Embed Size (px)
Citation preview

How I Treat Series
ACQUIRED HEMOLYTIC ANEMIA
How I treat warm autoimmune hemolytic anemiaWilma Barcellini1,* and Bruno Fattizzo1,2,*
1Hematology, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy; and 2Department ofOncology and Onco-hematology, University of Milan, Milan, Italy
Warm autoimmune hemolytic anemia (wAIHA) is caused by increased erythrocyte destruction by immunoglobulin G(IgG) autoantibodies, with or without complement activation. Antibody-dependent cell-mediated cytotoxicity bymacrophages/activated lymphocytes occurs in the lymphoid organs and spleen (extravascular hemolysis). The ability ofthe bonemarrow (BM) to compensate determines clinical severity. The different pathogenicmechanisms, their complexinterplay, and changes over time may explain wAIHA’s great clinical heterogeneity and unpredictable course. Thedisease may be primary, drug induced, or associated with lymphoproliferative neoplasms, autoimmune and infectiousdiseases, immunodeficiencies, solid tumors, or transplants. Therapeutic interventions include steroids, splenectomy,immunosuppressants, and rituximab; the latter is increasingly used in steroid-refractory cases based on evidence fromthe literature and a few prospective trials. We present 5 patient case studies highlighting important issues: (1) thediagnosis and proper use of steroid therapy, (2) the concerns about the choice between rituximab and splenectomy insecond-line treatment, (3) the need of periodical re-evaluation of the disease to assess the possible evolution ofrelapsed/refractory cases in myelodysplastic and BM failure syndromes, and (4) the difficulties in managing cases ofsevere/acute disease that are at high risk of relapse. Incorporating novel targeted therapies into clinical practice will bean exciting challenge in the future. (Blood. 2021;137(10):1283-1294)
IntroductionWarm autoimmune hemolytic anemia (wAIHA) is the mostprevalent form of autoimmune hemolytic anemia (AIHA), ac-counting for 60% to 70% of all cases. It is usually due to an im-munoglobulin G (IgG) autoantibody that may activate complement(C) if present at high titer or if IgG1 and IgG3 subclasses areprevalent. The cornerstone of diagnosis is the direct antiglobulintest (DAT), which may be positive with anti-IgG antisera (70% of allwAIHA) or anti-IgG plus C at low titer. Antibody-dependent cell-mediated cytotoxicity (ADCC) is the main driver of red blood cell(RBC) destruction, occurring in the lymphoid organs and spleen.Additional pathophysiologic mechanisms include autoreactivecellular effectors (T cells, macrophages), unbalanced CD41 regu-latory T cells and cytokines, and,most importantly, the ability of thebone marrow (BM) to compensate. All of these factors, as well astheir complex interplay and changes over time, may explain thegreat clinical heterogeneity and unpredictable course of AIHA.1-3
Moreover, wAIHAmay be primary, or associated with a variety ofconditions, such as lymphoproliferative neoplasms, autoimmuneand infectious diseases, immunodeficiencies, solid tumors,or transplants, or with drugs, including the novel checkpointinhibitors.3,4 Therapies used to treat wAIHA are based on limitedevidence derived from expert opinion5-9 and a small number ofprospective trials.10-12 Over the last 2 decades, steroids, sple-nectomy, and immunosuppressants have been the mainstays oftreatment. More recently, rituximab has increasingly been used,and experimental therapies directed at complement, protea-somes, and various kinases are under development.9,13 Below,
we present 5 patient case studies that highlight important issuesregarding diagnosis and first-line steroid therapy, the choice ofsecond-line treatment, the potential for relapsed or refractory(R/R) cases of AIHA to evolve into myelodysplastic syndromes(MDSs) and BM failure (BMF), and difficulties in the managementof severe or acute wAIHA.
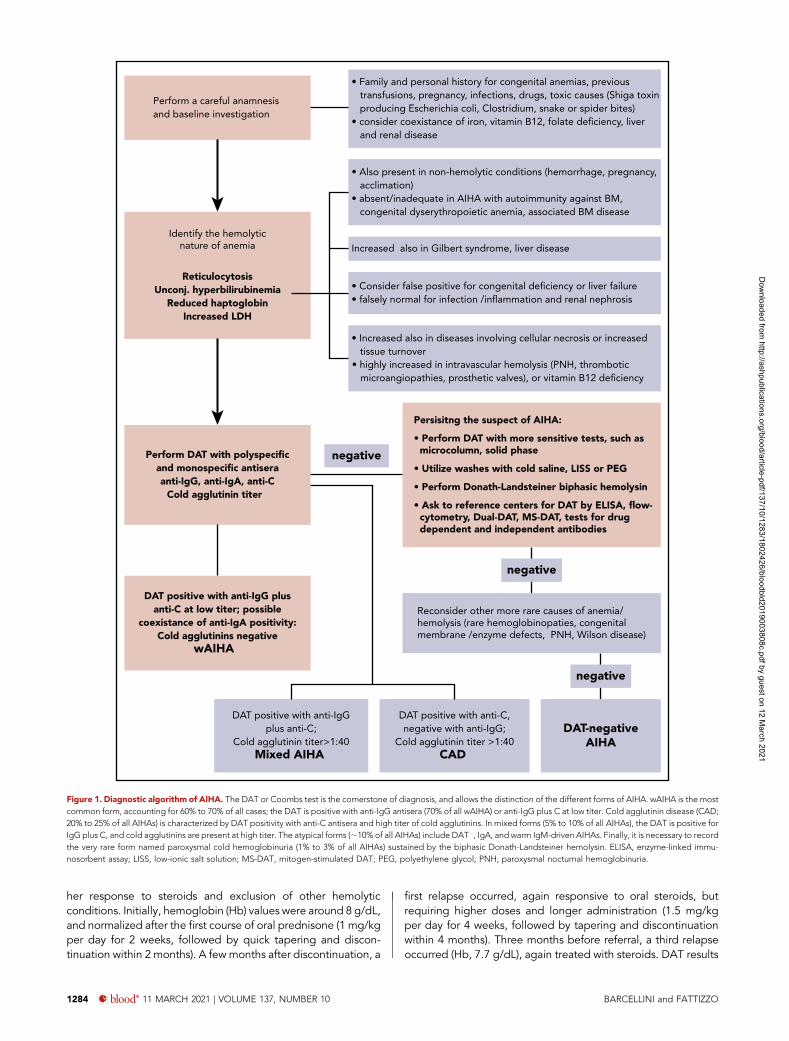
The pitfalls of diagnosis and first-linesteroid therapyThe DAT is the cornerstone of AIHA diagnosis. However, 5% to10% of patients with clear evidence of the disease may testnegative, despite extensive evaluation with methods moresensitive than the standard tube (microcolumn, solid phase,washings with cold or low-ionic salt solutions) (Figure 1).Moreover, it may be falsely positive in many healthy subjects,after some therapies (IV immunoglobulins, Rh immune globulins,antithymocyte globulins, daratumumab), in conditions withparaproteins or elevated serum globulins, or in recently trans-fused patients (alloantibodies), and may also remain positive inpatients with AIHA in remission.3,14,15 Thus, AIHA cannot bediagnosed from DAT positivity alone; there should be evidenceof hemolysis, and other congenital or acquired hemolytic dis-orders should be excluded in complex cases. Furthermore,confounding factors should always be considered (Figure 1).
Patient 1A 49-year-old woman was referred to our center with presumedDAT2 AIHA, present for 2 years, and diagnosed on the basis of
© 2021 by The American Society of Hematology blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 1283
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021
her response to steroids and exclusion of other hemolyticconditions. Initially, hemoglobin (Hb) values were around 8 g/dL,and normalized after the first course of oral prednisone (1 mg/kgper day for 2 weeks, followed by quick tapering and discon-tinuation within 2 months). A few months after discontinuation, a
first relapse occurred, again responsive to oral steroids, butrequiring higher doses and longer administration (1.5 mg/kgper day for 4 weeks, followed by tapering and discontinuationwithin 4 months). Three months before referral, a third relapseoccurred (Hb, 7.7 g/dL), again treated with steroids. DAT results
Identify the hemolyticnature of anemia
ReticulocytosisUnconj. hyperbilirubinemia
Reduced haptoglobinIncreased LDH
• Also present in non-hemolytic conditions (hemorrhage, pregnancy, acclimation)• absent/inadequate in AIHA with autoimmunity against BM, congenital dyserythropoietic anemia, associated BM disease
• Increased also in diseases involving cellular necrosis or increased tissue turnover • highly increased in intravascular hemolysis (PNH, thrombotic microangiopathies, prosthetic valves), or vitamin B12 deficiency
Perform a careful anamnesisand baseline investigation
Increased also in Gilbert syndrome, liver disease
• Consider false positive for congenital deficiency or liver failure• falsely normal for infection /inflammation and renal nephrosis
• Family and personal history for congenital anemias, previous transfusions, pregnancy, infections, drugs, toxic causes (Shiga toxin producing Escherichia coli, Clostridium, snake or spider bites)• consider coexistance of iron, vitamin B12, folate deficiency, liver and renal disease
Reconsider other more rare causes of anemia/hemolysis (rare hemoglobinopaties, congenitalmembrane /enzyme defects, PNH, Wilson disease)
DAT positive with anti-IgGplus anti-C;
Cold agglutinin titer>1:40Mixed AIHA
Perform DAT with polyspecificand monospecific antiseraanti-IgG, anti-IgA, anti-C
Cold agglutinin titer
Persisitng the suspect of AIHA:
• Perform DAT with more sensitive tests, such as microcolumn, solid phase
• Utilize washes with cold saline, LISS or PEG
• Perform Donath-Landsteiner biphasic hemolysin
• Ask to reference centers for DAT by ELISA, flow- cytometry, Dual-DAT, MS-DAT, tests for drug dependent and independent antibodies
negative
negative
DAT positive with anti-C,negative with anti-IgG;
Cold agglutinin titer >1:40CAD
DAT positive with anti-IgG plusanti-C at low titer; possible
coexistance of anti-IgA positivity:Cold agglutinins negative
wAIHA
DAT-negativeAIHA
negative
Figure 1. Diagnostic algorithm of AIHA. The DAT or Coombs test is the cornerstone of diagnosis, and allows the distinction of the different forms of AIHA. wAIHA is the mostcommon form, accounting for 60% to 70% of all cases; the DAT is positive with anti-IgG antisera (70% of all wAIHA) or anti-IgG plus C at low titer. Cold agglutinin disease (CAD;20% to 25% of all AIHAs) is characterized by DAT positivity with anti-C antisera and high titer of cold agglutinins. In mixed forms (5% to 10% of all AIHAs), the DAT is positive forIgG plus C, and cold agglutinins are present at high titer. The atypical forms (;10% of all AIHAs) include DAT2, IgA, and warm IgM-driven AIHAs. Finally, it is necessary to recordthe very rare form named paroxysmal cold hemoglobinuria (1% to 3% of all AIHAs) sustained by the biphasic Donath-Landsteiner hemolysin. ELISA, enzyme-linked immu-nosorbent assay; LISS, low-ionic salt solution; MS-DAT, mitogen-stimulated DAT; PEG, polyethylene glycol; PNH, paroxysmal nocturnal hemoglobinuria.
