Embed Size (px)
Citation preview

ww.sciencedirect.com
i n t e rn a t i o n a l j o u rn a l o f h y d r o g e n en e r g y x x x ( 2 0 1 4 ) 1e9
Available online at w
ScienceDirect
journal homepage: www.elsevier .com/locate/he
Hybrid functional calculations of potentialhydrogen storage material: Complex dimagnesiumiron hydride
Bakhtiar Ul Haq a, Mohammed Benali Kanoun b, Rashid Ahmed a,Mohamed Bououdina c,d, Souraya Goumri-Said b,*aDepartment of Physics, Faculty of Science, Universiti Teknologi Malaysia, UTM Skudai, 81310 Johor, Malaysiab Physical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal
23955-6900, Saudi ArabiacNanotechnology Centre, College of Science, University of Bahrain, PO Box 32038, Kingdom of BahraindDepartment of Physics, College of Science, University of Bahrain, PO Box 32038, Kingdom of Bahrain
a r t i c l e i n f o
Article history:
Received 2 December 2013
Received in revised form
27 March 2014
Accepted 2 April 2014
Available online xxx
Keywords:
Hydrides
DFT
Electronic structure
Optical properties
Storage capacity
* Corresponding author.E-mail addresses: mohammed.kanoun@k
edu.sa (S. Goumri-Said).
Please cite this article in press as: Ul HComplex dimagnesium iron hydridej.ijhydene.2014.04.014
http://dx.doi.org/10.1016/j.ijhydene.2014.04.00360-3199/Copyright ª 2014, Hydrogen Ener
a b s t r a c t
By employing the state of art first principles approaches, comprehensive investigations of a
very promising hydrogen storage material, Mg2FeH6 hydride, is presented. To expose its
hydrogen storage capabilities, detailed structural, elastic, electronic, optical and dielectric
aspects have been deeply analysed. The electronic band structure calculations demon-
strate that Mg2FeH6 is semiconducting material. The obtained results of the optical
bandgap (4.19 eV) also indicate that it is a transparent material for ultraviolet light, thus
demonstrating its potential for optoelectronics application. The calculated elastic proper-
ties reveal that Mg2FeH6 is highly stiff and stable hydride. Finally, the calculated hydrogen
(H2) storage capacity (5.47 wt.%) within a reasonable formation energy of �78 kJmol�1, at
room temperature, can be easily achievable, thus making Mg2FeH6 as potential material for
practical H2 storage applications.
Copyright ª 2014, Hydrogen Energy Publications, LLC. Published by Elsevier Ltd. All rights
reserved.
Introduction
Rapid lessening of limited conventional energy resources and
their noxious impacts on nature in the form of environmental
pollution has stimulated remarkable research interests to-
wards the search of alternative energy resources. Further-
more, the underlined subject of the ongoing research is that,
aust.edu.sa (M.B. Kanoun
aq B, et al., Hybrid fun, International Journa
14gy Publications, LLC. Publ
the novel energy resources must be sustainable, efficient,
cleaner and economical, as well as should be abundantly
available in nature. Hydrogen, being an efficient energy carrier
is of high importance for energy production as it accomplishes
all the mentioned required nuts and bolts of future energy
demand. However, the major challenge for its practical
application, especially exploitation of this unlimited energy
source for transportation, still unresolved because of its
), [email protected] (R. Ahmed), souraya.goumri-said@kaust.
ctional calculations of potential hydrogen storage material:l of Hydrogen Energy (2014), http://dx.doi.org/10.1016/
ished by Elsevier Ltd. All rights reserved.

i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n en e r g y x x x ( 2 0 1 4 ) 1e92
limited storage capacity and reversibility issue under specific
conditions. Though several methods have been proposed,
such as storing H2 as pressurised gas, liquid H2 under low
temperatures, metal (H2 atoms occupy interstices sites avail-
able within the lattice of the compounds) and complex hy-
drides (ionic hydrides) known as solid storage [1], etc. Among
these abundant available metal and complex hydrides
together with superior thermodynamic properties and high
reversible storage characteristics compared to both gas and
liquid storage, are considered themost appropriate in terms of
its cyclic ability and safety issues for their practical applica-
tions [1e7].
Recently synthesized Mg-based 3d-transition metal hy-
drideMg2FeH6 has attracted considerable attention because of
its appreciable H2 storage capacity (5.47wt.%), high volumetric
H2 density (double than liquid hydrogen) of 150 kgm�3 [8], and
particularly showing exceptional capability of storing thermo-
chemical energy up to 773 K and exhibiting its potential as
catalyst to improve the de/rehydrogenaton properties of other
hydrides [9]. The above characteristics of Mg2FeH6 adding to
that the low cost of its components Mg and Fe metals,
distinguish it from other complex hydrides (for instance
Mg2MHx (M¼Co, Ni)), however, there are still some issues that
need to be raised such as, slow absorption kinetics, high
dissociation temperature and instability, which impend its
practical applications.
To deal with these issues, some experimental and funda-
mental studies have been reported previously at different
levels, for example, crystal structure of Mg2FeH6 was for the
first time determined experimentally by Didisheim et al. [10],
followed by an investigation of its hydrogenation and dehy-
drogenation features by Ivanov et al. [11]. Thereafter, in-
vestigations on the synthesis and characterisation of Mg2FeH6
have been the subject of numerous studies [4,12e25],
including recent report of Retuerto [26], who designed a new
synthesis technique for the fabrication of Mg2FeH6 at rela-
tively high temperature of 750 �C and pressure 2 GPa [26]. More
recently, Danaie et al. [25] have adapted high energy ball
milling technique to synthesize Mg2FeH6 starting from a
mixture of Fe and MgH2 powders. In spite of all these efforts,
compared to MgeMeH (M¼Ni, Co) systems, MgeFeeH equi-
librium ternary alloy phase diagram does not reveal the for-
mation of stable binary alloys. Additionally, none the
conventional synthesis processes is showing potential in the
near future for the possibility of the fabrication of pure
Mg2FeH6 phase. Therefore, comprehensive understanding of
some important aspects regarding H2 storage process in
Mg2FeH6 within available experimental tools is very much
limited.
