Embed Size (px)
Citation preview

Volume 26, Number 4, 2010
ImmunohematologyJournal of Blood Group Serology and Education


ImmunohematologyJournal of Blood Group Serology and Education
Volume 26, Number 4, 2010
Contents
133 ReviewPolylactosamines, there’s more than meets the “Ii”: a review of the I systemL. Cooling
156 Original ReportAttempts to support an immune etiology in 800 patients with direct antiglobulin test–negative hemolytic anemiaR.M. Leger, A. Co, P. Hunt, and G. Garratty
161 ReviewThe pathophysiology and prevention of transfusion-related acute lung injury (TRALI): a review D.C. Mair and T. Eastlund
174 Original ReportComparison of gel test and conventional tube test for antibody detection and titration in D-negative pregnant women: study from a tertiary care hospital in North IndiaM.K. Thakur, N. Marwaha, P. Kumar, S.C. Saha, B. Thakral, R.R. Sharma, K. Saluja, H.K. Dhawan, and A. Jain
178 ReviewThe Rh and RhAG blood group systemsS.T. Chou and C.M. Westhoff
187 CommunicationLetter to the editorsAnti-Vel and cold-reactive autoantibody
188 CommunicationLetter from the editorsThank you to the contributors of the 2010 issues
193 Announcements
195 Advertisements
199 Instructions For Authors
189 Index — Volume 26, Nos. 1, 2, 3, 4, 2010

Immunohematology is published quarterly (March, June, September, and December) by the American Red Cross, National Headquarters, Washington, DC 20006.
Immunohematology is indexed and included in Index Medicus and MEDLINE on the MEDLARS system. The contents are also cited in the EBASE/Excerpta Medica and Elsevier BIOBASE/Current
Awareness in Biological Sciences (CABS) databases.
The subscription price is $40.00 (U.S.) and $50.00 (foreign) per year.
Subscriptions, Change of Address, and Extra Copies:
Immunohematology, P.O. Box 40325 Philadelphia, PA 19106
Or call (215) 451-4902
Web site: www.redcross.org/immunohematology
Copyright 2010 by The American National Red Cross ISSN 0894-203X
Editor-in-ChiefSandra Nance, MS, MT(ASCP)SBBPhiladelphia, Pennsylvania
Managing EditorCynthia Flickinger, MT(ASCP)SBBPhiladelphia, Pennsylvania
Senior Medical EditorGeralyn M. Meny, MDPhiladelphia, Pennsylvania
Technical EditorsChristine Lomas-Francis, MScNew York City, New York
Dawn M. Rumsey, ART (CSMLT)Glen Allen, Virginia
Associate Medical EditorsDavid Moolten, MDPhiladelphia, Pennsylvania
Ralph R. Vassallo, MDPhiladelphia, Pennsylvania
Editorial AssistantSheetal Patel
Copy EditorMary L. Tod
ProofreaderLucy Oppenheim
Production AssistantMarge Manigly
Electronic PublisherPaul Duquette
On Our Cover“Les yeux clos” (1889–1905), Odilon Redon
Odilon Redon created several versions of Les yeux clos (Closed Eyes) between 1889 and 1905. The striking radiance of the woman’s face hints at both the mystery and the significance of inner vision. With her eyes shut, she is unable to see the world, but because the eyes are so central to human identity, the world is also unable to truly see her. Is she asleep or dead? What are her dreams, her thoughts? The subject of the painting was Redon’s wife, although he may have also been inspired by a Michelangelo sculpture of a dying Roman slave. The Ii blood group is the subject of a review article in this issue, which provides insight into the clinical and basic science of polylactosamines.
——DaviD Moolten, MD
Editorial Board
Patricia Arndt, MT(ASCP)SBBPomona, California
James P. AuBuchon, MDSeattle, Washington
Martha R. Combs, MT(ASCP)SBBDurham, North Carolina
Geoffrey Daniels, PhDBristol, United Kingdom
Anne F. Eder, MDWashington, District of Columbia
George Garratty, PhD, FRCPathPomona, California
Brenda J. Grossman, MDSt. Louis, Missouri
Christine Lomas-Francis, MScNew York City, New York
Gary Moroff, PhDRockville, Maryland
John J. Moulds, MT(ASCP)SBBShreveport, Louisiana
Paul M. Ness, MDBaltimore, Maryland
Joyce Poole, FIBMSBristol, United Kingdom
Mark Popovsky, MDBraintree, Massachusetts
Marion E. Reid, PhD, FIBMSNew York City, New York
S. Gerald Sandler, MDWashington, District of Columbia
Jill R. Storry, PhD Lund, Sweden
David F. Stroncek, MDBethesda, Maryland
Emeritus Editorial BoardDelores Mallory, MT(ASCP) SBBSupply, North Carolina

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 133
Polylactosamines, there’s more than meets the “Ii”: a review of the I system
Review
L. Cooling
Key Words: polylactosamine, I blood group, cold agglutinins
HistoryThe unofficial birth of the I blood group system can
be traced to a seminal case report in 1956.1 Wiener and colleagues at New York University and Jewish Hospital reported a fatal case of cold agglutinin syndrome caused by a potent autoantibody they named “anti-I” for “individuality.” The patient was a 62-year-old woman with a 5-year history of anemia and episodes of acute life-threatening hemolysis. Although the patient’s serum was compatible with donor RBCs at 37°C, she had periodic acute hemolytic transfusion reactions with “compatible” RBCs, even after warming the blood before transfusion. In an attempt to provide compatible blood, the patient’s serum was eventually tested against family members, 22,000 blood donors, and an assortment of animals including rabbit, sheep, horse, and cow. A total of five compatible donors were eventually identified, and their RBCs were successfully transfused to the patient during the next 2 years. Because these donors appeared to lack I, their RBCs were designated i phenotype. In a desperate moment when “compatible” human blood was unavailable, the patient was transfused with cow blood, which had demonstrated only weak activity at 4°C. Not surprisingly, the transfusion was aborted within 10 minutes, after the patient developed anxiety, acute dyspnea, and feelings of “impending doom”!
The serologic relationship between I and i phenotypes was solidified with the identification of an anti-i by Dr. Lawrence Marsh.2 The antibody was strongly reactive with cord RBCs and iadult RBCs but not normal adult RBCs. More importantly, Marsh was able to demonstrate a progressive age-dependent decrease in i on infant RBCs, suggesting a developmental relationship between I and i.
Ii SystemAs noted by Marsh, I and i are developmentally regulated
antigens on RBCs. At birth, cord RBCs serologically type as I–i+. A discernible increase in I expression is observed by 3 months, accompanied by parallel decreases in i expression, with an adult-type I+ phenotype observed by 18 months.2 Increases in I mirror increases in ABO expression, which typically reach adult levels by 2 to 3 years of age (see section on HDFN).3 Using human anti-I sera, the number of available I sites is estimated to be anywhere from 30,000 to 140,000 on untreated adult RBCs,4–6 but increases nearly threefold after enzyme treatment (see section on Ii antibodies).6
The iadult phenotype is a rare, autosomal-recessive trait characterized by an absence of I on RBCs as a result of
mutations in the I gene, IGnT (GCNT2).7–10 Serologic studies of family members, who presumably are heterozygous, can have an intermediate I+i+ phenotype with elevated i and weakened I expression relative to normal controls.2,11 In certain kindreds, iadult is associated with congenital cataracts.12–14 Based on older serologic studies, the incidence of iadult ranges from 1 in 4400 to 1 in 17,000.1,15 The prevalence of mutant IGnT alleles in the general population is unknown but is presumably rare.
Increased i expression can be observed with several inherited and acquired hemolytic disorders as a sign of stressed erythropoiesis. Increased i expression is found on reticulocytes, and also occurs in megaloblastic anemia, erythroleukemia, thalassemia, paroxysmal nocturnal hemoglobinuria, and other hemolytic disorders.16–18 Increased i is also a common finding in hereditary erythroblastic multinuclearity with positive acidified serum lysis test (HEMPAS) disease, a rare hematopoietic disorder characterized by dyserythropoiesis, altered RBC glycosylation, and hemolysis.19 Unlike other hemolytic disorders, the increased i observed on HEMPAS RBCs reflects abnormal Golgi trafficking and glycosylation.20,21
BiochemistryIi Structure
I and i are structurally and biosynthetically related oligosaccharide chains, composed of repeating units of N-acetyllactosamine (Fig. 1; [Galβ14GlcNAcβ1-]n, LacNAc). I and i are ubiquitously expressed on human tissues and membrane structures. On RBC glycoproteins, polylactosamines are expressed as asparagine- or N-linked glycans, ranging from biantennary to large, polyvalent tetraantennary structures (Fig. 1B). On gut, leukocytes, and other tissues, polylactosamine may also be expressed as O-linked glycans via covalent linkages on serine and threonine residues (Fig. 1C). Polylactosamines are also found on membrane glycosphingolipids (Fig. 1A), including massive polyglycosylceramides ranging from 20 to 50 carbohydrate residues in size.22
Structurally, i is defined as a linear, unbranched type 2 chain structure bearing at least two successive LacNAc residues (Fig. 1A).23 I is a branched polylactosamine derived from i by the addition of a β1,6-linked polylactosamine side chain.23 Antibodies against I, therefore, must recognize GlcNAcβ16Galβ-R as part of the immune epitope.23,24 Although I is often depicted with binary, terminal β13 and β16 LacNAc epitopes, in reality, β1,6 branching can occur

134 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
L. Cooling
anywhere along the polylactosamine backbone, assuming the presence of at least two successive LacNAc residues (see later section). The best example of the latter is the massive N-glycan on Band 3 of adult RBCs, which contains 40 to 50 oligosaccharides including five, short β1,6 LacNAc side chains (see section on HDFN).25
Ii expression can be modified by enzyme treatment of RBCs. Endo-β-galactosidase from Bacteroides fragilis and Escherichia freundii cleave internal, unsubstituted Galβ14R linkages.23 The enzyme will readily cleave i-active oligosaccharides in the absence of branching or modification. Internal galactose residues bearing substitutions, such as fucose (ABH, LeX; Fig. 1D) or β16GlcNAc (I) are resistant to enzyme cleavage.26 As a result, endo-β-galactosidase will destroy i reactivity and reduce, but not eliminate, I because of the presence of substituted galactose at β1,6 branch points (Fig. 1A). In contrast, Ii expression tends to increase after digestion with proteases and neuraminidase.26 Proteases can increase accessibility to Ii expressed on glycolipids and polyglycosylceramides. Neuraminidase, on the other hand, can remove terminal sialic acid on sialylated type 2 glycans, resulting in a terminal LacNAc epitope (see Fig. 1D).
Polylactosamines serve as scaffold structures for the synthesis of other type 2 chain carbohydrate antigens
such as ABO, LeX, sLeX, LeY, and sialo-Ii activity (Fig. 1D).27 On human RBCs, more than 95 percent of all ABO antigens are type 2 chain oligosaccharides on N-glycans and glycolipids.28 Not surprisingly, some examples of anti-I/i have complex reactivity, reacting more strongly with RBCs of specific ABO types.18
Polylactosamine BiosynthesisThe Golgi and Glycosyltransferases
Proteins and lipids are initially synthesized in the endoplasmic reticulum (ER, Fig. 2A), followed by posttranslation glycosylation in the Golgi. The regulatory mechanisms directing the passage and modification of substrates through the Golgi is still not entirely understood owing to its inherent dynamic and structural complexity. Grossly, the Golgi is composed of stacked membranous cisternae arranged into three regions; cis-, medial-, and trans-Golgi. Cargo (proteins, lipids) is shuttled between the ER and various Golgi compartments by membrane budding, followed by targeting and fusion of vesicles to another Golgi compartment or plasma membrane. The process is dependent on GTP, coat complex proteins (COPI, COPII), SNARE proteins, membrane tethering proteins, and lipid transfer proteins.29 Mutations in COPII proteins
Fig. 1 Structures of Ii oligosaccharides on cell membranes. (A) Structure of i-active and I-active glycosphingolipid (N-acetyl ceramide). Endo-ß-endoceramidase can cleave i-active oligosaccharides; however, I-active oligosaccharides are resistant. (B) Structure of biantennary and tetraantennary glycans on N- or asparginine-linked glycoproteins. (C) O-glycan structures. (D) Terminal modification of Ii or type 2 chain oligosaccharides. Cer = ceramide; Gal = galactose; GalNAc = N-acetylgalactosamine; Glc = glucose; GlcNAc = N-acetylglucosamine; Man = mannose.
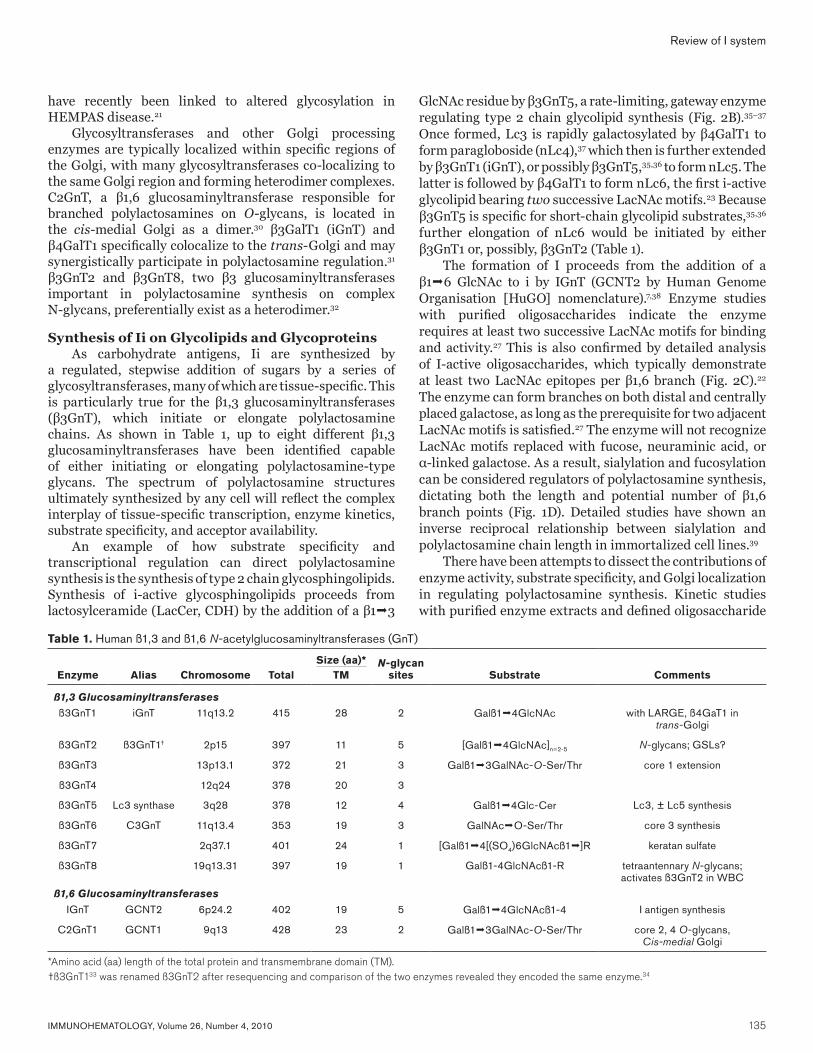
IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 135
Review of I system
have recently been linked to altered glycosylation in HEMPAS disease.21
Glycosyltransferases and other Golgi processing enzymes are typically localized within specific regions of the Golgi, with many glycosyltransferases co-localizing to the same Golgi region and forming heterodimer complexes. C2GnT, a β1,6 glucosaminyltransferase responsible for branched polylactosamines on O-glycans, is located in the cis-medial Golgi as a dimer.30 β3GalT1 (iGnT) and β4GalT1 specifically colocalize to the trans-Golgi and may synergistically participate in polylactosamine regulation.31 β3GnT2 and β3GnT8, two β3 glucosaminyltransferases important in polylactosamine synthesis on complex N-glycans, preferentially exist as a heterodimer.32
Synthesis of Ii on Glycolipids and GlycoproteinsAs carbohydrate antigens, Ii are synthesized by
a regulated, stepwise addition of sugars by a series of glycosyltransferases, many of which are tissue-specific. This is particularly true for the β1,3 glucosaminyltransferases (β3GnT), which initiate or elongate polylactosamine chains. As shown in Table 1, up to eight different β1,3 glucosaminyltransferases have been identified capable of either initiating or elongating polylactosamine-type glycans. The spectrum of polylactosamine structures ultimately synthesized by any cell will reflect the complex interplay of tissue-specific transcription, enzyme kinetics, substrate specificity, and acceptor availability.
An example of how substrate specificity and transcriptional regulation can direct polylactosamine synthesis is the synthesis of type 2 chain glycosphingolipids. Synthesis of i-active glycosphingolipids proceeds from lactosylceramide (LacCer, CDH) by the addition of a β13
GlcNAc residue by β3GnT5, a rate-limiting, gateway enzyme regulating type 2 chain glycolipid synthesis (Fig. 2B).35–37 Once formed, Lc3 is rapidly galactosylated by β4GalT1 to form paragloboside (nLc4),37 which then is further extended by β3GnT1 (iGnT), or possibly β3GnT5,35,36 to form nLc5. The latter is followed by β4GalT1 to form nLc6, the first i-active glycolipid bearing two successive LacNAc motifs.23 Because β3GnT5 is specific for short-chain glycolipid substrates,35,36 further elongation of nLc6 would be initiated by either β3GnT1 or, possibly, β3GnT2 (Table 1).
The formation of I proceeds from the addition of a β16 GlcNAc to i by IGnT (GCNT2 by Human Genome Organisation [HuGO] nomenclature).7,38 Enzyme studies with purified oligosaccharides indicate the enzyme requires at least two successive LacNAc motifs for binding and activity.27 This is also confirmed by detailed analysis of I-active oligosaccharides, which typically demonstrate at least two LacNAc epitopes per β1,6 branch (Fig. 2C).22 The enzyme can form branches on both distal and centrally placed galactose, as long as the prerequisite for two adjacent LacNAc motifs is satisfied.27 The enzyme will not recognize LacNAc motifs replaced with fucose, neuraminic acid, or α-linked galactose. As a result, sialylation and fucosylation can be considered regulators of polylactosamine synthesis, dictating both the length and potential number of β1,6 branch points (Fig. 1D). Detailed studies have shown an inverse reciprocal relationship between sialylation and polylactosamine chain length in immortalized cell lines.39
There have been attempts to dissect the contributions of enzyme activity, substrate specificity, and Golgi localization in regulating polylactosamine synthesis. Kinetic studies with purified enzyme extracts and defined oligosaccharide
Table 1. Human ß1,3 and ß1,6 N-acetylglucosaminyltransferases (GnT)
Enzyme Alias Chromosome TotalSize (aa)*
TMN-glycan
sites Substrate Comments
ß1,3 Glucosaminyltransferases
ß3GnT1 iGnT 11q13.2 415 28 2 Galß14GlcNAc with LARGE, ß4GaT1 in trans-Golgi
ß3GnT2 ß3GnT1† 2p15 397 11 5 [Galß14GlcNAc]n=2-5 N-glycans; GSLs?
ß3GnT3 13p13.1 372 21 3 Galß13GalNAc-O-Ser/Thr core 1 extension
ß3GnT4 12q24 378 20 3
ß3GnT5 Lc3 synthase 3q28 378 12 4 Galß14Glc-Cer Lc3, ± Lc5 synthesis
ß3GnT6 C3GnT 11q13.4 353 19 3 GalNAcO-Ser/Thr core 3 synthesis
ß3GnT7 2q37.1 401 24 1 [Galß14[(SO4)6GlcNAcß1]R keratan sulfate
ß3GnT8 19q13.31 397 19 1 Galß1-4GlcNAcß1-R tetraantennary N-glycans; activates ß3GnT2 in WBC
ß1,6 Glucosaminyltransferases
IGnT GCNT2 6p24.2 402 19 5 Galß14GlcNAcß1-4 I antigen synthesis
C2GnT1 GCNT1 9q13 428 23 2 Galß13GalNAc-O-Ser/Thr core 2, 4 O-glycans, Cis-medial Golgi
*Amino acid (aa) length of the total protein and transmembrane domain (TM).†ß3GnT133 was renamed ß3GnT2 after resequencing and comparison of the two enzymes revealed they encoded the same enzyme.34

136 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
L. Cooling
substrates are mixed.27 It is clear that the synthesis of β13 GlcNAc by β3GnT is a critical rate-limiting step, with many β3GnT displaying specific substrate specificities (Table 1). On N-glycans, LacNAc synthesis requires the involvement of at least three different β3GnTs: β3GnT1,40 β3GnT2,34,41–43 and β3GnT8.32,44 β3GnT1 reacts well with linear and branched acceptors although the recombinant enzyme may have slightly higher activity with β1,6 branched acceptors.27,40 β3GnT2, like β3GnT1, has broad acceptor reactivity, whereas β3GnT8 is quite specific for large tetraantennary substrates on N-glycans (Fig. 1).44 β3GnT8 and β3GnT2 may cooperatively enhance polylactosamine synthesis, particularly on leukocytes.32 In HL60 cells, β3GnT8 has been shown to activate β3GnT2, increasing the Vmax/Km ratio tenfold with a fourfold increase in β3GnT2 activity.32
β3GnT1 and β4GalT1 colocalize within the trans-Golgi, suggesting a possible coregulatory role for β4GalT1.31 Although β4GalT1 is not a rate-limiting step, studies indicate subtle enzyme preferences that may influence the length and degree of branching.27 With simple trisaccharide substrates, β4GalT1 preferentially reacts with GlcNAcβ1-3LacNAc (i type) over GlcNAcβ1-6LacNAc (I type), suggesting a concerted effort by β3GnT1 and β4GalT1 toward synthesis of long i-type chains. Slightly different results are obtained when β4GalT1 is tested against branched substrates offering both β1,3 GlcNAc and β1,6 GlcNAc termini.27 Unlike linear substrates, β4GalT1 will preferentially recognize the β1,6 GlcNAc branch early in the reaction, although this tended to inhibit further galactosylation of the remaining β1,3 branch. In contrast, initial galactosylation of the β1,3 GlcNAc permitted a second galactosylation to form an I-type structure with both β1,3 and β1,6 LacNAc termini. Like sialylation and fucosylation, a distal β1,6 branch near the nonreducing galactose terminus may inhibit further chain elongation.
Molecular BiologyThe i Gene
The gene generally referred to as iGnT is β3GnT1, a β1,3-N-acetylglucosaminyltransferase, which transfers a GlcNAc β13 to lactose and LacNAc termini.40 The gene resides on chromosome 11q13.2 and encodes a 415-amino acid, 47-kDa type II transmembrane protein. The enzyme contains two potential N-glycosylation sites and an unusually long, 28-amino acid transmembrane domain.40 The gene shares little sequence homology with other β1,3 glucosaminyltransferases.34
Fig. 2 Biosynthesis of Ii oligosaccharides. (A) Transport of proteins and lipids from the endoplasmic reticulum (ER) through the cis-, medial-, and trans-Golgi. Inset shows the orientation and topical structure of a typical glycosyltransferase. (B) Synthesis of i- and I-active glycosphingolipids. (C) The structure of erythroglycan, an RBC polylactosaminylceramide. Note that each ß1,6 branch point is preceded by at least two lactosamine motifs (LacNAc). Cer = ceramide; Gal = galactose; Glc = glucose; GlcNAc = N-acetylglucosamine.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 137
Review of I system
The enzyme is reportedly capable of both initiating the synthesis of polylactosamines and elongating existing polylactosamine oligosaccharides. As discussed earlier, β3GnT1 colocalizes with β4GalT1 in the trans-Golgi, where β4GalT1 may co-associate and stabilize β3GnT1 retention.31 β3GnT1 also complexes with LARGE, an unusual glucosaminyltransferase implicated in O-linked glycans on α-dystrophin.45 By Northern blot, the enzyme is ubiquitously expressed in most human tissue tested; however, expression is extremely weak in WBCs, thymus, neural tissue, lung, and liver.40 The relative absence of detectable RNA in WBCs is noteworthy: granulocytes and monocytes express primarily type 2 chain glycans on both glycoproteins and glycolipids.46,47
Seven additional β3GnTs have been cloned and characterized (Table 1). The literature regarding these β3GnTs is a bit confusing as a result of duplicate publication and name changes. All seven β3GnTs share significant homology with each other and are related to a larger family of β1,3 galactosyltransferases.34,44 β3GnT2 (originally named β3GnT133 to distinguish it from iGnT), was isolated from human uterus and bladder.33,34,41 Like β3GnT1, β3GnT2 was able to initiate and elongate poly-N-acetyllactosaminoglycans on a variety of substrates and displays wide expression.33 Data from β3GnT2 knockout mice indicate a major role for β3GnT2 in polylactosamine synthesis on N-linked glycans.42,43 β3GnT2 appears to physically associate with β3GnT8,32 an elongating β3GnT specific for the β1,6 branch on tetraantennary N-glycans.44 In HL60 cells, myeloid differentiation is accompanied by a parallel increase in β3GnT8 mRNA and polylactosamine chain length.32 β3GnT8 is highly expressed in bone marrow, spleen, pancreas, and small intestine with lower level expression in other tissues.44
β3GnT5 is specific for initiating type 2 chain synthesis on glycolipids,35,36 which contributes significantly to Ii and sialo agglutinin expression on human RBCs. β3GnT5 is upregulated during myeloid differentiation,36 coincident with the synthesis of type 2 chain glycolipids with LeX, sLeX, and VIM activity (Fig. 1D).46,48 Polylactosamine synthesis on O-linked glycans and keratan is directed by β3GnT3, β3GnT6, C2GnT, and β3GnT7 (Fig. 1C, Table 1).34,49,50 β3GnT3 and β3GnT6 are related glycosyltransferases responsible for initiating core 2 and core 3 O-glycan synthesis and are highly expressed in gastrointestinal tissues (Fig. 1C).34,49 Core 3 O-glycans can be further modified by C2GnT (Fig. 1C), an I-like β1,6 branching enzyme located in the cis-medial Golgi.30,51 β3GnT7 recognizes and elongates sulfated LacNAc oligosaccharides found on keratan sulfate and is highly expressed in placenta, colon, stomach, and small intestine.50,52
The I GeneI (IGnT, GCNT2) is located on chromosome 6p24.2
(Fig. 3A).38 It consists of five exons (E1A, E1B, E1C, E2, E3), which give rise to three related isoforms of the enzyme
depending on which exon 1 is used (Fig. 3B, 3C).7,8 IGnTA (GCNT2A, IGnT2), the most common transcript found in human tissues, is encoded by exons E1A, E2, and E3. The IGnT2B (GCNT2B) uses exon E1B and is the only mRNA transcript identified in purified human lens epithelium.8 It is also highly expressed in fetal brain and adult cerebellum.
IGnTC (GCNT2C), on the other hand, is nearly unique to human reticulocytes and is responsible for I antigen expression on RBCs.7,8 This has been confirmed by studies in hematopoietic stem cells.53 IGnT expression is extremely low in both adult and cord CD34+ cells. On erythroid differentiation, a parallel increase in IGnTC and I antigen expression is observed.53 Mutations in exons E1C, E2, and E3 have all been associated with the iadult RBC phenotype.7–10
Although encoded by the same gene, IGnTA, IGnTB, and IGnTIGnTC share only 66 to 73 percent homology because of sequence differences in exon 1, which may account for differences in enzyme activity among the three enzyme isoforms.8 In enzyme assays, IGnTC has nearly twofold higher activity than either the IGnTA or IGnTB
Fig. 3 Structure of the IGnT gene. (A) Location of IGnT on chromosome 6. (B) The structure of IGnT showing the arrangement and size (bp) for exons E1A, E1B, E1C, E2, and E3. The size of the intervening introns is also shown. (C) The composition of the three IGnT isoforms and their tissue distribution. Triangles denote mutations associated with iadult phenotype. Solid triangles affecting exons E2 and E3 result in iadult with congenital cataracts. (D) The IGnTC promoter region showing transcription binding sites for Oct 2, Sp1, and C/EBPa. Functional binding of C/EBPa protein to IGnTC promoter requires altered phosphorylation of serine and threonine residues.

138 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
L. Cooling
isoform.8 Exon E1 encodes nearly 77 percent of the active enzyme (Fig. 4), including the transmembrane domain, stem region, and part of the catalytic domain containing the nucleotide binding site. E2 and E3, which are shared by all three isoforms, encode the carboxy-terminal end of the enzyme.
Transcriptional ControlThe identification of three tissue-specific IGnT/GCNT2
isoforms strongly suggests transcriptional regulation of exon 1 by cis-regulatory sequences in the 5′ untranslated region (5′UTR) or promoter region. This possibility is also supported by the structure of IGnT, in which exons E1A, E1B, and E1C are each separated by relatively large intronic sequences that may each harbor binding sites for tissue-specific transcription factors.7,8 This was recently confirmed by studies in K562 cells, which identified a promoter sequence –318 to –251 base pairs upstream of the
translation initiation site.53 As shown, the IGnTC promoter in erythroid cells contains consensus sequences for Oct-2, Sp1, and C/EBPα transcription factors (Fig. 3D).53
Although both Oct-2 and Sp1 bind the IGnTC promoter, C/EBPα binding is critical for IGnTC transcriptional activation.53 How C/EBPα regulates IGnTC is complex, apparently involving posttranslational phosphorylation of C/EBPα protein. In K562 erythroleukemia cells, butyrate induces changes in C/EBPα phosphorylation, a prerequisite for functional C/EBPα binding to the IGnTC promoter (Fig. 3D). Once bound, the phosphorylated-C/EBPα drives IGnTC transcription irrespective of Oct-2 and Sp1 binding. Similar results can be observed during in vitro differentiation of adult and cord stem cells.53 Loss of phosphoserine residues may be critical for C/EBPα-mediated transcription activation: serine phosphorylation has been shown to repress C/EBPα during early hematopoiesis.54
Fig. 4 Structure-function of IGnT. The IGnTC isoform, expressed in reticulocytes, is a 402–amino acid glycoprotein protein, possessing up to five N-glycosylation sites and nine conserved cysteine residues. Exon 1, which shows only 65 percent homology among the three IGnT isoforms, encodes nearly 76 percent of the enzyme. Like all glycosyltransferases, the molecule contains a short 19-amino acid hydrophobic transmembrane region (TM domain) that anchors the molecule within the Golgi membrane, a stem region, and a large catalytic domain. IGnT shares three regions of significant homology with C2GnT, a related ß-1,6-N-acetylglucosaminyltransferase ( ) whose three-dimensional structure has been characterized. Using C2GnT as a template, the location of four potential disulfide bonds (- - -), nucleotide-binding site, and acceptor-binding sites were extrapolated. IGnTC and C2GnT share significant homology in ß1, ß2, ß3, and ß4 strands that incorporate the UDP or nucleotide donor–binding site. Of six nucleotides critical for acceptor binding by C2GnT (E243, K251, R254, E320, W356, Y358; ⇓, bold), only two homologous sequences were identified in IGnT (E298, W328). Conserved sequences, including a lysine, along the ß7 strand may play a role in binding phosphate of the UDP donor.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 139
Review of I system
IGnT (GCNT2) Protein Structure-FunctionThe translated protein varies from 400 (IGnTB) to 402
amino acids (IGnTA, IGnTC).7,38 Like all glycosyltransferases, the enzyme is a type 2 transmembrane glycoprotein possessing several potential N-glycosylation sites and a 16– to 19–amino acid hydrophobic transmembrane domain near the amino terminus (Fig. 4).38 It is presumed to possess significant tertiary structure based on exhibiting nine conserved cysteine residues.38,55
IGnT shares sequence homology with C2GnT, a related β1-6 N-acetylglucosaminyltransferase that transfers UDP-GlcNAc to Galβ1-3GalNAc-O-Ser/Thr, transforming simple core 1 and core 3 O-glycans to branched, core 2 and core 4 O-glycans, respectively (Fig. 1C).55,56 Mucin-type C2GnT (C2GnT-M) also displays IGnT-like activity with synthesis of I antigen on N-glycans.56 A comparison of IGnT and C2GnT reveals three regions sharing approximately 60 percent homology: a large 126–amino acid stretch incorporating the putative UDP-GlcNAc binding site and two smaller regions (32 and 22 amino acids) in the carboxy-terminal acceptor binding domain of the enzyme.57,58 Unlike most glycosyltransferases identified to date, neither IGnT nor C2GnT possesses a DXD motif upstream of the UDP-GlcNAc binding site.57 DXD motifs help coordinate interactions of the enzyme with the phosphate group of the nucleotide donor (UDP) and a divalent cation such as Mn2+ or Mg2+.57
The x-ray crystallography structure of mouse C2GnT was recently determined and identified C2GnT as a novel metal ion–independent GT-A glycosyltransferase.58 Like most GT-A type glycosyltransferases, C2GnT has an α/β/α structure with a mixed β-sheet containing seven topological β strands. Strands β1 through β4 bind the nucleotide (UDP) of the UDP-GlcNAc donor: acceptor binding occurs downstream of the UDP site and involves amino acids located within the β5, β6, α5, and β7 regions (Fig. 4). Critical amino acids necessary for acceptor binding include glutamic acid (E243, E320), lysine (K251), tryptophan (W256), and tyrosine (Y258). E320 corresponds to a structurally conserved catalytic base required for activity in GT-A glycosyltransferases. E243, E320, and Y258 have direct interactions with the Galβ13GalNAc acceptor, whereas K251 helps stabilize a stacking interaction between the acceptor and W256. Two additional basic amino acids, arginine 378 (R378) and lysine 401 (K401), are structurally aligned to interact with UDP-phosphate residues, possibly substituting for the DXD motif and divalent cation. C2GnT also contains four disulfide bonds.
Using the C2GnT sequence and crystallography data as a template, a potential structure-function model of IGnTC enzyme was constructed (Fig. 4). Like C2GnT, IGnT possesses nine, positionally conserved cysteine residues and is therefore capable of four disulfide bonds. IGnTC also possesses homologous sequences for β1, β2, β3, and β4 strands implicated in UDP binding. Not surprisingly, all
four β strands were localized to a 126–amino acid stretch in exon 1 that shares significant homology with C2GnT. Less conservation was observed in the acceptor binding domain. IGnT contained glutamic acids (E298, E367) and tryptophan (W328) corresponding to E320, E401, and W356 residues in C2GNT; however, no clear homologs for E243, K251, and K254 were identified. Interestingly, E243, K251, and K254 reside downstream of the C2GNT β5 strand, a region that shares minimal sequence homology between IGnT and C2GnT genes.
iadult Phenotype and CataractsThe iadult phenotype is a rare, autosomal-recessive
phenotype caused by a mutation in the IGnT/GCNT2 gene. The linkage of iadult with congenital cataracts was originally reported by Ogata et al.12 and primarily limited to Asian kindreds. The etiology behind both iadult and congenital cataracts was finally resolved after mapping of the IGnT gene and identification of three distinct IGnT isoforms (Fig. 3).7–10 Mutations in exon E1C, which is the predominant isoform expressed in erythroblasts and reticulocytes, leads to an iadult RBC phenotype without congenital cataracts. Although IGnT activity is missing in RBCs, IGnT activity is intact in lens and other tissues via IGnTA and IGnTB (Fig. 3C). In contrast, mutations in exons E2 and E3, which are common to all three IGnT isoforms, result in a tissue-wide loss of IGnT activity including RBCs and lens. Mutations in E2 and E3 are responsible for iadult with congenital cataracts. A summary of the specific mutations and phenotypes is listed in Table 2.
An I+i+ phenotype might occur in some heterozygous carriers of mutant IGnT alleles. Early family studies of iadult individuals reported decreased I expression among many first-degree family members.2,11 Although these relatives still typed as I+, titration scores with anti-I and -i sera were intermediate between those of normal and iadult RBCs, resulting in an “Iwi+-like” phenotype. Examples of haploinsufficiency and weak antigen expression have been observed with other glycosyltransferases, including the related C2GnT.59,60 Heterozygosity for a single missense mutation in C2GnT (S158C) results in a marked decrease in core 2 O-glycans on T cells, accompanied by resistance to galectin-mediated cell apoptosis.59
Table 2. IGnT mutations associated with iadult phenotype7–10
Mutation Phenotype
Exon Nucleotide Amino acid iadult RBC Cataracts
E1C 243T>A N81L + –
E1C 505G>A A169T + –
E1C 683G>A R228Q + –
E2 983G>A W328X + +
E2 1005G>A G336R + +
E3 1049G>A G350E + +
E3 1154G>A R385H + +
140 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
L. Cooling
Antibodies to Ii Anti-I
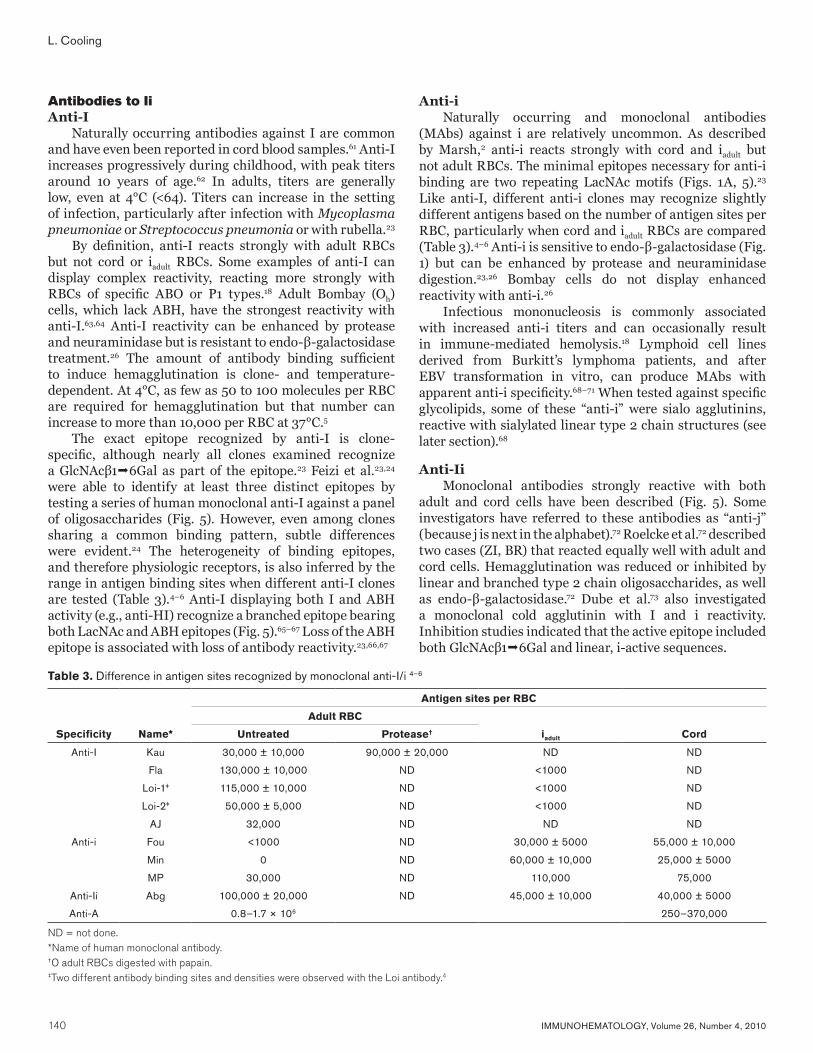
Naturally occurring antibodies against I are common and have even been reported in cord blood samples.61 Anti-I increases progressively during childhood, with peak titers around 10 years of age.62 In adults, titers are generally low, even at 4°C (<64). Titers can increase in the setting of infection, particularly after infection with Mycoplasma pneumoniae or Streptococcus pneumonia or with rubella.23
By definition, anti-I reacts strongly with adult RBCs but not cord or iadult RBCs. Some examples of anti-I can display complex reactivity, reacting more strongly with RBCs of specific ABO or P1 types.18 Adult Bombay (Oh) cells, which lack ABH, have the strongest reactivity with anti-I.63,64 Anti-I reactivity can be enhanced by protease and neuraminidase but is resistant to endo-β-galactosidase treatment.26 The amount of antibody binding sufficient to induce hemagglutination is clone- and temperature-dependent. At 4°C, as few as 50 to 100 molecules per RBC are required for hemagglutination but that number can increase to more than 10,000 per RBC at 37°C.5
The exact epitope recognized by anti-I is clone-specific, although nearly all clones examined recognize a GlcNAcβ16Gal as part of the epitope.23 Feizi et al.23,24 were able to identify at least three distinct epitopes by testing a series of human monoclonal anti-I against a panel of oligosaccharides (Fig. 5). However, even among clones sharing a common binding pattern, subtle differences were evident.24 The heterogeneity of binding epitopes, and therefore physiologic receptors, is also inferred by the range in antigen binding sites when different anti-I clones are tested (Table 3).4–6 Anti-I displaying both I and ABH activity (e.g., anti-HI) recognize a branched epitope bearing both LacNAc and ABH epitopes (Fig. 5).65–67 Loss of the ABH epitope is associated with loss of antibody reactivity.23,66,67
Anti-iNaturally occurring and monoclonal antibodies
(MAbs) against i are relatively uncommon. As described by Marsh,2 anti-i reacts strongly with cord and iadult but not adult RBCs. The minimal epitopes necessary for anti-i binding are two repeating LacNAc motifs (Figs. 1A, 5).23 Like anti-I, different anti-i clones may recognize slightly different antigens based on the number of antigen sites per RBC, particularly when cord and iadult RBCs are compared (Table 3).4–6 Anti-i is sensitive to endo-β-galactosidase (Fig. 1) but can be enhanced by protease and neuraminidase digestion.23,26 Bombay cells do not display enhanced reactivity with anti-i.26
Infectious mononucleosis is commonly associated with increased anti-i titers and can occasionally result in immune-mediated hemolysis.18 Lymphoid cell lines derived from Burkitt’s lymphoma patients, and after EBV transformation in vitro, can produce MAbs with apparent anti-i specificity.68–71 When tested against specific glycolipids, some of these “anti-i” were sialo agglutinins, reactive with sialylated linear type 2 chain structures (see later section).68
Anti-IiMonoclonal antibodies strongly reactive with both
adult and cord cells have been described (Fig. 5). Some investigators have referred to these antibodies as “anti-j” (because j is next in the alphabet).72 Roelcke et al.72 described two cases (ZI, BR) that reacted equally well with adult and cord cells. Hemagglutination was reduced or inhibited by linear and branched type 2 chain oligosaccharides, as well as endo-β-galactosidase.72 Dube et al.73 also investigated a monoclonal cold agglutinin with I and i reactivity. Inhibition studies indicated that the active epitope included both GlcNAcβ16Gal and linear, i-active sequences.
Table 3. Difference in antigen sites recognized by monoclonal anti-I/i 4–6
Antigen sites per RBC
Adult RBC
Specificity Name* Untreated Protease† iadult Cord
Anti-I Kau 30,000 ± 10,000 90,000 ± 20,000 ND ND
Fla 130,000 ± 10,000 ND <1000 ND
Loi-1‡ 115,000 ± 10,000 ND <1000 ND
Loi-2‡ 50,000 ± 5,000 ND <1000 ND
AJ 32,000 ND ND ND
Anti-i Fou <1000 ND 30,000 ± 5000 55,000 ± 10,000
Min 0 ND 60,000 ± 10,000 25,000 ± 5000
MP 30,000 ND 110,000 75,000
Anti-Ii Abg 100,000 ± 20,000 ND 45,000 ± 10,000 40,000 ± 5000
Anti-A 0.8–1.7 × 106 250–370,000
ND = not done.*Name of human monoclonal antibody.†O adult RBCs digested with papain.‡Two different antibody binding sites and densities were observed with the Loi antibody.4

