Embed Size (px)
Citation preview

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 1
Nephrology Division, Department of Medicine, Samsung Medical Centre, Samsung Biomedical Research Institute, Sungkyunkwan University School of Medicine, 81 Irwon-Ro Gangnam-gu, Seoul 135-710, South Korea (H.R.J.). Ross Building, Room 965, Nephrology Division, Department of Medicine, Johns Hopkins University School of Medicine, 720 Rutland Avenue, Baltimore, MD 21205, USA (H.R.).
Correspondence to: H.R. [email protected]
Immune cells in experimental acute kidney injuryHye Ryoun Jang and Hamid Rabb
Abstract | Acute kidney injury (AKI) prolongs hospital stay and increases mortality in various clinical settings. Ischaemia–reperfusion injury (IRI), nephrotoxic agents and infection leading to sepsis are among the major causes of AKI. Inflammatory responses substantially contribute to the overall renal damage in AKI. Both innate and adaptive immune systems are involved in the inflammatory process occurring in post-ischaemic AKI. Proinflammatory damage-associated molecular patterns, hypoxia-inducible factors, adhesion molecules, dysfunction of the renal vascular endothelium, chemokines, cytokines and Toll-like receptors are involved in the activation and recruitment of immune cells into injured kidneys. Immune cells of both the innate and adaptive immune systems, such as neutrophils, dendritic cells, macrophages and lymphocytes contribute to the pathogenesis of renal injury after IRI, and some of their subpopulations also participate in the repair process. These immune cells are also involved in the pathogenesis of nephrotoxic AKI. Experimental studies of immune cells in AKI have resulted in improved understanding of the immune mechanisms underlying AKI and will be the foundation for development of novel diagnostic and therapeutic targets. This Review describes what is currently known about the function of the immune system in the pathogenesis and repair of ischaemic and nephrotoxic AKI.
Jang, H. R. & Rabb, H. Nat. Rev. Nephrol. advance online publication 21 October 2014; doi:10.1038/nrneph.2014.180
IntroductionDespite remarkable advances in modern medicine, acute kidney injury (AKI) still remains a challenging condition that lacks specific tools for its early diagnosis and treat-ment. AKI worsens the overall clinical course of affected patients by causing uraemia, acid–base or electrolyte disturbances, and volume overload. The incidence of AKI has been reported to be as high as 5% of hospital-ized patients or 30% of critically ill patients.1 The risk of chronic kidney disease and end-stage renal disease is sub-stantially increased in patients with AKI.2 Most patients with AKI are diagnosed when injury is already estab-lished and, therefore, only conservative treatment includ-ing fluid therapy and dialysis is available. To improve the clinical outcome of AKI, novel diagnostic and therapeu-tic strat egies need to be developed. Understanding the pathophysi ology of AKI is, therefore, the cornerstone of exploration of novel diagnostic and therapeutic strategies.
Experimental models of AKI can be divided into several categories depending on the induction method (Figure 1). In models of septic AKI, the initial immune response against foreign antigens and innate triggers causes a complex secondary inflammatory response that facili-tates renal injury.3 Non-septic and septic AKI are known to have very different pathophysiological features. Septic AKI is a systemic manifestation of sepsis following exposure to foreign antigens such as bacteria or viruses; detailed discussion of septic AKI is beyond the scope of this review.
Immune mechanisms were not expected to have an important role in models of aseptic AKI, but numer-ous studies conducted over the past two decades have revealed that inflammatory processes mediated by the immune system are crucial in mediating renal injury.3 Immune mechanisms involved in the pathogenesis of renal injury have been studied most extensively in models of ischaemic AKI employing cold or warm ischaemia. Both types of ischaemia occur during organ transplanta-tion; cold ischaemia starts when the organ is cooled with cold perfusion solution after procurement, and lasts until the temperature of the organ reaches the physiologic tem-perature. Thereafter, warm ischaemia begins, and ends when perfusion is restored after completion of surgical anastomosis. Thus, two distinct periods of warm ischae-mia occur in the transplantation setting—during organ retrieval and implantation.4 Interestingly, the nephro toxi-city induced by cisplatin, a chemotherapeutic agent, has many pathophysiological features that overlap with those of ischaemia–reperfusion injury (IRI).
Both innate and adaptive immune systems are directly involved in the pathogenesis of ischaemic AKI. Various cellular and humoral immune system components con-tribute to AKI, some of which are also thought to be involved in the repair process following IRI.5,6 The healthy kidney produces hormones that influence the immune system, such as vitamin D (calcitriol) and erythropoi-etin,7 and the renal tubular epithelium expresses Toll-like receptors (TLRs), which critically contribute to activation of the complement system and recruitment of immune
Competing interestsThe authors declare no competing interests.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

2 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
cells in response to inflammatory stimuli.8,9 Several types of resident immune cells, such as dendritic cells, macro-phages, mast cells and lymphocytes are homeostati-cally maintained in the normal kidney, although these cells constitute a small population.10–13 Under normal conditions, the renal mononuclear phagocytes mainly comprise macrophages located in the renal medulla and capsule and renal dendritic cells found in the tubulo-interstitium.10,11,14 In mice, renal dendritic cells show a specific CD11c+CD11b+EMR1(F4/80)+CX3CR1 (CX3C-chemokine receptor)+CD8–CD205– phenotype, and have a similar transcriptome as dendritic cells residing in other nonlymphoid tissues.15,16 Dendritic cells are recruited to the kidney by a CX3CR1–CX3CL1 (CX3C-chemokine ligand 1, also known as fractalkine) chemokine pair,17 and have an important role in local injury or infection. Dendritic cells not only function as a potent source of other factors, such as neutrophil-recruiting chemokines and cytokines,12,18 but also present antigens to T cells.
Key points
■ Various components of the innate and adaptive immune systems are implicated in the pathogenesis and repair of acute kidney injury (AKI)
■ The roles of individual immune cell types have been most thoroughly investigated in models of ischaemic AKI
■ Various immune cells traffic into the post-ischaemic kidney and show changes in phenotypes and numbers depending on the time course after establishment of ischaemic AKI
■ The roles of macrophages, renal dendritic cells and T regulatory cells differ according to the pathogenesis of AKI
■ Although numerous studies in animal models of AKI show therapeutic potential for modulating immune cells, big hurdles must be overcome before applying these findings to patients
■ Functions and interactions of specific immune cell types and humoral factors in AKI differ between human disease and animal models, and depend on the type and stage of injury
Intrarenal macrophages exert homeostatic functions by phagocytosis of antigens in the kidney and undergo pheno typic changes that enable them to participate in both inflammatory and anti-inflammatory processes.14 Both dendritic cells and macrophages contribute substan-tially to homeostasis and regulation of immune responses (as resident renal mononuclear phagocytes) in the normal kidney. Mast cells also reside in the tubulointerstitium and mediate pathogenic processes in crescentic and other forms of glomerulonephritis. However, the exact roles of dendritic cells, macrophages and mast cells in the normal kidney are yet to be elucidated.19–21 Lymphocytes, including both T cells and B cells, have been found in normal mouse kidneys even after extensive exsanguin-ation and perfusion.22 Intrarenal resident T cells show distinctly different phenotypes from T cells in spleen and blood; those from normal mouse kidneys contain an increased percentage of CD3+CD4–CD8– double-negative T cells. Intrarenal T cells also show a high proportion of activated, effector and memory phenotypes, whereas a small percentage of regulatory T cells and natural killer (NK) T cells exist in perfused and exsanguinated mouse kidney.22
In this Review, we describe how immune cells partici-pate in the pathogenesis of AKI, focusing on ischaemic and nephrotoxic AKI. Immune system function in septic AKI is only outlined in this article, because the pathophysi-ology of septic AKI includes both immune responses to various foreign antigens and secondary systemic inflam-matory responses, which are distinctly different to the immune responses that occur in aseptic AKI.
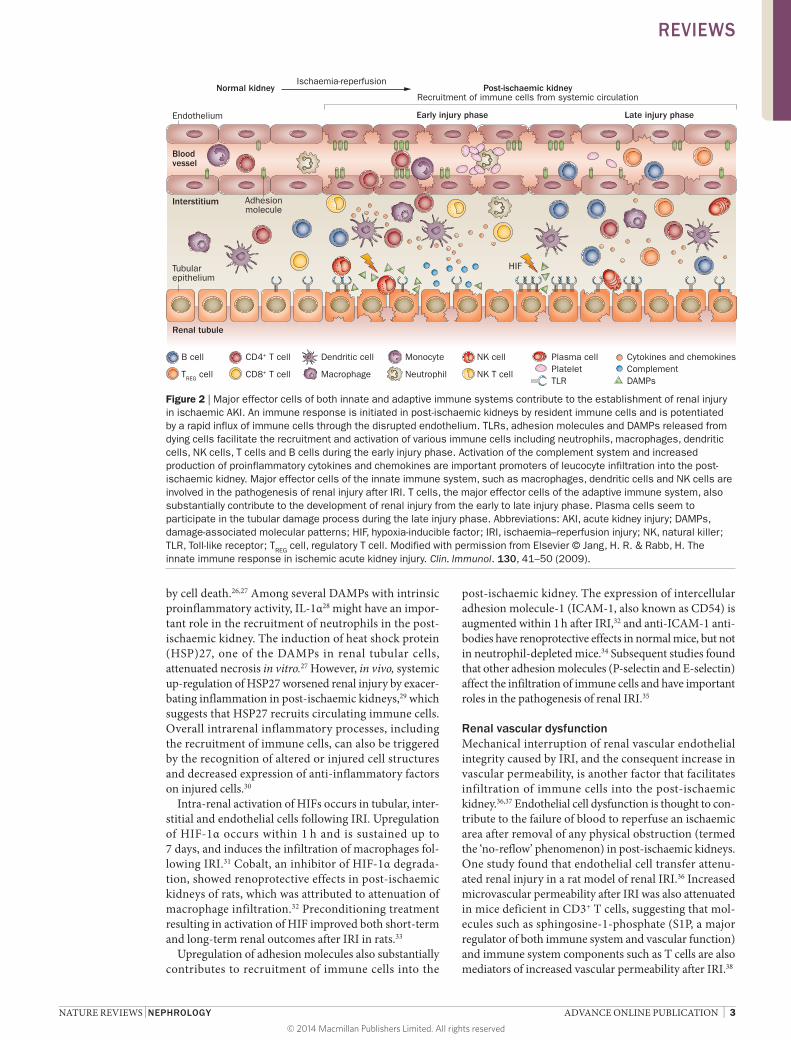
Aseptic ischaemic AKIRobust inflammatory responses mediated by the immune system start during the initial ischaemic insult and accel-erate upon reperfusion of the post-ischaemic kidney. However, post-ischaemic kidneys are not only targets of the immune system, but can also interact with systemic immune factors to recruit and activate immune cells. The mechanisms underlying activation and recruitment of immune cells in the post-ischaemic kidney involve proinflammatory damage-associated molecular pat-terns (DAMPs) in conjunction with hypoxia-inducible factors (HIFs) and adhesion molecules. These initiators of the inflammatory process cause permeability dysfunc-tion of the renal vascular endothelium and are associated with the release of proinflammatory chemokines and cytokines, and activation of TLRs (Figure 2). Various immune cells of both the innate and adaptive immune systems also have critical functions in the pathogenesis of renal injury following IRI (Tables 1 and 2).
