Embed Size (px)
Citation preview

Immunocytochemical Analysis of Human Muscular DystrophyC.A. SEWRY1,2*1Dubowitz Neuromuscular Centre, Department of Paediatrics and Neonatal Medicine, Imperial College School of Medicine,Hammersmith Hospital, London, United Kingdom W12 ONN2Department of Pathology, Robert Jones and Agnes Hunt Orthopaedic & District Hospital NHS Trust, Oswestry,SY10 7AG, United Kingdom
KEY WORDS dystrophin; sarcoglycans; laminin; emerin; utrophin; myosin
ABSTRACT Immunocytochemistry is an essential tool for the assessment of muscle biopsiesfrom patients with muscular dystrophy, especially the recessive forms. Antibodies can detectprimary defects when there is an alteration in expression, in particular in Xp21 musculardystrophies, Emery-Dreifuss muscular dystrophy, the limb-girdle dystrophies caused by abnormalexpression of the sarcoglycans, and in the form of congenital muscular dystrophy linked to the genefor laminin a2. Absence of a protein is easily observed and reduction in expression can be assessedprovided adequate controls and baselines are established. Assessment of secondary defects can alsobe of diagnostic value; they widen the understanding of pathology changes, and are helping in thedevelopment of therapeutic strategies. Microsc. Res. Tech. 48:142–154, 2000. r 2000 Wiley-Liss, Inc.
INTRODUCTIONImmunocytochemistry is used to visualise and loca-
lise specific proteins within a tissue. The principle isbased on the specific affinity of an antibody for itsantigen. With rapid advances in the identification ofprotein and gene defects in neuromuscular disorders,immunocytochemistry has become an essential tool forthe analysis of pathological muscle. Identification ofprimary defects has led to more accurate diagnosis anda better understanding of the clinical spectrum in somedisorders. In addition, some secondary defects in pro-tein expression are of diagnostic value; they contributeto our knowledge of the pathological response of muscle,and are useful in the development of therapeutic strate-gies. Immunocytochemistry is complementary to histo-chemistry and histology and all morphological tech-niques should be assessed in the light of clinical andbiochemical data.
Immunocytochemistry is particularly important inthe assessment of recessive conditions where alter-ations in protein expression are likely to be detected. Ithas a major role in identifying altered protein expres-sion in cases with small mutations that are difficult toidentify with molecular techniques. In dominant condi-tions, the value of immunocytochemstry is still uncer-tain, as both the normal and abnormal allele produceprotein, and alterations in expression may not bedetected. Antibodies to a wide variety of muscle pro-teins and their isoforms are now available, and manyhave been applied to human dystrophic muscle. Thisarticle aims to review the application of those mostrelevant to diagnosis and to the understanding of themuscular dystrophies, in particular the recessive condi-tions.
METHODSMuscle is obviously the tissue of choice, but as some
relevant proteins are also expressed in other tissues,such as skin, buccal cells, and chorionic villus samples,these can also be of diagnostic value (see below).
Frozen sections of muscle biopsies should always beused, and the fibres should be transversely orientated,if possible. Not only do frozen sections show bettermuscle architecture, but some antigens are onlyrecognised, or are only accessible, in unfixed tissue.With the advent of antigen retrieval methods, however,some immunocytochemical studies can be performed onfixed, wax embedded material. It is thus possible to usesome fixed, archival material, if only this is available.
Antibodies are visualised with conjugates to en-zymes, or to fluorochromes, and the use of these is oftena matter of choice. It is often easier to assess low levelsof expression, or identify small areas of antibody locali-sation, with fluorescent markers, where the positivelabelling is seen against a black background.
The use of controls is essential in all immunocyto-chemical studies. It is important to control not only forthe specificity of the antibody but also for the preserva-tion of the tissue, particularly the sarcolemma whenassessing muscular dystrophies. The preservation ofthe plasma membrane is best controlled by the use ofantibodies to b-spectrin, and that of the extracellularmatrix by antibodies to laminin g1. Necrotic, or verydamaged fibres, lose their plasma membrane or part ofit, and appear negative when labelled with antibodiesto most membrane proteins, such as dystrophin, spec-trin, and the sarcoglycans (Fig. 1). This also occurs inpost-mortem material and a false-negative result canbe obtained if preservation is poor. The extracellularmatrix, in contrast, is often retained, even in post-mortem material, but it may be absent on some necroticfibres. Studies of laminin a2 should therefore be con-trolled with parallel studies of laminin g1 to ensurethat the basal lamina is well preserved. Specificity of anantibody can be controlled by omission of the primaryantibody, and by assessing several biopsies in parallel.
*Correspondence to: Dr. C.A. Sewry, Dubowitz Neuromuscular Centre, Impe-rial College School of Medicine, Hammersmith Hospital, Du Cane Road, LondonW12 ONN, UK. E-mail: [email protected]
Received 1 November 1999; accepted in revised form 15 November 1999
MICROSCOPY RESEARCH AND TECHNIQUE 48:142–154 (2000)
r 2000 WILEY-LISS, INC.

In practice, a group of biopsies can be labelled inparallel and should always include one case that islikely to be positive for the proteins being assessed.This has the advantage that each sample will act ascontrol for the others, and technical problems can beidentified.
It is also important for each laboratory to establishits own baselines, particularly when trying to assessthe intensity of immunolabelling. Similarly, knowledgeof factors, such as development, that influence labellingmust be known. For example, regenerating fibres indystrophic muscle often have low expression of b-spec-trin and a7 integrin but highly express other proteinssuch as utrophin, laminin a5, and MHC class I antigens(Fig. 1). It is, therefore, important to identify regenerat-ing fibres with an antibody to fetal myosin so that anyabnormal expression can be differentiated from thenormal phenomenon related to immaturity (Fig. 1).
PRIMARY DEFECTSX-Linked Muscular Dystrophies
Dystrophin is the defective protein in Duchenne andBecker muscular dystrophy . Abnormalities in expres-
sion are due to mutations in the dystrophin gene onchromosome X p21. In normal muscle, dystrophin islocalised to the sarcolemma and each fibre showsuniform labelling (Fig. 2). In most cases of Duchennemuscular dystrophy (DMD), dystrophin is not apparenton most fibres, whereas in Becker muscular dystrophy(BMD) the fibres show reduced and/or uneven sarcolem-mal labelling (Fig. 2). This difference in labelling isexplained by deletions that disrupt the reading frameand give rise to a severe DMD phenotype, and thosethat maintain it and produce the milder phenotype ofBMD (see Brown and Lucy, 1997). Approximately 95%of cases conform to this dogma but there are severalexceptions. For example, deletion of exons 3–7 is aframe-shift deletion that should result in no expressionof dystrophin and a severe phenotype, but these casesoften have an intermediate phenotype and some dystro-phin is localised to the sarcolemma (Gangopadhya etal., 1992). Other exceptions involve exons around exon3, and cases with very large deletions (Muntoni et al.,1994). In general, most cases of DMD lack the C-terminal region and the region of the protein thatanchors dystrophin to b-dystroglycan in the plasmamembrane, while in most cases of BMD the C-terminusis preserved. Exceptions, however, have been reported(Clemens et al., 1992; Helliwell et al., 1992a).
