Embed Size (px)
Citation preview

JOURNAL OF VIROLOGY, Dec. 2010, p. 12599–12608 Vol. 84, No. 240022-538X/10/$12.00 doi:10.1128/JVI.01437-10Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Identification of Specific Determinants of Human APOBEC3F,APOBEC3C, and APOBEC3DE and African Green Monkey
APOBEC3F That Interact with HIV-1 Vif�
Jessica L. Smith and Vinay K. Pathak*Viral Mutation Section, HIV Drug Resistance Program, Center for Cancer Research, National Cancer Institute at Frederick,
Frederick, Maryland 21702
Received 9 July 2010/Accepted 4 October 2010
Human APOBEC3F (hA3F) and human APOBEC3G (hA3G) are potent anti-human immunodeficiency virus(anti-HIV) host factors that suppress viral replication by hypermutating the viral genome, inhibiting reversetranscription, and hindering integration. To overcome hA3F and hA3G, HIV-1 encodes Vif, which binds andtargets these host proteins for proteasomal degradation. Previously, we reported that the hA3F-Vif interactionsthat lead to hA3F degradation are located in the region comprising amino acids 283 to 300. We have nowperformed mutational analysis of this region and found that the 289EFLARH294 amino acids contribute tohA3F-Vif binding and are critical for A3F’s sensitivity to Vif. Mutants in which E289 is mutated significantlyincrease hA3F’s ability to inhibit viral infectivity in the presence of Vif, and coimmunoprecipitation assaysshow that binding of Vif to the E289K mutant is decreased. We examined the role of the EFLARH sequence inother A3 proteins, including human A3C (hA3C), human A3DE (hA3DE), African green monkey A3F(agmA3F), and rhesus macaque A3F (rhA3F). hA3C, hA3DE, and agmA3F were all susceptible to degradationinduced by HIV-1 Vif, while rhA3F was not. Mutagenesis of the glutamate in the EFLARH sites of hA3C,hA3DE, and agmA3F decreases the susceptibilities of these proteins to Vif-induced degradation. Together,these results indicate that the EFLARH region in hA3F, hA3C, hA3DE, and agmA3F interacts with HIV-1 Vifand that this interaction plays a role in the Vif-mediated proteasomal degradation of these A3 proteins. Thesestudies identify a conserved region in 3 of 7 human A3 proteins that is critical for degradation mediated byHIV-1 Vif and provide structural insights into the hA3F-Vif interactions that could facilitate the developmentof a novel class of anti-HIV agents.
Human cytidine deaminases APOBEC3F (hA3F) andAPOBEC3G (hA3G) are potent cellular defense proteins thatcounter Vif-deficient human immunodeficiency virus type 1(HIV-1) (27, 46, 56, 71). hA3F and hA3G inhibit HIV-1 bycytidine deamination resulting in G-to-A hypermutation of theviral genome (18, 24, 31, 51, 63, 67), inhibition of viral DNAsynthesis (1, 3, 14, 15, 20, 22, 26, 29, 34, 35, 40, 60), andinhibition of viral DNA integration and provirus formation(29, 34, 35).
To counter these cellular antiviral factors, the HIV-1 Vifprotein acts as a scaffold between a cellular E3 ubiquitinligase complex (consisting of cullin 5, elongin B, elongin C,and RING finger protein 1) and hA3F/hA3G (64), leadingto their polyubiquitination and degradation (7, 28, 33, 37,47, 50, 64). Because hA3F and hA3G are potent antiviralproteins, identification of the regions in Vif and hA3F/hA3G that are important for interactions between theseproteins could be used to design inhibitors to block hA3F/hA3G degradation.
Several domains in Vif have been identified as being importantfor its mechanism of inducing hA3G/hA3F ubiquitination anddegradation, including an 144SLQYLA149 domain which interacts
with elongin C and an 108H-X5-C-X17-18-C-X3-5-H139 domainwhich interacts with cullin 5 (2, 30, 36, 38, 65). We previouslyidentified distinct regions in Vif that are required for interactionswith hA3F (14DRMR17) or hA3G (40YRHHY44) (43). Otherstudies (recently reviewed in reference 49) also demonstrated thatcertain residues in Vif are required for hA3F or hA3G interac-tions, but not both, whereas other regions seem to be importantfor interactions with both hA3F and hA3G (6, 8, 10, 11, 19, 39, 42,43, 48, 52, 58).
Within the APOBEC3 proteins, two distinct regions inhA3G and hA3F that interact with Vif have been identified(44). In hA3G, we found that amino acids 126 to 132 areimportant for Vif binding. These results are consistent withother studies in which amino acids 128 to 130 were identifiedas being critical for Vif interactions (21). In hA3F, we showedthat amino acids 283 to 300 are important for binding to Vif aswell as Vif-induced degradation (44). Here, we have extendedour analysis of the hA3F protein to show that this region canbe further narrowed down to include only 289EFLARH294.While it has previously been shown that HIV-1 Vif-induceddegradation of A3G can be countered by a single amino acidchange in hA3G(D128K) (4, 32, 45, 57), such a single aminoacid change has not been identified for hA3F. Here, we showthat a single mutation in hA3F(E289K) can decrease bindingof hA3F to Vif and prevent its Vif-induced degradation. Fur-thermore, we found that the analogous residues in two otherhuman A3 proteins and African green monkey A3F (agmA3F)are also important for their Vif-induced degradation.
* Corresponding author. Mailing address: HIV Drug ResistanceProgram, National Cancer Institute—Frederick, P.O. Box B, Bldg.535, Rm. 334, Frederick, MD 21702-1201. Phone: (301) 846-1710. Fax:(301) 846-6013. E-mail: [email protected].
� Published ahead of print on 13 October 2010.
12599
on August 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

MATERIALS AND METHODS
Plasmid construction and cell culture. The pFLAG-A3F (43) plasmid and thepcDNA-HVif (41) DRMR�A4 and YRHHY�A5 mutants were described pre-viously (43); in the F binder Vif mutant, four alanine substitutions replaced14DRMR17, and in the G binder Vif mutant, five alanine substitutions replaced40YRHHY44. All APOBEC3 mutants were generated by site-directed mutagen-esis using a QuikChange II site-directed mutagenesis kit (Stratagene). Plasmidsexpressing N-terminally myc-tagged agmA3F and rhesus macaque A3F (rhA3F)were kindly provided by T. Hatziioannou (Aaron Diamond AIDS ResearchCenter). To generate plasmids that express human A3C (hA3C) and humanA3DE (hA3DE) proteins fused to the FLAG epitope, clones pFLAG-A3C andpFLAG-A3DE were constructed using pcDNA3.1-APOBEC3C-V5-6xHis andpcDNA3.1-APOBEC3DE-V5-6xHis, respectively, which were obtained throughthe NIH AIDS Research and Reference Reagent Program, Division of AIDS,NIAID, NIH, from B. Matija Peterlin and Yong-Hui Zheng (9, 71). The primersused for hA3C amplification were 5�-GATCGCGGCCGCTATGAATCCACAGATCAGAAACC-3� (FWD-FLAG-A3C) and 5�-GATCTCTAGATCACTGGAGACTCTCCCGTAGCC-3� (REV-FLAG-A3C). For amplification ofhA3DE, 5�-GATCGCGGCCGCTATGAATCCACAGATCAGAAATCC-3�(FWD-FLAG-A3DE) and 5�-GATCTCTAGATCACTGGAGAATCTCCCGTAG-3� (REV-FLAG-A3DE) were used.
The modified human embryonic kidney 293T cell line (59) and the HeLa-derived HIV-1 reporter cell line TZM-bl (12, 55), which has an HIV-1 Tat-drivenfirefly luciferase gene, were maintained in complete medium (CM) consisting ofDulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetalcalf serum, 1% penicillin-streptomycin, and 1% glutamine.