DAMPs, HIFs and adhesion moleculesDAMPs normally exist in the intracellular compart-ment and are concealed from the immune system by the plasma membrane.23,24 Proinflammatory DAMPs are released or exposed following hypoxic or anoxic cell injury, after which they can activate the innate immune system.25 Uric acid and nonmethylated CpG-rich DNA are DAMPs that contribute to the inflammation induced
Figure 1 | Experimental models of AKI. Models of AKI can be broadly categorized according to whether foreign antigens are involved (aseptic or septic AKI). Each category can be subdivided according to the method used to induce AKI. Ischaemic AKI is induced by ischaemia–reperfusion injury and by the type of ischaemia (warm or cold). Nephrotoxic AKI is induced by nephrotoxic agents, such as cisplatin. Abbreviation: AKI, acute kidney injury.
SepticImmune response against foreign antigens is followed by a complex secondary in�ammatory response leading to renal injury
Experimental models of AKI
AsepticRenal injury is driven by in�ammatory responses mediated by the immune system, in the abscence of direct alloantigen stimulation
Ischaemic AKIInduced by ischaemia –reperfusion injury
Nephrotoxic AKIInduced by nephrotoxic agents such as cisplatin
Induced by lipopolysaccharide injection
Induced by caecal ligation
Warm ischaemiaInduced in vivo by surgical clamping of renal pedicels under normothermic conditions
Cold ischaemiaInduced in vivo by surgical clamping of renal pedicels under University of Wisconsin solution �ow at 4°C
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved
NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 3
by cell death.26,27 Among several DAMPs with intrinsic proinflammatory activity, IL-1α28 might have an impor-tant role in the recruitment of neutrophils in the post-ischaemic kidney. The induction of heat shock protein (HSP)27, one of the DAMPs in renal tubular cells, attenuated necrosis in vitro.27 However, in vivo, systemic up-regulation of HSP27 worsened renal injury by exacer-bating inflammation in post-ischaemic kidneys,29 which suggests that HSP27 recruits circulating immune cells. Overall intrarenal inflammatory processes, including the recruitment of immune cells, can also be triggered by the recognition of altered or injured cell structures and decreased expression of anti-inflammatory factors on injured cells.30
Intra-renal activation of HIFs occurs in tubular, inter-stitial and endothelial cells following IRI. Upregulation of HIF-1α occurs within 1 h and is sustained up to 7 days, and induces the infiltration of macrophages fol-lowing IRI.31 Cobalt, an inhibitor of HIF-1α degrada-tion, showed renoprotective effects in post-ischaemic kidneys of rats, which was attributed to attenuation of macrophage infiltration.32 Preconditioning treatment resulting in activation of HIF improved both short-term and long-term renal outcomes after IRI in rats.33
Upregulation of adhesion molecules also substantially contributes to recruitment of immune cells into the
post-ischaemic kidney. The expression of intercellular adhesion molecule-1 (ICAM-1, also known as CD54) is augmented within 1 h after IRI,32 and anti-ICAM-1 anti-bodies have renoprotective effects in normal mice, but not in neutrophil-depleted mice.34 Subsequent studies found that other adhesion molecules (P-selectin and E-selectin) affect the infiltration of immune cells and have important roles in the pathogenesis of renal IRI.35
Renal vascular dysfunctionMechanical interruption of renal vascular endothelial integrity caused by IRI, and the consequent increase in vascular permeability, is another factor that facilitates infiltration of immune cells into the post-ischaemic kidney.36,37 Endothelial cell dysfunction is thought to con-tribute to the failure of blood to reperfuse an ischaemic area after removal of any physical obstruction (termed the ‘no-reflow’ phenomenon) in post-ischaemic kidneys. One study found that endothelial cell transfer attenu-ated renal injury in a rat model of renal IRI.36 Increased micro vascular permeability after IRI was also attenuated in mice deficient in CD3+ T cells, suggesting that mol-ecules such as sphingosine-1-phosphate (S1P, a major regulator of both immune system and vascular function) and immune system components such as T cells are also mediators of increased vascular permeability after IRI.38
Late injury phase
Post-ischaemic kidneyRecruitment of immune cells from systemic circulation
Early injury phaseEndothelium
Bloodvessel
Tubularepithelium
Plasma cellPlateletTLR
HIF
Cytokines and chemokinesComplementDAMPs
B cell
TREG cell
CD4+ T cell
CD8+ T cell
Dendritic cell
Macrophage
Monocyte
Neutrophil
NK cell
NK T cell
Renal tubule
Ischaemia-reperfusion
Adhesionmolecule
Normal kidney
Interstitium
Figure 2 | Major effector cells of both innate and adaptive immune systems contribute to the establishment of renal injury in ischaemic AKI. An immune response is initiated in post-ischaemic kidneys by resident immune cells and is potentiated by a rapid influx of immune cells through the disrupted endothelium. TLRs, adhesion molecules and DAMPs released from dying cells facilitate the recruitment and activation of various immune cells including neutrophils, macrophages, dendritic cells, NK cells, T cells and B cells during the early injury phase. Activation of the complement system and increased production of proinflammatory cytokines and chemokines are important promoters of leucocyte infiltration into the post-ischaemic kidney. Major effector cells of the innate immune system, such as macrophages, dendritic cells and NK cells are involved in the pathogenesis of renal injury after IRI. T cells, the major effector cells of the adaptive immune system, also substantially contribute to the development of renal injury from the early to late injury phase. Plasma cells seem to participate in the tubular damage process during the late injury phase. Abbreviations: AKI, acute kidney injury; DAMPs, damage-associated molecular patterns; HIF, hypoxia-inducible factor; IRI, ischaemia–reperfusion injury; NK, natural killer; TLR, Toll-like receptor; TREG cell, regulatory T cell. Modified with permission from Elsevier © Jang, H. R. & Rabb, H. The innate immune response in ischemic acute kidney injury. Clin. Immunol. 130, 41–50 (2009).
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved
4 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
Cytokines and chemokinesCytokines and chemokines are crucial mediators that regulate the infiltration of immune cells into post- ischaemic kidneys. Cytokine production is facilitated in the post-ischaemic kidney through interaction between cytokines and the transcriptional response induced directly by hypoxia. Intrarenal activation of transcrip-tion factors such as nuclear factor κB (NF-κB), heat shock factor protein 1 and HIF-1α occurs after IRI39,40 and stimulates the synthesis of a cascade of proinflam-matory cytokines, such as IL-1, IL-6 and tumour necro-sis factor (TNF).35,41,42 Splenectomy attenuated renal IRI by decreasing systemic production of inflammatory cytokines, including TNF, in rats.43 Chemokines are also direct mediators of chemotaxis and activation of immune cells: specifically, they guide neutrophils and
proinflammatory (M1) macrophages to the injury site.44,45 Previous studies showed that IL-8 (also known as C-X-C motif chemokine ligand 8, or CXCL8) induced neutrophil recruitment into the post-ischaemic kidney.46,47 The aug-mented expression of CXC receptor 3 (CXCR3) follow-ing IRI orchestrates recruitment of T helper type 1 (TH1) cells into the post-ischaemic kidney because this receptor is predominantly expressed on activated TH1 cells.48 The infiltration and activation of macrophages following IRI are enhanced by C-C motif chemokine 2 (also known as monocyte chemo attractant protein 1, or MCP-1) via C-C chemokine receptor type 2 (CCR2) signalling49 and C-X3-C motif chemokine receptor 1 (CX3CR1, also known as fractalkine receptor) signalling, which regulates the infiltration and phenotype change of macrophages, and affects renal interstitial fibrosis.50
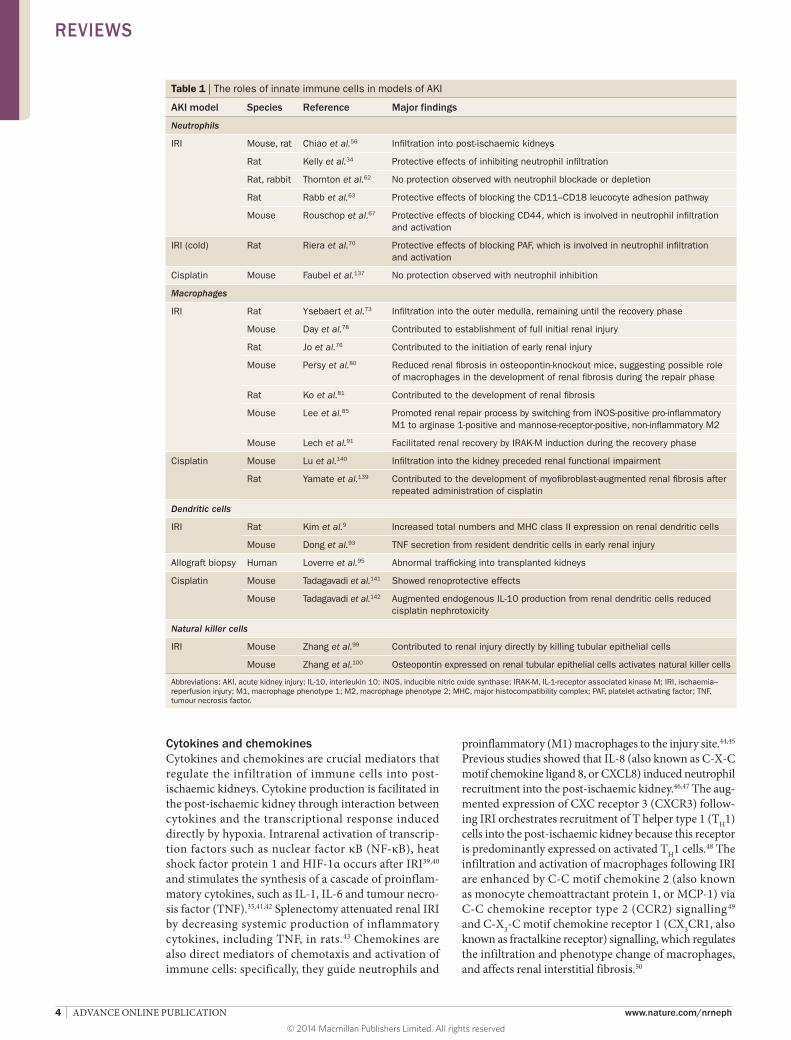
Table 1 | The roles of innate immune cells in models of AKI
AKI model Species Reference Major findings
Neutrophils
IRI Mouse, rat Chiao et al.56 Infiltration into post-ischaemic kidneys
Rat Kelly et al.34 Protective effects of inhibiting neutrophil infiltration
Rat, rabbit Thornton et al.62 No protection observed with neutrophil blockade or depletion
Rat Rabb et al.63 Protective effects of blocking the CD11–CD18 leucocyte adhesion pathway
Mouse Rouschop et al.67 Protective effects of blocking CD44, which is involved in neutrophil infiltration and activation
IRI (cold) Rat Riera et al.70 Protective effects of blocking PAF, which is involved in neutrophil infiltration and activation
Cisplatin Mouse Faubel et al.137 No protection observed with neutrophil inhibition
Macrophages
IRI Rat Ysebaert et al.73 Infiltration into the outer medulla, remaining until the recovery phase
Mouse Day et al.78 Contributed to establishment of full initial renal injury
Rat Jo et al.76 Contributed to the initiation of early renal injury
Mouse Persy et al.80 Reduced renal fibrosis in osteopontin-knockout mice, suggesting possible role of macrophages in the development of renal fibrosis during the repair phase
Rat Ko et al.81 Contributed to the development of renal fibrosis
Mouse Lee et al.85 Promoted renal repair process by switching from iNOS-positive pro-inflammatory M1 to arginase 1-positive and mannose-receptor-positive, non-inflammatory M2
Mouse Lech et al.91 Facilitated renal recovery by IRAK-M induction during the recovery phase
Cisplatin Mouse Lu et al.140 Infiltration into the kidney preceded renal functional impairment
Rat Yamate et al.139 Contributed to the development of myofibroblast-augmented renal fibrosis after repeated administration of cisplatin
Dendritic cells
IRI Rat Kim et al.9 Increased total numbers and MHC class II expression on renal dendritic cells
Mouse Dong et al.93 TNF secretion from resident dendritic cells in early renal injury
