Embed Size (px)
Citation preview

III/ Genome Rearrangements
1/ Evolution of matchings (cuts and joins), alternating permutations and labeled trees2/ Evolution of permutations (reversals) and a clicking game on F23/ Shuffling a deck of cards

Result of sequence searches
G1
G2
Genomes can be circular or linear, possibliy with several chromosomesBeware of the orientations, deduced from the double strand structure of genomes
Genes


Dotplot between 2 E. Coli strains

Genomes can be- signed strings- signed permutations- matchings on gene extremities
General reference for genome rearrangementsFertin et al, Combinatorics of genome rearrangements, 2009

1/ Genomes as matchings
2 more recent references on this part:Ouangraoua and Bergeron, JCB, 2010Miklos and Tannier, arxiv, 2013
Modelisation of a genome
Genes

Modelisation of a genome
Genes
Genome 1
adjacencies
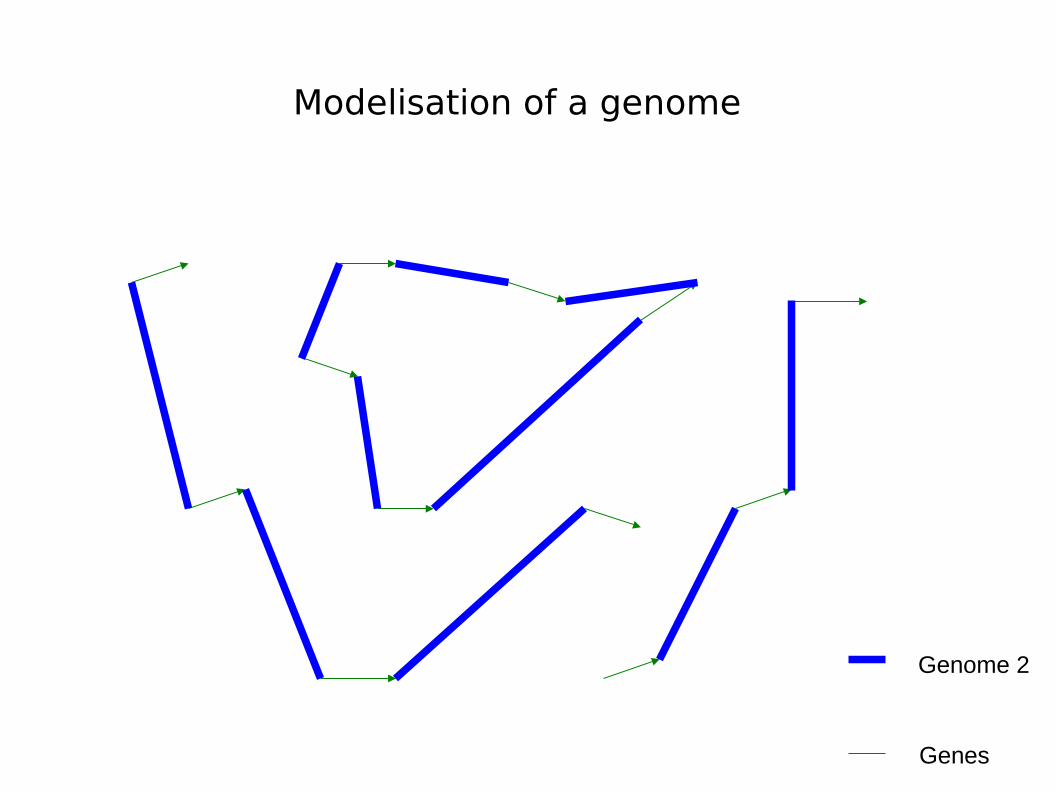
Modelisation of a genome
Genes
Genome 2

The comparison graph
Genes
Genome 1
Genome 2

The comparison graph
Genome 1
Genome 2

The comparison graph
Cycles, RedBlue paths, RedRed paths,BlueBlue paths

1.1 Single cut or joinAllowed operations: Remove an edge or add an edge
- How many operations are required (trivial)- How to count or sample solutions- The relation with alternating permutations

Alternating permutation (André, 1881):
c1,...,cn permutation of 1..n such thatc2i-1 < c2i and c2i> c2i+1
For example, 1,3,2,4 but not 1,3,4,2
An = number or alternating permutations

Transforming Red into Blue
The number of scenarios sorting an even (RedBlue) path with n edges is E(n)=An
The number of scenarios sorting an odd RedRed path is R(n)=An
The number of scenarios sorting an odd BlueBlue path is B(n)=An
The number of scenarios sorting a cycle is C(n)=1/2*n*An-1


Mix the paths and cycles with a multinomial coefficient

An inversion is 4 SCJ
We would like to model more realistic operations

Double cut-and-join (DCJ)
Fusion (SCJ et DCJ)
Fission (SCJ et DCJ)
Inversion (DCJ)
Reciprocal Translocation (DCJ)
Non reciprocal translocation (DCJ)

