Embed Size (px)
Citation preview

Host-Factor Enhancement of Therapy for Tuberculosis
By
Michael David Schump
A dissertation submitted in partial satisfaction of the
requirements for the degree of
Doctor of Philosophy
in
Infectious Diseases and Immunity
in the
Graduate Division
of the
University of California, Berkeley
Committee in charge:
Professor Lee W. Riley, Chair
Professor Martyn T. Smith
Adjunct Professor Sangwei Lu
Spring 2015

© 2015
Michael David Schump
All Rights Reserved

1
Abstract
Host-Factor Enhancement of Therapy for Tuberculosis
by
Michael David Schump
Doctor of Philosophy in Infectious Diseases and Immunity
University of California, Berkeley
Professor Lee W. Riley, Chair
Tuberculosis (TB) is a disease of major public health importance and improvements to its treatment could greatly benefit efforts aimed at eliminating the disease. Current treatment options for TB are limited in effectiveness and have numerous fundamental failings due to the necessarily lengthy duration of therapy and toxicity of the antimicrobial drugs deployed, among other issues. The studies described herein where undertaken with the goal of developing adjunctive treatments or modifications of existing treatments which could improve the treatment course, outcome, or both for standard TB antimicrobial chemotherapy.
Three areas of research are discussed beginning with adaptive immune augmentation through therapeutic vaccination, proceeding to investigations of innate immune adjuvant therapy and concluding with host environment mediated improvement of selectivity index of TB antimicrobial compounds.
A post-treatment, therapeutic vaccine was studied with the goal of developing a tool which could prevent relapse or reactivation disease. Though the project was a follow up to a study which demonstrated exceptional protection, the vaccine candidate did not demonstrate any detectable efficacy in three parallel murine infection experiments. Possible reasons and implications of this failure are discussed.
Because correlates of protection for adaptive immunity to TB are poorly understood and have not proven to be tractable for intervention, innate immune enhancement was investigated. Autophagy, a cell-intrinsic process with antimicrobial capabilities, was selected due to its well described tuberculocidal activity and pharmacologic manipulability. However, despite the apparent capacity of some test compounds to increase autophagic flux, none demonstrated robust restriction of mycobacterial growth in murine or human macrophages. That study did, however, lead to the serendipitous discovery that pH based drug partitioning can increase the selectivity index of antimicrobial drugs against M. tuberculosis inside cultured macrophages.

i
I dedicate this work to my parents, David and Elizabeth Schump – they made it all possible.

ii
Acknowledgements
Firstly, I would like to thank my mentor, Dr. Lee Riley, for believing in me from the
beginning. I am very grateful that he remained confident in me throughout the circuitous route
that my studies took.
I am also grateful for the many other supportive and vastly knowledgeable faculty members who
contributed to my professional development. I would specifically like to thank Dr. Sangwei Lu,
who had a hand in guiding me through nearly my entire time in the program and who was
generous enough as to serve both as my qualifying exam chair and subsequently on my
dissertation committee. Dr. Martyn Smith, too, should be recognized for generously agreeing to
oversee my work as a dissertation committee member. Additionally, I would like to thank Dr.
John Swartzberg for first introducing me to the instructor’s view of education and Dr. Paurene
Duramad for helping me take the next step and become more effective as a graduate student
instructor.
I owe a great deal to the fortuitous timing of the arrival of Dr. Sarah Stanley to our department. I
know that my work was considerably strengthened by the many helpful discussions I had with
her and also by the tools she was so kind as to share with me. Once her laboratory was
established, I benefited tremendously from working with the energetic and skilled scientists she
recruited. Jonathan Braverman, Kim Sogi, and Katie Lien, in particular, were friendly and
supportive biosafetly-level-three labmates.
Thanks are due, also, to Dr. Peter Gallagher at Eli Lilly and Co. for coordinating the transfer of
the fluoxetine analogs. It took over a year, so I am grateful for his perseverance.
I must gratefully acknowledge the contributions of Doug Fox, Sarah Weng and Julio Ortiz-
Canseco to my work. Indeed, they are the only colleagues who routinely helped with
experiments in a hands-on manner. When it was time to pick up a pipettor, I knew who I could
count on.
I would like to thank all the members –past and present– of the Riley lab for fostering a
welcoming and enjoyable work environment. They are too numerous to list and their
contributions to my work and overall experience are more numerous still, but I feel I must name
a few. Melaine Delcroix, Olivera Marjanovic, Amador Goodridge and Hillary Berman welcomed
me into the lab while Nicole Tarlton, Sheila Adams-Sapper, and Amelia Wallace gave me the
opportunity to pass on my working knowledge of how the lab operates.
In closing, I would like to acknowledge the seemingly boundless support of my girlfriend, Emma
Essock-Burns. Her perspective and continuous encouragement were vital to getting me through
these last five years.

iii
TABLE OF CONTENTS
Chapter 1 -------------------------------------------------------------------------------------- 1
Introduction -------------------------------------------------------------------------- 1
Tuberculosis – Pathogen and Disease -------------------------------------------- 2
Epidemiology -------------------------------------------------------------------------- 2
Treatment and Prevention ---------------------------------------------------------- 3
Microbial Niche ----------------------------------------------------------------------- 4
References ---------------------------------------------------------------------------------- 8
Chapter 2 ----------------------------------------------------------------------------------- 13
Trial of a Post-exposure, Therapeutic Vaccine for TB Based on the Mycobacterial Mce1a Protein ------------------------------------------------- 13
Tuberculosis – Control by Vaccination ----------------------------------------- 14
Mce1A Post-Exposure Vaccine Candidate ------------------------------------- 17
Cornell Model of Reactivation/Relapse Tuberculosis ----------------------- 17
Materials and Methods ------------------------------------------------------------ 18
Result - Mce1A Vaccine in Cornell Model with Wild Type Mtb ------------ 19
Discussion ---------------------------------------------------------------------------- 24
Conclusions -------------------------------------------------------------------------- 25
References -------------------------------------------------------------------------------- 28
Chapter 3 ----------------------------------------------------------------------------------- 33
Effect of mTOR-Independent Inducers of Autophagy on -------------- 33
Intracellular M. tuberculosis --------------------------------------------------- 33
Introduction ------------------------------------------------------------------------- 34
Immune Adjuvants ----------------------------------------------------------------- 34
Autophagy ---------------------------------------------------------------------------- 34
mTOR Independence --------------------------------------------------------------- 35
Adjunctive Therapy ---------------------------------------------------------------- 37
Materials and Methods ------------------------------------------------------------ 38

iv
Results and Conclusion ------------------------------------------------------------ 39
References -------------------------------------------------------------------------------- 45
Chapter 4 ----------------------------------------------------------------------------------- 48
Partitioning of Antimicrobial Compounds by pH Based Ion Trapping Can Increase Intra-Macrophage Selectivity Index Against Mycobacterium tuberculosis --------------------------------------------------- 48
Abstract ------------------------------------------------------------------------------- 49
Introduction ------------------------------------------------------------------------- 49
Materials and Methods ------------------------------------------------------------ 50
Results -------------------------------------------------------------------------------- 52
Discussion ---------------------------------------------------------------------------- 57
References --------------------------------------------------------------------------------- 60
Dissertation Summary and Conclusions ------------------------------------------ 63
Appendix A ---------------------------------------------------------------------------------- 64

v
TABLE OF FIGURES

1
CHAPTER 1
INTRODUCTION

2
TUBERCULOSIS – PATHOGEN AND DISEASE
Tuberculosis (TB) is an ancient disease of humans caused by the rod shaped bacteria of the
Mycobacterium tuberculosis (Mtb) complex, which continues to be a prolific killer of humans to
this day. Mtb kills more people each year than any other single infectious agent except HIV
(Lawn and Zumla 2011). The origin of the disease is still the subject of debate (Bos, Harkins et
al. 2014), but it has been known at least since the ancient Greek era when it was called phthisis
(Pease 1940). Although Mtb and several related mycobacterial species are capable of infecting a
wide range on animal hosts (O'Reilly and Daborn 1995), throughout this work TB will always
refer to the disease of humans, and unless otherwise specified, the causative agent may be
assumed to be Mtb.
In the medieval ages TB was called the White Plague or White Death and has been known more
recently as Consumption. TB is a wasting disease most commonly characterized by coughing
and weight loss, though all cell types and tissues can be infected (Harries and Dye 2006; Lawn
and Zumla 2011). In advanced cases with severe pathology, hemoptysis is often observed.
Practically all new infections occur by the inhalation of droplet nuclei containing Mtb expelled
from an individual suffering from active TB. Mtb was proven to be the definitive cause of
tuberculosis by Robert Koch in 1882 (Koch 1882).
In the absence of treatment, the case fatality rate is around 50% for a typical case of pulmonary
TB while disseminated disease is practically always fatal (Mitchison 2005). Many people –
probably the majority– who come into contact with the pathogen do not show signs of infection,
and even among those who do, 90-95% of infected individuals will contain the infection and
have what is known as a latent infection that never progresses to active disease (Harries and Dye
2006). The other 5-10% of individuals who carry a latent infection for a time will eventually
have the infection reactivate and become symptomatic and transmissible. HIV/AIDS and other
forms of immune suppression increase the likelihood of an infected person developing active
disease dramatically; among latently infected people, HIV infected individuals have a risk of
reactivation of around 8%-10% per year rather than 5-10% over a lifetime for HIV negative
people (Gordin and Masur 2012). The impact of HIV/AIDS co-infection with TB is enormous
and in the absence of well administered care, the prognosis is extremely bleak (Aaron, Saadoun
et al. 2004).
EPIDEMIOLOGY
The most recent report on the global epidemiology of TB from the World Health Organization
states that in 2013, “an estimated 9.0 million people developed TB and 1.5 million died from the
disease, 360,000 of whom were HIV-positive (WHO 2014).” It is estimated that around one third
of the world’s population is latently infected by Mtb (Dye, Scheele et al. 1999). Prevalence and
incidence of disease vary markedly with geographic location as well as with socio-economic
status in any one place (Dye and Williams 2010). See Figure 1.1 below which shows a map
detailing the estimated incidence of tuberculosis worldwide in 2013. Within the United States,
most cases are among foreign born individuals and are believed to be the result of reactivation of
a latent infection acquired abroad (Gordin and Masur 2012). The highest burden countries in
terms of overall number of cases are India, China, Nigeria and Pakistan. The highest incidence

3
rates of active disease are found in Swaziland, Lesotho, South Africa and Namibia (WHO 2014
pg. 33)
FIGURE 1-1: GLOBAL ESTIMATED TB INCIDENCE RATES PER 100,000
POPULATION IN 2013
SOURCE: (WHO 2014)
TREATMENT AND PREVENTION
The first major step toward preventing TB infections was the identification of the causative
microorganism by Robert Koch in 1882. Once the etiology of the disease was understood and the
primary mode of transmission (i.e., droplet nuclei expelled by coughing) known, the benefit of
simple interventions such as isolation of patients and improvement of ventilation in relevant
buildings was obvious. By the 1920’s, efforts to develop a vaccine had met with success and the
only currently licensed vaccine for the prevention of TB, known as BCG, dates from this period
(Liu, Tran et al. 2009). It is currently the most widely used vaccine worldwide. The efficacy of
BCG has been studied in depth and found to be highly variable; estimates range from zero to
eighty percent protection from pulmonary TB in adults, though efficacy preventing disseminated
disease in children is much more robust (Brewer 2000). Vaccination as a control strategy for
tuberculosis will be addressed in chapter two.