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 141
Review of I system
Fig. 5 Epitopes for examples of anti-Ii. Anti-i requires at least two LacNAc residues. Several different epitopes have been identified among anti-I (lines), although all include a GlcNAcß1→6Gal epitope. Examples of anti-Ii with HI and sialo agglutinin activity are also shown. Cer = ceramide; Fuc = fucose; Gal = galactose; GlcNAc = N-acetylglucosamine; NeuAc = neuraminic acid.

142 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
L. Cooling
Sialo AgglutininsSialo agglutinins represent a subgroup of I/i
antibodies that require a terminal sialic acid for activity. They are distinguished from anti-I and anti-i by their sensitivity to neuraminidase. Dieter Roelcke26 broadly subclassified the sialo agglutinins into two major classes, based on whether the antibodies recognize linear (Sia-l1) or branched (Sia-b1) sialooligosaccharides. Like anti-I, Sial-1b includes antibodies against branched structures with ABH activity (anti-FL).67 Sialo agglutinins with Sia-1b reactivity have been linked with both CMV and Mycoplasma infections.74,75
A third category of sialo agglutinins are Sia-lb1 and Sia-lb2, which recognize sialylated glycosphingolipids or gangliosides (Gd, or glycolipid-dependent).26 Sia-lb1,2 bind linear and branched type 2 chain gangliosides and are resistant to protease digestion. Inhibition studies indicate the minimal epitope for Sia-lb1,2 is the terminal mono-disaccharide incorporating sialic acid with or without galactose (Fig. 5). Not surprisingly, some Sia-1b1,2 antibodies also bind ganglioside GM3 (sialyl-lactose) and sialylparagloboside.26 As discussed earlier, anti-i produced by EBV-transformed human lymphocytes may recognize linear type 2 gangliosides.68,69
Sialylated lactosamines have been shown to be receptors for M. pneumoniae.76 Mycoplasma has been shown to bind sialyl-polylactosamines along the apical aspect of ciliated bronchial epithelium and microvilli.77 The latter could serve as a trigger for autoantibody formation and may contribute to the prevalence of sialo-I (Sia-1b) after Mycoplasma infection.74,78
Antibody Structure-FunctionCold Agglutinin Disease
MAbs from patients with cold agglutinin disease have been invaluable in dissecting the pathologic autoimmune response to Ii. Multiple independent investigators have confirmed that IgM binding to Ii is primarily mediated by the IgM heavy chain via the VH or variable heavy domain. Of the 123 to 129 heavy chain VH genes available, the VH4-34 gene segment (VH4-21 or VH4.21 in older papers) is almost exclusively used by monoclonal cold agglutinins with Ii autoreactivity (Fig. 6).79 VH4-34 is also overrepresented in many, but not all, B-cell lymphoproliferative disorders including follicular lymphoma, mantle cell lymphoma, and Waldenström’s macroglobulinemia.80 In contrast, VH4-34 is unusual in chronic lymphocytic leukemia.80,81 In normal individuals, VH4-34 is identified in 3 to 6 percent of adult and fetal B cells but constitutes less than 0.5 percent of the total circulating IgM and IgG.80 Antibodies bearing the VH4-34 gene segment can be identified with the anti-idiotypic antibody MAb 9G4.79,82
Detailed sequence analysis, coupled with construction of recombinant immunoglobulins, has uncovered the basis of Ii binding by VH4-34. Although only 110 to 120 amino acids in length, the VH region has distinct subdomains
determined to be important in antigen binding. As shown in Figure 6D, three hypervariable or complementarity determining regions (CDR) are flanked by so-called framework regions (FR). Among VH4-34 monoclonal cold agglutinins, binding to either I or i is mediated by the N-terminal framework region 1 (FR1). This has been verified by recombinant IgH clones using an FR1 from a different VH4 subgroup.79 The 9G4 epitope is located in FR1 (amino acids 23–25).79 Although MAb 9G4 can inhibit RBC agglutination,70 the 9G4-binding site lies outside the antigen-binding site.83 It is theorized MAb 9G4 may inhibit
Fig. 6 Structure-function of anti-Ii antibodies. (A) A schematic of human IgM showing five antigen binding sites per molecule. (B) The structure of an IgM heavy chain and light chain pair showing constant (CH, CL) and variable region (VH, VL) gene segments. The VH and VL regions encode the antigen binding site. The hinge region along CH2 is a site for F(ab´)2 autoantibodies. (C) Autoantibodies against Ii tend to use VH gene segments from specific families. Polyclonal benign anti-Ii tend to use VH gene segments from the VH3 and VH4 families, whereas the VH4-34 gene segment (historically VH4-21) accounts for the majority of monoclonal cold agglutinins. VH4-34 is also common among IgM anti-Rh monoclonal sera. (D) The VH gene segment consists of four framework regions (FR) and three complementarity determining regions (CDR). For VH4-34, FR1 is required for Ii binding. Sequence variation in CDRH3 may contribute to the fine specificity of the antibody (I or i). MAb 9G4 antibody recognizes an epitope in FR1 of VH4-34.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 143
Review of I system
binding by steric hindrance or induced conformational change.83
Recombinant IgH clones also revealed a role for CDR domains, mutational “hot spots” known to direct antigen binding and specificity. Of three CDR domains, evidence suggests that CDRH3, in concert with light chains, may define the fine specificity (anti-I or anti-i) of the antibody.79 The ability of CDRH3 to distinguish I from i, however, is not mediated by a specific sequence motif: individual B-cell clones show significant sequence variability in CDRH3.79,82 Crystallography studies of a human monoclonal anti-I show that CDRH3 and two light chain variable regions (VκCDRL1 and CDRL3) line the antigen-binding pocket of the antibody.83
The role of light chains in Ii specificity is less clear-cut. There is a preference for Vκ, particularly Vκ3, among examples of human monoclonal anti-I.82 Kappa light chain restriction is also observed among monoclonal anti-Pr cold sialo agglutinins, with Vκ4 and Vκ3 accounting for 61 percent of clones.84 Interestingly, Vκ4 was linked to recognition of sialo agglutinins bearing an α23 neuraminic acid.84 Monoclonal anti-i uses both Vκ and Vλ, with higher Vλ usage relative to polyclonal antibodies.82 Vλ usage is also relatively common in other autoimmune diseases, including antibodies against dsDNA, phospholipid, rheumatoid factor, and histone.85
Infection and Naturally Occurring AutoantibodiesUnlike monoclonal anti-I/i, polyclonal anti-I/i does
not display light or heavy chain restriction. Early studies using the 9G4 MAb showed that only a fraction of naturally occurring polyclonal anti-Ii carried the VH4-34 gene segment.82,86 This was also supported by the inability of MAb 9G4 to inhibit hemagglutination with polyclonal anti-I/i.86 Naturally occurring anti-Ii uses a mix of VH segments from the VH3 and VH4 families.86 VH3 and VH4 family genes account for 65 percent of the total B-cell repertoire and nearly 78.7 percent of all circulating immunoglobulin.85
Infection may increase the concentration of VH4-34 antibody.70 In patients with infectious mononucleosis and M. pneumoniae, there was an increase in VH4-34 antibody in the majority of patients. Antibody binding to RBCs was directly associated with total circulating VH4-34 antibody levels. When individual B-cell clones from infected patients were examined, only clones using the VH4-34 gene segment secreted anti-i IgM capable of RBC hemagglutination.70 There was no association between hemagglutination and light chain (κ, λ) usage.
The ability of infection to stimulate anti-I/i has been studied in a transgenic mouse model of cold agglutinin disease carrying the sequence for a human sialo agglutinin (Sia-1b).87,88 Normally, transgenic mice did not demonstrate autoimmune hemolytic anemia attributable to a depletion of “autoreactive” B cells in the bone marrow and periphery.87 Low-level anti-Siab1 was only produced by rare B cells
in the peritoneal cavity. Infection of mice with a murine Mycoplasma strain resulted in B-cell activation and loss of tolerance with markedly increased cold agglutinin synthesis, room temperature hemagglutination, and increased reticulocytosis.88
Cross-reactivity and molecular mimicry may also contribute to a rise in cold agglutinins after infection. The polysaccharide of S. pneumoniae is a linear trisaccharide sequence of Gal, GlcNAc, and Glc, with short, β1,4-linked Gal side chains that is similar to the I and i epitopes (Fig. 7).23 Lactosamine epitopes are also expressed on the lipooligosaccharide (LOS) of some Neisseria gonococcus and meningococcus strains and can stimulate antibodies capable of cross-reacting with Ii glycolipids on RBCs (Fig. 7).89 As discussed earlier, M. pneumoniae binding to branched sialo agglutinins on bronchial epithelium may stimulate cross-reactive antibodies.77,78
Monoclonal IgM Rh and Other AntibodiesNot surprisingly, other IgM MAbs using the VH4-34
gene segment often display anti-i activity.90,91 This has been well documented with several IgM anti-D MAbs. Thorpe et al.90 showed that 59 percent of all IgM anti-D MAbs carried the VH4-34 gene segment.90 Of these, 84 to 94 percent demonstrated anti-i activity when tested against papain-treated RBCs at 4°C.90 Similar “cold-agglutinin” activity has been demonstrated with other VH4-34–derived MAbs against endotoxin lipid A, DNA, and IgG (rheumatoid factor). Sequence analysis suggests that the apparent I/i reactivity of many VH4-34 anti-Rh MAbs is, in fact, charge-dependent.91 Most clones display a strong net positive charge at pH 7.5 and will react with RBCs, vimentin, and even cell nuclei at neutral pH and low ionic strength.
Fig. 7 Structure of the oligosaccharides on Streptococcus pneumoniae23 and Neisseria species89 (N. gonorrhea and N. meningitidis). Note that S. pneumoniae, a linear repeating trisaccharide, has cross-reactive motifs with structural similarity to i and I.23 Gal = galactose; Glc = glucose; GlcNAc = N-acetylglucosamine; Hep = heptose; KDO = 2-keto-3-deoxyoctulosonic acid.

144 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
L. Cooling
Regulatory Autoantibodies to Cold AgglutininsCold autoantibodies may be autoregulated by naturally
occurring anti-F(ab′)2 IgG antibodies directed against a short peptide epitope in the hinge-region.92,93 The Roelke laboratory demonstrated an inverse correlation between anti-F(ab′)2/hinge antibodies and monoclonal cold agglutinin titers. There was no correlation with hinge antibodies and polyclonal cold agglutinins. It is hypothesized that these hinge antibodies may suppress B-cell and autoantibody production. The relatively poor response of cold agglutinin disease to IVIG suggests that any downregulation by circulating hinge antibodies is moderate at best.
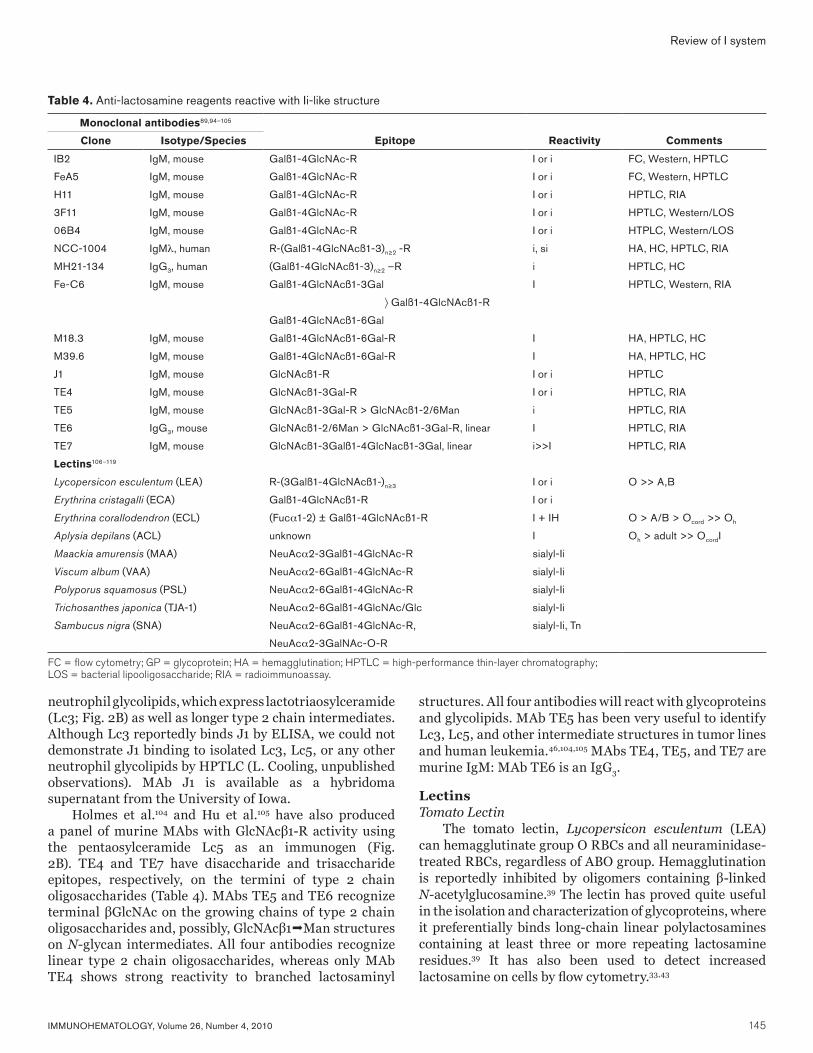
Reagents Against Lactosamine and Related EpitopesMonoclonal AntibodiesAnti-Ii
MAbs IB2, FeA5, and H11 are three murine IgM MAbs that recognize unmodified, terminal lactosamine residues on either linear or branched, I-active structures (Table 4).94–
96 MAbs H11 and 1B2 were raised against paragloboside and i-active glycolipids, whereas FeA5 was stimulated against murine testicular germ cells. In hemagglutination assays, MAbs FeA5 and 1B2 behave as an anti-I with little or no reactivity against unmodified cord cells. Both MAbs will strongly agglutinate adult and cord cells after neuraminidase digestion.94,97 No hemagglutination data are available for MAb H11.116 MAb 1B2 is available as cells from the American Type Culture Collection (ATCC, Manassas, VA). MAb FeA5 is available as a hybridoma supernatant from the Developmental Studies Hybridoma Bank held at the University of Iowa.
In addition to these antibodies, two MAbs developed against Neisseria gonococcus and meningococcus LOS also have lactosamine activity (Fig. 7).89 MAbs 3F11 and 06B4 recognize a paragloboside epitope expressed on most gonococcal LOS. In addition to paragloboside, both antibodies will recognize linear and branched polylactosamine epitopes on RBC glycolipids. MAbs 3F11 and 06B4 preferentially agglutinate adult RBCs at 4°C. Hemagglutination with both adult and cord RBCs can be observed after neuraminidase or trypsin digestion.
We have successfully used both 1B2 and FeA5 against glycoproteins and glycolipids by Western blotting, high-performance thin-layer chromatography (HPTLC), and flow cytometry.48,98 Although these antibodies are specific for Galβ14GlcNAcβ-R termini, they can be used to identify sialylated polylactosamines when coupled with neuraminidase.98
Anti-iTwo MAbs with anti-i specificity have been isolated by
heterohybridization of human B cells with murine myeloma cells.99,100 MAb NCC-1004 is a human IgMλ produced from
the B cells of a patient with metastatic adenocarcinoma of the lung.99 The antibody strongly and preferentially agglutinated cord RBCs at 4°C with no observable agglutination at 37°C. Typical of anti-I, epitope mapping confirmed that the antibody was specific for a linear oligosaccharide containing at least two LacNAc residues. The antibody would tolerate terminal substitutions, binding sialo-i and long-chain, linear A-active glycolipids (nor-Ac). When tested against human tissues, NCC-1004 recognized carcinoma of the thyroid, esophagus, lung, embryonal carcinoma, basal cell epithelium, mantle cell lymphocytes, and endothelium.
MAb MH21-134 is a human monoclonal IgG3 antibody derived from a patient with squamous cell carcinoma of the lung.100 The antibody specifically recognized glycolipids possessing a linear, polylactosamine structure. Unlike NCC-1004, MH21-134 will not bind sialylated i. In hemagglutination assays, MH21-134 will only weakly agglutinate neuraminidase-treated cord cells in the antiglobulin test: no agglutination was observed with unmodified cord and adult RBCs by immediate-spin. Among normal tissues, MH21-134 only reacted with granulocytes and bronchial and pancreatic epithelial cells. MH21-134 reacted with several gastrointestinal tract malignancies.
Anti-IThree anti-I murine MAbs are reported in the literature;
Fe-C6,95 M18.3, and M39.6.101 Initially identified as a stage-specific antigen on murine embryos, MAb Fe-C6 was subsequently shown to recognize a binary structure, requiring both an intact β1-3 and β1-6 terminal lactosamine epitope as shown in Table 4. Its reactivity is similar to the human MAbs Phi, Da, Sch, and Low (Fig. 5).24,95 MAb Fe-C6 is available as a hybridoma supernatant from the University of Iowa.
MAbs M18.3 and M39.6 were raised against human milk globule membranes as potential immune markers for breast tissue.101 Epitope mapping indicates that MAbs M18.3 and M39.6 primarily recognize the Galβ14GlcNAcβ13GlcNAc epitope, with evidence of β16 branching. Its reactivity is reminiscent of the human Step, Gra, Ver, and Ful antibodies (Fig. 5).24,95 MAbs 18.3 and M39.6 recognize breast epithelial cells: M39.6 may also recognize dendritic cells. In breast cancer, M18.3 staining is masked by increased sialylation.102
Anti-N-acetylglucosamine MAb J1 was developed against murine testicular cells
and recognizes a stage-specific differentiation antigen expressed on high molecular weight polylactosaminyl-glycans on sperm.103 Using a panel of glycolipids, J1 was shown to recognize terminal anti-N-acetylglucosamine (GlcNAc) residues on growing type 2 chain oligosaccharides (GlcNAcβ13/6Gal). J1 preferentially binds long-chain and multivalent epitopes. We have tested MAb J1 against
IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 145
Review of I system
neutrophil glycolipids, which express lactotriaosylceramide (Lc3; Fig. 2B) as well as longer type 2 chain intermediates. Although Lc3 reportedly binds J1 by ELISA, we could not demonstrate J1 binding to isolated Lc3, Lc5, or any other neutrophil glycolipids by HPTLC (L. Cooling, unpublished observations). MAb J1 is available as a hybridoma supernatant from the University of Iowa.
Holmes et al.104 and Hu et al.105 have also produced a panel of murine MAbs with GlcNAcβ1-R activity using the pentaosylceramide Lc5 as an immunogen (Fig. 2B). TE4 and TE7 have disaccharide and trisaccharide epitopes, respectively, on the termini of type 2 chain oligosaccharides (Table 4). MAbs TE5 and TE6 recognize terminal βGlcNAc on the growing chains of type 2 chain oligosaccharides and, possibly, GlcNAcβ1Man structures on N-glycan intermediates. All four antibodies recognize linear type 2 chain oligosaccharides, whereas only MAb TE4 shows strong reactivity to branched lactosaminyl
structures. All four antibodies will react with glycoproteins and glycolipids. MAb TE5 has been very useful to identify Lc3, Lc5, and other intermediate structures in tumor lines and human leukemia.46,104,105 MAbs TE4, TE5, and TE7 are murine IgM: MAb TE6 is an IgG3.
LectinsTomato Lectin
The tomato lectin, Lycopersicon esculentum (LEA) can hemagglutinate group O RBCs and all neuraminidase-treated RBCs, regardless of ABO group. Hemagglutination is reportedly inhibited by oligomers containing β-linked N-acetylglucosamine.39 The lectin has proved quite useful in the isolation and characterization of glycoproteins, where it preferentially binds long-chain linear polylactosamines containing at least three or more repeating lactosamine residues.39 It has also been used to detect increased lactosamine on cells by flow cytometry.33,43
Table 4. Anti-lactosamine reagents reactive with Ii-like structure
Monoclonal antibodies89,94–105
Clone Isotype/Species Epitope Reactivity Comments
IB2 IgM, mouse Galß1-4GlcNAc-R I or i FC, Western, HPTLC
FeA5 IgM, mouse Galß1-4GlcNAc-R I or i FC, Western, HPTLC
H11 IgM, mouse Galß1-4GlcNAc-R I or i HPTLC, RIA
3F11 IgM, mouse Galß1-4GlcNAc-R I or i HPTLC, Western/LOS
06B4 IgM, mouse Galß1-4GlcNAc-R I or i HTPLC, Western/LOS
NCC-1004 IgMλ, human R-(Galß1-4GlcNAcß1-3)n≥2 -R i, si HA, HC, HPTLC, RIA
MH21-134 IgG3, human (Galß1-4GlcNAcß1-3)n≥2 –R i HPTLC, HC
Fe-C6 IgM, mouse Galß1-4GlcNAcß1-3Gal I HPTLC, Western, RIA
〉 Galß1-4GlcNAcß1-R
Galß1-4GlcNAcß1-6Gal
M18.3 IgM, mouse Galß1-4GlcNAcß1-6Gal-R I HA, HPTLC, HC
M39.6 IgM, mouse Galß1-4GlcNAcß1-6Gal-R I HA, HPTLC, HC
J1 IgM, mouse GlcNAcß1-R I or i HPTLC
TE4 IgM, mouse GlcNAcß1-3Gal-R I or i HPTLC, RIA
TE5 IgM, mouse GlcNAcß1-3Gal-R > GlcNAcß1-2/6Man i HPTLC, RIA
TE6 IgG3, mouse GlcNAcß1-2/6Man > GlcNAcß1-3Gal-R, linear I HPTLC, RIA
TE7 IgM, mouse GlcNAcß1-3Galß1-4GlcNacß1-3Gal, linear i>>I HPTLC, RIA
Lectins106–119
Lycopersicon esculentum (LEA) R-(3Galß1-4GlcNAcß1-)n≥3 I or i O >> A,B
Erythrina cristagalli (ECA) Galß1-4GlcNAcß1-R I or i
Erythrina corallodendron (ECL) (Fuca1-2) ± Galß1-4GlcNAcß1-R I + IH O > A/B > Ocord >> Oh
Aplysia depilans (ACL) unknown I Oh > adult >> OcordI
Maackia amurensis (MAA) NeuAca2-3Galß1-4GlcNAc-R sialyl-Ii
Viscum album (VAA) NeuAca2-6Galß1-4GlcNAc-R sialyl-Ii
Polyporus squamosus (PSL) NeuAca2-6Galß1-4GlcNAc-R sialyl-Ii
Trichosanthes japonica (TJA-1) NeuAca2-6Galß1-4GlcNAc/Glc sialyl-Ii
Sambucus nigra (SNA) NeuAca2-6Galß1-4GlcNAc-R, sialyl-Ii, Tn
NeuAca2-3GalNAc-O-R
FC = flow cytometry; GP = glycoprotein; HA = hemagglutination; HPTLC = high-performance thin-layer chromatography; LOS = bacterial lipooligosaccharide; RIA = radioimmunoassay.

146 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
L. Cooling
Erythrina cristagalliErythrina cristagalli (ECA) is a lactosamine-
specific lectin from the seeds of the South American coral bean tree. It will agglutinate RBCs, regardless of ABO group. Agglutination is enhanced when tested against trypsin- or neuraminidase-treated RBCs.106 By thin-layer chromatography, ECA specifically recognizes glycolipids with terminal, unsubstituted LacNAc epitopes.107 Among I-active glycolipids, ECA will not bind sialo lactosamines, even if one terminal branch bears a bare, nonsialylated lactosamine terminus. Like E. corallodendron lectin (later), ECA can also bind type 2 chain H structures, although ECA does not display obvious HI activity when tested against RBCs.107
Erythrina corallodendronErythrina corallodendron (EcorL) is a galactophilic
lectin isolated from the seeds of the West Indian coral tree. In hemagglutination assays, the lectin displays both I and HI activity. EcorL strongly agglutinates adult O RBCs, with weaker activity against group O cord, iadult, A, and B RBCs.64 EcorL has little or no activity when tested against AB and Bombay (Oh) cells.64 The absence of agglutination with Bombay cells is a conundrum because Bombay cells should have the highest expression of lactosamine and sialo lactosamine glycans.
The I and HI-like activity have been confirmed by structural studies using well-defined oligosaccharide receptors, x-ray crystallography, and recombinant lectin mutants.107–109 Like ECL, EcorL will bind terminal LacNAc epitopes on paragloboside, type 2 chain H, linear and branched polylactosamine glycolipids. Fucose, however, is the only terminal modification accepted: EcorL will not recognize type 2 chain structures with a terminal sialic acid, Galα14 (P1 antigen), A or B epitope.107 In solid phase assays, EcorL binds the H-active glycolipid with higher affinity than paragloboside; however, no comparative binding studies were performed with i- or I-active, polylactosaminyl glycolipids.108 Crystallography indicates that LacNAc, not fucose, is the active ligand.109
Aplysia Gonad LectinAplysia gonad lectin (AGL) is a galactophilic lectin from
the gonads of Aplysia depilans, a fairly large sea slug found in the northeast Atlantic and Mediterranean Sea. In early studies, AGL was shown to agglutinate RBCs of humans and animals, particularly after papain digestion.110 A more recent study compared AGL, EcorL and human anti-I against adult, cord, and Bombay (Oh) RBCs.64 AGL reactivity was similar to human anti-I, preferentially binding adult RBCs. The strongest agglutination was observed with Bombay RBCs. Unfortunately, no information is available regarding the reactivity of AGL with neuraminidase-treated RBCs.
Maackia amurensisMaackia amurensis (MAA) is isolated from the seed
of the Amur maackia tree, a short, hardy tree common
to the midwest United States and Northeast China. The lectin has sialo agglutinin activity, recognizing a terminal NeuAcα23 lactosamine.111,112 It has been used successfully for the isolation of glycoproteins by immunoprecipitation and lectin affinity chromatography. It has also been used to identify sialylated polylactosamines by Western blotting, thin-layer chromatography, and flow cytometry. Of note, MAA does not bind glycophorin A and only weakly agglutinates human RBCs.111 We have used fluorescein-labeled MAA with ficin-treated RBCs when testing by flow cytometry.113
Viscum albumViscum album agglutinin (VAA) or mistletoe lectin
recognizes sialylated polylactosamines bearing a terminal α26 neuraminic acid.114 VAA has been used for Western blotting, thin-layer chromatography, and tissue culture. VAA strongly binds granulocytes and lymphocytes and has been shown to enhance phagocytic activity, stimulate cytokine secretion, and increase natural killer cells. VAA is a potent cytotoxin and has been studied as an adjuvant agent in cancer trials.114
Polyporus squamosusPSL agglutinin is a product of the polypore mushroom,
Polyporus squamosus, and is similar in activity to VAA.115–
117 Unlike SNA lectin (later), PSL is specific for sialylated lactosamines expressed on N-linked glycans: PSL will not bind Tn-like epitopes on O-linked glycans.115,116 In hemagglutination assays, PSL agglutinated formalin-fixed human and rabbit RBCs, irrespective of blood type.115 Hemagglutination was specifically inhibited by the trisaccharide NeuAcα26Galβ14GlcNAc, but not N-acetyllactosamine or its α23 sialo-derivative.116 PSL has been used for immunohistochemistry, Western blotting, and thin-layer chromatography.115–117
Sambucus nigraSambucus nigra agglutinin (SNA) is derived from
the bark of the elderberry tree. The lectin also recognizes α26 sialylated glycoconjugates, but has a broader acceptor profile than PSL or VAA.116,118 SNA readily precipitates both glycophorin A and human erythroglycan, indicating an ability to recognize NeuAcα26GalNAc and NeuAcα26Gal epitopes on O- and N-linked glycans, respectively.117–119 In hemagglutination assays, SNA agglutinates human RBCs regardless of ABO group or enzyme modification. SNA has been used for glycoprotein isolation, Western blotting, HPTLC-immunostaining, immunohistochemistry, and flow cytometry.112,113,116,117,119
Trichosanthes japonicaTJA-1 is one of two lectins isolated from the roots of
the snakegourd or Chinese cucumber plant, Trichosanthes japonica. TJA-II is specific for H-like epitopes, whereas

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 147
Review of I system
TJA-1 preferentially recognizes sialolactosamines.119 Like PSL and VAA, TJA-1 binds N-glycans bearing a terminal NeuAcα26Galβ14GlcNAc/Glc epitope. It has been used for the isolation and characterization of glycoproteins.119
Biologic RoleHemolytic Disease of the Fetus and Newborn
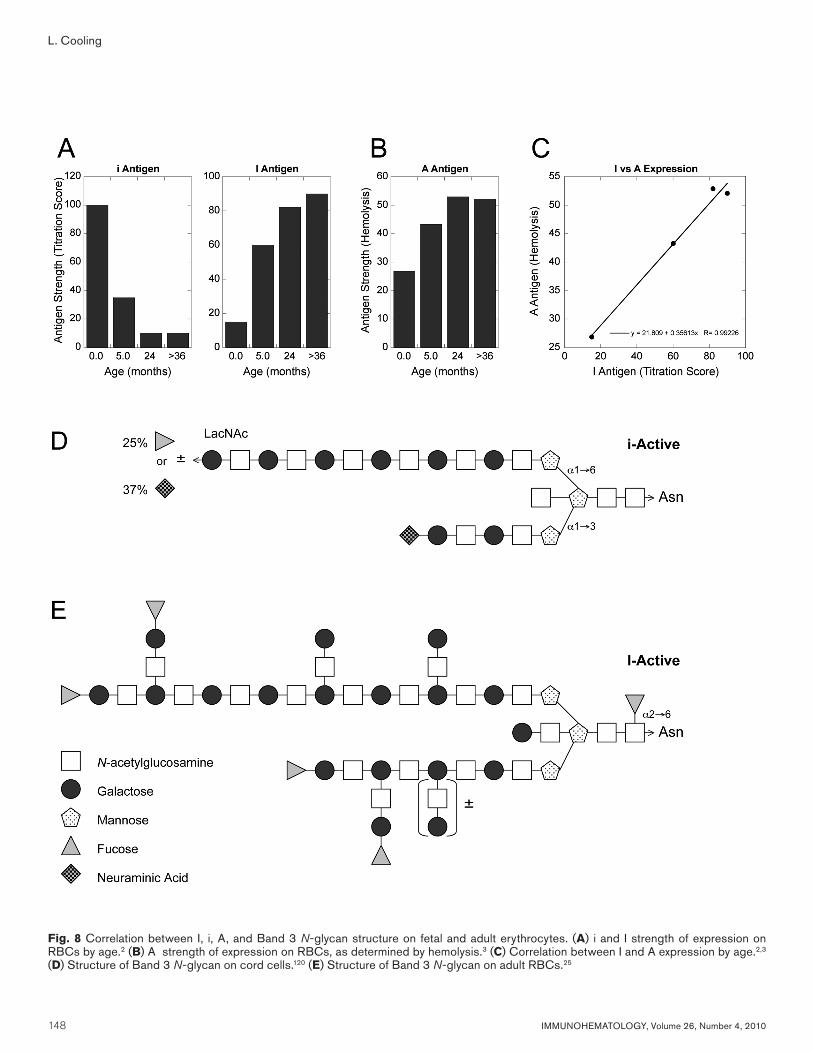
The delay in I synthesis until several months of age may play a protective role against hemolytic disease of the fetus and newborn (HDFN) caused by ABO incompatibility. Major maternal-fetal ABO incompatibility is relatively common, with up to 30 percent of infants demonstrating a positive DAT at birth.18 Nonetheless, the incidence of severe HDFN requiring exchange transfusion is quite rare (12 of 29,200 or 0.04%).18 The infrequency of clinically significant ABO-HDFN is believed to reflect low ABH levels on cord cells, which are 25 percent of adult levels (Table 3). The low density of ABH epitopes on fetal RBCs may be insufficient to support efficient complement activation by maternal IgG antibodies.18
A comparison of i, I, and A strength shows that there is a parallel increase in I and A in the first few months of life, with I and A reaching adult levels by 2 to 3 years of age (Fig. 8A–C).2,3 These results are consistent with detailed carbohydrate analysis of the lactosaminyl, biantennary N-glycan present on Band 3, which accounts for more than 50 percent of all ABH on RBCs (Fig 8D). On fetal RBCs, only a linear, i-active polylactosamine is synthesized, with approximately 25 percent bearing a terminal ABH epitope (fucose) on the long, α1-6 mannosyl branch.120 On adult RBCs, there is elongation and β1,6 branching along both α1,6 and α1,3 mannosyl branches, with up to four distal ABH epitopes.25 At one million molecules per RBC, Band 3 is capable of displaying 2 to 4 million ABH epitopes per adult RBC.
Animal Models
β3GnT2Two strains of mice lacking β3GnT2 have been bred
(β3GnT1 in some papers using the original citation by Zhou et al.33).42,43 β3GnT2–/– were shown to have altered neural development. There was a loss of olfactory sensory neurons early in embryonic development,42 decreased migration of gonadotropin-releasing hormone into the ventral forebrain,121 and decreased reproduction, possibly attributable to poor olfactory recognition of estrous females.122
β3GnT2–/– also experienced altered immune responses. There was a marked decrease in polylactosamine on N-glycans of CD28 and CD19, accompanied by evidence of T-cell, B-cell, and macrophage hyperresponsiveness. The results suggest a role for polylactosamines in regulating basal levels of immune reactivity.43
IGnTA murine model of IGnT (GCNT2) deficiency has been
constructed by Chen and colleagues.123 To produce an IGnT-deficient strain, the investigators deleted exon III
of the murine IGnT gene. As discussed earlier, mutations affecting exons E2 or E3 are associated with i phenotype and cataracts in humans (Table 2).
Contrary to expectations, IGnT-deficient mice had normal fertility and development, despite a decrease in embryoglycan and laminin adhesion on embryonic stem cells.123,124 More surprisingly, there was no increase in either the onset or incidence of cataracts in IGnT-deficient mice.123 IGnT-deficient mice did display subtle abnormalities, including a decrease in B cells, increased epidermoid cyst formation, and mild renal dysfunction, as evidenced by elevated blood urea nitrogen and creatinine and increased renal tubular vacuolization. The latter appears to represent an accumulation of autophagocytic vacuoles and diminished membrane repair as a consequence of decreases in N-glycosylated lysosomal proteins (LAMP-1, LAMP-2, synaptotagmin II, synaptotagmin IV). It is hypothesized that branched polylactosamines may play a role in stabilizing these lysoprotein proteins: inhibition of LAMP-1 glycosylation by tunicamycin shortens LAMP-1 half-life.123,125
CataractsThe association between the iadult phenotype and
congenital cataracts was initially described by Ogata et al.12 in 1979, who reported cataracts in 17 of 18 iadult individuals from 10 different Japanese families. Subsequent studies by other investigators, however, were conflicting, indicating that the iadult phenotype did not always predispose to congenital cataracts, particularly in non-Asian populations.13,14 The apparent discrepancy was finally resolved with cloning of the IGnT gene,38 followed by sequencing analysis of iadult individuals.7–10 As already described, the iadult phenotype without cataracts is the consequence of mutations affecting exon E1C, leading to an isolated loss of IGnT activity in erythroid cells (Table 2, Fig. 3). Conversely, mutations affecting E2 or E3 result in loss of IGnT activity in all tissues, including human lens.
It has been presumed that loss of branched type 2 glycans in human lens is responsible for congenital cataracts. Human lens epithelial cells do synthesize type 2 chain glycosphingolipids, including high molecular weight, long-chain sLeX gangliosides with repeating LeX motifs (Fig. 1D).48,126 In senile cataractous lens, there is an increase in LeX glycolipids, presumably caused by desialylation of sLeX structures.127 It is likely, therefore, that similar polylactosamines exist on lens glycoproteins. Although there are no published studies of glycoprotein oligosaccharides on human lens epithelial cells, lens tissue does express several N-linked glycoproteins, including α- and β-integrins, capable of I antigen expression.128,129 Altered glycosylation of β-integrins frequently accompanies cell differentiation, activation, and oncogenesis with changes in cell adhesion, motility, and signaling.130
The absence of congenital cataracts in IGnT-deficient mice,123 however, is perplexing and raises old questions
148 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
L. Cooling
Fig. 8 Correlation between I, i, A, and Band 3 N-glycan structure on fetal and adult erythrocytes. (A) i and I strength of expression on RBCs by age.2 (B) A strength of expression on RBCs, as determined by hemolysis.3 (C) Correlation between I and A expression by age.2,3 (D) Structure of Band 3 N-glycan on cord cells.120 (E) Structure of Band 3 N-glycan on adult RBCs.25