1.2 Double cut-and-join
- How many operations are required (easy)- How to count or sample solutions- The relation with parking functions, labeled trees, cycles in permutations- How to estimate the number of steps given the distance

d(Red,Blue) = n – (c + i/2)
Minimum number of DCJ
Number of genesNumber of cycles
Number of paths with anodd number of vertices(including isolated vertices)

n = number of genes, 2n = number of vertices
Two identical genomes only have size 2 cycles and size 1 paths
A size 2k cycle needs k-1 DCJA size 2k+1 path needs k DCJA size 2k path needs k DCJ
Good DCJs are transforming- 1 cycle into 2 cycles- 1 even path into 1 cycle and one even path- 1 odd path into 1 cycle and one odd path- 2 even paths into 2 odd paths

Number of ways to sort a cycle with 2n edges by DCJ =
Number of ways to decompose the permutation cycle (1..n) into transpositions

Number of ways to sort a cycle with 2n edges by DCJ =
Number of ways to decompose the permutation cycle (1..n) into transpositions =
Number of parking functions of length n-1 =
n^{n-2}
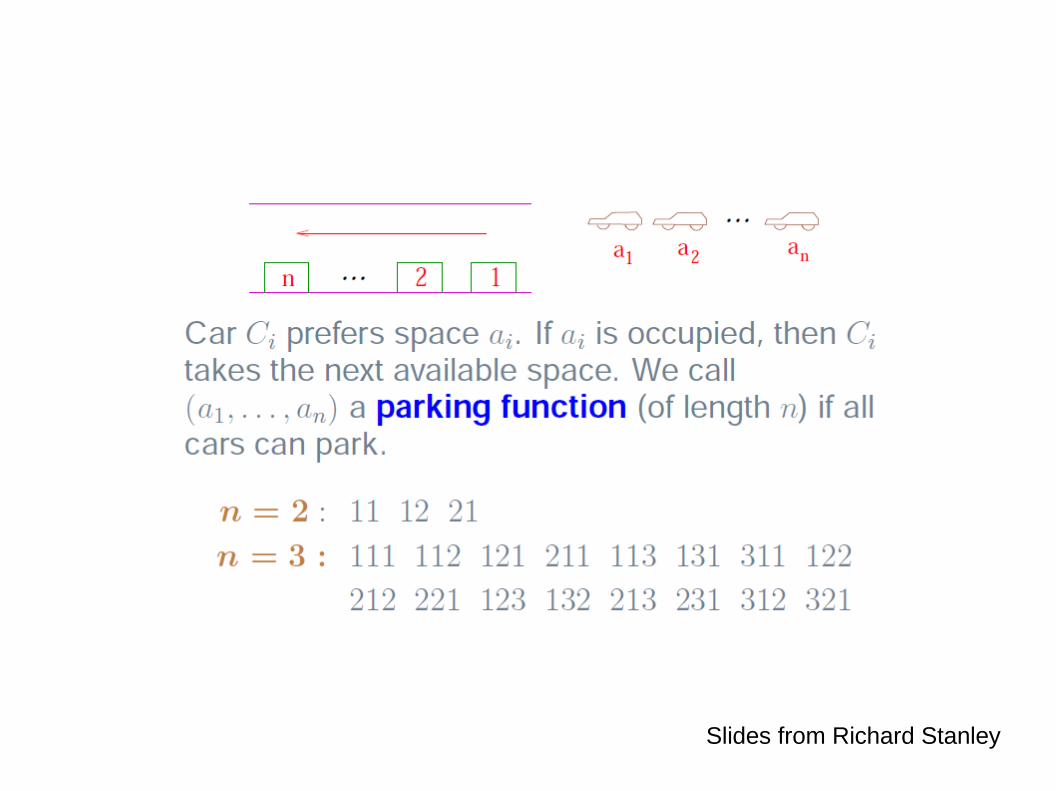
Slides from Richard Stanley