1284 blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 BARCELLINI and FATTIZZO
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021

were persistently negative. Upon referral, the patient was still ona high dose of steroids (0.7 mg/kg per day), with a partial re-sponse (Hb, 9.6 g/dL). Generalized Cushing habitus was pre-sent, with mild hyperglycemia and osteoporosis. The patientsought advice on a second-line therapy.
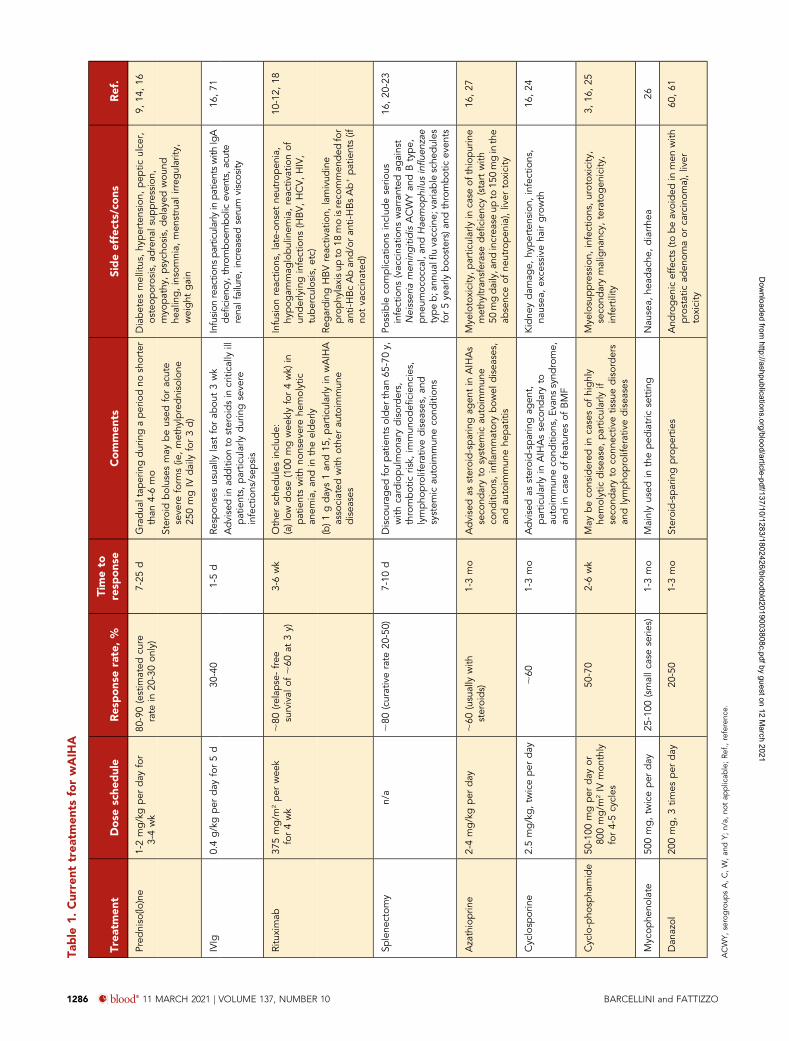
Commentary on patient 1It is well established that ;80% to 90% of wAIHA patients re-spond to a 3- to 4-week course of prednisone 1 to 2 mg/kgper day (Table 1). In steroid-responsive patients, the taper canbegin after 14 to 21 days, keeping in mind that relapse is morecommon if corticosteroids are tapered to#10 mg in,2 monthsand stopped in,6 months.9,14,16 Thus, it is important not only toconsider Hb values, but also to strictly monitor all hemolyticmarkers, and delay tapering if hemolysis is ongoing (Figure 2).
On the other hand, excessive and prolonged steroid therapy(.15 months) may result in significant side effects, particularlyosteoporosis, even at doses considered low by hematologists(prednisolone, ;7.5 mg per day for$3 months).17 Patient 1 hadnot received the recommended preventive actions (bone-mineral density screening, 800 IU of vitamin D daily, 700-1200 mgof daily calcium). Moreover, a quick tapering of steroids mayhave played some role in the first and second relapses. However,it is inappropriate to maintain a patient on steroids withoutconsidering second-line treatment; this cannot be fully justifiedby the uncertain diagnosis and persistently negative DAT.
Rituximab or splenectomy: whichsecond-line therapy for wAIHA?Rituximab is becoming the preferred second-line therapy forwAIHA, with an 80% overall response rate, a median responsetime of 3 to 6 weeks (with a range of 2 to 16 weeks), and asustained response rate of ;60% at 3 years (Table 1).18 It hasbeen thoroughly researched and has proven effective in primaryand most secondary forms of wAIHA, and retreatment appearssimilarly successful. Various doses have been used (375 mg/m2
per week for 4 weeks, most commonly; 100 mg weekly for4 weeks, mainly in nonsevere forms and in the elderly; 1 gon days 1 and 15, particularly in wAIHA associated with otherautoimmune diseases), with quite similar efficacy (response rate;80%, relapse-free survival of;60% at 3 years), although directcomparisons have not been made.9,10,19
An interesting approach is to use rituximab in first-line therapy: ina prospective randomized trial, rituximab with prednisoloneincreased the rate and duration of the response compared withprednisolone alone.11 Furthermore, a randomized double-blindtrial demonstrated the efficacy and safety of rituximab vs pla-cebo among newly diagnosed patients on standard prednisonetherapy.12
However, rituximab is not available worldwide, nor is it universallyavailable from public health services. Moreover, its significant sideeffects must be considered, particularly immunodeficiency (late-onset neutropenia, hypogammaglobulinemia) and reactivation ofunderlying infections (hepatitis B [HB] virus [HBV], hepatitis C [HCV],HIV, tuberculosis). Various tests (HB surface antigen [HBsAg], anti-HBs, anti-HBc, anti-HCV, anti-HIV, and quantiFERON-TB) are ad-visable before steroid therapy, but mandatory before rituximab.
Lamivudine prophylaxis for up to 18 months is recommendedfor anti-HBc and/or anti-HBs antibody-positive patients (if notvaccinated).
Splenectomy is as effective as rituximab, with somewhat longer-lasting remissions (Table 1).8,9,16,20-22 Use of splenectomy hasgradually declined, however, to ,10% of cases, mainly due tothe increased infection risk, particularly in the first year. This riskis not eliminated by vaccination and antibiotic prophylaxis.Notably, vaccination may result in a suboptimal response afterimmunosuppression, and is therefore recommended beforecommencing rituximab, in line with national guidelines.1,23
Patient 2A 39-year-old man with a 15-year history of wAIHA was referredto our clinic after his fifth relapse (Hb, 8.5 g/dL; lactate de-hydrogenase [LDH], 1.3 upper limit of normality [ULN]; reticu-locytes, 125 3 109/L; unconjugated bilirubin, 2.7 mg/dL; DAT,IgG1). He had been treated with steroids in various cycles andschedules, alone or in association with azathioprine, cyclo-phosphamide, or rituximab, and had repeatedly refused sple-nectomy. Upon referral, the patient sought an effective anddefinitive therapy. Poor compliance became apparent, pre-cluding his enrollment in trials of new drugs. After extensivediscussion, he agreed to undergo splenectomy, and pro-phylactic vaccination was performed. Laparoscopic splenectomywas converted to laparotomic, for surgical reasons, and wascomplicated by abdominal wall hematoma, but resulted in acomplete hematologic response. Four months after surgery, welearned that the patient died of overwhelming sepsis in a localhospital.
Commentary on patient 2The most feared complication of splenectomy is overwhelmingsepsis due to encapsulated bacteria, with a risk of 3% to 5% and amortality rate of up to 50%, even after the introduction ofvaccinations. The role and efficacy of antibiotic prophylaxis re-mains unclear, and not all experts recommend this approach.1,23
Given the patient’s poor compliance, continuous antibiotictherapy (although proposed) was probably not taken. Therefore,the patient’s level of education and awareness about compli-cations should always be strongly pursued.
One drawback of splenectomy is the lack of reliable predictors ofoutcome because its effectiveness has not been related todisease duration, response to steroids, or the extent of splenicsequestration. Splenectomy may be preferred in younger pa-tients, or in those who wish to become pregnant. Conversely, thefollowing factors make splenectomy inadvisable: age (.65-70years), cardiopulmonary disorder, previous history or seriousrisk of thrombosis, hepatitis C, underlying immunodeficiency,and lymphoproliferative and systemic autoimmune conditions.Moreover, splenectomy may be associated with surgical com-plications (pulmonary embolism, intra-abdominal bleeding,abdominal abscess, abdominal wall hematoma), although thelaparoscopic approach has reduced the risk comparedwith opensurgery.20,22
Unlike immune thrombocytopenia (ITP), in AIHA there is noconsensus on the timing of splenectomy, even if clinical prac-tice suggests a “neither too early nor too late” approach. Inpatient 2, long-standing immunosuppressionmay have hampered
HOW I TREAT WARM AUTOIMMUNE HEMOLYTIC ANEMIA blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 1285
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021
Table
1.Current
trea
tmen
tsforwAIH
A
Trea
tmen
tDose
sche
dule
Res
pons
erate,%
Timeto
resp
ons
eCommen
tsSideeffects/co
nsRef.