Allograft biopsy Human Loverre et al.95 Abnormal trafficking into transplanted kidneys
Cisplatin Mouse Tadagavadi et al.141 Showed renoprotective effects
Mouse Tadagavadi et al.142 Augmented endogenous IL-10 production from renal dendritic cells reduced cisplatin nephrotoxicity
Natural killer cells
IRI Mouse Zhang et al.99 Contributed to renal injury directly by killing tubular epithelial cells
Mouse Zhang et al.100 Osteopontin expressed on renal tubular epithelial cells activates natural killer cells
Abbreviations: AKI, acute kidney injury; IL-10, interleukin 10; iNOS, inducible nitric oxide synthase; IRAK-M, IL-1-receptor associated kinase M; IRI, ischaemia–reperfusion injury; M1, macrophage phenotype 1; M2, macrophage phenotype 2; MHC, major histocompatibility complex; PAF, platelet activating factor; TNF, tumour necrosis factor.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved
NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 5
TLRsTLR expression on renal tubular epithelial cells is an important contributor to the recruitment and activa-tion of immune cells, especially effector cells of the innate immune system. TLR2 and TLR4 are expressed on normal renal tubular epithelial cells and their expres-sion further increases after IRI.8,9,51 DAMPs such as histones or high-mobility-group protein B1 released from necrotic tubules activate TLRs on dendritic cells or macrophages and inflammasomes in the cytosol to trigger the secretion of proinflammatory cytokines and chemokines in the post-ischaemic kidney.51–55
NeutrophilsNeutrophils are important effector cells of the innate immune system that phagocytose pathogens and par-ticles, generate reactive oxygen and nitrogen species, and release antimicrobial peptides. Neutrophil infil-tration has been detected in post-ischaemic mouse kidneys56,57 and in biopsy samples from patients with early AKI.58,59 Neutrophils were, therefore, expected to have an important role in the pathogenesis of renal injury following IRI.
IL-17 produced by neutrophils regulates IFN-γ-mediated neutrophil migration into the post-ischaemic kidney,60 and warm ischaemia promotes neutrophil trafficking into the post-ischaemic kidney in mice.61 However, the precise role and kinetics of neutrophil trafficking into the post-ischaemic kidney after IRI remain controversial, despite many studies focusing on the role of neutrophils in renal IRI. In one study, renal injury was attenuated by inhibition of neutrophil infiltration or activity in rats,34 whereas others failed to find a protective effect of neutrophil blockade or deple-tion.62,63 Many factors that affect neutrophil infiltration or activation, including neutrophil elastase, tissue type plasminogen activator, hepatocyte growth factor and CD44 expression contribute to renal injury following IRI.64–67 Treatments that target several adhesion mol-ecules involved in migration of neutrophils (as well as other leucocytes), such as selectins, ICAM-1, and CD11a–CD18 (integrin αLβ2, also known as lympho-cyte function-associated antigen-1, LFA-1), exert partial protection in the post-ischaemic kidney in rodents.34,57,63 A phase I clinical trial of ICAM-1-blocking antibodies showed a reduced rate of delayed graft function fol-lowing kidney transplantation in the treated group.68 However, a randomized controlled trial of anti-ICAM-1 monoclonal antibody in recipients of cadaveric renal transplants failed to show a reduction in the rate of delayed graft function or acute rejection.69 Blockade of platelet-activating factor (PAF), which facilitates neutrophil adherence to the endothelium, also had a protective effect in a rat model of cold IRI.70
Despite conflicting results reported thus far, neutro-phils are likely to participate in the induction of renal injury, by obstructing the renal microvasculature and secreting oxygen free radicals and proteases. It is likely that neutrophils have a much less important role in renal IRI than they do in cardiac or skeletal muscle IRI.59,71,72
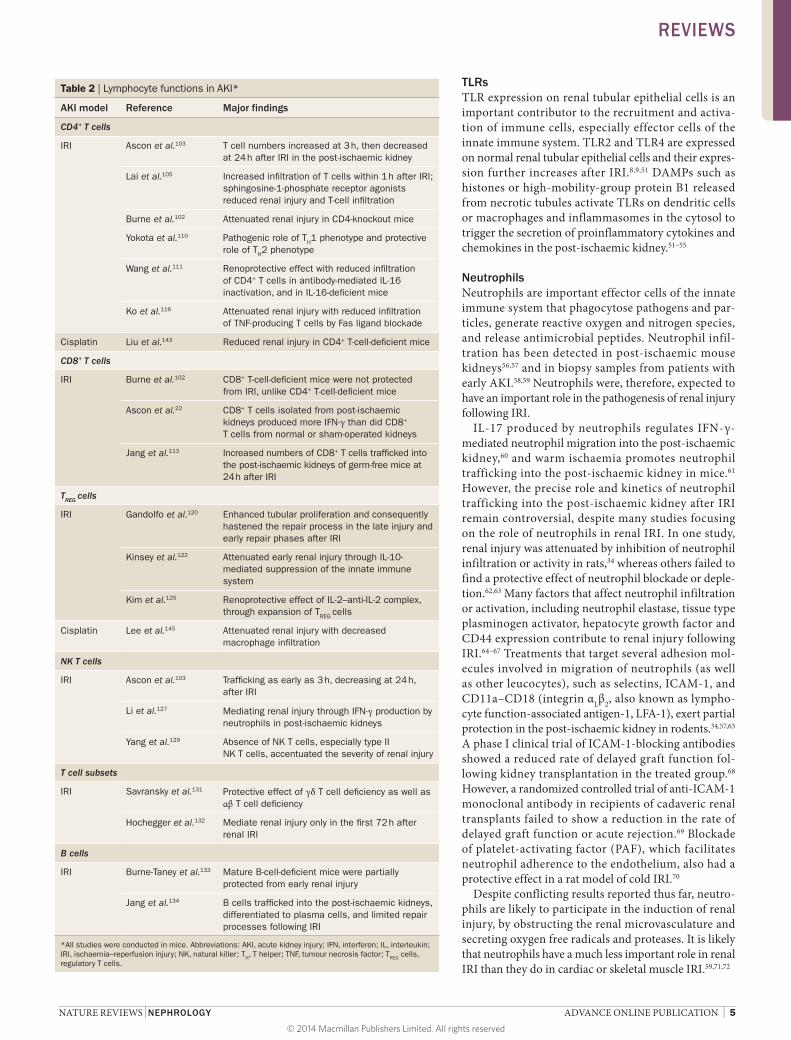
Table 2 | Lymphocyte functions in AKI*
AKI model Reference Major findings
CD4+ T cells
IRI Ascon et al.103 T cell numbers increased at 3 h, then decreased at 24 h after IRI in the post-ischaemic kidney
Lai et al.105 Increased infiltration of T cells within 1 h after IRI; sphingosine-1-phosphate receptor agonists reduced renal injury and T-cell infiltration
Burne et al.102 Attenuated renal injury in CD4-knockout mice
Yokota et al.110 Pathogenic role of TH1 phenotype and protective role of TH2 phenotype
Wang et al.111 Renoprotective effect with reduced infiltration of CD4+ T cells in antibody-mediated IL-16 inactivation, and in IL-16-deficient mice
Ko et al.116 Attenuated renal injury with reduced infiltration of TNF-producing T cells by Fas ligand blockade
Cisplatin Liu et al.143 Reduced renal injury in CD4+ T-cell-deficient mice
CD8+ T cells
IRI Burne et al.102 CD8+ T-cell-deficient mice were not protected from IRI, unlike CD4+ T-cell-deficient mice
Ascon et al.22 CD8+ T cells isolated from post-ischaemic kidneys produced more IFN-γ than did CD8+ T cells from normal or sham-operated kidneys
Jang et al.113 Increased numbers of CD8+ T cells trafficked into the post-ischaemic kidneys of germ-free mice at 24 h after IRI
TREG cells
IRI Gandolfo et al.120 Enhanced tubular proliferation and consequently hastened the repair process in the late injury and early repair phases after IRI
Kinsey et al.122 Attenuated early renal injury through IL-10-mediated suppression of the innate immune system
Kim et al.125 Renoprotective effect of IL-2–anti-IL-2 complex, through expansion of TREG cells
Cisplatin Lee et al.145 Attenuated renal injury with decreased macrophage infiltration
NK T cells
IRI Ascon et al.103 Trafficking as early as 3 h, decreasing at 24 h, after IRI
Li et al.127 Mediating renal injury through IFN-γ production by neutrophils in post-ischaemic kidneys
Yang et al.129 Absence of NK T cells, especially type II NK T cells, accentuated the severity of renal injury
T cell subsets
IRI Savransky et al.131 Protective effect of γδ T cell deficiency as well as αβ T cell deficiency
Hochegger et al.132 Mediate renal injury only in the first 72 h after renal IRI
B cells
IRI Burne-Taney et al.133 Mature B-cell-deficient mice were partially protected from early renal injury
Jang et al.134 B cells trafficked into the post-ischaemic kidneys, differentiated to plasma cells, and limited repair processes following IRI
*All studies were conducted in mice. Abbreviations: AKI, acute kidney injury; IFN, interferon; IL, interleukin; IRI, ischaemia–reperfusion injury; NK, natural killer; TH, T helper; TNF, tumour necrosis factor; TREG cells, regulatory T cells.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

6 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
MacrophagesMacrophages were expected to have an important func-tion in immune-mediated renal injury because these cells function as both effector cells and antigen-presenting cells, thereby connecting the innate and adaptive immune systems. Activated macrophages exert potent phagocytic activity and release several important cytokines, such as IL-1, IL-6, IL-8, IL-12 and TNF. Although the resident macrophages in normal kidneys are few, their number markedly increases in post-ischaemic kidneys (espe-cially in the outer medulla), soon after IRI.73 Monocytes adhere to the vasa recta 2 h after reperfusion, and most macrophage recruitment occurs around post-capillary venules in the outer medulla.74 IRI facilitated endothelial damage and modifications of heparin sulphate proteo-glycans in the microvascular basement membrane, which promoted their binding to L-selectin, as well as induc-tion of MCP-1. These changes induced the early influx of monocytes and macrophages into the post-ischaemic kidney.75
Macrophage influx upon reperfusion of the post-ischaemic kidney seems to facilitate the inflammatory cascade through secretion of cytokines, recruitment of neutrophils and induction of apoptosis, which contribute to the establishment of renal injury. Systemic depletion of monocytes and macrophages using liposomal clodronate attenuated early renal injury in a mouse model of renal IRI.76 Although IL-18 was suggested as a key mediator
of macrophage influx in the pathogenesis of IRI, a study of liposomal clodronate treatment in wild-type and cas-pase-1-knockout mice revealed that macrophages are not the source of the injurious IL-18 in ischaemic AKI.77 Although macrophages do have a role in injury occurring in the early phase of IRI,76,78 the augmented production of haem oxygenase 1 by infiltrated macrophages has been associated with the protective effects of statins in AKI.79
Macrophages are also suspected to have a role in renal repair following IRI (Figure 3). In one study, post- ischaemic kidneys of mice with knockout of osteopontin (a macrophage chemoattractant) had fewer infiltrating macrophages and less fibrosis than did post-ischaemic kidneys of wild-type mice.80 A few reports show that macrophages influence the development of renal fibro-sis during the recovery phase of IRI, which supports the concept that macrophages have an adverse effect on the repair of post-ischaemic kidneys.81,82 However, macrophage-specific deletion of transforming growth factor (TGF)-β1 did not halt the process of renal fibrosis following severe IRI.83 Colony-stimulating factor-1 pro-motes renal repair and attenuates interstitial fibrosis by inducing the expression of insulin-like growth factor-1 and anti-inflammatory genes in macrophages.84 One well-designed study showed that macrophages promote the renal repair process by switching from a proinflamma-tory M1 phenotype characterized by expression of indu-cible nitric oxide synthase, to an anti-inflammatory (M2)