Slides from Richard Stanley

Slides from Richard Stanley
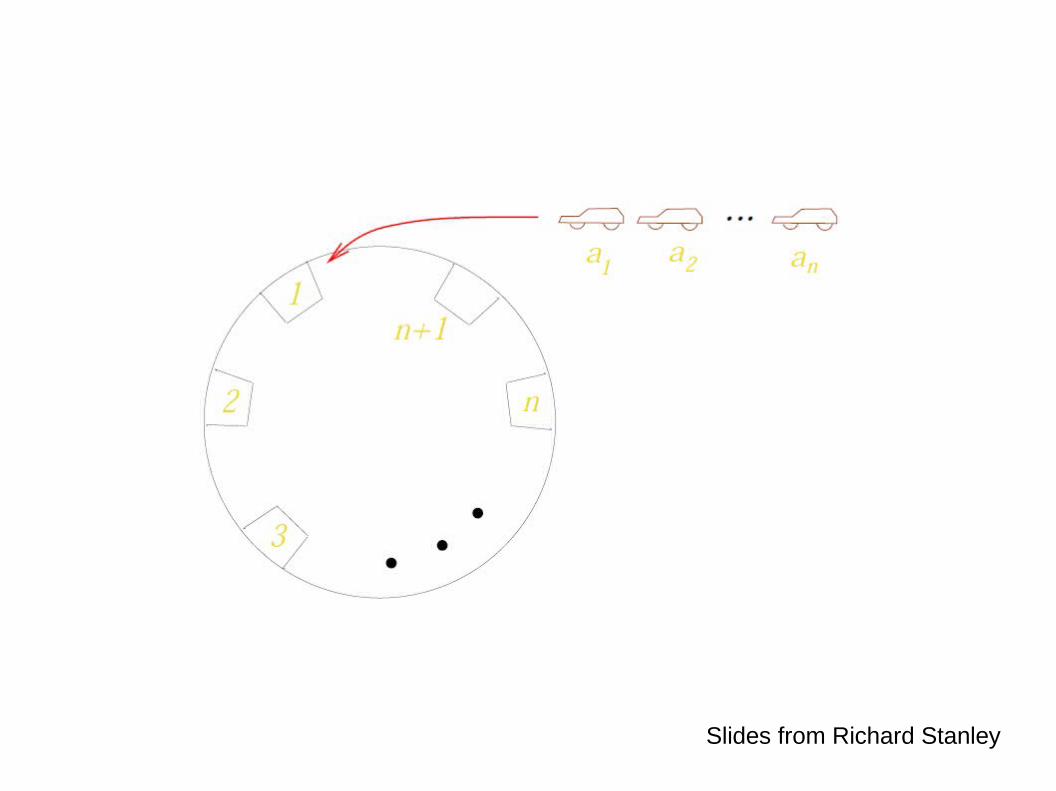
Slides from Richard Stanley

Slides from Richard Stanley

Identify a DCJ with a transposition (ab) a<bSay a is the base and b is the top
1/ The sequence of bases of a DCJ scenario sorting a cycle is a parking function(a number after the base cannot be reused after a base)
2/ The sequence of bases uniquely determines the DCJ scenario(a number at the top cannot be reused as a base)

1
2
3
4
5
6
7
8
9
1
2
3
4
6
7
8
5
(123456789)(45)(89)(18)(26)(27)(36)(28)(46)=(1)(2)(3)(4)(5)(6)(7)(8)(9)
Parking function: 48122324
8->94->54->63->62->62->72->81->8
Choose a top for each base, starting from the highest

Labeled trees (Cayley, 1889)
Number of unrooted node-labeled trees with n vertices =
Number of rooted edge-labeled trees with n vertices =
n^(n-2)
Relations in Stanley, 1997

For odd paths, it is still possible to enumerate
Use a multinomial to mix cycles and odd paths
Not in presence of even paths (open problem)

Final remarks on genomes as matching, and rearrangements as SCJ or DCJ
SCJ saturates faster, is less precise but is computationally feasible and close to the subtitution models.
Probabilistic models for DCJ need Monte Carlo methods to explore solution spaces, while for SCJ they could have analytical solutions.
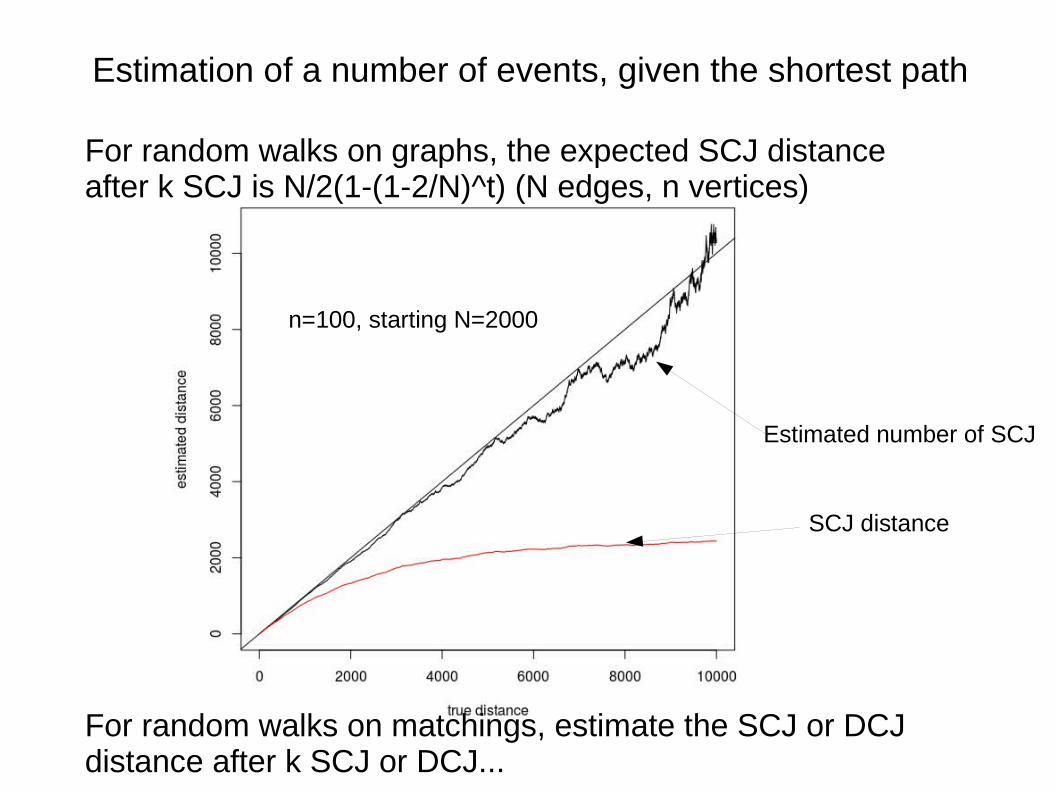
Estimation of a number of events, given the shortest path
For random walks on graphs, the expected SCJ distance after k SCJ is N/2(1-(1-2/N)^t) (N edges, n vertices)
For random walks on matchings, estimate the SCJ or DCJ distance after k SCJ or DCJ...
SCJ distance
Estimated number of SCJ
n=100, starting N=2000

