Embed Size (px)
Citation preview

Proc. Nati. Acad. Sci. USAVol. 88, pp. 9036-9040, October 1991Immunology
Identification of the Fc,, receptor class I binding site in human IgGthrough the use of recombinant IgG1/IgG2 hybrid andpoint-mutated antibodiesM. SUZANNE CHAPPEL*, DAVID E. ISENMAN*t, MARGARET EVERETT*, YUAN-YUAN XU*,KEITH J. DORRINGTON*, AND MICHEL H. KLEIN*tt§Departments of *Immunology and tBiochemistry, University of Toronto, Toronto, ON, Canada M5S 1A8; and tConnaught Centre for BiotechnologyResearch, Willowdale, ON, Canada M2R 3T4
Communicated by David R. Davies, June 24, 1991 (received for review May 9, 1991)
ABSTRACT To characterize the region on human IgG1responsible for its high-affinity interaction with the human Fclyreceptor class I (FcMRI), we have analyzed the binding prop-erties of a series of genetically engineered chimeric anti-dinitrophenyl antibodies with identical murine antibody com-bining sites and hybrid IgGl/IgG2 human constant (C) re-gions. In addition, we have investigated a panel of reciprocallypoint-mutated IgG1 and IgG2 chimeric antibodies to identifythe amino acid residues that confer cytophilic properties tohuman IgG1. Our data unambiguously indicate that cytophilicactivity of IgG1 is an intrinsic property of its heavy-chain Cregion 2 (CH2) domain. We report that the entire sequencespnning residues 234-237 (LLGG) is required to restore fullbinding activity to IgG2 and IgG4 and that individual aminoacid substitutions failed to render IgG2 active. Nevertheless,the reciprocal single point mutations in IgG1 either signifi-candy lowered its activity or abolished it completely. Finally,we observed that an IgG2 antibody containing the entireELLGGP sequence (residues 233-238) was more active thanwild-type IgG1. This finding suggests that in addition to theprimary contact site identified in the N terminus of the vl CH2domain, secondary sites involving residues from the C-terminalhalf of the domain may also contribute to the IgGl-Fc,$Iinteraction.
Fc receptors for human IgGs (FcR) form a family of integralmembrane proteins that specifically bind to immunoglobulinFc regions.O The interaction ofFcRs with IgG trigger criticalhost effector functions such as phagocytosis of immunecomplexes (1), antibody-dependent cell-mediated cytotoxic-ity (2-4), and the release of inflammatory mediators (5).Three classes of human FceR have been defined on the
basis of their reactivity with monoclonal antibodies (6). Thehigh-affinity FcRI (CD64) is a 72-kDa glycoprotein ex-pressed on mononuclear phagocytes (7) and interferon y-ac-tivated neutrophils (2). FcRI displays a hierarchy of affini-ties for monovalent human IgG subclasses. IgG1 and IgG3bind with high affinity (Ka 108-109 M-1), whereas IgG4 is10-fold less cytophilic and IgG2 is devoid of any significantbinding activity (8).
Several indirect approaches suggest that the primary sitethat mediates the IgG1-FcXRI interaction resides within theheavy-chain constant region 2 (CH2) domain. Monoclonalantibodies directed against a CH2 domain N-terminal epitopeincluding the isotype-specific residue K274, inhibited theIgG1-FcRI interaction (9). Furthermore, the CH2 domain-deleted IgG1 myeloma protein TIM failed to bind FcXRI (10).Results obtained with isolated CH2 domains remain contro-versial since dimeric, but not monomeric, domains possess
cytophilic activity (11, 12). In the context of a hybrid mole-cule, however, only a single cytophilicH chain is required forbinding activity (13). More recently, the functional analysis ofa panel of genetically engineered human IgG1/IgE hybridantibodies indicated that the FcRI binding site was essen-tially a property of the IgG1 CH2 domain (14).
Several observations suggest that the CH2 N terminusforms at least part of the IgG1 FcXRI recognition site.Comparative primary sequence analysis revealed that anLLGGP motif spanning residues 234-238 is conserved in allcytophilic IgGs (15). Site-directed mutagenesis of E235 innoncytophilic mouse IgG2b to L235 produced the LLGGPmotif on an IgG2b background, rendering this molecule fullyactive (16). This was not predicted to be the only criticalresidue in the human site since IgG1 and IgG4 are identicalat this position, yet IgG4 is 10-fold less active. Finally,whereas IgG1 Fc fragments obtained by digestion with papainretain cytophilic activity, those obtained with thermolysin,which cleaves between residues 234 and 235, do not (11). Thisindicates that FcXRI binding activity requires the presence ofresidues N-proximal to L235 but not that of the hingedisulfide bonds, since reduction and alkylation ofIgG1 has nosignificant effect on its binding affinity for FcXRI (15).We have engineered a panel of chimeric IgGl/IgG2 hybrid
molecules and a set of reciprocally point-mutated IgG1 andIgG2 antibodies to identify the region(s) and ultimately theamino acids that are essential for IgGl-FcXRI recognition andare sufficient to restore full activity to IgG2. We report thatIgG2 molecules containing only an IgG1 CH2 domain are ascytophilic as wild-type IgG1 and that the amino acids that arecritical for this activity are L234, L235, and G237.
