Embed Size (px)
Citation preview

Human myeloproliferative neoplasms:molecular mechanisms, diagnosis and
classification
Tony GreenCambridge Institute for Medical Research
University of Cambridge and Addenbrookes Hospital


1. Normal mammaryepithelial development
2. First driver mutation
3. Further driver mutationsand clonal expansions
4. Appearance of mostrecent common ancestor
5. Completion of lasttotal selective sweep
6. Final rate-limitingdriver mutation
7. Diagnosis
Nik-Zainal et al Cell 2012
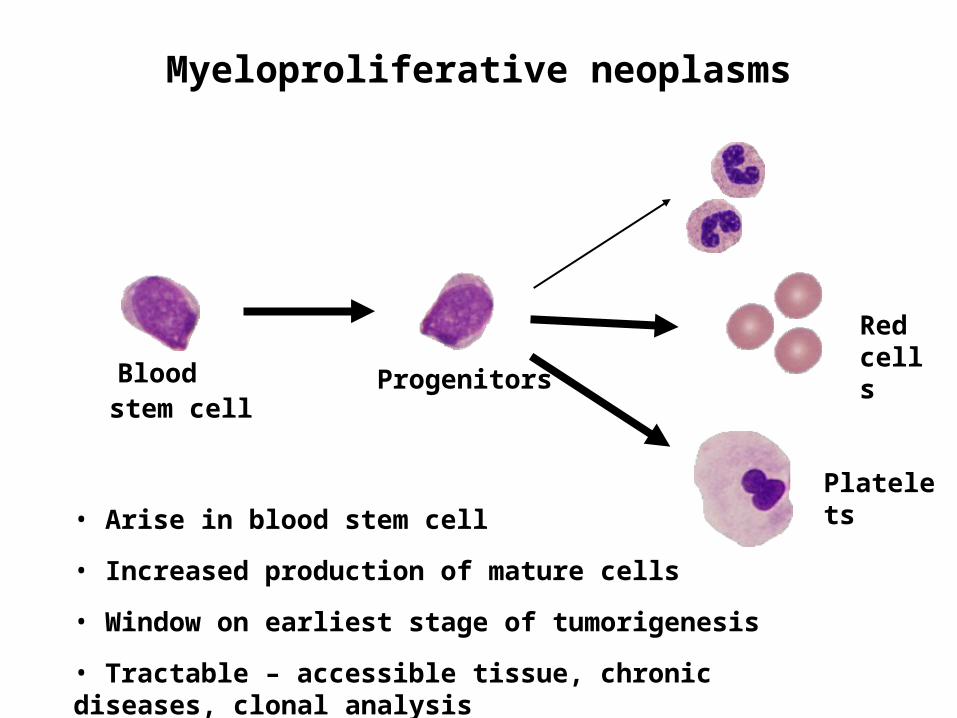
Myeloproliferative neoplasms
ProgenitorsBlood stem cell
Red cells
Platelets• Arise in blood stem cell
• Increased production of mature cells
• Window on earliest stage of tumorigenesis
• Tractable – accessible tissue, chronic diseases, clonal analysis

The BCR-ABL negative myeloproliferative neoplasms
Polycythaemia Vera (PV)
Essential thrombocythaemia
(ET)
Acute myeloid leukaemia
JAK2 V617F mutation
60%
95%

The JAK2 V617F mutation
James et al Nature 2005; Baxter et al Lancet 2005Levine et al Cancer Cell 2005; Kralovics et al NEJM 2005Scott et al NEJM 2007; Bercovich et al Lancet et al 2008
A T G T N T C T A T G T G T C T
Grans T cells
A T G T T T C T
Homozygous Mitoticrecombination
FERM SH2 JH2 Kinase
V617FExon 12
neg
JAK2
V617F mutation
Exon 16 mutations
Exon 12 mutations

“It all starts to make sense”
PI3K
pSTAT5
MAPK
P P
P P
PI3K
pSTAT5
MAPK
PP
P P
EPORTPOR
JAK2

Diagnosis
Appearance of mostrecent common ancestor

Rapid direct clinical impact
Identification of JAK2
mutation
PT-1 trials
Specialist MPN clinic
Recognition of
new disease
subtypes
Molecular testing in regional
diagnostic service
Therapeutic
JAK2 inhibitors
Harrison et al NEJM 2005
Baxter et al Lancet 2005
Campbell et al Lancet 2005
Scott et al Blood 2006
Campbell et al Blood 2006a
Campbell and Green NEJM 2006
Campbell et al Blood 2006b
Scott et al NEJM 2007
Wilkins et al Blood 2008
Beer et al Blood 2008
Zhao et al NEJM 2008
Campbell et al JCO 2009
Beer et al Blood 2010
Chen et al Cancer Cell 2010
Godfrey et al Blood 2012
Selected Green lab translational papers since 2005
2005 2010

A 1 Red cell mass >25% above predicted or Hct >60% male,
>56% female
2 No cause for 2o erythrocytosis
3 Palpable splenomegaly
4 Clonality marker
B 1 Platelet count >400 x109/l
2 Neutrophils >10 x109/L (>12.5 in smokers)
3 Splenomegaly on imaging
4 Endogenous erythroid colonies or reduced serum epo
A1 + A2 + any other A establishes diagnosis
A1 + A2 + two of B establish diagnosis
Old criteria for diagnosis of PV

