Embed Size (px)
Citation preview

Journal of the American Society of Nephrology 559
Idiopathic Hypercalciuria: The Contribution ofDr. Jacob Lemann, Jr.Fredric L. Coe1 and Joan H. Parks
FL. Coe, J.H. Parks, Nephrology Section, University ofChicago School of Medicine, Chicago, IL
(J. Am. Soc. Nephrol. 1994; 5:S59-S#{243}9)
ABSTRACTThe original contributions of Jacob Lemann to min-eral metabolism, especially calcium metabolism andIdiopathic hypercalciuria, are reviewed. One groupof studies concern acid base balance and calciumloss, showing that acid loads increase calcium loss inthe urine. Another group of studies concern the calci-uria of glucose or carbohydrate ingestion, with theobservation that stone patients, who as a populationare enriched with hypercalciuria, respond with moreexaggerated calciuria to glucose loads than do nor-mal people. Yet another body of work shows thatnormal men, when given noncalcemic loads of cal-citriol, exhibit two essential features of Idiopathic hy-percalciuria- hyperabsorptive hypercalciuria andbone mineral loss on a low-calcium diet. The finalgroup of studies presented worked on the problem ofthiazide hypocalciuric action, and where the calciumgoes that does not appear in the urine, as well as theeffects of potassium bicarbonate and sodium loadson mineral balance and acid base status.
Key Words: Mineral metabolism, idiopathic hypercalciuria.
acid base balance, carbohydrate hypercalciuria, calcitriol
I n his curriculum vitae, Jack Lemann lists 75 arti-
cles as original contributions. Of these, 41 seem, to
us, concerned with mineral metabolism, a fact that
shows as clearly as possible the importance of this
branch of research in his career. From the 41, we have
selected 19 that seem most germane to the problem of
Idiopathic hypercalciuria (IH) and nephrolithiasis.
One might ask why IH need be considered importantwhen selecting articles by this scientist, because al-
most none of his work actually used IH patients as
subjects. No doubt the answer lies in our interestsconjoined with the Immense importance of what JackLemann has done in furthering those interests; put
another way, no investigator has done as much as he
has to provide a scientific basis for understanding the
pathogenesis of IH, despite studying normal people
who do not have it.
1 Correspondence to Dr. F.L Coe. Nephrology Section, MC5100, University of
Chicago School of Medicine. 5841 South Maryland Ave., Chicago, 1160637.
1046�6673/0501-0S59SO3.OO/OJournal of the American Society of Nephrologycopyright © 1994 by the American Society of Nephrology
The IH problem lies at the center of mineral metab-olism, because the syndrome of normocalcemic hyper-
calciurla is not so much an ifiness as one extreme of
the distribution of normal urine calcium excretionrates. People at the upper end of the normal range
have nephrolithiasis at higher frequencies than dopeople with excretions at the lower or middle portionsof the range; therefore, students of stone disease are
invariably students of IH. However, being merely one
end of a range of normal, IH must arise from themechanisms that normally regulate calcium excre-
tion, and these include all factors regulating intestinal
calcium absorption, renal tubule calcium reabsorp-
tion, and bone calcium balance. Because it can only
be understood in terms of normal regulation, IH is
complex, and one excellent way to understand it is tostudy the normal regulation of calcium excretion. This
latter, Jack has done, with brilliance and technicalfluency, and we are honored to review his accomplish-
ments.
Studies of Acid Base Balance andCalcium Loss
One might not connect the two at first blush, but
hypercalciurla in response to acid load depicts in bold
outline the commonplace and more subtle loss of bonein response to dietary acid. Dietary acid causes at
least some of the daily urine calcium losses, and it isdaily calcium loss that, when high, gives us the diag-
nosis of IH. Just how much diet acid contributes to
calcium excretion in IH and whether exaggerated boneresponse to acid is a feature of IH are current research
questions of considerable interest. Their interest is
heightened by almost universal documentation of re-
duced bone mineral density values among patients
with IH, summarized elsewhere (1). So at the very
start, in defining bone buffering of acid loads, Jack
Lemann had already begun his contributions to anunderstanding of IH, although he certainly did not
give evidence of such understanding in the article
itself (2).
The enigma Jack faced (we simply help ourselves toa first name, as long association makes the formal“Dr. Lemann” seem absurd) was well known; despitechronic acid loading, blood bicarbonate falls to somestable level and remains there even though (Figure 1)acid retention is continuous and cumulative. Only
bone could be imagined as a buffer reservoir large
enough to offset the acid loading. The experiment ofammonlum chloride loading showed an expected loss
of bone mineral, but an unexpected kinetics, in thatmineral loss continued after loading stopped, and
bone never fully repaired itself during the rather longfollow-up periods used (Table 1). By the end, the total
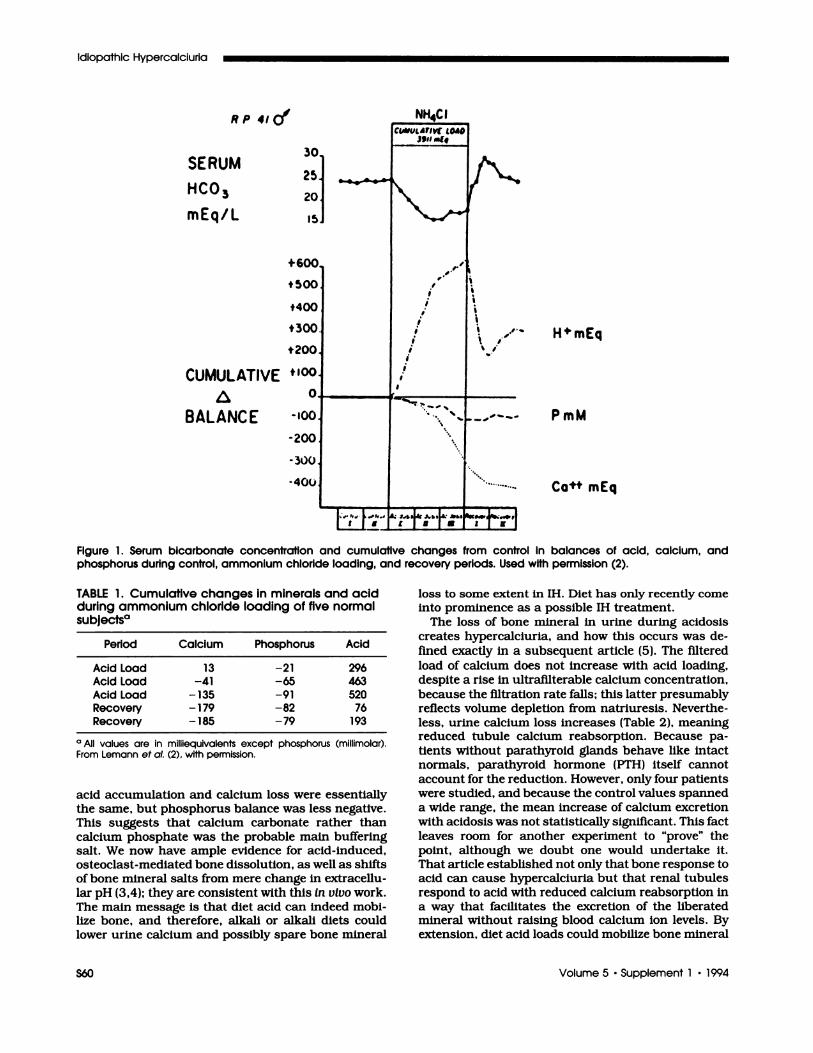
RP 41Q”
H’�mEq
PmM
Co+” mEq
Figure 1. Serum bicarbonate concentration and cumulative changes from control in baiances of acid, calcium, andphosphorus during control, ammonium chloride loading, and recovery periods. Used with permission (2).