MATERIALS AND METHODSCell Lines. The murine myeloma A-chain-producing mutant
cell line MOPC 315.26 was maintained as described (17).U937 cells were grown in RPMI 1640 medium supplementedwith 2% fetal calf serum, 2 mM glutamine, and antibiotics.Plasmid Constructs. The pSV2neoVH315 mammalian
expression vector has been described (18). The 3.2-kilobaseHindIII/BamHI IgG1 and IgG2 CH gene fragments werekindly provided by L. Hood (California Institute of Technol-ogy). All molecular cloning techniques were performed ac-cording to Sambrook et al. (19). The human IgG1, IgG2, andIgG4 C region genes were cloned into pEMBL-19, where all
Abbreviations: C, constant; H, H chain; FczRI, high-affinity Fcyreceptor class I.§To whom reprint requests should be addressed at: Department ofImmunology, Medical Sciences Building, University of Toronto,Toronto, ON, Canada M5S 1A8.Nomenclature Subcommittee, Oral Presentation, FASEB SummerResearch Conference on Fc Receptors & Immunoglobulin BindingFactors, June 15-19, 1987, Saxton's River, VT.
9036
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.

Proc. Natl. Acad. Sci. USA 88 (1991) 9037
HINGE
IgGl .1.1 I.1X
Hind III Pst I Xba I EcoR I BamH I
HINGE
IgG2.2.2.2
Hind IlIl Pst I Xba I EcoR I BamH I
IgG1.1.1., _I _ - IgG2.2.2.1
IgGI.l-2.1{_IgG2.2.1.2
IgIl.2.1.1{ n 3 ~ ~IgG2.1.2.2
no-m-m- I IgG2.2.1.1
IgGl.2.2.1 { IgG2.1.1.2
IgG1 IgG2
FIG. 1. Schematic representationof wild-type, hybrid IgG1/IgG2 (Left)and hybrid IgG2/IgG1 (Right) C re-gion gene constructs. Genes weresubcloned into pEMBL-19 betweenits HindIl1 and BamHI sites. UniqueXba I and EcoRI sites were intro-duced by site-directed mutagenesis inthe hinge-CH2 and the CH2-CH3 in-trons, respectively. Hybrid geneswere constructed by exchange of ex-ons excised with the appropriate re-striction enzymes.
subsequent genetic manipulations were done. Oligonucle-otide-directed mutagenesis was performed by the Ecksteinmethod (20) (Amersham) to introduce unique Xba I andEcoRI sites in the hinge-CH2 and CH2-CH3 introns, respec-tively, and to substitute amino acids. Hybrid IgGl/IgG2,IgG2/IgGl, and IgGl/IgG4 genes were constructed by re-ciprocally exchanging exons excised with the appropriaterestriction enzymes. The transfer of native and modifiedIgG1, IgG2, and IgG4 genes into the pSV2neoVH315 expres-sion vector was performed as described (18). The resultingexpression vector encodes an entire H chain composed of ahuman IgG C region and the murine MOPC 315 H-chainvariable region (VH) domain. The identity of each constructwas confirmed in both pEMBL-19 and in pSV2neoVH315 byrestriction map analysis and DNA sequencing.
Electroporation. The pSV2neoVH315 IgG1, IgG2, and IgG4H-chain constructs were transfected into the A-chain-producing cell line MOPC 315.26 by electroporation accord-ing to the method ofBaker et al. (21). The electroporated cellswere grown and selected in G418 (GIBCO) (17). Antibodyproduction was quantified by IgG-specific capture ELISA,using myeloma human IgG1 and IgG2 as standards.Assembly of Chimeric Molecules. The assembly of the
chimeric antibodies was evaluated by SDS/polyacrylamidegradient (4-15%) gel electrophoresis under nonreducing con-ditions. Culture supernatants containing -5 ,g of chimericimmunoglobulin were incubated with 50 ,ul of a 10%o suspen-sion of formalin-treated Staphylococcus aureus bacteria for1 hr at 4°C. The pellets were washed six times in phosphate-buffered saline (PBS) containing 0.5% Triton X-100, 0.25%deoxycholate, 0.5% SDS, 10 mM EDTA, and 2 mM phenyl-methylsulfonyl fluoride and then boiled for 3 min in 2xsample buffer containing 4% SDS and 1 mM iodoacetamideto prevent disulfide interchange.