Diagnostic criteria 20012
1 Raised Hb
2 JAK2 mutation
3 No cause for 20 erythrocytosis - normal art O2
- serum epo not high
McMullin et al BCSH guidelines BJH 2005, 2007

Idiopathic erythrocytosis or PV?
Scott et al NEJM 2007
FERM SH2 JH2 Kinase
V617FExon 12wt ATGGTGTTTCACAAAATCAGAAAT M V F H K I R N
mut ATGGTGTTTCAATTATTAATCAGAAAT M V F Q LQ L I R N
granulocytes
erythroid colony

PV variant with JAK2 exon 12 mutations
Glycophorin A
Scott et al NEJM 2007; Percy et al Haematologica 2007; Boyd et al BJH 2010
H&E
Multiple mutations
Isolated erythrocytosis
High Resolution Melt Analysis
granulocytes
erythroid colony
Can be low level in blood

Diagnosis of ET: JAK2 or MPL mutation-positive
WHO 2008
Sustained platelet count >450
Acquired mutation (JAK2 or MPL)
No other myeloid malignancy
Typical bone marrow appearances
(reticulin ≤ grade 2)
BCSH guidelines 2009
Sustained platelet count >450
Acquired mutation (JAK2 or MPL)
No other myeloid malignancy
BUT in absence of mutation still need to exclude reactive causes

Distinguishing different MPNs
ET PV PMF
Beer et al Blood 2011
diagnose and
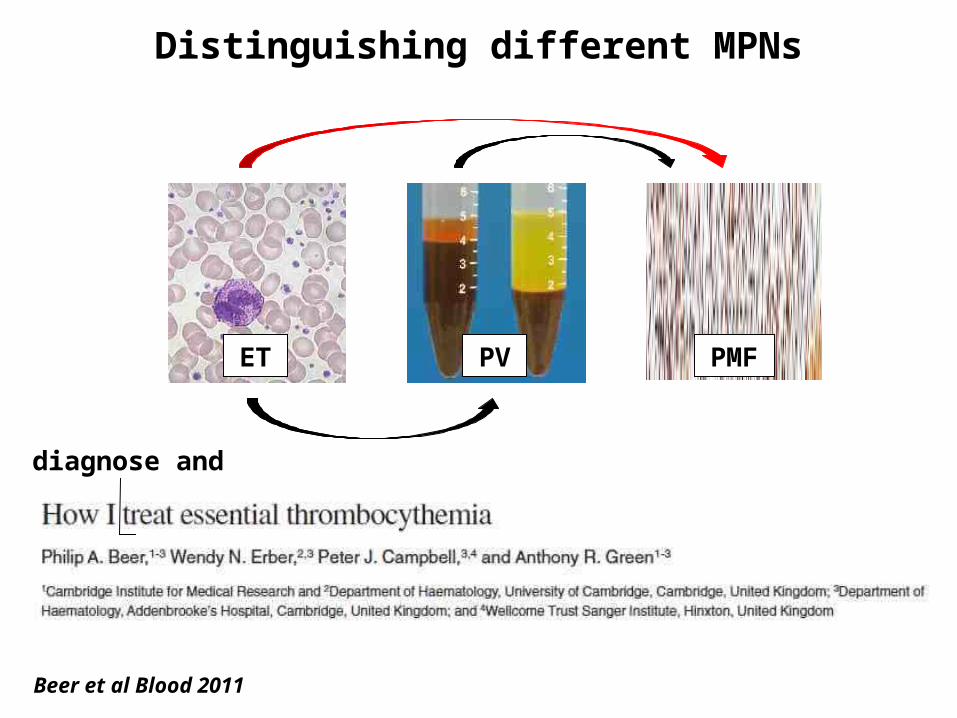
Distinguishing different MPNs
ET PV PMF
Beer et al Blood 2011
diagnose and
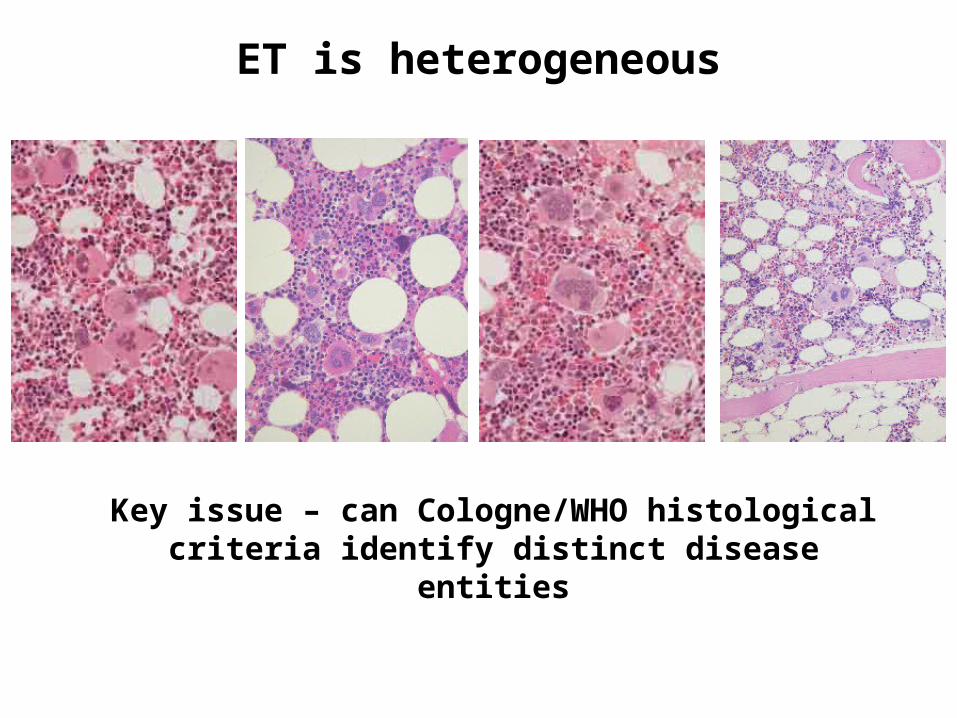
ET is heterogeneous
Key issue – can Cologne/WHO histological criteria identify distinct disease entities