Virus production and titration. For virus production, 293T cells were seededat 8 � 105 cells per well in six-well plates 1 day prior to transfection with thefollowing plasmids: 3.33 �g of the HIV-1 vector genome pHDV-EGFP (53), 0.67�g of vesicular stomatitis virus glycoprotein expression plasmid pHCMV-G (61),0.34 �g of wild-type (WT) or mutant pFLAG-A3F, and 4.5 �g of either WT ormutant pcDNA-HVif. To maintain equal amounts of DNA, pcDNA3.1noMCS(43) was used when needed. To test the antiviral function of hA3C(E106K),hA3DE(E302K), and agmA3F(E289K), viruses were produced as describedabove, except that 1.7 �g of A3 plasmid was used. All transfections were carriedout with polyethylenimine (PEI) (Sigma) by modification of a previously de-scribed procedure (5, 44). Briefly, 500 �l of DMEM was added to the DNAsamples, and 500 �l of a PEI (25 kDa)-DMEM solution (500 nM final concen-tration of PEI) was then added to the DNA-DMEM samples dropwise withgentle vortexing. Following a 20-min incubation of the DNA-PEI-DMEM sam-ples at room temperature, the samples were added to the cells after the CM wasremoved. The cells were incubated at 37°C for 3 to 4 h, and the DNA-PEI-DMEM samples were aspirated and replaced with CM (1 ml). After 48 h, thevirus-containing supernatant was filtered through a 0.45-�m filter and quantifiedusing a p24 CA enzyme-linked immunosorbent assay (Perkin-Elmer). TZM-blcells were seeded at 4 � 103 cells per well in 96-well plates 1 day prior toinfection. Virus samples were diluted with CM to have equivalent concentrationsof p24 CA (50 ng/ml), and the samples (100 �l) were used to infect the TZM-blcells. After incubation at 37°C for 3 to 4 h, another 100 �l of CM was added toeach well. The culture medium was removed 72 h later and replaced with 100 �lof DMEM without phenol red and 100 �l of Britelite luciferase solution (Perkin-Elmer). Luciferase enzyme activity was measured using a LUMIstar Galaxyluminometer.
Co-IP assays. Coimmunoprecipitation (co-IP) assays were done as previouslydescribed (43, 44). 293T cells were seeded at 4 � 106 cells per 100-mm-diameterdish, and 24 h later, the cells transfected with 6 �g of WT or mutant pFLAG-A3F, 9 �g of either WT or mutant pcDNA-HVif, and 1.2 �g pGreen Lantern-1(pGL), which expresses green fluorescent protein from a cytomegalovirus im-mediate-early promoter (GibcoBRL) as a positive control for transfections. Tomaintain equal amounts of DNA, pcDNA3.1noMCS was used when needed. PEIwas used for transfections as described above, except that 2.5 ml of PEI-DMEMwas added to 2.5 ml of DNA-DMEM. After 48 h, the cells were harvested andsamples for co-IP were prepared as previously described (44). Western blottingwas used to analyze the eluted complexes as well as the input cell lysates. TheAPOBEC3 proteins were detected using a rabbit anti-FLAG polyclonal antibody(Sigma) at a 1:5,000 dilution, followed by a horseradish peroxidase-labeled goatanti-rabbit secondary antibody (Sigma) at a 1:10,000 dilution; the Vif proteinswere detected using a rabbit anti-Vif polyclonal antibody (NIH AIDS Researchand Reference Reagent Program, Division of AIDS, NIAID, NIH; HIV-1HXB2
Vif antiserum from Dana Gabuzda) (13) at a 1:5,000 dilution and the samesecondary antibody at a 1:10,000 dilution. For input cell lysates, �-tubulin wasalso detected using mouse anti-�-tubulin antibody (Sigma) at a 1:5,000 dilution,
followed by a horseradish peroxidase-labeled goat anti-mouse secondary anti-body at a 1:10,000 dilution. Protein bands were visualized using a WesternLightning Chemiluminescence Reagent Plus kit (Perkin-Elmer).
APOBEC3 degradation. Western blot assays were performed to determineWT or mutant A3F protein levels in the presence of excess Vif. 293T cells wereseeded at 4 � 106 cells per 100-mm-diameter dish, and 24 h later, the cells weretransfected with 0.8 �g of WT or mutant pFLAG-A3F, 8 �g of either WT ormutant pcDNA-HVif, and 1.2 �g pGL. To maintain equal amounts of DNA,pcDNA3.1noMCS was used when needed. For analysis of A3F from otherprimates, the transfections were performed similarly, except that the expressionplasmids for myc-tagged agmA3F and rhA3F (66) were used. For the analysis ofhA3C and hA3DE, the pFLAG-A3C and pFLAG-A3DE expression plasmidswere used. For hA3C, cells were transfected with 0.8 �g of WT or mutantpFLAG-A3C, 8 �g of either WT or mutant pcDNA-HVif, and 1.2 �g pGL. ForhA3DE, cells were transfected with 6 �g of WT or mutant pFLAG-A3DE, 6 �gof either WT or mutant pcDNA-HVif, and 1.2 �g pGL. Transfected cell lysateswere harvested as previously described (44), and FLAG-APOBEC3, Vif, and�-tubulin were analyzed by Western blotting. For the detection of myc-taggedproteins, rabbit anti-c-Myc monoclonal antibody (Sigma) at a 1:5,000 dilutionwas used, followed by a horseradish peroxidase-labeled goat anti-rabbit second-ary antibody (Sigma) at a 1:10,000 dilution.
RESULTS
Identification of specific residues in hA3F involved in inhi-bition by HIV-1 Vif. Previously, amino acids 283 to 300 in theC-terminal half of A3F were shown to be important for itsinteraction with Vif, while amino acids within the region ofpositions 126 to 132 in the N-terminal half of hA3G are im-portant for its interactions with Vif (21, 44). To determine ifany of the residues in hA3F’s region comprising positions 283to 300 were particularly important for Vif interactions, a seriesof mutants were made in pFLAG-A3F by changing hA3F’sresidues to the corresponding residues in hA3G (Fig. 1A). Thefunctionality of these mutants was tested by examining theirability to inhibit the infectivity of vif-deleted HIV-1 in single-cycle assays (Fig. 1B). In addition, the susceptibility of thehA3F mutants to neutralization by WT Vif and an hA3F-specific mutant, called the F binder (43), was tested (Fig. 1B).The F binder Vif mutant has five alanine substitutions in aregion of Vif (40YRHHY44) that is critical for interactions withhA3G but not hA3F (43). Figure 1B shows that all mutantstested had antiviral activity in the absence of Vif, although theFGF5 mutant had about 4-fold-reduced inhibitory activity andabout 2-5-fold-reduced viral infectivity compared to the levelsfor the infection without A3F. The amino acid changes inFGF1(G285Q-V287M) did not affect this mutant’s ability toinhibit HIV-1 in the absence of Vif, nor did the mutationsaffect its susceptibility to WT Vif or the F binder. When twoadditional mutations were added to FGF1 to make FGF2, theability to rescue infectivity in the presence of this mutant wascompletely abolished for WT Vif and nearly completely abol-ished for the F binder. Three additional changes to the FGF2construct yielded the FGF3 construct. The ability of both WTVif and the F binder to rescue infectivity in the presence of thismutant was completely diminished. The results from theseexperiments indicate that residues 289 to 294 of hA3F areimportant for neutralization by Vif. Constructs FGF4 andFGF5 had all of the mutations of the previous constructs aswell as some additional mutations; rescue of infectivity by WTVif or the F binder was not observed in the presence of thesemutants.
To examine the Vif-induced degradation of the FGF con-structs, the steady-state levels of these proteins in the presence
12600 SMITH AND PATHAK J. VIROL.
on August 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

of excess Vif were examined by Western blotting (Fig. 1C).These results showed that WT hA3F and the FGF1 constructwere degraded by Vif but that the other FGF constructswere not. These results are consistent with the infectivitydata in Fig. 1B.
To determine whether the mutant constructs retained theirability to bind to Vif, co-IP experiments were performed. Forthese experiments, WT Vif, the F binder, and an hA3G-specificVif mutant called the G binder were used. The G binder Vifmutant has four alanine substitutions in a region of Vif(14DRMR17) which is critical for interactions with hA3F butnot hA3G (43). The FLAG-tagged WT and hA3F mutantswere immunoprecipitated, and Vif’s ability to coimmunopre-cipitate with these proteins was tested (Fig. 1D). As observedpreviously, WT Vif and the F binder, but not the G binder,were able to coimmunoprecipitate with WT hA3F (43, 44).Binding of the different Vif proteins to FGF1 was unaltered,while binding to FGF2 was reduced, and binding to FGF3,FGF4, and FGF5 was greatly reduced. The mutations in FGF2significantly reduced Vif sensitivity in addition to reducing Vif
binding, suggesting that the residues mutated in FGF2 contrib-ute to both Vif binding and Vif sensitivity.