In some biopsies of DMD, low levels of dystrophin canbe detected on several fibres, and a few fibres may havenear normal expression (Fig. 2). The latter are knownas ‘‘revertant’’ fibres (Fig. 2); immunolabelling is notalways even on the whole fibre periphery and inlongitudinal sections it is possible to see that only aportion of the fibre is positively labelled. This expres-sion of dystrophin in revertant fibres arises by restora-tion of the reading but the mechanism by which thisoccurs is uncertain. Studies of revertant fibres in themdx mouse have shown that various ‘‘exon skipping’’events occur (Qui Lu et al., 1998). The number ofrevertant fibres in a biopsy is variable; some may havenone, some a few isolated ones, and some have clusters.This number shows no apparent correlation with sever-ity (Fanin et al., 1992) but the problems of samplinghave to be considered. Removal of nonsense mutationsleading to an increase in the number of revertant fibreshas been proposed as a possible method of treatment ofDMD (Dunkley et al., 1998; Wilton et al., 1999).
Assessment of dystrophin should always be per-formed with more than one antibody that correspondsto more than one domain. It is now common practice touse antibodies to N-terminal, rod, and C-terminaldomains to avoid false-negative results, particularly incases with in-frame mutations. If an antibody with anepitope in a deleted region is used, the fibres will not belabelled, but a positive result may be obtained with anantibody that recognises a different epitope (Fig. 3).This can be used to advantage to identify small, orpoint, mutations using exon specific antibodies (Thanket al., 1995).
Carriers of Duchenne Muscular Dystrophy. Dys-trophin expression in carriers of DMD may be abnor-mal, and its analysis has become an essential tool indifferentiating between manifesting carriers of DMDand autosomal forms of muscular dystrophy. Symptom-atic carriers often show fibres that lack dystrophin,probably because of a skewed pattern of X-inactivation
Fig. 1. Serial sections immunolabelled with antibodies to (A)b-spectrin and (B) fetal myosin. Note the reduced expression ofb-spectrin in a group of regenerating fibres that express fetal myosin(arrow). Also note the loss of b-spectrin from some damaged fibres(arrowhead). Bar 5 50 µm.
143IMMUNOCYTOCHEMISTRY OF MUSCULAR DYSTROPHIES

in the muscle (Sewry et al., 1994a). The number of these‘‘negative’’ fibres is variable, but in transverse sectionsin young cases there is often a ‘‘mosaic’’ pattern ofpositive and negative fibres (Fig. 4). Some may onlyappear to be deficient in dystrophin, rather than nega-tive, and only part of the sarcolemma may be negative,as the pattern of expression varies along the length ofthe fibres. If dystrophin immunolabelling appears to benormal in a biopsy from a young female that is unequivo-cally dystrophic, it is unlikely that she is a manifestingcarrier. This, however, may not be the case in olderfemales, as the number of dystrophin-negative fibresmay decline, as occurs in the female carriers of dystro-phin deficient mice and dogs (Cooper et al., 1990;Karpati et al., 1990). The number of dystrophin-negative fibres may also vary between muscles (Mun-toni et al., 1992). Asymptomatic carriers usually showonly minor changes in dystrophin expression immuno-cytochemically, with perhaps occasional dystrophinnegative fibres, or uneven labelling. Abnormalities may,however, be more easily detected on immunoblots (Clerket al., 1991).
It is now known that some cases of X-linked cardiomy-opathy are caused by mutations in the dystrophin gene
(Ferlini et al., 1999). In these patients, the skeletalmuscle shows minimal weakness and there is appre-ciable expression of dystrophin. Cardiac muscle, how-ever, is preferentially affected and shows an absence offull-length dystrophin.
Emerin. This is the defective protein in X-linkedEmery Dreifuss muscular dystrophy (EDMD). It islocalised to the nuclear membrane (Manilal et al., 1996;Nagano et al., 1996). Emerin is detected in all nuclei innormal skeletal muscle but it is absent in most cases ofEDMD. A few mutations, however, result in detectableemerin (Manilal et al., 1998; Yates et al., 1999). Re-duced expression, in contrast to absent, emerin inskeletal muscle, detected immunocytochemically hasnot been reported and may be difficult to determine.
Although presentation of EDMD is often in adoles-cence, cases that present early without the typical signshave been reported (Muntoni et al., 1998a). Clinical andpathological indicators may make a definitive diagnosisdifficult, but the use of antibodies to emerin makes anaccurate diagnosis possible, without knowledge of themutation. Screening for emerin expression is a rapidmethod for identifying EDMD patients, and will detectthe majority of cases. It can also be used to detect the
Fig. 2. Sections from (A) a control showing normal sarcolemmal labelling of dystrophin on all fibres,(B) a case of Duchenne dystrophy with an absence of dystrophin, (C) a case of Becker dystrophy withuneven and reduced labelling of dystrophin, and (D) a case of Duchenne dystrophy showing a revertantfibre and very slight expression of dystrophin on most fibres. Bar 5 50 µm.
144 C.A. SEWRY

female carriers (Manilal et al., 1997; Sabatelli et al.,1998). As many tissues in addition to muscle expressemerin, these can be used to assess emerin expression(see below).
Recessive Limb-Girdle Muscular DystrophiesSarcoglycans. Dystrophin is associated with a
complex of proteins that are thought to link the extracel-lular matrix with the cytoskeleton (Lim and Campbell,1998; Osawa et al., 1998; Straub and Campbell, 1997).These include a and b-dystroglycan, the sarcoglycans(a,b,g.d,e), sarcospan, dystrobrevin, syntrophins, andneuronal nitric oxide synthase (nNOS), all of which areencoded by different genes. Primary defects have so faronly been found in a,b,g and d-sarcoglycan and theseare responsible for one group of recessive limb-girdlemuscular dystrophies, now often referred to as thesarcoglycanopathies. The sarcoglycans act as a com-plex, such that abnormal expression of one results in analteration in the others (Sewry et al., 1996b). Theexpression of each can vary from absence, to traces, to amild reduction in immunolabelling (Fig. 5). Carefulcomparison with controls labelled in parallel is impor-tant for the sarcoglycans as slight alterations may bedifficult to assess. Although only one antibody can beused to screen biopsies for possible defects, it is oftenbetter to rely on the results from at least two antibodiesto different sarcoglycans. Immunocytochemistry of the
Fig. 3. Labelling of dystrophin with (A) an antibody to the roddomain and (B) an antibody to the C-terminus in a case of Duchennedystrophy with a large deletion that included the epitope of the roddomain antibody. A false-positive result would have been obtained ifonly the antibody to the C-terminus had been used. Also note thatalthough the C-terminus is present, the phenotype of this child wassevere and that of Duchenne dystrophy. Bar 5 50 µm.