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 149
Review of I system
regarding the causal link between cataract formation and iadult phenotype. It is quite possible that IGnT deficiency plays no direct role in cataract formation but is closely linked to an unrelated mutant gene. Alternatively, there may be important species-specific differences in glycan expression on lens epithelial cells, making mice a poor animal model for studying the effect of IGnT deficiency on lens development. Indirect evidence for the latter explanation comes from knockout mice lacking α13 galactosyltransferase (α3GT-KO),131 an animal-specific glycosyltransferase responsible for linear B (Galα13Galβ1-R), a xenoantigen absent in humans and Old World apes.132,133 αGTKO mice develop congenital cataracts by day 36, suggesting a key role for α1,3-galactose in murine lens differentiation.131
HEMPASHEMPAS disease, or congenital dyserythropoietic
anemia type 2 (CAD2), is a congenital and acquired disorder of hematopoiesis. Patients with CAD2 have a chronic mild to severe hemolytic anemia, jaundice, and splenomegaly.19 Their bone marrow is striking for erythroid hyperplasia with 20 to 45 percent binucleated and multinucleated erythroblasts. Electron microscopy of marrow erythroblasts and erythrocytes shows “membrane duplication” as a result of accumulation and retention of fragmented endoplasmic reticulum.19 In the blood bank, CDAII or HEMPAS RBCs show increased i expression and hemolysis with acidified donor sera. The amount of i expressed on HEMPAS RBCs is significantly higher than that observed on cord RBCs.134
Detailed analysis of HEMPAS RBCs has shown altered glycosylation on RBC glycoproteins (ex, band 3, glycophorin) and glycolipids.20,21 Some aspects of HEMPAS can be duplicated in vitro by inhibiting N-glycan processing, leading to speculation that HEMPAS was a glycosyltransferase defect.21 In late 2009, the molecular basis for HEMPAS was finally resolved after extensive genetic studies in 33 individuals from 28 unrelated families.21 Schwarz and colleagues21 in Germany were able to show that all HEMPAS patients were heterozygous for mutations in SEC23B, a Golgi COPII component protein. COPII proteins facilitate and direct cargo vesicles among different Golgi compartments.29 It is possible that the defect in HEMPAS leads to disordered trafficking in the Golgi, with prolonged retention in some compartments and decreased trafficking to others. It is interesting to note that β4GalT1 and β3GnT1, which drive linear lactosamine synthesis, are colocalized in the trans-Golgi.31 Although the Golgi location for IGnT is unknown, C2GnT, which catalyzes β1,6 branching on O-glycans, is located within the cis-medial Golgi.30
Embryogenesis and NeoplasiaMany carbohydrate antigens are critical to normal
embryonic development and serve as oncofetal antigens during neoplastic transformation.135,136 In mice, I is expressed on unfertilized ovum up to the morulae.137 With
subsequent differentiation, strong expression of I can be observed in embryonic endoderm with apical polarization along differentiating epithelial cells.138 Increased Ii is also observed in murine teratocarcinomas and some human choriocarcinoma cell lines.137 Disseminated choriocarcinomatosis has been associated with elevated anti-i in some patients.139
Changes in Ii or their associated glycosyltransferases have been described in several tissues, but this topic is too extensive for a full discussion here. Among published reports are increased sialylation with masking of I expression in breast cancer.101,102 An increase in polylactosamines, β3GnT8 and GnTV, which catalyze synthesis of tetravalent N-glycans, is reported in colon cancer.44,100 Conversely, transitional cell carcinoma of the bladder is associated with an 11-fold decrease in β3GnT2.41 Particularly interesting is the possible role of β3GnT1 in promoting tumor metastasis.45 β3GnT1, in conjunction with LARGE glycosyltransferase, regulates the synthesis of laminin-binding glycans on α-dystroglycan. Decreased β1GnT1 could facilitate tumor migration and metastatic tumor formation in breast and prostate cancers.45
Gene Array DataFor several years, commercial “gene-chips” have been
available for high-throughput screening of tissues for thousands of genes, including many glycosyltransferases involved in blood group expression. Transcriptome studies are probably the most common analysis performed. In these studies total tissue mRNA is analyzed for global differences in gene expression. At this time, the NIH requires investigators to upload their findings into GenBank, a public access database, allowing individual gene expression to be queried directly by other investigators. The data are located in Entrez under GEO Profiles or the Gene Expression Omnibus.140 Of note, the system does support Boolean logic, limiting the search to human tissue. The latter is a useful trick to know: an initial search of GEO Profiles for IGnT resulted in 5654 entries, covering mouse, rat, and human arrays.
Among human studies, IGnT and iGnT array data are available for a wide range of studies, including these:
• Cancer• Cell differentiation• Organ transplantation• Heart failure, atrial fibrillation• Infection• Rheumatoid arthritis• Emphysema and cigarette smoking• Hormone replacement• Chemotherapy• Immunosuppression• Heavy metal exposure• Growth factors• Bipolar disorder• Social isolation (i.e., loneliness!)

150 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
A brief review did not identify any clear disease association between IGnT and β3GalT1 levels in the studies available. Interestingly, many tissues displayed significant normal variation in glycosyltransferase expression between individuals. An example of IGnT and β3GalT1 expression in fetal and adult human reticulocytes is shown in Figure 9.141
References 1. Wiener AS, Unger LJ, Cohen L, Feldman J. Type-
specific cold auto-antibodies as a cause of acquired hemolytic anemia and hemolytic transfusion reactions: biologic test with bovine red cells. Ann Internal Med 1956;44:221–40.
2. Marsh WL. Anti-i: a cold antibody defining the Ii relationship in human red cells. Br J Haematol 1961;7:200–9.
3. Grundbacher FJ. Changes in the human A antigen of erythrocytes with the individual’s age. Nature 1964;204:192–4.
4. Doinel C, Ropars C, Salmon C. Quantitative and thermodynamic measurements on I and i antigens of human red cells. Immunology 1976;30:289–97.
5. Oleson H. Thermodynamics of the cold agglutinin reaction. Scand J Clin Lab Invest 1966; 18:1–15.
6. Vigorito E, Robles A, Balter H, Nappa A, Goñi F. [125I]IgM (KAU) human monoclonal cold agglutinin: labelling and studies on its biological activity. Appl Radiat Isot 1995;46:975–9.
7. Inaba N, Hiruma T, Togayachi A, et al. A novel I-branching β-1,6-N-acetylglucosaminyltransferase involved in human blood group I antigen expression. Blood 2003;101:2870–6.
8. Yu L-C, Twu Y-C, Chou M-L, et al. The molecular genetics of the human I locus and molecular background explain the partial association of the adult i phenotype with congenital cataracts. Blood 2003;101:2081–8.
9. Lin M, Hou M-J, Yu L-C. A novel IGnT allele responsible for the adult i phenotype. Transfusion 2006;46: 1982–7.
10. Pras E, Raz J, Yahalom V, et al. A nonsense mutation in the glucosaminyl (N-acetyl) transferase 2 gene (GCNT2): association with autosomal recessive congenital cataracts. Invest Ophthalmol Vis Sci 2004; 45:1940–5.
11. Tippett P, Noades J, Sanger R, Race RR. Further studies of the I antigen and antibody. Vox Sang 1960; 5:107–121.
12. Ogata H, Okubo Y, Akabane T. Phenotype i associated with congenital cataract in Japanese. Transfusion 1979;19:166–8.
13. Marsh WL, DePalma H. Association between the Ii blood group and congenital cataract. Transfusion 1982;22:337–8.
14. McDonald EB, Douglas R, Harden PA. A Caucasian family with the i phenotype and congenital cataracts. Vox Sang 1983;44:322–5.
15. Jenkins WJ, Marsh WL, Noades J, Tippett P, Sanger R, Race RR. The I antigen and antibody. Vox Sang 1960;5:97–106.
16. Navonet JM, Muller JY, Blanchard D. Expression of blood group I antigen and fetal hemoglobin in paroxysmal nocturnal hemoglobinuria. Transfusion 1997;37:291–7.
17. Kolins J, Allgood JW, Burghardt DC, Klein HG, McGinniss MH. Modifications of B, I, i, and Lewis b antigens in a patient with DiGuglielmo’s erythroleukemia. Transfusion 1980;20:574–7.
18. Mollison PL, Engelfriet CP, Contreras M, eds. Blood transfusion in clinical medicine. 10th ed. Oxford, England: Blackwell Science, 1997.
19. Denecke J, Marquardt T. Congenital dyserythropoietic anemia type II (CDAII/HEMPAS): where are we now? Biochim Biophys Acta 2009;1792:915–20.
20. Zdebska E, Golaszewska E, Fabijańska-Mitek J, et al. Glycoconjugate abnormalities in patients with congenital dyserythropoietic anaemia type I, II and III. Br J Haematol 2001;114:907–13.
21. Schwarz K, Iolascon A, Verissimo F, et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet 2009;41:936–40.
22. Zdebska E, Krauze R, Kościelak J. Structure and blood-group I activity of poly(glycosyl)-ceramides. Carbohydr Res 1983;120:113–20.
23. Feizi T. The blood group Ii system: a carbohydrate antigen system defined by naturally monoclonal or
L. Cooling
Fig. 9 IGnT expression arrays. Individual data for ß3GnT1 (iGnT1) (A) and IGnT (C2GnT) (B) expression in human fetal and adult reticulocytes. Array data from Goh et al.141 as extracted and displayed on NCBI GEO Profiles.140

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 151
oligoclonal autoantibodies of man. Immunol Commun 1981;10:127–56.
24. Feizi T, Childs RA, Watanabe K, Hakomori S-I. Three types of blood group I specificity among monoclonal anti-I autoantibodies revealed by analogues of a branched erythrocyte glycolipid. J Exp Med 1979;149: 975–80.
25. Fukuda M, Dell A, Oates JE, Fukuda MN. Structure of a branched lactosaminoglycan, the carbohydrate moiety of Band 3 isolated from adult human erythrocytes. J Biol Chem 1984;259:8260–73.
26. Roelcke D. Sialic acid-dependent red blood cell antigens. In: Garratty G, ed. Immunobiology of transfusion medicine. New York, NY: Marcel-Dekker, 1994:69–95.
27. Renkonen O. Enzymatic in vitro synthesis of I-branches of mammalian polylactosamines: generation of scaffolds for multiple selectin-binding saccharide determinants. Cell Mol Life Sci 2000;57:1423–39.
28. Wilczyńska Z, Miller-Podraza H, Kościelak J. The contribution of different glycoconjugates to the total ABH blood group activity of human erythrocytes. FEBS Lett 1980;112:277–9.
29. Jackson CL. Mechanisms of transport through the Golgi complex. J Cell Sci 2009;122:443–52.
30. Skrincosky D, Kain R, El-Battari A, Exner M, Kerjaschki D, Fukuda M. Altered Golgi localization of core 2 beta-1,6-N-acetylglucosaminyltransferase leads to decreased synthesis of branched O-glycans. J Biol Chem 1997;272:22695–702.
31. Lee PL, Kohler JJ, Pfeffer SR. Association of β-1,3-N-acetylglucosaminyltransferase 1 and β-1,4-galactosyltransferase 1, trans-Golgi enzymes involved in coupled poly-N-acetyllactosamine synthesis. Glycobiology 2009;19:655–64.
32. Seko A, Yamashita K. Activation of β1,3-N-acetylglucosaminyltransferase-2 (β3Gn-T2) by β3Tn-T8: possible involvement of β3GnT8 in increasing poly-N-acetyllactosamine chains in differentiated HL-60 cells. J Biol Chem 2008;283:33094–100.
33. Zhou D, Dinter A, Gutiérrez Gallego R, et al. A β-1,3-N-acetylglucosaminyltransferase with poly-N-acetyllactosamine synthase activity is structurally related to β-1,3-galactosyltransferases. Proc Natl Acad Sci U S A 1999;96:406–11.
34. Shiraishi N, Natsume A, Togayachi A, et al. Identification and characterization of three novel β1,3-N-acetylglucosaminyltransferases structurally related to the β1,3-galactosyltransferase family. J Biol Chem 2001;276:3498–507.
35. Henion TR, Zhou D, Wolfer DP, Jungalwala FB, Hennet T. Cloning of a mouse β1,3 N-acetylglucosaminyltransferase GlcNAc(β1,3)Gal(β1,4)Glc-ceramide synthase gene encoding the
key regulator of lacto-series glycolipid biosynthesis. J Biol Chem 2001;276:30261–9.
36. Togayachi A, Akashima T, Ookubo R, et al. Molecular cloning and characterization of UDP-GlcNAc:lactosylceramide β1,3-N-acetylglucosaminyl-transferase (β3Gn-T5), an essential enzyme for the expression of HNK-1 and Lewis X epitopes on glycolipids. J Biol Chem 2001;276:22032–40.
37. Stults CLM, Macher BA. β1–3-N-acetylglucosaminyl-transferase in human leukocytes: properties and role in regulating neolacto glycosphingolipid biosynthesis. Arch Biochem Biophys 1993;303:125–33.
38. Bierhuizen MFA, Mattei M-G, Fukuda M. Expression of the developmental I antigen by a cloned human cDNA encoding a member of a β-1,6-N-acetylglucosaminyltransferase family. Genes Dev 1993;7:468–78.
39. Merkle RK, Cummings RD. Relationship of the terminal sequences to the length of poly-N-acetyllactosamine chains in asparagine-linked oligosaccharides from the mouse lymphoma cell line BW5147. Immobilized tomato lectin interacts with high affinity with glycopeptides containing long poly-N-acetyllactosamine chains. J Biol Chem 1987;262:8179–89.
40. Sasaki K, Kurata-Miura K, Ujita M, et al. Expression cloning of cDNA encoding a human β-1,3-N-acetylglucosaminyltransferase that is essential for poly-N-acetyllactosamine synthesis. Proc Natl Acad Sci U S A 1997;94:14294–9.
41. Gromova I, Gromov P, Celis JE. A novel member of the glycosyltransferase family, β3 Gn-T2, highly downregulated in invasive human bladder transitional cell carcinomas. Mol Carcinog 2001;32:61–72.
42. Henion TR, Raitcheva D, Grosholz R, et al. β1,3-N-acetylglucosaminyltransferase 1 glycosylation is required for axon pathfinding by olfactory sensory neurons. J Neurosci 2005;25:1894–903.
43. Togayachi A, Kozono Y, Ishida H, et al. Polylactosamine on glycoproteins influences basal levels of lymphocyte and macrophage activation. Proc Natl Acad Sci U S A 2007;104:15829–34.
44. Ishida H, Togayachi A, Sakai T, et al. A novel β1,3-N-acetylglucosaminyltransferase (β3Gn-T8), which synthesizes poly-N-acetyllactosamine, is dramatically upregulated in colon cancer. FEBS Lett 2005;579: 71–8.
45. Bao X, Kobayashi M, Hatakeyama S, et al. Tumor suppressor function of laminin-binding α-dystroglycan requires a distinct β3-N-acetylglu-cosaminyltransferase. Proc Natl Acad Sci USA 2009; 106:12109–14.
46. Symington FW, Hedges DL, Hakomori S-I. Glycolipid antigens of human polymorphonuclear neutrophils
Review of I system

152 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
and the inducible HL-60 myeloid leukemia line. J Immunol 1985;134:2498–506.
47. Cooling LLW, Zhang DS, Naides SJ, Koerner TAW. Glycosphingolipid expression in acute nonlymphocytic leukemia: common expression of shiga toxin and parvovirus B19 receptors on early myeloblasts. Blood 2003;101:711–21.
48. Cooling LW, De-Sheng Z, Koerner TAW. Lewis X and sialyl Lewis X glycosphingolipids. Trends Glycosci Glycotechnol 1997;9:191–209.
49. Iwai T, Inaba N, Naundorf A, et al. Molecular cloning and characterization of a novel UDP-GlcNAc: GalNAc-peptide β1,3-N-acetylglucosaminyltransferase (β3Gn-T6), an enzyme synthesizing the core 3 structure of O-glycans. J Biol Chem 2002;277:12802–9.
50. Kataoka K, Huh N. A novel β1,3-N-acetylglucos-aminyltransferase involved in invasion of cancer cells as assayed in vitro. Biochem Biophys Res Commun 2002;294:843–8.
51. Yeh J-C, Hiraoka N, Petryniak B, et al. Novel sulfated lymphocyte homing receptors and their control by a Core1 extension β1,3-N-acetylglucos-aminyltransferase. Cell 2001;105:957–69.
52. Seko A, Yamashita K. β1,3-N-Acetylglucosaminyl-transferase-7 (β3GnT7) acts efficiently on keratan sulfate-related glycans. FEBS Lett 2004;556:216–20.
53. Twu Y-C, Chen C-P, Hsieh C-Y, et al. I branching formation in erythroid differentiation is regulated by transcription factor C/EBPα. Blood 2007;110:4526–34.
54. Geest CR, Buitenhuis M, Laarhoven AG, et al. p38 MAP kinase inhibits neutrophil development through phosphorylation of C/EBPα on serine 21. Stem Cells 2009;27:2271–82.
55. Bierhuizen MFA, Fukuda M. Expression cloning of a cDNA encoding UDP-GlcNAc:Galβ1-3-GalNAc-R (GlcNAc to GalNAc) β1-6GlcNAc transferase by gene transfer into CHO cells expressing polyoma large tumor antigen. Proc Natl Acad Sci U S A 1992;89: 9326–30.
56. Yeh J-C, Ong E, Fukuda M. Molecular cloning and expression of a novel β-1,6-N-acetylglucos-aminyltransferase that forms core 2, core 4, and I branches. J Biol Chem 1999;274;3215–21.
57. Breton C, Snajdrová L, Jeanneau C, Koca J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology 2006;16:29R–37R.
58. Pak JE, Arnoux P, Zhou S, et al. X-ray crystal structure of leukocyte type core 2 β1,6-N-acetylglucosaminyl-transferase: evidence for a convergence of metal ion-independent glycosyltransferase mechanism. J Biol Chem 2006;281:26693–701.
59. Cabrera PV, Amano M, Mitoma J, et al. Haploinsufficiency of C2GnT-1 glycosyltransferase renders T lymphoma cells resistant to cell death. Blood 2006;108:2399–406.
60. Bengtson P, Larson C, Lundblad A, Larson G, Påhlsson P. Identification of a missense mutation (G329A;Arg110Gln) in the human FUT7 gene. J Biol Chem 2001;276:31575–82.
61. Adinolfi M. Anti-I antibody in normal human newborn infants. Immunology 1965;9:43–52.
62. Dube VE, Zuckerman L, Philipsborn HF Jr. Variation of cold agglutinin levels. Vox Sang 1978;34:71–6.
63. Doinel C. I antigenicity of the Bombay red cells [in French]. Rev Fr Transfus Immunohematol 1976;19: 185–91.
64. Gilboa-Garber N, Sudakevitz D, Levene C. A comparison of the Aplysia lectin anti-I specificity with human anti-I and several other I-detecting lectins. Transfusion 1999;39:1060–4.
65. Stults CLM, Sweeley CC, Macher BA. Glycosphingo-lipids: structure, biological source, and properties. Methods Enzymol 1989;179:167–214.
66. Gardas A. Studies on the I-blood-group-active sites on macro-glycolipids from human erythrocytes. Eur J Biochem 1976;68:185–91.
67. Kannagi R, Roelcke D, Peterson KA, Okada Y, Levery SB, Hakomori S-I. Characterization of an epitope (determinant) structure in a developmentally regulated glycolipid antigen defined by a cold agglutinin Fl, recognition of an α-sialosyl and α-L-fucosyl groups in a branched structure. Carbohydr Res 1983;120:143–57.
68. Nagatsuka Y, Watarai S, Yasuda T, Higashi H, Yamagata T, Ono Y. Production of human MAbs to i blood group by EBV-induced transformation: possible presence of a new glycolipid in cord red cell membranes and human hematopoietic cell lines. Immunol Lett 1995;46: 93–100.
69. Nagatsuka Y, Yamaki M, Watarai S, et al. The Epstein-Barr virus (EBV) alters the B cell glycolipid which is recognized by the human monoclonal antibody to i-blood group antigen. Virus Res 1996;43:57–68.
70. Chapman CJ, Spellerberg MB, Smith GA, Carter SJ, Hamblin TJ, Stevenson FK. Autoanti-red cell antibodies synthesized by patients with infectious mononucleosis utilize the VH4–21 gene segment. J Immunol 1993;151:1051–61.
71. Riboldi P, Gaidano G, Schettino EW, et al. Two acquired immunodeficiency syndrome-associated Burkitt’s lymphomas produce specific anti-i IgM cold agglutinins using somatically mutated VH4–21 segments. Blood 1994;83:2952–61.
72. Roelcke D, Kreft H, Hack H, Stevenson FK. Anti-j: human cold agglutinins recognizing linear (i) and branched (I) type 2 chains. Vox Sang 1994;67:216–21.
73. Dube VE, Kallio P, Tanaka M. Specificity of the monoclonal anti-I antibody (Hy). Mol Immunol 1986; 23:217–20.
74. Roelcke D, Kreft H, Northoff H, Gallasch E. Sia-β1 and I antigens recognized by Mycoplasma pneumoniae-
L. Cooling

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 153
induced human cold agglutinins. Transfusion 1991;31: 627–30.
75. Zilow G, Haffner D, Roelcke D. CMV-induced anti-Sia-β1 cold agglutinin in an immunocompromised patient. Beitr Infusionsther Transfusionsmed 1997;34: 180–4.
76. Loomes LM, Uemura K-I, Feizi T. Interaction of Mycoplasma pneumoniae with erythrocyte glycolipids of the I and i antigen types. Infect Immun 1985;47: 15–20.
77. Loveless RW, Feizi T. Sialo-oligosaccharide receptors for Mycoplasma pneumoniae and related oligosaccharides of poly-N-acetyllactosamine series are polarized at the cilia and apical-microvillar domains of the ciliated cells in the human bronchial epithelium. Infect Immun 1989;57:1285–9.
78. Feizi T, Loveless RW. Carbohydrate recognition by Mycoplasma pneumoniae and pathologic consequences. Am J Respir Crit Care Med 1996;154 (Suppl):S133–6.
79. Li Y, Spellerberg MB, Stevenson FK, Capra JD, Potter KN. The I binding specificity of human VH4-34 (VH4-21) encoded antibodies is determined by both VH framework region 1 and complementarity determining region 3. J Mol Biol 1996;256:577–89.
80. Stevenson FK, Spellerberg MB, Chapman CJ, Hamblin TJ. Differential usage of an autoantibody-associated VH gene, VH4-21, by human B-cell tumors. Leuk Lymphoma 1995;16:379–84.
81. Ishida F, Saito H, Kitano K, Kiyosawa K. Cold agglutinin disease by autoanti-i blood type antibody associated with B cell chronic lymphocytic leukemia. Int J Hematol 1998;67:69–73.
82. Silberstein LE, Jefferies LC, Goldman J, et al. Variable region gene analysis of pathologic human autoantibodies to the related I and i red blood cell antigens. Blood 1991;78:2372–86.
83. Cauerhff A, Braden BC, Carvalho JG, et al. Three-dimensional structure of the Fab from a human IgM cold agglutinin. J Immunol 2000;165:6422–8.
84. Leo A, Kreft H, Hack H, Kempf T, Roelcke D. Restriction in the repertoire of the immunoglobulin light chain subgroup in pathological cold agglutinins with anti-Pr specificity. Vox Sang 2004;86:141–7.
85. Foreman AL, Van de Water J, Gougeon M-L, Gershwin ME. B cells in autoimmune diseases: insights from analyses of immunoglobulin variable (Ig V) gene usage. Autoimmun Rev 2007;6:387–401.
86. Jefferies LC, Carchidi CM, Silberstein LE. Immunoglobulin gene use by naturally occurring cold agglutinins. Ann N Y Acad Sci 1995;764:433–5.
87. Havouis S, Dumas G, Avé P, et al. Negative regulation of autoreactive B cells in transgenic mice expressing a human pathogenic cold agglutinin. Eur J Immunol 2000;30:2290–9.
88. Havouis S, Dumas G, Chambaud I, et al. Transgenic B lymphocytes expressing a human cold agglutinin escape tolerance following experimental infection of mice by Mycoplasma pulmonis. Eur J Immunol 2002; 32:1147–56.
89. Mandrell RE, Griffiss JM, Macher BA. Lipooligosaccharides (LOS) of Neisseria gonorrhoeae and Neisseria meningitidis have components that are immunochemically similar to precursors of human blood group antigens. Carbohydrate sequence specificity of the mouse monoclonal antibodies that recognize crossreacting antigens on LOS and human erythrocytes. J Exp Med 1988;168:107–26.
90. Thorpe SJ, Boult CE, Stevenson FK, et al. Cold agglutinin activity is common among human monoclonal antibodies using the V4-34 heavy chain variable gene segment. Transfusion 1997;37:1111–16.
91. Thorpe SJ, Turner CE, Stevenson FK, et al. Human monoclonal antibodies encoded by the V4-34 gene segment show cold agglutinin activity and variable multireactivity which correlates with the predicted charge of the heavy-chain variable region. Immunology 1998;93:129–36.
92. Terness P, Kirschfink M, Navolan D, et al. Striking inverse correlation between IgG anti-F(ab′)2 and autoantibody production in patients with cold agglutination. Blood 1995;85:548–51.
93. Terness P, Kirschfink M, Navolan D, et al. Inverse correlation between IgG-antihinge region and antierythrocyte autoantibody in chronic benign and malignant cold agglutination. J Clin Immunol 1997; 17:220–7.
94. Young WW Jr, Portoukalian J, Hakomori S-I. Two monoclonal anticarbohydrate antibodies directed to glycosphingolipids with a lacto-N-glycosyl type II chain. J Biol Chem 1981;256:10967–72.
95. Fenderson BA, Nichols EJ, Clausen H, Hakomori S-I. A monoclonal antibody defining a binary N-acetyllactosaminyl structure in lactoisooctaosyl-ceramide (IV6 Galβ14GlcNAcnLc6): a useful probe for determining differential glycosylation patterns between normal and transformed human fibroblasts. Mol Immunol 1986;23:747–54.
96. Myoga A, Taki T, Arai K, et al. Detection of patients with cancer by monoclonal antibody directed to lactoneotetraosylceramide (paragloboside). Cancer Res 1988;48:1512–6.
97. Fenderson BA, Hahnel AC, Eddy EM. Immunohistochemical localization of two monoclonal antibody-defined carbohydrate antigens during early murine embryogenesis. Dev Biol 1983;100:318–27.
98. Cooling LLW, Zhang DS, Koerner TAW. Human platelets express gangliosides with LKE activity and ABH blood group activity. Transfusion 2001;41: 504–16.
Review of I system

154 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
99. Hirohashi S, Clausen H, Nudelman E, Inoue H, Shimosato Y, Hakomori S. A human monoclonal antibody directed to blood group i antigen: heterohybridoma between human lymphocytes from regional lymph nodes of a lung cancer patient and mouse myeloma. J Immunol 1986;136:4163–8.
100. Miyake M, Kohno N, Nudelman ED, Hakomori S-I. Human IgG3 monoclonal antibody directed to an unbranched repeating type 2 chain (Galβ14GlcNAcβ13Galβ14GlcNAcβ13Galβ1R) which is highly expressed in colonic and hepatocellular carcinoma. Cancer Res 1989;49:5689–95.
101. Gooi HC, Uemura K-I, Edwards PAW, Foster CS, Pickering N, Feizi T. Two mouse hybridoma antibodies against milk-fat globules recognise the I(Ma) antigenic determinant β-D-Galp-(14)-β-D-GlcpNAc-(16). Carbohydr Res 1983;120:293–302.
102. Foster CS, Neville AM. Monoclonal antibodies to the human mammary gland: monoclonal antibody LiCR-LON-M18 identifies impaired expression and excess sialylation of the I(Ma) cell-surface antigen by primary breast carcinoma cells. Hum Pathol 1984;15:502–13.
103. Symington FW, Fenderson BA, Hakomori S-I. Fine specificity of a monoclonal anti-testicular cell antibody for glycolipids with terminal N-acetyl-D-glucosamine structure. Mol Immunol 1984;21:877–82.
104. Holmes EH, Greene TG. Isolation and fine-structure characterization of four monoclonal antibodies reactive with glycoconjugates containing terminal GlcNAc residues: application to aspects of lacto-series tumor antigen biosynthesis. Arch Biochem Biophys 1991;288:87–96.
105. Hu J, Stults CLM, Holmes EH, Macher BA. Structural characterization of intermediates in the biosynthetic pathway of neolacto glycosphingolipids: differential expression in human leukaemia cells. Glycobiology 1994;4:251–7.
106. Iglesias JL, Lis H, Sharon N. Purification and properties of a D-galactose/N-acetyl-D-galactosamine-specific lectin from Erythrina cristagalli. Eur J Biochem 1982; 123:247–52.
107. Teneberg S, Angström J, Jovall P-A, Karlsson K-A. Characterization of binding of Galβ4GlcNAc-specific lectins from Erythrina cristagalli and Erythrina corallodendron to glycosphingolipids. Detection, isolation, and characterization of a novel glycosphingolipid of bovine buttermilk. J Biol Chem 1994;269:8554–63.
108. Moreno E, Teneberg S, Adar R, Sharon N, Karlsson K-A, Angström J. Redefinition of the carbohydrate specificity of Erythrina corallodendron lectin based on solid-phase binding assays and molecular modeling of native and recombinant forms obtained by site-directed mutagenesis. Biochemistry 1997;36:4429–37.
109. Elgavish S, Shaanan B. Structures of the Erythrina corallodendron lectin and of its complexes with mono- and disaccharides. J Mol Biol 1998;277:917–32.
110. Gilboa-Garber N, Susswein AJ, Mizrahi L, Avichezer D. Purification and characterization of the gonad lectin of Aplysia depilans. FEBS Lett 1985;181:267–70.
111. Knibbs RN, Goldstein IJ, Ratcliffe RM, Shibuya N. Characterization of the carbohydrate binding specificity of the leukoagglutinating lectin from Maackia amurensis: comparison with other sialic acid-specific lectins. J Biol Chem 1991;266:83–8.
112. Johansson L, Miller-Podraza H. Analysis of 3- and 6-linked sialic acids in mixtures of gangliosides using blotting to polyvinylidene difluoride membranes, binding assays, and various mass spectrometry techniques with application to recognition by Helicobacter pylori. Anal Biochem 1998;265:260–8.
113. Cooling LL, Hwang D, Gu Y. The LKE-negative and LKE-weak RBC phenotypes do not reflect decreased RBC sialylation [abstract]. Transfusion 2003;43:26.
114. Müthing J, Meisen I, Bulau P, et al. Mistletoe lectin I is a sialic acid-specific lectin with strict preference to gangliosides and glycoproteins with terminal Neu5Acα2-6Galβ1-4GlcNAc residues. Biochemistry 2004;43:2996–3007.
115. Mo H, Winter HC, Goldstein IJ. Purification and characterization of a Neu5Acα2-6Galβ1-4Glc/GlcNAc-specific lectin from the fruiting body of the polypore mushroom Polyporus squamosus. J Biol Chem 2000;275:10623–9.
116. Toma V, Zuber C, Winter HC, Goldstein IJ, Roth J. Application of a lectin from the mushroom Polysporus squamosus for the histochemical detection of the NeuAcα2,6Galβ1,4Glc/GlcNAc sequence of N-linked oligosaccharides: a comparison with the Sambucus nigra lectin. Histochem Cell Biol 2001;116:183–93.
117. Tateno H, Winter HC, Goldstein IJ. Cloning, expression in Escherichia coli and characterization of the recombinant Neu5Acα2,6Galβ1,4GlcNAc-specific high-affinity lectin and its mutants from the mushroom Polyporus squamosus. Biochem J 2004;382:667–75.
118. Shibuya N, Goldstein IJ, Broekaert WF, Nsimba-Lubaki M, Peeters B, Peumans WJ. The elderberry (Sambucus nigra L.) bark lectin recognizes the Neu5Ac(α2-6)Gal/GalNAc sequence. J Biol Chem 1987;262:1596–601.
119. Yamashita K, Umetsu K, Suzuki T, Ohkura T. Purification and characterization of a Neu5Acα2 6Galβ14GlcNAc and HSO3–6Galβ14GlcNAc specific lectin in tuberous roots of Trichosanthes japonica. Biochemistry 1992;31:11647–50.
120. Fukuda M, Dell A, Fukuda MN. Structure of fetal lactosaminoglycan. The carbohydrate moiety of Band 3 isolated from human umbilical cord erythrocytes. J Biol Chem 1984;259:4782–91.
L. Cooling

