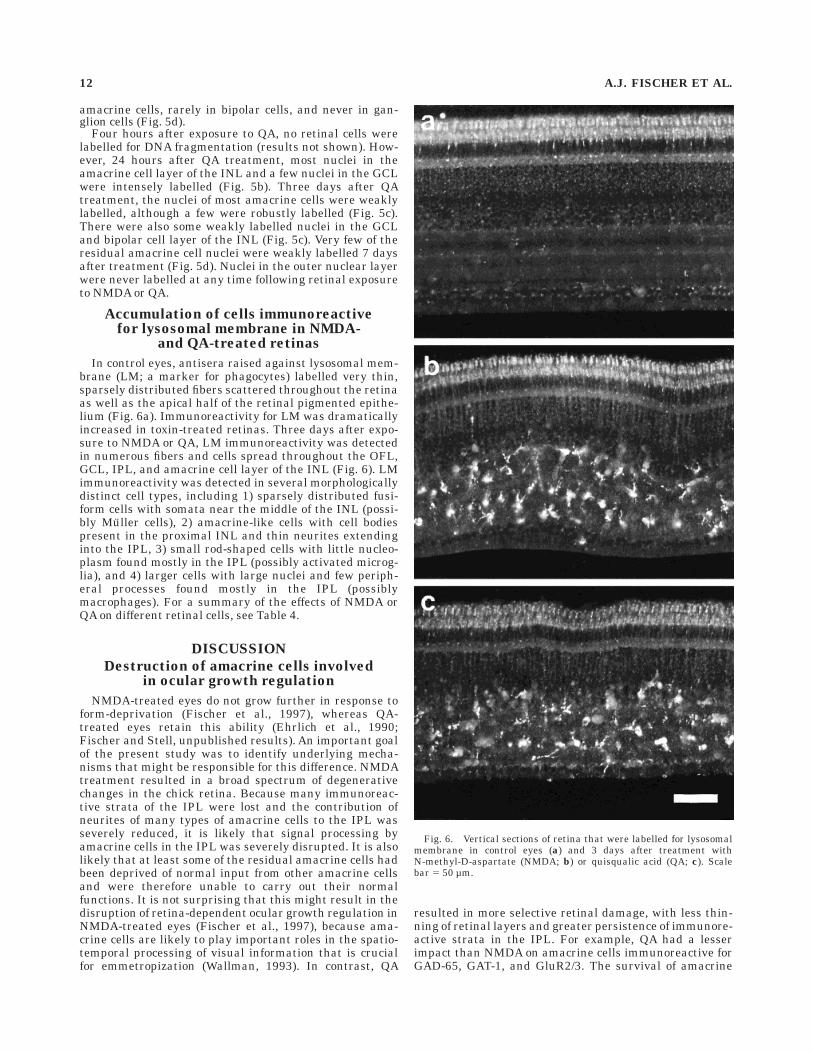
In control eyes, antisera raised against lysosomal mem-brane (LM; a marker for phagocytes) labelled very thin,sparsely distributed fibers scattered throughout the retinaas well as the apical half of the retinal pigmented epithe-lium (Fig. 6a). Immunoreactivity for LM was dramaticallyincreased in toxin-treated retinas. Three days after expo-sure to NMDA or QA, LM immunoreactivity was detectedin numerous fibers and cells spread throughout the OFL,GCL, IPL, and amacrine cell layer of the INL (Fig. 6). LMimmunoreactivity was detected in several morphologicallydistinct cell types, including 1) sparsely distributed fusi-form cells with somata near the middle of the INL (possi-bly Muller cells), 2) amacrine-like cells with cell bodiespresent in the proximal INL and thin neurites extendinginto the IPL, 3) small rod-shaped cells with little nucleo-plasm found mostly in the IPL (possibly activated microg-lia), and 4) larger cells with large nuclei and few periph-eral processes found mostly in the IPL (possiblymacrophages). For a summary of the effects of NMDA orQA on different retinal cells, see Table 4.