WHO book is like a country – with good aspects…

….. but some aspects less good

….. but some aspects less good

Three questions
• Is prefibrotic PMF really distinct
• Can WHO criteria be applied reproducibly
• Is prefibrotic MF a useful concept
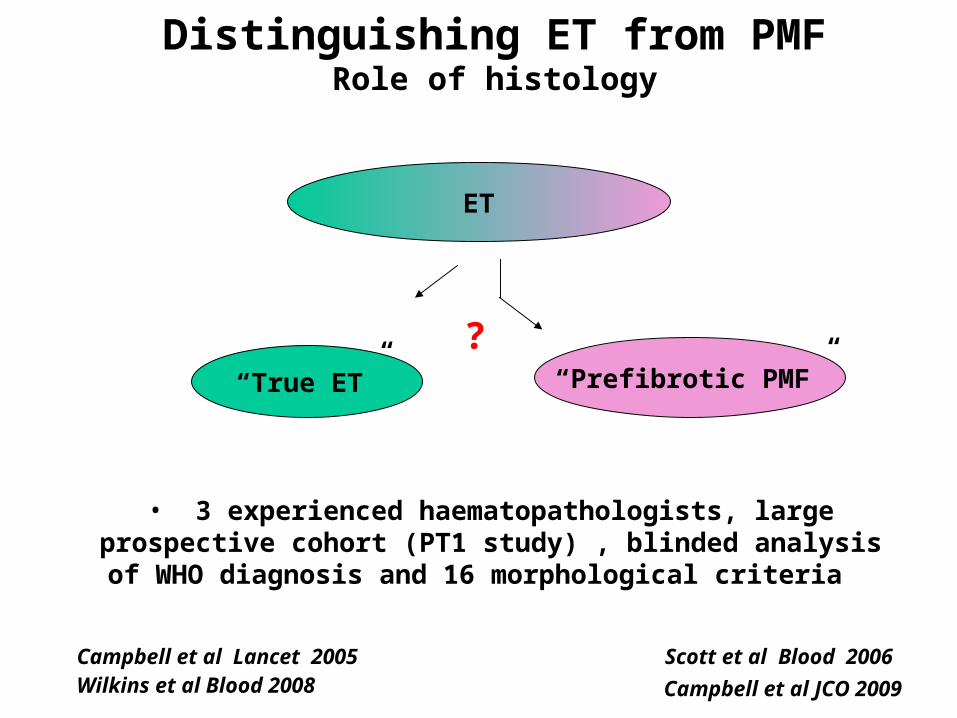
ET
Campbell et al Lancet 2005
Scott et al Blood 2006
Distinguishing ET from PMFRole of histology
“True ET” “Prefibrotic PMF”
Wilkins et al Blood 2008 Campbell et al JCO 2009
?
• 3 experienced haematopathologists, large prospective cohort (PT1 study) , blinded analysis of
WHO diagnosis and 16 morphological criteria

Even experienced haematopathologists don’t agree on what to call
“true ET” & “prefibrotic PMF”
Two alternative explanations:
- ‘Prefibrotic PMF’ not a distinct entity
- ‘Prefibrotic PMF’ exists but special training required
- even if true, questionable utility of criteria
the application of which is so difficult even
for highly experienced pathologists
Conclusions

Utility of prefibrotic PMF criteria (WHO 2008)
Thiele et al Blood 2011
Barbui, Thiele et al JCO 2011
Brousseau et al Histopathology 2010 “Distinction between ET and prefibrotic
PMF is of questionable clinical relevance”
Buhr, Kreipe et al Haematologica 2012 > 50% no agreement or unclassifiable -
“WHO criteria for discriminating ET from prefibrotic PMF are poorly to only moderately reproducible”
YES:
NO:
Concordance 73-88%

Barbui, Thiele et al JCO 2011
• Limitation of consensus approach to histology
• MF progression at 15 yrs: 10% vs 17% true ET vs prefib
MF • Leuk progression at 15 yrs 2% vs 11% - but no mention of therapy
• Even if real difference – prefibrotic PMF likely to represent later stage of same disease process