Idiopathic Hypercalciuria
S60 Volume 5 ‘Supplement 1 ‘1994
SERUM
HCO3
mEq/L
CUMULATIVE
BALANCE
TABLE 1. Cumulative changes in minerals and acidduring ammonium chloride loading of five normalsubjectsa
Period Calcium Phosphorus Acid
Acid Load 13 -21 296Acid Load -41 -65 463Acid Load -135 -91 520Recovery -179 -82 76Recovery -185 -79 193
0 All values are in milliequivalents except phosphorus (miliimolar).
From Lemann et a!. (2). with permission.
acid accumulation and calcium loss were essentially
the same, but phosphorus balance was less negative.
This suggests that calcium carbonate rather than
calcium phosphate was the probable main bufferingsalt. We now have ample evidence for acid-induced,osteoclast-mediated bone dissolution, as well as shiftsof bone mineral salts from mere change in extracellu-lar pH (3,4); they are consistent with this in vivo work.The main message is that diet acid can indeed mobi-
lize bone, arid therefore, alkali or alkali diets could
lower urine calcium and possibly spare bone mineral
loss to some extent in IH. Diet has only recently come
into prominence as a possible IH treatment.
The loss of bone mineral in urine during acidosis
creates hypercalciuria, and how this occurs was de-
fined exactly in a subsequent article (5). The ifiteredload of calcium does not increase with acid loading,
despite a rise in ultraffiterable calcium concentration,
because the filtration rate falls: this latter presumably
reflects volume depletion from natriuresis. Neverthe-less, urine calcium loss increases (Table 2), meaningreduced tubule calcium reabsorption. Because pa-
tients without parathyroid glands behave like intact
normals, parathyroid hormone (PTH) itself cannot
account for the reduction. However, only four patientswere studied, and because the control values spanned
a wide range, the mean increase of calcium excretionwith acidosis was not statistically significant. This factleaves room for another experiment to “prove” thepoint, although we doubt one would undertake It.
That article established not only that bone response to
acid can cause hypercalciuria but that renal tubules
respond to acid with reduced calcium reabsorption ina way that facifitates the excretion of the liberated
mineral without raising blood calcium ion levels. By
extension, diet acid loads could mobilize bone mineral
AU RI NECa
mmoUdoy
-+500
L�LJ V,mmol/day=Co
_3+1000027 AU�I +0.46Aod= 0.96
.
a
0
#{163}
- +100
-200 -100
.p*14c’OProIe,nl- 0aNoHCO3
o +ioo +2� +300
coe and Parks
Journal of the American Society of Nephrology 551
TABLE 2. Renal handling of calcium during acldosisIn humans#{176}
Experiment Control Acidosis Recovery
Normals, NH4CL (6)Filtered load 148 140 143ExcretIon 3.38 7.54 4.68FE (Ca)% 2.25 5.39 3.27
Normals,Acetazoiamide (3)
Filtered load 145 123 147ExcretIon 3.13 4.33 4.36FE (Ca)% 2.16 3.52 2,97
Hypoporathyroid,NH4CL (4)
Filtered load 90 92 88
Excretion 1.86 6.49 2.70FE (Ca)% 2.07 7.05 3,07
#{176}Valuesof filtered load and excretion are In micromoles per minute;numbers of subjects are In parentheses. From Lemann et a!. (5). withpermission. Values for calcium excretion and FE ca during acidosisdiffer from control and recovery values during ammonium chlorideacidosis and for allnormal subjects considered as a group, but not for
acetazolamlde. consIdered separately. The increase In calcium ex-cretion and FE ca in hypoporathyroldism, although large, included awide variation of control values and was not significant.
in a chronic manner and reduce calcium reabsorp-tion, thereby helping to set the level of calcium excre-tion in the steady state.
In passing, his group brought out a confirmatoryreport concerning the role of PTH and calcitriol in the
acidosis of hypercalciurla, even though the response
to acid in hypoparathyroid people essentially excluded
an important contribution of PTH to the process.
Mineral balances were done during acid loading (6)and reproduced hypercalciuria and negative calciumbalance; serum PTH fell slightly, and calcitriol levelswere unchanged. Of all people, Jack must have beenthe least surprised, given his own past results, butthat article certainly finished the Issue.
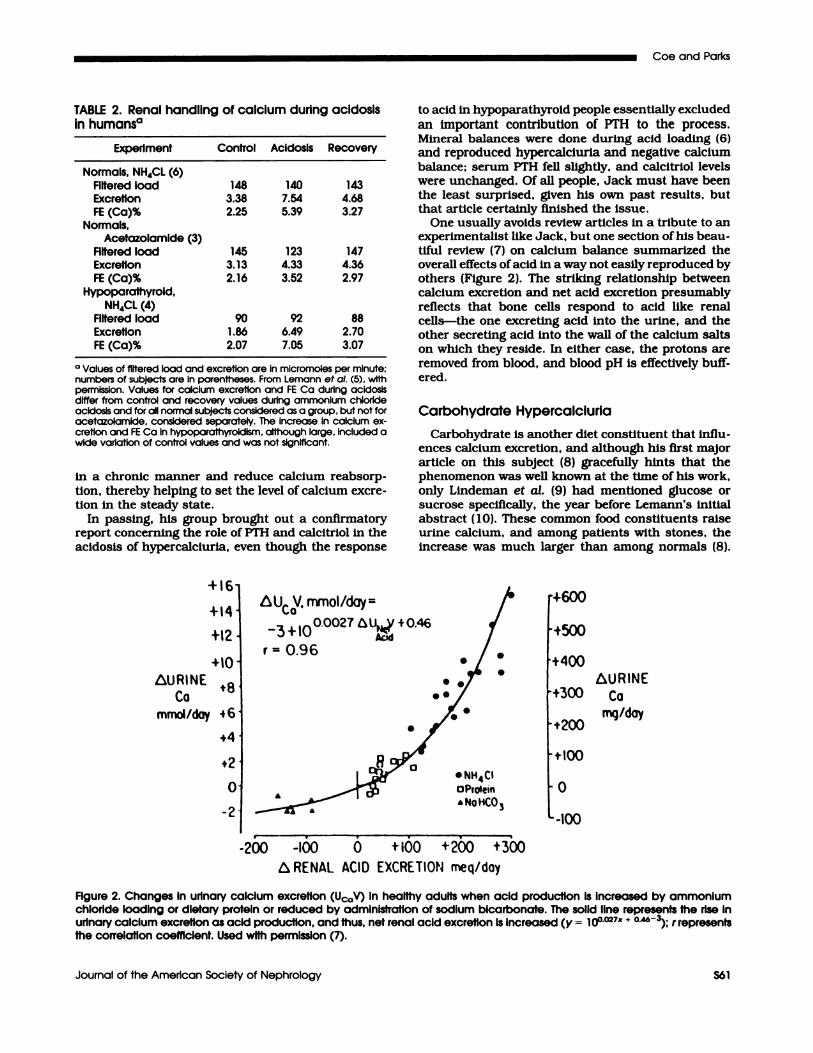
One usually avoids review articles in a tribute to anexperimentalist like Jack, but one section of his beau-tiful review (7) on calcium balance summarized theoverall effects of acid in a way not easily reproduced byothers (Figure 2). The striking relationship betweencalcium excretion and net acid excretion presumablyreflects that bone cells respond to acid like renalcells-the one excreting acid into the urine, and theother secreting acid into the wall of the calcium saltson which they reside. In either case, the protons are
removed from blood, and blood pH is effectively buff-ered.