To overcome upon the above encountered problems,
several research groups have adapted first principles compu-
tational modelling techniques [27e34], to investigate its
properties because of the competence of first principles cal-
culations in determining more precise results within a short
time and much cheaper resources without having prior
knowledge of experimental measurements. Most of these
studies are focused mainly on the electronic structure and
structural properties using density functional density (DFT)
within GGA and LDA approaches. Even then, there exist
several contentious reports regarding the bandgap nature
Please cite this article in press as: Ul Haq B, et al., Hybrid funComplex dimagnesium iron hydride, International Journaj.ijhydene.2014.04.014
(indirect/direct) of Mg2FeH6 as well as about the value of its
bandgap energy, as different reports present different values
in the literature, i.e. 1.74 eV [33], 1.73 eV [28] or 1.91 eV [34]
besides pointing out to its insulating or a semiconducting
nature with direct energy gap of 2.33 eV [35], 1.96 eV [32],
1.87 eV and 2.25 eV [29]. By reviewing more precisely the re-
sults of the above references, it is found that, most of these
first principles DFT studies have been performed at the level of
standard DFT. In fact, it is well known that standard DFT
calculations usually (calculations at the level of standard LDA
and GGA) underestimate the electronic and optical properties
very severely. To deal with such issues especially the calcu-
lations of electronic and other excited state properties of
complex materials, hybrid functionals for exchange correla-
tion energy/corresponding potential are successfully imple-
mented to reproduce appropriate results for the electronic,
structural, elastic and optical properties as well. To the best
knowledge of the authors, no study is reported yet beyond the
standard DFT approaches for the investigation of electronic
along with optical properties of Mg2FeH6 to obtain precise
results.
Therefore in this present study, the calculations are per-
formed on Mg2FeH6 by employing hybrid functionals (PBE0
and HSE06) and scrutinize their effect on the electronic as well
as optical properties, and in turn their effect on hydrogen
storage capabilities in comparison with standard DFT calcu-
lations. Moreover, in order to explore the real potential of
Mg2FeH6, a comprehensive understanding of its physical
properties, at quantummechanical scale is always in demand
and of great importance. As an example, the knowledge of
elastic properties of Mg2FeH6 is important. Similarly, some
hydride materials have been reported suitable as an alterna-
tive to transparent conducting oxides (TCO) for solar cell
technology. Accordingly, the study of the optical properties of
Mg2FeH6 will help to explore its potential for optoelectronic
applications as well. Taking the benefits of the first principles
approaches, the study was extended to structural, elastic,
electronic, optical and dielectric properties.
Computational details
Calculations in this work were carried out by employing pro-
jector augmented-wave (PAW) scheme of calculation [36,37]
as realized in Vienna ab-initio simulation package (VASP)
[38,39]. The PAWmethod was preferred over the full-potential
plane wave approach (FP-PW) because the former can opti-
mize large super cells in a reasonable time without compro-
mising on the quality of the results. For Mg and Fe, 3s2, 3d7and
4s1 states electrons were treated as valence whereas 1s1 for H
atoms. The exchange and correlations energy were treated
within the PerdeweBurkeeErnzerhof (PBE) GGA approxima-
tion in standard DFT calculations for the sake of comparison
with hybrid functional calculations [40]. For hybrid functional
calculations, the HeydeScuseriaeErnzerhof (HSE06) [41] and
PBE0 [42] hybrid energy functionals were used. PBE0 and
HSE06 approximations are somewhat similar, in a sense, as in
both approaches 25% of the exact screened HartreeeFock
exchange is mixed with the 75% of gradient-corrected PBE
exchange functional, but in both approaches long range
ctional calculations of potential hydrogen storage material:l of Hydrogen Energy (2014), http://dx.doi.org/10.1016/

Fig. 1 e (a) The unit cell structure of cubic K2PtCl6-type Mg2FeH6 and (b) polyhedral presentation.
i n t e rn a t i o n a l j o u rn a l o f h y d r o g e n en e r g y x x x ( 2 0 1 4 ) 1e9 3
interaction part of the exact exchange is treated differently. In
HSE functional, short-range part of the exact exchange is only
preserved and long-range part of exact exchange is cut off,
that made this approach more efficient. For both local and
non-local parts, the value of screening parameter (u) used is
0.2 A�1, which is identical to the value reported in reference
[41]. A 500 eV energy cut off was used to carry out the calcu-
lations. Brillouin zonewas sampled using theMonkhorst-Pack
(MP) method [43] with a maximum separation of 0.3 A be-
tween two points in the Brillouin zone. This separation gives
8� 8� 8 mesh for the four-atom unit cell. The SCF cycle was
stopped after the energy difference between two consecutive
iterations was less than 10�5 eV.
Results and discussion
Structure and elastic properties
The simulated unit cell of Mg2FeH6 has been schematically
shown in Fig. 1(a). The structural geometry of Mg2FeH6 is
analogous to that of cubic K2PtCl6 (fluorite-type) structure [10].
Mg2FeH6 has a lattice constant a¼ 6.443 A, space group Fm3m,
and contains 36 atoms per unit cell. Mg and Fe atoms have
fixed atomic coordinates and are, respectively, positioned at
tetrahedral and octahedral coordinates such as Mg (8c) (1/4, 1/
4, 1/4); and Fe (4a) (0, 0, 0). However H has variable position in
(24e) (x, 0, 0) [26,32]. Fig. 1(b) shows the polyhedral geometry of
Mg2FeH6, which is well analogous to the one schematized by
Retuerto et al. [26] in their experimental report. It is evident
that each Fe atom is surrounded by 6 hydrogen atoms that
make orthogonal planes to each Fe atom. Furthermore, the
polyhedral FeH6 complexes are tetrahedrally coordinated to
Mg atoms resulting in an fcc lattice in which the polyhedral
FeH6 resides at the centre. Unit cell structure of Mg2FeH6, with
the above details was fully relaxed. The calculated value for
lattice constant is reported in Table 1 alongwith experimental
data [10] for comparison. It can be noticed that with respect to
the experimental value, the calculated lattice parameter is
slightly underestimated within 0.51%. Using the hybrid
Please cite this article in press as: Ul Haq B, et al., Hybrid funComplex dimagnesium iron hydride, International Journaj.ijhydene.2014.04.014
calculations, the calculated equilibrium properties are also
found to be in good agreement with the experimental data [9].
Overall, it is observed that both hybrid functionals give
somewhat lower value of the lattice constant in comparison
with GGA-PBE. This might happen due the enhanced elec-
tronic localization, resulting into tight inter-atomic bonding.
In order to investigate the strengthandmechanical stability
of Mg2FeH6, the three independent elastic constants C11, C12
and C44, were calculated. The obtained values of C11, C12 and
C44 are 249, 88.4 and 65.5 GPa, respectively. As they satisfy the
BorneHuang mechanical stability criterion [44] defined for
cubic structures, i.e. (C11eC12)> 0, (C11þ 2C12)> 0 and C44> 0,
which reveal that Mg2FeH6 is a mechanically stable material.