Amino acid E289 in the EFLARH sequence of hA3F is crit-ical for HIV-1 Vif sensitivity. The results from the FGF mutants(Fig. 1) indicate that when mutations were introduced in theregion comprising amino acids 289 to 294, Vif’s ability to rescueinfectivity in the presence of hA3F and Vif-hA3F binding wasdecreased. To further investigate the roles of these amino acids inthe interactions with Vif, one or only a few residues within thisregion were mutated and tested. In the absence of Vif, each of themutants inhibited HIV-1 infectivity (Fig. 2A). Interestingly, for asingle hA3F(E289K) mutant or a double hA3F(E289K-L291I)mutant (called EL�KI), the abilities of WT Vif and the F binderto neutralize this mutant were completely abolished; however, asingle hA3F(L291I) mutant (Fig. 2A) or a hA3F(L291A) mutant(data not shown) were completely neutralized by WT Vif and theF binder. While the single mutants hA3F(A292S), hA3F(R293K),and hA3F(H294N) were neutralized by WT Vif and the Fbinder, the triple mutant hA3F(A292S-R293K-H294N) (calledARH�SKN) in this region could not be neutralized.
FIG. 1. Localization of the Vif interaction domain in hA3F. (A) Schematic representation of hA3F mutants used to determine the residues inhA3F which are important for interactions with HIV-1 Vif. The top line depicts amino acids 283 to 300 of WT hA3F, and the second line showsthe sequence within the same region in hA3G. The third line through the seventh line show the amino acid substitutions in hA3F for five FGFchimeras. Dots indicate identical amino acids. (B) Effect of hA3F and the FGF chimeras on HIV-1 infectivity in the presence and absence of HIV-1Vif. Single-cycle viruses were produced from 293T cells that were transfected with pHDV-eGFP, pHCMV-G, an hA3F expression plasmid, andeither no Vif, WT Vif, or the F binder. The p24 CA levels were determined and used to normalize the levels for virus samples prior to infectionof TZM-bl indicator cells. The infectivity of the viruses was determined by quantitation of luciferase activity. Data are plotted as relative infectivitylevels, with the level for the hA3F-free virus (not shown) set to 100%. Error bars indicate standard errors of the means (SEM) of results from sixinfectivity experiments performed with three independent virus stocks. (C) Effects of WT HIV-1 Vif on the degradation of WT hA3F or the FGFchimeras. Cotransfections of 293T cells were carried out with an hA3F expression plasmid and either no Vif or WT Vif expression plasmid. Celllysates were analyzed by Western blotting using anti-FLAG, anti-Vif, or anti-�-tubulin antibodies. The results are representative of twoindependent experiments. (D) Co-IP assays to determine binding of Vif to FGF chimeras. 293T cells were cotransfected with expression plasmidsfor FLAG-A3F or FLAG-FGF chimeras along with expression plasmids for WT Vif, the F binder, or the G binder. Cell lysates and immuno-precipitated proteins were analyzed by Western blotting using anti-FLAG and anti-Vif antibodies. The cell lysates were also analyzed for �-tubulinas a loading control. The results are representative of three independent experiments. WT, wild type; �, with; �, without.
VOL. 84, 2010 A3F, A3C, AND A3DE INTERACTIONS WITH HIV-1 Vif 12601
on August 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
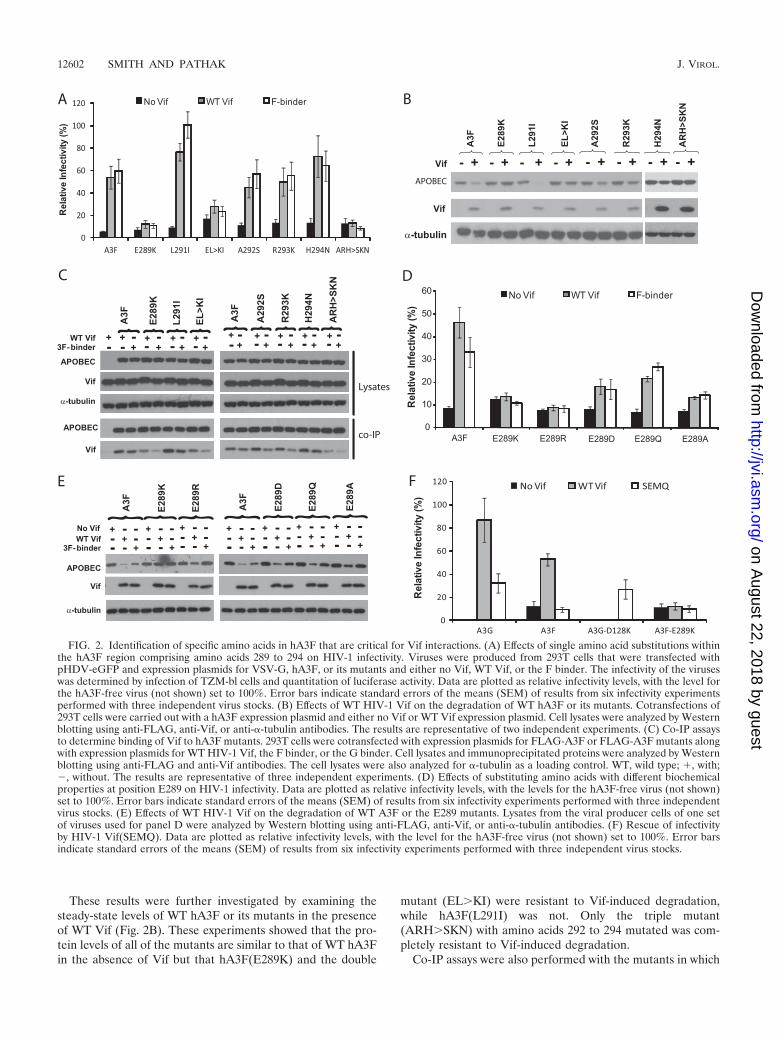
These results were further investigated by examining thesteady-state levels of WT hA3F or its mutants in the presenceof WT Vif (Fig. 2B). These experiments showed that the pro-tein levels of all of the mutants are similar to that of WT hA3Fin the absence of Vif but that hA3F(E289K) and the double
mutant (EL�KI) were resistant to Vif-induced degradation,while hA3F(L291I) was not. Only the triple mutant(ARH�SKN) with amino acids 292 to 294 mutated was com-pletely resistant to Vif-induced degradation.
Co-IP assays were also performed with the mutants in which
FIG. 2. Identification of specific amino acids in hA3F that are critical for Vif interactions. (A) Effects of single amino acid substitutions withinthe hA3F region comprising amino acids 289 to 294 on HIV-1 infectivity. Viruses were produced from 293T cells that were transfected withpHDV-eGFP and expression plasmids for VSV-G, hA3F, or its mutants and either no Vif, WT Vif, or the F binder. The infectivity of the viruseswas determined by infection of TZM-bl cells and quantitation of luciferase activity. Data are plotted as relative infectivity levels, with the level forthe hA3F-free virus (not shown) set to 100%. Error bars indicate standard errors of the means (SEM) of results from six infectivity experimentsperformed with three independent virus stocks. (B) Effects of WT HIV-1 Vif on the degradation of WT hA3F or its mutants. Cotransfections of293T cells were carried out with a hA3F expression plasmid and either no Vif or WT Vif expression plasmid. Cell lysates were analyzed by Westernblotting using anti-FLAG, anti-Vif, or anti-�-tubulin antibodies. The results are representative of two independent experiments. (C) Co-IP assaysto determine binding of Vif to hA3F mutants. 293T cells were cotransfected with expression plasmids for FLAG-A3F or FLAG-A3F mutants alongwith expression plasmids for WT HIV-1 Vif, the F binder, or the G binder. Cell lysates and immunoprecipitated proteins were analyzed by Westernblotting using anti-FLAG and anti-Vif antibodies. The cell lysates were also analyzed for �-tubulin as a loading control. WT, wild type; �, with;�, without. The results are representative of three independent experiments. (D) Effects of substituting amino acids with different biochemicalproperties at position E289 on HIV-1 infectivity. Data are plotted as relative infectivity levels, with the levels for the hA3F-free virus (not shown)set to 100%. Error bars indicate standard errors of the means (SEM) of results from six infectivity experiments performed with three independentvirus stocks. (E) Effects of WT HIV-1 Vif on the degradation of WT A3F or the E289 mutants. Lysates from the viral producer cells of one setof viruses used for panel D were analyzed by Western blotting using anti-FLAG, anti-Vif, or anti-�-tubulin antibodies. (F) Rescue of infectivityby HIV-1 Vif(SEMQ). Data are plotted as relative infectivity levels, with the level for the hA3F-free virus (not shown) set to 100%. Error barsindicate standard errors of the means (SEM) of results from six infectivity experiments performed with three independent virus stocks.