Fig. 4. Serial sections from a manifesting Duchenne carrier la-belled with antibodies to (A) dystrophin, (B) b-spectrin, and (C)utrophin. Note that the fibres with reduced or absent dystrophin havenormal expression of b-spectrin, and that both the dystrophin-negative and dystrophin-positive fibres express utrophin. Bar 550 µm.
145IMMUNOCYTOCHEMISTRY OF MUSCULAR DYSTROPHIES
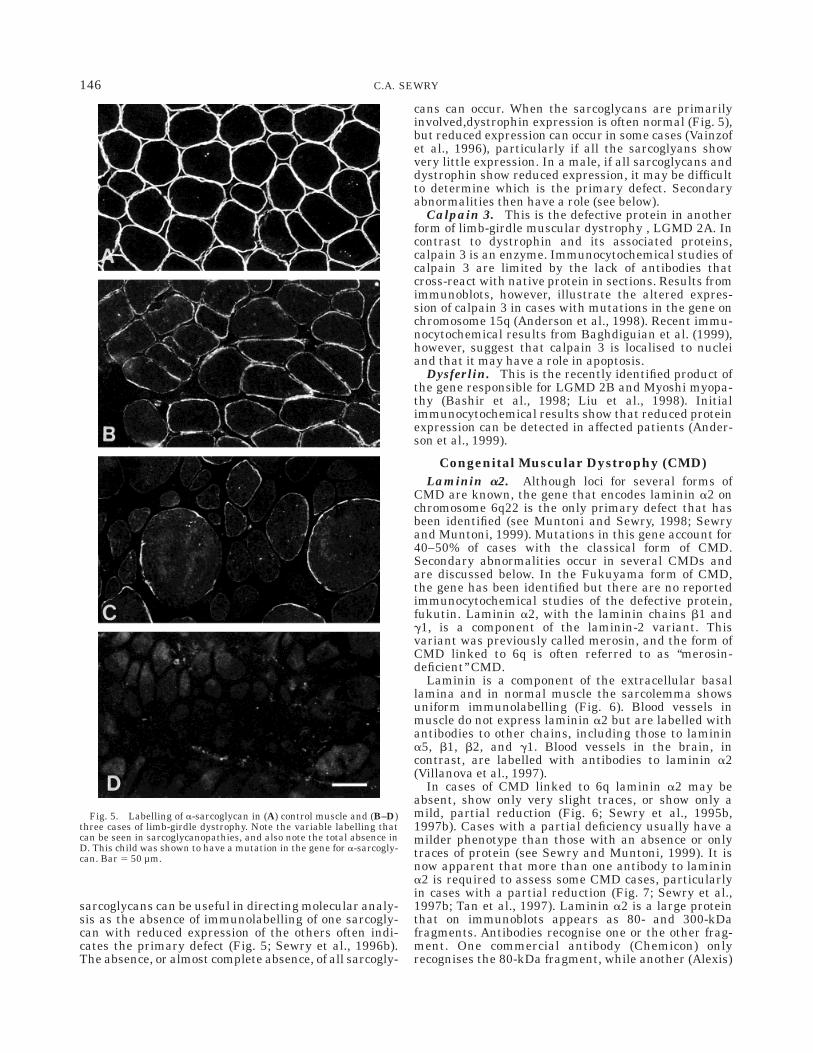
sarcoglycans can be useful in directing molecular analy-sis as the absence of immunolabelling of one sarcogly-can with reduced expression of the others often indi-cates the primary defect (Fig. 5; Sewry et al., 1996b).The absence, or almost complete absence, of all sarcogly-
cans can occur. When the sarcoglycans are primarilyinvolved,dystrophin expression is often normal (Fig. 5),but reduced expression can occur in some cases (Vainzofet al., 1996), particularly if all the sarcoglyans showvery little expression. In a male, if all sarcoglycans anddystrophin show reduced expression, it may be difficultto determine which is the primary defect. Secondaryabnormalities then have a role (see below).
Calpain 3. This is the defective protein in anotherform of limb-girdle muscular dystrophy , LGMD 2A. Incontrast to dystrophin and its associated proteins,calpain 3 is an enzyme. Immunocytochemical studies ofcalpain 3 are limited by the lack of antibodies thatcross-react with native protein in sections. Results fromimmunoblots, however, illustrate the altered expres-sion of calpain 3 in cases with mutations in the gene onchromosome 15q (Anderson et al., 1998). Recent immu-nocytochemical results from Baghdiguian et al. (1999),however, suggest that calpain 3 is localised to nucleiand that it may have a role in apoptosis.
Dysferlin. This is the recently identified product ofthe gene responsible for LGMD 2B and Myoshi myopa-thy (Bashir et al., 1998; Liu et al., 1998). Initialimmunocytochemical results show that reduced proteinexpression can be detected in affected patients (Ander-son et al., 1999).
Congenital Muscular Dystrophy (CMD)Laminin a2. Although loci for several forms of
CMD are known, the gene that encodes laminin a2 onchromosome 6q22 is the only primary defect that hasbeen identified (see Muntoni and Sewry, 1998; Sewryand Muntoni, 1999). Mutations in this gene account for40–50% of cases with the classical form of CMD.Secondary abnormalities occur in several CMDs andare discussed below. In the Fukuyama form of CMD,the gene has been identified but there are no reportedimmunocytochemical studies of the defective protein,fukutin. Laminin a2, with the laminin chains b1 andg1, is a component of the laminin-2 variant. Thisvariant was previously called merosin, and the form ofCMD linked to 6q is often referred to as ‘‘merosin-deficient’’ CMD.
Laminin is a component of the extracellular basallamina and in normal muscle the sarcolemma showsuniform immunolabelling (Fig. 6). Blood vessels inmuscle do not express laminin a2 but are labelled withantibodies to other chains, including those to laminina5, b1, b2, and g1. Blood vessels in the brain, incontrast, are labelled with antibodies to laminin a2(Villanova et al., 1997).
In cases of CMD linked to 6q laminin a2 may beabsent, show only very slight traces, or show only amild, partial reduction (Fig. 6; Sewry et al., 1995b,1997b). Cases with a partial deficiency usually have amilder phenotype than those with an absence or onlytraces of protein (see Sewry and Muntoni, 1999). It isnow apparent that more than one antibody to laminina2 is required to assess some CMD cases, particularlyin cases with a partial reduction (Fig. 7; Sewry et al.,1997b; Tan et al., 1997). Laminin a2 is a large proteinthat on immunoblots appears as 80- and 300-kDafragments. Antibodies recognise one or the other frag-ment. One commercial antibody (Chemicon) onlyrecognises the 80-kDa fragment, while another (Alexis)
Fig. 5. Labelling of a-sarcoglycan in (A) control muscle and (B–D)three cases of limb-girdle dystrophy. Note the variable labelling thatcan be seen in sarcoglycanopathies, and also note the total absence inD. This child was shown to have a mutation in the gene for a-sarcogly-can. Bar 5 50 µm.