DISCUSSION
Destruction of amacrine cells involvedin ocular growth regulation
NMDA-treated eyes do not grow further in response toform-deprivation (Fischer et al., 1997), whereas QA-treated eyes retain this ability (Ehrlich et al., 1990;Fischer and Stell, unpublished results). An important goalof the present study was to identify underlying mecha-nisms that might be responsible for this difference. NMDAtreatment resulted in a broad spectrum of degenerativechanges in the chick retina. Because many immunoreac-tive strata of the IPL were lost and the contribution ofneurites of many types of amacrine cells to the IPL wasseverely reduced, it is likely that signal processing byamacrine cells in the IPL was severely disrupted. It is alsolikely that at least some of the residual amacrine cells hadbeen deprived of normal input from other amacrine cellsand were therefore unable to carry out their normalfunctions. It is not surprising that this might result in thedisruption of retina-dependent ocular growth regulation inNMDA-treated eyes (Fischer et al., 1997), because ama-crine cells are likely to play important roles in the spatio-temporal processing of visual information that is crucialfor emmetropization (Wallman, 1993). In contrast, QA
resulted in more selective retinal damage, with less thin-ning of retinal layers and greater persistence of immunore-active strata in the IPL. For example, QA had a lesserimpact than NMDA on amacrine cells immunoreactive forGAD-65, GAT-1, and GluR2/3. The survival of amacrine
Fig. 6. Vertical sections of retina that were labelled for lysosomalmembrane in control eyes (a) and 3 days after treatment withN-methyl-D-aspartate (NMDA; b) or quisqualic acid (QA; c). Scalebar 5 50 µm.
12 A.J. FISCHER ET AL.

cells responsible for growth regulation would permit QA-treated eyes to respond to form-deprivation (Ehrlich et al.,1990; Fischer and Stell, unpublished results).
Pharmacological antagonists to opiate, VIP, and musca-rinic acetylcholine receptors suppress FDM (for review, seeWallman, 1993; Seltner et al., 1995; Seltner et al., 1997).The conventional interpretation of these findings would bethat the corresponding endogenous ligands (ENK, VIP,and ACh) released from retinal cells are responsible forFDM. However, QA-treated eyes grow excessively andbecome myopic in response to form-deprivation (Ehrlich etal., 1990; Fischer and Stell, unpublished result), despitethe absence of retinal cells that produce ENK, VIP andACh. Therefore, exogenous opiate, VIP, and muscarinicreceptor antagonists are unlikely to prevent FDM viaantagonism of the expected receptors, and may do so atextraretinal sites or via unknown receptor actions. Wehave found that the muscarinic antagonist atropine doessuppress FDM in QA-treated eyes (Fischer and Stell,unpublished results), and that opiate blockers of FDMprobably do not act via opiate receptors (Seltner, Fischerand Stell, unpublished result). This hypothesis has yet tobe tested for VIP.
Somehow, the retinal pathways that mediate form-deprivation-induced ocular growth remain intact after QAtreatment, despite the destruction of numerous subtypesof amacrine cell. It is possible that QA-insensitive ama-crine cells, including those that are immunoreactive forTH, are required for the progression of FDM or theprevention of excessive ocular enlargement during normalvision. For example, it has been reported that the destruc-tion of TH-IR cells by 6-hydroxydopamine prevents theprogression of FDM in chicks (Li et al., 1992; Schaeffel etal., 1994). Other candidates for growth-regulatory path-ways would include GABAergic and glucagon-IR amacrinecells, because a substantial fraction of these cells appearsto be spared by QA treatment.
Effects of NMDA and QA on subpopulationsof amacrine cells
NMDA and kainate have been shown to stimulate loss ofimmunoreactive neuroactive substances, including gluca-gon, neurotensin, and ENK, from specific amacrine cellpopulations in turtle retina in vitro (Yaqub and Eldred,1993). In chick retina, we observed that glucagon-IRamacrine cells were unaffected by both NMDA and QA,
whereas ENSLI amacrine cells were sensitive to both. Wefound that serotonergic amacrine cells in chick are sensi-tive to NMDA and QA, whereas serotonergic cells in theturtle retina were found to be affected only by kainate(Yaqub and Eldred, 1993). Thus, serotonergic amacrinecells in chick retina are sensitive to NMDA and QA,whereas those in turtle retina are not. In contrast, gluca-gonergic cells in the chick retina are sensitive to NMDA,unlike those in the turtle. It is possible, however, thatthese apparent differences are due at least in part todifferences in method (acute in vitro vs. chronic in situ)rather than differences in species.
Both QA and NMDA have been shown to evoke therelease of GABA and acetylcholine from isolated chickretina (Campochiaro et al., 1985; Zeevalk et al., 1989;Ferreira et al., 1994). Similarly, we have shown that bothGABAergic and cholinergic amacrine cells are destroyed insignificant numbers following exposure to NMDA or QA.These findings suggest that GABAergic and cholinergicamacrine cells express receptors that are activated byNMDA and QA. However, not all GABAergic and choliner-gic amacrine cells were destroyed by NMDA or QA treat-ments, suggesting that not all of these cells have receptorsfor NMDA or QA.