Figure 3 | Immune modulation during the repair phase of ischaemic AKI is a key factor in determining the outcome of AKI. TREG cells, B cells and macrophages have substantial roles in determining whether repair results in tubular regeneration or atrophy and interstitial fibrosis. TREG cells and M2 macrophages have important roles in tubular regeneration, whereas B cells enhance tubular atrophy and suppress tubular regeneration. Humoral factors, such as proinflammatory or anti-inflammatory cytokines and chemokines, also change the intrarenal microenvironment and affect phenotype switching of macrophages. The exact mechanisms by which these immune processes regulate tubular atrophy or regeneration are not yet known. Abbreviations: AKI, acute kidney injury; IL-10, interleukin-10; TGF-β, transforming growth factor β; TREG cells, regulatory T cells. Modified with permission from Elsevier © Jang, H. R. & Rabb, H. The innate immune response in ischemic acute kidney injury. Clin. Immunol. 130, 41–50 (2009).
Endothelium
Bloodvessel
Interstitium
Tubularepithelium
Renal tubule
Adhesionmolecule
Tubular regeneration Atrophy and �brosis
Plasma cell
Platelet
IL-10
TGF-β
B cell
TREG cell
CD4+ T cell
Monocyte
Dendritic cell
M2 macrophage
Fibrotic tissue
Repair phase
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 7
phenotype characterized by expression of arginase-1 and the mannose receptor.85 This report suggests that macrophages have a complex role in both IRI-induced inflammation and the subsequent repair process.
Switching of macrophages to an anti-inflammatory M2 phenotype seems to be induced by changes in the intrarenal microenvironment as well as by the phago-cytic uptake of apoptotic neutrophils by macrophages during the injury phase in post-ischaemic kidneys.45,86 In mouse models of renal IRI and selective proximal tubule injury induced by diphtheria toxin, the increased number of M2 phenotype macrophages resulted mainly from in situ proliferation of resident renal macrophages. Furthermore, genetic or pharmacological inhibition of macrophage colony-stimulating factor 1 (CSF-1) signal-ling blocked intrarenal proliferation of macrophages and dendritic cells, reduced M2 polarization, and inhibited renal recovery.87 Treatment of macrophages with netrin 1 suppressed the inflammatory response by inducing conversion to an M2 phenotype, which protected the kidney against subsequent IRI.88 IL-1 receptor-associated kinases (IRAKs) are involved in the IL-1 receptor–TLR–Myd88-dependent activation of NF-κB and are impor-tant regulators of macrophage phenotype polarization.89 IRAK-M selectively inhibits IRAK-4–mediated phos-phorylation of TNF receptor–associated factor 6, which is an essential step in this signalling pathway in mono-cytes and macrophages.90 IRAK-M induction during the recovery phase after renal IRI facilitates renal recovery by suppressing M1-macrophage-dependent renal inflam-mation, whereas IRAK-M inhibition (achieved by loss-of-function mutations or transient exposure to bacterial DNA) halted the repair process and induced persistent macrophage-related renal inflammation.91
Renal dendritic cellsThe basic function of dendritic cells is the presentation of antigens to T cells; thus, they act as messengers between the innate and adaptive immune systems. The results of several studies show that dendritic cells participate in ischaemic AKI. In a rat model of transplant-induced IRI, recipient leucocytes that expressed MHC class II antigens were trafficked into the transplanted kidney despite no signs of acute rejection, and some of them were identified as dendritic cells.92 The number of renal dendritic cells and their expression of MHC class II antigens increased after IRI.9 A subsequent study revealed that the population of resident dendritic cells predominantly consists of TNF-secreting cells in the early phase of AKI following IRI.93 Furthermore, binding of dendritic cells to the endo thelium and their migration seem to be facilitated during the initial inflammatory response following IRI.94 Trafficking of immature myeloid dendritic cells into the transplanted kidneys is also increased following IRI, resulting in an increased ratio of myeloid to plasmacytoid dendritic cells that might predispose to delayed graft function and acute rejection.95 In a study of syngeneic kidney transplantation from wild-type rats to transgenic rats expressing green fluorescent protein, cold IRI was associated with loss of graft-specific dendritic cells and progressive recruitment
of host dendritic cells and T cells.96 Contrary to previous studies, this report suggested that renal resident dendritic cells might have protective regulatory functions in the post-ischaemic kidney.
LymphocytesLymphocytes are key cells of the adaptive immune system. Lymphocytes were not expected to contribute to post-ischaemic AKI, given the traditional concept that lymphocytes respond to alloantigens or self-antigens in a delayed fashion. However, many studies performed during the past decade have revealed the substantial role of a diverse subset of lymphocytes in post-ischaemic and nephrotoxic AKI.
Natural killer cellsNK cells are a class of large, granular, cytotoxic lym-phocytes that lack T-cell and B-cell receptors. They kill infected cells directly and produce a variety of cytokines, including IFN-γ and TNF. NK cells were expected to have a role in inducing renal injury following IRI because in other organs, they secrete cytokines that facilitate the inflammatory process and activate macrophages and neutrophils.97,98 So far, few reports exist on the role of NK cells in AKI. NK cells were reported to contribute directly to renal injury following IRI by killing tubular epithelial cells, and in the same report, depletion of NK cells attenuated renal injury after IRI both functionally and structurally.99 A subsequent study by the same team reported that osteopontin expressed on renal tubular epithelial cells can directly activate NK cells to mediate apoptosis of tubular epithelial cells, and can also regulate chemotaxis of NK cells to the tubular epithelium.100
CD4+ and CD8+ T cellsSeveral research teams have reported that T cells, particu-larly CD4+ T cells, contribute both directly and indirectly to the establishment of renal injury in the early phase of IRI.74,101–105 T-cell-targeted medications such as tacroli-mus and mycophenolate mofetil substantially attenuated early renal injury following IRI.106,107 Blockade of the T-cell CD28–B7 co-stimulatory pathway with CTLA-4–Ig (a recombinant fusion protein containing CTLA-4, a structural homologue of CD28, fused to an IgG1 heavy chain), also substantially reduced early renal injury after cold IRI.108 Furthermore, CTLA-4–Ig treatment on the day of cold IRI and during the first week after cold IRI decreased proteinuria in uninephrectomized rats (a model of chronic, progressive proteinuria).109
Direct evidence of the pathophysiological role of T cells in ischaemic AKI was demonstrated in a mouse model of warm IRI.101 In this study, CD4,CD8 double-knockout mice were largely protected from early renal injury, and their T cells showed a twofold increase in adherence to renal tubular epithelial cells in vitro after hypoxia and reoxygenation. Another T-cell-knockout mouse strain, athymic Foxn1nu/nu mice, was also protected from IRI. Adoptive transfer of T cells into these mice restored renal injury following IRI, demonstrating that T-cell deficiency conferred renal protection from IRI.102 CD4-knockout
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

8 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
mice, but not CD8-knockout mice, were substantially less susceptible to renal IRI.102 Adoptive transfer of CD4+ T cells into CD4-knockout mice restored early renal injury following IRI.102 Both surface expression of CD28 and pro-duction of IFN-γ were key factors in the pathogenic effects of CD4+ T cells on the post-ischaemic kidney. Another study showed that the protective effects of adenosine 2A receptor agonists against IRI are mediated by specific actions on CD4+ T cells.104 A study of CD4+ T-cell subsets in a mouse model of renal IRI revealed that CD4+ T cells of the TH1 phenotype are pathogenic, whereas CD4+ T cells of the TH2 phenotype are protective. This work was per-formed using mice with targeted deletions in signal trans-ducer and activator of transcription (STAT)4 and STAT6, which regulate differentiation of T cells towards TH1 (IFN-γ-producing) or TH2 (IL-4-producing) phenotypes, respectively. STAT4-deficient mice were partially pro-tected from renal IRI, whereas STAT6-deficient mice had markedly impaired renal function and tubular injury.110 In this study, both IL-4-deficient mice and STAT6-deficient mice showed similar post-ischaemic injury, which sug-gested that IL-4 is a key mediator of STAT6-mediated renoprotection after IRI. The importance of CD4+ T cells in early renal injury after IRI is also supported by a report showing that inactivation of IL-16 (a T cell chemoattract-ant that is strongly expressed on renal tubules after IRI) using either blocking antibodies or IL-16 deficiency led to a reduction in CD4+ T-cell infiltration and attenuation of renal injury.111 Moreover, CD4+ T cells were reported to be involved in the splenic cholinergic anti- inflammatory pathway and to mediate the renoprotective effect of ultrasound pretreatment in renal IRI.112
Although the renal outcomes of CD8-knockout mice were similar to those of wild-type mice after IRI,102 the role of CD8+ T cells in ischaemic AKI is yet to be fully determined. Normal mouse kidneys contain more effec-tor memory CD4+CD8+ T cells (which also express CD44high and CD62Llow) than do either the spleen or blood.22 CD8+ T cells isolated from post-ischaemic kidneys produce more IFN-γ than do CD8+ T cells from normal or sham-operated kidneys.22 Moreover, renal injury after IRI was both functionally and struc-turally more severe in germ-free mice than in wild-type mice; furthermore, trafficking of CD8+ T cells into post- ischaemic kidneys was increased in germ-free mice at 24 h after IRI compared with their wild-type counter-parts.113 This report suggests that naive CD8+ T cells, which have a low chance of exposure to microbial stimuli, could infiltrate post-ischaemic kidneys and orchestrate the establishment of renal injury following IRI.