12602 SMITH AND PATHAK J. VIROL.
on August 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

amino acids 289 to 294 were mutated to examine their effectson Vif binding (Fig. 2C). The hA3F(E289K) mutant coimmu-noprecipitated smaller amounts of WT Vif and even less Fbinder Vif, indicating that this mutation decreased binding toVif. The hA3F(ARH�SKN) mutant also coimmunoprecipi-tated smaller amounts of WT Vif or F binder Vif. All otherhA3F single mutants with amino acids 292 to 294 mutated didnot have an impaired ability to bind WT Vif or the F binder.
Residue F290 is conserved between hA3F and hA3G and itis located next to the critical E289 residue in A3F. Therefore,we tested the effects of mutating this amino acid to tyrosine ortryptophan but saw no effect on the antiviral activity of thesemutations or their interactions with Vif (data not shown).
Because a single change in the E289 residue of A3F cancritically affect interactions with Vif, further analyses werefocused on this residue. To examine the effects of differentamino acids at the E289 position, various mutations were madeat this site and the mutants were tested in the single-cycleinfectivity assay for their ability to be neutralized by WT Vif orthe F binder. The results in Fig. 2D showed that, like lysine, thepositively charged arginine residue at the E289 position isresistant to Vif neutralization, since the hA3F(E289R) mutantretained its ability to inhibit viral infectivity in the presence ofWT Vif or the F binder. Although the hA3F(E289D) mutantretains the negative charge at this position, it has a shorter sidechain; neutralization of this mutant by Vif and the F binder wasincomplete. The hA3F(E289Q) mutant does not retain thenegative charge, but its side chain is similar in length to that ofglutamate; despite retaining a similar side chain length, thismutant was only partially neutralized by WT Vif and the Fbinder. Finally, the hA3F(E289A) mutant does not retain thecharge or the length of the side chain; this mutant was insen-sitive to Vif and was only partially neutralized by either WT Vifor the F binder. Together, these results suggest that both anegative charge and the length of the side chain at amino acid289 in hA3F are important for Vif’s ability to rescue viralinfectivity in the presence of hA3F. The lysates of the viralproducer cells were also examined by Western blot analysis(Fig. 2E). In the presence of WT Vif and F binder Vif, thelevels of WT A3F were substantially reduced, but the levels ofthe E289K mutant were not noticeably altered, confirming thatthis mutant was resistant to Vif-mediated degradation. TheE289R, E289D, E289Q, and E289A mutants were partiallysensitive to degradation induced by the Vif proteins.
To determine if a Vif mutant that has an overall negativecharge in the 14DRMR17 region could compensate for theswitch in charge and neutralize the hA3F(E289K) mutant,infectivity assays were performed with the Vif(SEMQ) mutant(43, 45), which has 14DRMR17 mutated to 14SEMQ17. Thismutant is able to rescue infectivity in the presence of WThA3G and a Vif-resistant hA3G(D128K) mutant but not in thepresence of WT hA3F (43, 45). The results in Fig. 2F show thatthe HIV-1 Vif(SEMQ) mutant partially rescues infectivity inthe presence of WT hA3G and hA3G(D128K) as expected butthat this mutant cannot rescue the infectivity in the presence ofWT hA3F or hA3F(E289K).
The Vif interaction sites of hA3F and hA3G are not func-tionally transferable. Two distinct Vif interaction domainshave been identified in hA3F and hA3G (44). To determinewhether these two domains could be functionally transferred
between these two proteins, mutants of hA3F and hA3G inwhich the hA3G-Vif binding sequence was added to A3F(called A3F-G125-131-F) and the hA3F-Vif binding sequencewas added to A3G (called A3G-F297-302-G) were made (Fig.3A). The abilities of the G binder Vif mutant to degradeA3F-G125-131-F and the F binder Vif mutant to degrade A3G-F297-302-G were tested. In each case, the mutant proteins werestably expressed in the absence of Vif, but both were suscep-tible to degradation induced by WT Vif (Fig. 3B). Like WThA3F, the A3F-G125-131-F protein was susceptible to the Fbinder but not the G binder. Furthermore, like WT hA3G, theA3G-F297-302-G protein was susceptible to the G binder butnot the F binder. These results indicated that the Vif interac-tion sites in hA3F and hA3G could not be easily transferred tothe other A3 protein.
Glutamates in the EFLARH sequences of hA3C and hA3DEare critical for HIV-1 Vif-mediated degradation. Sequencealignment results revealed that the EFLARH sequence that isimportant for hA3F-Vif interactions is present in other A3proteins, including hA3C and hA3DE (Fig. 4A). To determineif the glutamates in these sequences are important for HIV-1Vif-induced degradation, site-directed mutagenesis was per-formed on hA3C and hA3DE. The antiviral activity of theseproteins against HIV-1 is not as potent as that of hA3G andhA3F (9, 62, 71), but hA3C and hA3DE are susceptible toHIV-1 Vif-induced degradation (6, 9, 42, 62, 68, 69). There-fore, to assess the importance of the glutamate residue of
FIG. 3. Sensitivity of hA3F-G125-131-F to G binder Vif and hA3G-F297-302-F to F binder Vif. (A) Schematic representation of hA3F andhA3G with the amino acid substitutions used to generate the hA3F-G125-131-F and hA3G-F297-302-F constructs. The relative locations ofthe catalytic domains (CD) are indicated, and dots indicate identicalamino acids. (B) Effects of WT HIV-1 Vif, the 3F binder, and the 3Gbinder on the degradation of hA3F-G125-131-F and hA3G-F297-302-F.293T cells were cotransfected with hA3F, hA3F-G125-131-F, A3G, orhA3G-F297-302-G expression plasmids and either no Vif, WT Vif, Fbinder, or G binder expression plasmids. Cell lysates were analyzed byWestern blotting using anti-FLAG, anti-Vif, or anti-�-tubulin antibod-ies. The black triangle indicates removal of uninformative lanes froma single gel. The results are representative of two independent exper-iments.
VOL. 84, 2010 A3F, A3C, AND A3DE INTERACTIONS WITH HIV-1 Vif 12603
on August 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
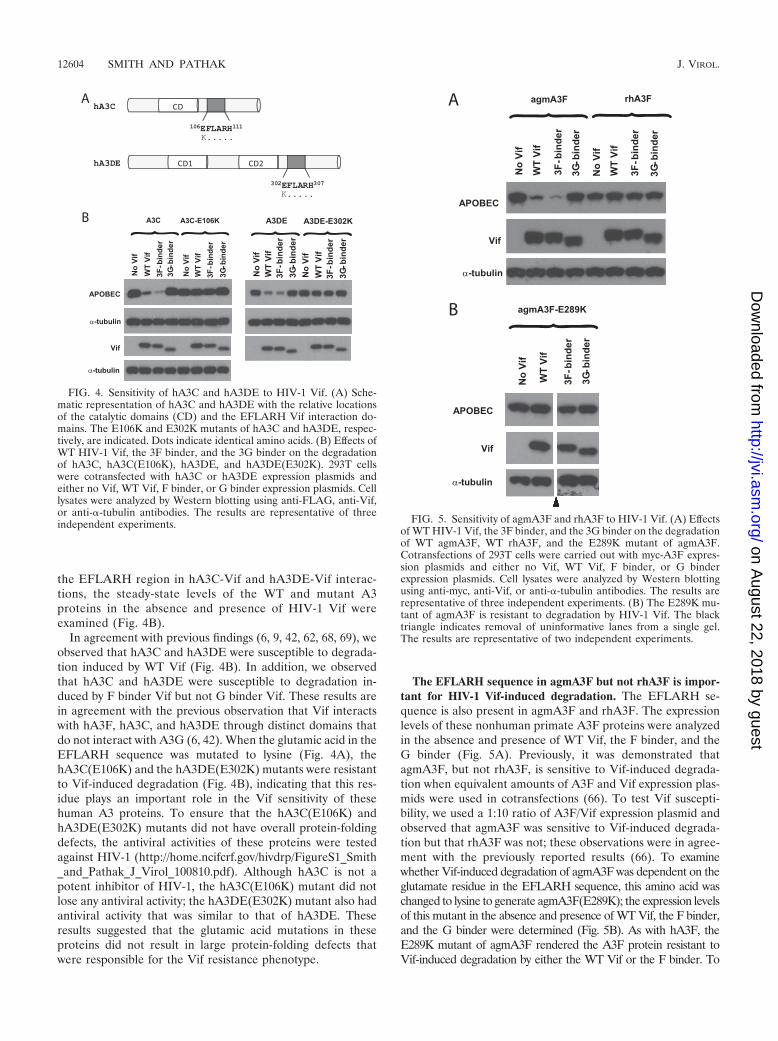
the EFLARH region in hA3C-Vif and hA3DE-Vif interac-tions, the steady-state levels of the WT and mutant A3proteins in the absence and presence of HIV-1 Vif wereexamined (Fig. 4B).