146 C.A. SEWRY

recognises the 300-kDa fragment. The precise epitopesof these antibodies are not known. Neither the epitopenor the fragment recognised by a third commercialantibody (Novocastra) is known. Partial expression oflaminin a2 is often easier to assess with the 300-kDaantibody (Alexis), although an indication of abnormal
expression is often apparent with the 80-kDa antibody.Similar results are obtained with the antibodies fromNovocastra and Alexis (Sewry et al., 1998). Cases thatshow an absence of laminin a2 with the antibody to the80-kDa fragment usually also show an absence with theother two antibodies, but cases with appreciable label-ling with the 80-kDa antibody may show a considerablereduction with the other two antibodies (Sewry et al.,1998). One case has been reported that is negative withthe 80-kDa antibody but a reduction with the others, asthe mutation is in the region coding for the globular80-kDa domain (Dubowitz, 1999).
Laminin a2 is expressed in several tissues other thanmuscle, some of which are useful for diagnosis (seebelow).
Other Muscular DystrophiesDefective gene products are also known in other
muscular dystrophies, including Bethlem myopathy(collagen VI), limb-girdle muscular dystrophy 1C (caveo-lin), and autosomal Emery-Dreifuss muscular dystro-phy/ limb-girdle muscular dystrophy 1B (nuclear laminA/C). Immunocytochemistry in these dominant disor-ders has revealed a few detectable differences but onlyin a few cases (Minetti et al., 1998). No differences inthe expression of collagen VI or lamin A in skeletalmuscle of cases with defects in the respective geneshave yet been reported.
The gene encoding a7 integrin on chromosome 12q13has been shown to be responsible for a congenitalmyopathy (see Sewry and Muntoni, 1999). Only a fewcases have so far been identified and these have arelatively mild myopathic disorder; whether cases thatresemble a muscular dystrophy occur has yet to beestablished. The cases with mutations show a completeabsence of the a7 integrin from the muscle fibres andblood vessels. Integrin a7 is developmentally regulatedand there is very little detectable expression on imma-ture fibres, in contrast to the laminins (Fig. 8). Immuno-cytochemical assessment of integrin a7 of muscle biop-sies, therefore, has to consider both the age of the caseand the presence of regenerating fibres. Blood vesselsexpress integrin a7, even in immature muscle, and willonly appear negative if there is a null mutation, as inthe cases reported. Assessment of blood vessels is,therefore, useful (Fig. 8).
SECONDARY DEFECTSDystrophin-Associated Proteins
Abnormal expression of dystrophin in DMD, BMD,and manifesting DMD carriers is accompanied by asecondary reduction of the proteins associated with it.This includes a- and b-dystroglycan, the sarcoglycans,dystrobrevin, syntrophin, and nNOS (Lim and Camp-bell, 1998; Metzinger et al., 1997; Osawa et al., 1998;Sewry et al., 1994a; Straub and Campbell, 1997).
UtrophinUtrophin is the homologue of dystrophin encoded by
a gene on chromosome 6 and there is considerableinterest in its possible therapeutic role. In normalmature muscle, antibodies to utrophin show it to belocalised to the neuromuscular junction, blood vessels,and myotendinous junction (Nguyen thi Man et al.,1991; Ohlendiek et al., 1991;Tanaka et al., 1991).
Fig. 6. Labelling of laminin a2 in (A) control muscle and (B–D)three cases of congenital muscular dystrophy known to link tochromosome 6q. Note the partial expression in B, the traces on somefibres in C, and the absence in D. Bar 5 50 µm.
147IMMUNOCYTOCHEMISTRY OF MUSCULAR DYSTROPHIES

Normal embryonic muscle and regenerating fibres inall disorders also express utrophin (Clerk et al., 1993;Helliwell et al., 1992b). In normal developing muscle,utrophin and dystrophin are expressed together on thesarcolemma until about 26 weeks gestation, whenutrophin becomes confined to the vascular tissue (Clerket al., 1993). As expression of utrophin is a normalphenomenon in developing muscle, it is important tocompare utrophin with the expression of fetal myosin indystrophic muscle. It is then possible to distinguish theabnormal from the normal expression.
In DMD and BMD, mature fibres express utrophin(Helliwell et al., 1992b). This is usually referred to asan up-regulation and it is age-related; very little utro-phin is detected on mature fibres in young cases ofDMD or BMD, but from about 2–3 years of age utrophinbecomes more apparent (Fig. 9; Taylor et al., 1997b). Inmanifesting carriers of DMD, both dystrophin-positiveand dystrophin-negative fibres express utrophin (seeFig. 4; Sewry et al., 1994a). Asymptomatic DMD carri-ers may show occasional fibres with utrophin and thiscan be a useful diagnositic aid (Sewry et al., 1994b).Similarly, cases of BMD show expression of utrophin,even if the reduction in dystrophin appears minimal.When expression of dystrophin and all the sarcoglycansappears reduced, utrophin can be useful in differentiat-ing between BMD and LGMD, as most cases of LGMDdo not show utrophin on mature fibres. Exceptions mayoccur, however, and some cases of BMD may not show
utrophin, while occasional sarcoglycanopathies mayshow utrophin (Sewry et al., 1994c).
Cases of X-linked cardiomyopathies that show only amild reduction of dystrophin in the skeletal muscle andan absence in cardiac muscle also show utrophin, bothin skeletal and cardiac muscle (Ferlini et al., 1999;Muntoni et al., 1995b).
Extracellular Matrix ProteinsThe laminin a2 chain shows a secondary reduction in
Fukuyama CMD and muscle eye brain disease (seeSewry and Muntoni, 1999). It is also reduced on somefibres in another form of CMD linked to chromosome 1q(Brockington et al., 1999; Muntoni et al., 1998b). Agroup of adult patients with a late onset musculardystrophy has also been reported that showed a markedreduction of laminin a2 on immunoblots, but not immu-nocytochemically on sections (Bushby et al., 1998). It isnow appreciated that changes in extracellular proteinsare associated with a wide clinical spectrum in theCMDs, and this is likely to broaden further as moredisease-related loci are identifed (Dubowitz, 1999; Sewryand Muntoni, 1999).
Laminin a5 is over-expressed in cases of CMD with aprimary abnormality in the expression of laminin a2(Sewry et al., 1995a; Tome et al., 1998). It may also beover-expressed in cases with a secondary reduction(Muntoni et al., 1998b). It was originally thought thatthis over-expression was due to laminin a1, but it is
Fig. 7. Labelling of laminin a2 in (A,B) control muscle and (C,D) an adult with a mutation in the genefor laminin a2 using two different antibodies that recognise (A,C) the 80-kDa fragment and (C,D) the300-kDa fragment. Note that the partial expression of laminin a2 is easier to observe with the 300-kDaantibody. Bar 5 25 µm (A,B), 50 µm (C,D).