Three questions
• Is prefibrotic PMF really distinct
• Can WHO criteria be applied reproducibly
• Is prefibrotic PMF a useful concept
? later stage
disease
NO
Nomenclature of “prefibrotic PMF” is flawed
“True ET”
PV PMF
Prefibrotic PMF?
Nomenclature of “prefibrotic PMF” is flawed
ET PV PMF
Prefibrotic PMF?
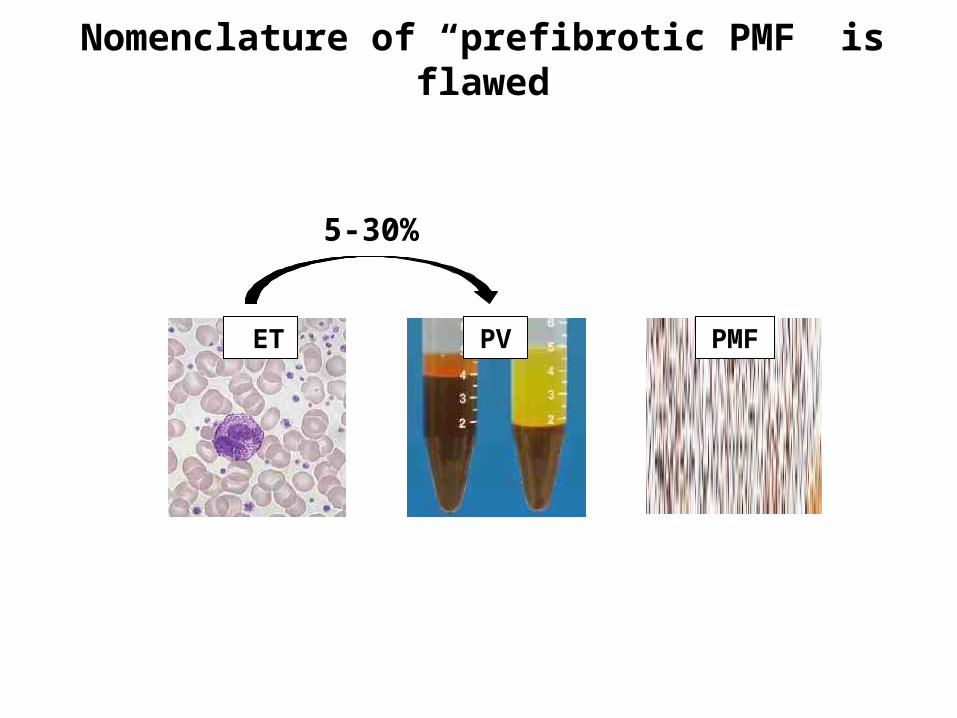
Nomenclature of “prefibrotic PMF” is flawed
ET PV PMF
5-30%

Nomenclature of “prefibrotic PMF” is flawed
ET PV PMF
Prepolycythaemic PV?

Concept of “prefibrotic PMF” is also potentially dangerous
For individual patient management - inappropriate therapy (eg BMT) for low risk patients
For the MPN field - patient cohorts will not be comparable

Three questions
• Is prefibrotic PMF really distinct
• Can WHO criteria be applied reproducibly
• Is prefibrotic PMF a useful concept
? later stage disease
NO
FlawedDangerous

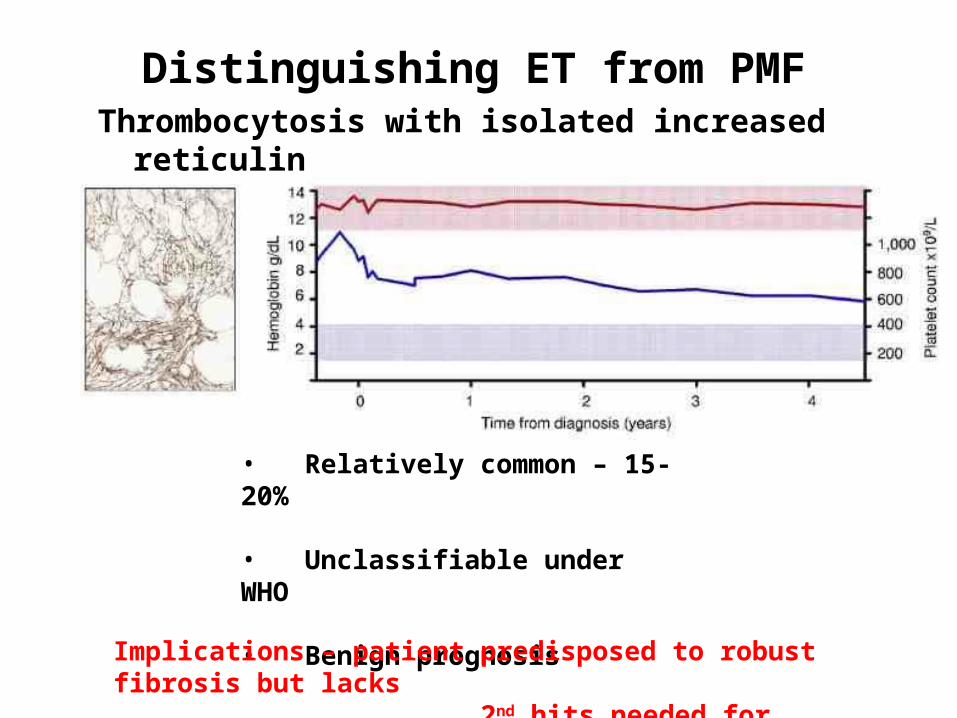
Thrombocytosis with isolated increased reticulin
Distinguishing ET from PMF
• Relatively common – 15-20%
• Unclassifiable under WHO
• Benign prognosisImplications – patient predisposed to robust fibrosis but lacks 2nd hits needed for evolution of clinical disease