2/ Genomes as permutations

Sorting by Reversals
0 7 5 3 -1 -6 -2 4 8
0 1 2 3 4 5 6 7 8
A permutation is a particular matchingAn inversion is a particular DCJ
Sometimes it is reasonable to consider only permutations and inversion

Sorting by Reversals
0 7 5 3 -1 -6 -2 4 8
0 1 2 3 4 5 6 7 8
0 1 -3 -5 -7 -6 -2 4 8

Sorting by Reversals
0 7 5 3 -1 -6 -2 4 8
0 1 2 3 4 5 6 7 8
0 1 -3 -5 -7 -6 -2 4 8
0 1 -3 -5 -4 2 6 7 8
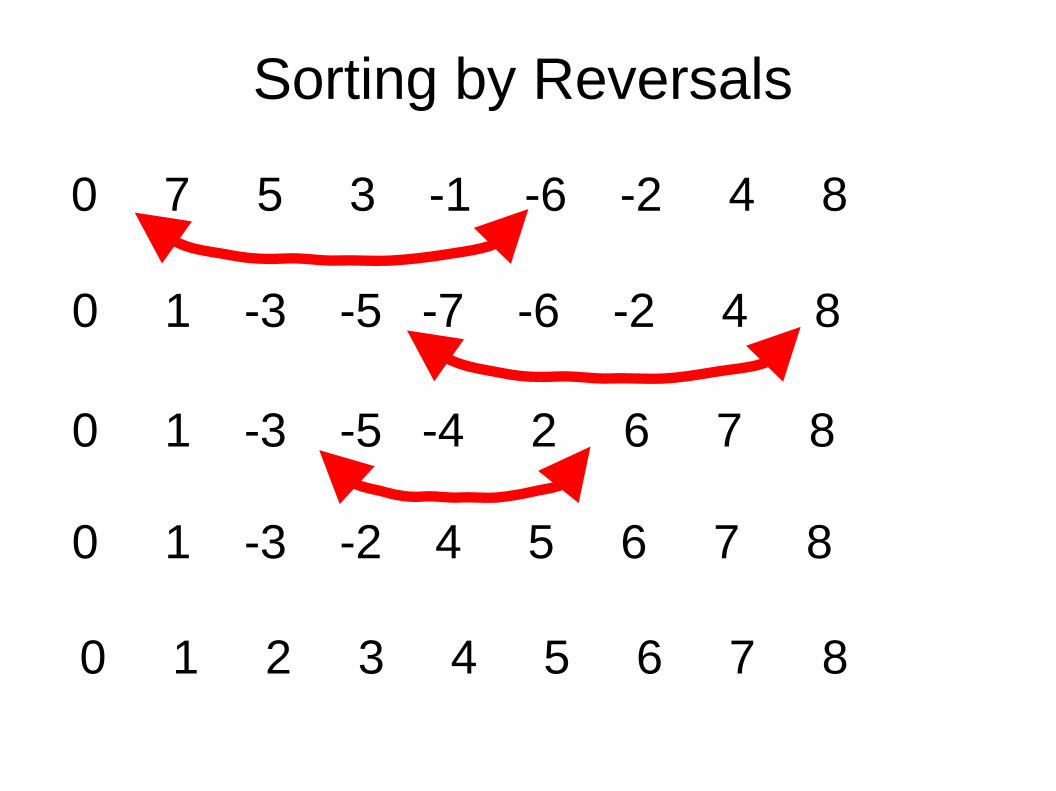
Sorting by Reversals
0 7 5 3 -1 -6 -2 4 8
0 1 2 3 4 5 6 7 8
0 1 -3 -5 -7 -6 -2 4 8
0 1 -3 -5 -4 2 6 7 8
0 1 -3 -2 4 5 6 7 8

Sorting by Reversals
0 7 5 3 -1 -6 -2 4 8
0 1 2 3 4 5 6 7 8
0 1 -3 -5 -7 -6 -2 4 8
0 1 -3 -5 -4 2 6 7 8
0 1 -3 -2 4 5 6 7 8

2/ Reversals
An example where the inversion distance is the DCJ distance (the previous example)
An example where it is not (reverse -3 -5 -7 -6 -2 in the previous example as a second step)