Preparation of Culture Supernatants for Binding InhibitionAssays. Culture supernatants were concentrated 10-fold usingCentriprep-30 microconcentrators (Amicon). The concen-trated culture supernatants were then subjected to ultracen-trifugation for 30 min at 100,000 x g at 4°C to removepotential IgG aggregates. The final concentration of chimericantibodies was assessed by triplicate ELISA determinations.Binding Inhibition Studies. Purified human IgG1 myeloma
protein was radioiodinated using Iodobeads (Pierce) to aspecific activity of 1 x 106 cpm/,ug. Bound 1251 was separatedfrom free material by gel filtration through a Biogel P4 spincolumn. IgG concentration was measured spectrophotomet-rically at 280 nm using an extinction coefficient E%1-"v of 1.4.U937 cells were washed once in PBS containing 0.02%sodium azide and bovine serum albumin (1 mg/ml) andresuspended in the same buffer. Direct binding and inhibitionassays were carried out in 96-well U-bottom microtiter platesessentially as described by Raychaudhuri et al. (22). In brief,
for the inhibition assay, 2.5 x 106 U937 cells were incubatedper well with 10 nM radiolabeled IgG1 and increasing con-centrations ofchimeric inhibitor for 2 hr at room temperatureand then spun through a dibutyl phthalate oil cushion; cellularpellets were assayed for bound radioactivity. Nonspecificbinding was determined from the residual bound radioactivityin the presence of 10 ,uM unlabeled IgG1 and never exceeded2% of total binding. All values were corrected for nonspecificbinding. The association constants of the chimeric antibodieswere estimated from the displacement of the inhibition curve(at 50%) obtained with chimeric inhibitor relative to thatobtained with myeloma IgG1 for which the Ka was deter-mined in a direct binding assay (23). Three independentdeterminations were obtained in triplicate for each chimeraspecies.
RESULTSConstruction, Expression, and FcRI Binding Properties of
Hybrid IgGl and IgG2 Antibodies. The role of individualdomains in the cytophilic activity ofhuman IgG isotypes wasanalyzed by exon-shuffling experiments. The panel of hybridgenes is depicted schematically in Fig. 1. An IgG1 moleculewith an IgG4 hinge was also constructed (data not shown).Hybrid molecules are described by four digit numbers indi-cating the subclass origin of the CH1, hinge, CH2, and CH3exons, respectively.Chimeric immunoglobulins from clones secreting in the
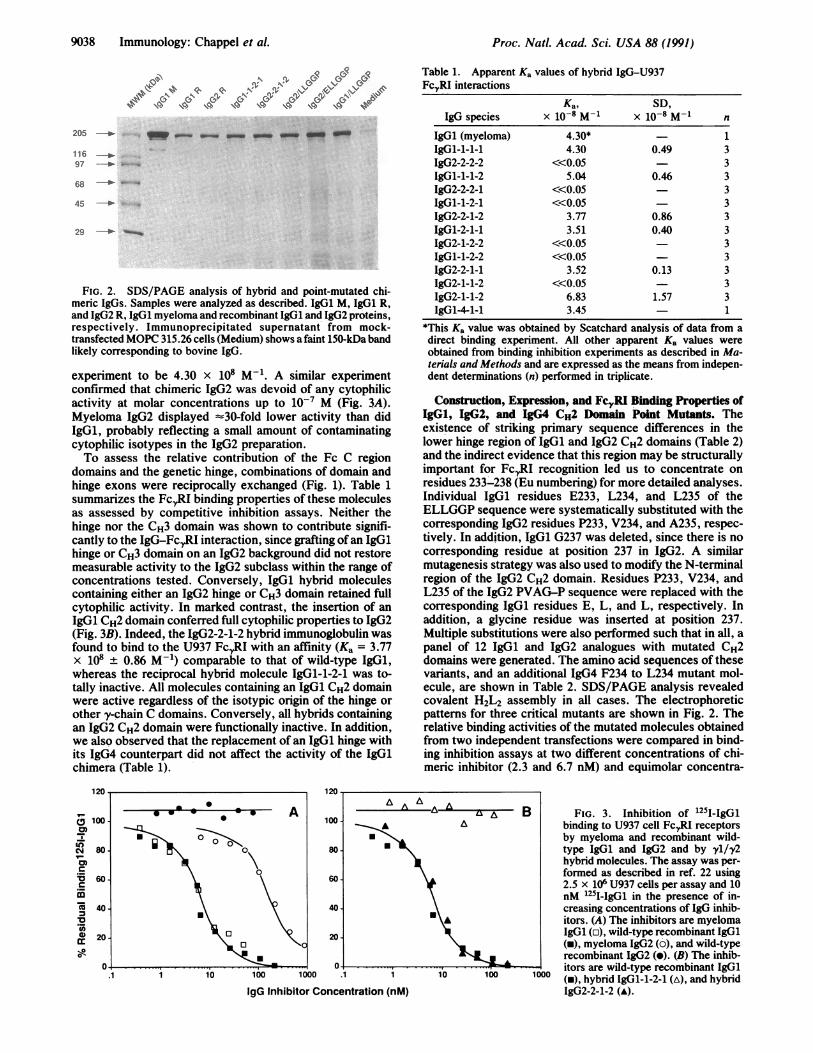
range of 5-10 ,ug of IgG per ml were affinity isolated with S.aureus cells and analyzed by SDS/PAGE. Nonreduced SDS/PAGE revealed that all molecules migrated as a single bandwith an apparent molecular mass of 150 kDa correspondingto a covalently assembled H2L2 molecule that could bedissociated into its constituent H and L chains upon reduc-tion (data not shown). Representative electrophoretic pat-terns obtained with wild-type chimeric IgG1 and IgG2 andhybrid IgGl-1-2-1, IgG2-2-1-2 molecules are shown in Fig.2.11The binding properties of the exon-shuffled antibodies