After obtaining elastic constants, the polycrystalline bulk
modulus (B), shearmodulus (G) and Young’s modulus (Y) were
also calculated by employing the VoigteReusseHill (VRH)
approximation [45]. The calculated polycrystalline elastic
constants are found to be 142, 71, and 182.7 GPa for B, G and Y,
respectively. It is predicted that Mg2FeH6 has a low bulk, Shear
and Young’s moduli, consistently indicating that it is a mate-
rial with low hardness. While comparing to the recent litera-
ture on hydrides [46], the calculated elastic constants and
mechanical properties, such as the bulk modulus, one can see
similar trends compared to some other relatedmetal hydrides
such as ε-ZrH2 [47], where DFT basedmethods have been used
to understand the relationship between structure and elastic
constants. Moreover, according to classical criteria of Pugh’s
modulus ratio B/G (as proposed by Pugh [48]), a material is
brittle (ductile) if the B/G ratio is less than 1.75 [48e51]. In our
case, for our considered compound, B/G value of 2.0 is larger
than 1.75, i.e. this material will behave in a ductile manner.
Electronic structure and related properties
To illustrate the electronic structure ofMg2FeH6, Fig. 2 displays
the total and partial density of states obtained at GGA-PBE,
HSE06 and PBE0 level of approximations. As shown in previ-
ous section, the calculated values of the lattice constant, at
the level of PBE0 and HSE06, are in close agreement with GGA-
PBE results, therefore to save calculations time, as input, GGA-
ctional calculations of potential hydrogen storage material:l of Hydrogen Energy (2014), http://dx.doi.org/10.1016/
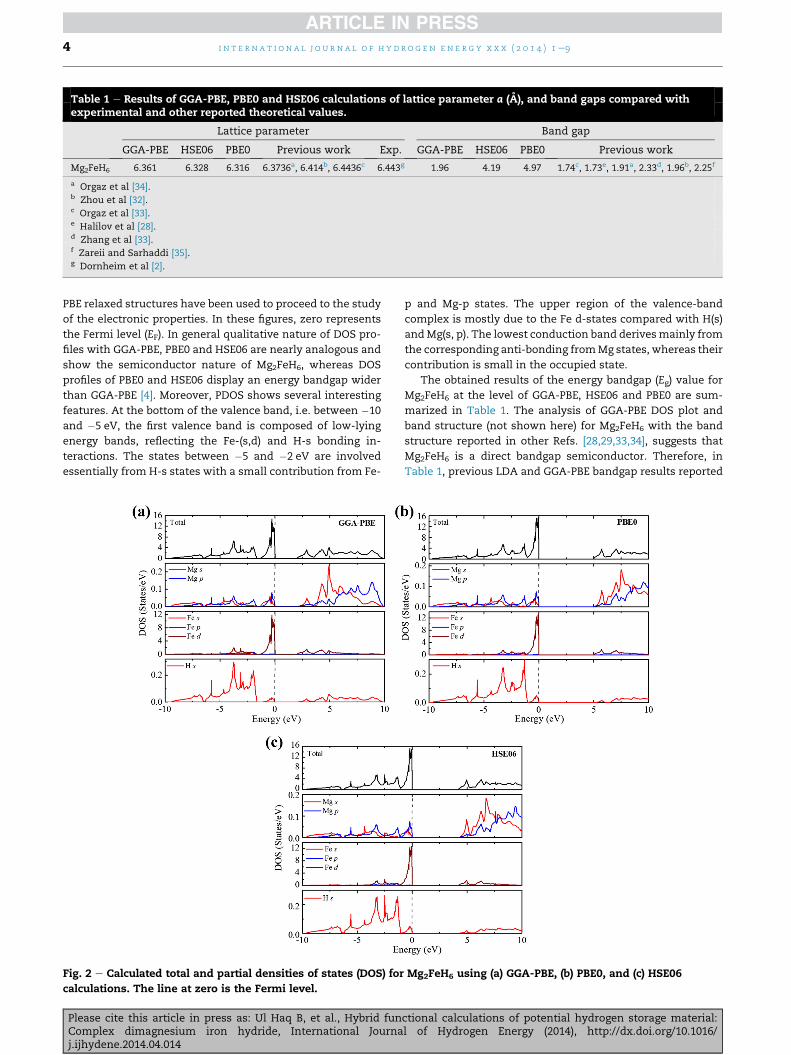
Table 1 e Results of GGA-PBE, PBE0 and HSE06 calculations of lattice parameter a (A), and band gaps compared withexperimental and other reported theoretical values.
Lattice parameter Band gap
GGA-PBE HSE06 PBE0 Previous work Exp. GGA-PBE HSE06 PBE0 Previous work
Mg2FeH6 6.361 6.328 6.316 6.3736a, 6.414b, 6.4436c 6.443g 1.96 4.19 4.97 1.74c, 1.73e, 1.91a, 2.33d, 1.96b, 2.25f
a Orgaz et al [34].b Zhou et al [32].c Orgaz et al [33].e Halilov et al [28].d Zhang et al [33].f Zareii and Sarhaddi [35].g Dornheim et al [2].
i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n en e r g y x x x ( 2 0 1 4 ) 1e94
PBE relaxed structures have been used to proceed to the study
of the electronic properties. In these figures, zero represents
the Fermi level (EF). In general qualitative nature of DOS pro-
files with GGA-PBE, PBE0 and HSE06 are nearly analogous and
show the semiconductor nature of Mg2FeH6, whereas DOS
profiles of PBE0 and HSE06 display an energy bandgap wider
than GGA-PBE [4]. Moreover, PDOS shows several interesting
features. At the bottom of the valence band, i.e. between �10
and �5 eV, the first valence band is composed of low-lying
energy bands, reflecting the Fe-(s,d) and H-s bonding in-
teractions. The states between �5 and �2 eV are involved
essentially from H-s states with a small contribution from Fe-
Fig. 2 e Calculated total and partial densities of states (DOS) for
calculations. The line at zero is the Fermi level.
Please cite this article in press as: Ul Haq B, et al., Hybrid funComplex dimagnesium iron hydride, International Journaj.ijhydene.2014.04.014
p and Mg-p states. The upper region of the valence-band
complex is mostly due to the Fe d-states compared with H(s)
andMg(s, p). The lowest conduction band derivesmainly from
the corresponding anti-bonding fromMg states, whereas their
contribution is small in the occupied state.