148 C.A. SEWRY

now known that the antibody used (4C7) recognises thelaminin a5 chain. Laminin a5 is developmentally regu-lated and is present on fetal and regenerating fibres(Sewry et al., 1995a,b). It is, therefore, again importantto correlate results with another antibody, such as fetalmyosin, to distinguish the abnormal expression fromthat related to regeneration, particularly in the muscu-lar dystrophies.
Cases of CMD linked to chromosome 6q, with aprimary reduction in laminin a2, also show a secondaryreduction of the laminin b2 chain and of the integrina7/b1D complex, a ligand for laminin a2. (see Burkinand Kaufmann, 1999; Cohn et al., 1998; Sewry andMuntoni, 1999).
The laminin b1 chain may also show a secondaryreduction in some muscular dystrophies (Patton et al.,1999). It has been reported in some cases of recessiveLGMD and EDMD (Mian et al., 1997; Patton et al.,1999) and also occurs in some dominant conditions,including autosomal EDMD/LGMD 1B and Bethlemmyopathy (Fig. 10; Merlini et al., 1999; Sewry et al.,1999; Taylor et al., 1997a). In the dominant cases, thereduction is only on the muscle fibres and expression on
the blood vessels is normal, indicating that there isprobably no mutation in the gene. In some conditions,the laminin b1 appears to be age related, as thereduction is only observed in adolescents and adults,not in paediatric cases (Sewry et al., 1999; Taylor et al.,1997a). Although it has been suggested that a reductionof laminin b1 is non-specific in the muscular dystro-phies (Patton et al., 1999), in the author’s experience itmost commonly occurs in dominant conditions. In allmuscular dystrophies, assessment of laminin b1 shouldbe compared with that of laminin g1 in serial sections,to ensure that the basal lamina is well preserved.
Major Histocompatibility Proteins (MHC)Adult skeletal muscle fibres are unusual in not
expressing detectable MHC class I or II (Appleyard etal., 1985; McDouall et al., 1989). MHC-class I, however,is present on regenerating fibres, but not fetal fibres;MHC-I is therefore detectable immunocytochemicallyin most of the muscular dystrophies. It has beenreported that fibres in biopsies from cases of DMD andBMD express MHC-I, but in these previous studiesmaturation was not taken into account. Some maturefibres in some cases of DMD do express MHC-I, but thisis not a consistent feature.
Fig. 8. Labelling of (A) integrin a7 and (B) laminin a2. Note thereduced expression of integrin a7 but normal expression of laminin a2in a group of regenerating fibres that also expressed fetal myosin (notshown). Note also the expression of integrin a7 but absence of laminina2 on capillaries Bar 5 50 µm.
Fig. 9. Expression of utrophin in (A) a 1-year-old and (B) an8-year-old case of Duchenne dystrophy. Note the low expression in theyoung case, except on the intensely labelled regenerating fibres, buthigh expression on all fibres in the older boy. Note also in B theutrophin at neuromuscular junctions (arrow) and on capillaries (arrow-head). Bar 5 50 µm.
149IMMUNOCYTOCHEMISTRY OF MUSCULAR DYSTROPHIES

Myosin IsoformsAntibodies to the fast and slow isoforms of myosin
reveal the characteristic checkerboard pattern of skel-etal muscle fibre types, and can be used as an alterna-tive to enzyme histochemistry. Immunocytochemistryhas the advantage that co-expression of different iso-forms in the same fibre can be assessed. In normalmuscle there is very little co-expression of fast and slowmyosin (Fig. 11) but in dystrophic muscle fast, slow, andfetal myosin isoforms can be present in various combi-nations in the same fibre, and this is a useful indicatorof abnormality (Fig. 12). Fibres with slow myosin tendto be predominant in most myopathic disorders andfibres with fetal myosin are common in dystrophicmuscle. Fetal myosin is present not only in basophilicfibres but also in regenerating fibres that are in theprocess of maturing (Fig. 13). Fetal myosin is a usefulindicator of regeneration, and, by inference, the degreeof muscle damage. The number and size of the fibres
and intensity of immunolabelling of fetal myosin arevariable. Cases of DMD, and the severe dystrophies,often have a large number of fibres with fetal myosinthat are of varying size (Fig. 13), while in some cases ofBMD only a few small fibres are labelled. In mild casesof CMD, with normal expression of laminin a2, fetalmyosin is often restricted to a population of very smallfibres that are not easily seen with routine histologicalstains (Fig. 13).
IMMUNOCYTOCHEMISTRY OFNON-MUSCLE TISSUES
As some of the proteins primarily affected in muscu-lar dystrophies are expressed not only in muscle butalso in other tissues, these can be useful for assessingexpression.
SkinLaminin a2 is present at the epidermal/dermal junc-
tion of skin, and skin biopsies are therefore an alterna-tive to muscle when muscle wasting is extensive, as insome severe cases of CMD (Fig. 14; Sewry et al., 1996a,
Fig. 10. Expression of laminin b1 in (A) control muscle and (B) anadult with autosomal dominant Emery-Dreifuss muscular dystrophy(ADEDMD) caused by a mutation in the gene for lamin A. Note theuniform labelling of both the fibres and the capillaries in the control,but reduced labelling of the fibres but not the capillaries in the case ofADEDMD. Bar 5 50 µm.
Fig. 11. Serial sections of control muscle labelled with antibodiesto A) slow myosin and B) fast myosin showing a normal two-fibrecheckerboard pattern with no co-expression of isoforms. Bar 5 50 µm.
150 C.A. SEWRY

1997a). The absence of laminin a2 is easily detected,and, by careful comparisons with control samples,reduced/partial expression can also be observed (Sewryet al., 1997a).
Skin and oral foliate cells can also be used to studythe presence or absence of emerin in X-linked EDMDpatients and female carriers (Manilal et al., 1997;Sabatelli et al., 1998). In carriers of EDMD a variablenumber of emerin-positive and negative cells can beseen. Caution in the use of oral foliate cells for carrierassessment is needed as a false-negative result can beobtained if the antibody does not penetrate all cells(personal observation).
Chorionic Villus SamplesMost laminin chains are present in chorionic villi
(CV) and can be used for prenatal diagnosis of 6q-linkedCMD (Muntoni et al., 1995a; Naom et al., 1997). Theabsence of laminin a2 is easily observed in CV samplesfrom fetuses affected by 6q-linked, ‘‘merosin-deficient’’CMD (Fig. 15). The CV from heterozygous fetusesshows normal immunolabelling of laminin a2 (Fig. 15),but the expression in cases with partial expression isnot known. Similarly, too few cases of CMD with asecondary deficieny of laminin a2 have been identifiedto know if immunolabelling has a role in prenataldiagnosis of these cases. As there is often appreciablelaminin a2 present on some muscle fibres in thesecases, a difference in expression in CV samples may be
Fig. 12. Serial sections labelled with antibodies to (A) fetal myosin,(B) fast myosin, and (C) slow myosin from a case of limb-girdlemuscular dystrophy. Note the co-expression of isoforms, in variouscombinations, in several fibres. Bar 5 50 µm.