Subtypes of cholinergic amacrine cells differed in sensi-tivity to NMDA and QA. Type I cholinergic amacrine cellswere the most tolerant to NMDA treatment, whereas typeII cells were the most tolerant to QA treatment. This mayhave resulted from a difference in the relative abundanceof GluR isoforms expressed by these cell types. For ex-ample, type I cells may express fewer NMDA receptors,whereas type II cells express fewer QA-selective receptors.Alternatively, different cell-death programs in these cellsmay be activated by NMDA- or QA-selective GluRs, therebyendowing them with different tolerances to NMDA or QA.
The receptor-specificity of NMDA- and QA-induced celldeath is a complicated issue. The high doses of drugs weapplied could decrease specificity for receptor subtypes,and the specificity of these drugs for GluRs and non-GluRsin chick tissues is not well characterized. Therefore, theresults presented in this study should be consideredprimarily as examples of excitotoxic rather than receptor-specific effects.
Survival of retinal cells after exposureto QA or NMDA
Most ganglion cells, photoreceptors, Muller cells (resultsnot shown), and bipolar cells survived exposure to eitherQA or NMDA. Similar results have been described in otherstudies on chick retina using a single dose of NMDA or QA(Sattayasai and Ehrlich, 1987; Barrington et al., 1989;Sheppard et al., 1991). There are several possibilities toexplain the tolerance of these cells. First, an absence or lowlevel of expression of relevant GluRs could render cellsinsensitive to NMDA or QA. Second, some cells thatexpress NMDA- and/or QA-selective receptors may pos-sess a greater ability than others to cope with sustaineddepolarization and ionic fluctuations. For example, excita-tory GluRs, including NMDA receptors, are expressed byganglion cells in the chick retina (Seltner et al., unpub-lished observation; present study), yet most of these cellssurvive exposure to toxic levels of kainate or NMDA(Ehrlich et al., 1990; Sheppard et al., 1991; present study).It has been suggested that the larger cytosolic volume inganglion cells might buffer the accumulation of intracel-
TABLE 4. Summary of the Effects of NMDA or QAUpon Different Retinal Cells1
Marker/cell type NMDA QA
TH/AC 2 1VIP/AC 111 1111Type I cholinergic AC 1 1111Type II cholinergic AC 111 111Type IIIa cholinergic AC 1111 1111Type IIIb cholinergic/ENK/AC 111 1111PKC/BP 2 2PKC/AC 111 111Glucagon/AC 2 2Serotonin/AC 111 1111Parvalbumin/AC 11 1111GABA/AC 11 1GAD/AC 11 1GAT-1/AC 111 1GluR1/AC and GC 11 1111GluR2-3/AC 111 11
1AC, amacrine cell; BP, bipolar cell; GC, ganglion cell; 2, little or no effect; 1, some lossof immunoreactivity; 11, moderate disruption; 111, substantial disruption; 1111,complete abalation.
QA- AND NMDA-INDUCED RETINAL EXCITOTOXICITY 13

lular Ca21, thereby rendering these cells less susceptibleto excitotoxicity than intrinsic retinal interneurons(Sheppard et al., 1991). Ganglion cells might also resistexcitotoxicity, because they are adapted to large ion fluxesassociated with bursts of action potentials or becausevoltage-gated calcium channels are absent from theirsomata.
Changes in the thickness of retinal layersfollowing treatment with NMDA or QA
Large changes in thickness of the IPL were seen aftertreatment with NMDA or QA. The swelling of the IPL 1day after treatment was likely caused by excessive depolar-ization of processes, build-up of ions (probably Cl2), andconsequent osmotic swelling. Such swelling of the IPL hasbeen seen in as little as 2 minutes following retinalexposure to kainate (Kleinschmidt et al., 1986a,b) and canbe prevented by pretreatment with chloride-channel block-ers (Zeevalk et al., 1989). There was no apparent swellingof the INL, even though swelling was observed in the IPL,which contains neurites from cells with somata located inthe INL. It is possible that, by 24 hours after treatment,substantial numbers of amacrine cells became pyknotic,whereas, conversely, others became swollen, resulting inlittle net change in the thickness of the INL. In fact, therewas a significant decrease in INL thickness 24 hours afterQA treatment (Fig. 3b), suggesting that some cells withinthe INL had already perished. This thinning of the INLmay result from the acute death of some horizontal andamacrine cells. This hypothesis is supported by the find-ings of Sattayasai and Ehrlich (1987), who reported thatQA caused swelling and death of horizontal cells andthinning of the INL 2 days after treatment.