4
The first powerful treatments for halting the growth of Mtb were para-amino salicylic acid,
discovered by Jorgen Lehmann in 1943 and thiosemicarbazone discovered by Gerhard Domagk
around 1945 (Daniel 2006), while the first antibiotic which could kill the bacterium was
streptomycin (Schatz, Bugle et al. 1944). The activity of streptomycin was demonstrated by the
clinical trials following the drug’s isolation which are also famous for being the first example of
rigorous randomization in clinical trial design (BMJ 1948). Unfortunately, Mtb began evolving
resistance to streptomycin immediately and resistant strains were described before the end of the
first trials (Youmans, Williston et al. 1946). Numerous other compounds were successfully
developed in the middle of the twentieth century for the treatment of TB, but by the beginning of
the twentieth century, resistance to exiting compounds was rapidly increasing while development
of new TB drugs was lacking(Gandhi, Nunn et al. 2010). The need for new therapeutics and
efforts to develop them, by modulating host functions and by leveraging elements of the host
environment, are the subjects of chapters three and four, respectively.
MICROBIAL NICHE
Mycobacterium tuberculosis generally gains access to its niche by inhalation into the lower
respiratory tract where even a single bacillus can cause infection. The most commonly cited
initial host cell is the tissue resident macrophage, but it has been shown that alveolar epithelial
cells can also be infected (Bermudez and Goodman 1996; Lin, Zhang et al. 1998) and a protein
which facilitates invasion of non-phagocytic cells by Mtb has been identified (Arruda, Bomfim et
al. 1993). Phagocytic host cells, such as macrophages and dendritic cells (Wolf, Linas et al.
2007) ingest bacteria with the help of several defined receptors (Ernst 1998). Prior to ingestion
by phagocytes, though, the initial contact of the bacillus with alveolar fluid on the mucosal lining
must be considered, and that fluid has been found to contain several important components
which govern the subsequent encounter of Mtb with immune cells including surfactants,
hydrolases, and complement (Arcos, Diangelo et al. 2015).
In a naïve host, no adaptive response is present, and the bacteria grow relatively unrestricted,
primarily within macrophages. While natural killer cells (Junqueira-Kipnis, Kipnis et al. 2003)
and innate immune effectors are present, they are generally unable to contain the infection until
the onset of adaptive immunity, which comes into force after two weeks of infection (Urdahl,
Shafiani et al. 2011). Nevertheless, innate immune sensors and effectors shape the immune
response (Korbel, Schneider et al. 2008). Interplay between bacillus and host is complex at this
point and mycobacterial avoidance of innate immune recognition involves numerous evasion
techniques including masking of immune-reactive bacterial components (Cambier, Takaki et al.
2014), effector molecule secretion (Stanley, Raghavan et al. 2003), and manipulation of
chemokine signaling (Slight and Khader 2013).
Once an adaptive response is active, a balance of inflammation and tissue repair is necessary for
the host to prevent unchecked bacterial growth on the one hand, while preventing excessive
tissue pathology on the other (Saunders and Britton 2007). For some years, the TH1/TH2
paradigm was applied to TB in the way it had been demonstrated to govern the outcome of

5
leishmaniasis (Mougneau, Bihl et al. 2011) and was thought to be analogous. Specifically, it was
thought that a TH1 response is protective while a TH2 response is deleterious (Demissie, Abebe
et al. 2004). It has become clear, however, that this view is overly simplistic (Jung, LaCourse et
al. 2002), and a more complicated balance of immune effectors appears to be necessary. Critical
components of a protective response have mostly been demonstrated by increased susceptibility
of gene knockout mice. In this way, interferon gamma, inducible nitric oxide synthase, tumor
necrosis factor alpha, CD4 and CD8 T cells, as well as other immune components, have been
demonstrated to be indispensable for control of mycobacterial infection (Flynn and Chan 2001).
When the full complement of immune cells are recruited to a focus of infection, they coalesce
into a structure called a granuloma which consists of concentric rings of various cell types and
debris. See Figure 1-2 for an overview of several commonly observed granuloma types. Other
types of granulomas than those depicted in the figure occur, too, such as calcified lesions.
Macrophages in the vicinity are often observed to increase in size and become laden with lipid
droplets. Such giant cells are referred to as foamy macrophages and they, along with the
infecting bacteria themselves, contribute a great deal of lipid rich material to the caseous core of
typical granulomas in humans (Russell, Cardona et al. 2009) The core of human granulomas are
often necrotic and hypoxic (Tsai, Chakravarty et al. 2006; Via, Lin et al. 2008). The kinetics of
formation and disintegration of granulomas have been studied in some detail using modern
imaging modalities (Egen, Rothfuchs et al. 2008).
While the structures themselves are reasonably well described, their function is a matter of some
debate (Russell 2007; Paige and Bishai 2010). Granulomas were long thought to represent a host
adaptation which limits infiltration, but they also allow for the persistence of bacteria, making it
less clear which side of the host-pathogen interaction is benefited more. While dissolution of
granulomas, or failure to form them in the first place, is associated with poor control, the bacteria
have been observed to more efficiently replicate and infect newly recruited macrophages within
established granulomas (Cosma, Humbert et al. 2008; Davis and Ramakrishnan 2009). Studies of
granulomas in latently infected non-human primates have further underscored the polymorphic
nature of these structures (Lin, Ford et al. 2013). Within an infected lung, granulomas can
behave independently, with some expanding while some contract or stay static. Consistent with
those observations are histologic examinations of human resected lung tissue from TB patients.
Such studies have demonstrated that “the lung of a chronic TB patient contains a diversity of
micro-anatomical niches created by the different immunological processes occurring
independently at these sites.” (Kaplan, Post et al. 2003) Importantly, though, even in the areas
with the greatest bacterial load –namely the luminal surface of cavities– bacteria are primarily
observed inside of macrophages.
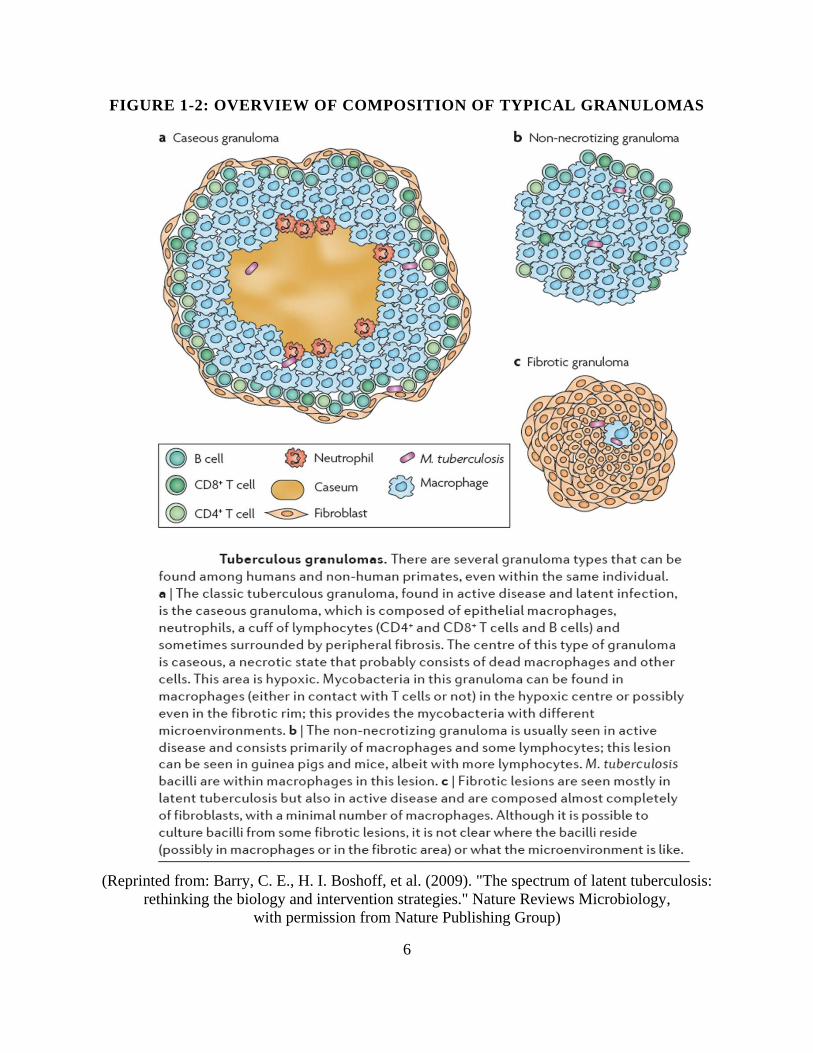
6
FIGURE 1-2: OVERVIEW OF COMPOSITION OF TYPICAL GRANULOMAS
(Reprinted from: Barry, C. E., H. I. Boshoff, et al. (2009). "The spectrum of latent tuberculosis:
rethinking the biology and intervention strategies." Nature Reviews Microbiology,
with permission from Nature Publishing Group)

7
The intracellular environment experienced by Mtb within macrophages has been the subject of
considerable research (Vergne, Chua et al. 2004; Ehrt and Schnappinger 2009; Cambier, Falkow
et al. 2014). Mycobacteria are generally thought to reside in partially acidified phagosomes
(Sturgill-Koszycki, Schlesinger et al. 1994) which are prevented from fusing with lysosomes
(Armstrong and Hart 1971). That Mtb resides in this intracellular niche is counterintuitive given
the primary role of macrophages in engulfing and destroying bacteria (Langermans, Hazenbos et
al. 1994). Indeed, macrophages deploy several systems for generating reactive oxygen and
nitrogen species (Bedard and Krause 2007) which have been demonstrated to be
mycobactericidal (Zahrt and Deretic 2002). Autophagy, a cell intrinsic mechanism which has
been reported to have anti-mycobacterial activity (Gutierrez, Master et al. 2004), is also inhibited
by the bacteria (Deretic, Singh et al. 2006). The manipulation of autophagy in host cells is the
subject of chapter three. There have been reports of Mtb found in the cytosol (van der Wel, Hava
et al. 2007), but the bacteria are generally thought to reside within a membrane bound
phagosome in an incompletely mature state which partially acidifies to around pH 6.4 (Pethe,
Swenson et al. 2004; Rohde, Yates et al. 2007). Like many intracellular pathogens, Mtb actively
competes with the host for acquisition of micronutrients such as iron (Agranoff and Krishna
2004).
After the establishment of granulomas, sufficient tissue destruction most occur for bacteria to
spill back out into airways in order to facilitate onward transmission. Matrix metalloproteinases
have been implicated in this progression (Parks, Wilson et al. 2004; Ong, Elkington et al. 2014)
and the primary driver of this pathology is thought to be the host immune response. That this
pathology is mediated by the host immune system is supported by the observation that
HIV/AIDS patients are actually less likely to transmit infection (Cauthen, Dooley et al. 1996)
despite evidence that they harbor higher bacterial loads (Diedrich and Flynn 2011). In the end,
then, it appears that the host is manipulated into opening the door for the bacteria to exit and
establish a new infection whereupon the cycle repeats itself.
The three chapters which follow describe efforts directed at breaking this cycle of transmission
by modifying the host response to infection using exogenously supplied antigenic, immune-
modulatory, or antimicrobial substances, respectively.

8
REFERENCES
Aaron, L., D. Saadoun, et al. (2004). "Tuberculosis in HIV-infected patients: a comprehensive
review." Clin Microbiol Infect 10(5): 388-398.
Agranoff, D. and S. Krishna (2004). "Metal ion transport and regulation in Mycobacterium
tuberculosis." Front Biosci 9: 2996-3006.
Arcos, J., L. E. Diangelo, et al. (2015). "Lung mucosa lining fluid modifies Mycobacterium
tuberculosis to reprogram human neutrophil killing mechanisms." J Infect Dis.
Armstrong, J. A. and P. D. Hart (1971). "Response of cultured macrophages to Mycobacterium
tuberculosis, with observations on fusion of lysosomes with phagosomes." J Exp Med 134(3 Pt
1): 713-740.
Arruda, S., G. Bomfim, et al. (1993). "Cloning of an M. tuberculosis DNA fragment associated
with entry and survival inside cells." Science 261(5127): 1454-1457.
Bedard, K. and K. H. Krause (2007). "The NOX family of ROS-generating NADPH oxidases:
physiology and pathophysiology." Physiol Rev 87(1): 245-313.
Bermudez, L. E. and J. Goodman (1996). "Mycobacterium tuberculosis invades and replicates
within type II alveolar cells." Infect Immun 64(4): 1400-1406.
BMJ, M. R. C.-. (1948). Streptomycin Treatment of Pulmonary Tuberculosis. A Medical
Research Council Investigation. 2: 769-782.
Bos, K. I., K. M. Harkins, et al. (2014). "Pre-Columbian mycobacterial genomes reveal seals as a
source of New World human tuberculosis." Nature 514(7523): 494-497.
Brewer, T. F. (2000). "Preventing tuberculosis with bacillus Calmette-Guerin vaccine: a meta-
analysis of the literature." Clin Infect Dis 31 Suppl 3: S64-67.
Cambier, C. J., S. Falkow, et al. (2014). "Host evasion and exploitation schemes of
Mycobacterium tuberculosis." Cell 159(7): 1497-1509.
Cambier, C. J., K. K. Takaki, et al. (2014). "Mycobacteria manipulate macrophage recruitment
through coordinated use of membrane lipids." Nature 505(7482): 218-222.
Cauthen, G. M., S. W. Dooley, et al. (1996). "Transmission of Mycobacterium tuberculosis from
tuberculosis patients with HIV infection or AIDS." Am J Epidemiol 144(1): 69-77.
Cosma, C. L., O. Humbert, et al. (2008). "Trafficking of superinfecting Mycobacterium
organisms into established granulomas occurs in mammals and is independent of the Erp and
ESX-1 mycobacterial virulence loci." J Infect Dis 198(12): 1851-1855.
Davis, J. M. and L. Ramakrishnan (2009). "The role of the granuloma in expansion and
dissemination of early tuberculous infection." Cell 136(1): 37-49.