In agreement with previous findings (6, 9, 42, 62, 68, 69), weobserved that hA3C and hA3DE were susceptible to degrada-tion induced by WT Vif (Fig. 4B). In addition, we observedthat hA3C and hA3DE were susceptible to degradation in-duced by F binder Vif but not G binder Vif. These results arein agreement with the previous observation that Vif interactswith hA3F, hA3C, and hA3DE through distinct domains thatdo not interact with A3G (6, 42). When the glutamic acid in theEFLARH sequence was mutated to lysine (Fig. 4A), thehA3C(E106K) and the hA3DE(E302K) mutants were resistantto Vif-induced degradation (Fig. 4B), indicating that this res-idue plays an important role in the Vif sensitivity of thesehuman A3 proteins. To ensure that the hA3C(E106K) andhA3DE(E302K) mutants did not have overall protein-foldingdefects, the antiviral activities of these proteins were testedagainst HIV-1 (http://home.ncifcrf.gov/hivdrp/FigureS1_Smith_and_Pathak_J_Virol_100810.pdf). Although hA3C is not apotent inhibitor of HIV-1, the hA3C(E106K) mutant did notlose any antiviral activity; the hA3DE(E302K) mutant also hadantiviral activity that was similar to that of hA3DE. Theseresults suggested that the glutamic acid mutations in theseproteins did not result in large protein-folding defects thatwere responsible for the Vif resistance phenotype.
The EFLARH sequence in agmA3F but not rhA3F is impor-tant for HIV-1 Vif-induced degradation. The EFLARH se-quence is also present in agmA3F and rhA3F. The expressionlevels of these nonhuman primate A3F proteins were analyzedin the absence and presence of WT Vif, the F binder, and theG binder (Fig. 5A). Previously, it was demonstrated thatagmA3F, but not rhA3F, is sensitive to Vif-induced degrada-tion when equivalent amounts of A3F and Vif expression plas-mids were used in cotransfections (66). To test Vif suscepti-bility, we used a 1:10 ratio of A3F/Vif expression plasmid andobserved that agmA3F was sensitive to Vif-induced degrada-tion but that rhA3F was not; these observations were in agree-ment with the previously reported results (66). To examinewhether Vif-induced degradation of agmA3F was dependent on theglutamate residue in the EFLARH sequence, this amino acid waschanged to lysine to generate agmA3F(E289K); the expression levelsof this mutant in the absence and presence of WT Vif, the F binder,and the G binder were determined (Fig. 5B). As with hA3F, theE289K mutant of agmA3F rendered the A3F protein resistant toVif-induced degradation by either the WT Vif or the F binder. To
FIG. 4. Sensitivity of hA3C and hA3DE to HIV-1 Vif. (A) Sche-matic representation of hA3C and hA3DE with the relative locationsof the catalytic domains (CD) and the EFLARH Vif interaction do-mains. The E106K and E302K mutants of hA3C and hA3DE, respec-tively, are indicated. Dots indicate identical amino acids. (B) Effects ofWT HIV-1 Vif, the 3F binder, and the 3G binder on the degradationof hA3C, hA3C(E106K), hA3DE, and hA3DE(E302K). 293T cellswere cotransfected with hA3C or hA3DE expression plasmids andeither no Vif, WT Vif, F binder, or G binder expression plasmids. Celllysates were analyzed by Western blotting using anti-FLAG, anti-Vif,or anti-�-tubulin antibodies. The results are representative of threeindependent experiments. FIG. 5. Sensitivity of agmA3F and rhA3F to HIV-1 Vif. (A) Effects
of WT HIV-1 Vif, the 3F binder, and the 3G binder on the degradationof WT agmA3F, WT rhA3F, and the E289K mutant of agmA3F.Cotransfections of 293T cells were carried out with myc-A3F expres-sion plasmids and either no Vif, WT Vif, F binder, or G binderexpression plasmids. Cell lysates were analyzed by Western blottingusing anti-myc, anti-Vif, or anti-�-tubulin antibodies. The results arerepresentative of three independent experiments. (B) The E289K mu-tant of agmA3F is resistant to degradation by HIV-1 Vif. The blacktriangle indicates removal of uninformative lanes from a single gel.The results are representative of two independent experiments.
12604 SMITH AND PATHAK J. VIROL.
on August 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

ensure that the agmA3F(E289K) mutant retained functionality, itsantiviral activity was tested against HIV-1 (http://home.ncifcrf.gov/hivdrp/FigureS1_Smith_and_Pathak_J_Virol_100810.pdf). TheagmA3F and agmA3F(E289K) proteins had similar effects oninhibition of HIV-1.
DISCUSSION
The results of these studies show that the 289EFLARH294
region of hA3F is critical for interactions with HIV-1 Vif thatinduce its proteasomal degradation. These studies further nar-row down the region of hA3F that is important for HIV-1 Vifinteraction from our previously identified region of amino ac-ids 283 to 300 (44) to six amino acids, of which only four appearto be important for binding to Vif. Furthermore, the aminoacids equivalent to E289 in hA3C, hA3DE, and agmA3F arecritical for the HIV-1 Vif-mediated degradation of these pro-teins, suggesting that the HIV-1 Vif interaction domain isconserved in three human and at least some nonhuman pri-mate A3 proteins. Although the structure of hA3F is not yetdetermined, we used the available structure of the C-terminaldomain of hA3G to gain insights into this Vif-interacting re-gion (Fig. 6A). Interestingly, the A3G residue K297, which isequivalent to E289 in hA3F, is exposed on the surface, consis-tent with its importance in interaction with HIV-1 Vif; simi-larly, the A3G residue I299, which is equivalent to L291 inhA3F, is buried within the protein, consistent with the obser-vation that the L291I mutation had little or no effect on HIV-1Vif-mediated degradation. The A3G residue K301, which isequivalent to the hA3F residue R293, is also surface exposed;thus, if hA3F and hA3G are highly similar in structure as ex-pected, the E289 and R293 residues should be adjacent to eachother on the exposed side of helix 3. We hypothesize that thesecharged residues bind to HIV-1 Vif through electrostatic interac-tions with the Vif DRMR region. Consistent with this hypothesis,the DRMR�SEMQ mutant of HIV-1 Vif failed to induce deg-radation of hA3F. However, extensive mutational analyses ofboth the EFLARH region of hA3F and the DRMR region ofHIV-1 Vif would be required to fully test this hypothesis.
Although the E289 residue and the EFLARH region appearto play a critical role in Vif interactions, other regions of hA3Fand HIV-1 Vif may interact and contribute to the stability ofbinding. In addition to the DRMR region (43), other aminoacids in HIV-1 Vif have been shown to be important for deg-radation of hA3F, including W11 (43, 52), Q12 (43), V25 (6,11), Y69 and L72 (19, 42, 58), E76 and W79 (19, 52, 58), and81LGxG84 and 171EDRW174 (8) (Fig. 6B). Thus, it is certainlypossible that A3F has multiple determinants that contribute toits interaction with Vif. In agreement with this, Lassen et al.recently reported that the N terminus of A3F has domains thatare involved in regulating Vif sensitivity (23).
Single amino acid changes in A3 proteins have previouslybeen shown to significantly affect interactions with HIV-1 Vif.The D128 residue of A3G was previously shown to renderhA3G resistant to degradation mediated by HIV-1 Vif (4, 32,45, 57). Subsequent studies have shown that A3G residues 126to 132 are involved in interaction with Vif (44). Mutations ofthe analogous amino acids in hA3F(E127) (28), hA3DE(D140)(9), and hA3C(Y140) (69) did not confer Vif resistance, sug-gesting that the Vif interaction mediated by this region of A3G
is not conserved. Recently, a single amino acid change in A3Hhaplotype I (K121E), which is different from the A3G regioncomprising amino acids 126 to 132, was shown to render thisprotein partially sensitive to HIV-1 Vif (25, 70).