were assessed by competitive inhibition assays. To validatethe use of chimeric IgGs as inhibitors, we confirmed that thecytophilic activity of the IgG1 chimera was indistinguishablefrom that of myeloma IgGI. Representative binding inhibi-tion curves obtained for these molecules are depicted in Fig.3A. The curves demonstrate a typical sigmoidal shape and aresuperimposable. This indicated that the Ka value for thechimeric IgG1 molecule was the same as that of myelomaIgGl, which was determined in an independent direct binding
11Example ofnomenclature for hybrid molecules: IgG1- 1-2-1 refers toCH1, hinge, and CH3 exons of IgG1 origin, and a CH2 exon of IgG2origin.
Immunology: Chappel et al.
IgGI.I.2."'d!--[::l
9038 Immunology: Chappel et al.
205 o o EPW
116 _97 _68 -
45 -0 a
29 -'_
FIG. 2. SDS/PAGE analysis of hybrid and point-mutated chi-meric IgGs. Samples were analyzed as described. IgG1 M, IgG1 R,and IgG2 R, IgG1 myeloma and recombinant IgG1 and IgG2 proteins,respectively. Immunoprecipitated supernatant from mock-transfected MOPC 315.26 cells (Medium) shows a faint 150-kDa bandlikely corresponding to bovine IgG.
experiment to be 4.30 x 108 M-1. A similar experimentconfirmed that chimeric IgG2 was devoid of any cytophilicactivity at molar concentrations up to 10-7 M (Fig. 3A).Myeloma IgG2 displayed -30-fold lower activity than didIgG1, probably reflecting a small amount of contaminatingcytophilic isotypes in the IgG2 preparation.To assess the relative contribution of the Fc C region
domains and the genetic hinge, combinations of domain andhinge exons were reciprocally exchanged (Fig. 1). Table 1summarizes the FcRI binding properties of these moleculesas assessed by competitive inhibition assays. Neither thehinge nor the CH3 domain was shown to contribute signifi-cantly to the IgG-FcRI interaction, since grafting ofan IgGlhinge or CH3 domain on an IgG2 background did not restoremeasurable activity to the IgG2 subclass within the range ofconcentrations tested. Conversely, IgGl hybrid moleculescontaining either an IgG2 hinge or CH3 domain retained fullcytophilic activity. In marked contrast, the insertion of anIgGl CH2 domain conferred full cytophilic properties to IgG2(Fig. 3B). Indeed, the IgG2-2-1-2 hybrid immunoglobulin wasfound to bind to the U937 Fc,,RI with an affinity (Ka = 3.77x 108 ± 0.86 M-1) comparable to that of wild-type IgG1,whereas the reciprocal hybrid molecule IgGl-1-2-1 was to-tally inactive. All molecules containing an IgG1 CH2 domainwere active regardless of the isotypic origin of the hinge orother y-chain C domains. Conversely, all hybrids containingan IgG2 CH2 domain were functionally inactive. In addition,we also observed that the replacement of an IgG1 hinge withits IgG4 counterpart did not affect the activity of the IgGlchimera (Table 1).