9
Demissie, A., M. Abebe, et al. (2004). "Healthy individuals that control a latent infection with
Mycobacterium tuberculosis express high levels of Th1 cytokines and the IL-4 antagonist IL-
4delta2." J Immunol 172(11): 6938-6943.
Deretic, V., S. Singh, et al. (2006). "Mycobacterium tuberculosis inhibition of phagolysosome
biogenesis and autophagy as a host defence mechanism." Cellular Microbiology 8(5): 719-727.
Diedrich, C. R. and J. L. Flynn (2011). "HIV/M. tuberculosis co-infection immunology: How
does HIV exacerbate TB?" Infect Immun.
Dye, C., S. Scheele, et al. (1999). "Consensus statement. Global burden of tuberculosis:
estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and
Monitoring Project." JAMA 282(7): 677-686.
Dye, C. and B. G. Williams (2010). "The population dynamics and control of tuberculosis."
Science 328(5980): 856-861.
Egen, J. G., A. G. Rothfuchs, et al. (2008). "Macrophage and T Cell Dynamics during the
Development and Disintegration of Mycobacterial Granulomas." Immunity 28(2): 271-284.
Ehrt, S. and D. Schnappinger (2009). "Mycobacterial survival strategies in the phagosome:
defence against host stresses." Cell Microbiol 11(8): 1170-1178.
Ernst, J. D. (1998). "Macrophage receptors for Mycobacterium tuberculosis." Infect Immun
66(4): 1277-1281.
Flynn, J. L. and J. Chan (2001). "Immunology of tuberculosis." Annu Rev Immunol 19: 93-129.
Gandhi, N. R., P. Nunn, et al. (2010). "Multidrug-resistant and extensively drug-resistant
tuberculosis: a threat to global control of tuberculosis." Lancet 375(9728): 1830-1843.
Gordin, F. M. and H. Masur (2012). "Current approaches to tuberculosis in the united states."
JAMA 308(3): 283-289.
Gutierrez, M. G., S. S. Master, et al. (2004). "Autophagy is a defense mechanism inhibiting BCG
and Mycobacterium tuberculosis survival in infected macrophages." Cell 119(6): 753-766.
Harries, A. D. and C. Dye (2006). "Tuberculosis." Ann Trop Med Parasitol 100(5-6): 415-431.
Jung, Y. J., R. LaCourse, et al. (2002). "Evidence inconsistent with a negative influence of T
helper 2 cells on protection afforded by a dominant T helper 1 response against Mycobacterium
tuberculosis lung infection in mice." Infect Immun 70(11): 6436-6443.
Junqueira-Kipnis, A. P., A. Kipnis, et al. (2003). "NK cells respond to pulmonary infection with
Mycobacterium tuberculosis, but play a minimal role in protection." J Immunol 171(11): 6039-
6045.

10
Kaplan, G., F. A. Post, et al. (2003). "Mycobacterium tuberculosis growth at the cavity surface: a
microenvironment with failed immunity." Infect Immun 71(12): 7099-7108.
Koch, R. (1882). Ueber die Aetiologie der Tuberculose, Berliner Medicinische Wochenschrift.
Korbel, D. S., B. E. Schneider, et al. (2008). "Innate immunity in tuberculosis: myths and truth."
Microbes and Infection 10(9): 995-1004.
Langermans, J. A., W. L. Hazenbos, et al. (1994). "Antimicrobial functions of mononuclear
phagocytes." J Immunol Methods 174(1-2): 185-194.
Lawn, S. D. and A. I. Zumla (2011). "Tuberculosis." Lancet.
Lin, P. L., C. B. Ford, et al. (2013). "Sterilization of granulomas is common in active and latent
tuberculosis despite within-host variability in bacterial killing." Nat Med.
Lin, Y., M. Zhang, et al. (1998). "Chemokine production by a human alveolar epithelial cell line
in response to Mycobacterium tuberculosis." Infect Immun 66(3): 1121-1126.
Liu, J., V. Tran, et al. (2009). "BCG vaccines: their mechanisms of attenuation and impact on
safety and protective efficacy." Hum Vaccin 5(2): 70-78.
Mitchison, D. A. (2005). "The diagnosis and therapy of tuberculosis during the past 100 years."
Am J Respir Crit Care Med 171(7): 699-706.
Mougneau, E., F. Bihl, et al. (2011). "Cell biology and immunology of Leishmania." Immunol
Rev 240(1): 286-296.
O'Reilly, L. M. and C. J. Daborn (1995). "The epidemiology of Mycobacterium bovis infections
in animals and man: a review." Tuber Lung Dis 76 Suppl 1: 1-46.
Ong, C. W., P. T. Elkington, et al. (2014). "Tuberculosis, pulmonary cavitation, and matrix
metalloproteinases." Am J Respir Crit Care Med 190(1): 9-18.
Paige, C. and W. R. Bishai (2010). "Penitentiary or penthouse condo: the tuberculous granuloma
from the microbe's point of view." Cell Microbiol 12(3): 301-309.
Parks, W. C., C. L. Wilson, et al. (2004). "Matrix metalloproteinases as modulators of
inflammation and innate immunity." Nat Rev Immunol 4(8): 617-629.
Pease, A. S. (1940). "Some Remarks on the Diagnosis and Treatment of Tuberculosis in
Antiquity." Isis 31(2): 380-393.
Pethe, K., D. L. Swenson, et al. (2004). "Isolation of Mycobacterium tuberculosis mutants
defective in the arrest of phagosome maturation." Proc Natl Acad Sci U S A 101(37): 13642-
13647.

11
Rohde, K., R. M. Yates, et al. (2007). "Mycobacterium tuberculosis and the environment within
the phagosome." Immunol Rev 219: 37-54.
Russell, D. G. (2007). "Who puts the tubercle in tuberculosis?" Nat Rev Microbiol 5(1): 39-47.
Russell, D. G., P.-J. Cardona, et al. (2009). "Foamy macrophages and the progression of the
human tuberculosis granuloma." Nature Immunology 10(9): 943-948.
Saunders, B. M. and W. J. Britton (2007). "Life and death in the granuloma: immunopathology
of tuberculosis." Immunol Cell Biol 85(2): 103-111.
Schatz, A., E. Bugle, et al. (1944). "Streptomycin, a Substance Exhibiting Antibiotic Activity
Against Gram-Positive and Gram-Negative Bacteria.∗†." Experimental Biology and Medicine
55(1): 66-69.
Slight, S. R. and S. A. Khader (2013). "Chemokines shape the immune responses to
tuberculosis." Cytokine & Growth Factor Reviews 24(2): 105-113.
Stanley, S. A., S. Raghavan, et al. (2003). "Acute infection and macrophage subversion by
Mycobacterium tuberculosis require a specialized secretion system." Proc Natl Acad Sci U S A
100(22): 13001-13006.
Sturgill-Koszycki, S., P. Schlesinger, et al. (1994). "Lack of acidification in Mycobacterium
phagosomes produced by exclusion of the vesicular proton-ATPase." Science 263(5147): 678-
681.
Tsai, M. C., S. Chakravarty, et al. (2006). "Characterization of the tuberculous granuloma in
murine and human lungs: cellular composition and relative tissue oxygen tension." Cell
Microbiol 8(2): 218-232.
Urdahl, K. B., S. Shafiani, et al. (2011). "Initiation and regulation of T-cell responses in
tuberculosis." Mucosal Immunol 4(3): 288-293.
van der Wel, N., D. Hava, et al. (2007). "M. tuberculosis and M. leprae translocate from the
phagolysosome to the cytosol in myeloid cells." Cell 129(7): 1287-1298.
Vergne, I., J. Chua, et al. (2004). "Cell biology of mycobacterium tuberculosis phagosome."
Annu Rev Cell Dev Biol 20: 367-394.
Via, L. E., P. L. Lin, et al. (2008). "Tuberculous granulomas are hypoxic in guinea pigs, rabbits,
and nonhuman primates." Infect Immun 76(6): 2333-2340.
WHO (2014). "Global tuberculosis report 2014. Geneva: World Health Organization,
2014. http://www.who.int/tb/publications/global_report/en/ (accessed Feb 26, 2015)."

12
Wolf, A. J., B. Linas, et al. (2007). "Mycobacterium tuberculosis Infects Dendritic Cells with
High Frequency and Impairs Their Function In Vivo." The Journal of Immunology 179(4): 2509-
2519.
Youmans, G. P., E. H. Williston, et al. (1946). "Increase in resistance of tubercle bacilli to
streptomycin; a preliminary report." Proc Staff Meet Mayo Clin 21: 126.
Zahrt, T. C. and V. Deretic (2002). "Reactive nitrogen and oxygen intermediates and bacterial
defenses: unusual adaptations in Mycobacterium tuberculosis." Antioxid Redox Signal 4(1): 141-
159.

13
CHAPTER 2
TRIAL OF A POST-EXPOSURE, THERAPEUTIC VACCINE FOR TB BASED ON THE MYCOBACTERIAL MCE1A PROTEIN

14
TUBERCULOSIS – CONTROL BY VACCINATION
Vaccines are among the most powerful tools for infectious disease prevention. Many diseases for
which effective vaccines exist have been drastically reduced in prevalence and one, smallpox,
has been altogether eradicated ([CDC] 2011). Historically, vaccines have been prophylactic
interventions to prevent infection by priming a host response in advance of exposure to a
pathogen. However in the case of tuberculosis, due to the enormous pool of already infected
individuals, estimated at one third of the global population (Dye, Scheele et al. 1999), a post-
exposure vaccine holds great potential for preventing the development of disease among those
already latently infected with the bacillus. Additionally, an effective therapeutic vaccine could
hypothetically reduce the relapse rate after treatment (Wallis, Doherty et al. 2009).
Unfortunately, despite the enormous toll tuberculosis exerts on the human population, as
described in chapter one, only one vaccine against it has been developed, Bacillus Calmette–
Guérin (BCG). Although safe, unfortunately, the vaccine does not robustly protect adults from
pulmonary tuberculosis (Brewer 2000), even if booster doses are given (Rodrigues, Pereira et al.
2005).
The protection afforded by BCG has proven to be highly variable depending on geographic
location (Black, Weir et al. 2002) and has been suggested to be dependent on many factors such
as concomitant helminth infection (Elias, Britton et al. 2008), which biases toward a non-
protective TH2 T-cell response, and exposure to cross-reactive mycobacterial antigens from non-
tuberculous mycobacteria (Lin, Reddy et al. 2009), which leads to underestimation of vaccine
activity due to unequal baseline immunity in comparator groups or interference with BCG
response (Brandt, Feino Cunha et al. 2002). Other factors such as the nutritional status of the
recipient and latitude (possibly mediated by vitamin D levels) have been suggested (Fine 1995).
The strain used for vaccination is yet another important variable, since deviations in handling
have led to differences between strains used in different parts of the world (Liu, Tran et al.
2009). While BCG does protect against childhood forms of the disease and disseminated
infection (Rodrigues, Diwan et al. 1993), these are not the source of most new infections, so
protection afforded in that setting does little to avert future cases of TB. An additional layer of
complexity is added by variability in the strain of Mtb actually encountered by individuals after
vaccination which has been shown to lead to different immunologic manifestations (Ordway,
Shang et al. 2011).
Vaccination against TB is particularly challenging, because unlike many other infectious
diseases like chickenpox or yellow fever, a resolved episode of TB does not lead to lifelong -or
even particularly powerful- immunity. Indeed, after successful treatment and resolution of TB
infection, only about a one log reduction in bacterial load in the lungs is seen in animal models
and infection rate is not affected (Russell, Barry et al. 2010). Therefore, the protection sought
from a vaccine needs to be better than that induced by a natural infection. Still, it has been
demonstrated that vaccination with subunits (i.e., part[s] of the bacterium) can impact immunity
to the disease since culture filtrate confers similar resistance to BCG (Andersen 1994). Current
approaches using whole cell, subunit, and viral vectored candidates have been reviewed
elsewhere (Kaufmann, Hussey et al. 2010; da Costa, Walker et al. 2015). The exact nature of the
necessary immune response to protect against clinical TB –much less asymptomatic Mtb
infection– is poorly defined (Bhatt, Verma et al. 2015). While both CD4 and CD8 T cells are