The hA3F Vif-interacting region appears to be conservedamong several human and nonhuman A3 proteins (Fig. 6C).The overall sequence similarities between the C-terminalhalves of hA3F and hA3C and between the whole hA3F andhA3DE proteins are �77% (9, 68), and each of these proteinshas the EFLARH sequence. It was previously reported thathA3C and hA3DE are susceptible to HIV-1 Vif (6, 9, 42, 62,68, 69); however, the specific amino acids in these proteins thatare important for Vif interactions have not previously beenidentified. Our results showed that mutation of the amino acidequivalent to E289 rendered these proteins resistant to Vif-mediated degradation, suggesting that the EFLARH region inthese proteins is involved in interactions with HIV-1 Vif thatmediate proteasomal degradation.
It was previously shown that a deletion mutation in theN-terminal region (residues 2 to 47) of hA3C before the activesite was not susceptible to HIV-1 or African green monkeysimian immunodeficiency virus (SIVagm) Vif, suggesting thatan important determinant for Vif-induced degradation is lo-cated in this region of A3C (69). Interestingly, we found thatthe E106 residue, located just after the active site (Fig. 4A), isimportant for HIV-1 Vif susceptibility. The results of these twostudies suggest that more than one region of hA3C may beinvolved in the Vif-mediated degradation of this protein.
The hA3F Vif-interacting region is also present in theagmA3F (Chlorocebus aethiops) and rhA3F (Macaca mulatta)proteins, which have high overall sequence similarity to hA3F(88% and 87% identity, respectively) (66). Our results showedthat agmA3F, but not rhA3F, is susceptible to HIV-1 Vif-mediated degradation, in agreement with previous reports (54,66). Mutation of E289 in agmA3F rendered this protein resis-tant to Vif-mediated degradation. It is not clear why rhA3Fremains resistant to HIV-1 Vif, but presumably, other struc-tural features of rhA3F interfere with HIV-1 Vif binding or asubsequent step that is essential for Vif-induced degradation.
The gorilla A3C (Gorilla gorilla) and the chimpanzee A3D(Pan troglodytes) proteins also contain the EFLARH region,but whether these proteins are sensitive to HIV-1 Vif has notbeen determined. Interestingly, rhA3D was previously shownto be sensitive to HIV-1 Vif (54). While A3D isoform 1 con-tains an EFLATH sequence, the rhA3D sequence that wasused to determine Vif sensitivity contained the EFLARH re-gion. In addition, rhA3C, which has a KFLARH sequence, wasfound to be resistant to HIV-1 Vif-induced degradation (54).These results are consistent with the prediction of our studies andsuggest that HIV-1 Vif interacts with rhA3D by use of the EFLARH region but cannot interact with rhA3C. The EFLARHregion is not present in hA3G, hA3B, hA3A, and hA3H.
We also determined whether EFLARH-like sequences werepresent in the N-terminal domains of A3 proteins that containtwo zinc-binding motifs; however, none of the human or non-human primate A3 proteins analyzed contained an exact matchto this sequence. The EFLAEH sequences that is present inhA3F and hA3DE are most likely not involved in neutraliza-tion by Vif, since the E289K mutation in hA3F and the anal-ogous mutation in hA3DE conferred Vif resistance to these
VOL. 84, 2010 A3F, A3C, AND A3DE INTERACTIONS WITH HIV-1 Vif 12605
on August 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

proteins. In addition, the EFLAEH region in rhA3F is also notlikely to be involved in neutralization by Vif, since rhA3F isnaturally resistant to Vif-mediated degradation.
In summary, we have demonstrated that residues 289EFLARH294
in hA3F are important for HIV-1 Vif interactions. In particular, theglutamate residue is critical for HIV-1 Vif-mediated degradation ofhA3F as well as other A3 proteins, such as hA3C, hA3DE, and
agmA3F. The identification of the HIV-1 Vif interaction region inhA3F may facilitate the development of novel antiviral drugs thattarget this region.
ACKNOWLEDGMENTS
We especially thank Wei-Shau Hu for intellectual input throughoutthe project. We also thank Gisela Heidecker, Chawaree Chaipan,
FIG. 6. Model structure of the HIV-1 Vif binding domain of hA3F. (A) Model structure of the C-terminal domain of hA3G (Protein Data Bankaccession number 2KEM) (17) is shown. hA3G amino acids 297 to 302 of �-helix 3, which are equivalent to hA3F amino acids 289 to 294, are shownin space-filling form, with color coding as indicated. The remaining hA3G protein is shown in ribbon form; the �-helices are shown in red, andthe -sheets are shown in cyan. The figure was generated using the Accelrys Discovery Studio Visualizer (version 2.5) software program.(B) Schematic representation of hA3F-Vif and hA3G-Vif interactions. The major determinants of hA3F-Vif interactions and hA3G-Vif are shown.The determinants that were used in this study are shown extending out from each protein. The residues shown are those that have been shownto decrease interactions with A3F (top) or A3G (bottom) upon mutagenesis but to retain binding to the other APOBEC3 protein or cullin 5.(C) Sequence alignments of seven human A3 proteins and the A3F, A3C, and A3DE proteins from several nonhuman primates. The sequencesare compared to the hA3F C-terminal EFLARH sequence and surrounding amino acids (left) and the N-terminal EFLAEH sequence andsurrounding amino acids (right). Protein alignments were done using the BioEdit Sequence Alignment Editor, version 7.0.5.3 (16). The GenBankaccession numbers of the sequences used in these comparisons were NP_660341.2 (A3F isoform a; Homo sapiens), AAH38808.1 (A3F; H.sapiens*), AAH11739.1 (A3C; H. sapiens), NP_689639.2 (A3D; H. sapiens), NP_068594.1 (A3G; H. sapiens), EAW60281.1 (A3B; H. sapiens),NP_663745.1 (A3A; H. sapiens), ACK77772.1 (A3H; H. sapiens), NP_001035832.1 (A3F; M. mulatta), XP_525658.2) (A3F; P. troglodytes),AAT44387.1 (A3C; G. gorilla), ABY85203.1 (A3C; C. aethiops), ABY85204.1 (A3C; M. mulatta), XP_001094328.2 (A3D; M. mulatta),XP_525657.2 (A3D; P. troglodytes), XP_001094452.2 (A3G; M. mulatta), and NP_001009001.1 (P. troglodytes). The sequence for A3F (C. aethiops)was provided in reference 66.
12606 SMITH AND PATHAK J. VIROL.
on August 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

Salim Manoharadas, and Narasimhan J. Venkatachari for critical com-ments during manuscript preparation.
This research was supported in part by the Intramural ResearchProgram of the NIH, National Cancer Institute, Center for CancerResearch.
The content of this publication does not necessarily reflect the viewsor policies of the Department of Health and Human Services, nor doesmention of trade names, commercial products, or organizations implyendorsement by the U.S. Government.
REFERENCES
1. Anderson, J. L., and T. J. Hope. 2008. APOBEC3G restricts early HIV-1replication in the cytoplasm of target cells. Virology 375:1–12.
2. Bergeron, J. R., H. Huthoff, D. A. Veselkov, R. L. Beavil, P. J. Simpson, S. J.Matthews, M. H. Malim, and M. R. Sanderson. 2010. The SOCS-box ofHIV-1 Vif interacts with ElonginBC by induced-folding to recruit its Cul5-containing ubiquitin ligase complex. PLoS Pathog. 6:e1000925.
3. Bishop, K. N., M. Verma, E. Y. Kim, S. M. Wolinsky, and M. H. Malim. 2008.APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog.4:e1000231.
4. Bogerd, H. P., B. P. Doehle, H. L. Wiegand, and B. R. Cullen. 2004. A singleamino acid difference in the host APOBEC3G protein controls the primatespecies specificity of HIV type 1 virion infectivity factor. Proc. Natl. Acad.Sci. U. S. A. 101:3770–3774.
5. Boussif, O., F. Lezoualc’h, M. A. Zanta, M. D. Mergny, D. Scherman, B.Demeneix, and J. P. Behr. 1995. A versatile vector for gene and oligonu-cleotide transfer into cells in culture and in vivo: polyethylenimine. Proc.Natl. Acad. Sci. U. S. A. 92:7297–7301.
6. Chen, G., Z. He, T. Wang, R. Xu, and X. F. Yu. 2009. A patch of positivelycharged amino acids surrounding the human immunodeficiency virus type 1Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J. Virol.83:8674–8682.
7. Conticello, S. G., R. S. Harris, and M. S. Neuberger. 2003. The Vif proteinof HIV triggers degradation of the human antiretroviral DNA deaminaseAPOBEC3G. Curr. Biol. 13:2009–2013.