Pred
niso(lo
)ne
1-2mg/kgper
day
for
3-4wk
80-90(estim
ated
cure
rate
in20
-30on
ly)
7-25
dGradua
ltap
eringduringaperiodno
shorter
than
4-6mo
Diabetes
mellitus,hy
pertension,
pep
ticulce
r,osteop
orosis,ad
rena
lsup
pression,
myo
pathy
,psych
osis,delayed
wou
ndhe
aling,insomnia,
men
strual
irreg
ularity
,weight
gain
9,14
,16
Steroidboluses
may
beused
foracute
seve
reform
s(ie
,methy
lprednisolone
250mgIV
daily
for3d)
IVIg
0.4g/kgper
day
for5d
30-40
1-5d
Resp
onsesusua
llylast
forab
out3wk
Infusion
reactio
nspa
rticularlyinpa
tientswith
IgA
deficien
cy,throm
boem
bolic
even
ts,a
cute
rena
lfailure,increased
serum
viscosity
16,71
Advisedin
addition
tosteroidsin
critically
illpatients,
partic
ularly
duringseve
reinfections/sep
sis
Rituximab
375mg/m
2per
wee
kfor4wk
;80
(relapse-free
survival
of;60
at3y)
3-6wk
Other
sche
dules
includ
e:(a)low
dose(100
mgwee
klyfor4wk)
inpatientswith
nonsev
erehe
molytic
anem
ia,an
din
theelderly
Infusion
reactio
ns,late-onset
neutropen
ia,
hypog
ammag
lobulinem
ia,reactiv
ationof
underlyinginfections
(HBV,HCV,HIV,
tubercu
losis,
etc)
10-12,
18
(b)1
gdays1an
d15
,partic
ularly
inwAIHA
associated
with
othe
rau
toim
mun
edisea
ses
RegardingHBVreactiv
ation,
lamivud
ine
prophy
laxisup
to18
moisreco
mmen
ded
for
anti-HBcAban
d/oran
ti-HBsAb1patients(if
notvaccinated
)
Splene
ctom
yn/a
;80
(curativerate
20-50)
7-10
dDisco
urag
edforp
atientsolder
than
65-70y,
with
cardiopulmon
arydisorders,
thrombotic
risk,
immun
odefi
cien
cies,
lympho
proliferativedisea
ses,
and
system
icau
toim
mun
eco
ndition
s
Possible
complications
includ
eserio
usinfections
(vaccina
tions
warranted
against
Neisseria
men
ingitidis
ACWYan
dBtype,
pne
umoc
occal,an
dHae
mop
hilusinflue
nzae
typeb;ann
ualfluvaccine;
varia
ble
sche
dules
for5ye
arly
boo
sters)an
dthrombotic
even
ts
16,20
-23
Azathioprin
e2-4mg/kgper
day
;60
(usually
with
steroids)
1-3mo
Advisedas
steroid-sparingag
entin
AIHAs
seco
ndaryto
system
icau
toim
mun
eco
ndition
s,inflam
matorybow
eldisea
ses,
andau
toim
mun
ehe
patitis
Mye
lotoxicity,p
artic
ularlyin
case
ofthiopurine
methy
ltran
sferasedefi
cien
cy(startwith
50mgdaily,and
increa
seup
to15
0mginthe
absenc
eof
neutropen
ia),liver
toxicity
16,27
Cyclosp
orine
2.5mg/kg,tw
iceper
day
;60
1-3mo
Advisedas
steroid-sparingag
ent,
partic
ularly
inAIHAsseco
ndaryto
autoim
mun
eco
ndition
s,Ev
anssynd
rome,
andin
case
offeatures
ofBMF
Kidne
ydam
age,
hypertension,
infections,
nausea
,ex
cessiveha
irgrowth
16,24
Cyclo-pho
spha
mide
50-100
mgper
day
or80
0mg/m
2IV
mon
thly
for4-5cycles
50-70
2-6wk
May
beco
nsidered
incasesof
highly
hemolytic
disea
se,partic
ularly
ifseco
ndaryto
conn
ectiv
etissuedisorders
andlympho
proliferativedisea
ses
Mye
losuppression,
infections,urotox
icity
,seco
ndarymaligna
ncy,
teratogen
icity
,infertility
3,16
,25
Mycop
heno
late
500mg,tw
iceper
day
25-100
(smallc
aseserie
s)1-3mo
Mainlyused
intheped
iatric
setting
Nau
sea,
head
ache
,diarrhe
a26
Dan
azol
200mg,3tim
esper
day
20-50
1-3mo
Steroid-sparingpropertie
sAnd
rogen
iceffects(to
beavoided
inmen
with
prostatic
aden
omaor
carcinom
a),liver
toxicity
60,61
ACWY,
serogroup
sA,C,W,an
dY;
n/a,
notap
plicab
le;Re
f.,referenc
e.
1286 blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 BARCELLINI and FATTIZZO
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021

immunocompetence, predisposing him to harmful infections.However, no reliable markers of immunocompetence areavailable, and even after vaccination, protective antibody titersare difficult to establish. As the number of patients subjected todifferent types of immunosuppression therapy for various con-ditions increases, large prospective studies to address thisknowledge gap are warranted. Historical immunosuppressants(azathioprine, cyclophosphamide, methotrexate, cyclosporine,mycophenolate mofetil) are still widely used in clinical practice,mostly in association with steroids and/or as steroid-sparingagents, with reported efficacy in 40% to 50% of cases(Table 1).16,24-28 Their long-term detrimental side effects arefrequently underestimated, and have never been investigated inprospective randomized-controlled trials.
Patient 3A 45-year-old woman was diagnosed with wAIHA (Hb, 7.6 g/dL;white blood cells [WBCs], 6.93 109/L; normal differential countsand platelets; LDH, 1.9 ULN; reticulocytes, 250 3 109/L; un-conjugated bilirubin, 2.8mg/dL; DAT IgG1) with additional positivityfor anti-nuclear antibodies (1/160 titer, homogeneous pattern),anti–b2-glycoprotein 1 IgG, anti-cardiolipin IgG and IgM, and lupus-like anticoagulant (2 positive of 3 tests). Clinical criteria for the di-agnosis of a definite systemic autoimmune disease (in particular,systemic lupus erythematosus andantiphospholipid syndrome)werenot met. An anti-HBs antibody (Ab) test was positive, with HBsAg,anti-HBc Ab, anti HBe Ab, HBV-DNA, and anti-HCV Ab2. Renal andliver function tests were normal. She was successfully treated withsteroids, but relapsed 6 months after discontinuation (Hb, 6.9 g/dL;WBCs, 8.9 3 109/L; lymphocytes, 5.2 3 106/mL; LDH, 2.2 ULN;reticulocytes, 205 3 109/L; unconjugated bilirubin, 2.9 mg/dL; andDAT confirmed positive for IgG alone). Flow cytometry was con-sistent with the diagnosis of B-cell chronic lymphocytic leukemia(CLL; B-CLL) (stage 0A, negative fluorescence in situ hybridization,unmutated IGHV status). BM evaluation (aspirate and trephine bi-opsy) showed erythroid hyperplasia, B-CLL infiltration (5% to 10%),and normal cytogenetics. A total-body computed tomography scanwas unremarkable. The patient was treated with a standard dose ofrituximab; steroid taperingwas held at 0.3mg/kg per day during the4 weeks of rituximab. A progressive amelioration of Hb and he-molytic markers was observed, with a complete response at month16 and subsequent progressive steroid withdrawal (month 19).The patient is still in remission after 2 years, and is on “watch-and-wait” follow-up for CLL.
Commentary on patient 3The first issue arising from this case is whether and when toperform BM evaluation in wAIHA. Although not strictly recom-mended at diagnosis in primary AIHA, it is advisable at firstrelapse, in order to identify any underlying conditions, and toconfirm a good erythroid reservoir.3,9,13 Moreover, it may beuseful in primary forms to examine the prevalent type of lym-phocyte infiltrate, to guide immunosuppressive therapy.29 No-tably, response to rituximab became evident 1 month from theend of treatment, emphasizing the time needed to achieve acomplete response.
It is generally thought that treatment of CLL-associated AIHAshould be the same as that for primary disease, provided thatCLL requires no treatment.4,9,30 Current and novel specifictherapies in the event of AIHA relapse after rituximab arereported in Table 2, including monoclonal antibodies, small
molecules, and combination therapies.4,30-47 Splenectomy wasexcluded given its poor efficacy in CLL-associated forms.9,30
Further concern about this option is its known thrombotic risk(10% to 20% of AIHA cases), which may have been particularlyhigh in patient 3 due to antiphospholipid positivity. Reportedthrombotic events in AIHA may be severe, and include pul-monary emboli, splanchnic thromboses, stroke, and cardiac is-chemia. In addition to splenectomy, active disease (high LDHand low Hb levels) is associated with an increased risk.48
Prophylactic anticoagulation may be considered, and is highlyrecommended in the presence of additional risk factors (age,.70 years; reduced mobility; acute infection; previousthrombosis; preexisting thrombophilic condition; recenttrauma and/or surgery; heart and/or respiratory failure; activecancer).1,8,9,16
With patient 3, one more reason to avoid splenectomy waspositive serology for systemic autoimmune conditions, whichmay precede overt disease. In fact, splenectomy has poor ef-fectiveness in AIHA secondary to systemic autoimmune disor-ders and immunodeficiencies.49 Recently, the availability ofmolecular tools has increased the chances of identifying theseforms early: mutations in KDM6A or KMT2D have been iden-tified in Kabuki syndrome, and mutations in genes implicated inprimary immunodeficiencies (TNFRSF6, CTLA4, STAT3, PIK3CD,CBL,ADAR1, LRBA, RAG1, and KRAS) have been reported in 40%of Evans syndrome patients, an association of AIHA and ITP.4,50
Whether these findings might influence therapies is an excitingsubject for future investigation.
Finally, there is increasing awareness of underlying subclinical orchronic infections that may be reactivated by immunosuppres-sive therapies. Other than lamivudine for HBV, no precise in-dications are available for other traditional treatments (steroids),newer therapies (complement, proteasome, and kinases inhibitors), orother infections (tuberculosis, HCV, herpes viruses, and others).1,3,9
Future studies are needed, and a case-by-case discussion with aninfectious disease expert is advisable in case of clinical suspicionand/or heavily treated patients.