Carbohydrate Hypercalciuria
Carbohydrate is another diet constituent that influ-ences calcium excretion, and although his first majorarticle on this subject (8) gracefully hints that thephenomenon was well known at the time of his work,
only Lindeman et al. (9) had mentioned glucose orsucrose specifically, the year before Lemann’s initialabstract (10). These common food constituents raiseurine calcium, and among patients with stones, theincrease was much larger than among normals (8).
+16-
+14’
+12
+10’
+8’
-.6
+4.
+2’
0’
-2’
A RENAL ACID EXCRETION meq/doy
AU R IN E
‘+300 Ca
-+200 mg/day
Figure 2. Changes In urinary calcium excretion (Uc0V) in healthy adults when acid production is increased by ammoniumchloride loading or dietary protein or reduced by administration of sodIum bicarbonate. The solid line represents the rise inurinary calcium excretion as acid production, and thus, net renal acid excretion is increased (y = 100o27�r + 0.46-3). r representsthe correlation coefficient. Used with permission (7).
NORMAL SUBJECTS CALCIUM STONE FORMERS RELATIVES OF CALCIUMSTONE FORMERSGLUCOSE GLUCOSE
OR SUCROSE OR SUCROSE
I I
GLUCOSE
OR SUCROSE
I
uca
mM/L
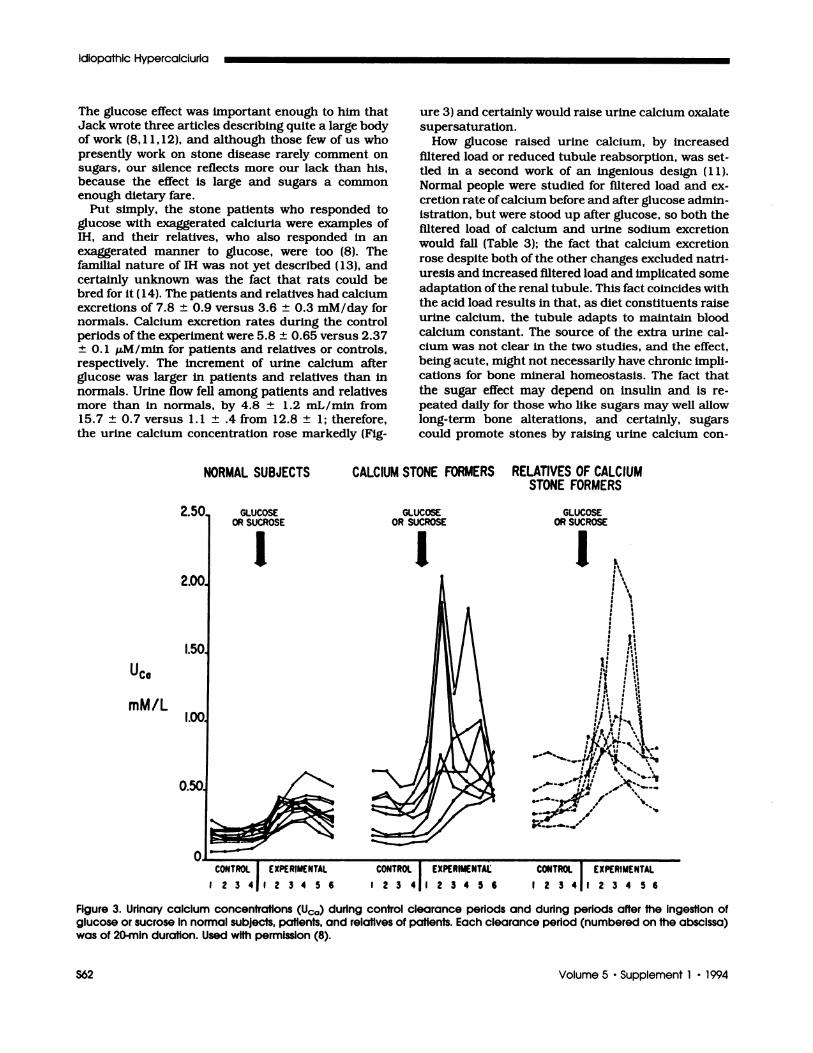
FIgure 3. Urinary calcium concentrations (Usa) during control clearance periods and during periods after the ingestion ofglucose or sucrose In normal subjects, patients, and relatives of patients. Each clearance period (numbered on the abscissa)was of 20-mm duration. Used with permission (8).
CONTROL EXPERIMENTAL CONTROL EXPERIMENTAL CONTROL EXPERIMENTAL
1234123456 1234123456 1234123456
Idiopathic Hypercalciuria
S62 Volume 5 ‘Supplement 1 ‘1994
The glucose effect was important enough to him that
Jack wrote three articles describing quite a large body
of work (8,11,12), and although those few of us who
presently work on stone disease rarely comment on
sugars, our silence reflects more our lack than his,
because the effect is large and sugars a common
enough dietary fare.
Put simply, the stone patients who responded to
glucose with exaggerated calciurla were examples of
IH, and their relatives, who also responded in anexaggerated manner to glucose, were too (8). The
familIal nature of IH was not yet described (13), and
certainly unknown was the fact that rats could be
bred for it (14). The patients and relatives had calcium
excretions of 7.8 ± 0.9 versus 3.6 ± 0.3 mM/day fornormals. Calcium excretion rates during the controlperiods of the experiment were 5.8 ± 0.65 versus 2.37
± 0.1 �M/min for patients and relatives or controls,
respectively. The increment of urine calcium afterglucose was larger in patients and relatives than in
normals. Urine flow fell among patients and relativesmore than in normals, by 4.8 ± 1.2 mL/min from
15.7 ± 0.7 versus 1.1 ± .4 from 12.8 ± 1; therefore,
the urine calcium concentration rose markedly (Fig-
ure 3) and certainly would raise urine calcium oxalate
supersaturation.
How glucose raised urine calcium, by increased
ifitered load or reduced tubule reabsorption, was set-
tled in a second work of an ingenious design (11).
Normal people were studied for filtered load and ex-
cretion rate of calcium before and after glucose admin-
istration, but were stood up after glucose, so both the
filtered load of calcium and urine sodium excretion
would fall (Table 3); the fact that calcium excretion
rose despite both of the other changes excluded natri-
uresis and increased ifitered load and implicated some
adaptation of the renal tubule. This fact coincides with
the acid load results in that, as diet constituents raise
urine calcium, the tubule adapts to maintain bloodcalcium constant. The source of the extra urine cal-
cium was not clear in the two studies, and the effect,
being acute, might not necessarily have chronic impli-
cations for bone mineral homeostasis. The fact that
the sugar effect may depend on insulin and is re-
peated daily for those who like sugars may well allowlong-term bone alterations, and certainly, sugars
could promote stones by raising urine calcium con-
I0
9
8
<0
7 40r
6
(/F Mq
4
3
E/FCo 2
(/F No
(OC� 0C5
Coe and Parks
Journal of the American Society of Nephrology S63
TABLE 3. Effects of glucose on calcium excretionduring standing#{176}
Measurement ControlStanding
+ Glucose
Sodium ExcretIon 168 ± 24 68 ± 14Filtered CalcIum 172 ± 9 150 ± 8Calcium Excretion 2.8 ± 0.5 3.6 ± 0.5
#{176} values are micromoles per minute; mean ± SE. From Lemann eta!. (11), wIth permission.
centrations. The fact that IH patients respond in anexaggerated manner is not at all explained to this day.