Distinguishing ET from PMFPMF as presentation in accelerated phase
of pre-existing MPN
• PMF and MF transformation
indistinguishable
• PMF patients may have prior thrombocytosis
• PMF exhibits features of late stage disease - high clonal burden – more cytogenetic abnormalities - increased progression to AML

Campbell and Green NEJM 2006
Molecular classification of ET and PMF
Chronic phase
Heterogeneous
(mostly Pl)
Includes MF transformn
+ some PMF and atypical
CML
Accel phase
WBC retic dysplasia
Leuk phase
JAK2 MPL
Beer et al Blood 2011
Mutation load
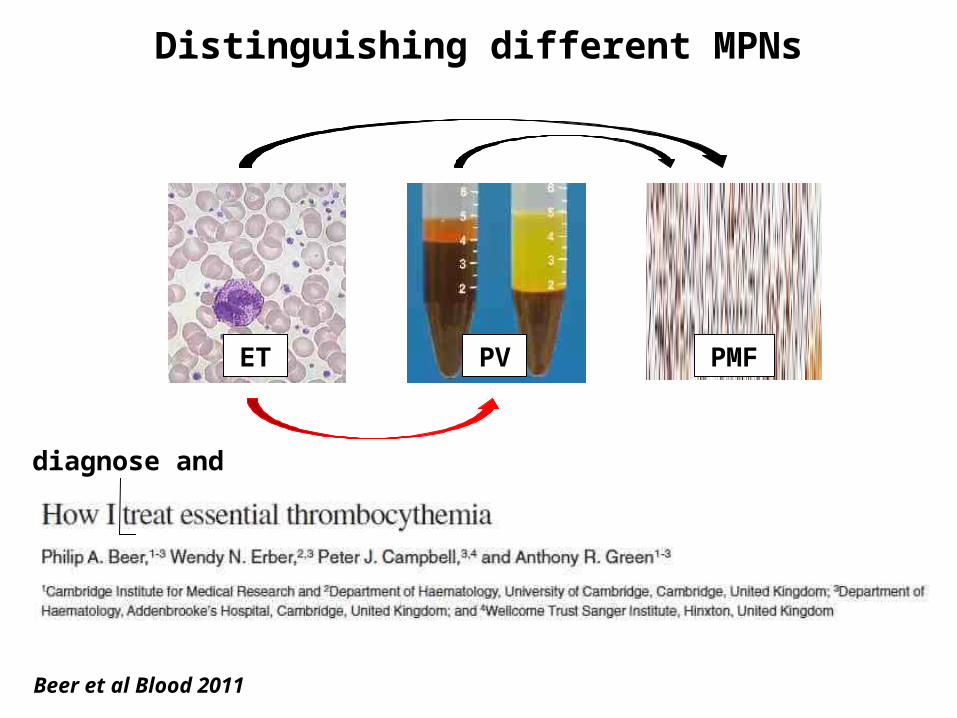
Distinguishing different MPNs
ET PV PMF
Beer et al Blood 2011
diagnose and

JAK2-positive ET is forme fruste of PV
809 ET patients in PT-1 trial
Campbell et al Lancet 2005 Scott et al Blood 2006
Distinguishing ET from PV
Higher Hb and WBCIncreased e’poiesis and g’poiesisMore venous thrombosisMore transformation to PV
JAK2 mut neg JAK2 mut pos
Campbell et al Lancet 2005
Harrison et al NEJM 2005
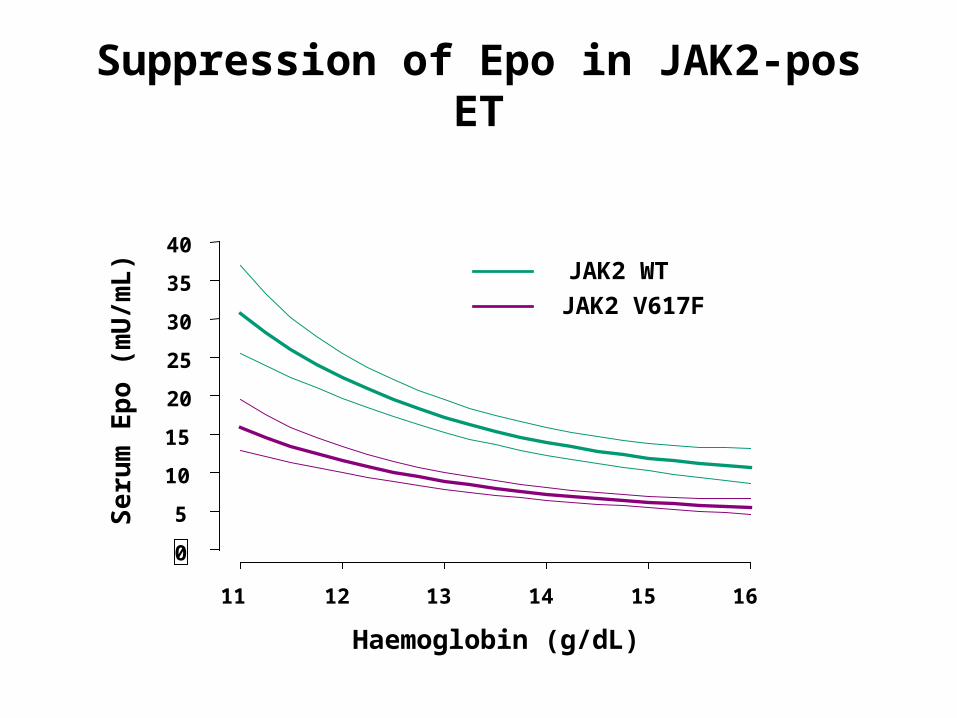
Suppression of Epo in JAK2-pos ET
Haemoglobin (g/dL)
Seru
m E
po (
mU
/mL)
0
5
10
15
20
25
30
35
40
11 12 13 14 15 16
JAK2 WT
JAK2 V617F
p<0.0001

