Embed Size (px)
Citation preview

An agency of the European Union
How are medicines evaluated at the EMA
Presented by: Nathalie Bere Patient interaction / Stakeholders and communication Division

1 1
Optimised utilisation of resources Harmonised scientific opinions
Harmonised information to healthcare professionals & patients
All Systems allow Centralised Procedure (via EMA)
Mutual Recognition/ Decentralised
Procedure
The European System

2
Centralised procedure

• 1 application
• 1 evaluation
• 1 authorisation for all EU
• 1 invented name
• 1 product information (SPC, Labelling, PL)
• All EU languages
Centralised procedure
The EMA is not responsible for pricing or reimbursement

4
The Agency is responsible for:
• The evaluation of marketing authorisation for human and veterinary applications submitted by pharmaceutical companies
• The coordination of European pharmacovigilance (supervision of the medicines on the market)
• The provision of scientific advice on the development of medicines
• The evaluation of applications for orphan designation in EU
• The evaluation of paediatric investigation plans (or waivers)
• The evaluation of arbitration and referral procedures
• The provision of good quality and independent information on the medicines it evaluates to patients and health
• The coordination of Member States’ inspections (GMP, GCP, GLP)
The various roles of the EMA

5
Eligibility: “Mandatory Scope”
Since May 08
Auto-immune diseases and Other immune dysfunctions
Viral diseases
AIDS Cancer Neurodegenerative disorders
Diabetes
Orphan medicines
Recombinant DNA technology Controlled gene expression Monoclonal AB Since Jan 95
Gene therapy products Somatic Cell therapy products Tissue engineered products
ADVANCED THERAPY MEDICINAL PRODUCTS:
Since Dec 08

6
Eligibility “Optional Scope”
Art 3(3) Generic of a product authorised via EMA
New Active Substances
Significant Innovation
(Therapeutic, &/or Scientific, &/or Technical)
Interest of Patients at
Community Level
OR
The centralised procedure attracts most innovative medicines. Decentralised and MRP mainly do generics and new indications for existing products

7
approval
Type of Approvals
Conditional Approval: • Comprehensive data not available; to be provided after approval • Must fulfil scope (orphan drugs, emergency threats, serious and life-threatening diseases)
Approval valid for 1 year, renewable
Exceptional Circumstances: • Comprehensive data not available and cannot be provided • Must meet criteria (rarity, medical ethics, state of scientific knowledge)
Normal: Comprehensive data

8
28 EEA Member States + 4,500 European experts
EU institutions: Commission - Parliament
Committee for Orphan Medicinal Products
(COMP)
Committee for Herbal Medicinal Products
(HMPC)
EMA
Secretariat
Committee for Veterinary Medicinal Products
(CVMP)
Management Board
Committee for Human
Medicinal Products (CHMP)
Committee for Advanced Therapies
(CAT)
Paediatric Committee (PDCO)
EMA-EU Network
Pharmacovigilance Risk Assessment Committee
(PRAC)

9
1 scientific expert member nominated by each MS + 1 alternate 5 co-opted members
Chairperson: Tomas Salmonson
CHMP

Other working parties Biosimilar
Biostatistics Blood Products Cardiovascular
Central Nervous System Infectious Diseases Oncology Working Pharmacogenomics Pharmacokinetics
Rheumatology/Immuno. Vaccines
10
Working Parties and other Groups
SAG diagnostics
SAG Neurology
SAG Psychiatry
SAG HIV /
Antiviral
SAG Oncology
SAG CVS
SAG Diabetes Endoc.
HCPWP Healthcare
professionals
SAG Anti-infectives SAG
Vaccines
ad-hoc expert groups
CMDh Co-ordination Group
for Mutual Recognition and Decentralised
Procedures
PCWP Patients and consumers
BWP Biologics
SWP Safety
QWP Quality
SAWP Scientific advice
GCP Inspectors Working group
QRD Working Group on Quality Review of
documents
Working Parties

Centralised procedure - product life-cycle
CHMP CAT
PRAC
CHMP SAWP
CHMP PRAC
Orphan Designation
Scientific Adv. Protocol assist.
Clinical trials
Paediatric investigation
Post Marketing Authorisation
MAA Evaluation
COMP PDCO
SAGs WPs
CAT
Patient input
SAGs WPs
Patient input
Patient input

Draft PI & RMP
Submission of application
Assessment of Risk
Management Plan
Assessment of Product Information
Assessment on need for post safety/efficacy
studies
Final Product
Information & RMP
Evaluation of benefit/risk
Preparation of RMP summary
CHMP
D1 Start D80 AR D120 LoQ D150 JAR D180 LoOI/OE D210 Opinion D277 CD