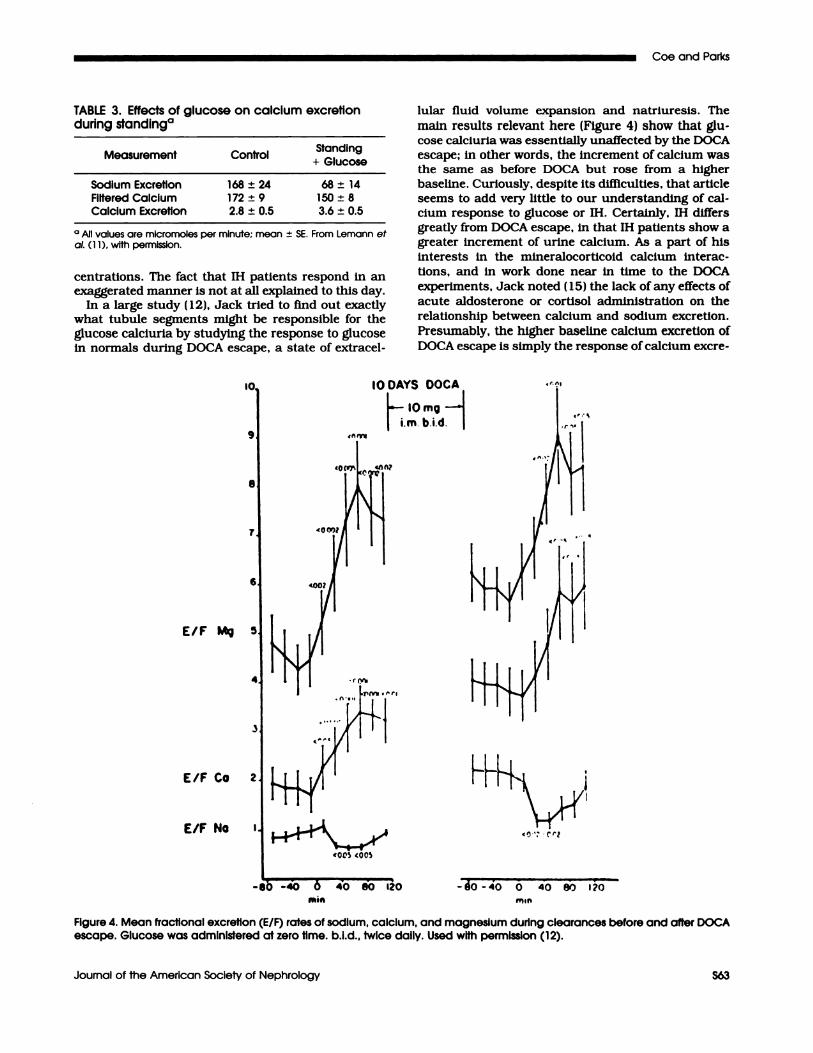
In a large study (12), Jack tried to find out exactlywhat tubule segments might be responsible for theglucose calciuria by studying the response to glucosein normals during DOCA escape, a state of extracel-
lular fluid volume expansion and natriuresis. The
main results relevant here (Figure 4) show that glu-
cose calciurla was essentially unaffected by the DOCAescape; in other words, the increment of calcium wasthe same as before DOCA but rose from a higherbaseline. Curiously, despite its difficulties, that article
seems to add very little to our understanding of cal-
cium response to glucose or IH. Certainly, IH differsgreatly from DOCA escape, in that IH patients show a
greater increment of urine calcium. As a part of his
interests in the mineralocorticoid calcium interac-
tions, and in work done near in time to the DOCAexperiments, Jack noted (15) the lack of any effects ofacute aldosterone or cortisol administration on the
relationship between calcium and sodium excretion.
Presumably, the higher baseline calcium excretion of
DOCA escape is simply the response of calcium excre-
tO DAYS DOCA
10mg
im bid
-80 -40 0 40 80120 -�O -40 0 40 �D 170
mm
Figure 4. Mean fractional excretion (ElF) rates of sodium, calcium, and magnesium durIng clearances before and after DOCAescape. Glucose was administered at zero time. b.i.d., twice daily. Used with permission (12).
0
ovvo,
7
‘0‘A
VA
,0 ,
0
010
/
/
‘0
0 a
0
0
/‘
V
NET INTESTINAL CALCIUM ABSORPTION, mg/day
Idiopathic Hypercalciuria
554 Volume 5 . Supplement 1 ‘1994
tion to sodium excretion, a well-established pattern
before Jack began his career. Altogether, sugars exag-gerate urine calcium excretion in IH and promotesupersaturation; how they do it is not known, nor whyIH patients respond more than normal people.
Calcitriol and Mineral Balance
If asked, we might say that among the most impor-
tant of the studies Jack contributed that bear on IH
are those concerning interactions between calcitriol
and calcium intake to affect urine calcium and bonemineral balance. Certainly, by 1976, one already had
in hand overwhelming evidence that IH could never be
simply a state of intestinal calcium hyperabsorption
alone, although absorption certainly Is always high
and high absorption Is always a main contributor to
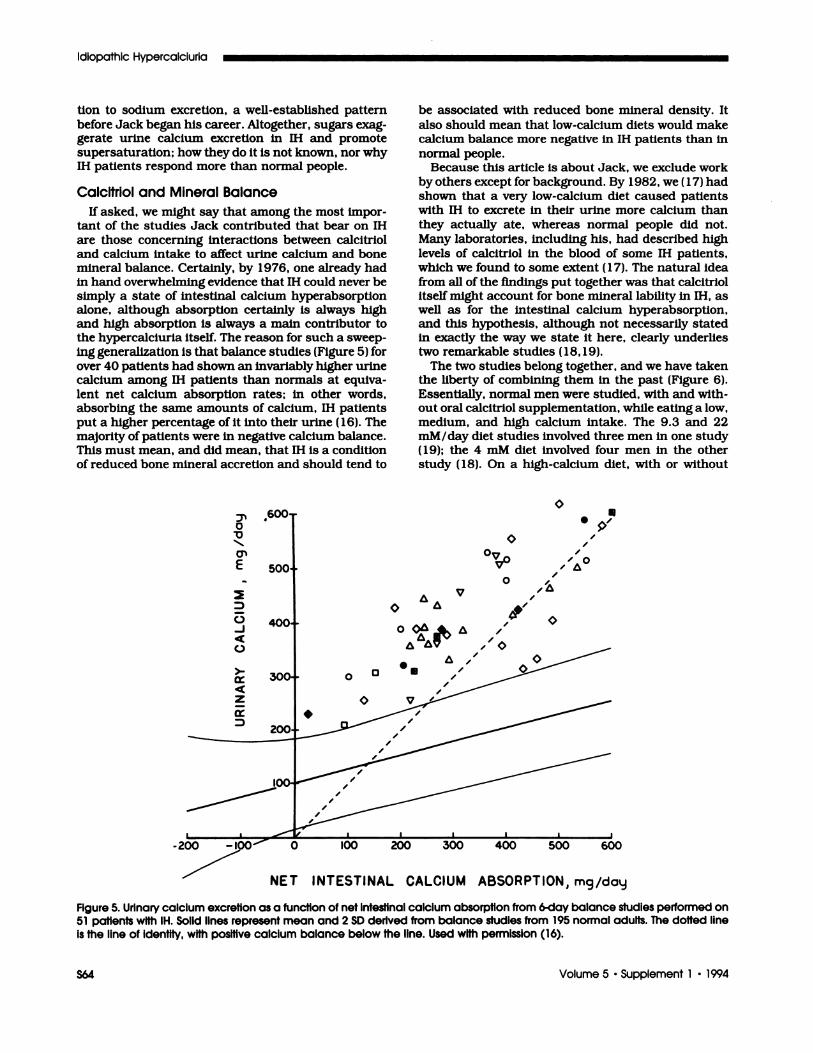
the hypercalclurla Itself. The reason for such a sweep-ing generalization Is that balance studIes (FIgure 5) forover 40 patIents had shown an invariably higher urine
calcium among IH patients than normals at equiva-
lent net calcium absorption rates; in other words,absorbing the same amounts of calcium, IH patients
put a higher percentage of it into their urine (16). The
majority of patients were in negative calcium balance.This must mean, and did mean, that IH is a conditionof reduced bone mineral accretion and should tend to
be associated with reduced bone mineral density. It
also should mean that low-calcium diets would makecalcium balance more negative in IH patients than in
normal people.Because this article is about Jack, we exclude work
by others except for background. By 1982, we (17) hadshown that a very low-calcium diet caused patients
with IH to excrete in their urine more calcium than
they actually ate, whereas normal people did not.Many laboratories, including his, had described high
levels of calcitriol in the blood of some IH patients,which we found to some extent (17). The natural Idea
from all of the findings put together was that calcitrlolItself might account for bone mineral lability in IH, aswell as for the intestinal calcium hyperabsorptlon,
and this hypothesis, although not necessarily stated
in exactly the way we state It here, clearly underlies
two remarkable studies (18,19).The two studies belong together, and we have taken
the liberty of combining them in the past (Figure 6).