CD3+CD4−CD8− double-negative T cells are expected to participate in the pathogenesis of renal IRI. These cells comprise a higher percentage of the T cell population in normal mouse kidneys than is observed in other organs.22 Treatment with mouse anti-thymocyte globulin did not attenuate renal injury following IRI despite profound depletion of CD4+ and CD8+ T cells from the kidneys as well as from peripheral blood.114 In this study, a substan-tial portion of CD3+CD4−CD8− double-negative T cells remained in the post-ischaemic kidneys of mice treated
with anti-thymocyte globulin, which suggested a possible pathogenic role of these cells in early renal injury after IRI.
Although T cells critically contribute to initial renal injury following IRI, whether antigen T-cell recep-tor (TCR) engagement followed by antigen-specific T-cell activation has an important role in such injury was unknown. TCR engagement contributed to estab-lishing full renal injury after IRI, though alloantigen-independent activation in AKI could also participate.103 A study using Foxn1nu/nu mice and DO11.10 transgenic mice (which have a limited TCR repertoire, resulting in T cells that only recognize chicken ova antigens) demon-strated that both TCR-repertoire-dependent and TCR-repertoire-independent factors mediate the pathogenic functions of T cells in renal IRI.115 Fas ligand (FasL) blockade attenuated renal injury with reduced infiltra-tion of TNF-producing T cells into the post- ischaemic kidney, suggesting that FasL on leucocytes has an important role in the pathogenesis of renal IRI.116
The roles of T cells seem to extend to the late injury or repair phase of IRI and are not confined to the early injury phase. Increased numbers of activated T cells and effector-memory T cells were found in the post-ischaemic kidneys of mice as late as 6 weeks after IRI, which suggests that T cells are also involved in long-term structural changes in post-ischaemic kidneys and possibly contribute to the transition from AKI to CKD (Figure 4).117
Despite numerous investigations regarding the role of T cells in renal IRI, further studies are required to elucidate the precise mechanisms underlying the role of T cells and the interaction between T cells and B cells in ischaemic AKI. T-cell depletion with thymectomy fol-lowed by administration of T-cell-depleting antibody attenuated renal injury following IRI;118 however, mice deficient in both T cells and B cells were not protected from renal IRI.119 This topic requires active investiga-tion because it has important translational potential for human AKI.
Regulatory T cellsRegulatory T (TREG) cells were expected to have a role in renal regeneration or renoprotection, on the basis of numerous reports suggesting their anti-inflammatory effect on immune responses (Figure 3). TREG cells promote tubular proliferation and consequently hasten repair processes in the late injury and early recovery phases after IRI.120 Trafficking of TREG cells is increased in the post-ischaemic kidney at 3 days and 10 days after IRI (Figure 4). TREG cell depletion using anti-CD25 antibodies, starting 1 day after IRI, exacerbated renal tubular damage, reduced tubular proliferation, and increased cytokine production by infiltrating T cells on day 3, and increased TNF generation by CD4+ T cells on day 10. By contrast, adoptive transfer of TREG cells on day 1 after IRI resulted in reduced production of IFN-γ by CD4+ T cells on day 3, as well as improved repair and reduced generation of pro-inflammatory cytokines by day 10. During the early repair phase, treatment with mycophenolate mofetil reduced the total number of kidney-infiltrating mononuclear
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 9
cells and specifically decreased the population of intra-renal TREG cells, which worsened tubular damage.121 In the early injury phase after renal IRI, TREG cells modu-lated injury through IL-10-mediated suppression of the innate immune system.122 In this study, partial depletion of TREG cells with an anti-CD25 monoclonal antibody after IRI potentiated renal injury in the post-ischaemic kidney by increasing infiltration of IFN-γ-producing acti-vated neutrophils and macrophages, and upregulating the expression of mRNAs that encode innate immune system cytokines such as IL-6, TNF and TGF-β.
Autocrine adenosine signalling is a key mechanism of TREG cell-mediated renoprotection.123 The synthetic S1P analogue, fingolimod, attenuated renal injury in the post-ischaemic kidney, in part through a TREG cell-mediated mechanism.124 IL-2–anti-IL-2 complexes also reduce the damage associated with renal IRI through expansion of the TREG cell population.125 After ischaemic precondi-tioning, numbers of TREG cells and mature CD11c+ cells (macro phages and dendritic cells) substantially increased, which was associated with renoprotective effects on the post-ischaemic kidney.126 However, the TREG cell popula-tion is very small, both in normal kidneys and during the early injury phase in post-ischaemic kidneys after IRI. There are many questions that need to be answered regarding the role of TREG cells in ischaemic AKI. Such questions include how TREG cells modulate inflamma-tory responses in the early injury phase, despite the small
number of cells. Effector cells of the innate and adaptive immune system are the major mediators of TREG cells, although the specific mediators are currently unknown.
Natural killer T cellsNK T cells are a unique lymphocyte population that express both NK receptors and TCRs, and exert regulatory functions by secreting several cytokines, including IL-4, IL-10 and IFN-γ. NK T cells traffic into post- ischaemic kidneys as early as 3 h after IRI, but their numbers decrease to a level that is similar to that of a normal, healthy kidney 24 h after IRI (Figure 4).103 NK T-cell acti-vation can be inhibited in three ways: treatment with anti-CD1d antibody; NK T-cell depletion using anti-NK1.1 antibody in wild-type mice; or using NK T-cell deficient mice. All three approaches conferred renal protection in the early IRI phase, with decreased trafficking of IFN-γ-producing neutrophils after IRI.127 The results of this study suggest that NK T cells contribute to the induc-tion of early renal injury by mediating neutrophil IFN-γ production. NK T cells are also involved in the renal pro-tection induced by isoflurane anaesthetic treatment in a mouse model of renal IRI.128 However, another report showed conflicting data, indicating that the absence of NK T cells (especially type II NK T cells) accentuated the severity of renal injury, whereas repletion of NK T cells attenuated renal injury.129 In this study, an endogenous glycolipid, 3-sulphated galactosylceramide (sulphatide),
Figure 4 | Important immune cells in each phase of renal IRI. Neutrophils and NK T cells infiltrate the post-ischaemic kidney in the early injury phase and contribute to initiation of the inflammatory cascade. NK cells also contribute to renal tissue injury in the early injury phase. Renal dendritic cells increase in number and are activated to mediate inflammation from the early to late injury phase. Macrophages have diverse roles throughout the pathogenesis of renal IRI. In the injury phase, M1 macrophages contribute to inflammation and tissue injury, whereas M2 macrophages exert anti-inflammatory functions in post-ischaemic kidneys and facilitate renal tubular regeneration during the recovery phase. T cells also show dynamic changes in number and phenotype depending on the phase of renal IRI. CD4+ T cells have a substantial role in inducing renal tissue damage in the early injury phase. TREG cells increase in the late injury phase and facilitate tubular regeneration in the recovery phase. B cells are activated and differentiate in the injury phase, and limit tubular regeneration in the recovery phase. Abbreviations: DAMPs, damage-associated molecular patterns; IRI, ischaemia–reperfusion injury; NK, natural killer; TLR, Toll-like receptor; TREG cells, regulatory T cells.
Endothelium
Bloodvessel
Interstitium
Tubularepithelium
Renal tubule
Late injury phaseEarly injury phase Recovery phase
Adhesionmolecule
Fibrosis
Plasma cellPlateletTLR
Cytokines and chemokinesComplementDAMPs
B cell
TREG cell
CD4+ T cell
Effector memoryT cell
M1 macrophage
M2 macrophage
Dendritic cell
Neutrophil
NK cell
NK T cell
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

10 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
was used to induce activation of type II NK T cells, which suggested that pharmacological modulation of type II NK T-cell activation could confer renal protection after IRI. As the population of NK T cells is relatively small compared to that of T cells or B cells in normal kidneys and post-ischaemic kidneys, further investigations are required to define the precise role and kinetics of infiltrated NK T cells in the post-ischaemic kidney.
Intraepithelial γδ T cellsMost T cells bear a TCR composed of one α and one β chain, whereas the TCR of γδ T cells consists of one γ and one δ chain. The γδ T cells are a minor subset of T cells that recognize phospholipids or intact proteins instead of peptide antigens carried on MHCs. The γδ T cells are divided into two highly distinct sets accord-ing to their primary residing location and function. One set is found in the lymphoid tissue of all vertebrates, and exhibits a highly diversified TCR similar to that of αβ T cells. By contrast, intraepithelial γδ T cells show differ-ences in their location and/or function between species although these cells are found in jawed vertebrates130 and express very limited TCR diversity, particularly in γδ T cells of the skin and the female reproductive tract of mice. Two reports suggest that mice deficient in γδ T cells were protected from early renal tubular injury after IRI to a similar extent as were mice deficient in αβ T cells.131,132 However, whether this small cell popula-tion is really involved in the pathogenesis of renal injury following IRI remains unclear.
B cellsOnly a few studies have investigated the role of B cells in renal IRI. B-cell deficiency in mice conferred renal protection in the early phase of renal IRI.133 B cells also traffic into post-ischaemic kidneys and differenti-ate into plasma cells during the repair phase of IRI.134 In this work, an increase in B-cell chemoattractant in the post-ischaemic kidney preceded B-cell trafficking. Post-ischaemic kidneys of B-cell-deficient mice showed higher expression of IL-10 and vascular endothelial growth factor, and exhibited more tubular proliferation and less tubular atrophy during the repair phase than did post-ischaemic kidneys from wild-type mice. Adoptive transfer of B cells into B-cell-deficient mice 24 h after renal IRI reduced tubular proliferation and increased tubular atrophy. These results suggest that B cells have adverse effects on the repair process after renal IRI and indicate the therapeutic potential of targeting B cells to enhance repair after IRI (Figure 3). Further studies are required to elucidate the precise role and kinetics of B-cell trafficking into the post-ischaemic kidney, especi-ally as B cells are gaining substantial attention as a factor affecting the long-term survival of renal allografts.