The obtained results of the energy bandgap (Eg) value for
Mg2FeH6 at the level of GGA-PBE, HSE06 and PBE0 are sum-
marized in Table 1. The analysis of GGA-PBE DOS plot and
band structure (not shown here) for Mg2FeH6 with the band
structure reported in other Refs. [28,29,33,34], suggests that
Mg2FeH6 is a direct bandgap semiconductor. Therefore, in
Table 1, previous LDA and GGA-PBE bandgap results reported
Mg2FeH6 using (a) GGA-PBE, (b) PBE0, and (c) HSE06
ctional calculations of potential hydrogen storage material:l of Hydrogen Energy (2014), http://dx.doi.org/10.1016/

i n t e rn a t i o n a l j o u rn a l o f h y d r o g e n en e r g y x x x ( 2 0 1 4 ) 1e9 5
by other theoretical calculations are listed. However, at this
stage, we did not find any experimental data in literature for
comparison. The calculated GGA-PBE band gap value is about
1.96 eV. Though the results obtained in this study, at the level
of GGA-PBE, are in agreement with other calculations,
whereas standard LDA and GGA-PBE methods sufficiently
underestimate the bandgap energy value for semiconductors
and insulators in addition to the erroneous prediction of
ground states properties of strongly correlated materials. As
PBE0 and HSE06 functionals have been reported to be suitable
to reproduce better values of the band gap energy [52],
therefore in addition to GGA-PBE, PBE0 and HSE06 functionals
have been implemented for band structure calculations to
improve the bandgap value of Mg2FeH6. The calculated
bandgap energy value with PBE0 is found to be slightly higher
than the bandgap value obtained with HSE06.Since there is no
experimental data available in literature on energy bandgap
related to Mg2FeH6, calculations based on GGA-PBE for pure a-
MgH2 as reported by Ahuja and co-workers [53], can be
considered as reference, i.e. Eg¼ 3.47 eV, which strongly un-
derestimates the value of Eg relative to the expected experi-
mental value (5.6 eV) [54]. However, it was found that PBE0 and
HSE06 functionals improve the value of the band gap of pure
a-MgH2; 4.58 and 5.31 eV, respectively, which are in better
agreement with the experimental value of 5.6 eV. Accordingly
and by analyzing other studies performed with PBE0 and
HSE06 on some other semiconductors available in literature, it
can be concluded that for electronic properties, hybrid func-
tionals are more appropriate for Mg2FeH6.
More importantly and for H2-storage, from PDOS, it can be
noticed that the s-state electrons of H atoms contribute more
near the Fermi level, thus, it is easier to release H atoms. A
particular emphasiswasdevoted to theenthalpyof energy (DH)
for the hydride system by considering the following reaction:
2Mgþ Feþ 3 H2/Mg2FeH6
The corresponding value of the enthalpy of energy can be
calculated using the following expression:
DH ¼ 13
�E�Mg2FeH6 � 2EðMgÞ � EðFeÞ � 3ðH2Þ
��
where the total energies (E) of Mg2FeH6, Fe and Mg were ob-
tained by geometry relaxations. To estimate the properties of
H2 molecule, a simple cubic unit cell was used by taking large
value of lattice constant. The enthalpy of energy of H2 for
Mg2FeH6 is found about �84.34 kJ (mol H2)�1 which is in good
agreement with the experimental value of �98 kJmol�1 of H2
[2]. The capacity of H2-storage in Mg2FeH6 was also estimated,
which is about 5.47 wt.%. These findings are very competitive
compared to the characteristics of MgH2, which is another
potential H2-storage material, with a formation energy of
�78 kJmol�1 and H-storage capacity 7.65 wt.% [2,7].
Optical properties
Comprehensive data about the optical properties of a material
can be derived using dielectric function ε(u)¼ ε1(u)þ iε2(u) over
the entire photon energies. For inter-band transitions, the
Please cite this article in press as: Ul Haq B, et al., Hybrid funComplex dimagnesium iron hydride, International Journaj.ijhydene.2014.04.014
dielectric function is computed using momentum representa-
tion. The dispersive (Real) part (ε1(u)) of the dielectric function
describes the propagation properties whereas the absorptive
(Imaginary) part(ε2(u)) is associated to the optical absorption in a
medium and is directly linked with the electronic structure.
The imaginary part (ε2(u)) can be derived from [3]:
ε2 ¼ 2e2pUε0
Xk;V;C
���jCKjbu$rjjV
K
���2d�ECk � EV
k � E�
where jCK and jk
C are the wave functions corresponding to
energies ECk and EV
k , respectively, ε0 represents the permittivity
of free space, the unit cell volume is defined by U, u de-
termines the polarization of the incident electric field, r and k
represent the vectors in the real and reciprocal lattice,
respectively. The knowledge of both real and imaginary parts
of the dielectric tensor will allow the calculation of other
important linear optical properties such as the reflectivity (R),
the refractive index (n) and the absorption coefficient (A).
Hereafter, only the calculations of the optical properties of
Mg2FeH6 using GGA-PBE and HSE06 functionals are reported,
because PBE0 is leading to the same results as HSE06. The
calculated macroscopic dielectric functions are plotted in
Fig. 3. It is clearly shown in Fig. 3(a) that GGA-PBE curve un-
derestimates the band edge because of its severe underesti-
mation of the electronic bandgap energy. However the nature
of the calculated band edge by HSE06 ismore or less analogous
with GGA-PBE. It means that the inclusion of the hybrid
functionals description does not affect the nature/character of
band edge though it reasonably improves the bandgap value
as compared to the standard GGA. The absorption peak ob-
tained via HSE06 is moved to higher energies compared to
GGA-PBE. As shown in Fig. 3(a), depiction of the direct and
indirect transitions is appropriately performed with all
methods. There is a pronounced sharp peak at about 5.33 eV
in the calculated ε2(u) spectrum close to the absorption edge.
Without taking into account light polarization effect, in the
imaginary part of the dielectric function, it is noted (above the
excitonic peak) the presence of three key spectral features
related to the peaks in the spectrum obtained using HSE06
functionals [3]. The second peak at around 6.38 eV mainly
corresponds to the transitions occurring from the uppermost
band of the valence bands to conduction band states.Whereas
the complicated third peak between 7 eV and 10 eV is pri-
marily originated by the transitions taking place from Fe3d
valence bands to conduction bands [3]. Furthermore, the peak
located at 10.8 eV is largely arising from Mg 2p orbitals of
Mg2FeH6. The real part of the dielectric function is obtained by
Kramer-Kronig transformation. The calculated real part of the
dielectric function for Mg2FeH6 with GGA-PBE and HSE06 are
shown in Fig. 3(b). In the case of HSE06, the real part Re(u)
shows again a sharp peak at about 5.33 eV, which is obviously
linked to the peak of the dielectric function’s imaginary
component. And then, the curves decrease to values close to
zero at 9.81 eV. For GGA-PBE, the curve of the real part of ε(u) is
incredibly analogous to the HSE06 hybrid functional. The
value of HSE06 dielectric constant is predicted to be ε(0)¼5.70 eV. Thus, it can be concluded that the calculated macro-
scopic static electronic dielectric constants by hybrid method
ameliorates the values of ε(0) than GGA-PBE (4.4 eV).