Distinguishing ET from PV
N
14 16 18 20 Hb
Normal ET PV
Inherent problem in using continuous variable (eg Hct or RCM) to make a binary distinction

Polycythaemia vera
Essential thrombocythaemia
Mitotic recombination
Rare Common
One mutation but two diseasesOne mutation but two diseases
Hypothesis – homozygosity for JAK2 mutation causes PV phenotype
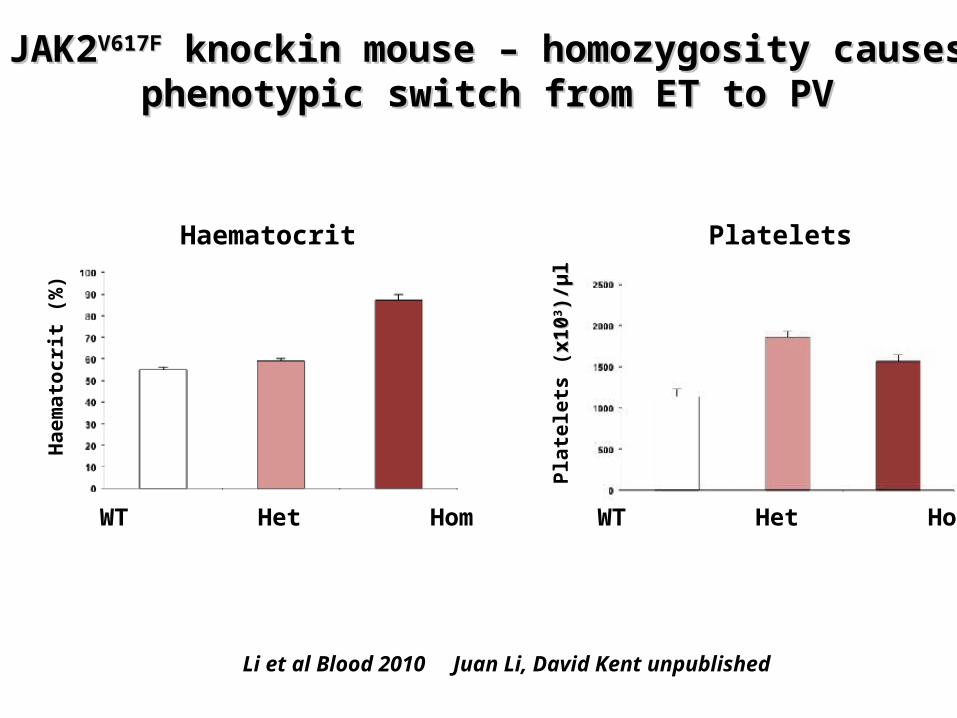
JAK2JAK2V617FV617F knockin mouse knockin mouse – homozygosity – homozygosity causes phenotypic switch from ET to PVcauses phenotypic switch from ET to PV
Li et al Blood 2010 Juan Li, David Kent unpublished
Pla
tele
ts (
x1
0x
1033 )
/)/µµ
ll
Ha
em
ato
cri
t (%
)
WT Het Hom WT Het Hom
Haematocrit Platelets
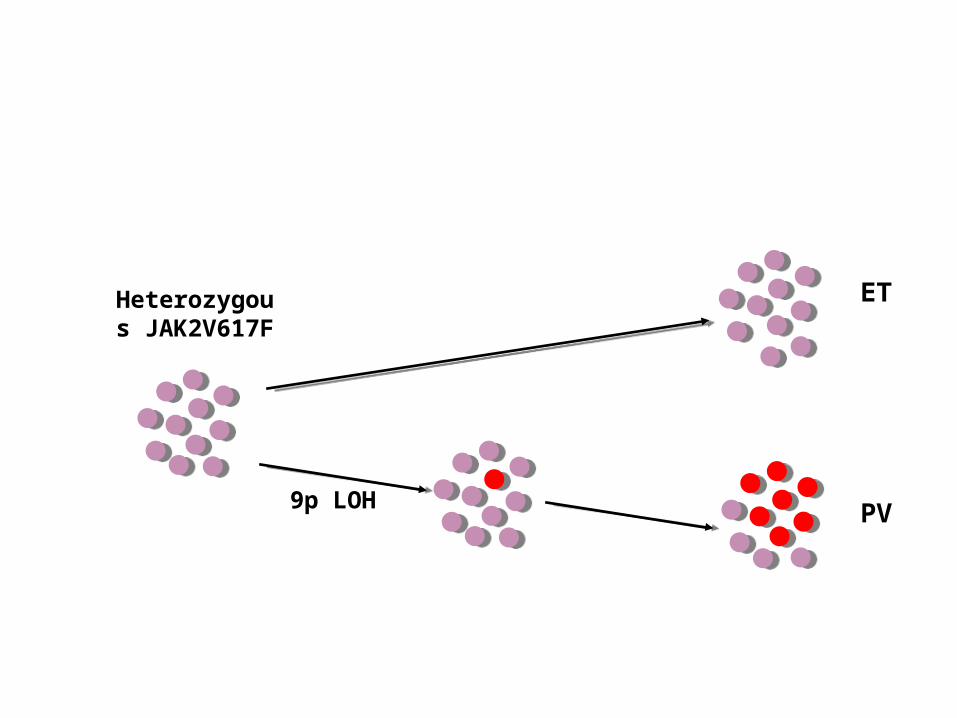
Heterozygous
JAK2V617F
9p LOH
ET
PV