B-1 cells are a minor subset of B cells that can be dis-tinguished from conventional B-2 cells by expression of the cell surface protein CD5 and their primary residing location in the peritoneal and pleural cavities. Although the production of natural antibodies (immunoglobulins that arise in the absence of specific antigenic stimulation)
is thought to be the primary role of B-1 cells, their precise role in AKI has yet to be defined. IgM natural antibodies from B-1 cells are involved in the initiation of injury in mouse models of intestinal IRI.135 In a mouse model of ischaemic AKI, the presence of B cells that express CD5 and IgM (surface markers similar to those of B-1 cells but with uncertain function) was increased in the post-ischaemic kidney 10 days after IRI, suggesting a possible role of these cells in the early repair phase.134 IgM anti-leucocyte natural autoantibodies also regulate excess inflammation, especially the inflammatory response mediated by TH17 cells that is not effectively suppressed by TREG cells in post-ischaemic kidneys.136
Nephrotoxic AKIThe roles of immune cells in the pathogenesis of nephro-toxic AKI have mostly been investigated using a mouse model of cisplatin-induced AKI. In this model, the infil-tration and activation of immune cells seems to be initi-ated by injured or dead cells, or the nephrotoxic agent itself as a sterile inflammatory stimuli.25 There is con-siderably less research in the pathophysiology and role of immune cells in nephrotoxic AKI.
NeutrophilsIn a mouse model of cisplatin-induced AKI, neutro-phil infiltration into the kidney and the expression of IL-1β, IL-18 and IL-6 were increased, although blocking either neutrophil infiltration or these cytokines did not prevent cisplatin-induced AKI.137 The activation of TLR4 on renal parenchymal cells activates p38 mitogen acti-vated protein kinase pathways, which increases both the production of proinflammatory cytokines such as TNF and infiltration of neutrophils, and mediates subsequent renal injury.138
MacrophagesThe role of macrophages in cisplatin-induced AKI is not well established, although rapid expansion of the macrophage population (probably resulting from cisplatin- induced myelosuppression) contributes to the development of myofibroblast-augmented renal fibrosis after repeated administration of cisplatin.139 In a mouse model of cisplatin-induced AKI, macrophage infiltration into the kidney preceded impairment of renal function, although inhibition of macrophage infiltration into the kidney was not sufficient to prevent AKI.140
Renal dendritic cellsIn a mouse model of cisplatin-induced AKI, dendritic cells showed renoprotective effects141 that were not observed in a mouse model of IRI.142 Mice depleted of dendritic cells before or during cisplatin treatment, but not those depleted at later stages, showed more severe renal dysfunction, tubular injury, neutrophil infiltra-tion and greater mortality compared with non-depleted mice.141 However, endogenous IL-10 production by renal dendritic cells was augmented in response to cisplatin administration, which led to reductions in both cispla-tin nephrotoxicity and inflammation.142 Further studies
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 11
are required to reveal the precise kinetics and roles of renal dendritic cells as antigen-presenting and effector cells in renal injury.
LymphocytesT cellsT cells, especially CD4+ T cells, are important mediators in the pathogenesis of renal injury in a mouse model of cisplatin-induced AKI.143 T-cell numbers were signifi-cantly increased in kidneys of wild-type mice as early as 1 h after cisplatin administration, peaked at 12 h, and declined by 24 h. CD4-deficient (and, to a lesser degree, CD8-deficient) mice showed decreased cisplatin-induced mortality and renal dysfunction compared with wild-type mice. The development of cisplatin-induced AKI seems to depend upon Fas-mediated apoptosis, which is driven by FasL expressed on renal tubular cells and infiltrating immune cells, especially T cells. However, the expression of FasL on renal tubular cells has a more sub-stantial role in the pathogenesis of cisplatin-induced AKI than does expression of FasL on T cells.144 Further studies are required to reveal the underlying immunologic mechanism of cisplatin-induced AKI in more detail.
Regulatory T cellsRenoprotective effects of TREG cells have also been found in models of nephrotoxic AKI. In a mouse model of cisplatin- induced AKI, adoptive transfer of TREG cells attenuated renal injury and decreased macrophage infiltration in both (mature-T-cell-deficient) Foxn1nu/nu mice and wild-type mice.145 TREG cells migrated to lymph nodes under the guidance of CC motif chemokine receptor 7 (CCR7). Wild-type but not CCR7-knockout TREG cells protect the kidney from nephrotoxic serum nephritis, which is induced by injection of rabbit anti-mouse glomerular base-ment membrane antibody and worsened by a preceding immunization against rabbit immunoglobulin.146
Relevance of animal models of AKINumerous investigations using animal models of AKI have achieved substantial advances in our understand-ing of the role of immune cells in AKI. Many agents have shown therapeutic potential over the past few decades for improvement of renal outcome by modulating the number of immune cells and their functions. However, most of the new therapeutic agents failed to show sig-nificant improvement of renal outcomes in patients with
established AKI. Species differences between humans and experimental animals are obviously an important factor in this lack of translation. The fact that many experimental agents were proven to be effective in genet-ically engineered animal models, or only at extremely high doses, might be other important causes of the dis-crepancies between successful experimental outcomes in animals and disappointing therapeutic outcomes in patients. These substantial challenges must be overcome before investigational findings can be safely applied to patients.
ConclusionsIn the past 25 years, numerous studies have explored inflammation during AKI. Various immune compo-nents of both innate and adaptive immune systems are now known to be important mediators of renal injury and repair after IRI. Various immune cells (including CD4+ T cells, B cells, NK T cells, macrophages and renal den-dritic cells) have a key function in renal injury following IRI. The latest research also suggests a complex role for immune cells in attenuating renal injury and facilitat-ing repair via tissue remodelling after IRI; for example, TREG cells and M2 macrophages are involved in both reno-protection and repair processes during the recovery phase of renal IRI. Although many groups have investigated the roles and kinetics of each cell type in immune responses occurring in the post-ischaemic kidney, the precise roles of immune cells at different time points after IRI are yet to be determined. Careful dissection of the role of specific immune cells at different time points in ischaemic AKI and nephrotoxic AKI, and improved understanding of the interactions between various immune cells will help elucidate the complicated immune mechanisms under-lying AKI, while contributing to the development of novel diagnostic and therapeutic tools for this condition.
Review criteria
Peer-reviewed research papers, written in English were selected using keyword searches of the PubMed database. Search terms included “ischaemic OR nephrotoxic AKI”, “immune response” and “immune cells”. Searches were largely restricted to articles published in the past 20 years. Only six of the 146 references were published more than 20 years ago. These references were selected based on their contribution to major subsequent reports or pre-existing knowledge.
1. Thadhani, R., Pascual, M. & Bonventre, J. V. Acute renal failure. N. Engl. J. Med. 334, 1448–1460 (1996).
2. Coca, S. G., Singanamala, S. & Parikh, C. R. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 81, 442–448 (2012).
3. Goncalves, G. M., Zamboni, D. S. & Camara, N. O. The role of innate immunity in septic acute kidney injuries. Shock 34, S22–S26 (2010).
4. Halazun, K. J., Al-Mukhtar, A., Aldouri, A., Willis, S. & Ahmad, N. Warm ischemia in transplantation: search for a consensus definition. Transplant. Proc. 39, 1329–1331 (2007).
5. Jang, H. R., Ko, G. J., Wasowska, B. A. & Rabb, H. The interaction between ischemia-reperfusion and immune responses in the kidney. J. Mol. Med. 87, 859–864 (2009).
6. Jang, H. R. & Rabb, H. The innate immune response in ischemic acute kidney injury. Clin. Immunol. 130, 41–50 (2009).
7. Kurts, C., Panzer, U., Anders, H. J. & Rees, A. J. The immune system and kidney disease: basic concepts and clinical implications. Nat. Rev. Immunol. 13, 738–753 (2013).
8. Wolfs, T. G. et al. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-γ and TNF-α mediated up-regulation during
inflammation. J. Immunol. 168, 1286–1293 (2002).
9. Kim, B. S. et al. Ischemia-reperfusion injury activates innate immunity in rat kidneys. Transplantation 79, 1370–1377 (2005).
10. Kaissling, B. & Le Hir, M. Characterization and distribution of interstitial cell types in the renal cortex of rats. Kidney Int. 45, 709–720 (1994).
11. Kruger, T. et al. Identification and functional characterization of dendritic cells in the healthy murine kidney and in experimental glomerulonephritis. J. Am. Soc. Nephrol. 15, 613–621 (2004).
12. Soos, T. J. et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

12 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
the entire kidney. Kidney Int. 70, 591–596 (2006).
13. Woltman, A. M. et al. Quantification of dendritic cell subsets in human renal tissue under normal and pathological conditions. Kidney Int. 71, 1001–1008 (2007).
14. Nelson, P. J. et al. The renal mononuclear phagocytic system. J. Am. Soc. Nephrol. 23, 194–203 (2012).
15. Guilliams, M. et al. From skin dendritic cells to a simplified classification of human and mouse dendritic cell subsets. Eur. J. Immunol. 40, 2089–2094 (2010).
16. Miller, J. C. et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 13, 888–899 (2012).
17. Kim, K. W. et al. In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood 118, e156–e167 (2011).
18. Tittel, A. P. et al. Functionally relevant neutrophilia in CD11c diphtheria toxin receptor transgenic mice. Nat. Methods 9, 385–390 (2012).
19. Timoshanko, J. R., Kitching, A. R., Semple, T. J., Tipping, P. G. & Holdsworth, S. R. A pathogenetic role for mast cells in experimental crescentic glomerulonephritis. J. Am. Soc. Nephrol. 17, 150–159 (2006).
20. Scandiuzzi, L. et al. Mouse mast cell protease-4 deteriorates renal function by contributing to inflammation and fibrosis in immune complex-mediated glomerulonephritis. J. Immunol. 185, 624–633 (2010).
21. Gan, P. Y. et al. Mast cells contribute to peripheral tolerance and attenuate autoimmune vasculitis. J. Am. Soc. Nephrol. 23, 1955–1966 (2012).
22. Ascon, D. B. et al. Normal mouse kidneys contain activated and CD3+CD4– CD8– double-negative T lymphocytes with a distinct TCR repertoire. J. Leukoc. Biol. 84, 1400–1409 (2008).
23. Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12, 991–1045 (1994).
24. Matzinger, P. The danger model: a renewed sense of self. Science 296, 301–305 (2002).
25. Rock, K. L., Latz, E., Ontiveros, F. & Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 28, 321–342 (2010).
26. Kono, H., Chen, C. J., Ontiveros, F. & Rock, K. L. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J. Clin. Invest. 120, 1939–1949 (2010).