Essentially, normal men were studied, with and with-out oral calcitrlol supplementation, while eating a low,medium, and high calcium intake. The 9.3 and 22mM! day diet studies involved three men in one study(19); the 4 mM diet involved four men in the otherstudy (18). On a high-calcium diet, with or without
0�0
0”
E
0
-J
C.)
>-
z
Figure 5. Urinary calcium excretion as a function of net intestinal calcium absorption from 6-day balance studies performed on51 patIents with IH. Solid lines represent mean and 2 SD derived from balance studies from 195 normal adults. The dotted lineis the line of identIty, with positive calcium balance below the line. Used with permission (16).
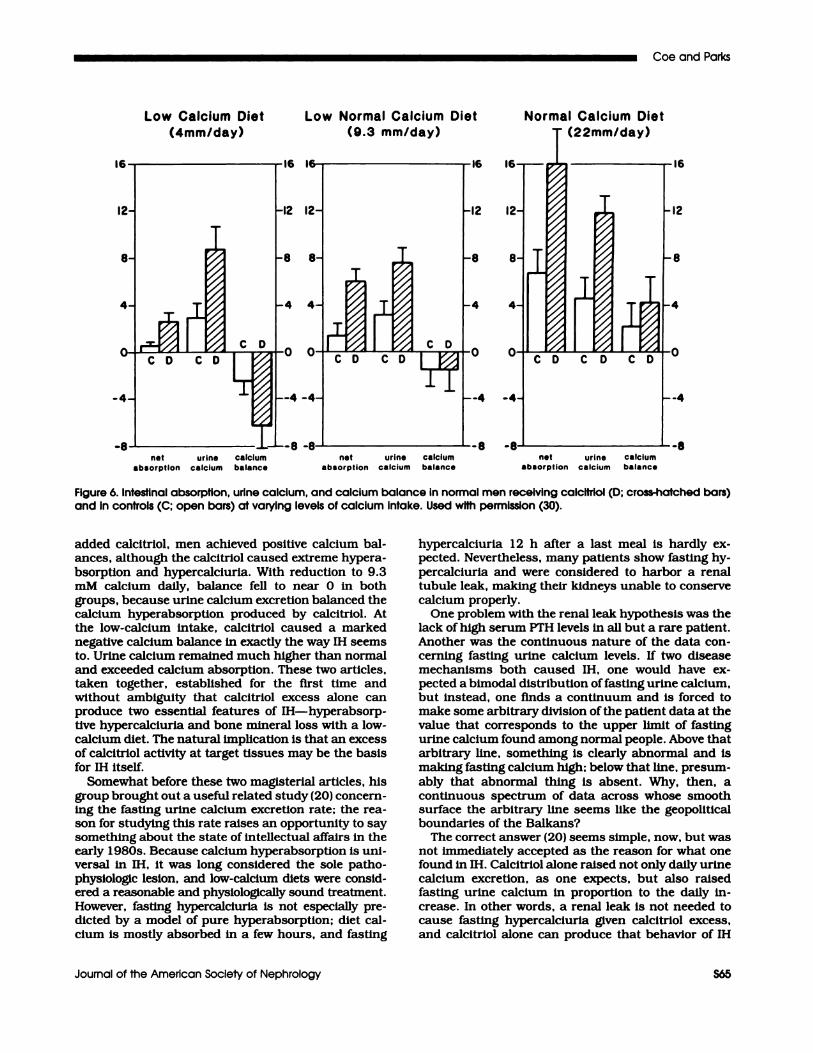
Low Calcium Diet Low Normal Calcium Diet
(4mm/day) (9.3 mm/day)
Normal Calcium Diet
(22mm/day)
net urine calcium net urine calcium net urine calciumabsorption calcium balance absorption calcium balance absorption calcium balance
Coe and Parks
Journal of the American Society of Nephrology 555
Figure 6. Intestinal absorption, urine calcium, and calcium balance in normal men receiving calcitriol (D; cross-hatched bars)and in controls (C: open bars) at varying levels of calcium Intake. Used with permission (30).
added calcitriol, men achieved positive calcium bal-
ances, although the calcitriol caused extreme hypera-bsorption and hypercalciuria. With reduction to 9.3mM calcium daily, balance fell to near 0 in both
groups, because urine calcium excretion balanced thecalcium hyperabsorption produced by calcitriol. At
the low-calcium intake, calcitriol caused a marked
negative calcium balance in exactly the way IH seems
to. Urine calcium remained much higher than normal
and exceeded calcium absorption. These two articles,
taken together, established for the first time andwithout ambiguity that calcitriol excess alone canproduce two essential features of IH-hyperabsorp-
five hypercalciuria and bone mineral loss with a low-calcium diet. The natural implication is that an excess
of calcitriol activity at target tissues may be the basis
for IH itself.
Somewhat before these two magisterial articles, his
group brought out a useful related study (20) concern-ing the fasting urine calcium excretion rate; the rea-
son for studying this rate raises an opportunity to say
something about the state of intellectual affairs in theearly 1980s. Because calcium hyperabsorptlon is uni-versal in IH, it was long considered the sole patho-physiologic lesion, and low-calcium diets were consid-ered a reasonable and physiologically sound treatment.
However, fasting hypercalciurla is not especially pre-
dicted by a model of pure hyperabsorptlon; diet cal-
cium is mostly absorbed in a few hours, and fasting
hypercalciuria 12 h after a last meal is hardly ex-pected. Nevertheless, many patients show fasting hy-
percalciuria and were considered to harbor a renal
tubule leak, making their kidneys unable to conserve
calcium properly.