Are R/R cases of wAIHA always“pure” AIHAs?The mechanisms involved in the occurrence of autoimmunediseases, and their subsequent evolution into chronic or re-lapsing disorders, are not completely understood, but involvegenetic background, environmental factors, and dysregulationof the complex immunologic pathways and effectors involved inthe recognition of self and nonself. Focusing on AIHA, auto-immunity is often triggered by molecular mimicry between self-antigens and foreign antigens for diseases associated withbacterial, mycoplasma, or viral infections.3 Likewise, neoantigensgenerated by drugs/chemicals may be responsible for triggeringdrug-induced AIHA.51,52
Finally, it is worth citing the emergence of “forbidden clones” inAIHA secondary to lymphoproliferative disorders, which maybe an important factor for relapse and chronicity. Regardingautoimmunity against BM, aplastic anemia is a typical example,being caused by an autoimmune attack against hemato-poietic stem cells. Moreover, there is evidence that autoimmune
HOW I TREAT WARM AUTOIMMUNE HEMOLYTIC ANEMIA blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 1287
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021

phenomena are present in several conditions other than overtand “classic” autoimmune diseases, such as myelofibrosisand myelodysplasia.53,54 In particular, the presence of anti-erythroblast antibodies, an overinflammatory cytokine milieu,and a proapoptotic pattern have been reported in 50% of pa-tients with low-risk MDS, suggesting a possible pathogenic role
in the ineffective erythropoiesis and an underlying unfavorablemarrow microenviroment.54
Patient 4A 69-year-old woman has been treated at our institution since2009. She was initially diagnosed with wAIHA IgG1 and primary
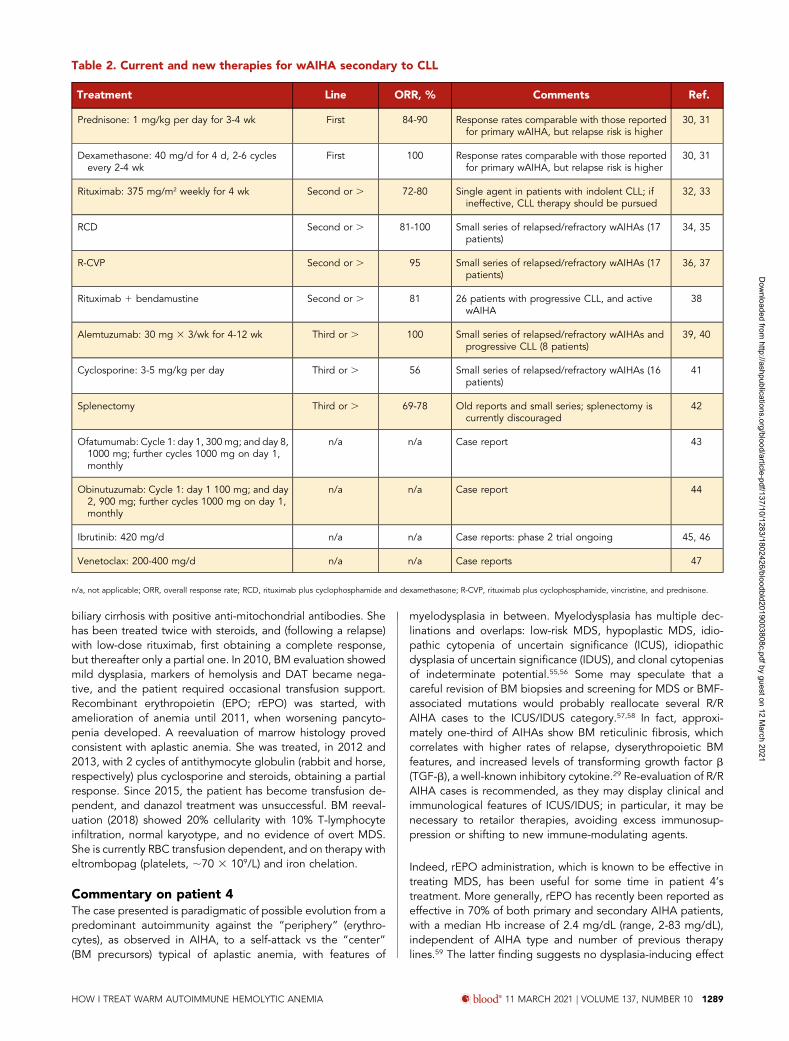
Acute and very severe onset • Hb<6 g/dL and/or
hemodinamic instability
• methylprednisolone 100-200 mg/day for 7-10 days or 250 to 1000 mg/day for 1-3 days, then follow the oral schedule of predniso(lo)ne (right panel)• Transfusion 1 blood unit/day, monitor post transfusion Hb and hemolysis• IvIg 0.4 g/kg/d x 5 d, particularly if infection is present• early rituximab 375 mg/kg/wk for 4 wk in case of no response in 1 wk• Consider PEX in case of no response in 7-10 days• Test endogenous EPO if reticulocytopenia• LMWH prophylaxis if no contraindications • Rituximab 100 mg /wk for 4 wks. Hold on the last dose
of steroids for the 4 rituximab wks
Relapse or no response
• Perform BM evaluation• Rituximab 375 mg/kg/wk for 4 wks. Hold on the last dose of steroids for the 4 wks• Test endogenous EPO if reticulocitopenia
Relapse or no response
Relapse or no response after 8-12 months from diagnosis
Splenectomy
• Perform BM evaluation• Consider enrolment in a clinical trial• mycophenolate mofetil, cyclosporine or cyclophosphamide as steroid sparing agents
Relapse or no response
• Consider enrolment in a clinical trial• mycophenolate mofetil, cyclosporine or cyclophosphamide as steroid sparing agents
Relapse (or no response) after more than1 year from diagnosis
• Perform BM evaluation, repeat DAT, endogenous EPO, search for secondary causes• Rituximab 375 mg/kg/wk for 4 wks (better if more than 2 years from previous administration)• Consider enrolment in a clinical trial• mycophenolate mofetil, cyclosporine or cyclophosphamide (if not previously used)
If very severe
Repeat BM evaluation, DAT, endogenous EPO, exclude secondary causes
Age>65 yrs, presence of comorbidities orthrombotic risks, patient’s refusal
age<65 yrs, no comorbidity or thrombotic risk
• predniso(lo)ne 1-1.5 mg/kg/d for 3-4 wks, then taper according to the following schedule: 10 mg/wk until 0.5 mg/kg/d, then 5 mg/wk until stop • Monitor blood counts, reticulocytes, LDH, unconj bilirubin every 1-2 wk, and hold on taper if Hb <10 mg/dL and hemolysis active
Non acute/severe onset• Hb 6-8 g/dL, anemia well tolerated, no comorbidity, and age <40 yrs• Hb 8-10, anemia well tolerated
Figure 2. Therapeutic flow-chart of wAIHA. This figure illustrates the therapy lines of wAIHA. Responses include complete responses (CR) and partial responses (PR); CR isdefined by normalization of Hb and hemolytic markers (unconjugated bilirubin, LDH, haptoglobin, and reticulocytes); PR is defined by Hb .10 g/dL or at least an increase by.2 g/dL, with or without biochemical resolution of hemolysis; lack of response or relapse is defined as Hb ,10 g/dL or at least 2 g/dL decrease with alteration of hemolyticmarkers. EPO, erythropoietin; LMWH, low-molecular-weight heparin; PEX, plasma exchange.
1288 blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 BARCELLINI and FATTIZZO
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021
biliary cirrhosis with positive anti-mitochondrial antibodies. Shehas been treated twice with steroids, and (following a relapse)with low-dose rituximab, first obtaining a complete response,but thereafter only a partial one. In 2010, BM evaluation showedmild dysplasia, markers of hemolysis and DAT became nega-tive, and the patient required occasional transfusion support.Recombinant erythropoietin (EPO; rEPO) was started, withamelioration of anemia until 2011, when worsening pancyto-penia developed. A reevaluation of marrow histology provedconsistent with aplastic anemia. She was treated, in 2012 and2013, with 2 cycles of antithymocyte globulin (rabbit and horse,respectively) plus cyclosporine and steroids, obtaining a partialresponse. Since 2015, the patient has become transfusion de-pendent, and danazol treatment was unsuccessful. BM reeval-uation (2018) showed 20% cellularity with 10% T-lymphocyteinfiltration, normal karyotype, and no evidence of overt MDS.She is currently RBC transfusion dependent, and on therapy witheltrombopag (platelets, ;70 3 109/L) and iron chelation.
Commentary on patient 4The case presented is paradigmatic of possible evolution from apredominant autoimmunity against the “periphery” (erythro-cytes), as observed in AIHA, to a self-attack vs the “center”(BM precursors) typical of aplastic anemia, with features of
myelodysplasia in between. Myelodysplasia has multiple dec-linations and overlaps: low-risk MDS, hypoplastic MDS, idio-pathic cytopenia of uncertain significance (ICUS), idiopathicdysplasia of uncertain significance (IDUS), and clonal cytopeniasof indeterminate potential.55,56 Some may speculate that acareful revision of BM biopsies and screening for MDS or BMF-associated mutations would probably reallocate several R/RAIHA cases to the ICUS/IDUS category.57,58 In fact, approxi-mately one-third of AIHAs show BM reticulinic fibrosis, whichcorrelates with higher rates of relapse, dyserythropoietic BMfeatures, and increased levels of transforming growth factor b(TGF-b), a well-known inhibitory cytokine.29 Re-evaluation of R/RAIHA cases is recommended, as they may display clinical andimmunological features of ICUS/IDUS; in particular, it may benecessary to retailor therapies, avoiding excess immunosup-pression or shifting to new immune-modulating agents.
Indeed, rEPO administration, which is known to be effective intreating MDS, has been useful for some time in patient 4’streatment. More generally, rEPO has recently been reported aseffective in 70% of both primary and secondary AIHA patients,with a median Hb increase of 2.4 mg/dL (range, 2-83 mg/dL),independent of AIHA type and number of previous therapylines.59 The latter finding suggests no dysplasia-inducing effect
Table 2. Current and new therapies for wAIHA secondary to CLL
Treatment Line ORR, % Comments Ref.