Fig. 13. Expression of fetal myosin in (A) a case of Duchennedystrophy (DMD), and (B) a case of congenital muscular dystrophy(CMD) with normal expression of laminin a2. Note that several fibresof varying size are labelled in the DMD, but only a few small fibres inthe mild case of CMD, reflecting differences in muscle degeneration/regeneration. Bar 5 50 µm.
151IMMUNOCYTOCHEMISTRY OF MUSCULAR DYSTROPHIES

difficult to observe. Immunocytochemistry is a powerfultool for prenatal diagnosis of CMD but it should alwaysbe performed in conjunction with linkage analysis andwith knowledge of the laminin a2 expression in theproband.
FibroblastsCultured skin fibroblasts can be used to study colla-
gen synthesis, for example in Bethlem myopathy. Also,fibroblasts from CV samples or skin can be transfectedwith MyoD to convert them to muscle cells and dystro-phin expression can then be studied.
CONCLUSIONSImmunocytochemistry is a powerful tool for assess-
ing protein expression, provided adequate controls areperformed and baselines established. It has a majordiagnostic role in the recessive muscular dystrophieswhere expression of defective proteins can vary fromabsence to variable degrees of partial expression. Therole of immunocytochemistry in assessing dominantmuscular dystrophies is not yet clear. Secondary changesin protein expression are also useful for diagnosis, and
for a wider understanding of the pathological changesin skeletal muscle.
REFERENCESAnderson LVB, Davidson K, Moss JA, Richard I, Fardeau M, Tome
FMS, Hubner C, Lasa A, Colomer J and Beckmann JS. 1998.Characterisation of monoclonal antibodies to calpain 3 and proteinexpression in muscle from patients with limb-girdle musculardystrophy type 2A. Am J Pathol 153:1169–1179.
Anderson LVB, Davidson K, Moss JA, Young C, Cullen MJ, Walsh J,Johnson MA, Bashir R, Britton S, Keers S, Argov Z, Mahjneh I,Fourgerousse F, Beckamann JS, Bushby KM. 1999. Dysferlin is aplasma membrane protein and is expressed early in human develop-ment. Hum Mol Genet 8:855–861.
Appleyard ST, Dunn MJ, Dubowitz V, Rose ML. 1985. Increasedexpression of HLA ABC class I antigens by muscle fibres inDuchenne muscular dystrophy, inflammatory myopathy and otherneuromuscular disorders. Lancet i:361–363.
Baghdiguian S, Martin M, Richardm I, Pons F, Astier C, Bourg N, HayRT, Chemaly R, Halaby G, Loiselet J, Anderson LVB, Lopez deMunain A, Fardeau M, Mangeat P, Beckmann JS, Lefranc G. 1999.Calpain 3 deficiency is associated with myonuclear apoptosis andprofound pertubation of the IkBa/NF-kB pathway in limb-girdlemuscular dystrophy type 2A. Nat Med 5:503–511.
Bashir R, Britton S, Strachan T, Keere S, Vafiadaki E, Lako M,Richard I, Marchand S, Bourg N, Argov Z, Sadeh M, Mahjneh I,Marconi G, Passo- Bueno MR, Moreira E de S, Zatz M, BeckmannJS, Bushby K. 1998. A gene related to Caenorhabditis elegansspermatogenenesis factor fer–1 is mutated in limb-girdle musculardystrophy type 2B. Nat Genet 20:37–42.
Brockington M, Sewry CA, Hermann R, Naom I, Dearlove A, RhodesM, Dubowitz V, Voit V, Muntoni F. 1999. Assignment of a form ofcongenital muscular dystrophy with secondary merosin deficiency tochromosome 1q42. Am J Genet (in press).
Brown SC, Lucy JA, editors. 1997. Dystrophin: gene, protein and cellbiology. Cambridge: Cambridge University Press.
Burkin DJ, Kaufmann SJ. 1999. The alpha7beta1integrin in muscledevelpoment and disease. Cell Tissue Res 296:183–190.
Fig. 14. A: Normal labelling of laminin a2 labelling of skin in acontrol. B: Absence of laminin a2 from the dermal/epidermal junctionand sensory nerves in a case of congenital muscular dystrophy linkedto chromosome 6q (merosin-deficient CMD). Bar 5 50 µm.
Fig. 15. A: Normal labelling of laminin a2 in a chorionic villussample in a heterozygote with a mutation in the gene for laminin a2 ononly one allele; B: absence of laminin a2 in a chorionic villus samplefrom a fetus affected by congenital muscular dystrophy linked tochromosome 6q (merosin-deficient CMD). Bar 5 50 µm.
152 C.A. SEWRY

Bushby K, Anderson LVB, Pollitt C, Naom I, Muntoni F, Bindoff L.1998. Abnormal merosin in adults. A new form of late onsetmuscular dystrophy not linked to chromosome 6q2. Brain 121:581–588.
Clemens PR, Ward PA, Caskey CT, Bulman DE, Fenwick RG. 1992.Premature chain termination causing Duchenne muscular dystro-phy Neurology 42:1755–1782.
Clerk A, Rodillo E, Heckmatt JZ, Dubowitz V, Strong PN, Sewry CA.1991. Characterisation of dystrophin in carriers of Duchenne muscu-lar dystrophy. J Neurol Sci 102:197–205.
Clerk A, Morris GE, Dubowitz V, Davies K, Strong PN, Sewry CA.1993. Dystrophin-related protein, utrophin, in normal and dystro-phic human foetal skeletal muscle. Histochem J 25:554–561.
Cohn RD, Hermann R, Sorokin L, Wewer UM, Voit T. 1998. Lamininalpha 2 chain-deficient congenital muscular dystrophy: Variableepitope expression in severe and mild cases. Neurology 51:94–100.
Cooper BJ, Gallagher EA, Smith CA, Valentine BA, Winand NJ. 1990.Mosaic expression of dystrophin in carriers of canine X-linkedmuscular dystrophy. Lab Invest 62:171–178.
Dubowitz V. 1999. 68th ENMC International Workshop on Congenitalmuscular dystrophy. Neuromusc Disord 9:446–454.
Dunkley M, Manoharan M, Villiet P, Eperon I, Dickson G. 1998.Modification of splicing in the dystrophin gene in cultured mdxmuscle cells by antisense oligoribonucleotides. Hum Mol Genet5:1083–1090.
Fanin M, Danieli G A, Vitiello L, Senter L, Angelini C. 1992.Prevelance of dystrophin positive fibers in 85 Duchenne musculardystrophy patients. Neuromusc Disord 2:41–45.
Ferlini A, Sewry C, Melis MA, Mateddu A, Muntoni F. 1999. X-linkeddilated cardiomyopathy and the dystrophin gene. Neuromusc Dis-ord 9:339–346.