Thinning of the IPL 7 days after treatment was probablydue to the destruction and removal of neurites of amacrinecells and possibly ganglion cells. This retinal degenerationclosely matches qualitative aspects of NMDA- or QA-induced IPL degeneration reported previously in the chickeye (Sattayasai and Erhlich, 1987; Barrington et al., 1989;Erhlich et al., 1990; Sheppard et al., 1991). Similarly,Dvorak and Morgan (1983) reported that kainic acid-induced retinal thinning due to the permanent removal ofneurons is stabilized 1 week after treatment.
Accumulation of cells in the IPL followingretinal exposure to NMDA or QA
The identity of cells that appeared within the IPL 3 daysafter treatment and the mechanisms underlying theirappearance remain somewhat uncertain. These cells con-tain immunoreactivity for LM and are therefore likelyphagocytic glial cells (activated microglia) and/or macro-phages that have entered the retina to remove cellulardebris. Attempts to further characterize and label thesecells with antisera directed against glial fibrillar acidicprotein (Sigma), vimentin (H5, AMF-17b, and 40E-C;Developmental Studies Hybridoma Bank, University ofIowa, Iowa City, IA), quail hematopoietic stem cells (QH-1;Developmental Studies Hybridoma Bank; Cuadros et al.,1992), NN-1 and NN-2 (Dr. P. Raymond), human lysozyme,human CD68 (Dako Corporation, Carpenteria, CA), andH386F and H381B4B5 (Dr. N. Tumosa) were unsuccessful,perhaps because of species-specific differences in epitopesfor these mostly mammalian microglial markers.
Regional retinal sensitivity to QA and NMDA
It remains unclear why less DNA fragmentation andmore type II cholinergic amacrine cells were detected intemporal retinal regions than in other regions in treatedretinas. It is possible that lesser amounts of QA andNMDA reached the temporal retina because of greaterdilution at a distance from the nasodorsal injection site. Itis also possible that temporal retinal regions are simplyless susceptible to excitotoxicity for unknown reasons.Indeed, Zeevalk et al. (1989) reported that temporal re-gions of isolated embryonic chick retina were affected lessthan nasal regions by exposure to kainate in vitro, suggest-ing that intrinsic rather than microenvironmental factorscould be responsible for these regional differences.
QA- and NMDA-induced DNA fragmentation
Most of the amacrine cells destroyed by NMDA or QAappear to undergo apoptosis, assuming that the DNAfragmentation detected in our study is representative ofprogrammed cell death. The number of retinal cells show-ing fragmentation of DNA was maximal at 24 hours andwas completed 3–7 days after treatment with either NMDAor QA. In cultured cerebellar granule cells, excitotoxin-induced apoptosis has been shown to produce DNA frag-mentation that is maximal at 4 hours and is completed by12 hours after treatment (Simonian et al., 1996), whereasapoptosis caused by low extracellular potassium is maxi-mal at 24 hours and is completed by 4 days after treatment(D’Mello et al., 1993). These results suggest that excito-toxin-induced apoptosis in cerebellar granule cells and celldeath in retinal amacrine cells occur at different rates andpossibly by different mechanisms. In addition, many reti-nal cells contained fragmented DNA only 4 hours afterexposure to NMDA, whereas QA elicited no such effect.This suggests that different cell death pathways, in whichDNA fragmentation begins soon after insult, are activatedby exposure to NMDA rather than QA in some populationsof retinal neurons.
DNA fragmentation was detected in the vast majority of,if not in all, amacrine cells following exposure to QA.However, it was clear that not all amacrine cells weredestroyed, because the thickness of the amacrine cell layerwas reduced by only 60% after QA. Similarly, althoughmany bipolar cells contained fragmented DNA followingexposure to NMDA, there was no detectable loss of PKC-IRbipolar cells. These results suggest that some cells dam-aged by QA or NMDA, although they were labelled by the38 nick-end labelling technique, were not committed toapoptosis. These nonapoptotic cells that contained frag-mented chromatin may have sustained limited DNA dam-age (not enough to become committed to apoptosis) or mayultimately have been rescued by DNA repair.