Table 1. Apparent Ka values of hybrid IgG-U937FcyRI interactions
IgG species
IgG1 (myeloma)IgGl-1-1-1IgG2-2-2-2IgGl-1-1-2IgG2-2-2-1IgGl-1-2-1IgG2-2-1-2IgGl-2-1-1IgG2-1-2-2IgGl-1-2-2IgG2-2-1-1IgG2-1-1-2IgG2-1-1-2IgGl-4-1-1
Ka, SD,X 10-8 M-1 X 10-8M-
4.30*4.30
<<0.055.04
<<0.05<<0.05
3.773.51
<<0.05<<0.05
3.52<<0.05
6.833.45
0.49
0.46
0.860.40
0.13
1.57
n
13333333333331
*This Ka value was obtained by Scatchard analysis of data from adirect binding experiment. All other apparent Ka values wereobtained from binding inhibition experiments as described in Ma-terials and Methods and are expressed as the means from indepen-dent determinations (n) performed in triplicate.
Construction, Expression, and Fc1,RI Binding Properties ofIgGl, IgG2, and IgG4 CH2 Domain Point Mutants. Theexistence of striking primary sequence differences in thelower hinge region of IgG1 and IgG2 CH2 domains (Table 2)and the indirect evidence that this region may be structurallyimportant for FcRI recognition led us to concentrate onresidues 233-238 (Eu numbering) for more detailed analyses.Individual IgG1 residues E233, L234, and L235 of theELLGGP sequence were systematically substituted with thecorresponding IgG2 residues P233, V234, and A235, respec-tively. In addition, IgGl G237 was deleted, since there is nocorresponding residue at position 237 in IgG2. A similarmutagenesis strategy was also used to modify the N-terminalregion of the IgG2 CH2 domain. Residues P233, V234, andL235 of the IgG2 PVAG-P sequence were replaced with thecorresponding IgG1 residues E, L, and L, respectively. Inaddition, a glycine residue was inserted at position 237.Multiple substitutions were also performed such that in all, apanel of 12 IgG1 and IgG2 analogues with mutated CH2domains were generated. The amino acid sequences of thesevariants, and an additional IgG4 F234 to L234 mutant mol-ecule, are shown in Table 2. SDS/PAGE analysis revealedcovalent H2L2 assembly in all cases. The electrophoreticpatterns for three critical mutants are shown in Fig. 2. Therelative binding activities of the mutated molecules obtainedfrom two independent transfections were compared in bind-ing inhibition assays at two different concentrations of chi-meric inhibitor (2.3 and 6.7 nM) and equimolar concentra-
100 1000 .1 1
IgG Inhibitor Concentration (nM)
FIG. 3. Inhibition of 1251-IgGlbinding to U937 cell FcXRI receptorsby myeloma and recombinant wild-type IgG1 and IgG2 and by yl/y2hybrid molecules. The assay was per-formed as described in ref. 22 using2.5 x 106 U937 cells per assay and 10nM 1251-IgGl in the presence of in-creasing concentrations of IgG inhib-itors. (A) The inhibitors are myelomaIgG1 (o), wild-type recombinant IgG1(i), myeloma IgG2 (0), and wild-typerecombinant IgG2 (e). (B) The inhib-itors are wild-type recombinant IgG1(-), hybrid IgGl-1-2-1 (A), and hybridIgG2-2-1-2 (A).
120
(D 100.0-
CMj 800)
Imc
-
C 40~0._'4) 20.
Proc. Natl. Acad. Sci. USA 88 (1991)

Proc. Natl. Acad. Sci. USA 88 (1991) 9039
Table 2. Apparent Ka values of the FcyR1I-gG interactionobtained with IgGi1 and IgG2 point mutants
SequenceIgG (amino acids Ka, SD,
background 231-238) x 10-8 M-1 x 10-8 M-1 n
IgG1WT APELLGGP 4.30 0.49 3IgG2 WT APPVAG-P 3IgGi APPLLGGP 3IgG2 APZVAG-P - 3IgGI APEVLGGP 2.26 0.36 3IgG2 APPLAG-P - - 3IgG1 APELAGGP 3IgG2 APPVLG-P 3IgG1 APELLG-P 3IgG2 APPVAGzP - - 3IgG1 APELAG-P 3IgG2 APPV.LGiP 3IgG2 APPLLGzP 5.17 0.39 4IgG2 APZLLGzP 16.5 5.51 4IgG4 APELLGGP 3.44 1IgG4 WT APEFLGGP 0.04 1
Underlined letters indicate point mutations. All apparent Ka valueswere obtained from binding inhibition experiments and are expressedas the means of independent determinations (n) performed in tripli-cate. WT, wild type.