One problem with the renal leak hypothesis was thelack of high serum PTH levels in all but a rare patient.Another was the continuous nature of the data con-cerning fasting urine calcium levels. If two diseasemechanisms both caused IH, one would have ex-pected a bimodal distribution of fasting urine calcium,but instead, one finds a continuum and is forced tomake some arbitrary division of the patient data at thevalue that corresponds to the upper limit of fastingurine calcium found among normal people. Above that
arbitrary line, something is clearly abnormal and is
making fasting calcium high; below that line, presum-
ably that abnormal thing is absent. Why, then, acontinuous spectrum of data across whose smoothsurface the arbitrary line seems like the geopoliticalboundaries of the Balkans?
The correct answer (20) seems simple, now, but wasnot immediately accepted as the reason for what onefound in 11-I. Calcitriol alone raised not only daily urinecalcium excretion, as one expects, but also raisedfasting urine calcium in proportion to the daily in-crease. In other words, a renal leak is not needed tocause fasting hypercalcluria given calcitrIol excess,and calcitriol alone can produce that behavior of IH

Idiopathic Hypercalciuria
556 Volume 5 ‘Supplement 1 ‘1994
just as it produces calcium hyperabsorption and bonemineral lability, with a low-calcium diet. In fact, this
study also showed levels of urine calcium during a 4mM/day calcium intake of 7.3 mM/day, clear evi-dence for bone mineral loss, although not with thebalance data, as in the later studies.
Almost in passing, although not with lack of due
respect, we note at least three studies related to their
wonderful work on mineral balances, but concerned
more with the regulation of calcitriol than its biology.
One, on vitamin D metabolites in anephric people, haslittle modern consequence, because newer methodsanswered directly what it attempted to learn at anearlier time (21); we mention it only for completeness.
Another, on calcitriol regulation (22), offered someearly proof that a low-calcium diet raises serum PTH
via lowered serum calcium and raises calcitriol levels,presumably because of the lower serum calcium and
high serum PTH. Of these, PTH seemed most impor-tant. This was correctly observed and has often been
shown elsewhere.Of special interest in this trilogy is an article on
low-phosphorus diet, which is instructive for showing
a marked sex difference in response and includessome instructive data concerning the pitfalls of bal-ance work (23). When challenged by inadequate dietphosphorus in a liquid diet, women raised their urinecalcium excretion, whereas men did not, and wentinto marked negative calcium balance. Both sexesshowed equal negative phosphorus balances. Duringrecovery, both sexes replaced a large portion of theirphosphorus deficits, but the women did not replacetheir lost calcium; the men, inexplicably, added addi-tional calcium, undoubtably to bone, despite neverhaving lost bone mineral during the phosphorus de-pletion phase.
The basis of the sex differences was not clear at thetime, but one should, in passing, note that the studywas flawed in a special sense. Liquid diets reducecalcitriol levels, and in men, control calcitriol levelswere taken while on normal diets, so any rise from a
low-phosphorus diet would be obscured. In a laterarticle (24), aluminum hydroxy gel was used for phos-phate depletion, and calcitriol levels rose in men. Theprior responses of urine calcium and calcium balancewere not reproduced; men showed the same negativecalcium balances as women had shown in the priorstudy. We quote this pair of articles because of whatthey can teach us about the extreme dIfficulties of
human balance work. It is also wonderful to see howthe group corrected Its own inadvertent error.
Acid Base Balance and Mineral Loss II
The last, and very great, studies from his career
returned to the theme that began it,although the first
article of the lot (25) seemed to be about the thla.zldehypocalciuric action. The fact that thIazide lowers
urine calcium and prevents kidney stone recurrence
in IH patients was well known to Jack, but he asked
where the calcium goes, ifnot into the urine. Insteadof IH patients, he used normals, either with or without
supplemental calcitriol, and either way, the resultswere the same. Thiazlde could reduce calcium absorp-tion, so the calcium goes into the stool, or it might not,so the calcium goes into bone instead of either stool orurine. The answer was that it goes into bone (Table 4).We have shown that what Jack demonstrated fornormals is true for IH patients (26), as might be
expected from the response when calcitriol was givento normals.
How thiazide might work was and is unclear. Thearticle proposes that acid base balance might playsome role. Thiazide caused its usual rise in blood pHand bicarbonate levels, and in an analysis from all ofthe balance studies he had ever performed, Jackshowed that calcium balance was always inverselyproportional to acid balance, in other words, positivewhen acid balance was negative and the obverse. In away, this correlation is unsurprising, because acid
administration certainly lowers calcium balance, andhis studies often used acid loading. The fact thatthiazide acts via acid base changes is not at all provedby the study. Nevertheless, the consequences of thia-
zlde are proved, and possibifitles for mechanisms are
raised. As well, the value of thiazide in protecting bonewas shown clearly and led directly to our proof of asimilar action when IH Is so treated (26).
Fasting urine calcium as an index of a renal leak
and a way to categorize IH patients aroused Jack on asecond occasion, when he asked if acid loads could
raise fasting urine calcium at steady calcitriol levelsand intestinal calcium absorption rates (27). Now,Jack had already shown increased fasting calcium
from acid loads, but he felt that the point neededemphasis and association with measurements of cal-citriol and PTH, to make clear how much fasting urinecalcium might reflect the state of acId balance. Thedose of ammonlum chloride was 3 mM / kg daily, ofmodest extent, and the acidemla was modest (Table5). Acid loading raised fasting urine calcium mark-edly, but the increment of fasting calcium after acalcium load, which reflects calcium absorption, wasunchanged.
TABLE 4. Effects of hydrochlorothiazide (25 mg)twice daily#{176}
Measurement Control Thiazide
Fecai Calcium 4.6 4.4Urine Calcium 4.3 2.9Calcium Balance -3.8 -2.2Urine Hydroxyproline uM/DAY 390 260Blood pH 7.36 7.41Blood Bicarbonate (mM/I) 26 28.7
#{176}Summarized from Reference 25 (Table 2). Subjects were men; dietcalcium averaged 5.1 mM/day. All values in table are millimoles per
day. unless otherwise noted. All values differed between control andthiazide. P < 0.05, except fecal calcium, which was unchanged.