15
believed to play important roles, and certain cytokines such as interferon gamma and tumor
necrosis factor alpha have been shown to be involved in containment of Mtb infection in the host
(Flynn and Chan 2001), the precise response that a highly effective vaccine must induce is not
understood (Dietrich and Doherty 2009).
Despite all of these challenges, the potential benefit of a highly effective vaccine against TB is
too great to ignore. Using a mathematical model of TB dynamics, the potential for effective
vaccines to reduce transmission and subsequent disease rates have been estimated (Young and
Dye 2006). Projections for various interventions involving pre- or post-exposure vaccines are
depicted in Figure 2-1 using South Asia as a case study. Though the projections are profoundly
dependent on the assumptions regarding vaccine effectiveness and deployment (in the model
below, the combined coverage and efficacy yield 70% protection of the target population), such
reductions in disease, if realized, would surely rank among the great public health triumphs of
human history.
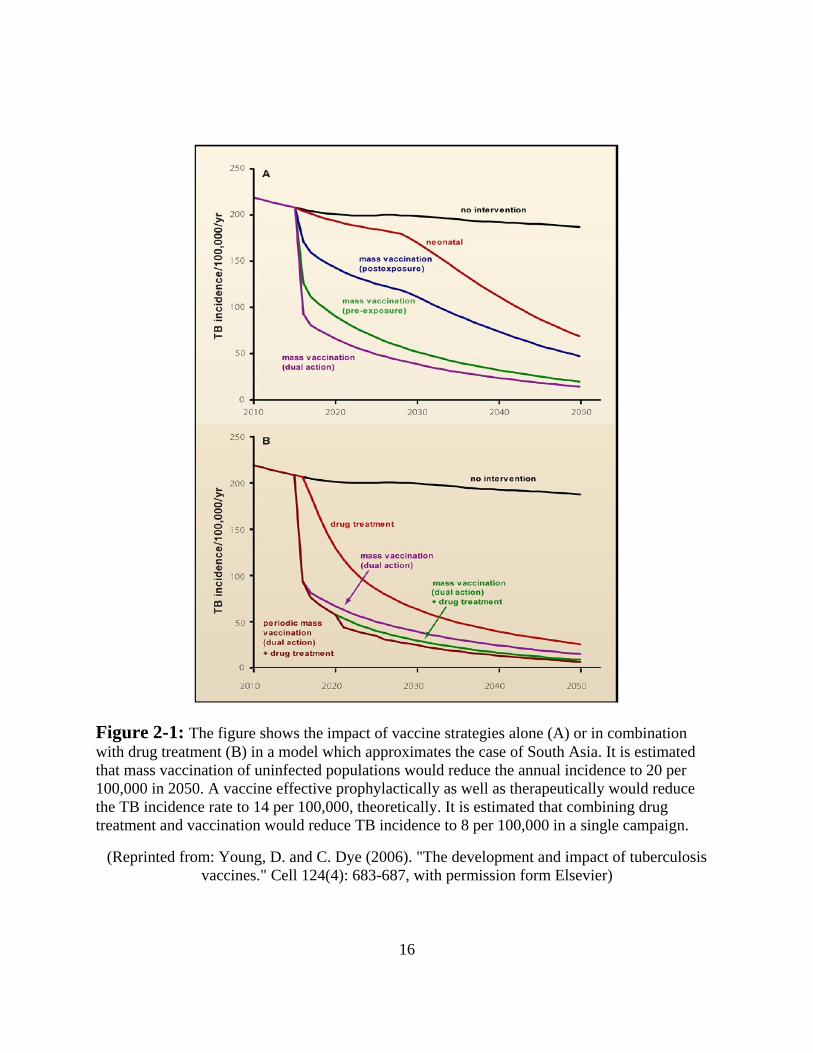
16
Figure 2-1: The figure shows the impact of vaccine strategies alone (A) or in combination
with drug treatment (B) in a model which approximates the case of South Asia. It is estimated
that mass vaccination of uninfected populations would reduce the annual incidence to 20 per
100,000 in 2050. A vaccine effective prophylactically as well as therapeutically would reduce
the TB incidence rate to 14 per 100,000, theoretically. It is estimated that combining drug
treatment and vaccination would reduce TB incidence to 8 per 100,000 in a single campaign.
(Reprinted from: Young, D. and C. Dye (2006). "The development and impact of tuberculosis
vaccines." Cell 124(4): 683-687, with permission form Elsevier)

17
Because as many as one third of the global population is already latently infected with Mtb and
because a very large proportion of disease and transmission stems from reactivation of latent
infection or relapse of incompletely sterilized infections (Lawn and Zumla 2011), a post
exposure vaccine would be particularly promising for use alongside chemotherapy as a means of
limiting TB disease and transmission.
MCE1A POST-EXPOSURE VACCINE CANDIDATE
The Mce1A vaccine candidate consists of a recombinant fragment of the Mce1A protein from
Mtb which spans amino acids 51-454 and includes a motif which has cell penetrating properties
(Arruda, Bomfim et al. 1993; Lu, Tager et al. 2006). The truncation of the protein was intended
to decrease its hydrophobicity and facilitate production and purification of the recombinant
protein in E. coli. Because of its cell penetrating properties, which may facilitate antigen uptake
and because it is known to be expressed in macrophages in-vivo (Uchida, Casali et al. 2007),
Mce1a was selected as a vaccine antigen. In a published study, (Miyata, Cheigh et al. 2012), it
was demonstrated that the Mce1a vaccine confers nearly completely sterilizing immunity in the
lungs of mice when administered intraperitoneally after drug treatment in a modified version of
the “Cornell mouse model.” The Cornell model is described below. The original study did not
make use of any adjuvant or excipient and was delivered in phosphate buffered saline (PBS).
Importantly, the challenge strain of Mtb in that study was a mutant with a targeted knock out of
the repressor of the mce1 operon (mce1R KO). Because the mce1R KO strain has been shown to
overexpress proteins of the mce1 operon (Casali, White et al. 2005), including mce1A, the
potential for the demonstrated efficacy to be an immunologic artifact due to overexpression of
the vaccine antigen was considered. The present study was undertaken to ascertain whether
similar protection would be generated against a wild type strain of Mtb.
CORNELL MODEL OF REACTIVATION/RELAPSE TUBERCULOSIS
Essentially, the Cornell model is meant to mimic reactivation or relapse TB where an initial
infection is brought under control though the use of antibiotics such that no viable bacteria can
be isolated from the lungs of mice for a period of time (McCune, Feldmann et al. 1966).
Subsequently, though, some mice spontaneously reactivate/relapse and have a measurable
bacterial load in their lungs (or other organs, such as spleen). The period after drug treatment is
intended to mimic the phenomenon of latency in humans, which is a state of low or undetectable
bacterial load (Chan and Flynn 2004). Bacterial outgrowth afterward is meant to mimic
reactivation, which can happen spontaneously in humans after a variable period of latency. In
practice though, it is also a model of relapse, which occurs when incomplete sterilization of an
infection leads to eventual treatment failure and bacterial recrudescence. Both of these are
undesirable outcomes with regard to Mtb infection, so a vaccine which can protect against either
of them would be valuable. A typical plot of bacterial load in the lungs and spleens of mice used
in this model is displayed in Figure 2-2.
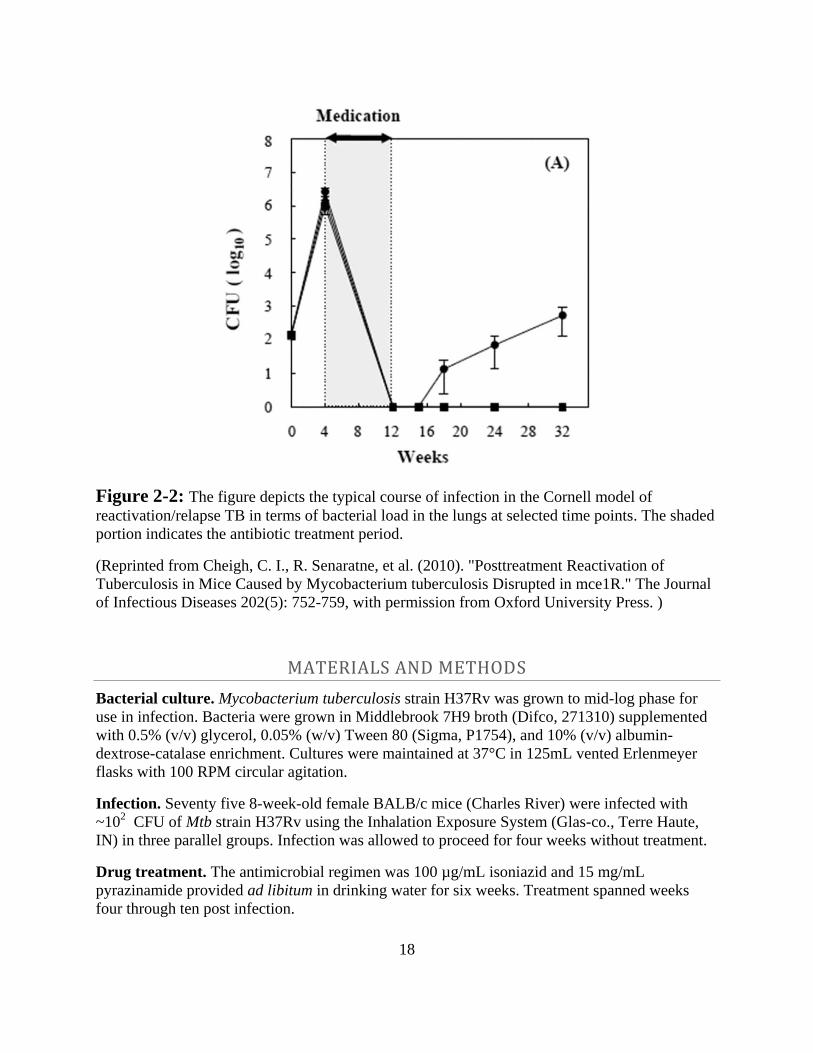
18
Figure 2-2: The figure depicts the typical course of infection in the Cornell model of
reactivation/relapse TB in terms of bacterial load in the lungs at selected time points. The shaded
portion indicates the antibiotic treatment period.
(Reprinted from Cheigh, C. I., R. Senaratne, et al. (2010). "Posttreatment Reactivation of
Tuberculosis in Mice Caused by Mycobacterium tuberculosis Disrupted in mce1R." The Journal
of Infectious Diseases 202(5): 752-759, with permission from Oxford University Press. )
MATERIALS AND METHODS
Bacterial culture. Mycobacterium tuberculosis strain H37Rv was grown to mid-log phase for
use in infection. Bacteria were grown in Middlebrook 7H9 broth (Difco, 271310) supplemented
with 0.5% (v/v) glycerol, 0.05% (w/v) Tween 80 (Sigma, P1754), and 10% (v/v) albumin-
dextrose-catalase enrichment. Cultures were maintained at 37°C in 125mL vented Erlenmeyer
flasks with 100 RPM circular agitation.
Infection. Seventy five 8-week-old female BALB/c mice (Charles River) were infected with
~102 CFU of Mtb strain H37Rv using the Inhalation Exposure System (Glas-co., Terre Haute,
IN) in three parallel groups. Infection was allowed to proceed for four weeks without treatment.
Drug treatment. The antimicrobial regimen was 100 µg/mL isoniazid and 15 mg/mL
pyrazinamide provided ad libitum in drinking water for six weeks. Treatment spanned weeks
four through ten post infection.