tions ofthe myeloma 125I-labeled IgG1 (1251-IgGl) tracer (Fig.4A). Single substitutions at positions 233 (P for E) or 235 (Afor L) completely abolished IgG1 cytophiic activity, as didthe deletion of G237. The relatively conservative V for Lsubstitution at position 234 yielded a molecule with reducedactivity. This molecule, when examined more closely in a fullbinding-inhibition assay, displayed a 2-fold decrease in ap-parent Ka (Fig. 4B; Table 2). Although single mutationsintroduced in IgGl between residues 233 and 238 reduced orabolished its functional activity, interestingly, none of thereciprocal mutations conferred any significant binding affin-ity to IgG2 (Fig. 4A; Table 2). Consequently, progressivemutations were sequentially introduced in IgG2 to graduallyreconstruct the IgG1 ELLGGP sequence. Only simultaneoussubstitutions of L at positions 234 and 235, along with theinsertion of G at position 237 (PLLGGP) restored bindingaffinity to a level comparable to that of wild-type IgG1. Arepresentative binding inhibition curve obtained for this triplemutant is shown in Fig. 4B. The Ka was estimated to be 5.17x 108 ± 0.39 M-1. Reconstruction of the IgG1-like ELLGGPsequence on an IgG4 background through the single F234 toL234 substitution yielded a cytophilic molecule with a bind-
ing affinity indistinguishable from that ofIgG1 (Fig. 4A; Table2). Finally, grafting of the entire IgG1 ELLGGP motif on anIgG2 background yielded a mutated molecule that was -4times more active than wild-type IgG1 (Ka = 1.65 x 109 ±0.55 M-1; Fig. 4B; Table 2).
DISCUSSIONHuman IgG subclasses differ with respect to their cytophilicproperties. The objective of this study was to identify theamino acid residues that play a critical role in the IgGl-Fc,,RIinteraction.As a first step, we engineered a panel of IgG1/IgG2
antibodies by reciprocal exon shuffling to identify the ho-mology region of IgG1 responsible for its interaction withFcXRI on U937 cells. We first demonstrated that the bindingaffinities of the chimeric IgG1 and IgG2 molecules wereindistinguishable from those of their human myeloma coun-terparts (8).The genetic hinges of IgG1 (15 residues) and IgG2 (12
residues), which include the upper and core regions of the"functional hinge" differ by three amino acids in length andby their primary sequences. However, reciprocal shuffling ofhinge regions between IgG1 and IgG2 subclasses and thegrafting of an IgG4 hinge onto an IgG1 background did notaffect the FcXRI binding properties of the parent molecules.These findings strongly suggest that neither the length of theupper hinge, which restricts segmental flexibility (24), nor theamino acid sequence of the core hinge modulates cytophilicactivity. In addition, it has been shown that this effectorfunction is not affected by reduction of the hinge disulfidebridges (15). However, it requires the presence of a spacerbetween the Fab arms and the Fc region since the hinge-deleted IgG1 paraprotein Dob does not bind to FcyRI onmonocytes (10).
Reciprocal exchange ofCH3 domains, which differ by onlyone conservative substitution, did not affect the bindingproperties of the native molecules. Our results are in agree-ment with the findings that noncovalent pFc' dimers (10) andmonoclonal anti-CH3 antibodies are unable to inhibit thebinding of IgG1 to the monocyte FcRI (9).
Reciprocal shuffling of CH2 domains between the IgG1 andIgG2 subclasses unambiguously revealed that the FcRIbinding site is an intrinsic property of the IgG1 CH2 domain,since all IgG2 hybrid molecules containing an IgG1 CH2domain are as active as wild-type IgG1. Furthermore, cyto-philic activity was abolished in all IgG1 hybrids containing anIgG2 CH2 domain. These results are consistent with indirectevidence that the CH2 domain is the primary site for FcyRI
url0)
c0
0_
I
._G);a
cr
_j IgG Inhibitor Concentration (nM)CJ> -i > _ > _ > >> >
.W0 0 0WW0 0.0.
5- -5,
FIG. 4. Inhibition of 1251-IgGlbinding to U937 FcyRI receptors byrecombinant native and point-mu-tated IgG1, IgG2, and IgG4 mole-cules. (A) Bar graph representation ofan experiment in which the concen-trations of both 1251-IgGI and recom-binant inhibitor were kept constant at6.67 nM. The inhibitor used in theassay is indicated at the base of its
1ooo corresponding bar. WT, wild type.(B) Binding inhibition curves using 10nM 125I-gG1 and increasing concen-trations of the following inhibitors:wild-type recombinant IgG1 (A), IgG1/EVLGGP (-), IgG2/PLLGGP (o),and IgG2/ELLGGP (A).