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 155
121. Bless E, Raitcheva D, Henion TR, Tobet S, Schwarting GA. Lactosamine modulates the rate of migration of GnRH neurons during mouse development. Eur J Neurosci 2006;24:654–60.
122. Biellmann F, Henion TR, Bürki K, Hennet T. Impaired sexual behavior in male mice deficient for the β1-3 N-acetylglucosaminyltransferase-I gene. Mol Reprod Dev 2008;75:699–706.
123. Chen GY, Muramatsu H, Kondo M, et al. Abnormalities caused by carbohydrate alterations in Iβ6-N-acetylglucosaminyltransferase-deficient mice. Mol Cell Biol 2005;25:7828–38.
124. Muramatsu H, Kusano T, Sato M, Oda Y, Kobori K, Muramatsu T. Embryonic stem cells deficient in I β1,6-N-acetylglucosaminyltransferase exhibit reduced expression of embyroglycan and the loss of a Lewis X antigen, 4C9. Glycobiology 2008;18:242–9.
125. Barriocanal JG, Bonifacino JS, Yuan L, Sandoval IV. Biosynthesis, glycosylation, movement through the Golgi system, and transport to lysosomes by an N-linked carbohydrate-independent mechanism of three lysosomal integral membrane proteins. J Biol Chem 1986;261:16755–63.
126. Ogiso M, Ohta M, Okinaga T, et al. Glycosphingolipids in cultured lens epithelial cells from dog and rhesus monkey. Glycobiology 1994;4:375–82.
127. Ogiso M, Komoto M, Okinaga T, Koyota S, Hoshi M. Age-related changes in ganglioside composition in human lens. Exp Eye Res 1995;60:317–23.
128. Nishi O, Nishi K, Akaishi T, Shirasawa E. Detection of cell adhesion molecules in lens epithelial cells of human cataracts. Invest Ophthalmol Vis Sci 1997;38:579–85.
129. McLean SM, Mathew MRK, Kelly JB, et al. Detection of integrins in human cataract lens epithelial cells and two mammalian lens epithelial cell lines. Br J Ophthalmol 2005;89:1506–9.
130. Bellis SL. Variant glycosylation: an underappreciated regulatory mechanism for β1 integrins. Biochim Biophys Acta 2004;1663:52–60.
131. Sørensen DB, Dahl K, Ersboll AK, Kirkeby S, d’Apice AJF, Hansen AK. Aggression in cataract-bearing α-1,3-galactosyltransferase knockout mice. Lab Anim 2008;42:34–44.
132. Galili U, Shohet SB, Kobrin E, Stults CLM, Macher BA. Man, apes, and Old World monkeys differ from other mammals in the expression of α-galactosyl epitopes on nucleated cells. J Biol Chem 1988;263:17755–62.
133. Joziasse DH, Shaper JH, Jabs EW, Shaper NL. Characterization of an α1-3-galactosyltransferase homologue on human chromosome 12 that is organized as a processed pseudogene. J Biol Chem 1991;266:6991–8.
134. Cooling L. Increased expression of i on HEMPAS red cells: a flow cytometric study. Transfusion 1997; 37:1102–3.
135. Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem 2004;73:491–537.
136. Hakomori S-I. Cancer-associated glycosphingolipid antigens: their structure, organization and function. Acta Anat (Basel) 1998;161:79–90.
137. Knowles BB, Rappaport J, Solter D. Murine embryonic antigen (SSEA-1) is expressed on human cells and structurally related human group antigen I is expressed on mouse embryos. Dev Biol 1982;93:54–8.
138. Kapadia A, Feizi T, Evans MJ. Changes in the expression and polarization of blood group I and i antigens in post-implantation embryos and teratocarcinomas associated with cell differentiation. Exp Cell Res 1981; 131:185–95.
139. Boughton BJ. Anti-i cold agglutinins in choriocarcinomatosis: trophoblastic i antigen. J Clin Pathol 1979;32:523–7.
140. GEO Profiles, National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov/sites/entrez?db=geo (Last accessed 4/2010).
141. Goh SH, Josleyn M, Lee YT, et al. The human reticulocyte transcriptome. Physiol Genomics 2007; 30:172–8.
Laura Cooling MD, MS, Associate Professor, Pathology, Associate Medical Director, Transfusion Medicine, University of Michigan Hospitals, 2F225 UH, Box 0054, 1500 East Medical Center Drive, Ann Arbor, MI 48109-0054.
Review of I system

156 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
Attempts to support an immune etiology in 800 patients with direct antiglobulin test–negative hemolytic anemia
Original Report
R.M. Leger, A. Co, P. Hunt, and G. Garratty
Clinical and hematologic evidence of warm autoimmune hemolytic anemia (AIHA) is present in some patients whose direct antiglobulin test (DAT) is negative. The most common causes for AIHA associated with a negative DAT are RBC-bound IgG below the sensitivity threshold of the DAT, RBC-bound IgA and IgM not detectable by routine reagents, and low-affinity IgG that dissociates during the testing process. Samples submitted from 800 patients with hemolytic anemia and a negative DAT were tested by an antiglobulin sera (AGS) panel of anti-IgG, anti-C3, anti-IgM, and anti-IgA by a routine DAT. Additional tests included a direct Polybrene test to detect small amounts of RBC-bound IgG, a cold-wash technique to detect low-affinity IgG, and a DAT by gel test with anti-IgG. A positive result was obtained with at least one method for 431 (54%) of 800 specimens tested. The AGS panel was positive for 400 (50%) of samples, with IgG or C3 or both accounting for reactivity in 48 percent. IgA alone was found on 2 percent of samples; IgM was never found alone. Low-affinity IgG was found on 37 (5%) samples. The direct Polybrene test was the only positive test for 15 (2%) samples. The gel anti-IgG test was never the only positive test. Clinical correlations for these data were not available; however, previously published correlations suggest a positive predictive value for tests that extend routine DAT methods in patients with DAT-negative AIHA. Immunohematology 2010;26:156–60.
Key Words: autoimmune hemolytic anemia, DAT-negative, Coombs-negative
Most patients with autoimmune hemolytic anemia (AIHA) have IgG or complement, or both, on their RBCs detectable by the routine DAT. However, approximately 2 to 11 percent of patients with hematologic and clinical findings suggestive of warm AIHA have a negative DAT by routine tube technique and have no detectable serum antibodies.1 Hematologists seeking evidence supporting immune-mediated hemolysis may request nonroutine and more sensitive tests for these patients.
It has been suggested that there are at least three causes for AIHA associated with a negative DAT: RBC-bound IgG below the threshold of detection of the routine antiglobulin test, low-affinity IgG that dissociates from RBCs during saline washes for the DAT, and RBC-bound IgM and IgA that are not detectable by routine reagents.1,2 The routine tube DAT can detect about 150 to 200 molecules of IgG per RBC. Gilliland and coworkers3,4 suggested that the inability to detect IgG on the RBCs of some patients with AIHA was caused by low levels of RBC-bound IgG that were below the threshold of detection by the routine DAT.
They developed a method, the complement-fixing antibody consumption (CFAC) test, capable of detecting this low level of RBC-bound IgG. The authors reported that RBCs from five of six patients with acquired hemolytic anemia and a negative DAT were found to have greater quantities of IgG per RBC (70–434 molecules) compared with 15 to 35 molecules of IgG per RBC found on the RBCs obtained from normal persons. Gilliland4 extended these results with an additional nine patients; six of these nine responded to corticosteroid therapy.
Petz and Garratty5 performed extensive tests, including the CFAC, on specimens obtained from 27 patients with a diagnosis of immune hemolytic anemia with a negative DAT, confirming that all of these patients had RBC-bound IgG detectable by the CFAC; 11 (41%) patients had RBC-bound C3 detectable by a potent anti-C3 prepared in their laboratory but not detected by the referring hospital. Because the CFAC is difficult to standardize and to perform consistently, other tests have been used to detect RBC-bound IgG, including enzyme-linked antiglobulin test (ELAT), radiolabeled anti-IgG, flow cytometry, monocyte monolayer assay (MMA), concentrated eluates, the direct Polybrene test, the direct polyethylene glycol test, solid phase, column agglutination, and the DAT using cold washes.2 When hematologists or referring laboratories contact our laboratory, they often ask for a “Super Coombs Test.” There is no such test. We believe the term originated as jargon to cover tests such as the ELAT and radiolabeled anti-IgG previously used to detect small amounts of RBC-bound IgG.
No one test has been shown to be optimal for investigating DAT-negative AIHA; we and others have found that a battery of tests appears to be the best approach.1,6–9 By 1995 we had decided that there was no cost benefit in performing time-consuming assays such as the ELAT, so we concentrated on a serologic approach. We transferred DAT-negative AIHA workups from our Immunohematology Research Laboratory to the Immunohematology Reference Laboratory (IRL). All samples that are submitted for a DAT-negative AIHA investigation are tested by tube test with an antiglobulin sera (AGS) panel consisting of anti-IgG, anti-C3, anti-IgM, and anti-IgA. When the AGS panel is not informative, additional tests performed include a direct Polybrene test to detect low amounts of immunoglobulin, a cold (4°C) LISS wash technique for preparation of RBCs to detect low-affinity IgG, and a column agglutination (gel) test for IgG.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 157
DAT-negative hemolytic anemia
We report the serologic results for a large series of patients’ samples that were submitted for a DAT-negative AIHA evaluation.
Material and MethodsThe AGS panel consisted of a rabbit anti-IgG (American
National Red Cross, Washington, DC; or Ortho Clinical Diagnostics, Raritan, NJ), anti-C3 (reagent prepared by injecting a rabbit with purified C3), rabbit anti-human IgM (CLB, distributed by Research Diagnostics, Flanders, NJ; or Sanquin, distributed by Accurate Chemical & Scientific Corporation, Westbury, NY), goat anti-human IgA (Tago, Burlingame, CA), and a 6% bovine serum albumin control. The anti-C3, anti-IgM, and anti-IgA were standardized for RBC agglutination in tube tests as previously described.1 Specimens were also tested from April through December 2008 with Gamma-clone Anti-IgG (murine monoclonal, Immucor, Norcross, GA). For the DAT, RBCs were washed four times with PBS (pH 7.0–7.4) and resuspended to achieve a 3 to 5% suspension; 1 drop was added to 2 drops of the antiglobulin reagent or control, centrifuged, and read immediately. Centrifugation and reading was repeated after incubation at room temperature if indicated by the standardization. A positive result with the 6% albumin control invalidated the test. When a positive result with the albumin control was obtained, repeat testing may have been performed after washing a fresh aliquot of RBCs with warm (37ºC) saline to remove cold autoagglutinins, or after treatment with 0.01 M dithiothreitol (DTT) to disperse IgM autoagglutinins.10
The cold LISS wash technique was performed by washing the RBCs four times with ice-cold (4°C) LISS11 using a refrigerated centrifuge.1 The RBCs were resuspended to a 3 to 5% suspension in cold LISS; 1 drop was added to 2 drops of cold anti-IgG or cold 6% albumin, centrifuged in a serologic centrifuge, and read immediately. The 6% albumin served as a control for the presence of a cold autoagglutinin; a positive result with the 6% albumin control invalidated the cold LISS wash test with anti-IgG.
The direct Polybrene test,1 a modification of the indirect Polybrene test,12 was performed as previously described. Briefly, 1 drop of 3 to 5% washed RBCs was added to 2 drops of 5% group AB inert plasma. In parallel, 3 drops of a 1.5% suspension of the patient’s unwashed RBCs suspended in autologous plasma were tested. One milliliter of the low-ionic medium was added to each tube, and the test was incubated at room temperature for 1 minute. Two drops of 0.05% Polybrene (hexadimethrine bromide, Sigma Chemical, Co., St. Louis, MO) in unbuffered saline were added, and the contents were mixed. After 15 seconds, the tubes were centrifuged at 1000 x g for 10 seconds. The supernatant was decanted, and 2 drops of sodium citrate resuspending solution were added. The tubes were gently shaken at a 45-degree angle and examined for agglutination. Results were compared with concurrent positive and negative
controls to ascertain the persistence of agglutination. One additional drop of resuspending solution was added plus 2 drops of group AB plasma (undiluted). After mixing, the tubes were washed three times and converted to an antiglobulin test with anti-IgG.
The gel test DAT was performed as described by the manufacturer (ID-MTS Anti-IgG Card, Ortho). Control tests were performed in parallel with the ID-MTS Buffered Gel Card.
ResultsEight hundred specimens submitted January 1997
through December 2008 (12 years) for suspected DAT-negative AIHA were evaluated. All 800 samples were tested by the AGS panel; 761 (95%) were tested by the 4ºC LISS wash technique, and 667 (83%) were tested by the direct Polybrene test. All three techniques were performed on 635 (79%) of the 800 specimens tested; the gel test was performed on 434 (54%) samples. When positive results were obtained by the AGS panel, especially with anti-IgG, the 4°C LISS wash or direct Polybrene test was not always performed. Positive results were obtained with at least one of the methods for 431 (54%) of the 800 specimens tested. Some specimens were reactive by one method but not by another; others were reactive by more than one method. Results for the battery of serologic tests used in the DAT-negative AIHA workup are shown in Table 1. Specimens that yielded an invalid result, i.e., a reactive negative control, were not included in the number of specimens tested for that method. The number of invalid results obtained for each method was as follows: AGS panel = 2, direct Polybrene test = 2, 4ºC LISS wash technique = 11, and DAT by gel test = 2. An initially reactive control for the AGS panel was resolved for 10 samples by washing the RBCs with warm (37ºC) saline (n = 7) or by treating them with DTT (n = 3).13
Our AGS panel provided a positive DAT result in 50 percent of cases. The distribution of the AGS panel reactivity is shown in Figure 1. Reactivity with anti-IgG or anti-C3 accounted for positive results in 48 percent of cases; most of this reactivity was weak (≤1+). All eight specimens that reacted with anti-IgM also reacted with anti-C3 or with anti-IgG plus anti-C3. IgA was detected on 13 (2%) specimens that were nonreactive with anti-IgG and anti-C3.
Table 1. Results for battery of serologic tests for DAT-negative autoimmune hemolytic anemia
Results No. tested* No. positive (%)
Any positive test 800 431 (54%)
Antiglobulin sera panel positive 800 400 (50%)
4ºC LISS wash anti-IgG positive, room temp wash anti-IgG negative 761 37 (4.9%)
Direct Polybrene test only positive 667 15 (2.2%)
Gel anti-IgG test only positive 434 0
*Number tested does not include invalid results.

158 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
R.M. Leger et al.
The direct Polybrene test was positive for 67 of 667 (10%) specimens tested, and was the only positive test for 15 specimens. The 4ºC LISS wash technique was used for 761 samples. Of these, low-affinity IgG was detected on 37 (4.9%) samples that did not react with anti-IgG using the routine room temperature wash in the DAT AGS panel; 21 of these 37 samples reacted with anti-C3 in the routine test. The anti-IgG reactivity after the 4ºC LISS wash varied from weak to 4+. Interestingly, three of these were readily detected (1+ to 2+) by the murine monoclonal anti-IgG (Immucor), but were nonreactive (n = 2) or only very weakly (microscopically, n = 1) reactive with the rabbit anti-IgG (Ortho).14
The anti-IgG gel test was positive in 57 of 434 (13%) tests, but was never the only positive test. The reactivity of ten samples that were positive by gel test (weak to 1+) but negative with anti-IgG by room temperature tube method is shown in Table 2. RBC-bound IgG was detected by the gel test in four samples that were nonreactive with anti-IgG by tube test using room temperature or 4ºC LISS wash;
these RBCs also reacted with anti-C3. The direct Polybrene test was also positive in the three samples that were tested. Eighteen of the 37 (49%) low-affinity IgG autoantibodies were tested by the gel test: five were reactive and 13 were nonreactive. We discontinued using the gel test in April 2005 as part of the DAT-negative AIHA workup.
DiscussionWe have previously reported on a 10-year series of 259
cases of hemolytic anemia in which an immune basis was suspected but the DAT was reported as negative.6 Similar to the results in the current series, 10 percent of the cases had positive DATs with commercial anti-IgG (e.g., used by hospitals) when tested in our laboratory. A battery of tests (the direct Polybrene test, direct polyethylene glycol test, DAT with cold washes, flow cytometry with fluorescein-labeled anti-IgG, direct ELAT, a concentrated eluate, and the MMA) was used to detect RBC-bound IgG on the remaining samples. Thirty-four percent of the 234 samples were positive by at least one of the additional tests. Although it was found that the direct Polybrene test was as efficient as the more-complex flow cytometry or ELAT methods, some samples that were positive by the latter were not positive by the direct Polybrene test and vice versa. It was concluded that no one test is optimal, and a battery of tests would be the most efficient approach for these DAT-negative AIHA cases. Subsequently, a battery of serologic tests that could be performed as a routine investigation in our IRL was adopted. In the current series of 12 years of data generated from this battery of tests, we were able to provide some serologic evidence of an immune etiology in slightly more than half of the cases submitted.
Our panel of AGS detected immunoglobulin or complement on RBCs of 50 percent of the samples that were DAT negative at the hospital. The 13 percent positive for IgG probably reflects differences in performing the DAT between the referring and the reference laboratory; weak agglutination can be easily shaken out and missed by less experienced technologists. Some of the 45 percent positive anti-C3 results may reflect a difference in technique plus the use of a stronger rabbit anti-C3. Most of the anti-C3 reactions were weak (≤1+), but 47 (13%) were moderately to strongly reactive. RBC-bound IgM can be difficult to detect by the antiglobulin test13; fortunately, IgM antibodies associated with AIHA characteristically bind complement. IgA autoantibodies are uncommon, and for them to occur as the only detectable immunoglobulin in a patient with AIHA occurs in only about 2 percent of AIHA.1
Low-affinity autoantibodies are uncommon, but they may sometimes cause a false-negative DAT result because of a loss of reactivity during the test process. These antibodies are susceptible to “elution” when the RBCs are washed with room temperature or 37ºC saline. Washing RBCs with cold (4ºC) saline or LISS has been shown to enhance detection of low-affinity antibodies.1,15 In the current series, washing
Fig. 1 Distribution of DAT antiglobulin sera panel positive results.
Table 2. Reactivity of ten samples of RBCs using anti-IgG by gel test but nonreactive with anti-IgG in the routine tube test
SampleGel
anti-IgG AGS panelAnti-IgG by 4ºC
LISS washDirect
Polybrene test
1 1+ C3 only Negative Positive
2 1+ C3 only Negative Positive
3 1+ C3 only 1+ NT
4 1+ C3 only 4+ NT
5 1+ C3 only Micro NT
6 1+ C3 only Negative NT
7 1+ Negative 1+ NT
8 Weak C3 only Negative Positive
9 1+ C3 only Invalid Positive
10 1+ C3 only 1+ NT
AGS = antiglobulin sera; NT = not tested.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 159
DAT-negative hemolytic anemia
with 4ºC LISS enabled detection of IgG in 37 (5%) samples when the routine DAT (i.e., washing with room temperature saline) was negative.
Column agglutination tests, e.g., gel test, have sometimes yielded a positive DAT for IgG on RBCs that are DAT-negative by routine tube test.16–20 This, however, may depend on the method and reagents used by the manufacturer as various techniques and antibody sources used in different countries are not identical. The increasing use of the gel test in the United States may change the failure rate of hospitals for detection of RBC-bound IgG because of poor technique when performing tube methods. Column agglutination tests theoretically would be helpful in detecting low-affinity antibodies as the washing phase has been eliminated.21 There have been a few reports of low-affinity IgG detected using a cold-wash technique that were also detected by column agglutination.17,22 In our study, two thirds of the low-affinity IgG detected using the 4ºC LISS wash were not detected by the ID-MTS gel anti-IgG test.
The direct Polybrene test is rapid, but the test is technique-dependent. Polybrene (hexadimethrine bromide) causes nonspecific aggregation and close approximation of normal RBCs permitting cross-linking of antibody molecules. Aggregates are dispersed by the addition of a sodium citrate solution, but antibody-mediated agglutination will persist after neutralization of the Polybrene effect. The test can then be converted to an antiglobulin test (anti-IgG) if antibody is not detected after the aggregates are dispersed. The direct Polybrene test has previously been shown to be almost as sensitive as flow cytometry, and as sensitive as the ELAT, for detecting low levels of RBC-bound IgG.1,6,23 Others have also found this test to be useful for DAT-negative AIHA samples.8,24
We had limited hematologic data and follow-up of the patients in this study. The data reflect the results of tests on samples from patients who were suspected to have AIHA but the hospital laboratory had reported a negative DAT. The hematologists were seeking some support for their suspected diagnosis and therapy. The current study was not designed to confirm earlier publications showing that such tests are useful in the diagnosis of DAT-negative AIHA. The data were reviewed to see how many times in 12 years we found RBC-bound proteins not detected in routine DATs performed at hospitals. The results in this large series show that there is value in using an IRL experienced in reading weak reactions, where special AGS are available, and where tests to detect low-affinity and small amounts of RBC-bound IgG can be performed.
References 1. Petz LD, Garratty G. Immune hemolytic anemias. 2nd
ed. Philadelphia: Churchill Livingstone, 2004. 2. Garratty G. Immune hemolytic anemia associated with
negative routine serology. Semin Hematol 2005;42: 156–64.
3. Gilliland BC, Leddy JP, Vaughan JH. The detection of cell-bound antibody on complement-coated human red cells. J Clin Invest 1970;49:898–906.
4. Gilliland BC. Coombs-negative immune hemolytic anemia. Semin Hematol 1976;13:267–75.
5. Petz LD, Garratty G. Acquired immune hemolytic anemias. New York: Churchill Livingstone, 1980.
6. Garratty G, Postoway N, Nance S, Arndt P. Detection of IgG on red cells of patients with suspected direct antiglobulin test negative autoimmune hemolytic anemia (AIHA). 1990 ISBT/AABB Book of Abstracts, p. 87.
7. Garratty G, Leger RM, Hunt P, Co A. Serological investigation of a large series of direct antiglobulin test-negative hemolytic anemias (abstract). Transfusion 2004;44(Suppl):121A–2A.
8. Rubino M, Kavitsky DM, Nance S. Serologic testing in autoimmune hemolytic anemia (AIHA) with negative direct antiglobulin test (DAT) (abstract). Transfusion 2002;42(Suppl):104S.
9. Fueger JT, Gottschall JL, Curtis BR, Johnson ST. DAT negative immune hemolytic anemia—the role of enhanced techniques (abstract). Transfusion 2003; 43(Suppl):103A.
10. Reid ME. Autoagglutination dispersal utilizing sulphydryl compounds. Transfusion 1978;18:353–5.
11. Löw B, Messeter L. Antiglobulin test in low-ionic strength salt solution for rapid antibody screening and cross-matching. Vox Sang 1974;26:53–61.
12. Lalezari P, Jiang AF. The manual Polybrene test: a simple and rapid procedure for detection of red cell antibodies. Transfusion 1980;20:206–11.
13. Arndt PA, Leger RM, Garratty G. Serologic findings in autoimmune hemolytic anemia associated with immunoglobulin M warm autoantibodies. Transfusion 2009;49:235–42.
14. Arndt PA, Hunt PP, Garratty G. Suspected autoimmune hemolytic anemia associated with RBC-bound IgG only detectable by 4C LISS wash method using certain anti-IgG reagents (abstract). Transfusion 2009;49(Suppl):129A.
15. Garratty G, Arndt P, Nance S, Postoway N. Low affinity autoantibodies—a cause of false negative direct antiglobulin test. 1990 ISBT/AABB Book of Abstracts, p. 87.
16. Kroll H, Salama A, Berghöfer H, Mueller-Eckhardt C. The direct antiglobulin test: clinical significance and prospective comparison of column agglutination technology, gel test and tube test in 163 patients (abstract). Vox Sang 1994;67(Suppl 2):65.
17. Lai M, Rumi C, D’Onofrio G, Voso MT, Leone G. Clinically significant autoimmune hemolytic anemia with a negative direct antiglobulin test by routine tube test and positive by column agglutination method. Immunohematology 2002;18:109–13.

160 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
18. Sajur J, Fournier Y, Sheridan B, Heddle NM. Comparison of gel and tube techniques for direct antiglobulin tests (abstract). Transfusion 1997;37(Suppl):27S.
19. Chaudhary R, Das SS, Gupta R, Khetan D. Application of flow cytometry in detection of red cell bound IgG in Coombs negative AIHA. Hematology 2006;11: 295–300.
20. Dittmar K, Procter JL, Cipolone K, Njoroge JM, Miller J, Stroncek DF. Comparison of DATs using traditional tube agglutination to gel column and affinity column procedures. Transfusion 2001;41:1258–62.
21. Lapierre Y, Rigal D, Adam J, et al. The gel test: a new way to detect red cell antigen-antibody reactions. Transfusion 1990;30:109–13.
22. Sokol RJ, Booker DJ, Stamps R, Jalihal S, Paul B. Direct Coombs test-negative autoimmune hemolytic anemia and low-affinity IgG class antibodies. Immunohematology 1997;13:115–18.
23. Garratty G, Postoway N, Nance S, Brunt D. The detection of IgG on the red cells of “Coombs Negative” autoimmune hemolytic anemias (abstract). Transfusion 1982;22:430.
24. Owen I, Hows J. Evaluation of the manual hexadimethrine bromide (Polybrene) technique in the investigation of autoimmune hemolytic anemia. Transfusion 1990;30:814–18.
Regina M. Leger, MSQA,MT(ASCP)SBB,CMQ/OE(ASQ) (corresponding author), Research Associate, Asuncion Co, MT(ASCP)SBB, Senior Reference Specialist, Penny Hunt, MT(ASCP)SBB, Senior Reference Specialist, and George Garratty, PhD, FRCPath, Scientific Director, American Red Cross Blood Services, Southern California, 100 Red Cross Circle, Pomona, CA 91768.
R.M. Leger et al.
ManuscriptsThe editorial staff of Immunohematology welcomes manuscripts pertaining to blood group serology and education for consideration for publication. We are especially interested in case reports, papers on platelet and white cell serology, scientific articles covering original investigations, and papers on new methods for use in the blood bank. Deadlines for receipt of manuscripts for consideration for the March, June, September, and December issues are the first weeks in November, February, May, and August, respectively. For instructions for scientific articles, case reports, and review articles, see Instructions for Authors in every issue of Immunohematology or on the Web at www.redcross.org/immunohematology. Include fax and phone numbers and e-mail address with all articles and correspondence. E-mail all manuscripts to [email protected].

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 161
Review
Transfusion-related acute lung injury (TRALI) is a clinically important complication of transfusion that is often difficult to diagnose, is probably underreported, and likely has a multifactorial origin that is incompletely understood, making it challenging to find effective treatments and preventative steps. The spectrum of its severity and clinical symptoms seems wide, but its pathogenesis is most likely associated with pulmonary damage from activated recipient neutrophils. Despite the pathogenesis of TRALI being unclear, many severe cases are related to transfusion of donor WBC antibodies, and preventive measures based on avoiding donations by multiparous donors have been implemented at some sites, with early reports showing benefits. This review will address some of the questions surrounding the etiology of this potentially fatal reaction and how measures, predicated on many severe cases being related to transfusion of plasma from multiparous donors, led to preventive steps to avoid these donations. Immunohematology 2010;26:161–73.
Transfusion-related acute lung injury (TRALI) has become one of the more frequently reported causes of transfusion-related morbidity and mortality in several countries including the United States.1,2 From 2005 to 2009, TRALI was the leading cause of patient death attributable to blood components, comprising 48 percent of all the fatal transfusion reactions reported to the Food and Drug Administration (FDA).3 Because TRALI is suspected to be underrecognized, underdiagnosed, and hence underreported, these documented cases likely represent just the tip of what has been called the TRALI iceberg.4 In other words, the true prevalence of TRALI is unknown, and consequently its mortality rate may be higher than the number of reports would suggest.
Because treatment for TRALI is basically supportive, and up to 10 percent of patients with it die even with aggressive therapy,5 implementation of effective preventive measures is urgently needed. However, preventing an illness usually requires a good understanding of its distinctive clinical characteristics and its causes, and the ready availability of diagnostic testing. At present the exact cause or causes of TRALI remain unclear. Its pathogenesis appears complicated, and its clinical presentation is similar to that of other illnesses and syndromes. In addition, there are no clear-cut diagnostic tests. Further studies into its pathogenesis are needed to develop more effective therapies than the general supportive measures that are available today.
There are two leading theories on the pathophysiology of TRALI, the antibody-mediated theory and a “two-event,
or two-hit model.” The former has served as the basis for some recent preventive interventions that appear to be decreasing the prevalence of TRALI in the United States and internationally.
In this review we will address the definitions of TRALI and the two commonly proposed pathogenetic mechanisms of TRALI along with the central role the neutrophil plays in causing it, and we will specifically look at data from recent animal models and clinical research. Finally, we will touch on how our current understanding of TRALI formed the foundation of the preventive measures that have already been implemented.
TRALI: Background and DefinitionA hallmark of TRALI is noncardiogenic pulmonary
edema. When a transfusion is accompanied by severe shortness of breath, causes other than TRALI are often the first to be considered, including hemolytic reactions, bacterially contaminated components, circulatory volume overload, bronchospastic “anaphylactoid” allergic pulmonary reactions, and coincidental underlying conditions such as pulmonary emboli, asthma, and anxiety that can occur during a transfusion.
Reports of posttransfusion, noncardiogenic pulmonary edema first surfaced in the 1950s.6,7 Since then two seminal papers by Popovsky et al.8,9 in the 1980s described pulmonary reactions in patients from the Mayo Clinic and linked them with donor leukocyte antibodies in what we now identify as a clinical entity called TRALI. The 1985 article depicted 36 patients with acute respiratory distress and new infiltrates on chest x-ray lacking an obvious cause such as circulatory overload or pneumonia.9 All of the reactions occurred within 6 hours of administering the blood, and most of them took place within 2 hours. Two of the patients died. In 89 percent of the cases a WBC antibody was identified in a donor, and in 6 percent the antibody was in the patient.
Since these landmark papers, TRALI has generally been considered to be mediated by the transfusion of components containing a donor antibody with specificity to the recipient’s WBCs. Although happening rarely, cases have been reported that seem to be related to a recipient antibody with specificity to donor WBCs contained in the transfusion. Because antibodies have not been identified in all cases of TRALI, there has been much interest in TRALI cases that do not seem to be antibody mediated.
The pathophysiology and prevention of transfusion-related acute lung injury (TRALI): a reviewD.C. Mair and T. Eastlund

162 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
D.C. Mair and T. Eastlund
Studies have shown that other substances in stored blood components may also cause TRALI, and this has been called the two-event, or two-hit, model of TRALI. In this model, endogenous inflammatory mediators produced in certain patients may result in priming and sequestration of neutrophils in the lung. Substances that develop and accumulate during blood component storage can activate the sequestered neutrophils of transfused recipients, causing damage and alveolar permeability. This two-event theory was proposed as a major alternative to the antibody-mediated model of TRALI’s pathogenesis. Both antibody-mediated TRALI and the two-event model will be discussed in more detail in a later section.
Since the 1980s, with improvements in infectious disease screening of donors and reduction of transfusion-transmitted infections, TRALI gradually became recognized as a leading cause of transfusion-related morbidity and mortality. Partly in response to the increasing number of TRALI fatalities reported to agencies like the FDA,3 British Blood Services,1 etc., the National Heart, Lung, and Blood Institute (NHLBI)10 and a Canadian consensus conference11 each convened experts to analyze the TRALI literature with the ultimate goal of improving patient safety. Because the cause of TRALI remains somewhat unclear, both forums concluded that decreasing the number of TRALI cases posed potentially significant challenges given the current state of knowledge and that additional studies were needed. To advance research on TRALI, a commonly accepted clinical description was viewed as indispensable. NHLBI and the Canadian panel produced similar definitions based on the American-European Consensus Conference (AECC) definition of acute lung injury (ALI),12 with one difference being the Canadian group created a separate entity called “possible TRALI” for patients who might have other risk factors for lung damage besides transfusion (Table 1). Both
groups selected the AECC definition for ALI versus the clinically more severe acute respiratory distress syndrome (ARDS), but added O2 saturation as another way to measure hypoxemia.10–13 This gave physicians the flexibility of using pulse oximetry to aid in the diagnosis of TRALI when an arterial blood gas had not been performed. It is also noteworthy that these TRALI definitions rely on clinical information, and although the presence of WBC antibodies in the donor or patient may support the diagnosis, neither definition required that testing be performed.
Less severe forms of posttransfusion dyspnea are often not investigated for a diagnosis of TRALI, and the NHLBI and Canadian Conference definitions did not address this less established entity called “mild TRALI.” Palfi et al.14 demonstrated that plasma from multiparous donors, which was more likely to contain antibody (although the investigators did not test for this), induced a small, yet clinically significant, drop in the partial pressure of arterial oxygen to fraction of inspired oxygen (PaO2/FIO2) ratio of 100 patients when compared with control plasma. The authors suggested this slight hypoxia may represent a milder form of TRALI. In an abstract published 3 years later, Palfi and associates15 noted the majority of TRALI reactions reported in south Sweden were considered to be mild. They also acknowledged the need for an accurate consensus definition. As research and our understanding of TRALI’s pathophysiology and incidence improve, hopefully milder forms of transfusion-associated respiratory distress will also be addressed.
Retrospective “lookback” studies of recipients who received transfusions containing donor WBC antibodies have provided evidence that donor antibodies frequently do not cause symptoms in transfused recipients and also suggested there may be more subtle versions of the reaction that qualify as mild TRALI.16,17 In these reports, an
Table 1. The American-European Consensus Conference (AECC) definition for ARDS/ALI and the National Heart Lung and Blood Institute and Canadian Consensus Conference definitions of TRALI
American-European Consensus Conference definition of ARDS and ALI12
National Heart, Lung, and Blood Institute definition of TRALI10
Canadian Consensus Conference definition of TRALI11
Acute onset
Hypoxemia PaO2/FIO2 ≤ 200 mm Hg = ARDS PaO2/FIO2 ≤ 300 mm Hg = ALI
Bilateral infiltrates on chest radiograph
PAOP ≤ 18 mm Hg when measured or no clinical evidence of left atrial hypertension
New-onset ALI (i.e., no ALI exists before transfusion) by AECC definition of ALI*
Clear temporal relationship to transfusion During or within 6 hours of completing the
transfusion
Patients may have other risk factors for ALI†
If risk factors other than transfusion are also present, patient’s clinical course may clarify whether ALI was caused by blood components or the alternative cause or both
New-onset ALI (i.e., no ALI exists before transfusion) by AECC definition of ALI*
Clear temporal relationship to transfusion During or within 6 hours of completing the
transfusion
No alternative risk factor for ALI
Possible TRALI New onset ALI with a temporal relationship
to a transfusion Lung injury also has a clear temporal
relationship to an alternative risk factor for ALI†
ALI = acute lung injury; ARDS = acute respiratory distress syndrome; FIO2 = fraction of inspired oxygen; PaO2= partial pressure of arterial oxygen; PAOP= pulmonary artery occlusion pressure.* NHLBI and the Canadian group expanded the criteria for hypoxemia to include O2 saturation of ≤ 90% on room air = ALI.† Causes of ALI other than transfusion may include aspiration, severe sepsis, pneumonia, shock, toxic inhalation, multiple trauma, lung contusion, burn injury,
near drowning, acute pancreatitis, cardiopulmonary bypass, and drug overdose.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 163
Review of TRALI
antibody-positive donor was identified in the investigation of a severe or fatal TRALI. Evaluating prior recipients of components from these antibody-positive donors revealed previously unreported cases of milder respiratory distress. A minority of the recipients also had severe reactions that were suspicious for TRALI and should have been reported to the hospital’s transfusion service, but were not.17 This latter finding supports the contention that TRALI is often unrecognized and probably underreported.
The Pathophysiology of TRALIThe Role of the Neutrophil in TRALI
The clinical presentation of ARDS/ALI has much in common with the features observed in TRALI patients. Beyond the signs and symptoms there is also similarity in the histopathology insofar as neutrophils are concerned. For example, microscopic examination of postmortem lung samples from TRALI victims has shown sequestration of neutrophils in the pulmonary vasculature and in the alveolar space, analogous to what has been observed in ARDS and ALI.18 Reviewing how the neutrophil functions in pulmonary inflammatory processes, such as infection or lung injury, would be beneficial in understanding its role in either the antibody-mediated or the two-event model.
The neutrophil is the first line of defense against bacterial infection, and it can generate a wide array of molecules that are bacteriocidal and initiate inflammation that can damage healthy cells and tissue in the same area. To more efficiently eradicate infectious agents, neutrophils ideally go through a stage of priming first. Primed neutrophils have enhanced microbial killing capabilities in response to a subsequent stimulus versus their unprimed counterparts.19,20 Priming agents such as platelet-activating factor (PAF) or tumor necrosis factor ́ (TNF )́ potentiate both the oxidative and granular microbicidal arsenal when the neutrophil is later exposed to an activating substance such as N-formyl-Met-Leu-Phe or fMLP (Figure 1).19 Priming starts when neutrophils home in on infected areas in response to chemoattractants that could be bacterially derived or released when local endothelial cells become activated. Increased expression of neutrophil adhesion molecules occurs on the surface of both the neutrophil and the endothelium.19,21,22 The neutrophils become less deformable, further slowing their transit through the narrow pulmonary vascular beds, allowing more time and increased contact between the leukocytes and the now more receptive endothelium, leading to further accumulation of neutrophils (sequestration) at the site of infection.22 At this point the neutrophils can be said to be primed.21,22 In an infection, the neutrophils then marginate into the tissue’s interstitium, where they can phagocytize the pathogens, resulting in neutrophil activation and destruction of the microbe through the release of reactive oxidative species and proteases.21 In this way, a primed neutrophil can clear an infection more effectively than it could in its
pre-primed state. In most cases, this response to the infection is short and limited to a level that is appropriate to remove the infecting agents without significant neutrophil-related damage to the surrounding tissues.22
This same sequence of neutrophil priming, sequestration, and activation is believed to occur in the lung in TRALI, except the activated neutrophils release their toxic substances onto the pulmonary capillary, disrupting the barrier between the endothelium and lung epithelium, and ending with edema in the alveolar space. In healthy people the lung normally stores a relatively large amount of the body’s available neutrophils (28%), and because this quantity increases even further with sequestration, the respiratory system may be more inclined to experience neutrophil-induced injury than other organs.22 Neutrophil priming and activating agents that cause TRALI may spare nonpulmonary tissue because of the relatively low neutrophil burden outside of the lung. These substances that trigger sequestration and cell damage for each of the two TRALI theories are discussed in a later section.
The Role of Antibodies in TRALIA causal relationship between the transfusion of WBC
antibodies and ALI has been suspected for more than 50 years. In 1957, Brittingham7 described a healthy research subject who exhibited symptoms of ALI after receiving a leukoagglutinin-containing blood component. Popovsky and colleagues8,9 in 1983 and 1985 crafted the term TRALI and emphasized the association between TRALI and the passive transfusion of donor HLA or granulocyte-specific antibodies into recipients with corresponding antigens on
α α
Fig. 1. Neutrophil priming by inflammatory mediators results in enhanced superoxide production. The dashed arrow between the unprimed and primed state of the neutrophil indicates that priming may be a reversible process. fMLP = N-formyl-methionine-leucine-phenylalanine; GM-CSF = granulocyte/macrophage colony stimulating factor; LPS = lipopolysaccharide; O2– = superoxide anion; PAF = platelet-activating factor; TNFα = tumor necrosis factor ´. Reproduced with permission from Condliffe et al.20α
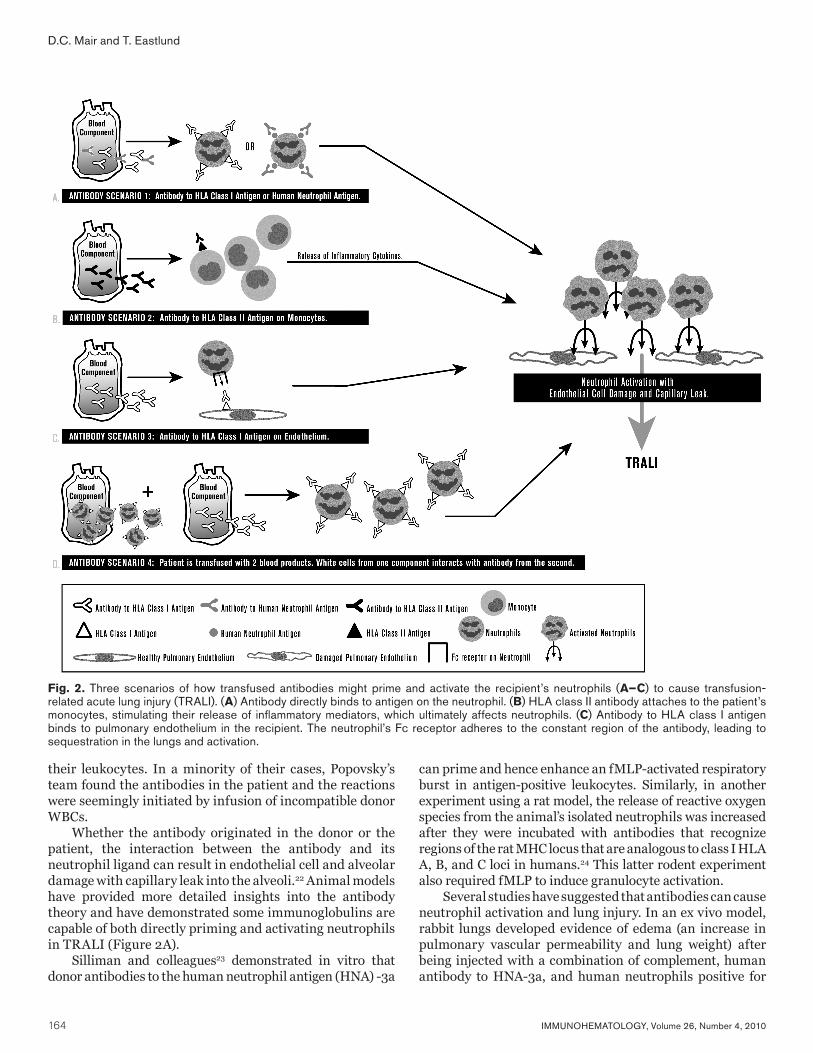
164 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
their leukocytes. In a minority of their cases, Popovsky’s team found the antibodies in the patient and the reactions were seemingly initiated by infusion of incompatible donor WBCs.
Whether the antibody originated in the donor or the patient, the interaction between the antibody and its neutrophil ligand can result in endothelial cell and alveolar damage with capillary leak into the alveoli.22 Animal models have provided more detailed insights into the antibody theory and have demonstrated some immunoglobulins are capable of both directly priming and activating neutrophils in TRALI (Figure 2A).
Silliman and colleagues23 demonstrated in vitro that donor antibodies to the human neutrophil antigen (HNA) -3a
can prime and hence enhance an fMLP-activated respiratory burst in antigen-positive leukocytes. Similarly, in another experiment using a rat model, the release of reactive oxygen species from the animal’s isolated neutrophils was increased after they were incubated with antibodies that recognize regions of the rat MHC locus that are analogous to class I HLA A, B, and C loci in humans.24 This latter rodent experiment also required fMLP to induce granulocyte activation.
Several studies have suggested that antibodies can cause neutrophil activation and lung injury. In an ex vivo model, rabbit lungs developed evidence of edema (an increase in pulmonary vascular permeability and lung weight) after being injected with a combination of complement, human antibody to HNA-3a, and human neutrophils positive for
Fig. 2. Three scenarios of how transfused antibodies might prime and activate the recipient’s neutrophils (A–C) to cause transfusion-related acute lung injury (TRALI). (A) Antibody directly binds to antigen on the neutrophil. (B) HLA class II antibody attaches to the patient’s monocytes, stimulating their release of inflammatory mediators, which ultimately affects neutrophils. (C) Antibody to HLA class I antigen binds to pulmonary endothelium in the recipient. The neutrophil’s Fc receptor adheres to the constant region of the antibody, leading to sequestration in the lungs and activation.
D.C. Mair and T. Eastlund