19
Vaccination regimen and treatment arms. Mice received three injections subcutaneously
spaced three weeks apart after the drug treatment period and an additional one week washout
period. The four treatment groups were: Mce1A protein alone (50 µg), Mce1A protein (5 µg)
plus 5 µg glucopyranosyl lipid A adjuvant in stable emulsion (GLAS-SE), placebo alone (BSA,
equimolar concentration to Mce1A), or placebo with GLA-SE. All injections were delivered in
200µL of PBS.
Assessment of bacterial load. Bacterial load was quantified by colony forming unit (CFU)
counts of lung and spleen homogenates serially diluted in Middlebrook 7H9 medium with 0.05%
Tween 80 and plated onto Middlebrook 7H11 agar plates. Agar plates were incubated for three
weeks at 37°C in a humidified incubator before enumeration.
Time points. One day after infection, three mice from each infection group were sacrificed to
confirm the bacterial inoculum delivered. At four weeks and ten weeks of infection, three mice
were sacrificed for CFU enumeration. At 32 weeks post infection seven mice from each
treatment group were sacrificed and bacterial quantified in the same way. Finally, at 52 weeks
post infection eight mice from each treatment group were sacrificed and assessed for bacterial
load.
RESULT - MCE1A VACCINE IN CORNELL MODEL WITH WILD TYPE MTB
The execution of the modified Cornell model was successful and the initial infection, four week
peak of untreated bacterial load and post drug treatment CFU counts were comparable with the
initial study. Disappointingly, however, bacterial loads in the lungs and spleens of mice at 35 and
52 weeks post infection were not different from placebo (BSA) control (all comparisons p >>
0.05 by unpaired, two tailed t-test). Time course graphs of the bacterial loads in the lungs and
spleens of mice are displayed as Figures 2-3 and 2-4, respectively. The individual results by
treatment arm at 35 and 52 weeks are given as Figures 2-5 and 2-6, for clarity (all relevant
comparisons p > 0.5 by two tailed, unpaired t-test). Beyond average bacterial load, the rate of
reactivation/relapse was also not different, as shown in Figures 2-7 and 2-8 which show the
proportion of mice with any bacteria recovered by organ and by treatment arm at the two time
points following vaccination. In all cases, the label “adj” denotes that a treatment group included
GLA-SE while “non-adj” indicates that no adjuvant was present.
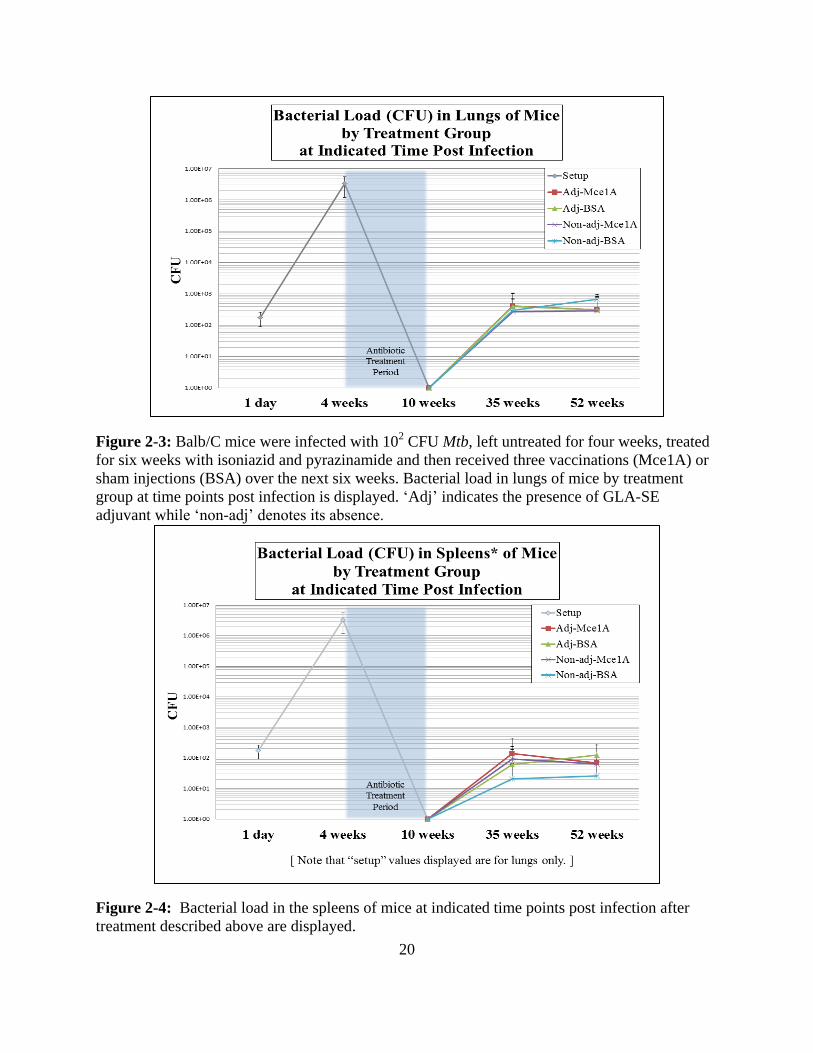
20
Figure 2-3: Balb/C mice were infected with 102 CFU Mtb, left untreated for four weeks, treated
for six weeks with isoniazid and pyrazinamide and then received three vaccinations (Mce1A) or
sham injections (BSA) over the next six weeks. Bacterial load in lungs of mice by treatment
group at time points post infection is displayed. ‘Adj’ indicates the presence of GLA-SE
adjuvant while ‘non-adj’ denotes its absence.
Figure 2-4: Bacterial load in the spleens of mice at indicated time points post infection after
treatment described above are displayed.
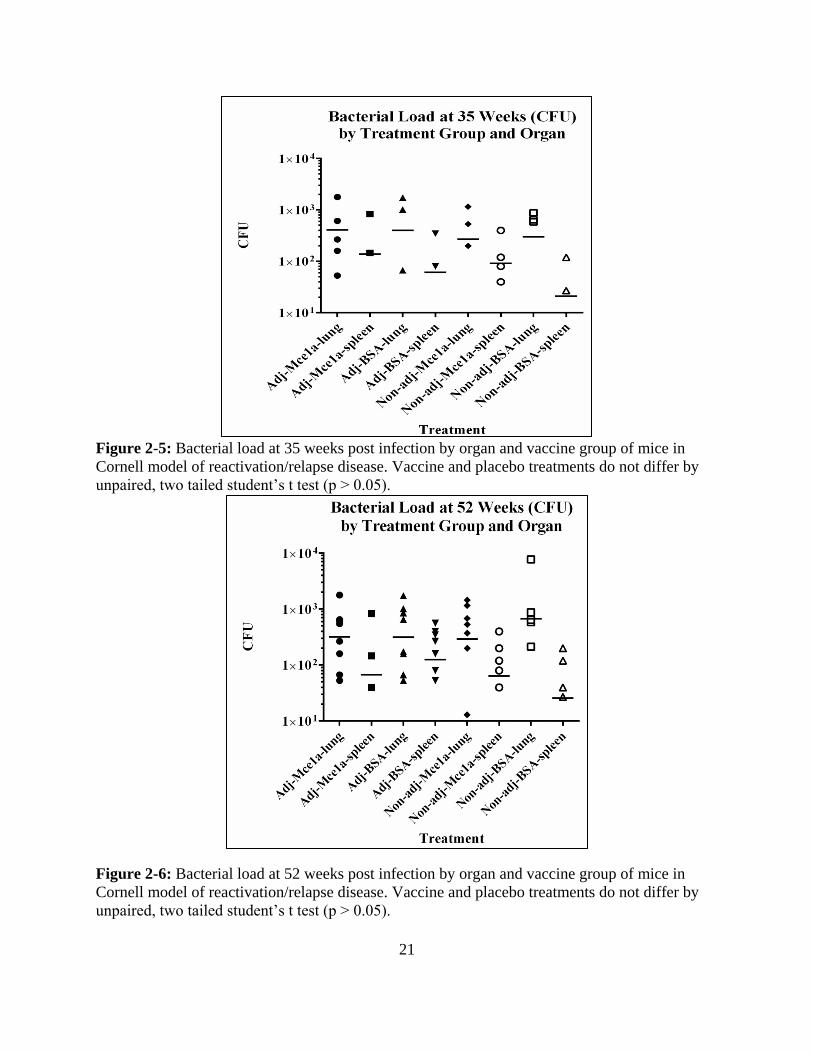
21
Figure 2-5: Bacterial load at 35 weeks post infection by organ and vaccine group of mice in
Cornell model of reactivation/relapse disease. Vaccine and placebo treatments do not differ by
unpaired, two tailed student’s t test (p > 0.05).
Figure 2-6: Bacterial load at 52 weeks post infection by organ and vaccine group of mice in
Cornell model of reactivation/relapse disease. Vaccine and placebo treatments do not differ by
unpaired, two tailed student’s t test (p > 0.05).
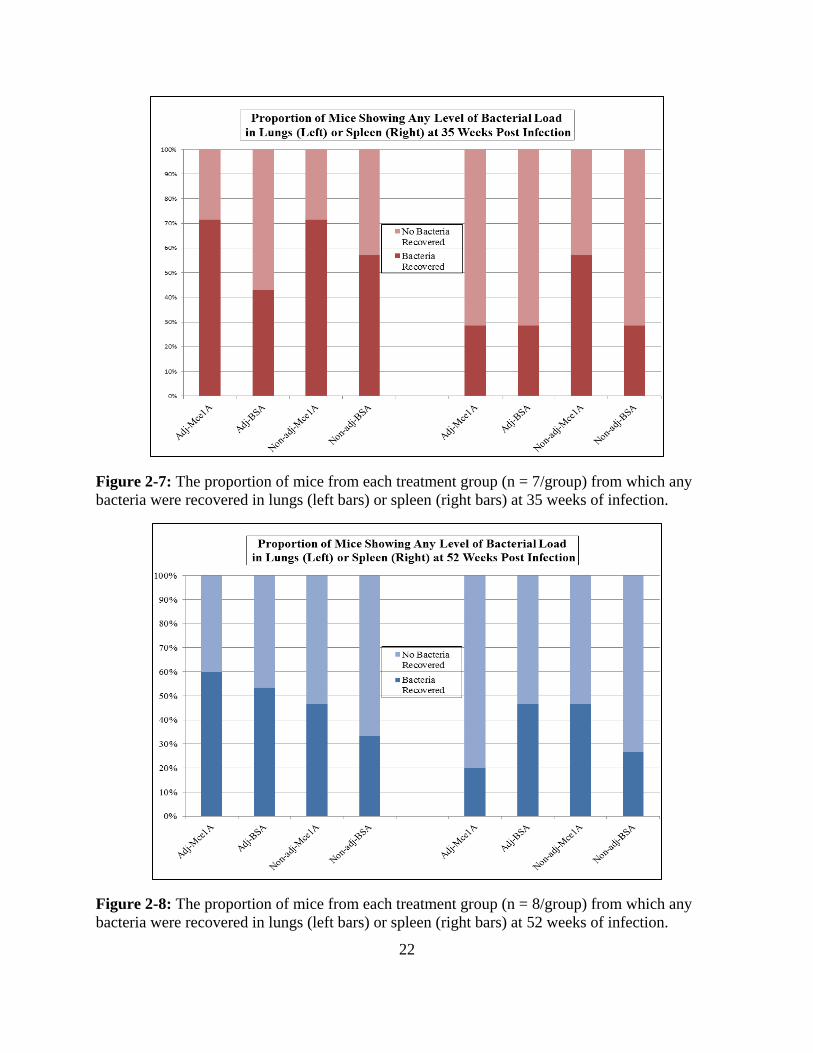
22
Figure 2-7: The proportion of mice from each treatment group (n = 7/group) from which any
bacteria were recovered in lungs (left bars) or spleen (right bars) at 35 weeks of infection.
Figure 2-8: The proportion of mice from each treatment group (n = 8/group) from which any
bacteria were recovered in lungs (left bars) or spleen (right bars) at 52 weeks of infection.

23
The weight of mice was tracked individually as a surrogate for overall health, but no major
differences in the average weight of mice in different treatment groups were observed Figure 2-
9. Error bars are omitted from Figure 2-9 for clarity; the range of standard deviation values
across all treatment groups was from 0.9 to 2.8 grams or 3.8-10.6% of the measured weight. The
lack of divergence of the groups by weight is consistent with the CFU data and supports the
conclusion that the Mce1A vaccine candidate lacked any detectable efficacy in this setting.
Beyond tracking of weight, all mice were monitored by veterinary technicians from the office of
laboratory animal care for general health. The work in this chapter was performed under animal
use protocol #R228-1211B.
Figure 2-9: The average weight of mice from each treatment group over time is shown.