Coe arid Parks
Journal of the American Society of Nephrology S67
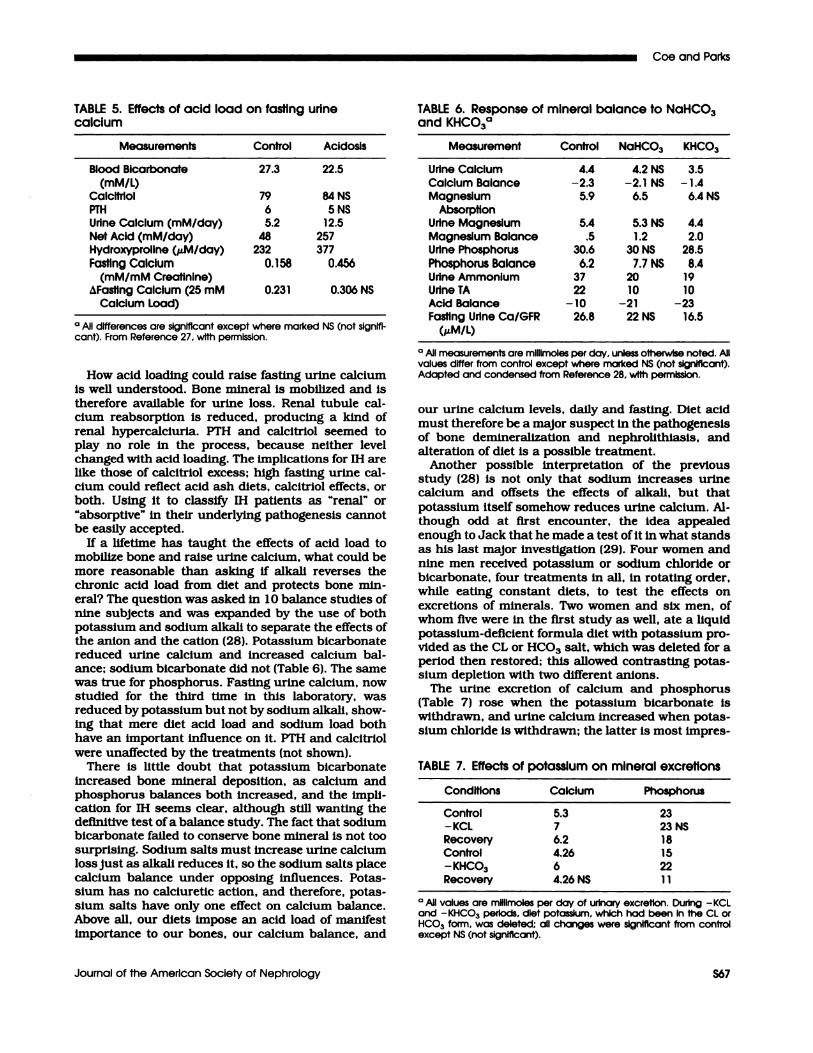
TABLE 5. Effects of acid load on fasting urinecalcium
Measurements Control Acidosis
Blood Bicarbonate 27.3 22.5(mM/L)
Calcitriol 79 84 NS
PTH 6 5NSUrine Calcium (mM/day) 5.2 12.5Net Acid (mM/day) 48 257Hydroxyproline (�M/day) 232 377Fasting Calcium 0.158 0.456
(mM/mM Crealinlne)i�Fasting Calcium (25 mM 0.23 1 0.306 NS
Calcium Load)
#{176}All differences are signIficant except where marked NS (not signS-cant). From Reference 27, wIth permission.
How acid loading could raise fasting urine calcium
Is well understood. Bone mineral is mobilized and is
therefore available for urine loss. Renal tubule cal-cium reabsorption is reduced, producing a kind of
renal hypercalciuria. PTH and calcitrIol seemed to
play no role in the process, because neither level
changed with acid loading. The implications for IH arelike those of calcitriol excess; high fasting urine cal-cium could reflect acid ash diets, calcitriol effects, or
both. Using It to classi1� IH patients as “renal” or
“absorptive” in their underlying pathogenesls cannot
be easily accepted.
If a lifetime has taught the effects of acid load tomobilize bone and raise urine calcium, what could be
more reasonable than asking if alkali reverses thechronic acid load from diet and protects bone min-
eral? The question was asked in 10 balance studies ofnine subjects and was expanded by the use of both
potassium and sodium alkali to separate the effects ofthe anion and the cation (28). Potassium bicarbonatereduced urine calcium and increased calcium bal-ance; sodium bicarbonate did not (Table 6). The samewas true for phosphorus. Fasting urine calcium, nowstudied for the third time in this laboratory, wasreduced by potassium but not by sodium alkali, show-ing that mere diet acid load and sodium load both
have an important influence on it. PTH and calcitrlolwere unaffected by the treatments (not shown).
There is little doubt that potassium bicarbonateincreased bone mineral deposition, as calcium andphosphorus balances both increased, and the impli-cation for IH seems clear, although still wanting thedefinitive test of a balance study. The fact that sodiumbicarbonate failed to conserve bone mineral is not toosurprising. Sodium salts must increase urine calciumloss just as alkali reduces It, so the sodium salts place
calcium balance under opposing influences. Potas-sium has no calciuretic action, and therefore, potas-sium salts have only one effect on calcium balance.Above all, our diets impose an acid load of manifestimportance to our bones, our calcium balance, and
TABLE 6. Response of mineral balance to NaHCO3and KHCO3#{176}
Measurement Control NaHCO3 KHCO3
Urine CalcIum 4.4 4.2 NS 3.5Calcium Balance -2.3 -2.1 NS -1.4Magnesium 5.9 6.5 6.4 NS
AbsorptionUrine Magnesium 5.4 5.3 NS 4.4Magnesium Balance .5 1.2 2.0Urine Phosphorus 30.6 30 NS 28.5Phosphorus Balance 6.2 7.7 NS 8.4Urine Ammonium 37 20 19UrIneTA 22 10 10Acid Balance -10 -21 -23Fasting Urine Ca/6FR 26.8 22 NS 16.5
(aiM/I)
#{176}All measurements are mlilimoles per day. unless otherwise noted. Allvalues differ from control except where marked NS (not significant).Adapted and condensed from Reference 28. with permission.
our urine calcium levels, daily and fasting. Diet acid
must therefore be a major suspect in the pathogenesis
of bone demineralization and nephrolithiasis, and
alteration of diet is a possible treatment.
Another possible interpretation of the previous
study (28) is not only that sodium increases urinecalcium and offsets the effects of alkali, but that
potassium itself somehow reduces urine calcium. Al-though odd at first encounter, the idea appealed
enough to Jack that he made a test of Itin what standsas his last major investigation (29). Four women and
nine men received potassium or sodium chloride orbicarbonate, four treatments in all, in rotating order,
while eating constant diets, to test the effects on
excretions of minerals. Two women and six men, ofwhom five were in the first study as well, ate a liquid
potassium-deficient formula diet with potassium pro-
vided as the CL or HCO3 salt, which was deleted for aperiod then restored; this allowed contrasting potas-sium depletion with two different anions.
The urine excretion of calcium and phosphorus(Table 7) rose when the potassium bicarbonate is
withdrawn, and urine calcium increased when potas-
sium chloride is withdrawn; the latter is most impres-
TABLE 7. Effects of potassium on mineral excretions
Conditions Calcium Phosphorus
Control 5.3 23-XCI 7 23NSRecovery 6.2 18Control 4.26 15-KHCO3 6 22Recovery 4.26 NS 11
#{176}All vaiues are millimoles per day of urinary excretion. During -KCLand - KHCO3 periods, diet potassium, which had been in the CL orHCOa form, was deleted; all changes were significant from controlexcept NS (not significant).

Idiopathic Hypercalciurla
558 Volume 5 ‘Supplement 1 1994
sive because the increase could not be merely another
example of alkali actions. Merely supplementing the
diet with potassium chloride, however, does not lower
urine calcium, whereas giving supplemental potas-sium bicarbonate does, the latter presumably a reflec-tion of alkali effects. Because potassium chloride didnot reduce urine calcium, the main thing one can say
about potassium itself is that, when withdrawn fromthe diet in the chloride form, urine calcium rises. In
other words, potassium is essential for proper calciumconservation by the kidneys and possibly bone.
This leads one to consider potassium depletion fromdiuretics as a possible offset against their hypocalci-
uric actions in IH patients and strengthens the casefor maintaining potassium stores intact. The fact that
the potassium alkali are more effective in loweringurine calcium than are sodium alkali salts is now well
proven, by this article and its antecedents. As for diet,
from what Jack has done, we need to consider thatnephrolithiasis and bone disease may be fostered not
only by acid loads but also by relative potassium
insufficiency.