CONCLUSIONS
NMDA-induced excitotoxicity resulted in widespreaddisruption of many subpopulations of retinal amacrinecells accompanied by general disruption of the IPL, whereasQA-induced excitotoxicity resulted in more specific destruc-tion of restricted subsets of retinal amacrine cells with lessloss of neurites from the IPL. Damage within the QA-treated IPL appeared to be restricted enough to allow FDMto develop, whereas damage within the NMDA-treatedIPL either was too widespread or was directed to critical
14 A.J. FISCHER ET AL.

cell types involved in the regulation of ocular growth.These results support the hypothesis that retinal control ofocular growth is dependent upon signal processing involv-ing amacrine cells in the IPL. We propose that amacrinecells containing acetylcholine, ENK, or VIP are not re-quired for the progression of FDM, whereas those contain-ing dopamine, GABA, and glucagon may be required. Weobserved that DNA fragmentation is evident in manyamacrine cells and in some bipolar cells exposed to QA orNMDA, but we conclude that it may not be an accuratecriterion for commitment to apoptosis.
ACKNOWLEDGMENTS
We thank Dr. I. Morgan for critical reading of the paper.This work was supported by a student fellowship inmemory of James Thurber from the Fight For SightResearch Division of Prevent Blindness America and bystudentships from the Neuroscience Research Group atthe University of Calgary, the Pharmaceutical and MedicalAssociation of Canada-Medical Research Council, and theAlberta Heritage Foundation for Medical Research toA.J.F. and by grants from The Medical Research Council ofCanada, the Roy Allen Fund, the Marigold Foundation,and the Edwin L. and John E. Gustus Endowment inVision Disorders at the University of Calgary to W.K.S.
LITERATURE CITED
Barrington, M., J. Sattayasai, J. Zappia, and D. Ehrlich (1989) Excitatoryamino acids interfere with normal eye growth in posthatch chicks. Curr.Eye Res. 8:781–792.
Brecha, N. (1983) Retinal neurotransmitters: Histochemical and biochemi-cal studies. In P.C Emson (ed): Chemical Neuroanatomy. New York:Raven Press, pp. 85–129.
Brecha, N., H.J. Karten, and C. Laverack (1979) Enkephalin containingamacrine cells in the avian retina: Immunohistological localization.Proc. Natl. Acad. Sci. USA 76:3010–3014.
Campochiaro, P., J.W. Ferkany, and J.T. Coyle (1985) Excitatory amino acidanalogs evoke release of endogenous amino acids and acetylcholinefrom chick retina in vitro. Vision Res. 25:1375–1378.
Cuadros, M.A., A. Moujahid, G. Martin-Partido, and J. Navascues (1992)Microglia in the mature and developing quail brain as revealed by amonoclonal antibody recognizing hemopoietic cells. Neurosci. Lett.148:11–14.
D’Mello, S.R., C. Galli, T. Ciotti, and P. Calissano (1993) Induction ofapoptosis in cerebellar granule neurons by low potassium: Inhibition ofdeath by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci.USA 90:10989–10993.
Dvorak, D.R. and I.G. Morgan (1983) Intravitreal kainic acid permanentlyeliminates OFF-pathways from chicken retina. Neurosci. Lett. 36:249–253.
Ehrlich, D., J. Sattayasai, J. Zappia, and M. Barrington (1990) Effects ofselective neurotoxins on eye growth in the young chick. In G.R. Bockand K. Widdows (eds): Myopia and the Control of Eye Growth; CibaFoundation Symposium, Vol. 155. Chichester, England: John Wiley andSons, pp. 63–84.
Ferreira, I.L., C.B. Duarte, P.F. Santos, C.M. Carvalho, and A.P. Carvalho(1994) Release of [3H]GABA evoked by glutamate receptor agonists incultured chick retina cells: Effect of Ca21. Brain Res. 664:252–256.
Fischer, A.J., R.L.P. Seltner, and W.K. Stell (1997) NMDA-induced excitotox-icity causes myopia in posthatch chicks. Can. J. Ophthalmol. 32:373–377.
Kiyama, H., Y. Katayama-Kumoi, J. Kimmel, H. Steinbusch, J.F. Powell,A.D. Smith, and M. Tohyama (1985) Three dimensional analysis of
retinal neuropeptides and amines in the chick. Brain Res. Bull.15:155–165.
Kleinschmidt, J., C.L. Zucker, and S. Yazulla (1986a) Neurotoxic action ofkainic acid in the isolated toad and goldfish retina: I. Description ofeffects. J. Comp. Neurol. 254:184–195.