Timelines dependent on specific procedure/medicine
RevisedPI
Submission of change
authorisation
Update of Product Information?
Assessment of safety update reports
Decision on need for new post safety
studies
Update of Product
Information /RMP
Re-evaluation of benefit/risk
Update of RMP summary?
EMA - CHMP - PRAC
Signal detection
Annual re-assessment / conditional renewal 5 yr -Renewal
Safety variations
Safety Referrals

Pharmacovigilance and Risk Management
14
What we know at the end of the clinical trial programme…
What we don’t know! . What happens when the medicinal product is used in
normal practice? . What is its adverse event
profile?

Pharmacovigilance Risk Assessment Committee (PRAC)
Assesses aspects of risk management (detection, assessment, minimisation and communication of risk of adverse reactions)
15
• 1 member (+ 1 alternate) per MS • + NO & IS
• 6 experts nominated by EC • 1 member (+ 1 alternate)
healthcare professionals • 1 member (+ 1 alternate)
patients organisations
Chair: Dr June Raine

OR
MAH NCA
Pharmacovigilance and Risk Management;
Data Collection and Management
Safety monitoring
Patient with ADR
Healthcare professional
ADR report
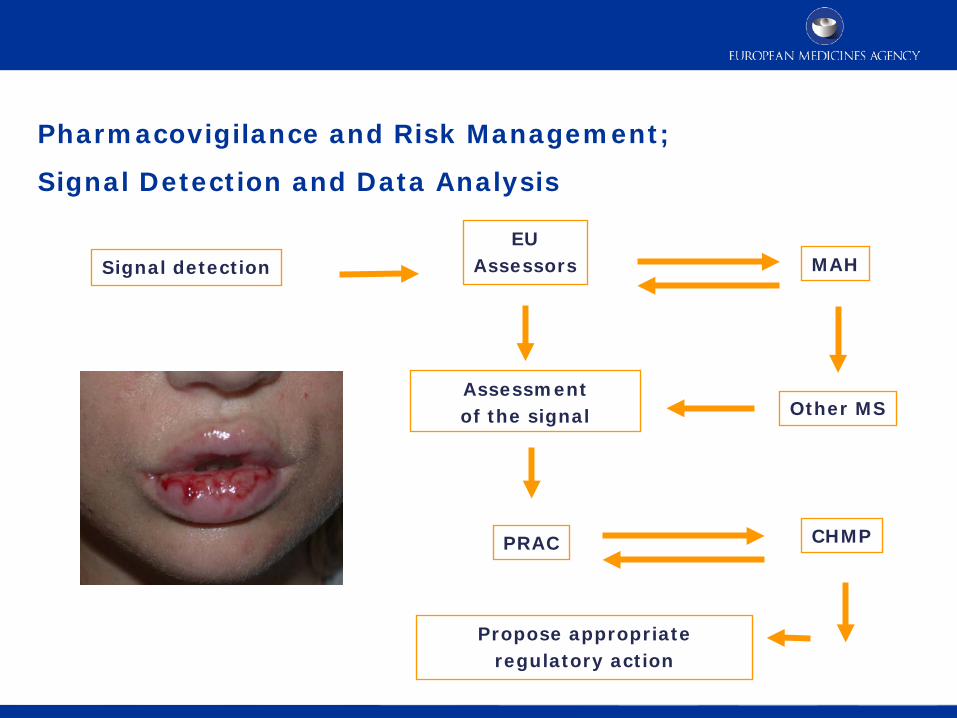
Pharmacovigilance and Risk Management;
Signal Detection and Data Analysis
MAH EU
Assessors Signal detection
Other MS
PRAC CHMP
Assessment of the signal
Propose appropriate regulatory action

Acronyms
• ADR = Adverse Reaction
• AR = Assessment Report
• CHMP = Committee for Medicinal Products for Human Use
• CD = Commission Decision
• D1, etc = Day 1 (procedural timeline)
• GCP – Good Clinical Practice
• GLP = Good Laboratory Practice
• GMP = Good Manufacturing Practice
• LoQ = List of Questions
• LoOIs = List of Outstanding Issues
18
• MAH = Marketing Authorisation Holder
• MS = Member State
• OE = Oral explanation
• PASS = Post Authorisation Safety Study
• PI = product information
• PRAC = Pharmacovigilance Risk Assessment Committee
• PSUR = Periodic Safety Update Report
• RMP – Risk Management Plan
• SmPC = Summary of Product Characteristics