Higher number of homozygous colonies in PV patients compared to ET
- homozygous clone has selective advantage in PV but not ET
- recurrent acquisition of homozygosity
PV 80% ET 52%
Homozygous mutant BFU-EHomozygous mutant BFU-E present in many patients with ETpresent in many patients with ET
Godfrey et al Blood 2012

44 5 4
Mb from telomere
4.0 D9S2884.8 D9S18106.2 D9S1852
14.8D9S235
18.3D9S925
19.7D9S162
27.6 D9S161
36.4D9S1791
38.3D9S2148
30.9 D9S43
33.9 D9S1817
102.1 D9S176
Cen
Tel
JAK2
HeterozygousLOH
125 130 135 125 130 135 125 130 135
A CB170 180 170 180 170 180
Recurrent acquisition of 9p LOH in patient with PV
Number of colonies
A B C

• Recurrent acquisition of homozygosity - in 5/8 PV patients and 2/2 ET patients tested - resolution limited (2.3 to 14.2 Mb) so number of
distinct clones may be underestimate
• PV distinguished from ET by presence of dominant homozygous clone ~10 fold larger than minor clones
- ? additional lesions
• Multiple clones arise in HSPC compartment - persist over time - detectable in sorted CMPs
Summary

Heterozygous
JAK2V617F
Recurrent 9p LOH
ET
PV
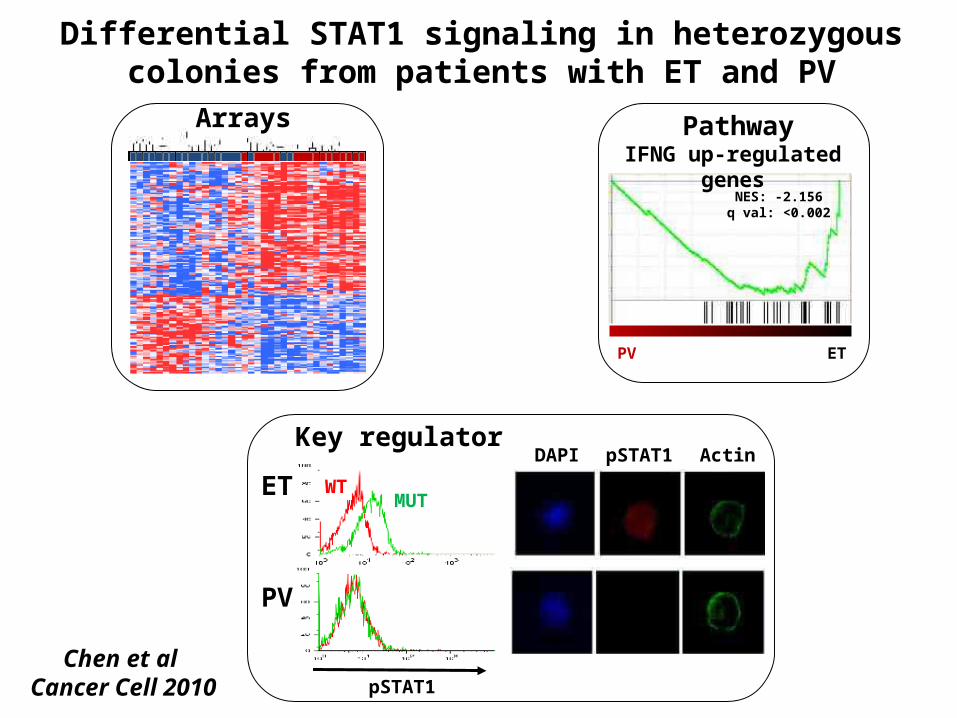
IFNG up-regulated genes
NES: -2.156q val: <0.002
PV ET
PV
MUTWTET
pSTAT1
DAPI pSTAT1 Actin
Differential STAT1 signaling in heterozygous colonies from patients with ET and PV
Arrays Pathway
Key regulator
Chen et al Cancer Cell 2010