FINAL REMARKS
During a long career, its shape and evident purpose
are blurred, if by nothing else, simple motion and
change, but at the end, everything becomes clear. Thepart of Jack’s work that will not be easily made
obsolete by technology and progress, in other words,
that portion partaking of universal interest and value,
surely is the balance studies. No one in our genera-
tion-for we share one with him-has contributed somany fine and pertinent ones as he has, and theresults, for acid base balance especially, are bedrock
for any Investigator. People seek antecedents, invari-ably. wondering from what root a branch has grown,and a quick answer for Jack would be the acid baseschool from which his earliest articles derive. How-
ever, we are not so sure, because the mineral studies
overshadow those concerning acid base balance per
Se, and the methods of procedure in balance work are
not native to such an origin. Further back in time
resides a better figure to choose as intellectual father
to this work, and ifAlbrlght did not instruct Jack, for
the two do not overlap in time, his work certainly did.The other question is about who comes after, and
here one also has the usual problem of determining.His articles name many distinguished colleagues, so
one need not look far for young people Jack has
influenced who can and have carried on his line ofwork. But some less evident beneficiaries may have
promulgated and extended his work, even more thansome closer to him. Among these, surely, are members
of our own research group, as it is now and as it was
10 years ago. We have studied his work in abstracts
and have often used it for our own beginning place,
and although Jack has not instructed us directly,most of the time, his work certainly did, one way or
another.
REFERENCES
1. Coe FL, Parks JH, Asplin JR: The pathogenesis andtreatment of kidney stones-medical progress. N Engl JMed 1992;327:1141-1 152.
2. Lemann J Jr. Litzow JR, Lennon EJ: The effects ofchronic acid loads in normal man: Further evidence forthe participation of bone mineral in the defense againstchronic metabolic acidosis. J Clin Invest 1966:45:1608-1614.
3. Bushinsky DA, Krieger NS, Geisser DI, Grossman EB,Coe FL: Effects of pH on bone calcium and proton fluxesin vitro. Am J Physiol 1983;245:F204-F209.
4. Bushinsky DA, Lechleider 1W: Mechanism of proton-induced bone calcium release: calcium carbonate-dissolution. Am J Physiol 1987;253:F998-F1005.
5. Lemann J Jr, Lltzow JR. Lennon EJ: Studies of themechanism by which chronic metabolic acidosis aug-ments urinary calcium excretion in man. J Clin Invest1967:46:1318-1328.
6. Adams ND, Gray RW, Lemann J Jr: The calciuria ofIncreased fixed acid production in humans: Evidenceagainst a role for parathyroid hormone and 1 .25-(OH)2-vitamin D. Calcif Tissue mt 1979;28:233-239.
7. Lemann J Jr. Adams ND, Gray RW: Urinasy calciumexcretion in human beings. N Engl J Med 1979;301 :535-541.
8. Lemann J Jr, Piering WF, Lennon EJ: Possible role ofcarbohydrate-induced calciuria In calcium oxalate kid-ney-stone formation. N EngI J Med 1969:280:232-237.
9. Lindeman RD. Adler 5, Yiengst MJ, Beard ES: Influenceof various nutrients on urinary divalent cation excretion.J Lab Clin Med 1967:70:236-245.
10. Pierlng WF, Lemann J Jr, Lennon LI: Effect of carbo-hydrate administration on urinary calcium and magne-sium excretion. Chin Res 1968:16:393.
11. Lemann J Jr. Lennon LI, Piering WF, Prien ELI,Ricanati ES: Evidence that glucose ingestion inhibits netrenal tubular reabsorption of calcium and magnesium inman. J Lab Chin Med 1970;75:578-585.
12. Lennon EJ, Lemann J Jr. Piering WF, Larson LS: Theeffect of glucose on urinary cation excretion duringchrolnic extraceilular volume expansion in normal man.J Chin Invest 1974:53:1424-1433.
13. Coe FL, Parks JH, Moore ES: Familial Idiopathic hyper-calciuria. N Engl J Med 1979:300:337-340.
14. Bushinsky DA, Favus MJ: Mechanism of hypercalciuriain genetic hypercalciuric rats. Inherited defect in intes-tinal calcium transport. J Chin Invest 1988:82:1585-159 1.
15. Lemann J Jr. Piering WF, Lennon Li: Studies of theacute effects of aldosterone and cortisol on the interre-lationship between renal sodium, calcium and magne-sium excretion in normal men. Nephron 1970; 7:117-130.
16. Coe FL, Favus MJ: Disorders of stone formation. In:Brenner BM, Rector PC Jr, Eds. The Kidney. Philadel-phia: WB Saunders: 1986:1403-1442.
17. Coe FL, Favus MJ, Crockett T, et at,: Effects of low-calcium diet on urine calcium excretion, parathyroidfunction and serum I ,25(OH)2D3 levels in patients withidiopathic hypercalciuria and in normal subjects. Am JMed 1982:72:25-32.
18. Malerhofer WJ, Gray RW, Adams ND: Bone resorptionstimulated by elevated serum 1 ,25-(OH)2-vltamin D con-centrations in healthy men. Kidney Int 1983:24:555-560.
19. Maierhofer WJ, Lemann J Jr. Gray RW, Cheung HS:Dietary calcium and serum 1 .25-(OH)2-vitamin D con-centrations as determinants of calcium balance inhealthy men. Kidney kit 1984:26:752-759.
20. Adams ND, Gray RW, Lemann J Jr. Cheung HS: Effectsof calcitriol administration on calcium metabolism Inhealthy men. Kidney Int 1982:21:90-97.
21. Gray RW, Weber HP, Dominguez JH, Lemann J Jr: Themetabolism of vitamin D3 and 25-hydroxyvitamin D3 Innormal and anephric humans. J Chin Endocrinol Metab1974:39:1045-1056.

Coe and Parks
Journal of the American Society of Nephroiogy Soc
22. Adams ND, Gray RW, Lemann J Jr: The effects of oralCaCo3 loading and dietary calcium deprivation onplasma 1 ,25-dihydroxyvitamin D concentration inhealthy adults. J Clin Endocrlnol Metab 1979:48:1008-1016.
23. Dominguez JR. Gray RW, Lemann J Jr: Dietary phos-phate deprivation in women and men: Effects on mineraland acid balances, parathyroid hormone and the metab-olism of 25-OH-vitamin D. J Clin Endocrinol Metab1976;43: 1056-1068.
24. Malerhofer WJ, Gray RW, Lemann J Jr: Phosphatedeprivation increases serum 1 ,25-(OH)2-vitamln D con-centrations in healthy men. Kidney kit 1984:25:571-575.
25. Lemann J Jr. Gray RW, Malerhofer WJ, Cheung HS:Hydrochlorothlazide inhibits bone resorbtion in mendespite experimentally elevated serum 1,25-dihy-droxyvitamin D concentrations. Kidney hit 1985;28:951-958.
26. Coe FL, Parks JR. Bushinsky DA, Langman CB, FavusMJ: Chlorthalidone promotes mineral retention in pa-tients with idiopathic hypercalciuria. Kidney hit 1988:33:1140-1 146.
27. Lemann J Jr. Gray RW, Malerhofer WJ, Cheung HS: Theimportance of renal net acid excretion as a determinantof fastIng urinary calcium excretion. Kidney hit 1986:29:743-746.
28. Lemann J Jr, Gray RW, Pleuss JA Potassium bicarbon-ate, but not sodium bicarbonate, reduces urinary cal-cium excretion and improves calcium balance in healthymen. Kidney bit 1989:35:688-695.
29. Lemann J Jr, Pleuss JA. Gray RW, Hoffmann RG:Potassium administration reduces and potassium depri-vation increases urinary calcium excretion in healthyadults. Kidney kit 199 1:39:973-983.
30. Coe FL, Parks JH: FamilIal (idiopathic hypercalciuria).Nephrolithiasis: Pathogenesis and Treatment. Chicago:Year Book Medical Publishers; 1988: 108-138.





