Prednisone: 1 mg/kg per day for 3-4 wk First 84-90 Response rates comparable with those reportedfor primary wAIHA, but relapse risk is higher
30, 31
Dexamethasone: 40 mg/d for 4 d, 2-6 cyclesevery 2-4 wk
First 100 Response rates comparable with those reportedfor primary wAIHA, but relapse risk is higher
30, 31
Rituximab: 375 mg/m2 weekly for 4 wk Second or . 72-80 Single agent in patients with indolent CLL; ifineffective, CLL therapy should be pursued
32, 33
RCD Second or . 81-100 Small series of relapsed/refractory wAIHAs (17patients)
34, 35
R-CVP Second or . 95 Small series of relapsed/refractory wAIHAs (17patients)
36, 37
Rituximab 1 bendamustine Second or . 81 26 patients with progressive CLL, and activewAIHA
38
Alemtuzumab: 30 mg 3 3/wk for 4-12 wk Third or . 100 Small series of relapsed/refractory wAIHAs andprogressive CLL (8 patients)
39, 40
Cyclosporine: 3-5 mg/kg per day Third or . 56 Small series of relapsed/refractory wAIHAs (16patients)
41
Splenectomy Third or . 69-78 Old reports and small series; splenectomy iscurrently discouraged
42
Ofatumumab: Cycle 1: day 1, 300 mg; and day 8,1000 mg; further cycles 1000 mg on day 1,monthly
n/a n/a Case report 43
Obinutuzumab: Cycle 1: day 1 100 mg; and day2, 900 mg; further cycles 1000 mg on day 1,monthly
n/a n/a Case report 44
Ibrutinib: 420 mg/d n/a n/a Case reports: phase 2 trial ongoing 45, 46
Venetoclax: 200-400 mg/d n/a n/a Case reports 47
n/a, not applicable; ORR, overall response rate; RCD, rituximab plus cyclophosphamide and dexamethasone; R-CVP, rituximab plus cyclophosphamide, vincristine, and prednisone.
HOW I TREAT WARM AUTOIMMUNE HEMOLYTIC ANEMIA blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 1289
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021
frommultitreatment, although this should be addressed in largerstudies. Moreover, endogenous EPO levels were reduced inAIHA, and correlated with ineffective erythropoiesis and in-flammatory cytokine tumor necrosis factor a levels. The patientwas also treated with danazol, a drug which has received recentattention for its use in BMF treatment, and telomeropathies.60-62
Finally, whether the presence of BMF and MDS-associatedmutations (SF3B1, SRSF2, TET2, DNMT3A, ASXL1, BCOR/BCORL1, TP53, RUNX1, PIGA) plays a role in the describedevolution will be a fascinating area to explore in the future.13
What to do in acute and severe cases?Acute and severe cases represent a challenge for clinicians, eventhose familiar with AIHA. Usually there is no time to test theefficacy of different treatments, and rapidly worsening organfailures lead to a burden of overlapping therapies. In such cases,mortality rates of up to 57% have been reported, despite in-tensive treatment, including transfusions, steroid boluses, IVimmunoglobulins (IVIgs), rituximab, rEPO, and plasma exchange(PEX).16,28,63,64 Likewise, a recent study of 44 AIHA cases ad-mitted to the intensive care unit for severe anemia reported 30%mortality.65 Associated conditions may also impact on diseaseseverity and prognosis, with higher mortality reported in AIHA
secondary to systemic lupus erythematosus, lymphoma, andcancer.3,9 Transfusion issues may also create challenges inmanaging AIHAs in an acute setting, as the principle “neverdeny but avoid if unnecessary” is not always easily applicableto transfusion decisions. Moreover, the presence of alloanti-bodies (reported in 15% to 40% of AIHA cases) may complicateand delay the selection of compatible units, as well as fuelacute hemolysis and/or increase delayed posttransfusionreactions.3,9,14
Patient 5A 31-year-old man presented at the emergency department inacute and severe wAIHA crisis (Hb, 2.8 g/dL; reticulocytes, 573109/L; unconjugated bilirubin, 4.9 mg/dL; LDH, 3.5 ULN; DAT1,IgG plus C), along with fever (102°F), tachycardia (105 bpm),tachypnea (24 bpm), leukocytosis (16.3 3 109/L), and hemo-globinuria. His past history was unremarkable except for anepisode of ITP 5 years before admission, successfully treatedwith steroids. Platelet counts, and liver and kidney functiontests, were within the normal range. A whole-body computedtomograpy scan was negative for splenomegaly or lymphnode enlargement, thrombosis, and infectious lesions. Nor-mal coagulation parameters and absence of schistocytes ex-cluded disseminated intravascular coagulation and other
Table 3. New drugs for R/R wAIHA
Treatment Dose scheduleResponse rate,
% Comments Ref.
Parsaclisib (PI3Ki) 1 to 2.5 mg/d n/a Open-label phase 2 study 72
Pegcetacoplan (C3i) 270 mg-360 mg/d 40-50 Open-label phase 2 study 73
Orilanolimab (FcRni) n/a n/a Open-label phase 1b/2 study: phase 2randomized, double-blind, placebo-controlled study
74, 75
Nipocalimab (FcRni) IV infusion every 2 or 4 wk n/a Phase 2 randomized, double-blind,placebo-controlled study
76
Fostamatinib (SyKi) 100 to 150 mg, twice daily 44 (time to response2-30 wk)
Open-label phase 2 study; phase 3randomized, double-blind, placebo-controlled study
77, 78
Bortezomib (proteasome-i) 1.3 mg/m2 subcutaneous 32/wkfor 2 wk
n/a Open-label phase 2 study in associationwith anti-CD20 MoAb
79
Interleukin-2 Cycle 1: 1.5 million IU per day for 9 wk;further cycles: 3 million IU per day for16 wk
n/a Open-label phase 1/2 study 80
Daratumumab (anti-CD38MoAb)
16 mg/kg week IV n/a Case reports in AIHA secondary to BMtransplant
81, 82
Sirolimus (mTORi) 2 mg/m2 (adjusted to achieve a steadystate of 5–10 ng/mL)
100 Cases reports of AIHA secondary tocombined liver, small bowel, andpancreas transplant
83
Abatacept (CTLA-4 Ig) 10 mg/kg monthly 100 Case reports in AIHA secondary to BMtransplant
84
Imlifidase (ADCCi) n/a n/a Studied in IgG-mediated diseases andtransplant rejection
85, 86
ADCCi, ADCC inhibitor; C3i, complement component C3 inhibitor; CTLA-4 Ig, fusion protein of CTLA-4 with the Fc region of IgG; FcRni, neonatal crystallizable fragment receptor inhibitor;MoAb, monoclonal antibody; mTORi, mammalian target of rapamycin inhibitor; PI3Ki, phosphatidylinositol-4,5-bisphosphate 3-kinase d type inhibitor; Proteasome-1, proteasome inhibitor;Syki, spleen tyrosine kinase inhibitor.
1290 blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 BARCELLINI and FATTIZZO
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021
microangiopathies. The patient was transfused and treated withsteroid boluses (methylprednisolone 250 mg IV daily for 3 days),IV antibiotics, and prophylactic heparin. Due to his septic state,IVIgs were started on day13. Hb levels stabilized around 4 g/dLwith a further 3 blood units, but reticulocytopenia and elevatedLDH persisted. On day 15 the patient developed acute renalfailure (creatinine, 4 mg/dL), and was transferred to the intensivecare unit. The patient received fluids and vasopressors withamelioration of renal function, and underwent 3 daily plasmaexchanges. Despite steroid treatment, anemia and retic-ulocytopenia did not significantly improve (day 110). After ex-clusion of underlying conditions at BM evaluation, rituximabstandard dose was started. On day 117, rEPO (epoietin a,40 000 IU per week) was added, with progressive amelioration ofHb values (8-10 g/dL) and the appearance of the reticulocytecrisis. After discharge, the patient continued steroid therapy withtapering and was able to discontinue rEPO after 6 weeks.