ctional calculations of potential hydrogen storage material:l of Hydrogen Energy (2014), http://dx.doi.org/10.1016/
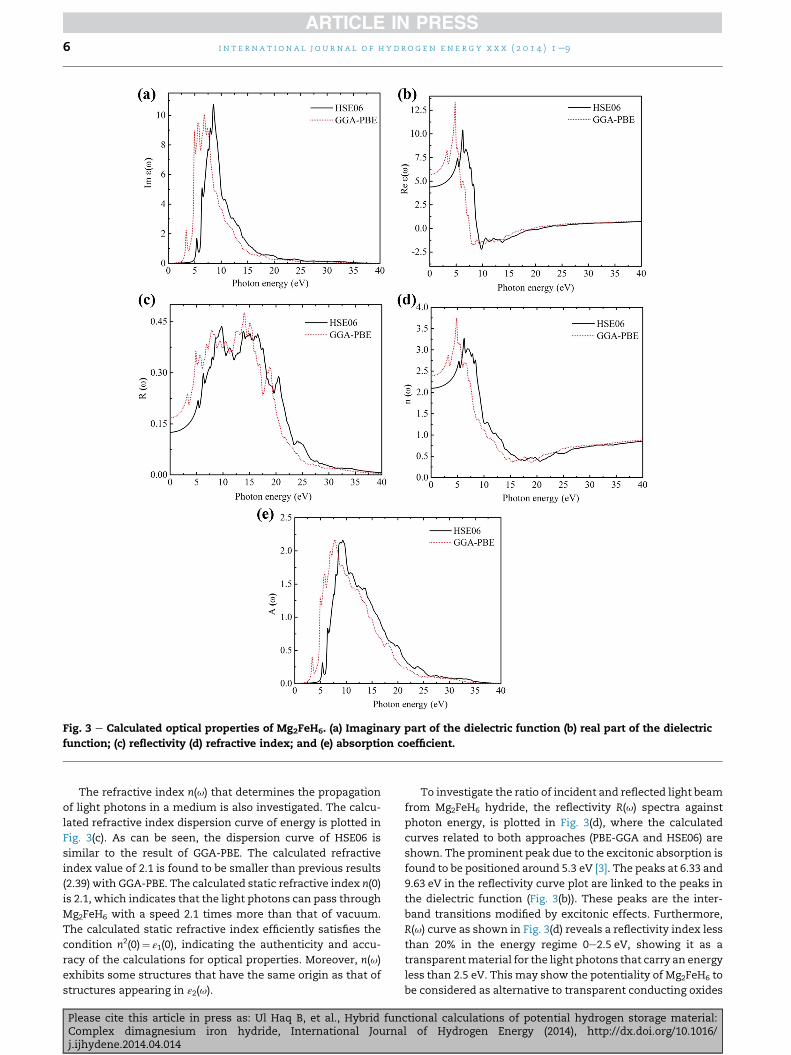
Fig. 3 e Calculated optical properties of Mg2FeH6. (a) Imaginary part of the dielectric function (b) real part of the dielectric
function; (c) reflectivity (d) refractive index; and (e) absorption coefficient.
i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n en e r g y x x x ( 2 0 1 4 ) 1e96
The refractive index n(u) that determines the propagation
of light photons in a medium is also investigated. The calcu-
lated refractive index dispersion curve of energy is plotted in
Fig. 3(c). As can be seen, the dispersion curve of HSE06 is
similar to the result of GGA-PBE. The calculated refractive
index value of 2.1 is found to be smaller than previous results
(2.39) with GGA-PBE. The calculated static refractive index n(0)
is 2.1, which indicates that the light photons can pass through
Mg2FeH6 with a speed 2.1 times more than that of vacuum.
The calculated static refractive index efficiently satisfies the
condition n2(0)¼ ε1(0), indicating the authenticity and accu-
racy of the calculations for optical properties. Moreover, n(u)
exhibits some structures that have the same origin as that of
structures appearing in ε2(u).
Please cite this article in press as: Ul Haq B, et al., Hybrid funComplex dimagnesium iron hydride, International Journaj.ijhydene.2014.04.014
To investigate the ratio of incident and reflected light beam
from Mg2FeH6 hydride, the reflectivity R(u) spectra against
photon energy, is plotted in Fig. 3(d), where the calculated
curves related to both approaches (PBE-GGA and HSE06) are
shown. The prominent peak due to the excitonic absorption is
found to be positioned around 5.3 eV [3]. The peaks at 6.33 and
9.63 eV in the reflectivity curve plot are linked to the peaks in
the dielectric function (Fig. 3(b)). These peaks are the inter-
band transitions modified by excitonic effects. Furthermore,
R(u) curve as shown in Fig. 3(d) reveals a reflectivity index less
than 20% in the energy regime 0e2.5 eV, showing it as a
transparentmaterial for the light photons that carry an energy
less than 2.5 eV. This may show the potentiality of Mg2FeH6 to
be considered as alternative to transparent conducting oxides
ctional calculations of potential hydrogen storage material:l of Hydrogen Energy (2014), http://dx.doi.org/10.1016/
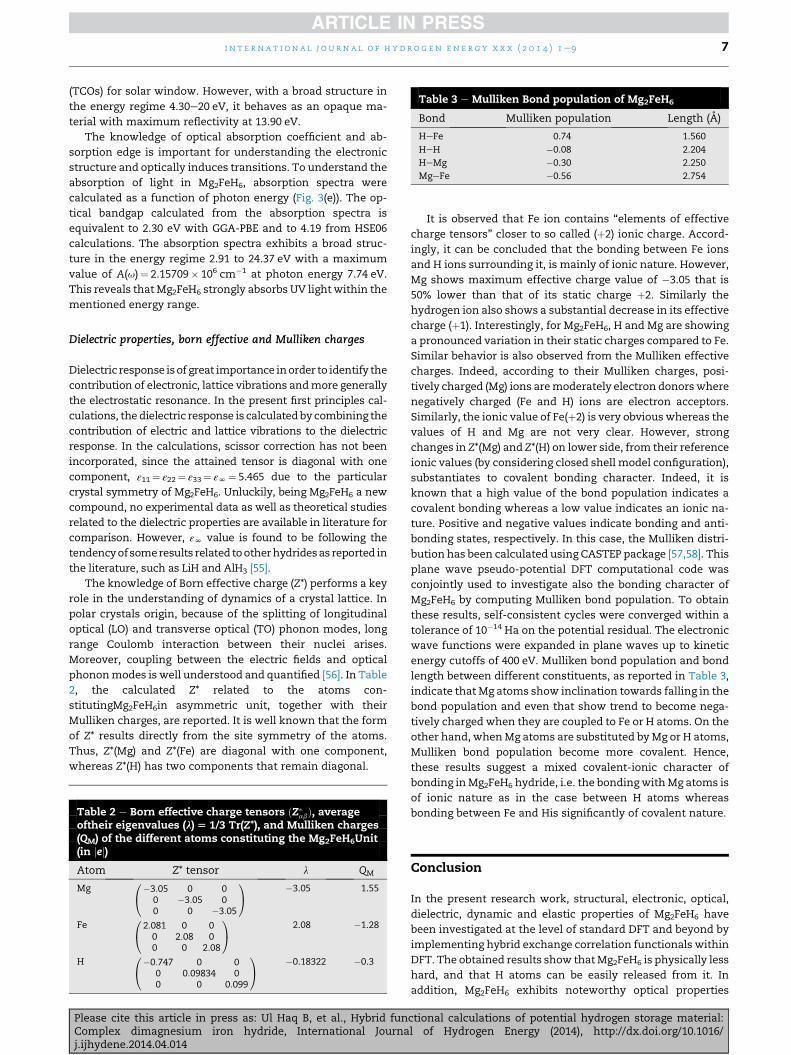
Table 3 e Mulliken Bond population of Mg2FeH6
Bond Mulliken population Length (A)
HeFe 0.74 1.560
HeH �0.08 2.204
HeMg �0.30 2.250
MgeFe �0.56 2.754
i n t e rn a t i o n a l j o u rn a l o f h y d r o g e n en e r g y x x x ( 2 0 1 4 ) 1e9 7
(TCOs) for solar window. However, with a broad structure in
the energy regime 4.30e20 eV, it behaves as an opaque ma-
terial with maximum reflectivity at 13.90 eV.