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 165
that antigen.25 No edema was seen in animals when HNA-3a–negative neutrophils were substituted or if complement was not added. In animals it seemed that anti-HNA-3a, in the presence of complement, activated antigen-positive human neutrophils and caused lung injury.
A study by Sachs et al.,26 using another ex vivo lung rodent model, showed the antibody-antigen reaction could cause lung injury independent of complement and that TRALI may be more complex than simply antibody-to-WBC antigen recognition alone. In these experiments, rats were infused with human HNA-2a–positive neutrophils and CD177 monoclonal antibody (an antibody that recognizes the HNA-2a antigen) without the addition of complement. A key variable here was that the quantity of HNA-2a antigen expression can vary dramatically among different antigen-positive persons. Using quantitative indirect immunofluorescence and flow cytometry, two different sources of neutrophils were identified for this study: one from individuals with a high percentage of HNA-2a–positive cells (defined as greater than 70% expression) and the second from low expressers (less than 30%). TRALI did not occur in these rats when the neutrophils came from individuals with a lower percentage of HNA-2a, unless it was followed by injection of fMLP as an activator. Conversely, HNA-2a antibody alone appeared to activate leukocytes and cause lung injury when cells from high expressers were used.26,27 On the basis of this study, neutrophil-activated TRALI reactions attributable to anti-HNA-2a appeared to be antigen dose–dependent (i.e., the amount of antigen present was also important to neutrophil activation), although this could be overcome by the later addition of another inflammatory mediator (e.g., fMLP).
Infusion of Donor-Derived Antibody Without Causing TRALI
The observation made by Sachs and colleagues,26 that infused antibody with specificity for recipient antigens may sometimes be insufficient to induce TRALI, has also been demonstrated in a number of lookback studies on transfused patients. In these publications donor antibody, directed against antigens that most patients would be expected to carry on their cells, was not harmful in the majority of recipients.
Toy et al.16 identified only one patient with TRALI from among 103 recipients of blood from a donor with multiple HLA antibodies. Fifty-four of these patients had a corresponding WBC antigen and no TRALI was observed. Forty-two of these 103 patients were neutropenic, an attribute that might have protected them against lung injury mediated by activated neutrophils. In another study, Win and associates28 found no cases of TRALI in a lookback analysis on 43 transfusions from six donors with leukocyte antibodies, again directed against either multiple HLA or neutrophil antigens having a relatively high frequency in the general population. Nicolle et al.29 reported two cases of
TRALI as a result of donor HLA antibodies and evaluated 18 other recipients of blood from the same donors. Of five plasma recipients only one was found to have undiagnosed TRALI, whereas another had an HLA type matching the donor antibody specificity but had no reaction. Of 13 RBC recipients, none developed a reaction. Kopko et al.17 initiated a lookback investigation after a fatal TRALI reaction was attributed to a granulocyte antibody directed against HNA-3a. This antigen is common throughout the population as it has been found in 89 percent to 99 percent of tested individuals.30 The lookback study evaluated 36 patients who previously received blood from that donor and identified 15 transfusion reactions in only 13 (36.1%) of the recipients, and just 2 of these individuals were initially diagnosed with TRALI by the treating physicians.17 Although an additional 7 of these 13 reacting patients had severe respiratory problems that possibly should have been called TRALI, most of the 36 recipients had been transfused without incident. Muniz et al.31 also identified a donor with an anti-HNA-3a whose blood had been given previously to 11 patients. One experienced TRALI, another had chills, but the remaining nine had no reactions. Fadeyi et al.32 described a donor-derived anti-HNA-2a (the corresponding antigen is present in 95–99% of the population) that induced dyspnea or leukopenia in 18 of 39 transfusion events, but no episodes of TRALI were found. As was discussed earlier, variation in HNA-2a expression among antigen-positive individuals might explain why many of the recipients were asymptomatic or did not experience more severe pulmonary compromise.
Each of these lookback studies suggests that even in the setting of donor antibody with specificity for recipient antigen, TRALI usually does not occur. Even taking into account potential TRALI cases that were initially missed, most recipients were asymptomatic. The reasons for this wide range of clinical responses are unclear, but may be related to individual variations in antibody titers or avidity, concentration of corresponding antigens on recipient cells, underlying diseases in recipients, variability in neutrophil reactivity to activated complement and cytokines, concentration of neutrophil adhesion molecules, and reactivity of the recipient’s pulmonary vessels and alveoli.
Antibodies to Class II HLA AntigensIn addition to antibodies directed at class I HLA and
HNA specificities, later work showed that donor antibodies to HLA class II antigens can also be associated with TRALI,33 and these antigens are not normally expressed on neutrophils.34 Granulocytes have exhibited HLA-DR (a class II antigen) on their surface after exposure to agents like granulocyte/macrophage colony-stimulating factor, interferon (IFN)-γ, and interleukin (IL)-3,34 but this antigen expression has not been documented in TRALI cases.22 Immunohistochemical stains performed on lung tissue from a TRALI fatality found HLA class II expression
Review of TRALI

166 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
D.C. Mair and T. Eastlund
was limited only to pulmonary macrophages, despite granulocyte accumulation in the capillaries and evidence the endothelium was activated.35 Activated pulmonary endothelium may also demonstrate HLA class II antigens, but pulmonary endothelium from the deceased TRALI patient did not express class II antigens despite being positive for CD34, an indicator of endothelial activation.35 Accordingly, some authors have postulated these antibodies act indirectly on neutrophils by first binding to monocytes, which in turn release cytokines that sequester and activate neutrophils to induce lung damage (Figure 2B).22 In an in vitro study in which Kopko et al.33 combined anti-HLA class II antibodies with human monocytes of a corresponding HLA type, the cells became activated and produced the inflammatory cytokines IL-1β and TNFα, as well as tissue factor, and each of these inflammatory cytokines is capable of attracting and stimulating neutrophils.36 When Nishimura and colleagues37 incubated antibodies to HLA-DR15 with antigen-positive monocytes and cultured human lung microvascular endothelium, high levels of the cytokine leukotriene B4 (a potent neutrophil attractant) were produced. Endothelial cells were also needed to interact with the WBCs and antibody to generate leukotriene B4 in this model. From this it may be inferred that endothelium is also a necessary part of the inflammatory process in TRALI.
Antibody Interaction With Lung Tissue and TRALIPresumably, the antigen-binding site of the offending
antibody is targeting a matching substance on either the neutrophil or the monocyte in TRALI.4 However, recent work by Looney et al.,38 using a murine model of lung injury, argued that the antigen binding sites may also adhere to HLA antigens on the endothelium instead and that the Fc receptor of the circulating neutrophil interacts with the antibody’s constant region to alter the neutrophil’s circulation, leading to its sequestration and activation (Figure 2C). The investigators were able to induce ALI (demonstrated by excess lung water and increased vascular permeability) in vivo by injecting the mice with a monoclonal antibody targeting an antigen in the murine class I MHC complex. Although flow cytometry showed that the monoclonal antibody recognized class I MHC antigen on neutrophils, this antibody to WBC antigen binding was not thought to have caused the lung injury because the antibody did not directly activate the cells. Microscopic examination of lung tissue from the affected mice confirmed there was marked pulmonary neutrophil sequestration, and immunohistochemistry revealed the monoclonal antibody preferentially bound to pulmonary endothelium over tissue in other organs. Despite other WBCs having Fc receptors, the mice did not exhibit lung injury if they were made neutropenic before being challenged with the class I MHC antibody. TRALI also did not occur when antigen-positive mice that lacked Fc receptors on their cells were given the antibody, but lung
injury did transpire if they first were infused with normal Fc receptor–positive neutrophils and then received the monoclonal antibody. These authors hypothesized that the infused class I MHC monoclonal antibody preferentially bound to antigen present on the pulmonary endothelium, and the antibody then sequestered circulating neutrophils via the Fc receptor interaction, resulting in neutrophil activation and lung injury. The presence of Fc receptors on neutrophils appeared to be a requisite feature for TRALI in this experiment.
Clinical evidence that TRALI can develop from transfusion of antibody with specificity to antigens on lung tissue came from a patient who experienced unilateral TRALI.39 Ten weeks after a single-lung transplant, the patient became dyspneic and exhibited pulmonary infiltrates in the single transplanted lung but not in the native lung after receiving a unit of RBCs containing antibodies with HLA-B44 specificity. The recipient was HLA-B44 negative, but the transplanted lung was B44 antigen positive. Although the lung donor’s B44-positive leukocytes may have been circulating in small numbers, the transfused antibody was presumed to have bound to the B44-positive transplanted lung itself.
The Two-Event Model and the Role of Biologically Reactive Mediators in TRALI
Although much of the transfusion medicine literature has focused on the antibody-mediated cause of TRALI, the two-event model of ARDS has attempted to answer some of the questions that remain about TRALI’s origin and especially how TRALI can develop in the absence of an antibody.40 The first event is a preexisting clinical condition in the patient (e.g., sepsis, recent surgery) whereby the patient’s illness produces endogenous generalized inflammation that affects the pulmonary endothelium. The endothelium can then become activated, expressing adherence molecules on its surface, resulting in binding of the neutrophils to the pulmonary vasculature, sequestration, and the neutrophils becoming primed.19,21,22 Neutrophil priming agents may be released from patient-derived sources in the postoperative period or by invading infectious agents. Potential neutrophil priming substances, which may be produced by dying cells, WBCs, or activated pulmonary endothelium, include PAF, TNFα, and IL-8.41–43 Lipopolysaccharides (LPS) from bacterial cell walls may also serve in this capacity.44
The proposed second event is transfusion of a cellular blood component containing various exogenous cytokines or lipid-based biologically reactive mediators (also known as biologic response modifiers or BRMs) that stimulate and activate the adherent neutrophils to release proteases, which, as in the antibody-mediated model, causes endothelial cell and alveolar damage with noncardiogenic pulmonary edema.40,45
These lipid-based substances seem to accumulate only in cellular components during storage and are not

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 167
Review of TRALI
found in frozen plasma components.46–49 CD40 ligand, a platelet-derived inflammatory mediator, as well as lysophosphatidylcholines (Lyso-PCs), IL-6, IL-8, and others, have all been proposed to function as BRMs in this model.46–50 Older platelet components and packed RBCs contain greater amounts of these BRMs and thus appear to pose a greater risk of causing TRALI.5,48,51
Wyman et al.19 exposed monolayers of human pulmonary endothelium to one potential priming agent: bacterial endotoxin (i.e., LPS). In response, the cells generated chemokines and expressed adhesion molecules. When neutrophils were later added, they became primed and adherent to the monolayer. Analogously to what is thought to happen in TRALI, the adherent neutrophils were then activated by biologically active lipids from stored packed RBCs (i.e., Lyso-PC) and damaged the endothelial cells. Blocking antibodies against the adhesion molecules on the neutrophils and the endothelium prevented injury to the monolayer. Also notable was that LPS and Lyso-PC used individually only primed the neutrophils, but adding them in sequence caused neutrophil activation and harm to the monolayer. This showed that using two priming agents together could result in activation.
BRMs have also been demonstrated in the posttransfusion serum of patients who exhibited TRALI reactions.40 When their post-TRALI serum was incubated with neutrophils, a significantly greater superoxide release was observed on activation than when neutrophils were incubated with the same patient’s pretransfusion serum or when the sera of recipients with febrile and urticarial reactions were used. This suggested the presence of a neutrophil priming agent in these TRALI patients that arose from the infusion of the blood component.
Although BRMs are not found in frozen plasma components,47–49 donor-derived antibodies can be found in these components. This explains the strong correlation between frozen plasma transfusion and the development of TRALI.52 Because antibodies can serve as the second event, they can play a causal role in either the antibody hypothesis or the two-event model, and hence these two TRALI theories are not mutually exclusive. A recent study using a rat model demonstrated that either antibodies or BRMs can cause TRALI.24 As the first event, rats were injected intraperitoneally with LPS versus control animals that were injected with an equal volume of saline solution. For the second event the rodents were injected with either an antibody directed against class I MHC antigens or the extract from a 42-day-old RBC. Both antibody and the RBC extract were able to produce pulmonary edema in the LPS-injected group but not in the control animals. Microscopic examination in the rats with pulmonary injury documented neutrophil infiltration in the lungs, again demonstrating the importance of neutrophils in ALI. Furthermore, inducing neutropenia in the LPS-primed rodents was protective against lung damage.
The investigators of a previously discussed murine model, which argued the importance of the Fc-receptor on neutrophils, surprisingly found in later work that the experiment was less likely to produce lung injury after the mice had been moved to barrier-protected, pathogen-free housing.53 The rodents from the prior study had been kept in a “dirtier” environment and had a higher neutrophil count than their counterparts in the more sterile setting, which the authors attributed to a more primed state of the immune system in the previous group. Exposing the animals from the pathogen-free environment to LPS (analogous to a first event) restored the model’s ability to cause TRALI. Looney et al.53 interpreted this as evidence their experiments also represented a two-event model of TRALI. Interestingly, they found not only that neutropenia was protective but that thrombocytopenia was as well. Mice pretreated with aspirin also did not experience lung injury, which suggests that anti-platelet therapy may have a role in the future treatment or prevention of TRALI.
One limitation of the two-event model is not all investigators accept the premise of TRALI being limited to the ill.54 For example, Brittingham’s7 healthy research subject, appearing to lack a first event, exhibited clinical findings consistent with TRALI (dyspnea, fever, cyanosis, hypotension, leukopenia, and marked bilateral infiltrates on chest x-ray) after receiving just 50 mL of blood from a patient with a strong leukoagglutinin. Recently, a theory known as the threshold model attempted to explain this discrepancy between the Brittingham case and the two-event theory, while proposing answers to other TRALI puzzles including the origin of mild TRALI and why antibody and corresponding WBC antigen does not produce TRALI in all cases.
The Threshold Model of TRALIMany investigators now accept that TRALI appears to
be multifactorial with neutrophil activation and lung injury representing a final common pathway that can be initiated by either an antibody or biologically active lipids.55
Along these lines, the threshold model put forth by Bux and Sachs56 attempts to merge the two TRALI theories. Consistent with its name, this model assumes neutrophils must exceed a certain hypothetical threshold of activation to cause lung injury. The first and second events (and again the latter can be passive donor antibody) have additive inflammatory effects toward surmounting this theoretical activation level needed to cause either a severe or a mild TRALI reaction56 (Figure 3). Presumably, a mild TRALI case has a lower threshold than a serious one and no reaction is observed if the degree of activation falls beneath the mild level.
This threshold model incorporates both of the leading theories and takes into account the observation that a broad variety of substances may be able to prime or activate neutrophils. The patient’s threshold could be reached by the transfusion of an extremely potent antibody from a

168 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
D.C. Mair and T. Eastlund
blood component (Figure 3: healthy research participant). Alternatively, the patient approaches the threshold by releasing his or her own endogenous inflammatory substances as a result of a preexisting disease state, and then transfusion of a less potent antibody or a moderate level of BRMs is sufficient to push the patient over the limit (Figure 3: patients 1 and 2).
This model also addresses some of the gaps in our understanding of this transfusion reaction. In the rare circumstance of a healthy person exhibiting TRALI, the recipient would be assumed to contribute no endogenously generated priming or activating agents to the process. Therefore, the blood component alone must have enough potency to push the recipient over the TRALI threshold without assistance from a preexisting illness. This may have caused the transfusion-associated respiratory distress in the research subject reported by Brittingham,7 and this uncommon event has been referred to by some as a “one-event” TRALI.4 From results of lookback studies, one could hypothesize that most patients did not experience TRALI as a result of some combination of their not being ill enough and the blood donor’s antibody not being potent enough. The dose dependency of the HNA-2a antigen in causing TRALI, found in the rat model used by Sachs et al.,26 may be another such example. When low expressers of HNA-2a antigen were used, antibody was insufficient to overcome the threshold and fMLP had to be added to push the neutrophils beyond the activation level needed to cause lung injury. Additional research would be needed to explore whether such variables truly play a role in the low reaction rates observed in these lookback cases.
TRALI PreventionBecause treatment of TRALI is currently supportive,
actions taken to prevent the reaction become the paramount means of decreasing the morbidity and mortality associated with it. A very important first step for prevention of TRALI is to avoid unneeded transfusions. Monitoring of use, enforcement of guidelines, and education about the judicious use of blood components should not be ignored. This is especially important for transfusions of plasma and platelets that contain a larger portion of donor plasma than other components.
Some of the more specific recommended approaches for preventing TRALI have included changes to donor screening practices such as gender-based exclusions or implementing new tests. Others have suggested different manipulations to the final component (e.g., washing), but regardless, all of the proposed interventions are based on the two leading theories of TRALI.
Preventive Measures for Frozen PlasmaBased on the antibody-mediated mechanism,
components with the most plasma and highest antibody content would be more likely to cause TRALI and
thus would make a logical target for preventive action. Previously pregnant female donors are more likely to have WBC antibodies and hence pose a greater risk than male donors of being associated with TRALI reactions. Several studies have supported preventing TRALI by restricting the use of plasma from parous donors. From 1996–2003 the United Kingdom’s hemovigilance program (Serious Hazards of Transfusion, or SHOT) analyzed 93 reported TRALI cases and found frozen plasma and platelets were respectively about 7 and 8 times more likely to be the implicated component than a relatively low plasma-containing component such as a packed RBC.57 Densmore et al.58 found that 14.6 percent of female apheresis donors who reported one or two pregnancies had HLA antibodies, and this rate increased to 26.3 percent among multiparous donors, defined as women with three or more pregnancies. These authors’ work was also recently confirmed in the much larger leukocyte antibody prevalence study (LAPS), which enrolled approximately 8000 donors, including about 5800 females. LAPS found the prevalence of HLA antibodies in women with zero, one, two, three, and four or more pregnancies was 1.7 percent, 11.2 percent, 22.5 percent, 27.5 percent, and 32.2 percent, respectively.59
In October 2003, the British implemented a predominantly male-donor plasma program. SHOT data published after taking this initiative showed that highly likely or probable cases of TRALI, attributed to plasma, decreased from their preprogram level of 29 to a total of 2.57 It should be noted, the editorial accompanying this publication commented that the SHOT description of a highly likely or probable case was not purely clinical and relied in part on the presence of a WBC antibody.60 This differed from the NHLBI and Canadian TRALI definitions, which did not require positive serology in the donor or patient. Accordingly, TRALI cases caused by the other mechanisms (e.g., the two-event model) and lacking antibody may not have been appropriately included or categorized.60 But given the reported success of the British male plasma initiative, and because TRALI was the leading cause of transfusion-related mortality in the United States, Americans considered adopting this strategy. Additionally, data from the American Red Cross was found to be consistent with the SHOT study’s findings. The American Red Cross, which provides more than 40 percent of the nation’s blood, analyzed 550 suspected TRALI cases, 72 of which were fatal, in the system for 2003 to 2005.52 In 71 percent of these fatalities, a female, WBC antibody–positive donor was believed to be the cause, and in 75 percent, plasma was the transfused component.52 AABB followed the United Kingdom’s lead in 2006 and instituted recommendations for TRALI reduction: blood collection facilities needed to implement processes that lessen the chance of high-plasma volume–containing components being collected from donors at higher risk of having WBC antibodies.61 These processes needed to be in place by the fall of 2007 for frozen plasma components (except cryoprecipitate, which was considered

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 169
Review of TRALI
a low-volume, hence low-risk, component) and apheresis platelets by fall of 2008.
As in Britain, preliminary data in the United States collected by the American Red Cross since establishment of a male-predominant plasma initiative revealed the project has already had a positive impact.62 Plasma distribution from male donors was 55 percent in 2006, 79 percent in 2007, and 95 percent in 2008, and the number of probable TRALI fatalities attributed to plasma in those years were 6, 5, and then 0, respectively, with the last year being a statistically significant drop from the preceding 2 years. The quantity of nonfatal TRALI cases attributable to plasma also decreased dramatically from 26 in 2006 to 7 in 2008.
An international forum published in 2007 revealed that most of its participants had implemented the production of plasma from males only or used a solvent detergent-treated (SD) component.2 The latter is suspected to diminish the risk of TRALI because it is manufactured from pools of more than a thousand donations, so
theoretically any single donor with a potentially harmful WBC antibody should have the immunoglobulin diluted below the level of clinical significance for a recipient.63 Studies have shown that no antibodies to HNA or HLA class I or II antigens are detectable in this component even when they were present in some of the units of starting plasma.64,65 SD-plasma is not available in the United States but has been used in Europe and no cases of TRALI have been reported after use of over 13 million units.65,66
Preventive Measures for PlateletsAlthough some were considering it at the time, none
of the countries with nationalized blood systems had yet implemented HLA or HNA antibody screening of their donors by the time of their participation in the 2007 international forum.2 Testing for granulocyte antibodies is generally considered to be labor intensive and does not readily lend itself to the high throughput needed for processing blood.67 On the other hand, solid-phase assays with shorter turnaround times and relevance to donor screening may become a reality in the near future now
Fig. 3. The threshold model.58 Shown are three blood component recipients and the hypothetical levels of neutrophil activation needed to exceed their thresholds for a transfusion-related acute lung injury (TRALI) reaction. The gray bar represents the relative contribution of their own inflammatory mediators before transfusion, and the black bar is the contribution of mediators in the blood component, such as biologically reactive mediators (BRMs) or antibodies. In both patients 1 and 2, the blood component alone had insufficient inflammatory potential to exceed the threshold, and therefore the occurrence of either a severe (patient 1) or mild (patient 2) TRALI reaction relied on contributions from the recipients’ preexisting inflammatory mediators. The severity of the reaction is determined by the magnitude of neutrophil response. Unlike the first two, the third recipient was healthy before transfusion and thus had no endogenous mediators (i.e., lacked a first event). In this setting, all of the TRALI- producing mediators were provided by a potent blood component alone (e.g., as a strong leukoagglutinin), and hence this bar is completely black. The double arrow between the resting and primed state of the neutrophils indicates that the cells may deprime if activation does not occur.20,22,58

170 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
that several neutrophil antigens have been characterized molecularly, including the clinically important HNA-3a.68,69
Platelet additive solutions, which have been licensed in some European countries, were also mentioned in the international forum, but because 100 mL of plasma may remain in the final component, the boon to TRALI reduction is still in question.2 Several countries admitted to a variety of different initiatives to decrease the risk of TRALI from this component, such as resuspending the pooled buffy coat platelets in the plasma from one of the male donors, whereas other countries restricted women with a history of pregnancy from donating whole blood–derived platelets (WBPs).2
Recently, a number of American blood centers opted to meet the AABB’s 2008 deadline for apheresis platelets by implementing HLA antibody screening, as this was technically less cumbersome than testing for granulocyte antibodies. Some use algorithms based on parity to more strategically select which donors to perform HLA antibody testing on, and this practice is supported by the LAPS paper that showed that asking about transfusion history brought no additional benefit as there was no statistical difference in the rate of antibody positivity among untransfused males, transfused males, and nontransfused females without a history of pregnancy.59 As a result, blood centers might decrease testing costs by choosing not to screen these three groups for HLA antibodies.58 The LAPS authors estimated that deferring previously pregnant, HLA antibody–positive donors from apheresis collections would result in a potentially tolerable 6 percent decrease in platelet apheresis donations.59 The AABB’s 2006 recommendations did not suggest interventions for WBPs because any antibody present was believed to be diluted by pooling.61
Preventing TRALI as a Result of BRMsTo date, most of the actions to avoid TRALI reactions
have concentrated on preventing antibody-mediated reactions. Interventions for decreasing BRMs infused into recipients have also been described, although not as popularly implemented because of potential downsides. Washing is one way to remove BRMs; however, because of the preparation time involved, washed RBCs and platelets would likely be used only for planned transfusions and would be impractical for emergency or massive-transfusion settings.67 Furthermore, little data exist on the effectiveness of lipid removal by washing.67 Theoretically, in a nonemergent setting, severely ill patients believed to be at the highest risk for TRALI (e.g., in the intensive care unit), could be scheduled to receive washed components, but this potential benefit would have to be weighed against the reduction in component quality and shelf life that is caused by washing.67
Since cellular blood components develop higher levels of biologically active lipids and cytokines during storage,47–49 some investigators believe that patients with a preexisting illness might benefit from receiving fresher blood (e.g., RBCs
that are less than 2 weeks old and platelets less than 3 days old).70,71 This might minimize accumulation of and exposure to BRMs that act as the second event in TRALI development. In the 2007 international forum, only Poland indicated they advise against giving blood older than 14 days to patients in severe clinical condition.2 However, concerns about blood availability and how to appropriately define fresh have likely limited this practice. None of the other participants, which included representatives from South America, Japan, the United States, and several other European countries, admitted to having interventions aimed at preventing TRALI as a result of BRMs. Continued research is needed to more fully understand the generation and accumulation of BRMs during storage of cellular blood components and to develop effective methods for their prevention or removal to reduce the risk of TRALI and other transfusion complications.
ConclusionsMany questions remain about the mechanism or likely
mechanisms behind TRALI. Until recently, clinical research on this reaction was believed to be limited by the lack of a commonly accepted definition. Despite these challenges, current data suggest that TRALI has a pathogenesis that is multifactorial. It appears that transfusion of either BRMs or antibodies can end in a final common pathway of lung injury as a result of neutrophil activation. Why some patients may be more susceptible to TRALI than others remains a mystery, but potential explanations include severity of their preexisting illness or possibly the amount of antigen expressed and therefore available to certain infused antibodies. The apparent, early success of preventive measures aimed at avoiding donations by multiparous donors suggests our understanding of TRALI may be improving. Now that similar clinical definitions of TRALI are available, and new insights have been provided by recent animal models, we can hope that forthcoming clinical and laboratory research will provide even better preventive steps in the future, along with treatments that are curative instead of just supportive.
References 1. Stainsby D, Jones H, Asher D, et al. Serious hazards
of transfusion: a decade of hemovigilance in the UK. Transfus Med Rev 2006;20:273–82.
2. Wendel S, Biagini S, Trigo F, et al. Measures to prevent TRALI. Vox Sang 2007;92:258–77.
3. U.S. Food and Drug Administration. Fatalities reported to FDA following blood collection and transfusion: annual summary for fiscal year 2009. Rockville, MD: U.S. Department of Health and Human Services, 2009. Available from: http://www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/ReportaProblem/TransfusionDonationFatalities/ucm204763.htm.
D.C. Mair and T. Eastlund

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 171
4. Boshkov LK. Transfusion-related acute lung injury and the ICU. Crit Care Clin 2005;21:479–95.
5. Boshkov LK. Transfusion-associated acute lung injury (TRALI): an evolving understanding of the role of anti-leukocyte antibodies. Vox Sang 2002;83(Suppl 1):299–303.
6. Barnard RD. Indiscriminate transfusion: a critique of case reports illustrating hypersensitivity reactions. N Y State J Med 1951;51:2399–402.
7. Brittingham TE. Immunologic studies on leukocytes. Vox Sang 1957;2:242–8.
8. Popovsky MA, Abel MD, Moore SB. Transfusion-related acute lung injury associated with passive transfer of antileukocyte antibodies. Am Rev Respir Dis 1983;128:185–9.
9. Popovsky MA, Moore SB. Diagnostic and pathogenetic considerations in transfusion-related acute lung injury. Transfusion 1985;25:573–7.
10. Toy P, Popovsky MA, Abraham E, et al. Transfusion-related acute lung injury: definition and review. Crit Care Med 2005;33:721–6.
11. Kleinman S, Caulfield T, Chan P, et al. Toward an understanding of transfusion-related acute lung injury: statement of a consensus panel. Transfusion 2004;44:1774–89.
12. Phua J, Stewart TE, Ferguson ND. Acute respiratory distress syndrome 40 years later: time to revisit its definition. Crit Care Med 2008;36:2912–21.
13. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000;342:1334–49.
14. Palfi M, Berg S, Ernerudh J, Berlin G. A randomized controlled trial of transfusion-related acute lung injury: is plasma from multiparous blood donors dangerous? Transfusion 2001;41:317–22.
15. Palfi M, Holthuis N. Does mild TRALI exist? Analysis of one year transfusion complications from south Sweden (abstract #M10.04). Vox Sang 2004;87 (Suppl 3):S4.
16. Toy P, Hollis-Perry KM, Jun J, Nakagawa M. Recipients of blood from a donor with multiple HLA antibodies: a lookback study of transfusion-related acute lung injury. Transfusion 2004;44:1683–8.
17. Kopko PM, Marshall CS, MacKenzie MR, Holland PV, Popovsky MA. Transfusion-related acute lung injury: report of a clinical look-back investigation. JAMA 2002;287:1968–71.
18. Dry SM, Bechard KM, Milford EL, Churchill WH, Benjamin RJ. The pathology of transfusion-related acute lung injury. Am J Clin Pathol 1999;112:216–21.
19. Wyman TH, Bjornsen AJ, Elzi DJ, et al. A two-insult in vitro model of PMN-mediated pulmonary endothelial damage: requirements for adherence and chemokine release. Am J Physiol Cell Physiol 2002;283:C1592–603.
20. Condliffe AM, Kitchen E, Chilvers ER. Neutrophil priming: pathophysiological consequences and under- lying mechanisms. Clin Sci (Lond) 1998;94:461–71.
21. Silliman CC, Kelher M. The role of endothelial activation in the pathogenesis of transfusion-related acute lung injury. Transfusion 2005;45(Suppl):109S–16S.
22. Fung YL, Silliman CC. The role of neutrophils in the pathogenesis of transfusion-related acute lung injury. Transfus Med Rev 2009;23:266–83.
23. Silliman CC, Curtis BR, Kopko PM, et al. Donor antibodies to HNA-3a implicated in TRALI reactions prime neutrophils and cause neutrophil-mediated damage to human pulmonary microvascular endothelial cells in a two-event in vitro model. Blood 2007;109:1752–5.
24. Kelher MR, Masuno T, Moore EE, et al. Plasma from stored packed red blood cells and MHC class 1 antibodies causes acute lung injury in a 2-event in vivo rat model. Blood 2009;113:2079–87.
25. Seeger W, Schneider U, Kreusler B, et al. Reproduction of transfusion-related acute lung injury in an ex vivo lung model. Blood 1990;76:1438–44.
26. Sachs UJH, Hattar K, Weissmann N, et al. Antibody-induced neutrophil activation as a trigger for transfusion-related acute lung injury in an ex vivo rat lung model. Blood 2006;107:1217–19.
27. Lögdberg LE, Vikulina T, Zimring JC, Hillyer CD. Animal models of transfusion-related acute lung injury. Transfus Med Rev 2009;23:13–24.
28. Win N, Ranasinghe E, Lucas G. Transfusion-related acute lung injury: a 5-year look-back study. Transfus Med 2002;12:387–9.
29. Nicolle AL, Chapman CE, Carter V, Wallis JP. Transfusion-related acute lung injury caused by two donors with anti-human leucocyte antigen class II antibodies: a look-back investigation. Transfus Med 2004;14:225–30.
30. Davoren A, Curtis BR, Shulman IA, et al. TRALI due to granulocyte-agglutinating human neutrophil antigen-3a (5b) alloantibodies in donor plasma: a report of 2 fatalities. Transfusion 2003;43:641–5.
31. Muniz M, Sheldon S, Schuller RM, et al. Patient-specific transfusion-related acute lung injury. Vox Sang 2008;94:70–3.
32. Fadeyi EA, De Los Angeles Muniz M, Wayne AS, Klein HG, Leitman SF, Stroncek DF. The transfusion of neutrophil-specific antibodies causes leukopenia and a broad spectrum of pulmonary reactions. Transfusion 2007;47:545–50.
33. Kopko PM, Paglieroni TG, Popovsky MA, Muto KN, MacKenzie MR, Holland PV. TRALI: correlation of antigen-antibody and monocyte activation in donor-recipient pairs. Transfusion 2003;43:177–84.
Review of TRALI