Sorting by Reversals
0 7 5 3 -1 -6 -2 4 8
0 1 2 3 4 5 6 7 8
0 1 -3 -5 -7 -6 -2 4 8
0 1 -3 -5 -4 2 6 7 8

Sorting by Reversals
0 7 5 3 -1 -6 -2 4 8
0 1 2 3 4 5 6 7 8
0 1 -3 -5 -7 -6 -2 4 8
0 1 2 6 7 5 3 4 8
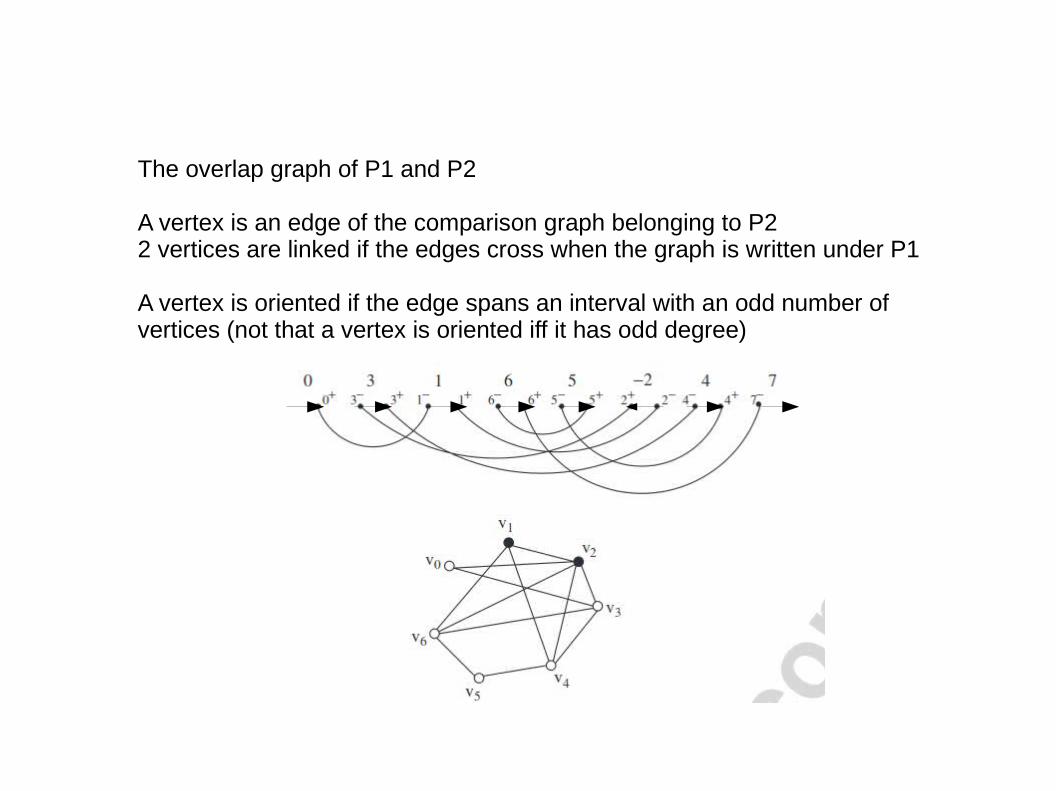
The overlap graph of P1 and P2
A vertex is an edge of the comparison graph belonging to P22 vertices are linked if the edges cross when the graph is written under P1
A vertex is oriented if the edge spans an interval with an odd number of vertices (not that a vertex is oriented iff it has odd degree)
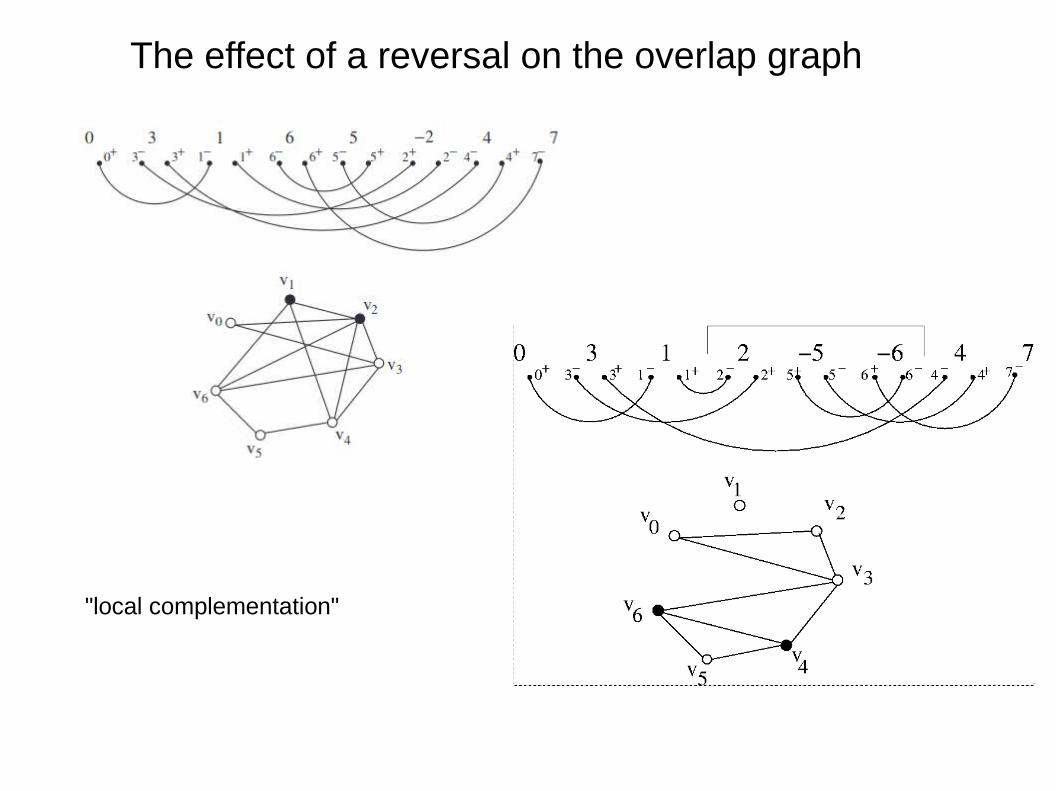
The effect of a reversal on the overlap graph
"local complementation"