8. Dang, Y., R. W. Davis, I. A. York, and Y. H. Zheng. 2010. Identification of81LGxGxxIxW89 and 171EDRW174 domains from human immunodefi-ciency virus type 1 Vif that regulate APOBEC3G and APOBEC3F neutral-izing activity. J. Virol. 84:5741–5750.
9. Dang, Y., X. Wang, W. J. Esselman, and Y. H. Zheng. 2006. Identification ofAPOBEC3DE as another antiretroviral factor from the human APOBECfamily. J. Virol. 80:10522–10533.
10. Dang, Y., X. Wang, I. A. York, and Y. H. Zheng. 2010. Identification of acritical T[Q./D/E]x5ADx2[I/L] motif from primate lentivirus Vif proteinsthat regulate APOBEC3G and APOBEC3F neutralizing activity. J. Virol.84:8561–8570.
11. Dang, Y., X. Wang, T. Zhou, I. A. York, and Y. H. Zheng. 2009. Identificationof a novel WxSLVK motif in the N terminus of human immunodeficiencyvirus and simian immunodeficiency virus Vif that is critical for APOBEC3Gand APOBEC3F neutralization. J. Virol. 83:8544–8552.
12. Derdeyn, C. A., J. M. Decker, J. N. Sfakianos, X. Wu, W. A. O’Brien, L.Ratner, J. C. Kappes, G. M. Shaw, and E. Hunter. 2000. Sensitivity of humanimmunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated bycoreceptor specificity defined by the V3 loop of gp120. J. Virol. 74:8358–8367.
13. Goncalves, J., P. Jallepalli, and D. H. Gabuzda. 1994. Subcellular localiza-tion of the Vif protein of human immunodeficiency virus type 1. J. Virol.68:704–712.
14. Guo, F., S. Cen, M. Niu, J. Saadatmand, and L. Kleiman. 2006. Inhibition offormula-primed reverse transcription by human APOBEC3G during humanimmunodeficiency virus type 1 replication. J. Virol. 80:11710–11722.
15. Guo, F., S. Cen, M. Niu, Y. Yang, R. J. Gorelick, and L. Kleiman. 2007. Theinteraction of APOBEC3G with human immunodeficiency virus type 1 nu-cleocapsid inhibits tRNA3Lys annealing to viral RNA. J. Virol. 81:11322–11331.
16. Hall, T. A. 1999. BioEdit: a user-friendly biological sequence alignmenteditor and analysis program for Windows 95/98/NT. Nucleic Acids Symp.Ser. 41:95–98.
17. Harjes, E., P. J. Gross, K. M. Chen, Y. Lu, K. Shindo, R. Nowarski, J. D.Gross, M. Kotler, R. S. Harris, and H. Matsuo. 2009. An extended structureof the APOBEC3G catalytic domain suggests a unique holoenzyme model.J. Mol. Biol. 389:819–832.
18. Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deami-nation mediates innate immunity to retroviral infection. Cell 113:803–809.
19. He, Z., W. Zhang, G. Chen, R. Xu, and X. F. Yu. 2008. Characterization ofconserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3Finteraction. J. Mol. Biol. 381:1000–1011.
20. Holmes, R. K., F. A. Koning, K. N. Bishop, and M. H. Malim. 2007.APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription
products in the absence of hypermutation. Comparisons withAPOBEC3G. J. Biol. Chem. 282:2587–2595.
21. Huthoff, H., and M. H. Malim. 2007. Identification of amino acid residues inAPOBEC3G required for regulation by human immunodeficiency virus type1 Vif and Virion encapsidation. J. Virol. 81:3807–3815.
22. Iwatani, Y., D. S. Chan, F. Wang, K. S. Maynard, W. Sugiura, A. M. Gronen-born, I. Rouzina, M. C. Williams, K. Musier-Forsyth, and J. G. Levin. 2007.Deaminase-independent inhibition of HIV-1 reverse transcription byAPOBEC3G. Nucleic Acids Res. 35:7096–7108.
23. Lassen, K. G., S. Wissing, M. A. Lobritz, M. Santiago, and W. C. Greene.2010. Identification of two APOBEC3F splice variants displaying HIV-1antiviral activity and contrasting sensitivity to Vif. J. Biol. Chem. 285:29326–29335.
24. Lecossier, D., F. Bouchonnet, F. Clavel, and A. J. Hance. 2003. Hypermu-tation of HIV-1 DNA in the absence of the Vif protein. Science 300:1112.
25. Li, M. M., L. I. Wu, and M. Emerman. 2010. The range of humanAPOBEC3H sensitivity to lentiviral Vif proteins. J. Virol. 84:88–95.
26. Li, X. Y., F. Guo, L. Zhang, L. Kleiman, and S. Cen. 2007. APOBEC3Ginhibits DNA strand transfer during HIV-1 reverse transcription. J. Biol.Chem. 282:32065–32074.
27. Liddament, M. T., W. L. Brown, A. J. Schumacher, and R. S. Harris. 2004.APOBEC3F properties and hypermutation preferences indicate activityagainst HIV-1 in vivo. Curr. Biol. 14:1385–1391.
28. Liu, B., P. T. Sarkis, K. Luo, Y. Yu, and X. F. Yu. 2005. Regulation ofApobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J. Virol. 79:9579–9587.
29. Luo, K., T. Wang, B. Liu, C. Tian, Z. Xiao, J. Kappes, and X. F. Yu. 2007.Cytidine deaminases APOBEC3G and APOBEC3F interact with humanimmunodeficiency virus type 1 integrase and inhibit proviral DNA forma-tion. J. Virol. 81:7238–7248.
30. Luo, K., Z. Xiao, E. Ehrlich, Y. Yu, B. Liu, S. Zheng, and X. F. Yu. 2005.Primate lentiviral virion infectivity factors are substrate receptors that as-semble with cullin 5-E3 ligase through a HCCH motif to suppressAPOBEC3G. Proc. Natl. Acad. Sci. U. S. A. 102:11444–11449.
31. Mangeat, B., P. Turelli, G. Caron, M. Friedli, L. Perrin, and D. Trono. 2003.Broad antiretroviral defence by human APOBEC3G through lethal editingof nascent reverse transcripts. Nature 424:99–103.
32. Mangeat, B., P. Turelli, S. Liao, and D. Trono. 2004. A single amino aciddeterminant governs the species-specific sensitivity of APOBEC3G to Vifaction. J. Biol. Chem. 279:14481–14483.
33. Marin, M., K. M. Rose, S. L. Kozak, and D. Kabat. 2003. HIV-1 Vif proteinbinds the editing enzyme APOBEC3G and induces its degradation. Nat.Med. 9:1398–1403.
34. Mbisa, J. L., R. Barr, J. A. Thomas, N. Vandegraaff, I. J. Dorweiler, E. S.Svarovskaia, W. L. Brown, L. M. Mansky, R. J. Gorelick, R. S. Harris, A.Engelman, and V. K. Pathak. 2007. Human immunodeficiency virus type 1cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 81:7099–7110.
35. Mbisa, J. L., W. Bu, and V. K. Pathak. 2010. APOBEC3F and APOBEC3Ginhibit HIV-1 DNA integration by different mechanisms. J. Virol. 84:5250–5259.
36. Mehle, A., J. Goncalves, M. Santa-Marta, M. McPike, and D. Gabuzda.2004. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 18:2861–2866.
37. Mehle, A., B. Strack, P. Ancuta, C. Zhang, M. McPike, and D. Gabuzda.2004. Vif overcomes the innate antiviral activity of APOBEC3G by promot-ing its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem.279:7792–7798.
38. Mehle, A., E. R. Thomas, K. S. Rajendran, and D. Gabuzda. 2006. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J. Biol.Chem. 281:17259–17265.
39. Mehle, A., H. Wilson, C. Zhang, A. J. Brazier, M. McPike, E. Pery, and D.Gabuzda. 2007. Identification of an APOBEC3G binding site in humanimmunodeficiency virus type 1 Vif and inhibitors of Vif-APOBEC3G bind-ing. J. Virol. 81:13235–13241.
40. Newman, E. N., R. K. Holmes, H. M. Craig, K. C. Klein, J. R. Lingappa,M. H. Malim, and A. M. Sheehy. 2005. Antiviral function of APOBEC3G canbe dissociated from cytidine deaminase activity. Curr. Biol. 15:166–170.