172 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
D.C. Mair and T. Eastlund
34. Gosselin EJ, Wardwell K, Rigby WF, Guyre PM. Induction of MHC class II on human polymorphonuclear neutrophils by granulocyte/macrophage colony-stimulating factor, IFN-gamma, and IL-3. J Immunol 1993;151:1482–90.
35. Kao GS, Wood IG, Dorfman DM, Milford EL, Benjamin RJ. Investigations into the role of anti-HLA class II antibodies in TRALI. Transfusion 2003;43:185–91.
36. Bux J, Sachs UJH. Pathophysiology of TRALI. In: Kleinman SH, Popovsky MA, eds. TRALI: mechanisms, management and prevention. Bethesda, MD: AABB Press, 2008:43–69.
37. Nishimura M, Hashimoto S, Takanashi M, Okazaki H, Satake M, Nakajima K. Role of anti-human leucocyte antigen class II alloantibody and monocytes in development of transfusion-related acute lung injury. Transfus Med 2007;17:129–34.
38. Looney MR, Su X, Van Ziffle JA, Lowell CA, Matthay MA. Neutrophils and their Fcγ receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Invest 2006;116:1615–23.
39. Dykes A, Smallwood D, Kotsimbos T, Street A. Transfusion-related acute lung injury (TRALI) in a patient with a single lung transplant. Br J Haematol 2000;109:674–6.
40. Silliman CC, Paterson AJ, Dickey WO, et al. The association of biologically active lipids with the development of transfusion-related acute lung injury: a retrospective study. Transfusion 1997;37:719–26.
41. Vercellotti GM, Yin HQ, Gustafson KS, Nelson RD, Jacob HS. Platelet-activating factor primes neutrophil responses to agonists: role in promoting neutrophil-mediated endothelial damage. Blood 1988;71:1100–7.
42. Berkow RL, Wang D, Larrick JW, Dodson RW, Howard TH. Enhancement of neutrophil superoxide production by preincubation with recombinant human tumor necrosis factor. J Immunol 1987;139:3783–91.
43. Daniels RH, Finnen MJ, Hill ME, Lackie JM. Recombinant human monocyte IL-8 primes NADPH-oxidase and phospholipase A2 activation in human neutrophils. Immunology 1992;75:157–63.
44. Guthrie LA, McPhail LC, Henson PM, Johnston RB Jr. Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide. Evidence for increased activity of the superoxide-producing enzyme. J Exp Med 1984;160:1656–71.
45. Silliman CC, Bjornsen AJ, Wyman TH, et al. Plasma and lipids from stored platelets cause acute lung injury in an animal model. Transfusion 2003;43:633–40.
46. Silliman CC, Voelkel NF, Allard JD, et al. Plasma and lipids from stored packed red blood cells cause acute lung injury in an animal model. J Clin Invest 1998;101:1458–67.
47. Silliman CC, Thurman GW, Ambruso DR. Stored blood components contain agents that prime the neutrophil
NADPH oxidase through the platelet-activating-factor receptor. Vox Sang 1992;63:133–6.
48. Silliman CC, Johnson CA, Clay KL, Thurman GW, Ambruso DR. Compounds biologically similar to platelet activating factor are present in stored blood components. Lipids 1993;28:415–18.
49. Silliman CC, Clay KL, Thurman GW, Johnson CA, Ambruso DR. Partial characterization of lipids that develop during the routine storage of blood and prime the neutrophil NADPH oxidase. J Lab Clin Med 1994; 124:684–94.
50. Khan SY, Kelher MR, Heal JM, et al. Soluble CD40 ligand accumulates in stored blood components, primes neutrophils through CD40, and is a potential cofactor in the development of transfusion-related acute lung injury. Blood 2006;108:2455–62.
51. Silliman CC, Dickey WO, Paterson AJ, et al. Analysis of the priming activity of lipids generated during routine storage of platelet concentrates. Transfusion 1996;36:133–9.
52. Eder AF, Herron R, Strupp A, et al. Transfusion-related acute lung injury surveillance (2003–2005) and the potential impact of the selective use of plasma from male donors in the American Red Cross. Transfusion 2007;47:599–607.
53. Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest 2009;119:3450–61.
54. Engelfriet CP, Reesink HW, Brand A, et al. Transfusion-related acute lung injury (TRALI). Vox Sang 2001;81:269–83.
55. Swanson K, Dwyre DM, Krochmal J, Raife TJ. Transfusion-related acute lung injury (TRALI): current clinical and pathophysiologic considerations. Lung 2006;184:177–85.
56. Bux J, Sachs UJH. The pathogenesis of transfusion-related acute lung injury (TRALI). Br J Haematol 2007;136:788–99.
57. Chapman CE, Stainsby D, Jones H, et al. Ten years of hemovigilance reports of transfusion-related acute lung injury in the United Kingdom and the impact of preferential use of male donor plasma. Transfusion 2009;49:440–52.
58. Densmore TL, Goodnough LT, Ali S, Dynis M, Chaplin H. Prevalence of HLA sensitization in female apheresis donors. Transfusion 1999;39:103–6.
59. Triulzi DJ, Kleinman S, Kakaiya RM, et al. The effect of previous pregnancy and transfusion on HLA alloimmunization in blood donors: implications for a transfusion-related acute lung injury risk reduction strategy. Transfusion 2009;49:1825–35.
60. Hume HA. TRALI: moving toward prevention. Transfusion 2009;49:402–5.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 173
Notice to ReadersImmunohematology, Journal of Blood Group Serology and Education, is printed on acid-free paper.
Review of TRALI
61. Strong DM, Lipton KS. Transfusion-related acute lung injury. American Association of Blood Banks Bulletin#06-07, Nov 3, 2006.
62. Eder AF, Herron RM Jr, Strupp A, et al. Effective reduction of transfusion-related acute lung injury risk with male-predominant plasma strategy in the American Red Cross (2006–2008). Transfusion 2010 Apr 30 [Epub ahead of print].
63. Chapman CE, Williamson LM. UK hemovigilance and the preferential use of male plasma. In: Kleinman SH, Popovsky MA, eds. TRALI: mechanisms, management and prevention. Bethesda, MD: AABB Press, 2008:143–60.
64. Sinnott P, Bodger S, Gupta A, et al. Presence of HLA antibodies in single-donor-derived fresh frozen plasma compared with pooled, solvent detergent-treated plasma (Octaplas). Eur J Immunogenet. 2004; 31:271-4.
65. Sachs UJ, Kauschat D, Bein G. White blood cell-reactive antibodies are undetectable in solvent/detergent plasma. Transfusion. 2005;45:1628-31.
66. Triulzi DJ. Transfusion-related acute lung injury: current concepts for the clinician. Anesth Analg 2009; 108:770–6.
67. Mair DC, Hirschler N, Eastlund T. Blood donor and component management strategies to prevent
transfusion-related acute lung injury (TRALI). Crit Care Med 2006;34(Suppl):S137–43.
68. Greinacher A, Wesche J, Hammer E, et al. Characterization of the human neutrophil alloantigen-3a. Nat Med 2010;16:45–8.
69. Curtis BR, Cox NJ, Sullivan MJ, et al. The neutrophil alloantigen HNA-3a (5b) is located on choline transporter-like protein 2 and appears to be encoded by an R>Q154 amino acid substitution. Blood 2010 115:2073–6.
70. Silliman CC, Ambruso DR, Boshkov LK. Transfusion-related acute lung injury. Blood 2005;105:2266–73.
71. Goldman M, Webert KE, Arnold DM, Freedman J, Hannon J, Blajchman MA; TRALI Consensus Panel. Proceedings of a consensus conference: towards an understanding of TRALI. Transfus Med Rev 2005;19: 2–31.
David C. Mair, MD (corresponding author), American Red Cross-North Central Region, 100 South Robert Street, St. Paul, MN 55107, and Ted Eastlund, MD, University of New Mexico School of Medicine, Department of Pathology, Transfusion Medicine, Albuquerque NM.
Attention: State Blood Bank Meeting OrganizersIf you are planning a state meeting and would like copies of Immunohematology for distribution, please send request, 4 months in advance, to [email protected].
For information concerning Immunohematology, Journal of Blood Group Serology and Education, or the Immunohematology Methods and Procedures manual, contact us by e-mail at [email protected].

174 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
Original Report
Conventional tube testing was used for antibody screening and titration in D– pregnant women in our hospital until the recent introduction of the gel test. In this study we assessed the sensitivity of the gel test in our setup and tried to establish a correlation between these tests for determining antibody titer. We collected 652 blood samples from 223 antenatal D– women during a span of 1 year. The samples were tested separately by the conventional tube technique and the gel test for antibody detection and titration. The tube test detected 84 (12.8%) positive samples as compared with 93 (14.2%) by gel test, indicating the latter to be more sensitive (p < 0.01). The gel test picked up weakly reactive anti-D that the tube test missed. We did not use any enhancing media such as LISS in titration studies performed by either method in an effort to establish a correlation. However, much higher titers (one- to fivefold) were obtained by the gel test with no clear correlation with the corresponding tube values. When comparing the titer values to the finding of hydrops on ultrasound and Liley’s chart OD reading on amniocentesis, a value of less than 128 (i.e., 64) by gel test corresponded to normal results. Through this study, we thus conclude that the gel test is more sensitive for antibody detection, although a linear correlation could not be established for titers. Clinical correlation may point toward a critical titer of 64 for the gel test, but further studies need to be done to support this finding. Immunohematology 2010;26:174–77.
Key Words: alloantibodies, antibody screening, titration, gel test, D-negative women
Hemolytic disease of the fetus and newborn (HDFN) was first reported in 1609,1 although the discovery of the Rh system was made in 1939 and the implication of D antigen in its pathogenesis in 1940.2 However, to the present day, researchers and clinicians alike have worked to unravel its pathogenesis, effects, prevention, and treatment.
Anti-D is by far the most common cause of HDFN although its incidence has drastically fallen with antenatal D immunoglobulin use as prophylaxis. Other antibodies are also implicated, and their prevalence has been studied in the Western world but such data are lacking in the Indian population. The antibodies can be identified as well as semiquantitated from the mother’s serum. Titer determination of anti-D by tube test helps in the clinical decision to proceed with an invasive procedure such as amniocentesis.
The conventional tube test has stood the test of time in both antibody detection and titration. The gel test
introduced by Lapierre et al.3 in 1990 has gained popularity as a result of its standardized performance, technical ease, stable end point, and versatility of methodology. There have been many studies to show its superiority versus the tube test for antibody detection. The blood transfusion services in our country are gradually introducing gel technology for grouping, compatibility testing, and alloantibody detection owing to the many advantages. However, in some situations such as HDFN not only the specificity of alloantibody but also its titer has a direct impact on the fetus. Therefore, we evaluated the gel test and the conventional tube test for antibody screening and titration to determine if there is any correlation between the results obtained by using both technologies and to find out whether critical levels of alloantibody could be determined. We further studied obstetric management such as ultrasonography and intrauterine transfusion. We tried to correlate the titers with these clinical interventions.
Material and MethodsThe duration of study was from July 2005 to June 2006,
during which a total of 652 clotted samples were collected from 223 antenatal D– women. The study was conducted after approval from the Institute Ethics Committee. A written informed consent was taken from the patients for the study.
Sample CollectionA 3-mL clotted sample was collected, which was then
centrifuged and the serum separated. Serum was stored in a frozen state at –80°C in two aliquots each until further testing. The first sample was taken in the first trimester or at the time of the first visit. Subsequent samples were taken in every trimester and at 28 weeks before the administration of anti-D immunoglobulin. Immunized women were followed up every 3 to 4 weeks.
Antibody DetectionThe serum was thawed to room temperature before
being tested. Antibody detection was done in parallel by the conventional tube technique and the gel test. A commercially available three-cell screening panel (DiaScreen; DiaMed, Cressier sur Morat, Switzerland) was used. For tube testing,
Comparison of gel test and conventional tube test for antibody detection and titration in D-negative pregnant women: study from a tertiary-care hospital in North IndiaM.K. Thakur, N. Marwaha, P. Kumar, S.C. Saha, B. Thakral, R.R. Sharma, K. Saluja, H.K. Dhawan, and A. Jain

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 175
Comparison of gel test and tube test
a 3% suspension in normal saline was used, and for gel testing, a 0.8% suspension in LISS was used.
Tube IATOne drop of RBC suspension was added to two drops of
serum to be tested into labeled test tubes. The tubes were incubated in a water bath at 37°C for 60 minutes. After three washes with normal saline solution, polyspecific anti-human globulin was added to the RBC button and the tube was centrifuged at 3000 x g for 15 seconds in an appropriate centrifuge. The tubes were examined for agglutination. The reactions were graded and recorded as per the AABB Technical Manual.4
Gel TestWe dispensed 50 µL of RBCs into labeled gel cards
and added 25 µL of the serum to be tested. The cards were incubated for 15 minutes at 37°C in specially designed incubators. They were then centrifuged at 1050 rpm for 10 minutes. The reactions were graded and recorded. Antibody identification was done using commercially available panels (DiaMed) according to the manufacturer’s instructions.
Antibody TitrationTitration was performed on those samples that were
positive on antibody screening. Serial twofold dilutions were made in normal saline solution in clean test tubes. The dilutions were tested in parallel for both tests. In-house prepared R1R1 RBCs from a single donor were used for D antibodies, whereas RBCs with heterozygous expression were used for Lea and Leb antibodies. The RBCs were washed three times in normal saline solution and resuspended to a final concentration of 3% and 0.8% in normal saline solution for tube testing and gel testing, respectively. Critical titer was taken as 16 by the tube technique followed in our institute.
Tube TestThe method used was the same as that for antibody
detection.
Gel TestThe RBCs used were suspended in normal saline solution
to a final concentration of 0.8%, and the cards incubated at 37°C for 60 minutes. The remainder of the procedure was the same as for antibody detection. Titers were taken as the highest dilution that gave 1+ agglutination. The gel test had been modified from the manufacturer’s guidelines for titration. LISS was not used in the process, and the time of incubation was increased from 15 minutes to 60 minutes. We tested several samples in parallel with the standard method and found that deviating from the original method did not miss any antibodies.
The women who were positive for alloantibodies were investigated with ultrasonography for features of hydrops.
The findings were compared with the titer values obtained by both methods. All women with titer values of 16 or above by the conventional tube method were further evaluated by amniocentesis. The OD values were plotted on Liley’s chart5 and compared with the titer results by both methods.
Statistical analysis was performed using SPSS software version 13.0 (SPSS, Inc., Chicago, IL).
ResultsOn antibody screening, the tube technique detected 84
(12.8%) positive results, whereas the gel test detected 93 (14.2%) positive results individually. On comparison, 83 of the 84 positive by tube were also positive by gel, whereas 1 of the 84 was missed by gel. On the other hand 10 samples that were positive by gel had been missed by the tube technique. On identification the sample missed by gel was anti-Lea in specificity, and all samples missed by tube were anti-D in specificity. Both the tests showed a positive correlation in antibody detection that was significant with a Spearman’s correlation of +0.842 (p < 0.01). However, the gel technique proved to be more sensitive than the tube technique (p < 0.01). Table 1 shows the profile of antibodies detected.
Table 2 compares the titer values obtained by both methods for D antibodies. The Lewis antibody titer values are compared in Table 3. The titers on gel were generally higher than those obtained by the tube technique, and as the tube titers increased, the gel titers also increased. The titer increase with the gel test was higher when compared with the tube test. The values varied from onefold to fivefold (mean, 1.6-fold). The observed differences were onefold in 21 sera, twofold in 32, threefold in 18, fourfold in 3, and fivefold in 1.
Obstetric StudiesUltrasonography
The titers were compared with the fetal findings on ultrasonography, i.e., normal vs. hydropic. All the women with alloantibodies were investigated. The mean titer value for those who had normal ultrasonographic findings was 62.39 by tube technique with a standard deviation of 114.5 and titers ranging from 1 to 512. By the gel technique, the
Table 1. Comparison of antibodies detected by both methods
Sample tested Tube Gel
Total 652 652
Tests positive 84 93
Anti-D (all) 73 83
Anti–D (passive) 11 21
Anti-C* 2 2
Anti Lea 6 5
Anti Leb 5 5
*Anti-C was found in association with anti-D.
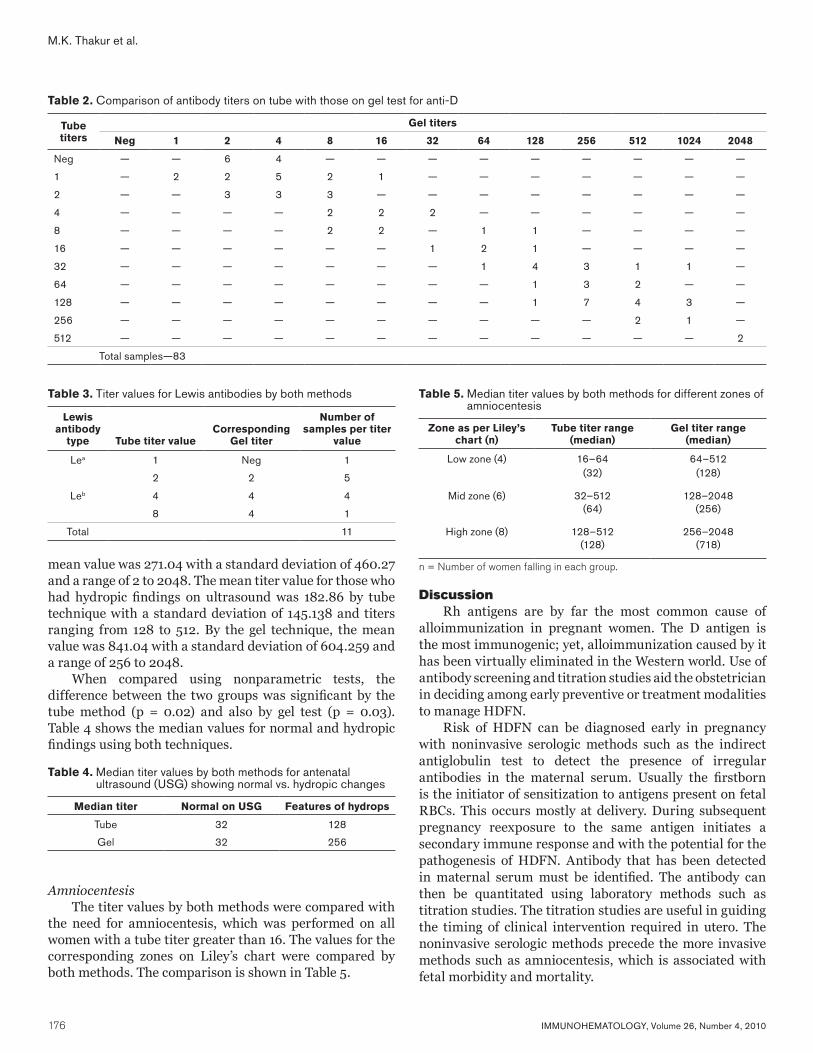
176 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
M.K. Thakur et al.
mean value was 271.04 with a standard deviation of 460.27 and a range of 2 to 2048. The mean titer value for those who had hydropic findings on ultrasound was 182.86 by tube technique with a standard deviation of 145.138 and titers ranging from 128 to 512. By the gel technique, the mean value was 841.04 with a standard deviation of 604.259 and a range of 256 to 2048.
When compared using nonparametric tests, the difference between the two groups was significant by the tube method (p = 0.02) and also by gel test (p = 0.03). Table 4 shows the median values for normal and hydropic findings using both techniques.
AmniocentesisThe titer values by both methods were compared with
the need for amniocentesis, which was performed on all women with a tube titer greater than 16. The values for the corresponding zones on Liley’s chart were compared by both methods. The comparison is shown in Table 5.
DiscussionRh antigens are by far the most common cause of
alloimmunization in pregnant women. The D antigen is the most immunogenic; yet, alloimmunization caused by it has been virtually eliminated in the Western world. Use of antibody screening and titration studies aid the obstetrician in deciding among early preventive or treatment modalities to manage HDFN.
Risk of HDFN can be diagnosed early in pregnancy with noninvasive serologic methods such as the indirect antiglobulin test to detect the presence of irregular antibodies in the maternal serum. Usually the firstborn is the initiator of sensitization to antigens present on fetal RBCs. This occurs mostly at delivery. During subsequent pregnancy reexposure to the same antigen initiates a secondary immune response and with the potential for the pathogenesis of HDFN. Antibody that has been detected in maternal serum must be identified. The antibody can then be quantitated using laboratory methods such as titration studies. The titration studies are useful in guiding the timing of clinical intervention required in utero. The noninvasive serologic methods precede the more invasive methods such as amniocentesis, which is associated with fetal morbidity and mortality.
Table 2. Comparison of antibody titers on tube with those on gel test for anti-D
Tube titers
Gel titers
Neg 1 2 4 8 16 32 64 128 256 512 1024 2048
Neg — — 6 4 — — — — — — — — —
1 — 2 2 5 2 1 — — — — — — —
2 — — 3 3 3 — — — — — — — —
4 — — — — 2 2 2 — — — — — —
8 — — — — 2 2 — 1 1 — — — —
16 — — — — — — 1 2 1 — — — —
32 — — — — — — — 1 4 3 1 1 —
64 — — — — — — — — 1 3 2 — —
128 — — — — — — — — 1 7 4 3 —
256 — — — — — — — — — — 2 1 —
512 — — — — — — — — — — — — 2
Total samples—83
Table 3. Titer values for Lewis antibodies by both methods
Lewis antibody
type Tube titer valueCorresponding
Gel titer
Number of samples per titer
value
Lea 1 Neg 1
2 2 5
Leb 4 4 4
8 4 1
Total 11
Table 4. Median titer values by both methods for antenatal ultrasound (USG) showing normal vs. hydropic changes
Median titer Normal on USG Features of hydrops
Tube 32 128
Gel 32 256
Table 5. Median titer values by both methods for different zones of amniocentesis
Zone as per Liley’s chart (n)
Tube titer range (median)
Gel titer range (median)
Low zone (4) 16–64 64–512(32) (128)
Mid zone (6) 32–512 128–2048(64) (256)
High zone (8) 128–512 256–2048(128) (718)
n = Number of women falling in each group.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 177
Comparison of gel test and tube test
The higher sensitivity and versatility of the gel test compared with the tube test have been reported in various studies.6,7 A recent study comparing the conventional tube test with the gel technique for crossmatch also showed the latter to be more sensitive.8 Other positive points of the gel test include smaller volume of sample required, elimination of washing steps, stable results, and easy readability. The tube test in our study missed 10 examples of anti-D that the gel test detected. However, the anti-D in these samples were all attributable to antenatal anti-D immunoprophylaxis and they became undetectable after 4 to 6 weeks. Thus, the tube test did not miss any of the significant antibodies. Data are also presented that indicate the propensity of the gel test to miss clinically insignificant antibodies like Lea.7 The gel test in our study missed one sample of anti-Lea. Nine samples of anti-Lea and anti-Leb were, however, detected by both methods. The titration of antibodies by both methods showed variable results. Similar data have been published by Novaretti et al.9 in which they tested gel and tube titers for anti-D. They found the titers to be threefold to eightfold higher. Our study showed the titers to be one- to fivefold higher. The gel titers tended to be higher when compared with the tube titers. However, no correlation between the two methods could be found.
The recommended titration study by Judd10 is a saline antiglobulin procedure with 60-minute incubation at 37°C. In our study, we did not use LISS for gel titration, which is a known enhancing medium, in an attempt to establish a correlation, if any exists, between the conventional tube technique and the gel technique. Judd also stated that until substantial data are available that show correlation between gel microcolumn assay and saline tube antiglobulin titers IgG gel column technology should not be used for prenatal antibody titration. The critical tube titers of 16 corresponded to gel titers ranging from 32 to 128. Hence, it is difficult to arrive at a definitive conclusion of critical gel titers when a direct correlation is attempted. When the titers were compared with ultrasonography for hydrops and amniocentesis OD values as per Liley’s chart, a gel value of less than 128 (i.e., 64) corresponded with normal ultrasonography and low zone OD values. However, this value should be interpreted with caution until more studies support it.
Based on the above observations, we conclude that the gel test is a better method than the conventional tube test for antibody detection because of its higher sensitivity and technical safety. Titration by gel, however, should not be considered for antenatal HDFN management as gel titers do not show linear correlation with tube titers, which predict fetal outcome in RhD sensitized women. Developing countries work under resource constraints, and such studies would optimize cost-effective use of this technology.
References 1. Bowman J. Thirty-five years of Rh prophylaxis.
Transfusion 2003;43:1661–6. 2. Landsteiner K, Wiener AS. An agglutinable factor in
human blood recognized by immune sera for rhesus blood. Proc Soc Exp Biol Med 1940;43:223–224.
3. Lapierre Y, Rigal D, Adam J, et al. The gel test: a new way to detect red cell antigen-antibody reactions. Transfusion 1990;30:109–13.
4. Brecher ME. Antibody detection, identification, and compatibility testing. In: Roback JD, Combs MR, Grossman BJ, Hillyer CD, eds. Technical manual. 16th ed. Maryland: American Association of Blood Banks, 2008: 899-917.
5. Liley AW. Liquor amnil analysis in the management of the pregnancy complicated by rhesus sensitization. Am J Obstet Gynecol 1961;82:1359–70.
6. Judd WJ, Steiner EA, Knafl PC. The gel test: sensitivity and specificity for unexpected antibodies to blood group antigens. Immunohematology 1997;13:132–5.
7. Delaflor-Weiss E, Chizhevsky V. Implementation of gel testing for antibody screening and identification in a community hospital, 3-year experience. Lab Med 2005;36:489–92.
8. Lange J, Selleng K, Heddle NM, Traore A, Greinacher A. Coombs’ crossmatch after negative antibody screening—a retrospective observational study comparing the tube test and the microcolumn technology. Vox Sang 2010;98:e269–75.
9. Novaretti MC, Jens E, Pagliarini T, Bonifácio SL, Dorlhiac-Llacer PE, Chamone DA. Comparison of conventional tube test with DiaMed gel microcolumn assay for anti-D titration. Clin Lab Haematol 2003;25: 311–15.
10. Judd WJ; Scientific Section Coordinating Committee of the AABB. Practice guidelines for prenatal and perinatal immunohematology, revisited. Transfusion 2001;41;1445–52.
Manish K. Thakur, MD (corresponding author), Senior Resident, and Neelam Marwaha, MD, Professor and Head, Department of Transfusion Medicine, Praveen Kumar, MD, DM(Neonatology), Additional Professor, Department of Pediatrics, Subhash C. Saha, MD, Associate Professor, Department of Obstetrics and Gynecology, and Beenu Thakral, MD, Assistant Professor, Ratti Ram Sharma, MD, Assistant Professor, Karan Saluja, MD, Assistant Professor, Hari Krishan Dhawan, MD, Senior Resident, Department of Transfusion Medicine, and Ashish Jain, MD, Senior Resident, Department of Transfusion Medicine, Post Graduate Institute of Medical Education and Research, Sector 12, Chandigarh, India 160012.