The knowledge of optical absorption coefficient and ab-
sorption edge is important for understanding the electronic
structure and optically induces transitions. To understand the
absorption of light in Mg2FeH6, absorption spectra were
calculated as a function of photon energy (Fig. 3(e)). The op-
tical bandgap calculated from the absorption spectra is
equivalent to 2.30 eV with GGA-PBE and to 4.19 from HSE06
calculations. The absorption spectra exhibits a broad struc-
ture in the energy regime 2.91 to 24.37 eV with a maximum
value of A(u)¼ 2.15709� 106 cm�1 at photon energy 7.74 eV.
This reveals that Mg2FeH6 strongly absorbs UV light within the
mentioned energy range.
Dielectric properties, born effective and Mulliken charges
Dielectric response isof great importance inorder to identify the
contribution of electronic, lattice vibrations andmore generally
the electrostatic resonance. In the present first principles cal-
culations, the dielectric response is calculatedby combining the
contribution of electric and lattice vibrations to the dielectric
response. In the calculations, scissor correction has not been
incorporated, since the attained tensor is diagonal with one
component, ε11¼ ε22¼ ε33¼ εN¼ 5.465 due to the particular
crystal symmetry of Mg2FeH6. Unluckily, being Mg2FeH6 a new
compound, no experimental data as well as theoretical studies
related to the dielectric properties are available in literature for
comparison. However, εN value is found to be following the
tendencyofsomeresults related tootherhydridesas reported in
the literature, such as LiH and AlH3 [55].
The knowledge of Born effective charge (Z*) performs a key
role in the understanding of dynamics of a crystal lattice. In
polar crystals origin, because of the splitting of longitudinal
optical (LO) and transverse optical (TO) phonon modes, long
range Coulomb interaction between their nuclei arises.
Moreover, coupling between the electric fields and optical
phononmodes is well understood and quantified [56]. In Table
2, the calculated Z* related to the atoms con-
stitutingMg2FeH6in asymmetric unit, together with their
Mulliken charges, are reported. It is well known that the form
of Z* results directly from the site symmetry of the atoms.
Thus, Z*(Mg) and Z*(Fe) are diagonal with one component,
whereas Z*(H) has two components that remain diagonal.
Table 2 e Born effective charge tensors ðZ�abÞ, average
oftheir eigenvalues (l)[ 1/3 Tr(Z*), and Mulliken charges(QM) of the different atoms constituting the Mg2FeH6Unit(in jej)Atom Z* tensor l QM
Mg0@�3:05 0 0
0 �3:05 00 0 �3:05
1A �3.05 1.55
Fe0@2:081 0 0
0 2:08 00 0 2:08
1A 2.08 �1.28
H0@�0:747 0 0
0 0:09834 00 0 0:099
1A �0.18322 �0.3
Please cite this article in press as: Ul Haq B, et al., Hybrid funComplex dimagnesium iron hydride, International Journaj.ijhydene.2014.04.014
It is observed that Fe ion contains “elements of effective
charge tensors” closer to so called (þ2) ionic charge. Accord-
ingly, it can be concluded that the bonding between Fe ions
and H ions surrounding it, is mainly of ionic nature. However,
Mg shows maximum effective charge value of �3.05 that is
50% lower than that of its static charge þ2. Similarly the
hydrogen ion also shows a substantial decrease in its effective
charge (þ1). Interestingly, for Mg2FeH6, H and Mg are showing
a pronounced variation in their static charges compared to Fe.
Similar behavior is also observed from the Mulliken effective
charges. Indeed, according to their Mulliken charges, posi-
tively charged (Mg) ions aremoderately electron donorswhere
negatively charged (Fe and H) ions are electron acceptors.
Similarly, the ionic value of Fe(þ2) is very obviouswhereas the
values of H and Mg are not very clear. However, strong
changes in Z*(Mg) and Z*(H) on lower side, from their reference
ionic values (by considering closed shell model configuration),
substantiates to covalent bonding character. Indeed, it is
known that a high value of the bond population indicates a
covalent bonding whereas a low value indicates an ionic na-
ture. Positive and negative values indicate bonding and anti-
bonding states, respectively. In this case, the Mulliken distri-
bution has been calculated using CASTEP package [57,58]. This
plane wave pseudo-potential DFT computational code was
conjointly used to investigate also the bonding character of
Mg2FeH6 by computing Mulliken bond population. To obtain
these results, self-consistent cycles were converged within a
tolerance of 10�14 Ha on the potential residual. The electronic
wave functions were expanded in plane waves up to kinetic
energy cutoffs of 400 eV. Mulliken bond population and bond
length between different constituents, as reported in Table 3,
indicate that Mg atoms show inclination towards falling in the
bond population and even that show trend to become nega-
tively charged when they are coupled to Fe or H atoms. On the
other hand, whenMg atoms are substituted byMg or H atoms,
Mulliken bond population become more covalent. Hence,
these results suggest a mixed covalent-ionic character of
bonding inMg2FeH6 hydride, i.e. the bondingwithMg atoms is
of ionic nature as in the case between H atoms whereas
bonding between Fe and His significantly of covalent nature.