Kleinschmidt, J., C.L. Zucker, and S. Yazulla (1986b) Neurotoxic action ofkainic acid in the isolated toad and goldfish retina: II. Mechanism ofaction. J. Comp. Neurol. 254:196–208.
Li, X.X, F. Schaeffel, K. Kohler, and E. Zrenner (1992) Dose-dependenteffects of 6-hydroxy-dopamine on deprivation myopia, electroretino-grams, and dopaminergic amacrine cells in chickens. Vis. Neurosci.9:483–492.
Millar, T.J., I. Ishimoto, I.W. Chubb, M.L. Epstein, C.D. Johnson, and I.G.Morgan (1987) Cholinergic amacrine cells of the chicken retina: A lightand electron microscope immunocytochemical study. J. Neurosci. 21:725–743.
Millar, T.J., C. Winder, I. Isimoto, and I.G. Morgan (1988) Putativeserotonergic bipolar and amacrine cells in the chick retina. Brain Res.439:77–87.
Morgan, I.G. (1987) AMPA is a powerful neurotoxin in the chicken retina.Neurosci. Lett. 79:267–271.
Morgan, I.G., J.W. Wellard, and M.K. Boelen (1994) A role for theenkephalin-immunoreactive amacrine cells of the chicken retina inadaptation to light and dark. Neurosci. Lett. 174:64–66.
Negishi, K., S. Kato, and T. Teranishi (1988) Dopamine and rod bipolar cellscontain protein kinase C-like immunoreactivity in some vertebrateretinas. Neurosci. Lett. 94:247–252.
Sattayasai, J. and D. Ehrlich (1987) Morphology of quisqualate-inducedneurotoxicity in the chick retina. Invest. Ophthalmol. Vis. Sci. 28:106–117.
Schaeffel, F., G. Hagel, M. Bartmann, K. Kohler, and E. Zrenner (1994)6-Hydroxy dopamine does not affect lens-induced refractive errors butsuppresses deprivation myopia. Vision Res. 34:143–149.
Seltner, R.L.P. and W.K. Stell (1995) The effect of vasoactive intestinalpeptide on development of form-deprivation myopia in the chick: Apharmacological and immunocytochemical study. Vision Res. 35:1265–1270.
Seltner, R.L.P., B. Rohrer, V. Grant, and W.K. Stell (1997) Endogenousopiates in the chick retina and their role in form-deprivation myopia.Vis. Neurosci. 14:801–809.
Sheppard, A.M., M. Konopka, S.R. Robinson, I.G. Morgan, and P.L. Jeffrey(1991) Thy-1 antigen is specific to ganglion cells in chicks. Neurosci.Lett. 123:87–90.
Simonian, N.A., R.L. Getz, J.C. Leveque, C. Konrad, and J.T. Coyle (1996)Kainate induces apoptosis in neurons. Neuroscience 74:675–683.
Su, Y.Y.T. and C.B. Watt (1987) Interaction between enkephalin anddopamine in the avian retina. Brain Res. 243:63–70.
Tung, N.N., I.G. Morgan, and D. Ehrlich (1990) A quantitative analysis ofthe effects of excitatory neurotoxins on retinal ganglion cells in thechick. Vis. Neurosci. 4:217–223.
Wallman, J. (1993) Retinal control of eye growth and refraction. Progr. Ret.Res. 12:133–153.
Watt, C.B. and W.J. Florack (1994) A triple-label analysis demonstratingthat enkephalin-, somatostatin- and neurotensin-like immunoreactivi-ties are expressed by a single population of amacrine cells in thechicken retina. Brain Res. 634:310–316.
Watt, C.B., H.B. Li, and D.M.K. Lam (1985) The presence of threeneuroactive peptides in putative glycinergic amacrine cells of an avianretina. Brain Res. 348:187–191.
Wildsoet, C.F. and J.D. Pettigrew (1988) Kainic acid-induced eye enlarge-ment in chickens: Differential effects on anterior and posterior seg-ments. Invest. Ophthalmol. Vis. Sci. 29:311–319.
Yaqub, A. and W.D. Eldred (1993) Effects of excitatory amino acids onimmunocytochemically identified populations of neurons in turtle retina.J. Neurocytol. 22:644–662.
Zeevalk, G.D., A.G. Hyndman, and W.J. Nicklas (1989) Excitatory aminoacid-induced toxicity in chick retina: Amino acid release, histology, andeffects of chloride channel blockers. J. Neurochem. 53:1610–1619.
QA- AND NMDA-INDUCED RETINAL EXCITOTOXICITY 15