24
DISCUSSION
Only the 35 week time point is comparable, in terms of time-post-infection, with the original
experiment described in (Miyata, Cheigh et al. 2012), since a 21 week time point was not
conducted in the present study and the original study did not follow mice out to 52 weeks. The
differing results in this experiment at 35 weeks post infection versus the previous study at 32
weeks are clear; nearly complete protection in that study versus no detectable effect in this study.
While it cannot be known whether such a difference between this study and the original one
existed at 21 weeks post infection, is seems unlikely that the protection observed in the original
study would have been observed, given later results. Even if protection at 21 weeks had been
observed in the present study, it would have represented transient protection, which would not be
sufficient to drive interest in the vaccine candidate and would be of questionable relevance.
Whether the protection observed in Miyata et al. would have persisted at 52 weeks cannot be
known form available information.
There are numerous potential reasons for the discrepancy in the results presented here versus
those published in Miyata et al. Differences in preparation or delivery of the antigen would be
the first to consider, chronologically.
The same plasmid was used to facilitate production of the protein in both studies, so the encoded
protein should be the same and was confirmed by SDS-PAGE of induced and uninduced (IPTG)
bacterial lysates. The identity of the protein was confirmed by western blot using a rabbit
polyclonal anti-Mce1A antibody. However, the exact nature of the protein purification was not
the same for both preparations of vaccine. Both were captured using a polyhistidine tag and
recovered in denaturing conditions, but the antigen for the current vaccine study had been
lyophilized whereas the protein in the pilot study was not. In neither case was the exact quantity
of LPS (lipopolysaccharide, also known as endotoxin) in the final vaccine preparation
determined. In the absence of rigorous efforts to remove LPS, a large but unknown quantity
would be expected to remain associated with any protein of bacterial origin. It seems possible
that part of the apparent success of the vaccine in Miyata et al., or the failure of the current study,
involved the unintentional co-administration of some quantity of LPS.
In fact, LPS stimulation of TLR4 has been suggested to be a promising target for adjuvant action
in vaccine development (Casella and Mitchell 2008) and the adjuvant used in the study described
here, GLA-SE, also acts by binding TLR4 and has demonstrated activity inducing expression of
cytokines known to be important for containment of Mtb infection, such as interferon gamma
and tumor necrosis factor alpha (Coler, Bertholet et al. 2011). One TB vaccine using this
adjuvant, “ID93+GLA-SE,” is in clinical trials currently (da Costa, Walker et al. 2015). That is
in contrast to TLR2, which is a natural target of binding by Mtb derived lipoproteins, but is
dispensable for adaptive immunity to TB (McBride, Bhatt et al. 2011). However, the fact that the
addition of TLR4 agonist adjuvant did not improve outcomes of those treatment arms where it
was included relative to those where it was omitted suggests that either TLR4 signaling is not

25
beneficial in this context or that the threshold for such an effect was exceeded and addition of
ligand above that level is of no, or deleterious, effect.
Among other potentially divergent aspects of the two studies could be the quantity of antigen
delivered and its condition upon administration. Although both vaccines were produced in E. coli
and purified yielding a final, denatured protein of the same truncated length, handling differences
could leave varying quantities of protein adsorbed to various glass –and to a greater degree,
plastic- ware used in the preparation of the vaccines, since proteins are known to adhere to such
surfaces, reducing the quantity passed onward in each handling step (Andrade and Hlady 1986).
This would be especially salient given the lack of any additional excipients in the described
vaccines which could otherwise have decreased loss. Additionally, as mentioned above, the
protein used in the current study had been lyophilized prior to preparation of the final vaccine
formulation and loading into syringes. The composition of the syringes, duration of time the
solutions were loaded in them, as well as temperature to which the vaccines were exposed, could
all have potentially affected the physical composition of the vaccine at the time of delivery. The
route of vaccination being changed could also potentially have been a factor in the differing
results. Route of antigen delivery has been suggested to modify the response to TB vaccines due
to the impact on the population of T cells that are exposed to the antigen (Horvath and Xing
2013). However, in Miyata et al, it is mentioned that subcutaneous vaccination was effective
against the mce1R KO strain. It seems unlikely, then, that the subcutaneous route of
administration was a major factor in the failure of the follow up study to recapitulate the
protection seen in the earlier study.
The most likely explanation for the disparate results of the two trials is the difference in strain of
Mtb used in the infections. The fact that the mce1R KO strain overexpresses the antigen which
was vaccinated against in (Miyata, Cheigh et al. 2012), whereas the follow up experiment
utilized an unmodified wild type strain, is probably the most meaningful difference between the
two trials and likely the primary driver of the different outcomes. While the mce1R KO strain
could potentially be used to test whole cell or subunit vaccines composed of parts of Mtb not
encoded or substantially affected by the mce1 operon, its use when the antigen of choice is so
composed apparently gives results which are not reproducible against wild type Mtb.
CONCLUSIONS
Failure of post-exposure vaccination in the Cornell model has been described previously; both
BCG and M. vaccae, another mycobacterial species, fail to protect against reactivation TB in the
Cornell model (Dhillon and Mitchison 1994). One reportedly successful treatment of established
Mtb infection using DNA vaccination after a similar setup to the Cornell model (Lowrie, Tascon
et al. 1999) was challenged in a subsequent report by a group which was unable to reproduce
those findings (Taylor, Turner et al. 2003) and actually observed worsening of pathology in
vaccinated animals. Indeed, the potential for exacerbating, rather than improving, outcomes by
vaccination of Mtb infected individuals has been a concern for some time (Anderson 1891;
Daniel 2006). The mechanism of such reactions has been partially elucidated and is believed to
be IL17 driven (Cruz, Fraga et al. 2010).

26
No vaccine has currently shown convincingly better efficacy than BCG in the nearly 100 years
since that vaccine was produced by empirically testing serially passaged M. bovis until it lost
sufficient pathogenicity to be used as a vaccine (Liu, Tran et al. 2009). A few candidates have
shown modest protection similar to BCG in animal models (Bertholet, Ireton et al. 2010;
Aagaard, Hoang et al. 2011) but such protection must be considered an incremental success. As
of the time of writing, robustly predictive correlates of acquired protection have not been well
defined (Ellner, Hirsch et al. 2000; Bhatt, Verma et al. 2015). The most advanced vaccine
candidate to date, MVA85A, recently failed to prevent infection with Mtb as well as progression
to TB disease in a large clinical trial (Tameris, Hatherill et al. 2013). Despite having been
successful in animal studies and good evidence that it elicits the TH1/TH17 response previously
thought to be most likely to offer protection against TB, the vaccine did not show efficacy
against incident infection or disease. This underscores the gaps in understanding correlates of
protection against tuberculosis and also suggests that current animal models of TB are imperfect.
Although the protection afforded by the Mce1A vaccine in (Miyata, Cheigh et al. 2012) was not
replicated with a wild-type Mtb strain, the modified Cornell model gave the expected
experimental parameters and the result obtained is believed to be valid. Refinement of this model
using the six week antibiotic treatment period seems to have produced precisely the desired setup
and should allow testing of vaccine candidates against the H37Rv wild type Mtb strain in
BALB/c mice, although there is some suggestion that more resistant mice (for example
C57BL/6) would generally show greater protection after vaccination (Dannenberg 2010).
Another potential for these studies to provide insight for future vaccine development is in the
further analysis of the response generated by the Mce1A vaccine in the mce1R KO strain. That
is, if the protection shown in Miyata et al. can be replicated against that strain, it may be worth
characterizing that response –even if it is not directly reproducible in other strains–to glean some
insight into what an effective response against Mtb infection might look like. Artificial as it may
be, this protection is the most powerful described to date, so it is likely worthwhile to
characterize it in detail in the pursuit of correlates of protection that could prove to be durable in
other contexts.
Such a correlate of protection from reactivation or relapse would be exceedingly valuable since
no other immune-modulatory approach to adjunctive TB treatment has shown such a profound
effect. If a robust correlate of protection against relapse and reactivation disease were identified,
it could be used as a biomarker for vaccine development which could lead to an effective vaccine
to prevent disease in the enormous population of latently infected individuals. Such a vaccine
could also potentially be given alongside conventional TB chemotherapy where it could
theoretically reduce the rate of relapse upon completion of treatment.
Overall, the results presented here suggest that empiricism is still most likely the most powerful
route for TB vaccine development. Either significant efforts must be directed to generating a
better understanding of the correlates of immunity to TB, or sufficient resources must be
allocated to generating an improved vaccine by some modern empiric or partially empiric testing
method (for example, using numerous candidates with many based upon known attenuation
mechanisms). Approaches could include testing myriad Mtb knockout strains, strains which
overexpress potentially protective antigens or numerous candidate subunit vaccines with an

27
assortment of adjuvants. Even then, it is not clear that success in any of the available animal
models would translate to improved protection afforded to humans.
Because such efforts are not feasible in a university setting without exceptional outside support,
alternatives to vaccination for improving TB treatment were pursued. Specifically, the process of
autophagy has recently been reported by several groups to be both capable of impacting Mtb
growth in infected cells or animals (Gutierrez, Master et al. 2004; Singh 2006; Alonso, Pethe et
al. 2007; Biswas, Qureshi et al. 2008; Kumar, Nath et al. 2010; Ponpuak, Davis et al. 2010; Zullo
and Lee 2012), and also tractable for pharmacologic intervention (Floto, Sarkar et al. 2007;
Sarkar, Perlstein et al. 2007; Zhang, Yu et al. 2007; Williams, Sarkar et al. 2008; Balgi, Fonseca
et al. 2009; Rose, Menzies et al. 2010; Hundeshagen, Hamacher-Brady et al. 2011; Stanley,
Barczak et al. 2014). Chapter three explores yet another approach to enhance host response to
control or reduce latent infection. The application of pharmacologic autophagy induction as an
immune-adjuvant treatment for tuberculosis is examined.

28
REFERENCES
[CDC], C. f. D. C. P. (2011). "Ten Great Public Health Achievements — Worldwide, 2001-
2010." Morbidity and Mortality Weekly Report 60(24): 814-818.
Aagaard, C., T. Hoang, et al. (2011). "A multistage tuberculosis vaccine that confers efficient
protection before and after exposure." Nature Medicine 17(2): 189-194.
Alonso, S., K. Pethe, et al. (2007). "Lysosomal killing of Mycobacterium mediated by ubiquitin-
derived peptides is enhanced by autophagy." Proc Natl Acad Sci U S A 104(14): 6031-6036.
Andersen, P. (1994). "Effective vaccination of mice against Mycobacterium tuberculosis
infection with a soluble mixture of secreted mycobacterial proteins." Infect Immun 62(6): 2536-
2544.
Anderson, M. (1891). "ON KOCH'S TREATMENT.1." The Lancet 137(3525): 651-652.
Andrade, J. D. and V. Hlady (1986). Protein adsorption and materials biocompatibility: A
tutorial review and suggested hypotheses. Biopolymers/Non-Exclusion HPLC, Springer Berlin
Heidelberg. 79: 1-63.
Arruda, S., G. Bomfim, et al. (1993). "Cloning of an M. tuberculosis DNA fragment associated
with entry and survival inside cells." Science 261(5127): 1454-1457.
Balgi, A. D., B. D. Fonseca, et al. (2009). "Screen for chemical modulators of autophagy reveals
novel therapeutic inhibitors of mTORC1 signaling." PLoS ONE 4(9): e7124.
Bertholet, S., G. C. Ireton, et al. (2010). "A Defined Tuberculosis Vaccine Candidate Boosts
BCG and Protects Against Multidrug-Resistant Mycobacterium tuberculosis." Science
Translational Medicine 2(53): 53ra74-53ra74.
Bhatt, K., S. Verma, et al. (2015). "Quest for Correlates of Protection against Tuberculosis." Clin
Vaccine Immunol 22(3): 258-266.
Biswas, D., O. S. Qureshi, et al. (2008). "ATP-induced autophagy is associated with rapid killing
of intracellular mycobacteria within human monocytes/macrophages." BMC Immunol 9: 35.
Black, G. F., R. E. Weir, et al. (2002). "BCG-induced increase in interferon-gamma response to
mycobacterial antigens and efficacy of BCG vaccination in Malawi and the UK: two randomised
controlled studies." Lancet 359(9315): 1393-1401.