27. Imaeda, A. B. et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Invest. 119, 305–314 (2009).
28. Eigenbrod, T., Park, J. H., Harder, J., Iwakura, Y. & Nunez, G. Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 α released from dying cells. J. Immunol. 181, 8194–8198 (2008).
29. Chen, S. W. et al. Mice that overexpress human heat shock protein 27 have increased renal injury following ischemia reperfusion. Kidney Int. 75, 499–510 (2009).
30. Thurman, J. M. Triggers of inflammation after renal ischemia/reperfusion. Clin. Immunol. 123, 7–13 (2007).
31. Rosenberger, C. et al. Cellular responses to hypoxia after renal segmental infarction. Kidney Int. 64, 874–886 (2003).
32. Matsumoto, M. et al. Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J. Am. Soc. Nephrol. 14, 1825–1832 (2003).
33. Bernhardt, W. M. et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J. Am. Soc. Nephrol. 17, 1970–1978 (2006).
34. Kelly, K. J. et al. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J. Clin. Invest. 97, 1056–1063 (1996).
35. Takada, M., Nadeau, K. C., Shaw, G. D., Marquette, K. A. & Tilney, N. L. The cytokine-adhesion molecule cascade in ischemia/reperfusion injury of the rat kidney. Inhibition by a soluble P-selectin ligand. J. Clin. Invest. 99, 2682–2690 (1997).
36. Brodsky, S. V. et al. Endothelial dysfunction in ischemic acute renal failure: rescue by transplanted endothelial cells. Am. J. Physiol. Renal Physiol. 282, F1140–F1149 (2002).
37. Sutton, T. A. et al. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am. J. Physiol. Renal Physiol. 285, F191–F198 (2003).
38. Liu, M. et al. Effect of T cells on vascular permeability in early ischemic acute kidney injury in mice. Microvasc. Res. 77, 340–347 (2009).
39. Eickelberg, O. et al. Functional activation of heat shock factor and hypoxia-inducible factor in the kidney. J. Am. Soc. Nephrol. 13, 2094–2101 (2002).
40. Cao, C. C. et al. In vivo transfection of NF-kappaB decoy oligodeoxynucleotides attenuate renal ischemia/reperfusion injury in rats. Kidney Int. 65, 834–845 (2004).
41. Donnahoo, K. K. et al. Early kidney TNF-α expression mediates neutrophil infiltration and injury after renal ischemia-reperfusion. Am. J. Physiol. 277, R922–R929 (1999).
42. Daha, M. R. & van Kooten, C. Is the proximal tubular cell a proinflammatory cell? Nephrol. Dial. Transplant. 15, S41–S43 (2000).
43. Hiroyoshi, T. et al. Splenectomy protects the kidneys against ischemic reperfusion injury in the rat. Transpl. Immunol. 27, 8–11 (2012).
44. Anders, H. J., Vielhauer, V. & Schlondorff, D. Chemokines and chemokine receptors are involved in the resolution or progression of renal disease. Kidney Int. 63, 401–415 (2003).
45. Swaminathan, S. & Griffin, M. D. First responders: understanding monocyte-lineage traffic in the acutely injured kidney. Kidney Int. 74, 1509–1511 (2008).
46. Miura, M., Fu, X., Zhang, Q. W., Remick, D. G. & Fairchild, R. L. Neutralization of Gro α and macrophage inflammatory protein-2 attenuates renal ischemia/reperfusion injury. Am. J. Pathol. 159, 2137–2145 (2001).
47. Araki, M. et al. Expression of IL-8 during reperfusion of renal allografts is dependent on ischemic time. Transplantation 81, 783–788 (2006).
48. Fiorina, P. et al. Role of CXC chemokine receptor 3 pathway in renal ischemic injury. J. Am. Soc. Nephrol. 17, 716–723 (2006).
49. Furuichi, K. et al. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J. Am. Soc. Nephrol. 14, 2503–2515 (2003).
50. Furuichi, K., Gao, J. L. & Murphy, P. M. Chemokine receptor CX3CR1 regulates renal interstitial fibrosis after ischemia-reperfusion injury. Am. J. Pathol. 169, 372–387 (2006).
51. Wu, H. et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Invest. 117, 2847–2859 (2007).
52. Allam, R. et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J. Am. Soc. Nephrol. 23, 1375–1388 (2012).
53. Wu, H. et al. HMGB1 contributes to kidney ischemia reperfusion injury. J. Am. Soc. Nephrol. 21, 1878–1890 (2010).
54. Leemans, J. C. et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Invest. 115, 2894–2903 (2005).
55. Iyer, S. S. et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl Acad. Sci. USA 106, 20388–20393 (2009).
56. Chiao, H. et al. α-melanocyte-stimulating hormone protects against renal injury after ischemia in mice and rats. J. Clin. Invest. 99, 1165–1172 (1997).
57. Nemoto, T. et al. Small molecule selectin ligand inhibition improves outcome in ischemic acute renal failure. Kidney Int. 60, 2205–2214 (2001).
58. Solez, K., Morel–Maroger, L. & Sraer, J. D. The morphology of “acute tubular necrosis” in man: analysis of 57 renal biopsies and a comparison with the glycerol model. Medicine (Baltimore) 58, 362–376 (1979).
59. Friedewald, J. J. & Rabb, H. Inflammatory cells in ischemic acute renal failure. Kidney Int. 66, 486–491 (2004).
60. Li, L. et al. IL-17 produced by neutrophils regulates IFN-γ-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J. Clin. Invest. 120, 331–342 (2010).
61. Fukuzawa, N. et al. High renal ischemia temperature increases neutrophil chemoattractant production and tissue injury during reperfusion without an identifiable role for CD4 T cells in the injury. Transpl. Immunol. 22, 62–71 (2009).
62. Thornton, M. A., Winn, R., Alpers, C. E. & Zager, R. A. An evaluation of the neutrophil as a mediator of in vivo renal ischemic-reperfusion injury. Am. J. Pathol. 135, 509–515 (1989).
63. Rabb, H. et al. Role of CD11a and CD11b in ischemic acute renal failure in rats. Am. J. Physiol. 267, F1052–F1058 (1994).
64. Hayama, T. et al. Benefical effect of neutrophil elastase inhibitor on renal warm ischemia-reperfusion injury in the rat. Transplant. Proc. 38, 2201–2202 (2006).
65. Roelofs, J. J. et al. Tissue-type plasminogen activator modulates inflammatory responses and renal function in ischemia reperfusion injury. J. Am. Soc. Nephrol. 17, 131–140 (2006).
66. Mizuno, S. & Nakamura, T. Prevention of neutrophil extravasation by hepatocyte growth factor leads to attenuations of tubular apoptosis and renal dysfunction in mouse ischemic kidneys. Am. J. Pathol. 166, 1895–1905 (2005).
67. Rouschop, K. M. et al. Protection against renal ischemia reperfusion injury by CD44 disruption. J. Am. Soc. Nephrol. 16, 2034–2043 (2005).
68. Haug, C. E. et al. A phase I trial of immunosuppression with anti-ICAM-1 (CD54) mAb in renal allograft recipients. Transplantation 55, 766–772 (1993).
69. Salmela, K. et al. A randomized multicenter trial of the anti-ICAM-1 monoclonal antibody (enlimomab) for the prevention of acute rejection and delayed onset of graft function in cadaveric renal transplantation: a report of the European Anti-ICAM-1 Renal Transplant Study Group. Transplantation 67, 729–736 (1999).
70. Riera, M. et al. Neutrophils accentuate renal cold ischemia-reperfusion injury. Dose-dependent protective effect of a platelet-activating factor receptor antagonist. J. Pharmacol. Exp. Ther. 280, 786–794 (1997).
71. Vinten–Johansen, J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 61, 481–497 (2004).
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

NATURE REVIEWS | NEPHROLOGY ADVANCE ONLINE PUBLICATION | 13
72. Crinnion, J. N., Homer-Vanniasinkam, S. & Gough, M. J. Skeletal muscle reperfusion injury: pathophysiology and clinical considerations. Cardiovasc. Surg. 1, 317–324 (1993).
73. Ysebaert, D. K. et al. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol. Dial. Transplant. 15, 1562–1574 (2000).
74. De Greef, K. E. et al. Anti-B7-1 blocks mononuclear cell adherence in vasa recta after ischemia. Kidney Int. 60, 1415–1427 (2001).
75. Celie, J. W. et al. Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. Am. J. Pathol. 170, 1865–1878 (2007).
76. Jo, S. K., Sung, S. A., Cho, W. Y., Go, K. J. & Kim, H. K. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrol. Dial. Transplant. 21, 1231–1239 (2006).
77. He, Z. et al. Macrophages are not the source of injurious interleukin-18 in ischemic acute kidney injury in mice. Am. J. Physiol. Renal Physiol. 296, F535–F542 (2009).
78. Day, Y. J., Huang, L., Ye, H., Linden, J. & Okusa, M. D. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am. J. Physiol. Renal Physiol. 288, F722–F731 (2005).
79. Gueler, F. et al. Statins attenuate ischemia-reperfusion injury by inducing heme oxygenase-1 in infiltrating macrophages. Am. J. Pathol. 170, 1192–1199 (2007).
80. Persy, V. P., Verhulst, A., Ysebaert, D. K., De Greef, K. E. & De Broe, M. E. Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int. 63, 543–553 (2003).
81. Ko, G. J., Boo, C. S., Jo, S. K., Cho, W. Y. & Kim, H. K. Macrophages contribute to the development of renal fibrosis following ischaemia/reperfusion-induced acute kidney injury. Nephrol. Dial. Transplant. 23, 842–852 (2008).
82. Vinuesa, E. et al. Macrophage involvement in the kidney repair phase after ischaemia/reperfusion injury. J. Pathol. 214, 104–113 (2008).
83. Huen, S. C., Moeckel, G. W. & Cantley, L. G. Macrophage-specific deletion of transforming growth factor-β1 does not prevent renal fibrosis after severe ischemia-reperfusion or obstructive injury. Am. J. Physiol. Renal Physiol. 305, F477–F484 (2013).
84. Alikhan, M. A. et al. Colony-stimulating factor-1 promotes kidney growth and repair via alteration of macrophage responses. Am. J. Pathol. 179, 1243–1256 (2011).
85. Lee, S. et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 22, 317–326 (2011).
86. Anders, H. J. & Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 80, 915–925 (2011).
87. Zhang, M. Z. et al. CSF-1 signaling mediates recovery from acute kidney injury. J. Clin. Invest. 122, 4519–4532 (2012).
88. Ranganathan, P. V., Jayakumar, C. & Ramesh, G. Netrin-1-treated macrophages protect the kidney against ischemia-reperfusion injury and suppress inflammation by inducing M2 polarization. Am. J. Physiol. Renal Physiol. 304, F948–F957 (2013).
89. Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
90. Suzuki, N. et al. Severe impairment of interleukin-1 and Toll-like receptor signalling
in mice lacking IRAK-4. Nature 416, 750–756 (2002).
91. Lech, M. et al. Macrophage phenotype controls long-term AKI outcomes—kidney regeneration versus atrophy. J. Am. Soc. Nephrol. 25, 292–304 (2014).
92. Penfield, J. G. et al. Transplant surgery injury recruits recipient MHC class II-positive leukocytes into the kidney. Kidney Int. 56, 1759–1769 (1999).
93. Dong, X. et al. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 71, 619–628 (2007).
94. Schlichting, C. L., Schareck, W. D. & Weis, M. Renal ischemia-reperfusion injury: new implications of dendritic cell-endothelial cell interactions. Transplant. Proc. 38, 670–673 (2006).
95. Loverre, A. et al. Ischemia-reperfusion injury-induced abnormal dendritic cell traffic in the transplanted kidney with delayed graft function. Kidney Int. 72, 994–1003 (2007).
96. Ozaki, K. S. et al. The loss of renal dendritic cells and activation of host adaptive immunity are long-term effects of ischemia/reperfusion injury following syngeneic kidney transplantation. Kidney Int. 81, 1015–1025 (2012).
97. Shau, H., Roth, M. D. & Golub, S. H. Regulation of natural killer function by nonlymphoid cells. Nat. Immun. 12, 235–249 (1993).
98. Chiche, L. et al. The role of natural killer cells in sepsis. J. Biomed. Biotechnol. 2011:986491 (2011).
99. Zhang, Z. X. et al. NK cells induce apoptosis in tubular epithelial cells and contribute to renal ischemia-reperfusion injury. J. Immunol. 181, 7489–7498 (2008).
100. Zhang, Z. X. et al. Osteopontin expressed in tubular epithelial cells regulates NK cell-mediated kidney ischemia reperfusion injury. J. Immunol. 185, 967–973 (2010).
101. Rabb, H. et al. Pathophysiological role of T lymphocytes in renal ischemia-reperfusion injury in mice. Am. J. Physiol. Renal Physiol. 279, F525–F531 (2000).
102. Burne, M. J. et al. Identification of the CD4(+) T cell as a major pathogenic factor in ischemic acute renal failure. J. Clin. Invest. 108, 1283–1290 (2001).
103. Ascon, D. B. et al. Phenotypic and functional characterization of kidney-infiltrating lymphocytes in renal ischemia reperfusion injury. J. Immunol. 177, 3380–3387 (2006).
104. Day, Y. J. et al. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: the role of CD4+ T cells and IFN-γ. J. Immunol. 176, 3108–3114 (2006).
105. Lai, L. W., Yong, K. C., Igarashi, S. & Lien, Y. H. A sphingosine-1-phosphate type 1 receptor agonist inhibits the early T-cell transient following renal ischemia-reperfusion injury. Kidney Int. 71, 1223–1231 (2007).
106. Sakr, M. et al. The protective effect of FK506 pretreatment against renal ischemia/reperfusion injury in rats. Transplantation 53, 987–991 (1992).
107. Jones, E. A. & Shoskes, D. A. The effect of mycophenolate mofetil and polyphenolic bioflavonoids on renal ischemia reperfusion injury and repair. J. Urol. 163, 999–1004 (2000).
108. Takada, M., Chandraker, A., Nadeau, K. C., Sayegh, M. H. & Tilney, N. L. The role of the B7 costimulatory pathway in experimental cold ischemia/reperfusion injury. J. Clin. Invest. 100, 1199–1203 (1997).
109. Chandraker, A. et al. CD28-b7 blockade in organ dysfunction secondary to cold ischemia/
reperfusion injury. Kidney Int. 52, 1678–1684 (1997).
110. Yokota, N., Burne-Taney, M., Racusen, L. & Rabb, H. Contrasting roles for STAT4 and STAT6 signal transduction pathways in murine renal ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 285, F319–F325 (2003).
111. Wang, S. et al. Decreased renal ischemia-reperfusion injury by IL-16 inactivation. Kidney Int. 73, 318–326 (2008).
112. Gigliotti, J. C. et al. Ultrasound prevents renal ischemia-reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J. Am. Soc. Nephrol. 24, 1451–1460 (2013).
113. Jang, H. R. et al. Early exposure to germs modifies kidney damage and inflammation after experimental ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 297, F1457–F1465 (2009).
114. Jang, H. R., Gandolfo, M. T., Ko, G. J., Racusen, L. & Rabb, H. The effect of murine anti-thymocyte globulin on experimental kidney warm ischemia-reperfusion injury in mice. Transpl. Immunol. 22, 44–54 (2009).
115. Satpute, S. R. et al. The role for T cell repertoire/antigen-specific interactions in experimental kidney ischemia reperfusion injury. J. Immunol. 183, 984–992 (2009).
116. Ko, G. J. et al. Blocking Fas ligand on leukocytes attenuates kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol. 22, 732–742 (2011).
117. Ascon, M. et al. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. 75, 526–535 (2009).
118. Yokota, N., Daniels, F., Crosson, J. & Rabb, H. Protective effect of T cell depletion in murine renal ischemia-reperfusion injury. Transplantation 74, 759–763 (2002).
119. Park, P. et al. Injury in renal ischemia-reperfusion is independent from immunoglobulins and T lymphocytes. Am. J. Physiol. Renal Physiol. 282, F352–F357 (2002).
120. Gandolfo, M. T. et al. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 76, 717–729 (2009).
121. Gandolfo, M. T. et al. Mycophenolate mofetil modifies kidney tubular injury and Foxp3+ regulatory T cell trafficking during recovery from experimental ischemia-reperfusion. Transpl. Immunol. 23, 45–52 (2010).
122. Kinsey, G. R. et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol. 20, 1744–1753 (2009).
123. Kinsey, G. R. et al. Autocrine adenosine signaling promotes regulatory T cell-mediated renal protection. J. Am. Soc. Nephrol. 23, 1528–1537 (2012).
124. Kim, M. G. et al. CD4+ CD25+ regulatory T cells partially mediate the beneficial effects of FTY720, a sphingosine-1-phosphate analogue, during ischaemia/reperfusion-induced acute kidney injury. Nephrol. Dial. Transplant. 26, 111–124 (2011).
125. Kim, M. G. et al. IL-2/anti-IL-2 complex attenuates renal ischemia-reperfusion injury through expansion of regulatory T cells. J. Am. Soc. Nephrol. 24, 1529–1536 (2013).
126. Cho, W. Y. et al. The role of Tregs and CD11c(+) macrophages/dendritic cells in ischemic preconditioning of the kidney. Kidney Int. 78, 981–992 (2010).
127. Li, L. et al. NKT cell activation mediates neutrophil IFN-γ production and renal ischemia-reperfusion injury. J. Immunol. 178, 5899–5911 (2007).
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved

14 | ADVANCE ONLINE PUBLICATION www.nature.com/nrneph
128. Lee, H. T. et al. Isoflurane protects against renal ischemia and reperfusion injury and modulates leukocyte infiltration in mice. Am. J. Physiol. Renal Physiol. 293, F713–F722 (2007).
129. Yang, S. H. et al. Sulfatide-reactive natural killer T cells abrogate ischemia-reperfusion injury. J. Am. Soc. Nephrol. 22, 1305–1314 (2011).
130. Matsunaga, T. Did the first adaptive immunity evolve in the gut of ancient jawed fish? Cytogenet. Cell Genet. 80, 138–141 (1998).
131. Savransky, V. et al. Role of the T-cell receptor in kidney ischemia-reperfusion injury. Kidney Int. 69, 233–238 (2006).
132. Hochegger, K. et al. Role of α/β and γ/δ T cells in renal ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 293, F741–F747 (2007).
133. Burne-Taney, M. J. et al. B cell deficiency confers protection from renal ischemia reperfusion injury. J. Immunol. 171, 3210–3215 (2003).
134. Jang, H. R. et al. B cells limit repair after ischemic acute kidney injury. J. Am. Soc. Nephrol. 21, 654–665 (2010).
135. Fleming, S. D. et al. Mice deficient in complement receptors 1 and 2 lack a tissue injury-inducing subset of the natural antibody repertoire. J. Immunol. 169, 2126–2133 (2002).
136. Lobo, P. I. et al. Natural IgM anti-leukocyte autoantibodies attenuate excess inflammation mediated by innate and adaptive immune
mechanisms involving Th-17. J. Immunol. 188, 1675–1685 (2012).
137. Faubel, S. et al. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1β, IL-18, IL-6, and neutrophil infiltration in the kidney. J. Pharmacol. Exp. Ther. 322, 8–15 (2007).
138. Zhang, B., Ramesh, G., Uematsu, S., Akira, S. & Reeves, W. B. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J. Am. Soc. Nephrol. 19, 923–932 (2008).
139. Yamate, J. et al. Immunohistochemical study of rat renal interstitial fibrosis induced by repeated injection of cisplatin, with special reference to the kinetics of macrophages and myofibroblasts. Toxicol. Pathol. 24, 199–206 (1996).
140. Lu, L. H. et al. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J. Pharmacol. Exp. Ther. 324, 111–117 (2008).
141. Tadagavadi, R. K. & Reeves, W. B. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J. Am. Soc. Nephrol. 21, 53–63 (2010).
142. Tadagavadi, R. K. & Reeves, W. B. Endogenous IL-10 attenuates cisplatin nephrotoxicity: role of dendritic cells. J. Immunol. 185, 4904–4911 (2010).
143. Liu, M. et al. A pathophysiologic role for T lymphocytes in murine acute cisplatin
nephrotoxicity. J. Am. Soc. Nephrol. 17, 765–774 (2006).
144. Linkermann, A. et al. Renal tubular Fas ligand mediates fratricide in cisplatin-induced acute kidney failure. Kidney Int. 79, 169–178 (2011).
145. Lee, H. et al. CD4+CD25+ regulatory T cells attenuate cisplatin-induced nephrotoxicity in mice. Kidney Int 78, 1100–1109 (2010).
146. Eller, K. et al. CCR7 deficiency exacerbates injury in acute nephritis due to aberrant localization of regulatory T cells. J. Am. Soc. Nephrol. 21, 42–52 (2010).
AcknowledgementsH.R.J. is supported by a grant from the Korean Health Technology Research & Development Project, Ministry of Health & Welfare, Republic of Korea (HI13C-1263-020,013), a grant from Samsung Biomedical Research Institute (GL1B22612), and a grant from the Korean Society of Nephrology (PHX1130221). H.R. is supported by the NIH, US National Kidney Foundation, and generous research support from Rogelio Miro, Anne Segerson and Stoney Stanfil.
Author contributionsH.R.J. wrote the manuscript and researched data for the article; H.R.J. and H.R. contributed equally to discussion of content, review and/or editing the manuscript before submission.
REVIEWS
© 2014 Macmillan Publishers Limited. All rights reserved
