Gangopadhyay SB, Sherratt TG, Heckmatt J Z, Dubowitz V, Miller G,Shokeir M, Ray PN, Strong PN, Worton RG. 1992. Dystrophin inframe shift deletion patients with Becker muscular dystrophy. Am JHum Genet 51:562–570.
Helliwell TR, Ellis JM, Mountford RC, Appleton RE, Morris GE.1992a. A truncated dystrophin lacking the C-terminal domain islocalized at the muscle membrane. Am J Hum Genet 50:508–514.
Helliwell TR, Nguyen thi Man, Morris GE, Davies KE. 1992b. Thedystrophin-related protein, utrophin, is expressed on the sarco-lemma of regenerating human skeletal muscle fibres in dystrophiesand inflammatory myopathies. Neuromusc Disord 2:177–184.
Karpati G, Zubrzycka-Gaarn EE, Carpenter S, Bulman DE, Ray PN,Worton RG. 1990. Age-related conversion of dystrophin-negative topositive fiber segments of skeletal but not cardiac muscle fibres inheterozygote mdx mice. J NeuropatholExp Neurol 49:96–105.
Lim LE, Campbell KP. 1998. The sarcopglycan complex in linmb-girdlemuscular dystrophy. Curr Opin Neurol 11:443–452.
Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C,Urtizberea JA, Hentati F, Hamida MB, Bohlega S, Culper EJ,Amato AA, Bossie K, Oeltjen J, Bejaoui K, McKenna-Yasek D,Hosler BA, Schurr E, Arahata K, de Jong PJ, Brown RH Jr. 1998.Dysferlin, a novel skeletal muscular gene, is mutated in Miyoshimyopathy and limb-girdle muscular dystrophy. Nat Genet 21:31–36.
Manilal S, Nguyen thi Man, Sewry C, Morris GE. 1996. The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear mem-brane protein. Hum Mol Genet 5:801–808.
Manilal S, Sewry CA, Nguyen thi Man, Muntoni F, Morris GE. 1997.Diagnosis of X-linked Emery Dreifuss muscular dystrophy byprotein analysis of leukocytes and skin. Neuromusc Disord 7:63–66.
Manilal S, Recan D, Sewry CA, Hoeltzenbein M, Llense S, Leturcq F,Deburgrave N, Barbot J-C, Nguyen thi Man, Muntoni F, Wehnert M,Kaplan J-C, Morris GE. 1998. Mutations in Emery-Dreifuss muscu-lar dystrophy and their effects on emerin protein expression. HumMol Genet 7:855–864.
McDouall RM, Dunn MJ, Dubowitz V. 1989. Expression of class I andII MHC antigens in neuromuscular diseases. J Neurol Sci 89:213–226.
Merlini L, Villanova M, Sabatelli P, Malandrini A, Maraldi NM. 1999.Decreased exppression of laminin b1 in chromosome 21-linkedBethlem myopathy. Neuromusc Disord 9:326–329.
Metzinger L, Blake DJ, Squier M, Anderson LVB, Deconinck AE,Nawrotzki R, Hilton-Jones D, Davies KE. 1997. Dystrobrevindeficiency at the sarcolemma of patients with muscular dystrophy.Hum Mol Genet 6:1185–1191.
Mian L, Dickson DW, Spiro AJ. 1997. Abnormal expression of lamininb1 chain in skeletal muscle of adult-onset limb-girdle musculardystrophy. Arch Neurol 54:1457- 1461.
Minetti C, Sotgia F, Bruno C, Scartezzini P, Bado M, Cordone G,Bricarelli FD, Lisanti MP, Zara F. 1998. Autosomal dominant
limb-girdle muscular dystrophy with caveolin–3 deficiency causedby mutation in the caveolin–3 gene. Muscle Nerve Suppl 7: S135.
Muntoni F, Sewry CA. 1998. From rags to riches. Neurology 51:14–16.Muntoni F, Mateddu A, Marrosu M G, Cau M, Congiu R, Melis MA,
Cao A, Cianchetti C. 1992. Variable dystrophin expression indifferent muscles of a Duchenne muscular dystrophy carrier. ClinGenet 42:35–38.
Muntoni F, Gobbi P, Sewry C, Sherratt T, Taylor J, Sandhu S, Abbs S,Roberts R, Hodgson SV, Bobrow M, Dubowitz V. 1994. Deletions inthe ‘5 region of dystrophin and resulting phenotypes. J Med Genet31:843–847.
Muntoni F, Sewry C, Wilson L, Angelini C, Trevisan CP, Brambati B,Dubowitz V. 1995a. Prenatal diagnosis in congenital musculardystrophy. Lancet 345:591.
Muntoni F, Wilson L, Marrosu G, Marrosu GM, Cianchetti C, MestroniL, Ganau A, Dubowitz V, Sewry C. 1995b. A mutation in thedystrophin gene selectively affecting dystrophin expression in theheart. J Clin Invest 96:693–699.
Muntoni F, Lichtarowicz-Krynska E.J, Sewry CA, Manilal S, Recan D,Llense S, Taylor J, Morris GE, Dubowitz V. 1998a. Early presenta-tion of X-linked Emery-Dreifuss muscular dystrophy resemblinglimb-girdle muscular dystrophy. Neuromusc Disord 8:72–76.
Muntoni F, Taylor J, Sewry CA, Naom I, Dubowitz V. 1998b. An earlyonset muscular dystrophy with diaphragmatic involvement, earlyrespiratory failure and secondary a2 laminin deficiency unlinked tothe lama2 locus on 6q22. Eur. J Paediatr Neurol 2:19–26.
Nagano A, Koga R, Ogawa M, Kurano Y, Kawada J, Okada R, HayashiYK, Tsukahara T, Arahata K. 1996. Emerin deficiency at the nuclearmembrane in patients with Emery-Dreifuss muscular dystrophy.Nat Genet 2:254–259.
Naom I, Sewry C, D’Alessandro M, Topaloglu H, Ferlini A, Dubowitz V,Muntoni F. 1997. Prenatal diagnosis of merosin deficient congenitalmuscular dystrophy: the role of linkage and immunocytochemicalanalysis. Neuromusc Disord 7:176–179.
Nguyen thi Man, Ellis J M, Love DR, Davies KE, Gatter KC, DicksonG, Morris GE. 1991. Localisation of the DMDL gene-encodeddystrophin-related protein using a panel of nineteen monoclonalantibodies: presence at neuromuscular junctions, in the sarcolemmaof dystrophic skeletal muscle, in vascular and other smooth muscles,and in proliferating brain cell lines. J Cell Biol 115:1695–1700.
Ohlendieck K, Ervasti JM, Matsumara K, Kahl SD, Leveille CJ,Campbell KP. 1991. Dystrophin-related protein is localised to neuro-muscular junctions of adult skeletal muscle. Neuron 7:499 –508.