41. Nguyen, K. L., M. llano, H. Akari, E. Miyagi, E. M. Poeschla, K. Strebel, andS. Bour. 2004. Codon optimization of the HIV-1 vpu and vif genes stabilizestheir mRNA and allows for highly efficient Rev-independent expression.Virology 319:163–175.
42. Pery, E., K. S. Rajendran, A. J. Brazier, and D. Gabuzda. 2009. Regulationof APOBEC3 proteins by a novel YXXL motif in human immunodeficiencyvirus type 1 Vif and simian immunodeficiency virus SIVagm Vif. J. Virol.83:2374–2381.
43. Russell, R. A., and V. K. Pathak. 2007. Identification of two distinct humanimmunodeficiency virus type 1 Vif determinants critical for interactions withhuman APOBEC3G and APOBEC3F. J. Virol. 81:8201–8210.
44. Russell, R. A., J. Smith, R. Barr, D. Bhattacharyya, and V. K. Pathak. 2009.Distinct domains within APOBEC3G and APOBEC3F interact with sepa-
VOL. 84, 2010 A3F, A3C, AND A3DE INTERACTIONS WITH HIV-1 Vif 12607
on August 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

rate regions of human immunodeficiency virus type 1 Vif. J. Virol. 83:1992–2003.
45. Schrofelbauer, B., D. Chen, and N. R. Landau. 2004. A single amino acid ofAPOBEC3G controls its species-specific interaction with virion infectivityfactor (Vif). Proc. Natl. Acad. Sci. U. S. A. 101:3927–3932.
46. Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolationof a human gene that inhibits HIV-1 infection and is suppressed by the viralVif protein. Nature 418:646–650.
47. Sheehy, A. M., N. C. Gaddis, and M. H. Malim. 2003. The antiretroviralenzyme APOBEC3G is degraded by the proteasome in response to HIV-1Vif. Nat. Med. 9:1404–1407.
48. Simon, V., V. Zennou, D. Murray, Y. Huang, D. D. Ho, and P. D. Bieniasz.2005. Natural variation in Vif: differential impact on APOBEC3G/3F and apotential role in HIV-1 diversification. PLoS Pathog. 1:e6.
49. Smith, J. L., W. Bu, R. C. Burdick, and V. K. Pathak. 2009. Multiple ways oftargeting APOBEC3-virion infectivity factor interactions for anti-HIV-1drug development. Trends Pharmacol. Sci. 30:638–646.
50. Stopak, K., C. de Noronha, W. Yonemoto, and W. C. Greene. 2003. HIV-1Vif blocks the antiviral activity of APOBEC3G by impairing both its trans-lation and intracellular stability. Mol. Cell 12:591–601.
51. Suspene, R., P. Sommer, M. Henry, S. Ferris, D. Guetard, S. Pochet, A.Chester, N. Navaratnam, S. Wain-Hobson, and J. P. Vartanian. 2004.APOBEC3G is a single-stranded DNA cytidine deaminase and functionsindependently of HIV reverse transcriptase. Nucleic Acids Res. 32:2421–2429.
52. Tian, C., X. Yu, W. Zhang, T. Wang, R. Xu, and X. F. Yu. 2006. Differentialrequirement for conserved tryptophans in human immunodeficiency virustype 1 Vif for the selective suppression of APOBEC3G and APOBEC3F.J. Virol. 80:3112–3115.
53. Unutmaz, D., V. N. KewalRamani, S. Marmon, and D. R. Littman. 1999.Cytokine signals are sufficient for HIV-1 infection of resting human T lym-phocytes. J. Exp. Med. 189:1735–1746.
54. Virgen, C. A., and T. Hatziioannou. 2007. Antiretroviral activity and Vifsensitivity of rhesus macaque APOBEC3 proteins. J. Virol. 81:13932–13937.
55. Wei, X., J. M. Decker, H. Liu, Z. Zhang, R. B. Arani, J. M. Kilby, M. S. Saag,X. Wu, G. M. Shaw, and J. C. Kappes. 2002. Emergence of resistant humanimmunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20)monotherapy. Antimicrob. Agents Chemother. 46:1896–1905.
56. Wiegand, H. L., B. P. Doehle, H. P. Bogerd, and B. R. Cullen. 2004. A secondhuman antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 andHIV-2 Vif proteins. EMBO J. 23:2451–2458.
57. Xu, H., E. S. Svarovskaia, R. Barr, Y. Zhang, M. A. Khan, K. Strebel, andV. K. Pathak. 2004. A single amino acid substitution in human APOBEC3Gantiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc. Natl. Acad. Sci. U. S. A. 101:5652–5657.
58. Yamashita, T., K. Kamada, K. Hatcho, A. Adachi, and M. Nomaguchi. 2008.Identification of amino acid residues in HIV-1 Vif critical for binding andexclusion of APOBEC3G/F. Microbes Infect. 10:1142–1149.
59. Yang, S., R. Delgado, S. R. King, C. Woffendin, C. S. Barker, Z. Y. Yang, L.Xu, G. P. Nolan, and G. J. Nabel. 1999. Generation of retroviral vector forclinical studies using transient transfection. Hum. Gene Ther. 10:123–132.
60. Yang, Y., F. Guo, S. Cen, and L. Kleiman. 2007. Inhibition of initiation ofreverse transcription in HIV-1 by human APOBEC3F. Virology 365:92–100.
61. Yee, J. K., T. Friedmann, and J. C. Burns. 1994. Generation of high-titerpseudotyped retroviral vectors with very broad host range. Methods CellBiol. 43(Pt. A):99–112.
62. Yu, Q., D. Chen, R. Konig, R. Mariani, D. Unutmaz, and N. R. Landau. 2004.APOBEC3B and APOBEC3C are potent inhibitors of simian immunodefi-ciency virus replication. J. Biol. Chem. 279:53379–53386.
63. Yu, Q., R. Konig, S. Pillai, K. Chiles, M. Kearney, S. Palmer, D. Richman,J. M. Coffin, and N. R. Landau. 2004. Single-strand specificity ofAPOBEC3G accounts for minus-strand deamination of the HIV genome.Nat. Struct. Mol. Biol. 11:435–442.
64. Yu, X., Y. Yu, B. Liu, K. Luo, W. Kong, P. Mao, and X. F. Yu. 2003. Inductionof APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCFcomplex. Science 302:1056–1060.
65. Yu, Y., Z. Xiao, E. S. Ehrlich, X. Yu, and X. F. Yu. 2004. Selective assemblyof HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex througha novel SOCS box and upstream cysteines. Genes Dev. 18:2867–2872.
66. Zennou, V., and P. D. Bieniasz. 2006. Comparative analysis of the antiret-roviral activity of APOBEC3G and APOBEC3F from primates. Virology349:31–40.
67. Zhang, H., B. Yang, R. J. Pomerantz, C. Zhang, S. C. Arunachalam, and L.Gao. 2003. The cytidine deaminase CEM15 induces hypermutation in newlysynthesized HIV-1 DNA. Nature 424:94–98.
68. Zhang, W., G. Chen, A. M. Niewiadomska, R. Xu, and X. F. Yu. 2008.Distinct determinants in HIV-1 Vif and human APOBEC3 proteins arerequired for the suppression of diverse host anti-viral proteins. PLoS One3:e3963.
69. Zhang, W., M. Huang, T. Wang, L. Tan, C. Tian, X. Yu, W. Kong, and X. F.Yu. 2008. Conserved and non-conserved features of HIV-1 and SIVagm Vifmediated suppression of APOBEC3 cytidine deaminases. Cell. Microbiol.10:1662–1675.
70. Zhen, A., T. Wang, K. Zhao, Y. Xiong, and X. F. Yu. 2010. A single aminoacid difference in human APOBEC3H variants determines HIV-1 Vif sen-sitivity. J. Virol. 84:1902–1911.
71. Zheng, Y. H., D. Irwin, T. Kurosu, K. Tokunaga, T. Sata, and B. M. Peterlin.2004. Human APOBEC3F is another host factor that blocks human immu-nodeficiency virus type 1 replication. J. Virol. 78:6073–6076.
12608 SMITH AND PATHAK J. VIROL.
on August 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from


















![Basic and depression specific emotion identification in ...webdocs.cs.ualberta.ca/~zaiane/postscript/CICLING19-1.pdf · label emotion mining from text [2,11,17]. With the increasing](https://img.pdfslide.us/doc/110x75/5f4a6fb08857b1069945e82f/basic-and-depression-speciic-emotion-identiication-in-zaianepostscriptcicling19-1pdf.jpg)