Polycythaemia vera
Essential thrombocythaemia
JAK2 V617F JAK2 V617F
STAT1 defect
JAK2 homozyg
Clonal expansion

• Unexpected complexity in early phase of “simple” malignancy
• Questions - how does clone expand given HSC defect - what drives recurrent mitotic recombination - what drives expansion of dominant homozygous
clone in PV - cause and effects of pSTAT1 defect in heterozygous
PV cells
• Exomes coming
Conclusions and questions

AcknowledgementsSanger Institute
Peter Campbell, Mike StrattonAndy Futreal, Elli Papaemmanuil
Cambridge UniversityAnne Ferguson-Smith
Carol Edwards
Nick CrossAmy Jones
Claire Harrison
Mary-Frances McMullin
Adam MeadSten-Eirik Jacobsen
Alessandro Vannucchi
Eva Hellstom
Ghulam Mufti
Jean Jacques Kiladjian
Green labMaria AhnAthar AzizPhilip BeerEdwin ChenJyoti Evans
Anna GodfreyTina Hamilton
David KentJuan Li
Steve LoughranCharlie Massie
June ParkDean Pask
Yvonne SilberRachel Sneade
Addenbrookes/BRCMike Scott
Joanna Baxter/Anthony Bench
MPD clinic, TRL team
NCRI MPN Study GroupPT-1 trial team
The myeloproliferative neoplasms
PrimaryMyelofibrosis
Polycythaemia Vera (PV)
Essential thrombo- cythaemia (ET)
Acute myeloid leukaemiaThrombosis
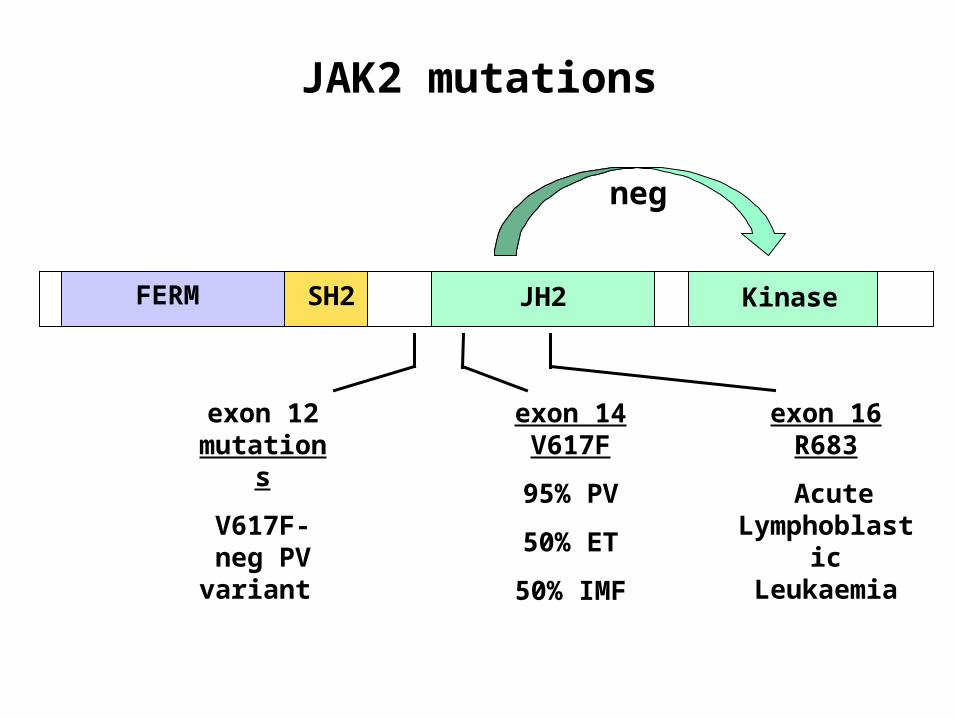
JAK2 mutations
FERM SH2 JH2 Kinase
exon 14
V617F
95% PV
50% ET
50% IMF
neg
exon 12 mutation
s
V617F-neg PV variant
exon 16 R683
Acute Lymphobla
stic Leukaemia

Difference between mutation pos ET and PV
JAK2Mutation
Normal Hb
Erythrocytosis
Depletediron stores
Genetic modifiers
Gender
Campbell et al Lancet 2005
Acquired mutations
Scott et al NEJM 2007


Nuclear JAK2 signaling directly regulates chromatin structure
JAK2 is a histone H3 kinase
H3Y41
H3Y54 H3Y99Dawson et al Nature
2009Collab with Kouzarides
lab
JAK2 signaling to chromatin mediates ES cell self-renewal
Griffiths et al Nat Cell Biol 2011
Collab with Gottgens lab
H3Y41
P H3Y41
P Nanog





