Commentary on patient 5Patient 5 had been diagnosed with Evans syndrome, with verysevere/acute AIHA, and previous ITP (known to occur together
or separately). Profound autoimmunity was present, sustained byautoantibodies that activate complement, causing intravascu-lar hemolysis, and associated with reticulocytopenia, possi-bly due to antibodies against erythroblasts that hamper BMcompensation.3,16,28,66 The various therapies administered weredirected at various pathogenetic mechanisms: steroids and rit-uximab to decrease inflammation and antibody-producing Blymphocytes, IVIg to mask ADCC and help against concomitantinfection, PEX to remove excess immunemediators, and rEPO toovercome a “shocked” BM.59
Rituximab is usually recommended as a second-line therapy;however, in urgent situations, the exact time of steroid failure isnot easy to define. In AIHAs following hematopoietic stem celltransplantation, which are generally severe, refractory to ste-roids, and fatal, rituximab is recommended either as a frontlineor early second-line therapy.67-70 This has led to improvedoutcomes, suggesting that this approach might work in all acutesettings. PEX is clearly a temporizing measure until specifictreatments become effective. Although evidence for its efficacyis limited and controversial, the removal of pathogenic immune
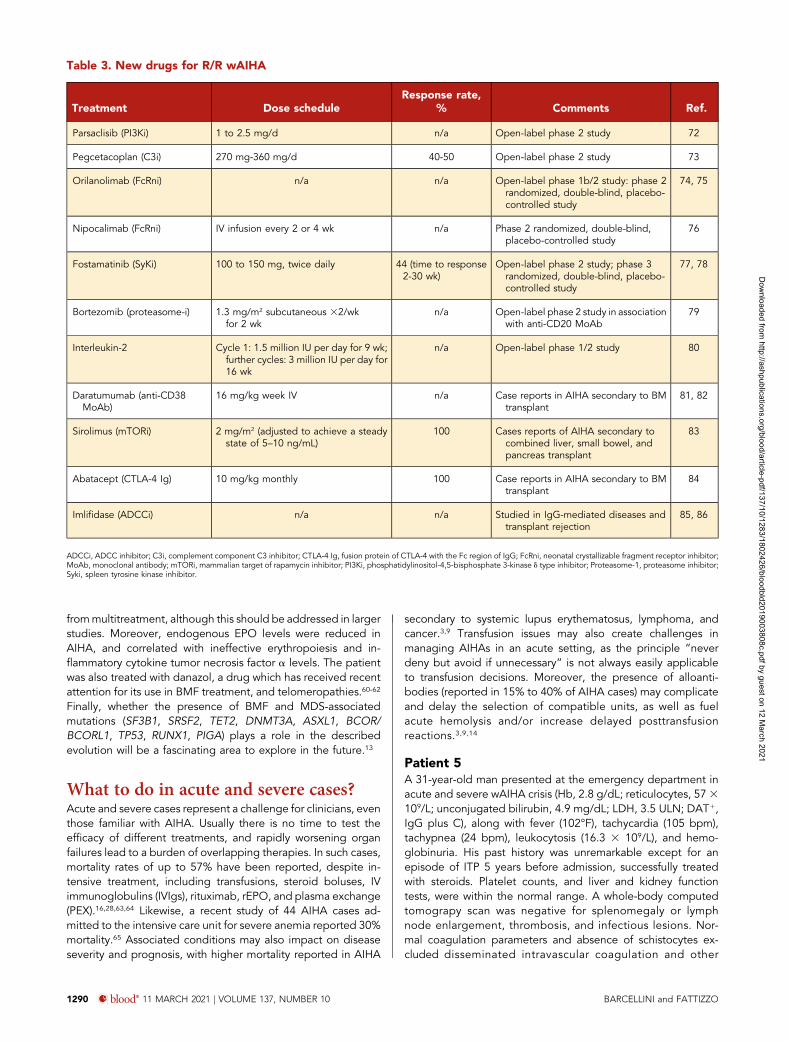
Macrophage/APC
Immune activationcytokine secretion
plasmacellT lymphocyte
B lymphocyte
Spleen (ADCC)Liver(CDC)
C3
MAC
C5
Complementcascade
Coagulationcascade
BM compensa on(re culocytosis)
Standard therapies
Target therapies
steroids azathioprine, cyclosporine, cyclophosphamide splenectomy
Alemtuzumab (anti-CD52)MMF (purine synthesis inhibitor)Sirolimus (anti-mTOR)
Rituximab (anti-CD20)Belimumab (BAFF-April system inhibitor)Bortezomib (proteosome inhibitor)Idelalisib (PI3Kd inhibitor)Daratumumab (anti-CD38)Ibrutinib (BTK inhibitor)Venetoclax (anti-Bcl2 )
Fostamatinib (Syk inhibitor)SYNT001/ALXN1830/M281 (anti-FcRn)
EPODanazolLuspatercet
ANX005 (C1q inhibitor)APL-2 (C3 inhibitor)
RBC lysis
Anti-RBC andanti-erythroblast
antibodies
RBC phagocytosisADCC/ CDC
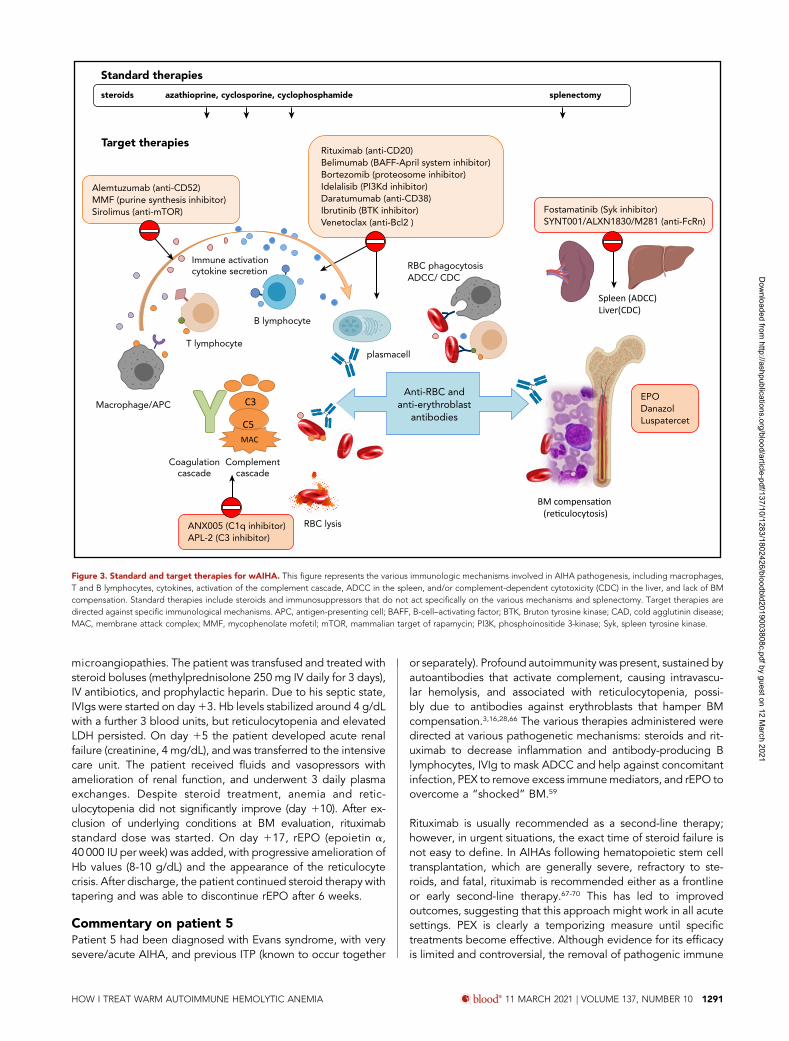
Figure 3. Standard and target therapies for wAIHA. This figure represents the various immunologic mechanisms involved in AIHA pathogenesis, including macrophages,T and B lymphocytes, cytokines, activation of the complement cascade, ADCC in the spleen, and/or complement-dependent cytotoxicity (CDC) in the liver, and lack of BMcompensation. Standard therapies include steroids and immunosuppressors that do not act specifically on the various mechanisms and splenectomy. Target therapies aredirected against specific immunological mechanisms. APC, antigen-presenting cell; BAFF, B-cell–activating factor; BTK, Bruton tyrosine kinase; CAD, cold agglutinin disease;MAC, membrane attack complex; MMF, mycophenolate mofetil; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; Syk, spleen tyrosine kinase.
HOW I TREAT WARM AUTOIMMUNE HEMOLYTIC ANEMIA blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 1291
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021

complexes, circulating autoantibodies, and activated comple-ment may be useful in acute and rapidly deteriorating cases.
The exact mechanism by which IVIg acts is not fullyunderstood.7,9,71 Recently, it has been suggested that IVIg ad-ministration can determine saturation of the neonatal Fc re-ceptor (FcRn), which is responsible for the salvage of IgG fromcatabolism, thus resulting in accelerated clearance of endoge-nous IgG (including pathogenic autoantibodies). New drugs thatblock FcRn are under development, hopefully providing a PEX-like strategy for the acute setting in the future. Likewise, com-plement inhibitors that are close to being approved for coldagglutinin disease (CAD; where complement activation is themain pathogenic mechanism) are under development in wAIHAand are likely to be useful in IgG-plus-C forms.7
Figure 2 summarizes the different approaches in acute and verysevere cases, compared with nonsevere ones, including optionsbeyond second-line therapy. In fact, patient 5 is at risk of furtherrelapse, given the AIHA type and severity, and the diagnosis ofEvans syndrome. Hazard ratios for relapse increased from 1.61 to1.74 and 1.98 for Hb values 8.1 to 10 g/dL, 6.1 to 8 g/dL, and,6 g/dL, respectively; also, taking into account Hb as a con-tinuous variable, each gram of reduction resulted in 7% higherrelapse risk.28 Moreover, Evans syndrome, acute renal failure,and infection (all present in our patient) have been associ-ated with increased mortality (hazard ratios of 8, 6.3, and 4,respectively).
Therefore, it is essential to plan a future therapeutic strategy.Splenectomy, provided patient consent, is a valid option, but it isprudent to wait ;1 year from diagnosis before adopting thisirreversible and not definitively curative option. Rituximab hasalready been used, and retreatment, although feasible, wouldprobably be ineffective in the event of an early relapse (within8-12 months of diagnosis). Meanwhile, the patient might bemanaged with steroids and classic immunosuppressants assteroid-sparing agents (Table 1). Among future therapeuticoptions, enrollment in a clinical trial is highly advisable. Table 3shows new treatments and ongoing studies with tyrosine ki-nases, complement, proteasome, FcRn, and ADCC inhibitors, aswell as case reports describing efficacy of daratumumab, siro-limus, and abatacept.13,72-86
ConclusionwAIHA is a greatly heterogeneous disease with an unpredictableclinical course, due to its multiple immunologic mechanisms,and their variable role over time. This unpredictability should
always be considered, and re-evaluation of DAT, hemolyticparameters, and BM characteristics is recommended, particu-larly in R/R cases. Complete BM evaluation is recommended forpatients with suspected underlying disease or those who haverelapsed after first-line steroid therapy. Consequently, therapiesshould ideally be used to target the prevailing immunopatho-genic mechanism (Figure 3). Our main clinical recommendationsinclude proper use of steroids (not for ,6 months, includingtapering; never for .1 year), early use of rituximab or evenretreatment, careful consideration of splenectomy at an ap-propriate time, and accurate detection of a possible evolution toICUS/IDUS in R/R patients. Likewise, careful investigation forsecondary forms is highly advisable, as this may change thetherapeutic strategy quite considerably. Given the lack of evi-dence beyond the second line, and the decline of splenectomy,enrollment in clinical trials should be pursued strongly in R/RwAIHA cases. As new target therapies become available,identifying the optimal choice, sequence, and combination ofdrugs, with the aim of “curing” wAIHA, will be an increasinglycomplex but fascinating challenge.
AcknowledgmentThis work was supported by research funding from Fondazione IRCCSCa’Granda Ospedale Maggiore Policlinico (RC 2018).
AuthorshipContribution: W.B. and B.F. conceived and wrote the manuscript.
Conflict-of-interest disclosure: W.B. provided consultancy services toAgios, Alexion, Apellis, Biocryst, Bioverativ, Incyte, Momenta, andNovartis, and received lecture fees/congress support from Alexion,Incyte, Novartis, and Sanofi. B.F. provided consultancy services toApellis, Momenta, and Novartis, and received lecture fees/congresssupport from Alexion and Apellis.
ORCID profiles: W.B., 0000-0003-1428-9944; B.F., 0000-0003-0857-8379.
Correspondence: Wilma Barcellini, Hematology, Fondazione IRCCS Ca’Granda Ospedale Maggiore Policlinico, Via F. Sforza 35, 20122 Milan,Italy; e-mail: [email protected].
FootnotesSubmitted 23 June 2020; accepted 28 August 2020; prepublished onlineon Blood First Edition 21 January 2021. DOI 10.1182/blood.2019003808.
*W.B. and B.F. contributed equally.
REFERENCES1. Hill QA, Stamps R, Massey E, Grainger JD,
Provan D, Hill A; British Society for Haema-tology. The diagnosis and management ofprimary autoimmune haemolytic anaemia. BrJ Haematol. 2017;176(3):395-411.