Osawa E, Noguchi S, Mizuno Y, Hagiwara Y, Yoshida M. 1998. Fromdystrophinopathy to sarcoglycanopathy:evolution of a concept ofmuscular dystyrophy. Muscle Nerve 21:421–438.
Patton BL, Connolly AM, Martin PT, Cunningham JM, Mehta S,Pestronk A, Miner JH, Sanes JR. 1999. Distribution of ten lamininchains in dystrophic and regenerating muscles. Neuromusc Disord9:423–433.
Qui Lu, Morris GE, Wilton SD, Strong P, Partridge T. 1998. Massiveidiosyncratic exon loss corrects and produces functional revertantfibres by clonal expression. Muscle Nerve Suppl 7:S192.
Sabatelli P, Squarzoni S, Petrini S, Capanni C, Ognibene A, CartegniL, Cobianchi F, Merlini L, Toniolo D, Maraldi NM. 1998. Oralexfoliative cytology for the non-invasive diagnosis in Emery-Dreifuss muscular dystrophy patients and carriers. NeuromuscDisord 8:67–71.
Sewry CA, Muntoni F. 1999. Inherited disorders of the extracellularmatrix. Curr Opin Neurol 12:519–526.
Sewry CA, Matsumura K, Campbell KP, Dubowitz V. 1994a. Expres-sion of dystrophin-associated glycoprotein and utrophin in carriersof Duchenne muscular dystrophy. Neuromusc Disord 4:401–409.
Sewry CA, Muntoni F, Sansome A, Philpot J, Dubowitz V. 1994b.Sarcolemmal expression of utrophin in diverse neuromusculardisorders. Muscle Nerve Suppl 1:S103.
Sewry CA, Sansome A, Matsumura K, Campbell KP, Dubowitz V.1994c. Deficiency of the 50 kDA dystrophin-associated glycoproteinand abnormal expression of utrophin in two south asian cousinswith variable expression of severe childhood autosomal recessivemuscular dystrophy. Neuromusc Disord 4 :121–129.
Sewry CA, Chevallay M, Tome FMS. 1995a. Expression of lamininsubunits in human fetal skeletal muscle. Histochem J 27:497–504.
Sewry CA, Philpot J, Mahony D, Wilson LA, Muntoni F, Dubowitz V.1995b. Expression of laminin subunits in congenital musculardystrophy. Neuromusc Disord 5:307–316.
Sewry CA. Philpot J, Sorokin L, Wilson LA, Naom I, Goodwin F,D’Alessandro M, Dubowitz V, Muntoni F. 1996a. Diagnosis ofmerosin (laminin a2)-deficient congenital muscular dystrophy byskin biopsy. Lancet 347:582–584.
153IMMUNOCYTOCHEMISTRY OF MUSCULAR DYSTROPHIES

Sewry CA, Taylor J, Anderson LVB, Ozawa E, Pogue R, Piccolo F,Bushby K, Dubowitz V, Muntoni F. 1996b. Abnormalities in a, b andg sarcoglycan in patients with limb girdle muscular dystrophy.Neuromusc Disord 6:467–474.
Sewry CA, D’Alessandro M, Wilson LA, Sorokin LM, Naom I, Bruno S,Dubowitz V, Muntoni F. 1997a. Expression of laminin chains in skinin merosin-deficient congenital muscular dystrophy. Neuropediat-rics 28:217–222.
Sewry CA, Naom I, D’Alessandro M, Sorokin L, Bruno S, Wilson LA,Dubowitz V, Muntoni F. 1997b. Variable phenotype in merosin-deficient congenital muscular dystrophy and differential immunola-belling of two fragments of the laminin a2 chain. Neuromusc Disord7:169–175.
Sewry CA, Anderson LV.B, Bushby K, Muntoni F. 1998. Comparison of3 antibodies to laminin a2 for the diagnosis of partial expression incongenital muscular dystrophy. Muscle Nerve Suppl 7:S109.
Sewry CA, Mercuri E, Muntoni F. 1999. Reduced laminin b1 expres-sion in dominant myopathies. Neuromusc Disord 9:510.
Straub V, Campbell KP. 1997. Muscular dystrophies and the dystro-phin-glycoprotein complex. Curr Opin Neurol 10:168–175.
Tanaka H, Ishiguro T, Eguchi C, Saito K, Ozawa E. 1991. Expressionof dystrophin-related protein associated with the skeletal musclecell membrane. Histochem J 96:1–5.
Tan E, Topaloglu H, Sewry C, Zorlu Y, Naom I, Sevin E, D’AlessandroM, Muntoni F, Dubowitz V. 1997. Late onset muscular dystrophywith cerebral white matter changes due to partial merosin defi-ciency. Neuromusc Disord 7:85–90.
Thank LT, Nguyen thi Man, Hori S, Sewry CA, Dubowitz V, Morris GE.1995. Characterization of internally-deleted dystrophins in Beckermuscular dystrophy using a new panel of exon-specific mononclonal
antibodies against a deletion-prone region of dystrophin. Am J MedGenet 58:177–186.
Taylor J, Muntoni F, Dubowitz V, Sewry CA. 1997a. Early onsetautosomal dominant myopathy; a role for laminin b1?. NeuromuscDisord 7:211–216.
Taylor J, Muntoni F, Dubowitz V, Sewry CA. 1997b. Abnormalexpression of utrophin in Duchenne and Becker muscular dystrophyis age-related. Neuropath Appl Neurobiol 23:399–405.
Tome FMS, Guicheney P, Fardeau M. 1998. In: Emery AEH, editor.Neuromuscular disorders: clinical and molecular perspectives.Chichester: John Wiley. p 21–58.
Vainzof M, Passos-Bueno MR, Canovas M, Moreira ES. PavaanelloRC, Marie SK, Anderson LVB, Bonneman CG, McNally EM, Nigro V,Kunkel LM, Zatz M. 1996. The sarcoglycan complex in the sixautosomal recessive limb-girdle (AR-LGMD) muscular dystrophies.Hum Mol Genet 5:1963–1969.
Villanova M, Sewry CA, Malandrini A, Sabatelli P, Toti Y, Sqarzoni S,Torelli S, Muntoni F, Six J, Palma L, Scarfo G, Maraldi NM, GuazziGC. 1997. Localisation of several laminin chains in the normalhuman central and peripheral nervous system. J Submicrosc CytolPathol 29:409–413.
Wilton SD, Lloyd F, Carville K, Fletcher S, Honeyman K, Agrawai S,Kole R. 1999. Specific removal of the nonsense mutation from themdx dystrophin mRNA using antisense oligonucleotides. Neuro-musc Disord 9:330–338.
Yates JRW, Bagshaw, J, Aksmanovic VM, Coomber E, McMahon R,Whittaker JL, Morrison PJ, Kendrick-Jones J, Ellis JA. 1999.Genotype-phenotype analysis in X-linked Emery-Dreifuss musculardystrophy and identification of a missense mutation associated witha milder phenotype. Neuromusc Disord 9:159–165.
154 C.A. SEWRY