Immunology: Chappel et al.

Proc. Natl. Acad. Sci. USA 88 (1991)
recognition. Covalent IgG1 CH2 domain dimers retain 85% ofthe cytophilic activity of the parent molecule (11), whereasthe CH2 domain-deleted paraprotein IgG1 TIM is devoid ofactivity (10). Furthermore, monoclonal anti-CH2 antibodiesinhibit the IgG1-FcXRI interaction (9). Recently, reciprocaldomain and hinge exchange experiments between humanIgG1 and IgE showed that both the IgG1 CH2 and CH3domains were required to restore full FcXRI binding activityto IgE (14). Based on free energy calculations, it was con-cluded that the IgG1 CH2 domain contributes -75% of thefree energy gain associated with the IgGl-FcXRI interaction.
Several lines of evidence suggest that the activity of theFcRI binding site depends on its conformation. Grafting anIgG1 CH2 domain onto an IgE background did not restoreFcRI binding activity to IgE (14) despite its free energycontribution to the FcrRi interaction. The affinity of bispe-cific mouse IgG2a/IgG2b hybrid antibodies suggests thatonly one cytophilic H chain is required for the IgGl-FcRIinteraction (13). However, the findings that only dimericisolated CH2 domains are cytophilic (12) and that the CH3domain-deleted IgGi paraprotein SIZ fails to bind FcXRI (10)infer that quaternary interactions between the domains arerequired to stabilize the active conformation of the IgG1effector site. Furthermore, inactive aglycosylated IgG1 dis-plays significantly enhanced sensitivity to pepsin and chy-motrypsin (25). Since thesle enzymes cleave within the CH2domain, these findings suggest that carbohydrates maintainits conformation and stabilize the FcXRI binding site (8).The most striking primary sequence differences between
IgG1 and IgG2 CH2 domains are clustered in the lower hingeregion (residues 233-238) encoded by the CH2 exon. Aconserved LLGGP motif between positions 234 and 238 isfound in all cytophilic IgG subclasses. The functional impor-tance of the crystallographically defined "upper region" ofthe CH2 domain is supported by the finding that inactiveaglycosylated IgG3 Fc fragments show structural perturba-tions, as assessed by 1H NMR, in the vicinity of the H268reporter group located in the spatial proximity of the lowerhinge (25). In addition, full Fc,,RI binding activity wasrestored to the inactive mouse IgG2b subclass by recon-structing the LLGGP motif with a single L for E substitutionat position 235(16). Moreover, recent reports (26, 27) showedthat the binding activity of human IgG3 was lost in a panel ofsingle point mutants where L234, L235, and G237 werereplaced with A residues. Together, these observations led usto engineer a panel of IgGi and IgG2 chimeras containingsingle and sequential point mutations in this region. Fullbinding activity was restored to an IgG2 molecule in which Gwas inserted at position 237, and V234 and A235 werereplaced with their IgG1 counterparts L234 and L235 (IgG2/PLLGGP), respectively. Interestingly, within the limits ofthe binding-inhibition assay, none of the individual mutationscould restore even partial activity to IgG2, although the singlereciprocal mutations in IgG1 significantly diminished orabolished it completely. Our results confirm the predictionthat the LLGGP motif is essential for FcrRI recognition (15).Indeed, an IgG4 mutant containing the ELLGGP motif wasfound to be as active as wild-type IgG1. Since only a singleH chain is required for the FcRI interaction (13), it is likelythat at least the "docking" portion of the binding site islocated on a loop or a single flexible strand at the N terminusof the CH2 domain. Unfortunately, the protein segmentcontaining the LLGGP sequence yielded no electron densityin the x-ray crystallographic structure of human IgG1 Fc (28).
Interestingly, grafting of the ELLGGP motif onto IgG2resulted in a molecule 4-fold more active than native IgGi.Although both the PLLGGP and the ELLGGP motifs re-stored activity to IgG2, only the ELLGGP sequence con-ferred full binding activity to IgG1. To rationalize theseobservations, it is necessary to propose the existence of
"secondary" IgG2 FcXRI binding sites, which are eithernonexistent in IgG1 or are more active than their IgG1counterparts. These sites probably reside in the IgG2 CH2domain, since none ofthe shuffled IgGi constructs displayedenhanced binding activity. IgG1 and IgG2 CH2 domainsdiverge at five residues in their C-terminal halves. Whetherthese divergent residues contribute to a "secondary" inter-action site between CH2 and FcXRI remains to be investi-gated.