2. Barcellini W. New insights in the pathogenesisof autoimmune hemolytic anemia. TransfusMed Hemother. 2015;42(5):287-293.
3. Barcellini W, Giannotta J, Fattizzo B.Autoimmune hemolytic anemia in adults:primary risk factors and diagnostic
procedures. Expert Rev Hematol. 2020;13(6):585-597.
4. Fattizzo B, Barcellini W. Autoimmune cyto-penias in chronic lymphocytic leukemia: focuson molecular aspects. Front Oncol. 2020;9:1435.
5. Kalfa TA. Warm antibody autoimmune he-molytic anemia.Hematology Am Soc HematolEduc Program. 2016;2016:690-697.
6. Go RS, Winters JL, Kay NE. How I treat au-toimmune hemolytic anemia. Blood. 2017;129(22):2971-2979.
7. Barcellini W, Fattizzo B, Zaninoni A. Currentand emerging treatment options for autoim-mune hemolytic anemia. Expert Rev ClinImmunol. 2018;14(10):857-872.
8. Brodsky RA. Warm autoimmune hemolyticanemia. N Engl J Med. 2019;381(7):647-654.
9. Jager U, Barcellini W, Broome CM, et al.Diagnosis and treatment of autoimmunehemolytic anemia in adults: recommenda-tions from the First International Con-sensus Meeting. Blood Rev. 2020;41:100648.
1292 blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 BARCELLINI and FATTIZZO
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021

10. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idio-pathic autoimmune hemolytic anemia: clinicalefficacy and biologic studies. Blood. 2012;119(16):3691-3697.
11. Birgens H, Frederiksen H, Hasselbalch HC,et al. A phase III randomized trial comparingglucocorticoid monotherapy versus gluco-corticoid and rituximab in patients with au-toimmune haemolytic anaemia. BrJ Haematol. 2013;163(3):393-399.
12. Michel M, Terriou L, Roudot-Thoraval F, et al.A randomized and double-blind controlledtrial evaluating the safety and efficacy of rit-uximab for warm auto-immune hemolyticanemia in adults (the RAIHA study). AmJ Hematol. 2017;92(1):23-27.
13. Barcellini W, Fattizzo B. The changing land-scape of autoimmune hemolytic anemia. FrontImmunol. 2020;11:946.
14. Petz LD, Garratty G. Immune HemolyticAnemias. 2nd ed. Philadelphia, PA: ChurchillLivingstone; 2004.
15. Barcellini W, Fattizzo B. Clinical applications ofhemolytic markers in the differential diagnosisand management of hemolytic anemia. DisMarkers. 2015;2015:635670.
16. Barcellini W, Fattizzo B, Zaninoni A, et al.Clinical heterogeneity and predictors of out-come in primary autoimmune hemolytic ane-mia: a GIMEMA study of 308 patients. Blood.2014;124(19):2930-2936.
17. Buckley L, Guyatt G, Fink HA, et al. 2017American College of Rheumatology guidelinefor the prevention and treatment ofglucocorticoid-induced osteoporosis. ArthritisCare Res (Hoboken). 2017;69(8):1095-1110.
18. ReynaudQ, Durieu I, Dutertre M, et al. Efficacyand safety of rituximab in auto-immune he-molytic anemia: a meta-analysis of 21 studies.Autoimmun Rev. 2015;14(4):304-313.
19. Fattizzo B, Zaninoni A, Pettine L, Cavallaro F,Di Bona E, Barcellini W. Low-dose rituximab inautoimmune hemolytic anemia: 10 years after.Blood. 2019;133(9):996-998.
20. Patel NY, Chilsen AM, Mathiason MA, KalliesKJ, Bottner WA. Outcomes and complicationsafter splenectomy for hematologic disorders.Am J Surg. 2012;204(6):1014-1019.
21. Roumier M, Loustau V, Guillaud C, et al.Characteristics and outcome of warm auto-immune hemolytic anemia in adults: new in-sights based on a single-center experiencewith 60 patients. Am J Hematol. 2014;89(9):E150-E155.
22. Ho G, Brunson A, Keegan THM, Wun T.Splenectomy and the incidence of venousthromboembolism and sepsis in patients withautoimmune hemolytic anemia. Blood CellsMol Dis. 2020;81:102388.
23. Thomsen RW, SchoonenWM, Farkas DK, et al.Risk for hospital contact with infection in pa-tients with splenectomy: a population-basedcohort study. Ann Intern Med. 2009;151(8):546-555.
24. Emilia G, Messora C, Longo G, Bertesi M.Long-term salvage treatment by cyclosporin inrefractory autoimmune haematological disor-ders. Br J Haematol. 1996;93(2):341-344.
25. Moyo VM, Smith D, Brodsky I, Crilley P, JonesRJ, Brodsky RA. High-dose cyclophospha-mide for refractory autoimmune hemolyticanemia. Blood. 2002;100(2):704-706.
26. Howard J, Hoffbrand AV, Prentice HG, MehtaA. Mycophenolate mofetil for the treatment ofrefractory auto-immune haemolytic anaemiaand auto-immune thrombocytopenia purpura.Br J Haematol. 2002;117(3):712-715.
27. Newman K, Owlia MB, El-Hemaidi I, AkhtariM. Management of immune cytopenias inpatients with systemic lupus erythematosus -old and new. Autoimmun Rev. 2013;12(7):784-791.
28. Barcellini W, Zaninoni A, Fattizzo B, et al.Predictors of refractoriness to therapy andhealthcare resource utilization in 378 patientswith primary autoimmune hemolytic anemiafrom eight Italian reference centers. AmJ Hematol. 2018;93(9):E243-E246.
29. Fattizzo B, Zaninoni A, Gianelli U, et al.Prognostic impact of bone marrow fibrosisand dyserythropoiesis in autoimmune hemo-lytic anemia. Am J Hematol. 2018;93(4):E88-E91.
30. Visco C, Barcellini W, Maura F, Neri A,Cortelezzi A, Rodeghiero F. Autoimmunecytopenias in chronic lymphocytic leukemia.Am J Hematol. 2014;89(11):1055-1062.
31. Rogers KA, Ruppert AS, Bingman A, et al.Incidence and description of autoimmunecytopenias during treatment with ibrutinib forchronic lymphocytic leukemia. Leukemia.2016;30(2):346-350.
32. Narat S, Gandla J, Hoffbrand AV, Hughes RG,Mehta AB. Rituximab in the treatment of re-fractory autoimmune cytopenias in adults.Haematologica. 2005;90(9):1273-1274.
33. D’Arena G, Laurenti L, Capalbo S, et al.Rituximab therapy for chronic lymphocyticleukemia-associated autoimmune hemolyticanemia. Am J Hematol. 2006;81(8):598-602.
34. Gupta N, Kavuru S, Patel D, et al. Rituximab-based chemotherapy for steroid-refractoryautoimmune hemolytic anemia of chroniclymphocytic leukemia. Leukemia. 2002;16(10):2092-2095.
35. Rossignol J, Michallet AS, Oberic L.Rituximab-cyclophosphamide-dexamethasonecombination in the management of autoim-mune cytopenias associated with chroniclymphocytic leukemia. Leukemia. 2011;25(3):473-478.
36. Bowen DA, Call TG, Shanafelt TD, et al.Treatment of autoimmune cytopenia compli-cating progressive chronic lymphocytic leu-kemia/small lymphocytic lymphoma withrituximab, cyclophosphamide, vincristine, andprednisone. Leuk Lymphoma. 2010;51(4):620-627.
37. Kaufman M, Limaye SA, Driscoll N, et al. Acombination of rituximab, cyclophosphamideand dexamethasone effectively treats immunecytopenias of chronic lymphocytic leukemia.Leuk Lymphoma. 2009;50(6):892-899.
38. Quinquenel A, Willekens C, Dupuis J, et al.Bendamustine and rituximab combination inthe management of chronic lymphocyticleukemia-associated autoimmune hemolyticanemia: a multicentric retrospective study of
the French CLL intergroup (GCFLLC/MW andGOELAMS). Am J Hematol. 2015;90(3):204-207.
39. Karlsson C, Hansson L, Celsing F, Lundin J.Treatment of severe refractory autoimmunehemolytic anemia in B-cell chronic lympho-cytic leukemia with alemtuzumab (humanizedCD52 monoclonal antibody). Leukemia. 2007;21(3):511-514.
40. Laurenti L, Tarnani M, Efremov DG, et al.Efficacy and safety of low-dose alemtuzumabas treatment of autoimmune hemolytic ane-mia in pretreated B-cell chronic lymphocyticleukemia. Leukemia. 2007;21(8):1819-1821.
41. Cortes J, O’Brien S, Loscertales J, et al.Cyclosporin A for the treatment of cytopeniaassociated with chronic lymphocytic leukemia.Cancer. 2001;92(8):2016-2022.
42. Cusack JC Jr., Seymour JF, Lerner S, KeatingMJ, Pollock RE. Role of splenectomy in chroniclymphocytic leukemia. J Am Coll Surg. 1997;185(3):237-243.
43. Nader K, Patel M, Ferber A. Ofatumumab inrituximab-refractory autoimmune hemolyticanemia associated with chronic lymphocyticleukemia: a case report and review of litera-ture. Clin Lymphoma Myeloma Leuk. 2013;13(4):511-513.
44. Pejsa V, Lucijanic M, Vrkljan Vuk A, et al.Prolonged methylprednisolone premed-ication prior to obinutuzumab in patients withchronic lymphocytic leukemia. Leuk Lym-phoma. 2020;61(4):934-939.
45. Montillo M, O’Brien S, Tedeschi A, et al.Ibrutinib in previously treated chronic lym-phocytic leukemia patients with autoimmunecytopenias in the RESONATE study [letter].Blood Cancer J. 2017;7(2):e524.
46. US National Library of Medicine:ClinicalTrials.gov. Ibrutinib in Steroid Refractory Autoim-mune Hemolytic Anemia (ISRAEL). https://clinicaltrials.gov/ct2/show/record/NCT03827603. Accessed 7 June 2020.
47. Lacerda MP, Guedes NR, Yamakawa PE, et al.Treatment of refractory autoimmune hemo-lytic anemia with venetoclax in relapsedchronic lymphocytic leukemia with del(17p).Ann Hematol. 2017;96(9):1577-1578.