178 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
Review
HistoryRh is the most well-recognized blood group system
after ABO because of the immunogenicity of the principal antigen, D. Discovered more than 70 years ago, the D antigen is well known as the target in severe and potentially fatal hemolytic disease of the fetus and newborn (HDFN).1 Today, HDFN caused by anti-D is much less frequently observed in developed countries owing to prevention by administration of Rh immunoglobulin (RhIgG; anti-D IgG) after delivery of a D+ infant.2,3 Children of women of European extraction are more likely affected as D– is most common in Caucasians (15–17%), less common in African Blacks (3–5%), and uncommon or rare in Asians (<0.1%).4 Thus, the D antigen status is not routinely tested for in some parts of Asia, principally China, where D– is considered a rare blood type. HDFN caused by anti-D is still seen where women have limited access to prenatal care and in countries where RhIG administration is not standard practice. Anti-D during pregnancy is also more often seen if RhIG is not administered during the third trimester of pregnancy in addition to after delivery. The policy of giving antepartum RhIG to all D– women has not been adopted in all countries.
The Rh system is a complex RBC antigen system that includes more than 50 different antigenic specificities (Table 1). “Rh-positive” and “Rh-negative” refer to the presence or absence of the D antigen. The five principal Rh antigens—D, C, c, E, and e—are responsible for the majority of clinically significant antibodies. Some of the others represent compound specificities, i.e., ce or f is an epitope on the Rhce protein that is not present on RBCs with RhcE or RhCe. Many of the antigens are found primarily in a population group or specific ethnic group; for example CX has the highest prevalence in Finns, and V and VS are found primarily in African Black ethnic backgrounds (summarized in Reid and Lomas-Francis5).
Our understanding of the Rh system has been greatly advanced since the genes were cloned in the early 1990s. The sequence of RHCE was first reported in 1990,6,7 and RHD was subsequently sequenced in 1992.6,8 The different RHCE alleles responsible for the C or c and E or e antigens were clarified in 1994.9 In the last decade, molecular genotyping has revealed that the genetic diversity of the RH locus greatly exceeds estimates predicted by serology. More than 170 RHD and more than 70 different RHCE alleles have been described, and new alleles are still being discovered. A directory of RHD alleles is maintained on the RhesusBase website, and RHCE alleles are found on the National Center for Biotechnology Information (NCBI) human blood group mutation Web site.10 In an effort to standardize terminology,
the International Society of Blood Transfusion (ISBT) Working Party on Red Cell Immunogenetics and Blood Group Terminology maintains a list of alleles that encode blood group antigens (see Web Resources).
Terminology and NomenclatureThe term “Rh” for Rhesus resulted from work by
Landsteiner and Wiener, who found that antiserum produced by immunizing guinea pigs and rabbits with RBCs of the Rhesus macaque agglutinated 85 percent of human RBCs. Initially, they believed that a common factor “Rh” had been identified in animals and humans.11 The antibody actually detected the LW antigen, which is present on D+ RBCs in greater amounts than on D– RBCs. Although misnomers, the terms Rh factor and anti-Rh continued in common usage to describe the human D antigen and the D antibody. Although the use of “Rhesus” is now discouraged, it has been pointed out (WA Flegel, personal communication) that “Rh” can be difficult to translate into other languages.
Over the years, four Rh system nomenclatures have been introduced (Table 1). Terminology established by Fisher-Race in England was based on the premise that three closely linked genes, C/c, E/e, and D, were responsible for the antigens, whereas the Wiener nomenclature (Rh-Hr) was based on the belief that a single gene encoded several blood group factors. The Rh system is encoded by two genes, RHD and RHCE, as predicted by Tippett.12 Rosenfeld gave each Rh antigen a number based on the order of its discovery or assignment to the Rh system. The ISBT Working Party on Terminology for Red Cell Surface Antigens adopted a six-digit number for each RBC antigen.13 The first three numbers represent the system (the number 004 was assigned to the Rh system), and the remaining three digits refer to the antigenic specificity (Table 1).
Current terminology distinguishes the genes and the proteins from the antigens. Capital letters, with or without italics, are used when referring to the RH genes, RHD and RHCE. Alleles of RHCE are designated after an asterisk, i.e. RHCE*ce, RHCE*Ce, RHCE*cE, RHCE*CE, according to which antigens they encode. Alleles of RHD are designated RHD*DVI, RHD*DIIIa, RHD*weak D type 2, and so on, according to the partial or weak D encoded, and the alleles have also been given numbers by the ISBT Working Party on Red Cell Immunogenetics and Blood Group Terminology (see Table 2 for examples and Web Resources). Lastly, the Rh proteins are indicated without italics and with the “h” in lowercase, as RhD or RhCE, or according to the specific antigens they carry, Rhce, RhCe, RhcE, or RhCE.
The Rh and RhAG blood group systemsS.T. Chou and C.M. Westhoff
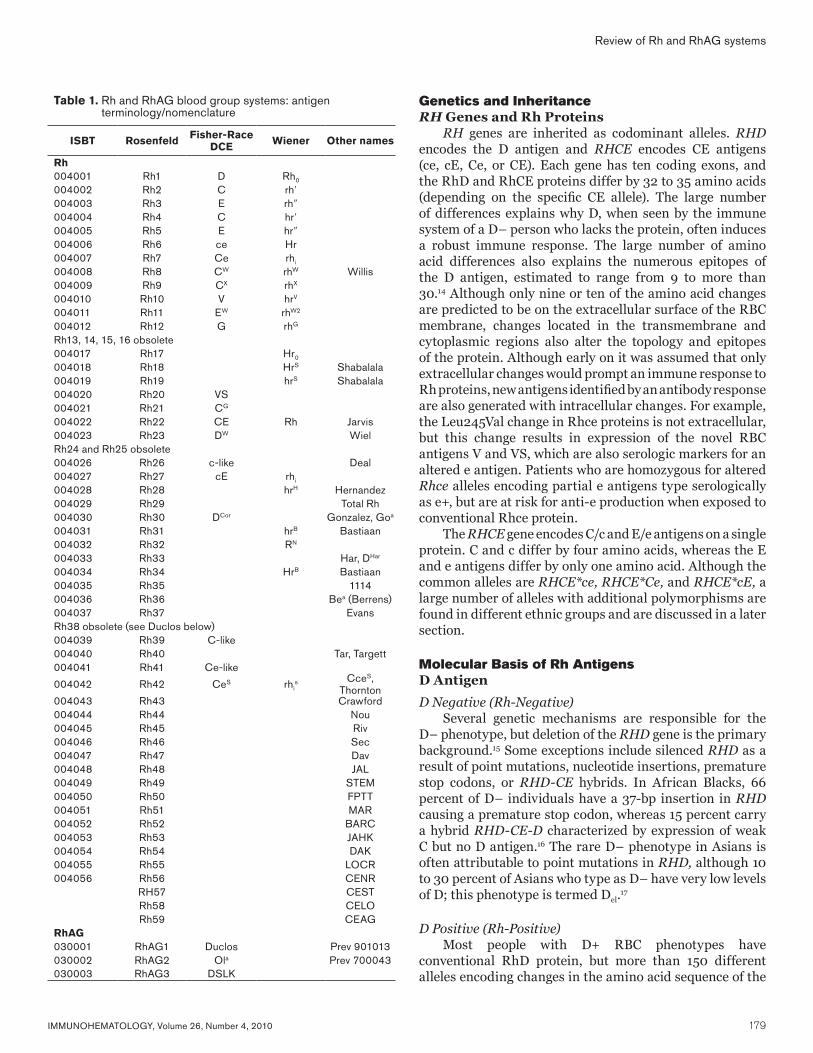
IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 179
Review of Rh and RhAG systems
Genetics and InheritanceRH Genes and Rh Proteins
RH genes are inherited as codominant alleles. RHD encodes the D antigen and RHCE encodes CE antigens (ce, cE, Ce, or CE). Each gene has ten coding exons, and the RhD and RhCE proteins differ by 32 to 35 amino acids (depending on the specific CE allele). The large number of differences explains why D, when seen by the immune system of a D– person who lacks the protein, often induces a robust immune response. The large number of amino acid differences also explains the numerous epitopes of the D antigen, estimated to range from 9 to more than 30.14 Although only nine or ten of the amino acid changes are predicted to be on the extracellular surface of the RBC membrane, changes located in the transmembrane and cytoplasmic regions also alter the topology and epitopes of the protein. Although early on it was assumed that only extracellular changes would prompt an immune response to Rh proteins, new antigens identified by an antibody response are also generated with intracellular changes. For example, the Leu245Val change in Rhce proteins is not extracellular, but this change results in expression of the novel RBC antigens V and VS, which are also serologic markers for an altered e antigen. Patients who are homozygous for altered Rhce alleles encoding partial e antigens type serologically as e+, but are at risk for anti-e production when exposed to conventional Rhce protein.
The RHCE gene encodes C/c and E/e antigens on a single protein. C and c differ by four amino acids, whereas the E and e antigens differ by only one amino acid. Although the common alleles are RHCE*ce, RHCE*Ce, and RHCE*cE, a large number of alleles with additional polymorphisms are found in different ethnic groups and are discussed in a later section.
Molecular Basis of Rh AntigensD Antigen
D Negative (Rh-Negative)Several genetic mechanisms are responsible for the
D– phenotype, but deletion of the RHD gene is the primary background.15 Some exceptions include silenced RHD as a result of point mutations, nucleotide insertions, premature stop codons, or RHD-CE hybrids. In African Blacks, 66 percent of D– individuals have a 37-bp insertion in RHD causing a premature stop codon, whereas 15 percent carry a hybrid RHD-CE-D characterized by expression of weak C but no D antigen.16 The rare D– phenotype in Asians is often attributable to point mutations in RHD, although 10 to 30 percent of Asians who type as D– have very low levels of D; this phenotype is termed Del.17
D Positive (Rh-Positive)Most people with D+ RBC phenotypes have
conventional RhD protein, but more than 150 different alleles encoding changes in the amino acid sequence of the
Table 1. Rh and RhAG blood group systems: antigen terminology/nomenclature
ISBT Rosenfeld Fisher-Race DCE Wiener Other names
Rh004001 Rh1 D Rh0
004002 Rh2 C rh′004003 Rh3 E rh″004004 Rh4 C hr′004005 Rh5 E hr″004006 Rh6 ce Hr004007 Rh7 Ce rhi
004008 Rh8 CW rhW Willis004009 Rh9 CX rhX
004010 Rh10 V hrV
004011 Rh11 EW rhW2
004012 Rh12 G rhG
Rh13, 14, 15, 16 obsolete004017 Rh17 Hr0
004018 Rh18 HrS Shabalala004019 Rh19 hrS Shabalala004020 Rh20 VS004021 Rh21 CG
004022 Rh22 CE Rh Jarvis004023 Rh23 DW WielRh24 and Rh25 obsolete004026 Rh26 c-like Deal004027 Rh27 cE rhi
004028 Rh28 hrH Hernandez004029 Rh29 Total Rh004030 Rh30 DCor Gonzalez, Goa
004031 Rh31 hrB Bastiaan004032 Rh32 RN
004033 Rh33 Har, DHar
004034 Rh34 HrB Bastiaan004035 Rh35 1114004036 Rh36 Bea (Berrens)004037 Rh37 EvansRh38 obsolete (see Duclos below)004039 Rh39 C-like004040 Rh40 Tar, Targett004041 Rh41 Ce-like
004042 Rh42 CeS rhis CceS,
Thornton004043 Rh43 Crawford004044 Rh44 Nou004045 Rh45 Riv004046 Rh46 Sec004047 Rh47 Dav004048 Rh48 JAL004049 Rh49 STEM004050 Rh50 FPTT004051 Rh51 MAR004052 Rh52 BARC004053 Rh53 JAHK004054 Rh54 DAK004055 Rh55 LOCR004056 Rh56 CENR
RH57 CESTRh58 CELORh59 CEAG
RhAG030001 RhAG1 Duclos Prev 901013030002 RhAG2 Ola Prev 700043030003 RhAG3 DSLK
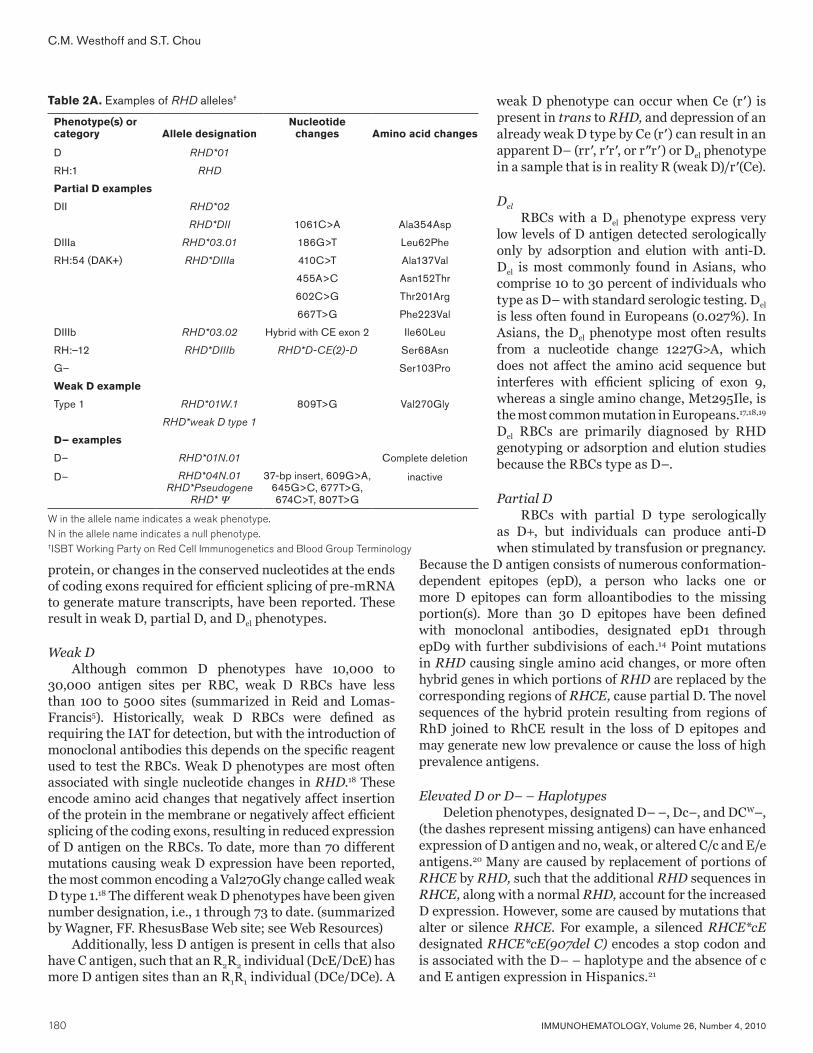
180 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
C.M. Westhoff and S.T. Chou
protein, or changes in the conserved nucleotides at the ends of coding exons required for efficient splicing of pre-mRNA to generate mature transcripts, have been reported. These result in weak D, partial D, and Del phenotypes.
Weak DAlthough common D phenotypes have 10,000 to
30,000 antigen sites per RBC, weak D RBCs have less than 100 to 5000 sites (summarized in Reid and Lomas-Francis5). Historically, weak D RBCs were defined as requiring the IAT for detection, but with the introduction of monoclonal antibodies this depends on the specific reagent used to test the RBCs. Weak D phenotypes are most often associated with single nucleotide changes in RHD.18 These encode amino acid changes that negatively affect insertion of the protein in the membrane or negatively affect efficient splicing of the coding exons, resulting in reduced expression of D antigen on the RBCs. To date, more than 70 different mutations causing weak D expression have been reported, the most common encoding a Val270Gly change called weak D type 1.18 The different weak D phenotypes have been given number designation, i.e., 1 through 73 to date. (summarized by Wagner, FF. RhesusBase Web site; see Web Resources)
Additionally, less D antigen is present in cells that also have C antigen, such that an R2R2 individual (DcE/DcE) has more D antigen sites than an R1R1 individual (DCe/DCe). A
weak D phenotype can occur when Ce (r′) is present in trans to RHD, and depression of an already weak D type by Ce (r′) can result in an apparent D– (rr′, r′r′, or r″r′) or Del phenotype in a sample that is in reality R (weak D)/r′(Ce).
Del
RBCs with a Del phenotype express very low levels of D antigen detected serologically only by adsorption and elution with anti-D. Del is most commonly found in Asians, who comprise 10 to 30 percent of individuals who type as D– with standard serologic testing. Del is less often found in Europeans (0.027%). In Asians, the Del phenotype most often results from a nucleotide change 1227G>A, which does not affect the amino acid sequence but interferes with efficient splicing of exon 9, whereas a single amino change, Met295Ile, is the most common mutation in Europeans.17,18,19 Del RBCs are primarily diagnosed by RHD genotyping or adsorption and elution studies because the RBCs type as D–.
Partial DRBCs with partial D type serologically
as D+, but individuals can produce anti-D when stimulated by transfusion or pregnancy.
Because the D antigen consists of numerous conformation-dependent epitopes (epD), a person who lacks one or more D epitopes can form alloantibodies to the missing portion(s). More than 30 D epitopes have been defined with monoclonal antibodies, designated epD1 through epD9 with further subdivisions of each.14 Point mutations in RHD causing single amino acid changes, or more often hybrid genes in which portions of RHD are replaced by the corresponding regions of RHCE, cause partial D. The novel sequences of the hybrid protein resulting from regions of RhD joined to RhCE result in the loss of D epitopes and may generate new low prevalence or cause the loss of high prevalence antigens.
Elevated D or D– – HaplotypesDeletion phenotypes, designated D– –, Dc–, and DCW–,
(the dashes represent missing antigens) can have enhanced expression of D antigen and no, weak, or altered C/c and E/e antigens.20 Many are caused by replacement of portions of RHCE by RHD, such that the additional RHD sequences in RHCE, along with a normal RHD, account for the increased D expression. However, some are caused by mutations that alter or silence RHCE. For example, a silenced RHCE*cE designated RHCE*cE(907del C) encodes a stop codon and is associated with the D– – haplotype and the absence of c and E antigen expression in Hispanics.21
Table 2A. Examples of RHD alleles†
Phenotype(s) or category Allele designation
Nucleotide changes Amino acid changes
D RHD*01
RH:1 RHD
Partial D examples
DII RHD*02
RHD*DII 1061C>A Ala354Asp
DIIIa RHD*03.01 186G>T Leu62Phe
RH:54 (DAK+) RHD*DIIIa 410C>T Ala137Val
455A>C Asn152Thr
602C>G Thr201Arg
667T>G Phe223Val
DIIIb RHD*03.02 Hybrid with CE exon 2 Ile60Leu
RH:–12 RHD*DIIIb RHD*D-CE(2)-D Ser68Asn
G– Ser103Pro
Weak D example
Type 1 RHD*01W.1 809T>G Val270Gly
RHD*weak D type 1
D– examples
D– RHD*01N.01 Complete deletion
D– RHD*04N.01RHD*Pseudogene
RHD* Ψ
37-bp insert, 609G>A, 645G>C, 677T>G, 674C>T, 807T>G
inactive
W in the allele name indicates a weak phenotype.N in the allele name indicates a null phenotype.†ISBT Working Party on Red Cell Immunogenetics and Blood Group Terminology
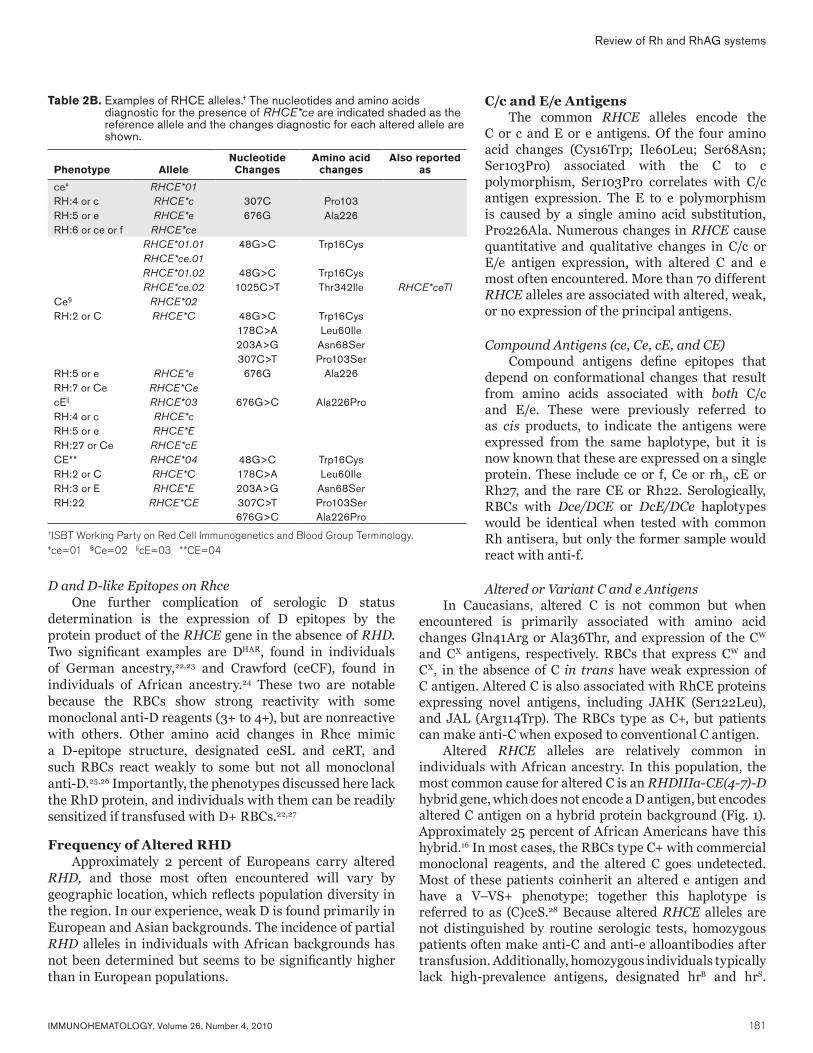
IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 181
D and D-like Epitopes on RhceOne further complication of serologic D status
determination is the expression of D epitopes by the protein product of the RHCE gene in the absence of RHD. Two significant examples are DHAR, found in individuals of German ancestry,22,23 and Crawford (ceCF), found in individuals of African ancestry.24 These two are notable because the RBCs show strong reactivity with some monoclonal anti-D reagents (3+ to 4+), but are nonreactive with others. Other amino acid changes in Rhce mimic a D-epitope structure, designated ceSL and ceRT, and such RBCs react weakly to some but not all monoclonal anti-D.25,26 Importantly, the phenotypes discussed here lack the RhD protein, and individuals with them can be readily sensitized if transfused with D+ RBCs.22,27
Frequency of Altered RHDApproximately 2 percent of Europeans carry altered
RHD, and those most often encountered will vary by geographic location, which reflects population diversity in the region. In our experience, weak D is found primarily in European and Asian backgrounds. The incidence of partial RHD alleles in individuals with African backgrounds has not been determined but seems to be significantly higher than in European populations.
C/c and E/e AntigensThe common RHCE alleles encode the
C or c and E or e antigens. Of the four amino acid changes (Cys16Trp; Ile60Leu; Ser68Asn; Ser103Pro) associated with the C to c polymorphism, Ser103Pro correlates with C/c antigen expression. The E to e polymorphism is caused by a single amino acid substitution, Pro226Ala. Numerous changes in RHCE cause quantitative and qualitative changes in C/c or E/e antigen expression, with altered C and e most often encountered. More than 70 different RHCE alleles are associated with altered, weak, or no expression of the principal antigens.
Compound Antigens (ce, Ce, cE, and CE)Compound antigens define epitopes that
depend on conformational changes that result from amino acids associated with both C/c and E/e. These were previously referred to as cis products, to indicate the antigens were expressed from the same haplotype, but it is now known that these are expressed on a single protein. These include ce or f, Ce or rhi, cE or Rh27, and the rare CE or Rh22. Serologically, RBCs with Dce/DCE or DcE/DCe haplotypes would be identical when tested with common Rh antisera, but only the former sample would react with anti-f.
Altered or Variant C and e AntigensIn Caucasians, altered C is not common but when
encountered is primarily associated with amino acid changes Gln41Arg or Ala36Thr, and expression of the CW and CX antigens, respectively. RBCs that express CW and CX, in the absence of C in trans have weak expression of C antigen. Altered C is also associated with RhCE proteins expressing novel antigens, including JAHK (Ser122Leu), and JAL (Arg114Trp). The RBCs type as C+, but patients can make anti-C when exposed to conventional C antigen.
Altered RHCE alleles are relatively common in individuals with African ancestry. In this population, the most common cause for altered C is an RHDIIIa-CE(4-7)-D hybrid gene, which does not encode a D antigen, but encodes altered C antigen on a hybrid protein background (Fig. 1). Approximately 25 percent of African Americans have this hybrid.16 In most cases, the RBCs type C+ with commercial monoclonal reagents, and the altered C goes undetected. Most of these patients coinherit an altered e antigen and have a V–VS+ phenotype; together this haplotype is referred to as (C)ceS.28 Because altered RHCE alleles are not distinguished by routine serologic tests, homozygous patients often make anti-C and anti-e alloantibodies after transfusion. Additionally, homozygous individuals typically lack high-prevalence antigens, designated hrB and hrS.
Review of Rh and RhAG systems
Table 2B. Examples of RHCE alleles.† The nucleotides and amino acids diagnostic for the presence of RHCE*ce are indicated shaded as the reference allele and the changes diagnostic for each altered allele are shown.
Phenotype AlleleNucleotide Changes
Amino acid changes
Also reported as
ce‡ RHCE*01RH:4 or c RHCE*c 307C Pro103RH:5 or e RHCE*e 676G Ala226RH:6 or ce or f RHCE*ce
RHCE*01.01 48G>C Trp16CysRHCE*ce.01RHCE*01.02 48G>C Trp16CysRHCE*ce.02 1025C>T Thr342Ile RHCE*ceTI
Ce§ RHCE*02RH:2 or C RHCE*C 48G>C Trp16Cys
178C>A Leu60Ile203A>G Asn68Ser307C>T Pro103Ser
RH:5 or e RHCE*e 676G Ala226RH:7 or Ce RHCE*CecE|| RHCE*03 676G>C Ala226ProRH:4 or c RHCE*cRH:5 or e RHCE*ERH:27 or Ce RHCE*cECE** RHCE*04 48G>C Trp16CysRH:2 or C RHCE*C 178C>A Leu60IleRH:3 or E RHCE*E 203A>G Asn68SerRH:22 RHCE*CE 307C>T Pro103Ser
676G>C Ala226Pro†ISBT Working Party on Red Cell Immunogenetics and Blood Group Terminology.‡ce=01 §Ce=02 ||cE=03 **CE=04

182 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
C.M. Westhoff and S.T. Chou
Anti-hrB and anti-hrS can be clinically significant,29,30 and finding compatible blood can be very difficult because of differing RH gene backgrounds.31,32 As an additional complication, altered RHCE*ce are often inherited with partial RHD (DIII, DAU, DAR, etc.). The inheritance of partial D with altered RHCE*ce may lead to production of antibodies sometimes identified as anti-HrB (RH34) and anti-Hr (RH18).
G AntigenThe G antigen is found on RBCs expressing C or D
with a 103Ser residue present on RhD, RhCe, or RhCE. Serologically, anti-G reacts as though it were anti-D plus anti-C. Anti-G explains the D– person transfused with D–C+ blood, or the D– woman who delivered a D–C+ child, who subsequently appears to have made anti-D. To distinguish anti-D, -C, and -G, adsorption and elution studies can be performed.33 Their results determine the need for RhIG prophylaxis for obstetric patients with anti-G to prevent additional immunization and formation of anti-D.
Antibodies in the Rh SystemMost Rh antigens are immunogenic and have the
potential to cause clinically significant HDFN or transfusion reactions. Rh alloimmunization occurs by exposure to foreign RBCs during pregnancy or after transfusion and may persist for many years. Even if serum antibody drops below detectable levels, subsequent exposure to the antigen can produce a rapid secondary immune response. Outside of the ABO system, the D antigen is the most immunogenic antigen, followed by c and E. Anti-c can cause severe HDFN, whereas anti-C, -E, and -e are associated with mild HDFN. Because Rh alloimmunization is IgG-mediated, transfusion reactions are primarily extravascular. The patient may have fever, jaundice, icteric sclera, dark urine, or a drop in hemoglobin level. Antigen-negative RBCs should be provided to any patient with a history of Rh antibody sensitization.
Rh antibodies are often found together. It is not unusual for a patient with a single Rh antibody to become alloimmunized to additional Rh antigens if further exposed. For example, a DCe/DCe (R1R1) patient with anti-E has usually been exposed to the c antigen as well. Anti-c may be present at the time anti-E is identified, but may be weak and
undetectable. Therefore, to prevent delayed transfusion reactions, some advocate avoiding c+ blood for patients alloimmunized to the E antigen. In contrast, pursuing anti-E in serum containing anti-c is not necessary as the patient will likely have been exposed to c without being exposed to E. Moreover, the vast majority of c– donor blood will be negative for E.
Rh Antibodies in Patients with Sickle Cell DiseaseSome sickle cell transfusion programs perform Rh typing
to match patients and donors for D, C, and E (in addition to K) to prevent alloimmunization.34,35 The prevalence of RH alleles that encode altered or variant D, C, and e antigens in African ethnic groups often underlies the production of complex Rh alloantibody specificities in chronically transfused patients with sickle cell disease (SCD). Many altered Rh protein epitopes cannot be distinguished serologically, and are only identified by RH genotyping once a patient develops apparent autoantibodies to Rh antigens. Some common examples include the partial C antigen encoded by the r′S allele, i.e. (C)ceS, and partial DIIIa; the RBCs react 3+–4+ with monoclonal reagents and are not detected as altered. Anti-hrS, -hrB, -HrB, and -Hr can be difficult to identify and can cause clinically significant hemolytic transfusion reactions including transfusion fatalities.30
Other Rh AntibodiesIn addition to the SCD population, D– – and Rhnull
individuals also develop alloantibodies to high-prevalence Rh antigens with Rh17 and Rh29 specificity, respectively. Lastly, autoantibodies to high-prevalence Rh antigens often occur in the sera of patients with warm autoimmune hemolytic anemia and in some cases of drug-induced autoimmune hemolytic anemia.36 The biologic explanation for this apparent specificity or cross-reactivity remains to be determined.
Clinical ConsiderationsDetermination of D status
The AABB Standards for Blood Banks and Transfusion Services37 requires donor blood to be tested for weak D expression and to be labeled as Rh-positive if the test is positive. Hospital transfusion services are also required to confirm the D-negative status before the unit is released for transfusion to avoid transfusion of D+ RBCs to D– recipients. It is recognized that some very weak D antigens are not detected, and standard typing procedures (including IAT) do not detect Del RBCs. Isolated cases have been reported in which very weak D and Del donor units have caused anti-D alloimmunization in D– recipients.38–41
When determining the D type of a patient, detecting weak D expression is not necessary unless testing RBCs of an infant of a D– mother at risk for D immunization. DVI is the most common partial D found in Caucasians, and anti-D produced by women with partial DVI has resulted in
Figure 1: Representation of the RH gene associated with altered C antigen. The RHD and RHCE genes are shown as 10 boxes representing the coding exons. Amino acid changes are indicated by their single letter designation and the position of the amino acid in the protein is given.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 183
Review of Rh and RhAG systems
fatal hemolytic disease.42 Therefore, current FDA-licensed monoclonal IgM reagents are selected to be nonreactive with partial DVI RBCs in direct tests. Performing only a direct test and eliminating the indirect test for female children and women of child-bearing age avoids the risk of sensitization by classifying DVI as D– for transfusion and RhIG prophylaxis.
Patients who have RBCs expressing the common type 1, 2, or 3 weak D phenotype usually do not make anti-D and can safely receive D+ blood. Patients with the rare weak D types 11 and 15 have become alloimmunized, suggesting they have altered D epitopes.38 The risk for anti-D production in other uncommon weak D types is unknown. In contrast to weak D, patients with partial D RBCs form alloanti-D directed at the missing D epitopes and should receive D– blood and RhIG when indicated. Unfortunately, serologic reagents frequently do not distinguish partial D status. Many partial D RBCs, including DIIIa, the most common partial D in African Americans, type strongly D-positive in direct tests and are only recognized after producing anti-D.
Sickle Cell Disease and Altered RH AllelesRBC alloimmunization remains a serious complication
of transfusion for patients with SCD, with more than 25 to 30 percent of chronically transfused SCD patients developing RBC antibodies. Altered D, C, and e antigens often underlie the complex RH alloantibodies SCD patients exhibit after transfusion. Although randomized controlled trials have not been performed, extended RBC antigen-matching (including D, C, E, and K) has been shown in numerous single-institutional and prospective multicenter experiences to significantly reduce the incidence of alloanti-body production in SCD.43–45 Therefore, many centers determine the pretransfusion RBC serologic phenotype and provide RBCs that are antigen-matched for D, C, E, and K. Because the high alloimmunization rates are also believed to be caused by antigenic disparity between African Americans and Caucasians, some programs actively recruit African American donors to supply blood specifically for patients with SCD.34,35 Although these approaches reduce the incidence of alloantibody production, some patients still become sensitized to Rh antigens (D, C, e, and hrB, HrB, hrS, HrS), indicating they were not Rh antigen matched.
RH GenotypingRBC genotyping methods were introduced to
transfusion medicine a decade ago after cloning of the genes made genetic testing for blood groups possible. Genotyping methods include amplification of target gene sequences by PCR followed by analysis using RFLP, real-time PCR, or sequence-specific primer PCR. More recently, mass-scale genotyping technologies have been developed by several manufacturers to perform high-throughput blood group prediction.46,47 There is nearly complete concordance between the genotype and serologic phenotype, and studies
have included samples from diverse backgrounds. However, the complexities and numerous alleles in the Rh blood group system have precluded high-throughput genotyping for RH other than for the common nonvariant forms of C/c and E/e antigens. The detection of numerous silencing mutations is required for accurate typing; several regions of the genes must be sampled to detect multiple alleles, and new alleles are continuously being identified.48
Currently, RH genotyping is primarily restricted to specialized immunohematology laboratories. One application of RH genotyping is to find compatible donors in the American Rare Donor Program (ARDP) for patients with antibodies to high-prevalence Rh antigens.32 Genotyping can also be used to determine the RHD status of a fetus by amniocentesis or chorionic villus sampling. More recently, noninvasive techniques have been used to collect cell-free, fetal-derived DNA from maternal plasma for RHD genotype prediction with a high-throughput method.49,50 Once high-throughput platforms encompassing all the Rh variants are developed and are cost effective, RH genotyping can complement serologic testing for typing transfused patients, RHD zygosity determination, fetal D typing, resolution of D status, and finding compatible blood for patients with SCD.
RhAG SystemRHAG is the ancestral gene from which RHCE and
RHD arose. It resides on chromosome 6 and encodes the Rh-associated glycoprotein and the antigens of the RhAG blood group system. The RhAG protein consists of 409 amino acids and associates with the Rh proteins in the membrane to form the Rh-core complex.51
AntigensThe RhAG protein does not express the common Rh
antigens, but it has recently been shown to carry several antigens,52 and it has been assigned system 30 by ISBT (Table 1). Two high-frequency antigens, Duclos and DSLK, and one low-frequency antigen, Ola, had serologic characteristics suggestive of expression on RhAG. This was confirmed by gene sequencing and expression of recombinant RhAG in HEK 293 cells. The Duclos-negative patient was homozygous for a nucleotide 316C>G change, encoding Gln106Glu. The DSLK-negative patient was homozygous for nucleotide 490A>C, encoding Lys164Gln. Two Ol(a+) family members of a Norwegian family were heterozygous for 680C>T encoding Ser227Leu. Duclos is RHAG1, Ola is RHAG2, and DSLK is provisionally RHAG3.
Rhnull
RhAG protein must be present for the expression of Rh antigens. Rhnull RBCs lack expression of Rh antigens, and the phenotype most often results from mutations in RHAG, previously termed “regulator” Rhnull. Less often, Rhnull individuals have inactivating or silencing mutations in RHCE and deletion of RHD previously referred to as “amorph” Rhnull.33

184 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
The Rh-core complex also interacts with band 3, glycophorins A and B, LW, and CD47 and is believed to contribute to the RBC membrane structure because Rhnull RBCs demonstrate abnormal morphology. The function of the Rh blood group proteins is not known, but RhAG is an ammonia transporter, and the recent crystalization of the human RhAG homolog from the kidney, RhCG, confirms the relationship of human Rh proteins to ammonia transporters.53
Summary PerspectiveThe Rh/RhAG blood group systems are some of the most
complex, and in the past two decades major insights have been gained into the molecular basis of the Rh system. The genetic information has confirmed many of the predictions of the serologists, whose primary (and often only) tools were the antibodies made by immunized individuals. Exploiting adsorption and elution approaches, along with selected RBC testing strategies, they uncovered many of the details concerning the specificity and complexity of the Rh system. The prediction that Rh antigens are encoded by two genes, not one or three, was adeptly forecast by Patricia Tippett based solely on serologic observations.12 Their work is the foundation of our understanding today, as new genetic information builds on the serologic backbone. Serologic reactivity is still the basis for blood groups and blood transfusion practice because serology defines an antigen. That will not change as we use DNA-based testing methods because it is important to remember that without a serologic relationship, a variation in a blood group allele at the DNA level is just another SNP (single nucleotide polymorphism), and these occur once in every 100 to 300 bp in the human genome. These polymorphisms are only of academic interest until associated with a phenotype and found to be relevant to transfusion medicine by stimulation of an antibody.
Molecular testing will be a powerful adjunct to serologic methods and improve transfusion safety and outcomes, particularly for the chronically transfused population. Which of the numerous alleles in the Rh system are clinically significant is yet to be fully determined. Ultimately, with the development of mass-scale RH genotyping technology, genetically matching donor units with the patients could significantly reduce alloimmunization, and RHD genotyping will provide a definitive D classification for donors and patients. The availability of cost-effective, reliable high-throughput genotyping platforms is needed for this to become incorporated into clinical transfusion medicine practice.
References 1. Levine P, Burnham L, Katzin WM, Vogel P. The
role of isoimmunization in the pathogenesis of erythroblastosis fetalis. Am J Obstet Gynecol 1941;42: 925–37.
2. Freda VJ, Gorman JG, Pollack W. Rh factor: prevention of isoimmunization and clinical trial on mothers. Science 1966;151:828–30.
3. Mollison PL, Hughes-Jones NC, Lindsay M, Wessely J. Suppression of primary RH immunization by passively-administered antibody. Experiments in volunteers. Vox Sang 1969;16:421–39.
4. Race RR, Sanger R. Blood groups in man. 6th ed. Oxford, England: Blackwell, 1975.
5. Reid ME, Lomas-Francis C. The blood group antigen factsbook. 2nd ed. San Diego, CA: Academic Press, 2004.
6. Le van Kim C, Mouro I, Chérif-Zahar B, et al. Molecular cloning and primary structure of the human blood group RhD polypeptide. Proc Natl Acad Sci U S A 1992;89:10925–9.
7. Chérif-Zahar B, Bloy C, Le Van Kim C, et al. Molecular cloning and protein structure of a human blood group Rh polypeptide. Proc Natl Acad Sci U S A 1990;87:6243–7.
8. Arce MA, Thompson ES, Wagner S, Coyne KE, Ferdman BA, Lublin DM. Molecular cloning of RhD cDNA derived from a gene present in RhD-positive, but not RhD-negative individuals. Blood 1993;82: 651–5.
9. Simsek S, de Jong CA, Cuijpers HT, et al. Sequence analysis of cDNA derived from reticulocyte mRNAs coding for Rh polypeptides and demonstration of E/e and C/c polymorphisms. Vox Sang 1994;67:203–9.
10. Blumenfeld OO, Patnaik SK. Allelic genes of blood group antigens: a source of human mutations and cSNPs documented in the Blood Group Antigen Gene Mutation Database. Hum Mutat 2004;23:8–16.
11. Landsteiner K, Wiener AS. An agglutinable factor in human blood recognized by immune sera for rhesus blood. Proc Soc Exp Biol Med 1940;43:223–4.
12. Tippett P. A speculative model for the Rh blood groups. Ann Hum Genet 1986;50:241–7.
13. Daniels G, Castilho L, Flegel WA, et al. International Society of Blood Transfusion Committee on terminology for red blood cell surface antigens: Macao report. Vox Sang 2009;96:153–6.
14. Scott ML, Voak D, Liu W, Jones JW, Avent ND. Epitopes on Rh proteins. Vox Sang 2000;78(Suppl 2):117–20.
15. Colin Y, Chérif-Zahar B, Le Van Kim C, Raynal V, Van Huffel V, Cartron JP. Genetic basis of the RhD-positive and RhD-negative blood group polymorphism as determined by Southern analysis. Blood 1991;78: 2747–52.
16. Singleton BK, Green CA, Avent ND, et al. The presence of an RHD pseudogene containing a 37 base pair duplication and a nonsense mutation in Africans with the Rh D-negative blood group phenotype. Blood 2000;95:12–18.
C.M. Westhoff and S.T. Chou

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 185
17. Shao CP, Maas JH, Su YQ, Köhler M, Legler TJ. Molecular background of Rh D-positive, D-negative, D(el) and weak D phenotypes in Chinese. Vox Sang 2002;83:156–61.
18. Wagner FF, Gassner C, Müller TH, Schönitzer D, Schunter F, Flegel WA. Molecular basis of weak D phenotypes. Blood 1999;93:385–93.
19. Sun CF, Chou CS, Lai NC, Wang WT. RHD gene polymorphisms among RhD-negative Chinese in Taiwan. Vox Sang 1998;75:52–7.
20. Daniels G. Human blood groups. 2nd ed. Cambridge, MA: Wiley-Blackwell, 2002.
21. Westhoff CM, Vege S, Nickle P, Hue-Roye K, Reid ME. A single nucleotide deletion in RHCE*cE responsible for the D- - haplotype in the Hispanic population. Vox Sang 2010:99 (Suppl):376.
22. Beckers EA, Porcelijn L, Ligthart P, et al. The RoHar antigenic complex is associated with a limited number of D epitopes and alloanti-D production: a study of three unrelated persons and their families. Transfusion 1996;36:104–8.
23. Wallace M, Lomas-Francis C, Tippett P. The D antigen characteristic of RoHar is a partial D antigen. Vox Sang 1996;70:169–72.
24. Flegel WA, Wagner FF, Chen Q, Schlanser G, Frame T, Westhoff CM, Moulds MK. The RHCE allele ceCF: the molecular basis of Crawford (RH43). Transfusion 2006:46:1334-42
25. Chen Q, Hustinx H, Flegel WA. The RHCE allele ceSL: the second example for D antigen expression without D-specific amino acids. Transfusion 2006;46:766–72.
26. Wagner FF, Ladewig B, Flegel WA.The RHCE allele ceRT: D epitope 6 expression does not require D-specific amino acids. Transfusion. 2003;43:1248-54.
27. Westhoff CM. Review: the Rh blood group D antigen... dominant, diverse, and difficult. Immunohematology 2005;21:155–63.
28. Daniels GL, Faas BH, Green CA, et al. The VS and V blood group polymorphisms in Africans: a serologic and molecular analysis. Transfusion 1998;38:951–8.
29. Brumit MC, Carnahan GE, Stubbs JR, Storry JR, Reid ME. Moderate hemolytic disease of the newborn (HDN) due to anti-Rh17 produced by a black female with an e variant phenotype. Immunohematology 2002;18:40–2.
30. Noizat-Pirenne F, Lee K, Pennec PY, et al. Rare RHCE phenotypes in black individuals of Afro-Caribbean origin: identification and transfusion safety. Blood 2002;100:4223–31.
31. Reid ME, Storry JR, Issitt PD, et al. Rh haplotypes that make e but not hrB usually make VS. Vox Sang 1997;72:41–4.
32. Vege S, Westhoff CM. Molecular characterization of GYPB and RH in donors in the American Rare Donor Program. Immunohematology 2006;22:143–7.
33. Issit P, Anstee, DJ. Applied blood group serology. 4th ed. Durham, NC: Montgomery Scientific Publications, 1998.
34. Afenyi-Annan A, Bandarenko N. Transfusion practices for patients with sickle cell disease at a major academic medical center. Immunohematology 2006;22:103–7.
35. Sesok-Pizzini DA, Friedman DF, Smith-Whitley K, Nance SJ. Transfusion support of patients with sickle cell disease at the Children’s Hospital of Philadelphia. Immunohematology 2006;22:121–5.
36. Petz LD. Review: evaluation of patients with immune hemolysis. Immunohematology 2004;20:167–76.
37. Price TH, ed. Standards for blood banks and transfusion services. 26th ed. Bethesda, MD: AABB 2009:29.
38. Denomme GA, Wagner FF, Fernandes BJ, Li W, Flegel WA. Partial D, weak D types, and novel RHD alleles among 33,864 multiethnic patients: implications for anti-D alloimmunization and prevention. Transfusion 2005;45:1554–60.
39. Yasuda H, Ohto H, Sakuma S, Ishikawa Y. Secondary anti-D immunization by Del red blood cells. Transfusion 2005;45:1581–4.
40. Flegel WA, Khull SR, Wagner FF. Primary anti-D immunization by weak D type 2 RBCs. Transfusion 2000;40:428–34.
41. Mota M, Fonseca NL, Rodrigues A, Kutner JM, Castilho L. Anti-D alloimmunization by weak D type 1 red blood cells with a very low antigen density. Vox Sang 2005;88:130–5.
42. Lacey PA, Caskey CR, Werner DJ, Moulds JJ. Fatal hemolytic disease of a newborn due to anti-D in an Rh-positive Du variant mother. Transfusion 1983;23: 91–4.
43. Vichinsky EP, Luban NL, Wright E, et al. Prospective RBC phenotype matching in a stroke-prevention trial in sickle cell anemia: a multicenter transfusion trial. Transfusion 2001;41:1086–92.
44. Ambruso DR, Githens JH, Alcorn R, et al. Experience with donors matched for minor blood group antigens in patients with sickle cell anemia who are receiving chronic transfusion therapy. Transfusion 1987;27: 94–8.
45. Tahhan HR, Holbrook CT, Braddy LR, Brewer LD, Christie JD. Antigen-matched donor blood in the transfusion management of patients with sickle cell disease. Transfusion 1994;34:562–9.
46. Hashmi G, Shariff T, Zhang Y, et al. Determination of 24 minor red blood cell antigens for more than 2000 blood donors by high-throughput DNA analysis. Transfusion 2007;47:736–47.
47. Hopp K, Weber K, Bellissimo D, Johnson ST, Pietz B. High-throughput red blood cell antigen genotyping using a nanofluidic real-time polymerase chain reaction platform. Transfusion 2010;50:40–6.
Review of Rh and RhAG systems

186 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
48. Hillyer CD, Shaz BH, Winkler AM, Reid M. Integrating molecular technologies for red blood cell typing and compatibility testing into blood centers and transfusion services. Transfus Med Rev 2008;22: 117–32.
49. Lo YM, Hjelm NM, Fidler C, et al. Prenatal diagnosis of fetal RhD status by molecular analysis of maternal plasma. N Engl J Med 1998;339:1734–8.
50. Finning K, Martin P, Summers J, Massey E, Poole G, Daniels G. Effect of high throughput RHD typing of fetal DNA in maternal plasma on use of anti-RhD immunoglobulin in RhD negative pregnant women: prospective feasibility study. BMJ 2008;336:816–18.
51. Matassi G, Chérif-Zahar B, Raynal V, Rouger P, Cartron JP. Organization of the human RH50A gene (RHAG) and evolution of base composition of the RH gene family. Genomics 1998;47:286-93.
52. Tilley L, Green C, Poole J, et al. A new blood group system, RHAG: three antigens resulting from amino acid substitutions in the Rh-associated glycoprotein. Vox Sang 2010;98:151–9.
53. Gruswitz F, Chaudhary S, Ho JD, et al. Function of human Rh based on structure of RhCG at 2.1 A. Proc Natl Acad Sci U S A 2010;107:9638–43.
Web ResourcesInternational Society of Blood Transfusion (ISBT) Working
Party on Red Cell Immunogenetics and Blood Group Terminology. http://ibgrl.blood.co.uk/ISBTPages/ISBTHome.htm. Accessed 1/4/2011.
RhesusBase. http://www.uni-ulm.de/~fwagner/RH/RB/. Accessed 1/4/2011.
NCBI Blood Group Mutation Database. http://www.ncbi.nlm.nih.gov/gv/mhc/xslcgi.cgi?cmd=bgmut/home. Accessed 1/4/2011.
Stella T. Chou, MD, Assistant Professor of Pediatrics, Division of Hematology, The Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine, 3615 Civic Center Boulevard, ARC 316D, Philadelphia, PA 19104, and Connie M. Westhoff, SBB, PhD, Director of Immunohematology and Genomics, New York Blood Center, 310 East 67th Street, New York, NY 10065.
C.M. Westhoff and S.T. Chou
Attention: State Blood Bank Meeting Organizers
If you are planning a state meeting and would like copies of Immunohematology for distribution, please send request, 4 months in advance,
For information concerning Immunohematology, Journal of Blood Group Serology and Education, or the Immunohematology Methods and Procedures manual, contact us by e-mail at [email protected]
Notice to Readers
All articles published, including communications and book reviews, reflect the opinions of the authors and do not necessarily reflect the official policy of the American Red Cross.
Attention: SBB and BB Students
You are eligible for a free 1-year subscription to
Immunohematology.
Ask your education supervisor to submit the name and
complete address for each student and the inclusive dates
of the training period to [email protected].