Conclusion
In the present research work, structural, electronic, optical,
dielectric, dynamic and elastic properties of Mg2FeH6 have
been investigated at the level of standard DFT and beyond by
implementing hybrid exchange correlation functionals within
DFT. The obtained results show thatMg2FeH6 is physically less
hard, and that H atoms can be easily released from it. In
addition, Mg2FeH6 exhibits noteworthy optical properties
ctional calculations of potential hydrogen storage material:l of Hydrogen Energy (2014), http://dx.doi.org/10.1016/

i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n en e r g y x x x ( 2 0 1 4 ) 1e98
thereby showing its potential as an alternative to TCOs for
solar window, therefore it can be exploited as base materials
for optoelectronic devices. Overall, first principles in-
vestigations reveal that Mg2FeH6 hydride, is one of the most
promising material among H-storage complex 3d-transition
metal hydrides. Further investigations at all levels with novel
experimental researchwork aswell as theoretical calculations
for establishing its structure-to-property relation in order to
expose its practical potential for hydrogen energy as well as in
optoelectronics, are of great importance and need to be
accomplished.
Acknowledgements
Authors from Universiti Teknologi Malaysia would like to
thank the financial support from the Ministry of Higher Edu-
cation (MOHE) Malaysia/Universiti Teknologi Malaysia
(UTM) of this research work through grant number
Q.J13000.7126.00J33. Moreover great thanks to the research
computing service at KAUST for the access to computational
resources.
r e f e r e n c e s
[1] Zuttel A. Materials for hydrogen storage. Mater Today2003;6(9):24e33.
[2] Dornheim M, Doppiu S, Barkhordarian G, Boesenberg U,Klassen T, Gutfleisch O, et al. Hydrogen storage inmagnesium-based hydrides and hydride composites. ScrMater 2007;56(10):841e6.
[3] Orimo S, Fujii H. Materials science of MgeNi-based newhydrides. Appl Phys A 2001;72(2):167e86.
[4] Zhang X, Yang R, Qu J, Zhao W, Xie L, Tian W, et al. Thesynthesis and hydrogen storage properties of purenanostructured Mg2FeH6. Nanotechnology 2010;21(9):095706.
[5] Huot J, Pelletier J, Liang G, Sutton M, Schulz R. Structure ofnanocomposite metal hydrides. J Alloys Compd2002;330:727e31.
[6] Bobet J, Akiba E, Nakamura Y, Darriet B. Study of Mg-M(M¼Co, Ni and Fe) mixture elaborated by reactivemechanical alloying hydrogen sorption properties. Int JHydrogen Energy 2000;25(10):987e96.
[7] Zarshenas M, Ahmed R, Kanoun MB, Ul Haq B, Mat Isa AR,Goumri-Said S. First principle investigations of the physicalproperties of hydrogen-rich MgH2. Phys Scr 2013;88:065704.
[8] Zuttel A, Wenger P, Rentsch S, Sudan P, Mauron P,Emmenegger C. LiBH4 a new hydrogen storage material. JPower Sources 2003;118(1):1e7.
[9] Deng S, Xiao X, Han L, Li Y, Li S, Ge H, et al. Hydrogen storageperformance of 5LiBH4þMg2FeH6 composite system. Int JHydrogen Energy 2012;37(8):6733e40.
[10] Didisheim J, Zolliker P, Yvon K, Fischer P, Schefer J,Gubelmann M, et al. Dimagnesium iron (II) hydride, Mg2FeH6,containing octahedral FeH6-anions. Inorg Chem1984;23(13):1953e7.
[11] Ivanov E, Konstanchuk I, Stepanov A, Boldyrev V.Magnesium mechanical alloys for hydrogen storage. J LessCommon Met 1987;131(1):25e9.
[12] Huot J, Boily S, Akiba E, Schulz R. Direct synthesis of Mg2FeH6
by mechanical alloying. J Alloys Compd 1998;280(1):306e9.
Please cite this article in press as: Ul Haq B, et al., Hybrid funComplex dimagnesium iron hydride, International Journaj.ijhydene.2014.04.014
[13] Castro F, Gennari F. Effect of the nature of the startingmaterials on the formation of Mg2FeH6. J Alloys Compd2004;375(1):292e6.
[14] Huot J, Hayakawa H, Akiba E. Preparation of the hydridesMg2FeH6 and Mg2CoH5 by mechanical alloying followed bysintering. J Alloys Compd 1997;248(1):164e7.
[15] Li X, Chiba A, Takahashi S. Preparation and magneticproperties of ultrafine particles of Fe-Ni alloys. J Magn MagnMater 1997;170(3):339e45.
[16] Li S, Varin R, Morozova O, Khomenko T. Controlledmechano-chemical synthesis of nanostructured ternarycomplex hydride Mg2FeH6 under low-energy impact modewith and without pre-milling. J Alloys Compd2004;384(1):231e48.
[17] Sai Raman S, Davidson D, Bobet J-L, Srivastava O.Investigations on the synthesis, structural andmicrostructural characterizations of Mg-based K 2PtCl 6 type(Mg2FeH6) hydrogen storage material prepared bymechanical alloying. J Alloys Compd 2002;333(1):282e90.
[18] Herrich M, Ismail N, Lyubina J, Handstein A, Pratt A,Gutfleisch O. Synthesis and decomposition of Mg2FeH6
prepared by reactive milling. Mater Sci Eng B2004;108(1):28e32.
[19] Gennari F, Castro F, Andrade Gamboa J. Synthesis ofMg2FeH6 by reactive mechanical alloying: formation anddecomposition properties. J Alloys Compd2002;339(1):261e7.
[20] Polanski M, Nielsen TK, Cerenius Y, Bystrzycki J, Jensen TR.Synthesis and decomposition mechanisms of Mg2FeH6
studied by in-situ synchrotron X-ray diffraction and high-pressure DSC. Int J Hydrogen Energy 2010;35(8):3578e82.
[21] Li Q, Liu J, Chou K-C, Lin G-W, Xu K-D. Synthesis anddehydrogenation behavior of MgeFeeH system preparedunder an external magnetic field. J Alloys Compd2008;466(1):146e52.
[22] Zaidi W, Bonnet J-P, Zhang J, Cuevas F, Latroche M,Couillaud S, et al. Reactivity of complex hydrides Mg2FeH6,Mg2CoH5 and Mg2NiH4 with lithium ion: far from equilibriumelectrochemically driven conversion reactions. Int JHydrogen Energy 2013;38(11):4798e808.
[23] Polanski M, Płoci�nski T, Kunce I, Bystrzycki J. Dynamicsynthesis of ternary Mg2FeH6. Int J Hydrogen Energy2010;35(3):1257e66.
[24] Wang Y, Cheng F, Li C, Tao Z, Chen J. Preparation andcharacterization of nanocrystalline Mg2FeH6. J Alloys Compd2010;508(2):554e8.
[25] Danaie M, Asselli AAC, Huot J, Botton GA. Formation ofthe ternary complex hydride Mg2FeH6 from magnesiumhydride (b-MgH2) and iron: an electron microscopy andenergy-loss spectroscopy study. J Phys Chem C2012;116(49):25701e14.