29
Brandt, L., J. Feino Cunha, et al. (2002). "Failure of the Mycobacterium bovis BCG vaccine:
some species of environmental mycobacteria block multiplication of BCG and induction of
protective immunity to tuberculosis." Infect Immun 70(2): 672-678.
Brewer, T. F. (2000). "Preventing tuberculosis with bacillus Calmette-Guerin vaccine: a meta-
analysis of the literature." Clin Infect Dis 31 Suppl 3: S64-67.
Casali, N., A. M. White, et al. (2005). "Regulation of the Mycobacterium tuberculosis mce1
Operon." Journal of Bacteriology 188(2): 441-449.
Casella, C. R. and T. C. Mitchell (2008). "Putting endotoxin to work for us: monophosphoryl
lipid A as a safe and effective vaccine adjuvant." Cell Mol Life Sci 65(20): 3231-3240.
Chan, J. and J. Flynn (2004). "The immunological aspects of latency in tuberculosis." Clin
Immunol 110(1): 2-12.
Coler, R. N., S. Bertholet, et al. (2011). "Development and Characterization of Synthetic
Glucopyranosyl Lipid Adjuvant System as a Vaccine Adjuvant." PLoS ONE 6(1): e16333.
Cruz, A., A. G. Fraga, et al. (2010). "Pathological role of interleukin 17 in mice subjected to
repeated BCG vaccination after infection with Mycobacterium tuberculosis." J Exp Med 207(8):
1609-1616.
da Costa, C., B. Walker, et al. (2015). "Tuberculosis Vaccines – state of the art, and novel
approaches to vaccine development." International Journal of Infectious Diseases 32(0): 5-12.
Daniel, T. M. (2006). "The history of tuberculosis." Respiratory Medicine 100(11): 1862-1870.
Dannenberg, A. M., Jr. (2010). "Perspectives on clinical and preclinical testing of new
tuberculosis vaccines." Clin Microbiol Rev 23(4): 781-794.
Dhillon, J. and D. A. Mitchison (1994). "Effect of vaccines in a murine model of dormant
tuberculosis." Tuber Lung Dis 75(1): 61-64.
Dietrich, J. and T. M. Doherty (2009). "Interaction of Mycobacterium tuberculosis with the host:
consequences for vaccine development." APMIS 117(5-6): 440-457.
Dye, C., S. Scheele, et al. (1999). "Consensus statement. Global burden of tuberculosis:
estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and
Monitoring Project." JAMA 282(7): 677-686.
Elias, D., S. Britton, et al. (2008). "Poor immunogenicity of BCG in helminth infected
population is associated with increased in vitro TGF-[beta] production." Vaccine 26(31): 3897-
3902.
Ellner, J. J., C. S. Hirsch, et al. (2000). "Correlates of protective immunity to Mycobacterium
tuberculosis in humans." Clin Infect Dis 30 Suppl 3: S279-282.

30
Fine, P. E. M. (1995). "Variation in protection by BCG: implications of and for heterologous
immunity." The Lancet 346(8986): 1339-1345.
Floto, R. A., S. Sarkar, et al. (2007). "Small molecule enhancers of rapamycin-induced TOR
inhibition promote autophagy, reduce toxicity in Huntington's disease models and enhance
killing of mycobacteria by macrophages." Autophagy 3(6): 620-622.
Flynn, J. L. and J. Chan (2001). "Immunology of tuberculosis." Annu Rev Immunol 19: 93-129.
Gutierrez, M. G., S. S. Master, et al. (2004). "Autophagy is a defense mechanism inhibiting BCG
and Mycobacterium tuberculosis survival in infected macrophages." Cell 119(6): 753-766.
Horvath, C. N. and Z. Xing (2013). "Immunization strategies against pulmonary tuberculosis:
considerations of T cell geography." Adv Exp Med Biol 783: 267-278.
Hundeshagen, P., A. Hamacher-Brady, et al. (2011). "Concurrent detection of autolysosome
formation and lysosomal degradation by flow cytometry in a high-content screen for inducers of
autophagy." BMC Biol 9: 38.
Kaufmann, S. H. E., G. Hussey, et al. (2010). "New vaccines for tuberculosis." The Lancet
375(9731): 2110-2119.
Kumar, D., L. Nath, et al. (2010). "Genome-wide Analysis of the Host Intracellular Network that
Regulates Survival of Mycobacterium tuberculosis." Cell 140(5): 731-743.
Lawn, S. D. and A. I. Zumla (2011). "Tuberculosis." Lancet.
Lin, M. Y., T. B. K. Reddy, et al. (2009). "Cross-Reactive Immunity to Mycobacterium
tuberculosis DosR Regulon-Encoded Antigens in Individuals Infected with Environmental,
Nontuberculous Mycobacteria." Infection and Immunity 77(11): 5071-5079.
Liu, J., V. Tran, et al. (2009). "BCG vaccines: their mechanisms of attenuation and impact on
safety and protective efficacy." Hum Vaccin 5(2): 70-78.
Lowrie, D. B., R. E. Tascon, et al. (1999). "Therapy of tuberculosis in mice by DNA
vaccination." Nature 400(6741): 269-271.
Lu, S., L. Tager, et al. (2006). "A cell-penetrating peptide derived from mammalian cell uptake
protein of Mycobacterium tuberculosis." Analytical Biochemistry 353(1): 7-14.
McBride, A., K. Bhatt, et al. (2011). "Development of a secondary immune response to
Mycobacterium tuberculosis is independent of Toll-like receptor 2." Infect Immun 79(3): 1118-
1123.
McCune, R. M., F. M. Feldmann, et al. (1966). "Microbial persistence. I. The capacity of
tubercle bacilli to survive sterilization in mouse tissues." J Exp Med 123(3): 445-468.

31
Miyata, T., C. I. Cheigh, et al. (2012). "An adjunctive therapeutic vaccine against reactivation
and post-treatment relapse tuberculosis." Vaccine 30(2): 459-465.
Ordway, D. J., S. Shang, et al. (2011). "Mycobacterium bovis BCG-Mediated Protection against
W-Beijing Strains of Mycobacterium tuberculosis Is Diminished Concomitant with the
Emergence of Regulatory T Cells." Clin Vaccine Immunol 18(9): 1527-1535.
Ponpuak, M., A. S. Davis, et al. (2010). "Delivery of Cytosolic Components by Autophagic
Adaptor Protein p62 Endows Autophagosomes with Unique Antimicrobial Properties."
Immunity 32(3): 329-341.
Rodrigues, L. C., V. K. Diwan, et al. (1993). "Protective effect of BCG against tuberculous
meningitis and miliary tuberculosis: a meta-analysis." Int J Epidemiol 22(6): 1154-1158.
Rodrigues, L. C., S. M. Pereira, et al. (2005). "Effect of BCG revaccination on incidence of
tuberculosis in school-aged children in Brazil: the BCG-REVAC cluster-randomised trial."
Lancet 366(9493): 1290-1295.
Rose, C., F. M. Menzies, et al. (2010). "Rilmenidine attenuates toxicity of polyglutamine
expansions in a mouse model of Huntington's disease." Hum Mol Genet 19(11): 2144-2153.
Russell, D. G., C. E. Barry, 3rd, et al. (2010). "Tuberculosis: what we don't know can, and does,
hurt us." Science 328(5980): 852-856.
Sarkar, S., E. O. Perlstein, et al. (2007). "Small molecules enhance autophagy and reduce
toxicity in Huntington's disease models." Nat Chem Biol 3(6): 331-338.
Singh, S. B. (2006). "Human IRGM Induces Autophagy to Eliminate Intracellular
Mycobacteria." Science 313(5792): 1438-1441.
Stanley, S. A., A. K. Barczak, et al. (2014). "Identification of Host-Targeted Small Molecules
That Restrict Intracellular <italic>Mycobacterium tuberculosis</italic> Growth." PLoS Pathog
10(2): e1003946.
Tameris, M. D., M. Hatherill, et al. (2013). "Safety and efficacy of MVA85A, a new tuberculosis
vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b
trial." The Lancet 381(9871): 1021-1028.
Taylor, J. L., O. C. Turner, et al. (2003). "Pulmonary Necrosis Resulting from DNA Vaccination
against Tuberculosis." Infection and Immunity 71(4): 2192-2198.
Uchida, Y., N. Casali, et al. (2007). "Accelerated immunopathological response of mice infected
with Mycobacterium tuberculosis disrupted in the mce1 operon negative transcriptional
regulator." Cellular Microbiology 9(5): 1275-1283.
Wallis, R. S., T. M. Doherty, et al. (2009). "Biomarkers for tuberculosis disease activity, cure,
and relapse." Lancet Infect Dis 9(3): 162-172.

32
Williams, A., S. Sarkar, et al. (2008). "Novel targets for Huntington's disease in an mTOR-
independent autophagy pathway." Nat Chem Biol 4(5): 295-305.
Young, D. and C. Dye (2006). "The development and impact of tuberculosis vaccines." Cell
124(4): 683-687.
Zhang, L., J. Yu, et al. (2007). "Small molecule regulators of autophagy identified by an image-
based high-throughput screen." Proc Natl Acad Sci U S A 104(48): 19023-19028.
Zullo, A. J. and S. Lee (2012). "Mycobacterial induction of autophagy varies by species and
occurs independently of mTOR inhibition." J Biol Chem.

33
CHAPTER 3
EFFECT OF MTOR-INDEPENDENT INDUCERS OF AUTOPHAGY ON
INTRACELLULAR M. TUBERCULOSIS

34
INTRODUCTION
The line of investigation described in this chapter was undertaken with the goal of developing an
adjunctive immune-modulatory treatment for tuberculosis (TB). Because Mtb is most often
observed inside of macrophages, even in high burden infection (Kaplan, Post et al. 2003), it was
hypothesized that augmenting innate immune effectors in macrophages could lead to reduced
bacterial load, reduced pathology or both. The approach pursued was based on the induction of
increased flux through the autophagy pathway. The rationale of this approach was based on
reports that autophagy is capable of restricting the intracellular growth of Mtb while
simultaneously suppressing inflammation (Castillo, Dekonenko et al. 2012).
Such restriction of growth, if achieved, could lead to the development of interventions for
preventing reactivation in latently infected individuals by boosting their anti-mycobacterial
immune function, or to interventions to be deployed alongside conventional chemotherapeutic
agents to prevent relapse disease in the same way. Autophagy induction has been described to
both directly affect bacterial load and to boost the adaptive immune response to mycobacterial
infection by upregulating antigen expression (Jagannath, Lindsey et al. 2009).
IMMUNE ADJUVANTS
The potential of host-directed, immune-modulatory treatment of TB has been demonstrated by
several different approaches including cytokine supplementation, DNA vaccination, M. vaccae
exposure, therapeutic subunit vaccination, and siRNA treatment (Condos, Rom et al. 1997;
Lowrie, Tascon et al. 1999; Johnson, Kamya et al. 2000; Aagaard, Hoang et al. 2011; Rosas-
Taraco, Higgins et al. 2011). Some of these approaches have also proven effective against drug
resistant Mtb (Condos, Rom et al. 1997; Okada, Kita et al. 2009; Bertholet, Ireton et al. 2010).
Host directed small molecule treatments have shown promise recently with inhibitors of
phosphodiesterases (PE) type four (Koo, Manca et al. 2011), PE types three and five (Maiga,
Agarwal et al. 2012), ABL kinase (Napier, Rafi et al. 2011), and HMG-CoA reductase (Parihar,
Guler et al. 2013) all demonstrating reduction in bacterial load in various models of
mycobacterial infection.
AUTOPHAGY
The process of autophagy is a bulk degradation pathway, the function of which is to remove
constituents of eukaryotic cells which are too large for proteosomal degradation (Yang and
Klionsky 2010). It is involved in numerous cell homeostatic processes, notably as the main
pathway leading to lysosomal destruction of organelles and long lived proteins (Mizushima and
Komatsu 2011). The removal of damaged mitochondria (i.e., “mitophagy”) is another important
function of autophagy (Youle and Narendra 2011). Metabolically, autophagy is a key mechanism
because it is regulated by several sensors of cellular energy stores (e.g., AMPK) and when
induced, has the effect of liberating metabolizable biomolecules to provide energy for the cell
(Mihaylova and Shaw 2011). In agreement with this function of autophagy is the well-