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 187
COMMUNICATIONS
To the Editors:
Anti-Vel and cold-reactive autoantibody
In Immunohematology Volume 26, Number 1, 2010, Linz and colleagues present a case report titled “Role for serial prenatal anti-Vel quantitative serologic monitoring with 2-ME serum during pregnancy.” The report includes a statement that anti-Vel can simulate the serologic profile of a cold-reactive autoantibody. The authors did not quote a previous paper with similar conclusions that further support this premise. Mechanic et al. describe a study in which eight samples of stored anti-Vel were adsorbed with rabbit erythrocyte stroma (RESt, Immucor/Gamma, Norcross GA). (Transfusion 2002;42(9):1180–3). The antibodies were reactive at various testing phases. The adsorbed samples were tested at phases selected based on the test results of the neat samples. The results indicated that anti-Vel was either partially or completely adsorbed by RESt. These results further support the idea that one must be thorough when investigating these types of antibodies.
Joan L Maurer BS, SBB(ASCP)American Red Cross Blood Services Penn Jersey Region
National Reference Laboratory for Blood Group Serology700 Spring Garden Street
Philadelphia, PA 19123
Sandra Taddie Nance, MS, MT, (ASCP)SBBAmerican Red Cross Biomedical Services
Immunohematology Reference Laboratories and Heritage Division700 Spring Garden Street
Philadelphia, PA 19123
Immunohematology is on the Web!
www.redcross.org/immunohematology
For more information, send an e-mail
For information concerning the National Reference
Laboratory for Blood Group Serology, including the American
Rare Donor Program, contact Sandra Nance, by phone at
(215) 451-4362, by fax at (215) 451-2538, or by e-mail at
Manuscripts
The editorial staff of Immunohematology welcomes manuscripts pertaining to blood group serology and education for consideration for publication. We are especially interested in case reports, papers on platelet and white cell serology, scientific articles covering original investigations, and papers on new methods for use in the blood bank. Deadlines for receipt of manuscripts for consideration for the March, June, September, and December issues are the first weeks in November, February, May, and August, respectively. For instructions for scientific articles, case reports, and review articles, see Instructions for Authors in every issue of Immunohematology or on the Web at www.redcross.org/immunohematology. Include fax and phone numbers and e-mail address with all manuscripts and correspondence. E-mail all manuscripts to [email protected]

188 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
COMMUNICATIONS
Letter from the Editors:
To Contributors to the 2010 Issues
The journal depends on readers, authors, editorial board, peer reviewers, and our Penn-Jersey staff. We wish we could thank all of you personally, but doing so is not practical. Instead, we thank each of you as members of an honored group.
First and foremost, we thank the authors for their reviews, scientific articles, case reports, book reviews, and letters to the editors that come not only from the United States but from many countries of the world. This has given the journal an international flavor.
Our editorial board is a prestigious one and we depend on its members, not only for peer reviews, but for guidance in policy and suggestions for improvements. Special thanks go to our medical editors, who review every article for medical content, and to our technical editors, who read every article for technical content. The current board is listed by name in the front of each issue of the journal.
Our peer reviewers did a wonderful job in 2010. They are listed below; our thanks to each.
Theresa Boyd, MDJohn W. Burch, MDKerry Burright-Hittner, MT(ASCP)SBBGail E. Coghlan, RT, BScMartha Rae Combs, MT(ASCP)SBBGeoffrey Daniels, PhDRichard Davey, MDGreg Denomme, PhDWilly Flegel, MDJanis R. Hamilton, MS, MT(ASCP)SBBJay Herman, MD Maryann Keashen-SchnellJoanne Kosanke, MT(ASCP)SBBMary Kowalski, MT(ASCP)SBBGeralyn M. Meny, MDDawn Michelle, MT(ASCP)SBB
Monique Mohammad John J. Moulds, MT(ASCP)SBBPamela Nickle, MT(ASCP)SBBJohn Nobiletti, MDJim Perkins, MDMelissa Pessin-Minsley, MD Joyce Poole, FIBMSNurJehan Quraishy, MDSavita SinghJill R. Storry, PhD, FIBMSDavid Stroncek, MDJeff Trimble, BSc.,ART(CSMLS) MLS(ASCP)Ralph R. Vassallo, MDSunitha Vege, MSJames Westra, MD
We also want to thank Marge Manigly, our production assistant, and Sheetal Patel, our editorial assistant, for their help in preparing the journal for press. We also thank Christine Lomas-Francis and Dawn Rumsey, our technical editors, Mary Tod, our copy editor, Lucy Oppenheim, our proofreader; and Wilson Tang, our electronic publisher.
Finally, thanks go to our readers, whose enthusiasm and interest in the journal make it all worthwhile.
Sandra Nance Editor-in-Chief
Cindy Flickinger Managing Editor

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 189
Auto antibodiesAuto-anti-D—diagnostic evidence . . . . . . . . . . . . . . 26(1):27Auto-anti-D—transfusion guidance . . . . . . . . . . . . . 26(1):27Auto-anti-Ge . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):60Auto-anti-I—association with infection . . . . . . . . 26(4):133Auto-anti-I—naturally occurring. . . . . . . . . . . . . . 26(4):133Auto-anti-LW—diagnostic evidence. . . . . . . . . . . . . 26(1):27Auto-anti-LW—transfusion guidance . . . . . . . . . . . . 26(1):27Comparison of methods for detection
and identification . . . . . . . . . . . . . . . . . . . . . . . 26(3):104Correlation with DAT in patients with
thalassemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):87Low affinity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):156
Autoimmune hemolytic anemia (AIHA)Anti-Ge specificity . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):60DAT-negative . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4)156
BiochemistryBiochemical physiology of the Colton glycoprotein 26(1):22Duffy—glycoproteins and chemokines . . . . . . . . . . .26(2):51Gerbich—glycophorin . . . . . . . . . . . . . . . . . . . . . . . . 26(2):60Ii—structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):133Ii—glycolipids and glycoproteins . . . . . . . . . . . . . . 26(4):133Lectins and I. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):133Polylactosamine biosynthesis . . . . . . . . . . . . . . . . 26(4):133Structure and function of the C4 protein . . . . . . . . .26(1):30
Blood componentsFFP—association with TRALI . . . . . . . . . . . . . . . . . 26(4):161
Blood group antibodiesAnti-D—caused by weak D type 21 . . . . . . . . . . . . . . 26(1):27Anti-Fya and Fyb. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):51Anti-I—review . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):133Anti-Ku—absence of HDFN . . . . . . . . . . . . . . . . . . .26(3):119Anti-Oka . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):124Anti-Vel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):8Cromer antibodies: Cra, Dra, Esa, TCa, Wesb,
UMC . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):109Rh system—review . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):178Sialo agglutinins . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):133Specificities of DAT-associated antibodies
in thalassemia patients . . . . . . . . . . . . . . . . . . . 26(3):87
Blood group systems/antigensChido/Rodgers—review . . . . . . . . . . . . . . . . . . . . . . .26(1):30Colton—review . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):22
Cromer—review . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):109Dombrock—review. . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):71Duffy—review. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):51Gerbich—review. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):60I—review . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):CoolKnops—review . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):2Ok—review . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):124Rh and RhAG—review . . . . . . . . . . . . . . . . . . . . . . .26(4):178
Case reportAbsence of HDFN—maternal high-titer IgG
anti-Ku . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(3):119Alloimmunization of D with weak D type 21 . . . . . . 26(1):27Anti-Vel—monitoring with 2-ME serum. . . . . . . . . . .26(1):8Comparison of gel test and conventional tube
test for antibody detection and titration in D-negative pregnant women. . . . . . . . . . . . .26(4):174
DAT-negative hemolytic anemia . . . . . . . . . . . . . . .26(4):156Traditional method and modified PEG
adsorption method—a comparison . . . . . . . . 26(3):104
Disease associationsAcute respiratory distress syndrome . . . . . . . . . . . 26(4):161Antibody-mediated pathogenesis of TRALI. . . . . . 26(4):161Capillary angioma of small intestine. . . . . . . . . . . 26(3):109Cold agglutinin disease. . . . . . . . . . . . . . . . . . . . . . 26(4):133Congenital cataracts and iadult phenotype . . . . . . . 26(4):133DARC and malaria. . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):51DARC and renal disease . . . . . . . . . . . . . . . . . . . . . . .26(2):51Disease associations with low levels of C4/complement. . . .
26(1):30Enteropathy—protein losing. . . . . . . . . . . . . . . . . . 26(3):109HEMPAS. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):133Paroxysmal nocturnal hemoglobinuria . . . . . . . . 26(3):109Positive DAT in thalassemia patients . . . . . . . . . . . 26(3):87Pneumonia and I . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):133Rh antibodies in patients with sickle cell
disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):178TRALI—granulocyte serology . . . . . . . . . . . . . . . . . . 26(1):11TRALI—noncardiogenic pulmonary edema. . . . . . 26(4):161
DonorsD and Del donors . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):178WBC antibodies in TRALI . . . . . . . . . . . . . . . . . . . . 26(4):161
GeneticsC4 and Chido/Rodgers . . . . . . . . . . . . . . . . . . . . . . . .26(1):30
Subject IndexImmunohematology—Volume 26, Nos. 1, 2, 3, 4, 2010

190 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
Colton—AQP1 locus . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):22Consortium for Blood Group Genes:
2009 report . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):47D1*01 and D1*02—comparison of phenotyping with
hemagglutination, PCR-SSP, and real-time PCR. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):66
DAF gene. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):109Diego—genotype and alleles in Brazilian
donors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):66Duffy—phenotypes, prevalence, and inherited
alleles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):51Ge phenotype—nucleotide and amino acid
changes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):60Gene array data . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):133Granulocyte genotyping . . . . . . . . . . . . . . . . . . . . . . . 26(1):11I gene . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):133Knops—CR1 gene. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):2Rh genotyping . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):178RhAG protein . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):178RHD and RHCE primers . . . . . . . . . . . . . . . . . . . . . .26(2):57
Hemolytic disease of the fetus and newborn (HDFN)Absence of HDFN despite maternal high-titer
IgG anti-Ku . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(3):119Antibody detection and titration in D-negative
pregnant women . . . . . . . . . . . . . . . . . . . . . . . . .26(4):174Anti-Ge3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):60Biologic role of I . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):133Rh immunoprophylaxis . . . . . . . . . . . . . . . . . . . . . . 26(3):92
Laboratory managementHLA laboratories—future direction . . . . . . . . . . . . . 26(1):11Rh immunoprophylaxis—laboratory methods. . . . 26(3):92Obstetric management of antenatal D–
women . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):174
MethodsAGS panel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):156Anti-HbF method in FMH . . . . . . . . . . . . . . . . . . . . 26(3):92Anti-IgG test. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):156Column agglutinin. . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):156Direct Polybrene test . . . . . . . . . . . . . . . . . . . . . . . . .26(4):156ELAT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):156Eluate HbF, elution assay . . . . . . . . . . . . . . . . . . . . . 26(3):92Enzyme-linked antiglobulin test . . . . . . . . . . . . . . . 26(3):92Flow cytometry—analysis of fetomaternal
hemorrahage . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):92Gel agglutinin cards . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):92Granulocyte agglutination test. . . . . . . . . . . . . . . . . . 26(1):11Granulocyte immunofluorescence test . . . . . . . . . . . 26(1):11Kleihauer-Betke acid-elution assays . . . . . . . . . . . . 26(3):92Microscopic weak D test . . . . . . . . . . . . . . . . . . . . . . 26(3):92
Monoclonal antibody immobilization of granulocyte antigens . . . . . . . . . . . . . . . . . . . . . . 26(1):11
Qualitative screens for fetal RBCs in maternal blood. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):92
Rh immunoprophylaxis . . . . . . . . . . . . . . . . . . . . . . 26(3):92Rosette screen for D+ RBCs . . . . . . . . . . . . . . . . . . . 26(3):92Serologic findings for maternal sample with
anti-Ku . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(3):119Traditional method and modified PEG
adsorption method—a comparison . . . . . . . . 26(3):104
MolecularChido/Rodgers phenotypes—prevalence in
random samples . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):30Colton-null phenotype—ancestry of the
probands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):22Colton—AQP1 locus . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):22Consortium for Blood Group Genes (CBGG):
2009 report . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):47CR1-1 protein bearing Knops antigen in SCR 25 . . . .26(1):2D antigen. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):178Diego genotyping—real-time PCR and melting
curve analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):66Doa (DO1) and Dob(DO2)—polymorphic
antigens . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):71Duffy—antigen receptor for chemokines . . . . . . . . .26(2):51IGnT protein structure-function . . . . . . . . . . . . . . 26(4):133Inab phenotype . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):109Knops antigen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):2Partial c antigen—DNA and RNA isolation,
RT-PCR, sequencing, and cloning. . . . . . . . . . . .26(2):57
Platelets/white cellsDonor leukocyte antibodies . . . . . . . . . . . . . . . . . . . 26(4):161Granulocyte antibody detection and antigen
typing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(1):11Granulocyte genotyping . . . . . . . . . . . . . . . . . . . . . . . 26(1):11ISBT working party on granulocyte
immunobiology . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(1):11Neutropenia
—alloimmune neonatal —autoimmune —drug induced . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(1):11
Role of neutrophil in TRALI . . . . . . . . . . . . . . . . . . 26(4):161
QualityConsortium for blood group genes (CBGG):
2009 report . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):47ISBT Working Party on Granulocyte
Immunobiology. . . . . . . . . . . . . . . . . . . . . . . . . . . 26(1):11
ReviewsChido/Rodgers blood group system . . . . . . . . . . . . .26(1):30Colton blood group system . . . . . . . . . . . . . . . . . . . . .26(1):22Cromer blood group system . . . . . . . . . . . . . . . . . . 26(3):109

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 191
Dombrock blood group system . . . . . . . . . . . . . . . . .26(2):71Duffy blood group system. . . . . . . . . . . . . . . . . . . . . .26(2):51Gerbich blood group system. . . . . . . . . . . . . . . . . . . 26(2):60Granulocyte serology—current concepts and
clinical significance . . . . . . . . . . . . . . . . . . . . . . . 26(1):11Knops blood group system . . . . . . . . . . . . . . . . . . . . . .26(1):2Laboratory methods for Rh
immunoprophylaxis . . . . . . . . . . . . . . . . . . . . . . 26(3):92Ok blood group system . . . . . . . . . . . . . . . . . . . . . . 26(3):124Polylactosamines, there’s more than meets
the Ii . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):133Pathophysiology and prevention of transfusion-
related acute lung injury (TRALI) . . . . . . . . . . 26(4):161The Rh system (Rh and RhAG) . . . . . . . . . . . . . . . .26(4):178
Rh blood group systemAlloimmunization to the D antigen by a patient
with weak D type 21 . . . . . . . . . . . . . . . . . . . . . . . 26(1):27Antibody detection and titration in D-negative
pregnant women . . . . . . . . . . . . . . . . . . . . . . . . .26(4):174Laboratory methods for Rh
immunoprophylaxis . . . . . . . . . . . . . . . . . . . . . . 26(3):92RHCE*ceAR encodes a partial c (RH4) antigen. . . .26(2):57The Rh system (Rh and RhAG) . . . . . . . . . . . . . . . .26(4):178
SerologyAntibody detection and titration in D-negative
pregnant women—a comparison of gel test and conventional tube test. . . . . . . . . . . . . . . . .26(4):174
Anti-Vel—quantitative serologic monitoring . . . . . . .26(1):8Attempts to support an immune etiology in
800 patients with DAT-negative hemolytic anemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):156
Granulocyte serology—current concepts and clinical significance . . . . . . . . . . . . . . . . . . . . . . . 26(1):11
Transfusion PracticesAnti-Vel serologic monitoring with 2-ME
serum treatment . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):8Etiology in 800 patients with DAT-negative
hemolytic anemia . . . . . . . . . . . . . . . . . . . . . . . .26(4):156Gel test and tube test for antibody detection
and titration . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):174Pathophysiology and prevention of TRALI . . . . . . 26(4):161Significance of a positive DAT in thalassemia
patients . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):87Transfusion of patients with D+ RBCs and
serum anti-D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(1):27
Free Classified Ads and Announcements
Immunohematology will publish classified ads and
announcements (SBB schools, meetings, symposia, etc.)
without charge. Deadlines for receipt of these items are
as follows:
Deadlines
1st week in January for the March issue
1st week in April for the June issue
1st week in July for the September issue
1st week in October for the December issue
E-mail or fax information to [email protected]
or (215) 451-2538
192 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
AAllen, S. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(1):8Arinsburg, S.A.. . . . . . . . . . . . . . . . . . . . 26(3):87
BBachowski, G.J. . . . . . . . . . . . . . . . . . . . . .26(1):11
CCampbell-Lee, S. . . . . . . . . . . . . . . . . 26(3):119Castilho, L. . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):47Castro, V. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):47Chamone, D.A.F.. . . . . . . . . . . . . . . . . . 26(2):66Chou, S.T.. . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):178Clay, M.E. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):11Co, A. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):156Cooling, L. . . . . . . . . . . . . . . . . . . . . . . . . .26(4):133Cushing, M.M.. . . . . . . . . . . . . . . . . . . . . 26(3):87
D, EDhawan, H.K.. . . . . . . . . . . . . . . . . . . . . 26(4):174Denomme, G.A. . . . . . . . . . . . . . . . . . . . .26(2):47Dorlhiac-Llacer, P.E. . . . . . . . . . . . . 26(2):66Eastlund, T.. . . . . . . . . . . . . . . . . . . . . . . . 26(4):161Etem, M.E.. . . . . . . . . . . . . . . . . . . . . . . . 26(3):104
FFigueroa, D. . . . . . . . . . . . . . . . . . . . . . . 26(3):104 Fuchisawa, A.. . . . . . . . . . . . . . . . . . . . . . .26(2):57Fueger, J.T.. . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(1):8
GGarratty, G. . . . . . . . . . . . . . . . . . . . . . . . .26(4):156Giardina, P.J. . . . . . . . . . . . . . . . . . . . . . . 26(3):87
HHalverson, G.R. . . . . . . . . . . . . . . . . . . . .26(1):22Halter Hipsky, C. . . . . . . . . . . . . . . . . . .26(2):57
Hedl, J.J. . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):104Holub, M.P. . . . . . . . . . . . . . . . . . . . . . . . 26(3):104Howard-Menk, C. . . . . . . . . . . . . . . . 26(3):119Hunt, P. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .26(4):156
I,J,KJain, A. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):174Johnson, S.T.. . . . . . . . . . . . . . . . . . . . . . . . . 26(1):8Kakaiya, R.M. . . . . . . . . . . . . . . . . . . . . 26(3):119Kleinert, D. . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):87Kumar, P. . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):174
LLaird-Fryer, B. . . . . . . . . . . . . . . . . . . 26(3):104Leger, R.M. . . . . . . . . . . . . . . . . . . . . . . . .26(4):156Linz, W.J. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(1):8Lomas-Francis, C. . . . . . . . . . . . . . . . .26(2):57Lomas-Francis, C. . . . . . . . . . . . . . . . . 26(2):71
MMair, D.C. . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):161Malecki, Z. . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):119Mallory, D. . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):39Mallory, D. . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):40Marwaha, N. . . . . . . . . . . . . . . . . . . . . . . 26(4):174McGann, H. . . . . . . . . . . . . . . . . . . . . . . . . . 26(1):27Mougey, R. . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):30Moulds, J.M. . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):2Meny, G.M. . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):51
N, ONovaretti, M.C.Z.. . . . . . . . . . . . . . . . . 26(2):66
P, QPapari, M. . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):119Peyrard, T. . . . . . . . . . . . . . . . . . . . . . . . . . . .26(1):22
RRami, J. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):119Reid, M.E.. . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):47Reid, M.E.. . . . . . . . . . . . . . . . . . . . . . . . . . . .26(2):57Reid, M.E.. . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):60Reid, M.E.. . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):71Reid, M.E.. . . . . . . . . . . . . . . . . . . . . . . . . 26(3):109Ruiz, A.S.. . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):66
S, TSaha, S.C.. . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):174Saluja, K. . . . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):174Sandler, S.G. . . . . . . . . . . . . . . . . . . . . . . . 26(3):92Sathiyamoorthy, S. . . . . . . . . . . . . . . 26(3):92Schuller, R.M. . . . . . . . . . . . . . . . . . . . . . . .26(1):11Sharma, R.R. . . . . . . . . . . . . . . . . . . . . . 26(4):174Skerrett, D.L. . . . . . . . . . . . . . . . . . . . . . . 26(3):87Smart, E.A. . . . . . . . . . . . . . . . . . . . . . . . .26(3):124St-Louis, M. . . . . . . . . . . . . . . . . . . . . . . . . .26(2):47Storry, J.R.. . . . . . . . . . . . . . . . . . . . . . . . 26(3):109Storry, J.R.. . . . . . . . . . . . . . . . . . . . . . . . .26(3):124Symington, D.B. . . . . . . . . . . . . . . . . 26(3):104Thakral, B. . . . . . . . . . . . . . . . . . . . . . . . . . 26(4):174Thakur, M.K.. . . . . . . . . . . . . . . . . . . . . . 26(4):174
V, W, YWalker, P.S. . . . . . . . . . . . . . . . . . . . . . . . . . 26(2):60Wenk, R.E.. . . . . . . . . . . . . . . . . . . . . . . . . . . 26(1):27Westhoff, C.M. . . . . . . . . . . . . . . . . . . . . .26(2):47Westhoff, C.M. . . . . . . . . . . . . . . . . . . . 26(4):178Whaley, A. . . . . . . . . . . . . . . . . . . . . . . . . . 26(3):119Yazer, M.H. . . . . . . . . . . . . . . . . . . . . . . . 26(3):109
Contributor IndexImmunohematology—Volume 26, Nos. 1, 2, 3, 4, 2010

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 193
Notice to Readers
Immunohematology, Journal of Blood Group Serology and Education, is printed on acid-free paper.
Important Notice About Manuscripts for Immunohematology
Please e-mail all manuscripts to [email protected]
Monoclonal antibodies available at no charge:
The New York Blood Center has developed a wide range of
monoclonal antibodies (both murine and humanized) that are
useful for donor screening and for typing RBCs with a positive
DAT. These include anti-A1, -M, -s, -U, -D, -Rh17, -K, -k, -Kpa,
-Jsb, -Fya, -Fy3, -Fy6, Wrb, -Xga ,-CD99, -Dob, -H, -Ge2, -Ge3,
-CD55 (both SCR2/3 and SCR4), -Oka, -I, and anti-CD59. Most
of the antibodies are murine IgG and require the use of anti-
mouse IgG for detection (Anti-K, k, and -Kpa). Some are directly
agglutinating (Anti-A1, -M, -Wrb and -Rh17) and a few have been
humanized into the IgM isoform (Anti-Jsb). The antibodies are
available at no charge to anyone who requests them. Please
visit our Web site for a complete list of available monoclonal
antibodies and the procedure for obtaining them.
For additional information, contact: Gregory Halverson, New
York Blood Center, 310 East 67th Street, New York, NY 10021/
e-mail: [email protected] (phone 212-570-
3026, Fax: 212-737-4935) or visit the Web site at http://www.
nybloodcenter.org >research >immunochemistry >current list of
monoclonal antibodies available.
Specialist in Blood Bank (SBB) Program
The Department of Transfusion Medicine, National Institutes
of Health, is accepting applications for its 1-year Specialist
in Blood Bank Technology Program. Students are federal
employees who work 32 hours/week. This program
introduces students to all areas of transfusion medicine
including reference serology, cell processing, HLA, and
infectious disease testing. Students also design and conduct
a research project. NIH is an Equal Opportunity Organization.
• Application deadline is December 31, 2010 for the July
2011 class.
• See www.cc.nih.gov/dtm > education for brochure and
application.
• For further information contact Karen M. Byrne at
(301)451-8645 or [email protected]
Announcements
The Clinical and Laboratory Standards Institute (www.clsi.org) recently published the guideline “Validation of Automated Systems for Immunohematological Testing Before Implementation; Approved Guideline” (I/LA33-A). This document provides guidance to the end user and laboratory for validation of automated systems used in immunohematologic testing before implementation. The link to this document is:
http://www.clsi.org/source/orders/Product_Display.cfm?section=Shop&task=3&CATEGORY=IL&PRODUCT_TYPE=SALES&SKU =ILA33A&DESCRIPTION=&FindSpec=&CFTOKEN=19503848&continue=1&SEARCH_TYPE
The link to the press release is:
http://www.clsi.org/Content/NavigationMenu/NewsandEvents/PressReleases/Jan10_ILA33A_2.pdf
For information on an upcoming teleconference from CLSI-APHL titled:
New Guidance for Laboratories: Validating Automated Systems for Immunohematological Testing (588-608-10), contact the Web site at www.aphl.org/clsi
For additional information, contact the Web site at www.clsi.org or Amanda Cushman Holm at (610) 688-0100 ext. 129 or [email protected].
Announcement

194 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
Masters (MSc) in Transfusion and Transplantation Sciences at
The University of Bristol, England
Applications are invited from medical or science graduates for the Master of Science (MSc) degree in Transfusion and Transplantation Sciences at the University of Bristol. The course starts in October 2011 and will last for 1 year. A part-time option lasting 2 or 3 years is also available. There may also be opportunities to continue studies for PhD or MD following the MSc. The syllabus is organized jointly by The Bristol Institute for Transfusion Sciences and the University of Bristol, Department of Pathology and Microbiology. It includes:
• Scientific principles of transfusion and transplantation
• Clinical applications of these principles
• Practical techniques in transfusion and transplantation
• Principles of study design and biostatistics
• An original research project
Application can also be made for Diploma in Transfusion and Transplantation Science or a Certificate in Transfusion and Transplantation Science.
The course is accredited by the Institute of Biomedical Sciences.
Further information can be obtained from the Web site:http://www.blood.co.uk/ibgrl/MscHome.htm
For further details and application forms please contact:
Dr Patricia Denning-KendallUniversity of BristolPaul O’Gorman Lifeline CentreDepartment of Pathology and MicrobiologySouthmead HospitalWestbury-on-Trym, Bristol BS10 5NB, EnglandFax +44 1179 595 342, Telephone +44 1779 595 455, e-mail: [email protected].
Announcements, con’t.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 195
Antibody identification and problem resolution
HLA-A, B, C, and DR typing
HLA-disease association typing
Paternity testing/DNA
For information, contact:
Mehdizadeh Kashi
at (503) 280-0210
or write to:
Pacific Northwest Regional Blood Services
ATTENTION: Tissue Typing Laboratory
American Red Cross
3131 North Vancouver
Portland, OR 97227
Reference and Consultation Services
CLIA licensed, ASHI accredited
IgA and anti-IgA testing is available to do the
following:
• Identify IgA-deficient patients
• Investigate anaphylactic reactions
• Confirm IgA-deficient donors
Our ELISA for IgA detects protein to 0.05 mg/dL.
For additional information contact:
Cindy Flickinger
at (215) 451-4909,
or e-mail:
or write to:
American Red Cross Blood Services
Musser Blood Center
700 Spring Garden Street
Philadelphia, PA 19123-3594
ATTN: Cindy Flickinger
IgA/Anti-IgA Testing
CLIA licensed
• Effective tool for screening large volumes of donors
• Gel diffusion test that has a 15-year proven track record:
Approximately 90 percent of all donors identified as IgA
deficient by are confirmed by the more sensitive testing
methods
For additional information:
Kathy Kaherl
at (860)678-2764
e-mail:
or write to:
Reference Laboratory
American Red Cross Biomedical Services
Connecticut Region
209 Farmington Ave.
Farmington, CT 06032
Donor IgA Screening
CLIA licensed
Immunohematology Reference LaboratoryAABB, ARC, New York State, and CLIA licensed
24-hour phone number: (215) 451-4901
Fax: (215) 451-2538
American Rare Donor Program24-hour phone number:
(215) 451-4900Fax:
(215) [email protected]
ImmunohematologyJournal of Blood Group Serology and Education
Phone, business hours: (215) 451-4902
Fax: (215) 451-2538
Quality Control of Cryoprecipitated–AHFPhone, business hours:
(215) 451-4903Fax:
(215) 451-2538
National Reference Laboratory for Blood Group Serology
Advertisements

196 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
Diagnostic testing for:• Neonatal alloimmune thrombocytopenia (NAIT)• Posttransfusion purpura (PTP)• Refractoriness to platelet transfusion• Heparin-induced thrombocytopenia (HIT)• Alloimmune idiopathic thrombocytopenia purpura (AITP)
Medical consultation available
Test methods:• GTI systems tests — detection of glycoprotein-specific platelet antibodies — detection of heparin-induced antibodies (PF4 ELISA)• Platelet suspension immunofluorescence test (PSIFT)• Solid phase red cell adherence (SPRCA) assay• Monoclonal immobilization of platelet antigens (MAIPA)• Molecular analysis for HPA-1a/1b
For further information, contact:
Platelet Serology Laboratory (215) 451-4205
Maryann Keashen-Schnell (215) 451-4041 office [email protected]
Sandra Nance (215) 451-4362 [email protected]
American Red Cross Blood ServicesMusser Blood Center
700 Spring Garden StreetPhiladelphia, PA 19123-3594
National Platelet Serology Reference Laboratory
CLIA licensed
Our laboratory specializes in granulocyte antibody detection and granulocyte antigen typing.
Indications for granulocyte serology testing include:• Alloimmune neonatal neutropenia (ANN)• Autoimmune neutropenia (AIN)• Transfusion related acute lung injury (TRALI)
Methodologies employed:• Granulocyte agglutination (GA)• Granulocyte immunofluorescence by flow cytometry (GIF)• Monoclonal antibody immobilization of neutrophil antigens
(MAINA)
TRALI investigations also include:• HLA (PRA) Class I and Class II antibody detection
For further information, contact:
Neutrophil Serology Laboratory (651) 291-6797
Randy Schuller (651) 291-6758 [email protected]
American Red Cross Blood ServicesNeutrophil Serology Laboratory
100 South Robert StreetSt. Paul, MN 55107
National Neutrophil Serology Reference Laboratory
CLIA licensed
Advertisements, cont.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 197
Ordering InformationThe book, which costs $25, can be ordered in two ways:
Order online from the publisher •at: www.sbbpocketbook.com
Order from the authors, who •will sign the book. Send a check, made payable to “New York Blood Center” and indicate “Pocketbook” on the memo line, to:
Marion Reid Laboratory of Immunochemistry New York Blood Center 310 East 67th Street New York, NY 10065
Please include the recipient’s complete mailing address.
Blood Group Antigens & AntibodiesA guide to clinical relevance & technical tipsby Marion E. Reid & Christine Lomas-Francis
Pocketbook Education FundThe authors are using royalties generated from the sale of this pocketbook for educational purposes to mentor people in the joys of immunohematology as a career. They will accomplish this in the following ways:
Sponsor workshops, seminars, and lectures•Sponsor students to attend a meeting•Provide copies of the pocketbook•
(See www.sbbpocketbook.com for details to apply for funds)
This compact “pocketbook” from the authors of the Blood Group Antigen FactsBook is a must for anyone who is involved in the laboratory or bedside care of patients with blood group alloantibodies.
The book contains clinical and technical information about the nearly 300 ISBT recognized blood group antigens and their corresponding antibodies. The information is listed in alphabetical order for ease of finding—even in the middle of the night. Included in the book is information relating to:
Clinical significance of antibodies in transfusion and HDN.•
Number of compatible donors that would be expected •to be found in testing 100 donors. Variations in different ethnic groups are given.
Characteristics of the antibodies and optimal technique(s) •for their detection.
Technical tips to aid their identification.•
Whether the antibody has been found as an autoantibody.•
Advertisements, cont.

198 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010
What is a certified Specialist in Blood Banking (SBB)?• Someone with educational and work experience qualifications who successfully passes the American Society for Clinical Pathology (ASCP)
board of registry (BOR) examination for the Specialist in Blood Banking.• This person will have advanced knowledge, skills, and abilities in the field of transfusion medicine and blood banking.
Individuals who have an SBB certification serve in many areas of transfusion medicine: • Serve as regulatory, technical, procedural and research advisors• Perform and direct administrative functions • Develop, validate, implement, and perform laboratory procedures• Analyze quality issues preparing and implementing corrective actions to prevent and document issues• Design and present educational programs• Provide technical and scientific training in blood transfusion medicine• Conduct research in transfusion medicine
Who are SBBs?Supervisors of Transfusion Services Managers of Blood Centers LIS Coordinators EducatorsSupervisors of Reference Laboratories Research Scientists Consumer Safety Officers Quality Assurance Officers Technical Representatives Reference Lab Specialist
Why be an SBB?Professional growth Job placement Job satisfaction Career advancement
How does one become an SBB?• Attend a CAAHEP-accredited Specialist in Blood Bank Technology Program OR • Sit for the examination based on criteria established by ASCP for education and experienceHowever: In recent years, a greater percentage of individuals who graduate from CAAHEP-accredited programs pass the SBB exam.
Conclusion:The BEST route for obtaining an SBB certification is …to attend a CAAHEP-accredited Specialist in Blood Bank Technology Program
Contact the following programs for more information:
Program Contact NamePhone Contact Email Contact Website
Onsite or Online Program
Walter Reed Army Medical Center William Turcan 202-782-6210 [email protected] www.militaryblood.dod.mil On site
American Red Cross, Southern California Region
Michael Coover 909-859-7496 [email protected] none On site
ARC-Central OH Region Joanne Kosanke 614-253-2740 x 2270
[email protected] none On site
Blood Center of Southeastern Wisconsin
Lynne LeMense 414-937-6403 [email protected] www.bcw.edu On site
Community Blood Center/CTS Dayton, Ohio
Nancy Lang 937-461-3293 [email protected] http://www.cbccts.org/education/sbb.htm
On line
Gulf Coast School of Blood Bank Technology
Clare Wong 713-791-6201 [email protected] www.giveblood.org/education/distance/htm
On line
Hoxworth Blood Center, University of Cincinnati
Susan Wilkinson 513-558-1275 [email protected] www.hoxworth.org On site
Indiana Blood Center Jayanna Slayten 317-916-5186 [email protected] www.indianablood.org On line
Johns Hopkins Hospital Jan Light 410-955-6580 [email protected] http://pathology2.jhu/department/divisions/tranfusion/sbb.cfm
On site
Medical Center of Louisiana Karen Kirkley 504-903-3954 [email protected] none On site
NIH Clinical Center Department of Transfusion Medicine
Karen Byrne 301-496-8335 [email protected] www.cc.nih.gov/dtm On site
Rush University Veronica Lewis 312-942-2402 [email protected] www.rushu.rush.edu/health/dept.html On line
Transfusion Medicine Center at Florida Blood Services
Marjorie Doty 727-568-5433 x 1514
[email protected] www.fbsblood.org On line
University of Texas Health Science Center at San Antonio
Linda Myers 210-731-5526 [email protected] www.uthscsa.edu On site
University of Texas Medical Branch at Galveston
Janet Vincent 409-772-3055 [email protected] www.utmb.edu/sbb On line
University of Texas SW Medical Center
Barbara Laird-Fryer 214-648-1785 [email protected] http://telecampus.utsystem.edu On line
Becoming a Specialist in Blood Banking (SBB)
Additional Information can be found by visiting the following Web sites: www.ascp.org, www.caahep.org and www.aabb.org Revised August 2007
Advertisements, cont.

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 199
I. GENERAL INSTRUCTIONSBefore submitting a manuscript, consult current issues of Immunohematology for style. Double-space throughout the manuscript. Number the pages consecutively in the upper right-hand corner, beginning with the title page.
II. SCIENTIFIC ARTICLE, REVIEW, OR CASE REPORT WITH LITERATURE REVIEW
A. Each component of the manuscript must start on a new page in the following order:1. Title page2. Abstract3. Text4. Acknowledgments5. References6. Author information7. Tables8. Figures
B. Preparation of manuscript 1. Title page
a. Full title of manuscript with only first letter of first word capitalized (bold title)
b. Initials and last name of each author (no degrees; all CAPS), e.g., M.T. JONES, J.H. BROWN, AND S.R. SMITH
c. Running title of ≤40 characters, including spacesd. Three to ten key words
2. Abstracta. One paragraph, no longer than 300 wordsb. Purpose, methods, findings, and conclusion of study
3. Key wordsa. List under abstract
4. Text (serial pages): Most manuscripts can usually, but not necessarily, be divided into sections (as described below). Survey results and review papers may need individualized sectionsa. Introduction — Purpose and rationale for study, including
pertinent background referencesb. Case Report (if indicated by study) — Clinical and/or hematologic
data and background serology/molecularc. Materials and Methods — Selection and number of subjects,
samples, items, etc. studied and description of appropriate controls, procedures, methods, equipment, reagents, etc. Equipment and reagents should be identified in parentheses by model or lot and manufacturer’s name, city, and state. Do not use patient’s names or hospital numbers.
d. Results — Presentation of concise and sequential results, referring to pertinent tables and/or figures, if applicable
e. Discussion — Implication and limitations of the study, links to other studies; if appropriate, link conclusions to purpose of study as stated in introduction
5. Acknowledgments: Acknowledge those who have made substantial contributions to the study, including secretarial assistance; list any grants.
6. Referencesa. In text, use superscript, Arabic numbers.b. Number references consecutively in the order they occur in the
text.7. Tables
a. Head each with a brief title; capitalize the first letter of first word (e.g., Table 1. Results of . . .) use no punctuation at the end of the title.
b. Use short headings for each column needed and capitalize first letter of first word. Omit vertical lines.
c. Place explanation in footnotes (sequence: *, †, ‡, §, ¶, **, ††).8. Figures
a. Figures can be submitted either by e-mail or as photographs (5″×7″ glossy).
b. Place caption for a figure on a separate page (e.g. Fig. 1 Results of...), ending with a period. If figure is submitted as a glossy, place first author’s name and figure number on back of each glossy submitted.
c. When plotting points on a figure, use the following symbols if possible: l l s s n n.
9. Author informationa. List first name, middle initial, last name, highest degree,
position held, institution and department, and complete address (including ZIP code) for all authors. List country when applicable.
III. EDUCATIONAL FORUM
A. All submitted manuscripts should be approximately 2000 to 2500 words with pertinent references. Submissions may include:1. An immunohematologic case that illustrates a sound investigative
approach with clinical correlation, reflecting appropriate collaboration to sharpen problem solving skills
2. Annotated conference proceedings
B. Preparation of manuscript1. Title page
a. Capitalize first word of title.b. Initials and last name of each author (no degrees; all CAPs)
2. Texta. Case should be written as progressive disclosure and may
include the following headings, as appropriatei. Clinical Case Presentation: Clinical information and
differential diagnosisii. Immunohematologic Evaluation and Results: Serology and
molecular testingiii. Interpretation: Include interpretation of laboratory results,
correlating with clinical findingsiv. Recommended Therapy: Include both transfusion and
nontransfusion-based therapiesv. Discussion: Brief review of literature with unique features of
this casevi. Reference: Limited to those directly pertinentvii. Author information (see II.B.9.)viii. Tables (see II.B.7.)
IV. LETTER TO THE EDITOR
A. Preparation1. Heading (To the Editor)2. Title (first word capitalized)3. Text (written in letter [paragraph] format)4. Author(s) (type flush right; for first author: name, degree, institution,
address [including city, state, Zip code and country]; for other authors: name, degree, institution, city and state)
5. References (limited to ten)6. Table or figure (limited to one)
Send all manuscripts by e-mail to [email protected]
ImmunohematologyJournal of Blood Group Serology and Education
Instructions for Authors

200 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010

IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010 201

202 IMMUNOHEMATOLOGY, Volume 26, Number 4, 2010


FIRST CLASSU.S. POSTAGE
PAIDSOUTHERN MD
PERMIT NO. 4144Musser Blood Center700 Spring Garden StreetPhiladelphia, PA 19123-3594
(Place Label Here)





