We would like to thank Dr. Leroy Hood for providing the originalyl, y2, and $4 C region gene segments and Cathy Home forengineering the EcoRI site in the CH2-CH3 introns. We are alsograteful to Dr. Aline Rinfret for providing the pSV2neoVH315expression vector and William Bradley for synthesizing the oligo-nucleotides used in this study. We also thank Dr. Alex Marks fordonating the MOPC 315.26 variant cell line. This work was supportedby Grant MT-4259 from the Medical Research Council of Canada.D.E.I. is the recipient of a Medical Research Council Grant (MT-7081) and M.S.C. is the recipient of an Ontario Graduate Scholar-ship.
1. Silverstein, S. C., Steinman, R. M. & Cohn, Z. A. (1977)Annu. Rev. Biochem. 46, 699-722.
2. Shen, L., Guyre, P. M. & Fanger, M. W. (1987) J. Immunol.139, 534-538.
3. Graziano, R. F. & Fanger, M. W. (1987) J. Immunol. 139,3536-3541.
4. Karpovsky, B., Titus, J. A., Stephany, D. A. & Segal, D. M.(1984) J. Exp. Med. 160, 1686-1701.
5. Rouzer, C. A., Scott, W. A., Kempe, J. & Cohn, Z. A. (1980)Proc. Nat!. Acad. Sci. USA 77, 4279-4282.
6. Anderson, C. L. & Looney, R. J. (1986) Immunol. Today 7,381-405.
7. Anderson, C. L. (1982) J. Exp. Med. 156, 1794-1806.8. Burton, D. R. (1985) Mol. Immunol. 22, 161-206.9. Partridge, L. J., Woof, J. M., Jefferis, R. & Burton, D. R.
(1986) Mol. Immunol. 23, 1365-1375.10. Woof, J. M., Nik Jaafar, M. I., Jefferis, R. & Burton, D. R.
(1984) Mol. Immunol. 21, 523-527.11. Ratcliffe, A. & Stanworth, D. R. (1983) Immunology 50, 93-
100.12. Barnett Foster, D. E., Dorrington, K. & Painter, R. (1980) J.
Immunol. 124, 2186-2190.13. Koolwijk, P., Spierenburg, G. T., Frasa, H., Boot, J. H., van
de Winkel, J. G. J. & Bast, B. J. (1989) J. Immunol. 143,1656-1662.
14. Shopes, B., Weetal, M., Holowaka, D. & Baird, B. (1990) J.Immunol. 14S, 3842-3848.
15. Woof, J. M., Partridge, L. J., Nik Jaafar, M. I., Jefferis, R. &Burton, D. R. (1986) Mol. Immunol. 23, 319-330.
16. Duncan, A. R., Woof, J. M., Partridge, L. J., Burton, D. R. &Winter, G. (1988) Nature (London) 332, 563-566.
17. Mosmann, T. R., Baumal, R. & Williamson, A. R. (1979) Eur.J. Immunol. 9, 511-517.
18. Rinfret, A., Home, C., Dorrington, K. & Klein, M. (1989) Mol.Immunol. 26, 431-434.
19. Maniatis, T., Fritsch, E. F. & Sambrook, J. (1982) MolecularCloning: A Laboratory Manual, (Cold Spring Harbor Lab.,Cold Spring Harbor, NY).
20. Taylor, J. W., Ott, J. & Eckstein, F. (1985) Nucleic Acids Res.13, 8764-8785.
21. Baker, M. D., Pennell, N., Bosnoyan, L. & Shulman, M. (1988)Proc. Nat!. Acad. Sci. USA 85, 6432-6436.
22. Raychaudhuri, G., McCool, D. & Painter, R. (1985) Mol.Immunol. 22, 1009-1019.
23. Berzofsky, J. & Schechter, A. N. (1981) Mol. Immunol. 18,751-763.
24. Tan, L. K., Shopes, B. J., Oi, V. T. & Morrison, S. L. (1990)Proc. Nat!. Acad. Sci. USA 87, 162-166.
25. Lund, J., Tanaka, T., Takahashi, N., Sarmay, G., Arata, Y. &Jefferis, R. (1990) Mol. Immunol. 27, 1145-1153.
26. Woof, J. M. (1990) Biochem. Soc. Trans. 18, 217-218.27. Jefferis, R., Lund, J. & Pound, J. (1990) Mol. Immunol. 27,
1237-1240.28. Deisenhofer, J. (1981) Biochemistry 20, 2361-2370.
9040 Immunology: Chappel et al.





