48. Audia S, Bach B, Samson M, et al. Venousthromboembolic events during warm auto-immune hemolytic anemia. PLoS One. 2018;13(11):e0207218.
49. Hall S, McCormick JL Jr., Greipp PR, MichetCJ Jr., McKenna CH. Splenectomy does notcure the thrombocytopenia of systemic lupuserythematosus. Ann Intern Med. 1985;102(3):325-328.
50. Hadjadj J, Aladjidi N, Fernandes H, et al;members of the French Reference Centerfor Pediatric Autoimmune Cytopenia(CEREVANCE). Pediatric Evans syndrome isassociated with a high frequency of potentiallydamaging variants in immune genes. Blood.2019;134(1):9-21.
51. Arndt PA, Garratty G. The changing spectrumof drug-induced immune hemolytic anemia.Semin Hematol. 2005;42(3):137-144.
52. Garratty G. Immune hemolytic anemia causedby drugs. Expert Opin Drug Saf. 2012;11(4):635-642.
HOW I TREAT WARM AUTOIMMUNE HEMOLYTIC ANEMIA blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 1293
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021

53. Barcellini W, Iurlo A, Radice T, et al. Increasedprevalence of autoimmune phenomena inmyelofibrosis: relationship with clinical andmorphological characteristics, and with im-munoregulatory cytokine patterns. Leuk Res.2013;37(11):1509-1515.
54. Zaninoni A, Imperiali FG, Cattaneo A, et al.Detection of erythroblast antibodies inmitogen-stimulated bone marrow culturesfrom patients with myelodysplastic syn-dromes. Transfusion. 2016;56(8):2037-2041.
55. Valent P. ICUS, IDUS, CHIP and CCUS: di-agnostic criteria, separation from MDS andclinical implications. Pathobiology. 2019;86(1):30-38.
56. Steensma DP. Does clonal hematopoiesisexplain unexplained anemia? Blood. 2020;135(14):1080-1082.
57. Barcellini W. The relationship between idio-pathic cytopenias/dysplasias of uncertainsignificance (ICUS/IDUS) and autoimmunity.Expert Rev Hematol. 2017;10(7):649-657.
58. Barcellini W, Fattizzo B, Zaninoni A, et al.Clinical evolution of autoimmune cytopeniasto idiopathic cytopenias/dysplasias of un-certain significance (ICUS/IDUS) and bonemarrow failure syndromes. Am J Hematol.2017;92(3):E26-E29.
59. Fattizzo B, Michel M, Zaninoni A, et al. Efficacyof recombinant erythropoietin in autoimmunehaemolytic anaemia: a multicentre in-ternational study [published online ahead ofprint 30 April 2020]. Haematologica. doi:10.3324/haematol.2020.250522.
60. Ahn YS, Harrington WJ, Mylvaganam R, AyubJ, Pall LM. Danazol therapy for autoimmunehemolytic anemia. Ann Intern Med. 1985;102(3):298-301.
61. Pignon JM, Poirson E, Rochant H. Danazol inautoimmune haemolytic anaemia. BrJ Haematol. 1993;83(2):343-345.
62. Townsley DM, Dumitriu B, Liu D, et al. Danazoltreatment for telomere diseases. N EnglJ Med. 2016;374(20):1922-1931.
63. Rattarittamrong E, Eiamprapai P, TantiworawitA, et al. Clinical characteristics and long-termoutcomes of warm-type autoimmune hemo-lytic anemia. Hematology. 2016;21(6):368-374.
64. Fattizzo B, Zaninoni A, Nesa F, et al. Lessonsfrom very severe, refractory, and fatal primaryautoimmune hemolytic anemias. AmJ Hematol. 2015;90(8):E149-E151.
65. Lafarge A, Bertinchamp R, Pichereau C, et al.Prognosis of autoimmune hemolytic anemia incritically ill patients. Ann Hematol. 2019;98(3):589-594.
66. Michel M, Chanet V, Dechartres A, et al. Thespectrum of Evans syndrome in adults: newinsight into the disease based on the analysisof 68 cases. Blood. 2009;114(15):3167-3172.
67. Wang M, Wang W, Abeywardane A, et al.Autoimmune hemolytic anemia after alloge-neic hematopoietic stem cell transplantation:analysis of 533 adult patients who underwenttransplantation at King’s College Hospital.Biol Blood Marrow Transplant. 2015;21(1):60-66.
68. Gonzalez-Vicent M, Sanz J, Fuster JL, et al.Autoimmune hemolytic anemia (AIHA) fol-lowing allogeneic hematopoietic stem celltransplantation (HSCT): a retrospective anal-ysis and a proposal of treatment on behalf ofthe Grupo Español De Trasplante de MedulaOsea en Niños (GETMON) and the GrupoEspañol de Trasplante Hematopoyetico(GETH) [published online ahead of print 3March 2018]. Transfus Med Rev. doi:10.1016/j.tmrv.2018.02.005.
69. Kruizinga MD, van Tol MJD, Bekker V, et al.Risk factors, treatment, and immune dysre-gulation in autoimmune cytopenia afterallogeneic hematopoietic stem cell trans-plantation in pediatric patients. Biol BloodMarrow Transplant. 2018;24(4):772-778.
70. Barcellini W, Fattizzo B, Zaninoni A.Management of refractory autoimmune he-molytic anemia after allogeneic hematopoi-etic stem cell transplantation: currentperspectives. J Blood Med. 2019;10:265-278.
71. Flores G, Cunningham-Rundles C, NewlandAC, Bussel JB. Efficacy of intravenous immu-noglobulin in the treatment of autoimmunehemolytic anemia: results in 73 patients. AmJ Hematol. 1993;44(4):237-242.
72. US National Library of Medicine:ClinicalTrials.gov. A Study of INCB050465 in ParticipantsWith Autoimmune Hemolytic Anemia. https://clinicaltrials.gov/ct2/show/NCT03538041.Accessed 7 June 2020.
73. US National Library of Medicine:ClinicalTrials.gov. Study to Assess the Safety, Tolerability,Efficacy and PK of APL-2 in Patients withWarmType Autoimmune Hemolytic Anemia(wAIHA) or Cold Agglutinin Disease (CAD).https://clinicaltrials.gov/ct2/show/NCT03226678. Accessed 7 June 2020.
74. US National Library of Medicine:ClinicalTrials.gov. A Safety Study of SYNT001 in Participantswith Warm Autoimmune Hemolytic Anemia(WAIHA). https://clinicaltrials.gov/ct2/show/NCT03075878. Accessed 7 June 2020.
75. US National Library of Medicine:ClinicalTrials.gov. ALXN1830 in Patients with WarmAutoimmune Hemolytic Anemia. https://clinicaltrials.gov/ct2/show/NCT04256148.Accessed 7 June 2020.
76. US National Library of Medicine:ClinicalTrials.gov. Efficacy and Safety ofM281 in Adults withWarm Autoimmune Hemolytic Anemia.https://clinicaltrials.gov/ct2/show/NCT04119050. Accessed 7 June 2020.
77. US National Library of Medicine:ClinicalTrials.gov. A Safety and Efficacy Study of R935788 inthe Treatment of Warm Antibody Autoim-mune Hemolytic Anemia (AIHA) (SOAR).https://clinicaltrials.gov/ct2/show/NCT02612558. Accessed 7 June 2020.
78. US National Library of Medicine:ClinicalTrials.gov. A Phase 3, Multi-Center, Randomized,Double-Blind, Placebo-Controlled, Study ofFostamatinib Disodium in the Treatment ofwAIHA. https://clinicaltrials.gov/ct2/show/NCT03764618. Accessed 7 June 2020.
79. US National Library of Medicine:ClinicalTrials.gov. Single-dose Anti-CD20 Antibody WithBortezomib for Relapsed Refractory Autoim-mune Hemolytic Anemia (RRAIHA01). https://clinicaltrials.gov/ct2/show/NCT04083014.Accessed 7 June 2020.
80. US National Library of Medicine:ClinicalTrials.gov. Evaluating the Interest of Interleukine-2for Patients With Active Warm HemolyticAnemia Resistant to Conventional Treatment(ANEMIL). https://clinicaltrials.gov/ct2/show/NCT02389231. Accessed 7 June 2020.
81. Schuetz C, Hoenig M, Moshous D, et al.Daratumumab in life-threatening autoimmunehemolytic anemia following hematopoieticstem cell transplantation. Blood Adv. 2018;2(19):2550-2553.
82. Even-Or E, Naser Eddin A, Shadur B, et al.Successful treatment with daratumumab forpost-HSCT refractory hemolytic anemia.Pediatr Blood Cancer. 2020;67(1):e28010.
83. Acquazzino MA, Fischer RT, Langnas A,Coulter DW. Refractory autoimmune hemo-lytic anemia after intestinal transplantresponding to conversion from a calcineurin tomTOR inhibitor. Pediatr Transplant. 2013;17(5):466-471.
84. Hess J, Su L, Nizzi F, et al. Successful treat-ment of severe refractory autoimmune he-molytic anemia after hematopoietic stem celltransplant with abatacept. Transfusion. 2018;58(9):2122-2127.
85. Allhorn M, Briceño JG, Baudino L, et al. TheIgG-specific endoglycosidase EndoS inhibitsboth cellular and complement-mediated au-toimmune hemolysis. Blood. 2010;115(24):5080-5088.
86. Ge S, Chu M, Choi J, et al. Imlifidase inhibitsHLA antibody-mediated NK cell activationand antibody-dependent cell-mediated cy-totoxicity (ADCC) in vitro. Transplantation.2020;104(8):1574-1579.
1294 blood® 11 MARCH 2021 | VOLUME 137, NUMBER 10 BARCELLINI and FATTIZZO
Dow
nloaded from http://ashpublications.org/blood/article-pdf/137/10/1283/1802426/bloodbld2019003808c.pdf by guest on 12 M
arch 2021
















![HISK 10 ANemia HEMOLITIK.ppt [Read-Only] - ocw.usu.ac.idocw.usu.ac.id/course/download/1110000096-hematology-and-immunology... · Autoimmune hemolytic anemia caused byAutoimmune hemolytic](https://img.pdfslide.us/doc/110x75/5c7c94c409d3f23a2a8b4fbf/hisk-10-anemia-read-only-ocwusuacidocwusuacidcoursedownload1110000096-hematology-and-immunology.jpg)