The effect of a reversal on the adjacency matrix of the overlap graph
0 0 1 1 0 0 00 1 1 0 1 0 11 1 1 1 1 0 11 0 1 0 1 0 1 = A0 1 1 1 0 1 00 0 0 0 1 0 10 1 1 1 0 1 0
0 0 1 1 0 0 00 0 0 0 0 0 01 0 0 1 0 0 01 0 1 0 1 0 1 = A + v1v1^T0 0 0 1 1 1 10 0 0 0 1 0 10 0 0 1 1 1 1

A component is oriented if it has a black vertex, unoriented otherwise
Sorting an unoriented component is done in as many reversals as the rank of the adjacency matrix of the overlap graph over F2
Rank is n-c, where n is the size of the matrix and c the number of cycles. (A cycle has rank n-1, as in a matroid)
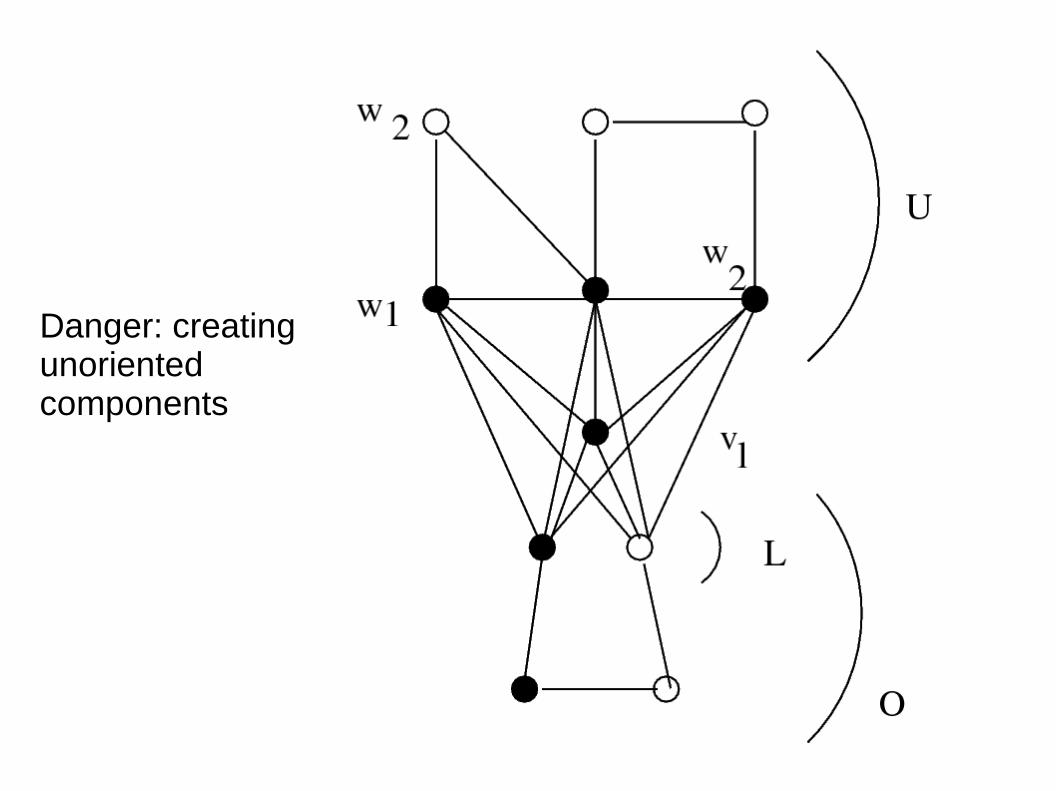
Danger: creating unoriented components

There is always a black vertex such that clicking on it does not create unoriented components.
Proof: Take the oriented vertex v which maximizesNumber of unoriented neighbors – Number of oriented neighbors
It does not create unoriented components. Indeed, if it does create one unoriented component C, C has an oriented vertex w adjacent to v. Calculate its score: an unoriented neighbor of v is a neighbor of w, and an oriented neighbor of w is a neighbor of v.