[26] Retuerto M, Sanchez-Benıtez J, Rodrıguez-Canas E,Serafini D, Alonso J. High-pressure synthesis of Mg2FeH6
complex hydride. Int J Hydrogen Energy 2010;35(15):7835e41.[27] Zhang J, Zhou D, Peng P, Liu J. First-principles investigation
of Mg2THy (T¼Ni, Co, Fe) complex hydrides. Phys B CondensMatter 2008;403(23):4217e23.
[28] Halilov S, Singh D, Gupta M, Gupta R. Stability and electronicstructure of the complex K2 PtCl2-structure hydrides DMH6(D¼Mg, Ca, Sr; M¼ Fe, Ru, Os). Phys Rev B2004;70(19):195117.
[29] Zareii S, Sarhaddi R. Structural, electronic properties andheat of formation of Mg2FeH6 complex hydride: an ab initiostudy. Phys Scr 2012;86(1):015701.
[30] Zhou D-W, Peng P, Liu JS, Chen L, Hu Y-J. First-principlesstudy on structural stability of 3d transition metal alloyingmagnesium hydride. Trans Nonferrous Met Soc China2006;16(1):23e32.
ctional calculations of potential hydrogen storage material:l of Hydrogen Energy (2014), http://dx.doi.org/10.1016/

i n t e rn a t i o n a l j o u rn a l o f h y d r o g e n en e r g y x x x ( 2 0 1 4 ) 1e9 9
[31] Song Y, Zhang W, Yang R. Stability and bonding mechanismof ternary (Mg, Fe, Ni)H2 hydrides from first principlescalculations. Int J Hydrogen Energy 2009;34(3):1389e98.
[32] Zhou H, Yu Y, Zhang H, Gao T. Structural, vibrational andthermodynamic properties of Mg2FeH6 complex hydride. EurPhys J B 2011;79(3):283e8.
[33] Orgaz E, Gupta M. The electronic properties of intermetallichydrides with the K2PtCl6 structure. J Phys Condens Matter1993;5(36):6697.
[34] Orgaz E, Aburto A. Electronic structure of ternary ruthenium-based hydrides. J Phys Chem C 2008;112(39):15586e94.
[35] Zhang J, Huang Y, Long C, Zhou D, Liu J. Density functionalstudy of Mg2FeH6 complex hydride. Mater Sci Poland2010;28(1).
[36] Blochl PE. Projector augmented-wave method. Phys Rev B1994;50(24):17953.
[37] Kresse G, Joubert D. From ultrasoft pseudopotentials to theprojector augmented-wave method. Phys Rev B1999;59(3):1758.
[38] Kresse G, Hafner J. Ab initio molecular dynamics for liquidmetals. Phys Rev B 1993;47(1):558.
[39] Kresse G, Furthmuller J. Efficient iterative schemes for abinitio total-energy calculations using a plane-wave basis set.Phys Rev B 1996;54(16):11169.
[40] Perdew JP, Burke K, Ernzerhof M. Generalizedgradient approximation made simple. Phys Rev Lett1996;77(18):3865.
[41] Heyd J, Scuseria GE, Ernzerhof M. Hybrid functionals basedon a screened Coulomb potential. J Chem Phys2003;118:8207.. J Chem Phys 2006;124:219906.
[42] Ernzerhof M, Scuseria GE. Assessment of the Perdewe BurkeeErnzerhof exchange-correlation functional. J Chem Phys1999;110:5029e36.
[43] Monkhorst HJ, Pack JD. Special points for Brillouin-zoneintegrations. Phys Rev B 1976;13(12):5188e92.
[44] Born M, Huang K. Dynamical theory and experiments I.Berlin: Springer Verlag Publishers; 1982.
[45] Hill R. Proc Phys Soc Lond A 1952;65:349.[46] Wang B-T, Zhang P, Song H, Shi H, Li D, Li W-D. Structural,
mechanical, thermodynamic, and electronic properties ofthorium hydrides from first-principles. J Nucl Mater2010;401(1):124e9.
Please cite this article in press as: Ul Haq B, et al., Hybrid funComplex dimagnesium iron hydride, International Journaj.ijhydene.2014.04.014
[47] Blomqvist J, Olofsson J, Alvarez A-M, Bjerken C. Structureand thermodynamical properties of zirconium hydridesfrom first-principle; 2012. arXiv preprint arXiv:1211.0858.
[48] Pugh SF. Relations between the elastic moduli and the plasticproperties of polycrystalline pure metals. Philos Mag1953;45:823.
[49] Varshney D, Shriya S. Elastic, mechanical andthermodynamic properties at high pressures andtemperatures of transition metal monocarbides. J RefractMetals Hard Mater 2013;41:375e401.
[50] Varshney D, Shriya S. Pressure and temperature dependentelastic, mechanical and thermodynamical properties ofnuclear fuel: UO2 and UN2. J Nucl Mater 2013;440:344e65.
[51] Kanoun MB, Goumri-Said S, Reshak AH. Theoretical study ofmechanical, electronic, chemical bonding and opticalproperties of Ti2SnC, Zr2SnC, Hf2SnC and Nb2SnC. ComputMater Sci 2009;47:491e500.
[52] Matsushita Y-I, Nakamura K, Oshiyama A. Comparativestudy of hybrid functionals applied to structural andelectronic properties of semiconductors and insulators. PhysRev B 2011;84(075205).
[53] Maark TA, Hussain T, Ahuja R. Structural, electronic andthermodynamic properties of Al- and Si-doped a-, b-, and g-MgH2: density functional and hybrid density functionalcalculations. Int J Hydrogen Energy 2012;37:9112e22.
[54] Isidorsson J, Giebels IAME, Arwin H, Griessen R. Opticalproperties of MgH2 measured in situ by ellipsometry andspectrophotometry. Phys Rev B 2003;68:115112e24.
[55] Van Setten MJ, Popa VA, de Wijs GA, Brocks G. Electronicstructure and optical properties of lightweight metalhydrides. Phys Rev B 2007;75(035204).
[56] Hermet P, Goumri-Said S, Kanoun MB, Henrard L. First-Principles investigations of the physical properties ofmagnesium nitridoboride. J Phys Chem C2009;113:4997e5003.
[57] Segall M, Lindan PJ, Probert M, Pickard C, Hasnip P, Clark S,et al. First-principles simulation: ideas, illustrations and theCASTEP code. J Phys Condens Matter 2002;14(11):2717.
[58] Vanderbilt D. Soft self-consistent pseudopotentials in ageneralized eigenvalue formalism. Phys Rev B1990;41(11):7892.
ctional calculations of potential hydrogen storage material:l of Hydrogen Energy (2014), http://dx.doi.org/10.1016/





