Sorting unoriented components: hurdles and fortresses
Hurdles: minimal unoriented components.
One inversion cuts 2 hurdles (c-1, h-2) or 1 hurdle (c,h-1)
Fortress: odd number of hurdles but additional unoriented components

Sorting unoriented components: hurdles and fortresses
Fortress
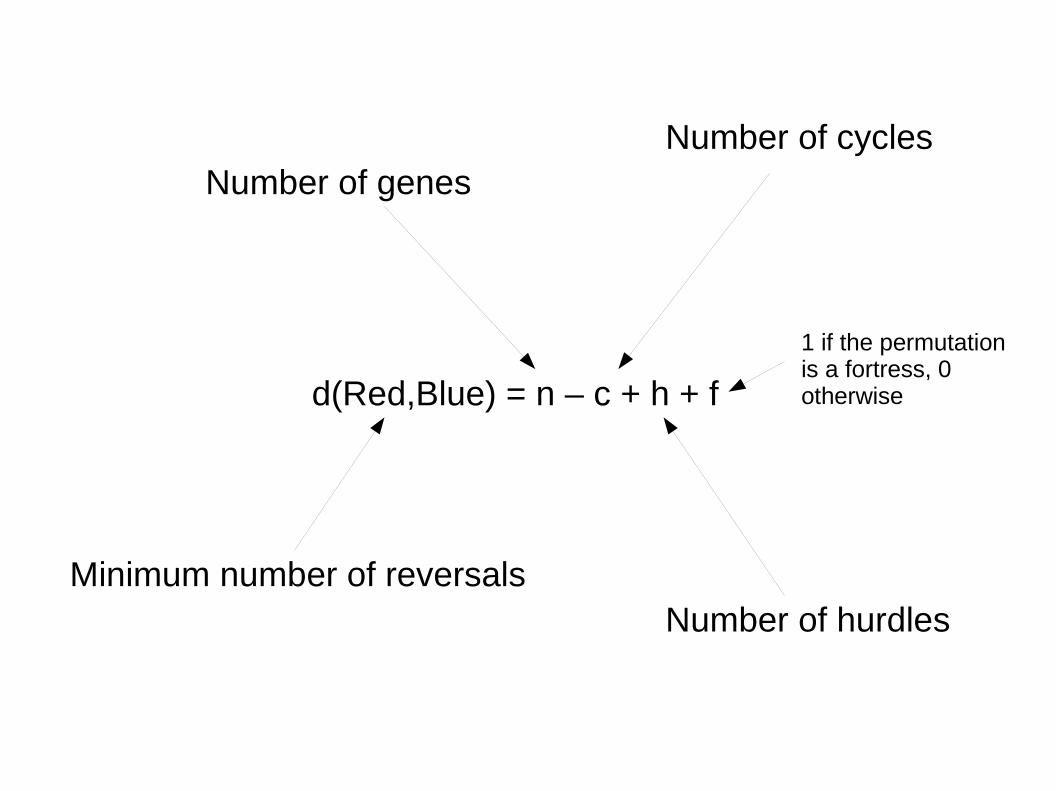
d(Red,Blue) = n – c + h + f
Minimum number of reversals
Number of genesNumber of cycles
Number of hurdles
1 if the permutation is a fortress, 0 otherwise

Counting, sampling or enumeratingsorting by reversals scenarios is almost open
Estimating the expected number of reversals given the distance is a possible homework subject.

The use of sorting by reversals in a controversial study: refuting a « random breakage model ».
Argument: any scenario has to break at least 2d times on n breakpoints. If c is low, d is close to n and each breakpoint is used twice.
Relating the breakpoint sizes and the total intergene size, it is seen to be too distant from the result of a uniform random model.

3/ Shuffling genomes or cards
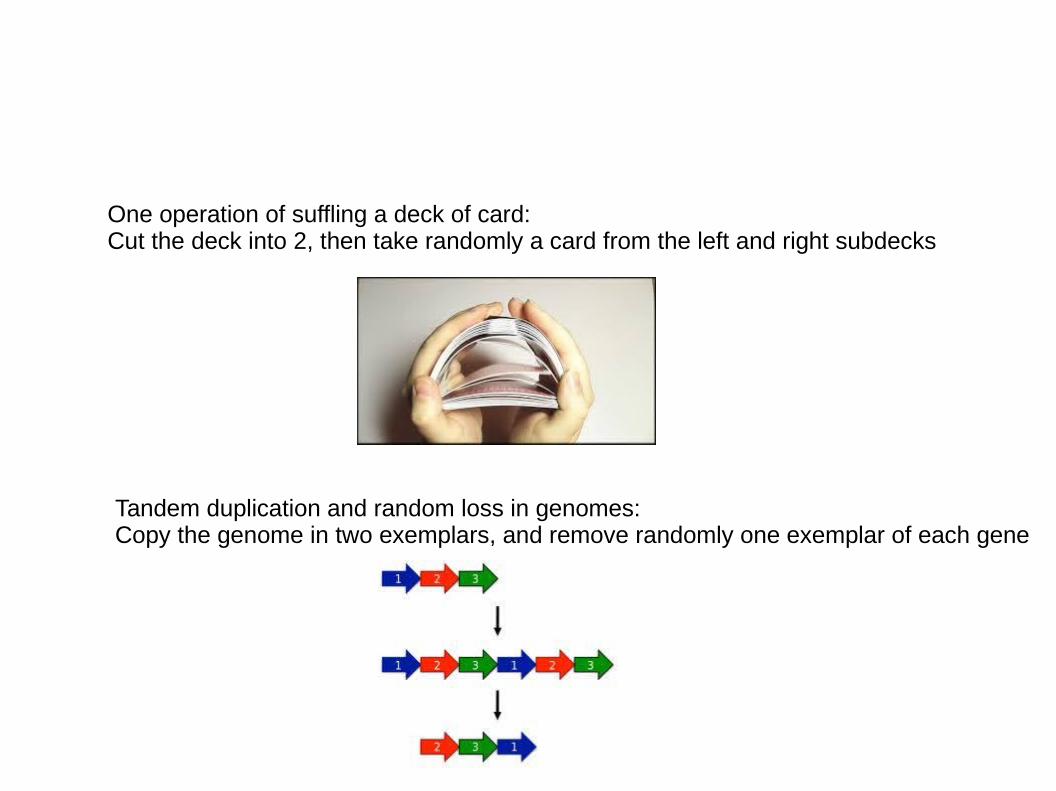
One operation of suffling a deck of card:Cut the deck into 2, then take randomly a card from the left and right subdecks
Tandem duplication and random loss in genomes:Copy the genome in two exemplars, and remove randomly one exemplar of each gene

(3 7 1 5 8 2 6 4)
(3 7 1 5 8 2 6 4 3 7 1 5 8 2 6 4)
(1 5 2 6 3 7 8 4)
(1 5 2 6 3 7 8 4 1 5 2 6 3 7 8 4)
(1 2 3 4 5 6 7 8)
(3 7 1 5 8 2 6 4)
(1 5 2 6)(3 7 8 4)
(1 5 2 6 3 7 8 4)
(1 2 3 4)(5 6 7 8)
(1 2 3 4 5 6 7 8)
Tandem duplication and losses
Riffle shuffle

A chain in a permutation is a maximal subsequence of consecutive numbers
(3 7 1 5 8 2 6 4) has 4 chains 12,34,56,78
A k-TDL is an operation copying a permutation k times and applying losses (usual TDL is 2-TDL)
Observation: A permutation is sorted in 1 k-TDL iff k is at least the number of chains

Theorem 1: if c<=k is the number of chains of a permutation p, then there are choose(n+k-c,n) ways to sort p with one k-TDL
Proof: (123456789...), the identity permutation, has to be cut into k pieces, each piece corresponding to a copy of p (we have to place k-1 cuts). c-1 cuts are compulsory. Ex: if p=(371582649), then 2 and 3 cannot go to the same copy, so Id is cut into (12|34|56|789).There remains k-c cuts to place, and repetition is allowed, yielding the result.

Theorem 2: There are as many ways to sort a permutation with one k1*k2-TDL as ways to sort a permutation with one k1-TDL followed by one k2-TDL.
Proof: (=>) k=k1*k2. Take a k-scenario, and label all k copies by coordinates ab, a in (1,..,k2) and b in (1,...,k1) in increasing lexicographic order. Each element of p is labeled by (a,b). Make k1 copies and sort according to the b coordinate. Then make k2 copies and sort according to the a coordinate. (<=) any k1-TDL scenario followed by a k2-scenario produces a coordinate system, which translates into a k-TDL

Corollaries: The minimum number of TDL to sort a permutation with c chains is ┌log_2(c)┐ The number of minimum size scenarios is choose(n+2^(┌log_2(c)┐)-c,n)

Note that the TDL distance is not symmetric.
It is the first non symmetric distance we have encountered so far.
This forces to go back to the evolutionary principle: define a TDL problem if two permutations are extant genomes.

General notes on rearrangements in bioinformatics
Computational problems are far more complex than with substitutions and indels (compare the distance computation and estimation between two sequences)
Very often the evolutionary studies use the parsimony principle, and are limited to few genomes with few genes, while statistical models of sequence evolution with subsitutions can handle hundreds of complete genomes with more accuracy (see part 3)

General notes on rearrangements in bioinformatics
In evolutionary studies with real data, most often DCJ or SCJ are used, and accessorily TDRL for some animal mitochondria.
Computationally, there exists a lot of variants of the rearrangement problem : sort a permutation, or a sequence, with an allowed operation, or a combination of operations - transpositions - pancakes - gains, losses, duplications - block interchanges - whole genome duplications - ...



![Sorting Signed Circular Permutations by Super Short Reversals · the genome rearrangement approach [4] in order to estimate the evolutionary distance. In genome rearrangements, one](https://img.pdfslide.us/doc/110x75/5f049a707e708231d40ec9e1/sorting-signed-circular-permutations-by-super-short-reversals-the-genome-rearrangement.jpg)

