35
established observation that nutrient starvation induces autophagy (Mizushima, Yoshimori et al.
2010). The centrality of autophagy as a cellular homeostatic system is demonstrated by its
evolutionary conservation back to yeast (Hughes and Rusten 2007). While autophagy has
sometimes been suggested to be a mechanism of cell death, in the vast majority of cases it is
likely an effect, and not a cause, of initiating cell death pathways (Kroemer and Levine 2008).
The rationale for manipulation of autophagy in the context of Mtb infection derived from studies
in which that process was shown to reduce mycobacterial survival (Gutierrez, Master et al. 2004;
Singh 2006; Alonso, Pethe et al. 2007; Biswas, Qureshi et al. 2008; Kumar, Nath et al. 2010;
Ponpuak, Davis et al. 2010; Zullo and Lee 2012). The role of autophagy in resistance to TB has
been underscored by genome wide association studies showing that mutations in autophagy
related genes correlate with increased incidence of active TB in humans (Intemann, Thye et al.
2009; Che, Li et al. 2010). Beyond that, an siRNA screen in human cells for host factors that
affect mycobacterial load discovered a dense network of interactions surrounding key autophagy
regulators (Kumar, Nath et al. 2010). These findings are further supported by the observation
that autophagy deficient (ATG5 cKO) mice are markedly more susceptible to TB (Castillo,
Dekonenko et al. 2012; Watson, Manzanillo et al. 2012). Also consistent with an important role
for autophagy in mycobacterial control is the observation that Th2 cytokines, long known to be
deleterious to the control of TB, suppress autophagy (Harris, De Haro et al. 2007).
Autophagy induction subsequently allows for a more robust immune response by making
additional pathogen derived peptides available for antigen presentation (Munz 2009).
Importantly, this has been demonstrated with regard to mycobacteria specifically (Jagannath,
Lindsey et al. 2009). To biologists familiar with the propensity of natural systems to optimize for
multi-functionality, it should not come as a surprise that a host cell would turn a system capable
of powerful catabolic activity on invading pathogens (Ponpuak, Davis et al. 2010). The resulting
downstream augmentation of the adaptive immune response, taken together with its
demonstrated direct bactericidal effect on mycobacteria (Gutierrez, Master et al. 2004; Singh
2006; Alonso, Pethe et al. 2007; Biswas, Qureshi et al. 2008; Kumar, Nath et al. 2010; Ponpuak,
Davis et al. 2010; Zullo and Lee 2012), clearly indicates that autophagy is a potentially powerful
target for immune-modulatory intervention. While there is evidence that Mtb has at least one
mechanism for inhibiting autophagy in human primary macrophages (Petruccioli, Romagnoli et
al. 2012), this resistance may be indicative of evolutionary efforts to combat a potent selective
pressure.
MTOR INDEPENDENCE
The standard method for autophagy induction, mTOR inhibition, is not an acceptable strategy in
the context of infectious disease because even very specific inhibitors of this kinase, such as
rapamycin, strongly suppresses the adaptive immune system (Araki, Ellebedy et al. 2011). This
has been demonstrated both in animal models (Calne, Collier et al. 1989), and more recently by
the clinical use of rapamycin after human organ transplantation to prevent rejection (Watson,
Friend et al. 1999). Furthermore, mTOR antagonists have documented lung toxicity (Duran, Siu
et al. 2006).

36
Pharmacologic induction of autophagy without detrimental effects on the adaptive immune
system has been made possible by the discovery of mTOR independent inducers of autophagy
(Sarkar, Floto et al. 2005; Sarkar, Davies et al. 2007; Zhang, Yu et al. 2007; Williams, Sarkar et
al. 2008). Preliminary data suggested that mTOR independent autophagy may be capable of
destroying intracellular mycobacteria (Floto, Sarkar et al. 2007).
A further advantage of many of the compounds screened for this effect in those studies is that
they have safety records in humans, albeit in a different context. Many are FDA approved for
other indications (Williams, Sarkar et al. 2008), are present in foods (resveratrol), or are
pharmaceutical excipients (trehalose). See Appendix A for a summary of relevant characteristics
of compounds which were considered for inclusion in these studies. Two of these drugs,
carbamazepine (CBZ) and fluoxetine (FLX), were hits in screens for compounds that restrict
mycobacterial viability inside macrophages (Sundaramurthy, Barsacchi et al. 2013; Stanley,
Barczak et al. 2014). Unfortunately, CBZ has some known interactions with anti-TB drugs.
However these interactions are not outright contraindications, but rather factors to be considered
with regard to dosing (Zolezzi 2002). Oxcarbazepine, an analog of CBZ which has fewer
metabolic side effects, was also a hit in the same screen as CBZ, albeit with a slightly lower
phenoscore (Sundaramurthy, Barsacchi et al. 2013). There is also evidence that oxcarbazepine
can exert effects through PI3K enzymes but in a way which apparently does not affect AKT, an
mTOR pathway regulator (Simao, Zamin et al. 2009).
Several of the compounds identified by other groups and which the present studies utilized
indirectly inhibit calpains as their mechanism for inducing autophagy (Williams, Sarkar et al.
2008). Calpain activity is necessary for hyper-secretion of inflammatory cytokines by
macrophages in autophagy deficient mice in the context of mycobacterial infection (Castillo,
Dekonenko et al. 2012), so this approach appeared to be particularly promising. Also, calpains
mediate a switch from pro-autophagic to pro-apoptotic phenotype via ATG5 (Yousefi, Perozzo
et al. 2006) so their blockade could be beneficial by preventing excessive host cell death, though
this remains speculative.
Resveratrol, a polyphenol with a good safety profile, has been reported to induce autophagy
(Mauthe, Jacob et al. 2011) in an mTOR independent manner (Scarlatti, Maffei et al. 2008).
Moreover, this has been shown to occur in-vivo (Lekli, Ray et al. 2010).
Some of the selected mTOR independent autophagy inducing compounds are also voltage gated
calcium channel inhibitors (see Appendix A). The channels they target have been reported to be
active in modulating immune cells, including lymphocytes, dendritic cells, NK cells and
neutrophils (Suzuki, Inoue et al. 2010), as well as affecting mycobacterial clearance in dendritic
cells in-vitro (Gupta, Salam et al. 2009). Additionally, rapamycin and its analogs have been
reported to bind these channels, so some portion their effect against mycobacterial infection may
be due to that interaction (Ruan, Pong et al. 2008). The role of calcium flux in Mtb persistence
has been reviewed (Kusner 2005). Essentially, calcium flux is not necessary for phagocytosis,
but is necessary for the respiratory burst and phagosomal fusion with lysosomes. Calcium flux
appears to be inhibited by Mtb and its inhibition correlates with increased survival. This
inhibition of calcium flux and its effects on Mtb bacterial load are reversible with ionophore
treatment or calcium chelation (Malik, Denning et al. 2000).

37
The exact mechanism of action by which most of the drugs in this class induce autophagy is
controversial; the role of calcium, in particular, is contested and has been reviewed elsewhere
(Decuypere, Bultynck et al. 2011). The activity of hVps34 is reported to be calcium independent,
however (Yan, Flinn et al. 2009), and calcium flux is not necessary for autophagy mediated by
inositol triphosphate receptor inhibition (Criollo, Maiuri et al. 2007). This mechanism, which
most of the drugs tested converge upon in their extended pathways (Renna, Jimenez-Sanchez et
al. 2010), is the most commonly identified mTOR independent target in high throughput screens
(Zhang, Yu et al. 2007; Williams, Sarkar et al. 2008). Because this induction is apparently
upstream of, and dependent on, Atg5, Atg10, Atg12, Beclin-1 and hVps34, it is expected to
operate along the canonical autophagy pathway (Criollo, Maiuri et al. 2007). The regulation of
autophagy by inositol derivatives is discussed in a salient review (Criollo, Vicencio et al. 2007)
ADJUNCTIVE THERAPY
When drug susceptible TB is treated with an effective antibiotic regimen, most of the culturable
bacteria are eliminated relatively quickly; a patient’s sputum is typically culture negative after
two months when three effective antibiotics are used (Gelband 2000), but treatment of even
uncomplicated cases is recommended to continue for a totally of six months (Mitchison 2005).
The additional time beyond apparent sterilization is intended to lower the rate of relapse disease,
or recrudescence of the infection (Gelband 2000). If the bacteria which persist within
macrophages after the initial intensive phase of killing by antibiotics could be eliminated by
autophagy induction, a reduction in relapse rates may be achieved. Alternatively, treatment
duration may be reduced by co-treatment with autophagy inducing drugs, which would be
expected to limit noncompliance and selection of drug resistance.
A study investigating antimicrobial tolerance phenotypes (Adams, Takaki et al. 2011) found that
one drug reported to induce autophagy independently of mTOR, verapamil (Williams, Sarkar et
al. 2008), also has the effect of limiting the development of antibiotic tolerance by mycobacteria.
Suppression of antimicrobial tolerance in this way, in addition to direct killing of mycobacteria
with antimicrobial agents, would hypothetically decrease relapse rates and drug resistance further
than autophagy induction plus antimicrobials compounds alone, so treatment with that compound
was pursued.
Additional synergy could be expected in-vivo from the generation of interferon gamma (IFN-γ)
by T cells, a cytokine which has itself been shown to induce autophagy (Singh, Davis et al.
2006). Relatedly, the autophagic destruction of internalized mycobacteria has been shown to up-
regulate antigen presentation and T cell activation leading to the secretion of IFN-γ (Jagannath,
Lindsey et al. 2009). It follows that the induction of autophagy and subsequent production of an
autophagy inducing cytokine would generate a feed forward cycle.

38
MATERIALS AND METHODS
Macrophage cell culture. All experiments, unless otherwise noted, were performed on resting
macrophages at a density of 5x104/well in 200µL of medium in 96 well cluster plates.
Macrophages used were the RAW 264.7 murine cell line (ATCC TIB-71), THP-1 human
monocytic cell line (ATCC TIB-202) or primary murine bone marrow derived macrophages
(BMDM). THP-1 cells were maintained in RPMI medium with 10% fetal bovine serum (FBS)
while RAW and primary murine macrophages were maintained in DMEM with 10% FBS. When
THP-1 cells were used, they were treated with 12-O-tetradecanoylphorbol 13-acetate (PMA) at
100nM for 48 hours before infection, after which time PMA was no longer used. For all
macrophage media, glutamine was supplied as Glutamax (L-alanyl-L-glutamine, Invitrogen) at
2mM. Macrophages were maintained at 37°C, 5% CO2 in a humidified incubator and handling
steps were executed as quickly as safely possible to minimize exposure to ambient conditions.
Autophagic flux assays. Fluorescence microscopy and western blot methods were used to
monitor autophagic flux using LC3 as a marker [For a review of the interpretation of autophagic
flux data, see (Mizushima, Yoshimori et al. 2010)]. For IF studies, RAW 264.7 macrophages
carrying the pROW2 plasmid (encoding CMV-GFP-LC3) were used as described (Watson,
Manzanillo et al. 2012). Confirmation of results was achieved using a well validated antibody
directed against LC3 (L8918, Sigma) according to manufacturer’s instructions. Images were
collected on an epi-fluorescence wide-field microscope (Zeiss Axio Observer D1) and scored
using a script for ImageJ software from NIH. Western blots were conducted essentially as
described (Menzies, Moreau et al. 2001) and quantified by ChemiDoc MP with ImageLab 4.0.1
software (Bio-Rad).
Bacterial culture. All experiments were conducted with Mycobacterium tuberculosis strain
Erdman. Bacteria were grown in 50mL of Middlebrook 7H9 broth supplemented with 0.5% (v/v)
glycerol, 0.05% Tween 80 (w/v), and 10% (v/v) oleate-albumin-dextrose-catalase (BBL
Middlebrook OADC Enrichment, catalog no. 212351). Cultures were maintained at 37°C in 1L
roller bottles.
Infection. Bacterial cultures were grown to mid log phase for use in infections. Bacteria were
washed thrice in PBS, resuspended and centrifuged at 50g to pellet clumps. Infection medium,
consisting of appropriate cell culture medium for the macrophage type used but with horse serum
substituted for FBS, was made to contain enough bacteria to achieve a multiplicity of infection of
one in 100µL. For example, for a typical experiment with 5x104 macrophages per well, 100µL of
infection medium at 5x105 CFU/mL would be applied. Phagocytosis was allowed to proceed for
four hours, after which macrophages were washed twice with warm PBS and returned to 200µL
of normal medium. For plating experiments, macrophages were lysed by the addition of 50µLof
0.5% Triton X-100, yielding a final concentration of 0.1% detergent. Serial dilutions were
produced in complete 7H9 medium and plated on Middlebrook 7H10 agar plates containing 10%
OADC and no antibiotics. Plates were enumerated after 17-21 days of incubation at 37°C in a
humidified incubator.
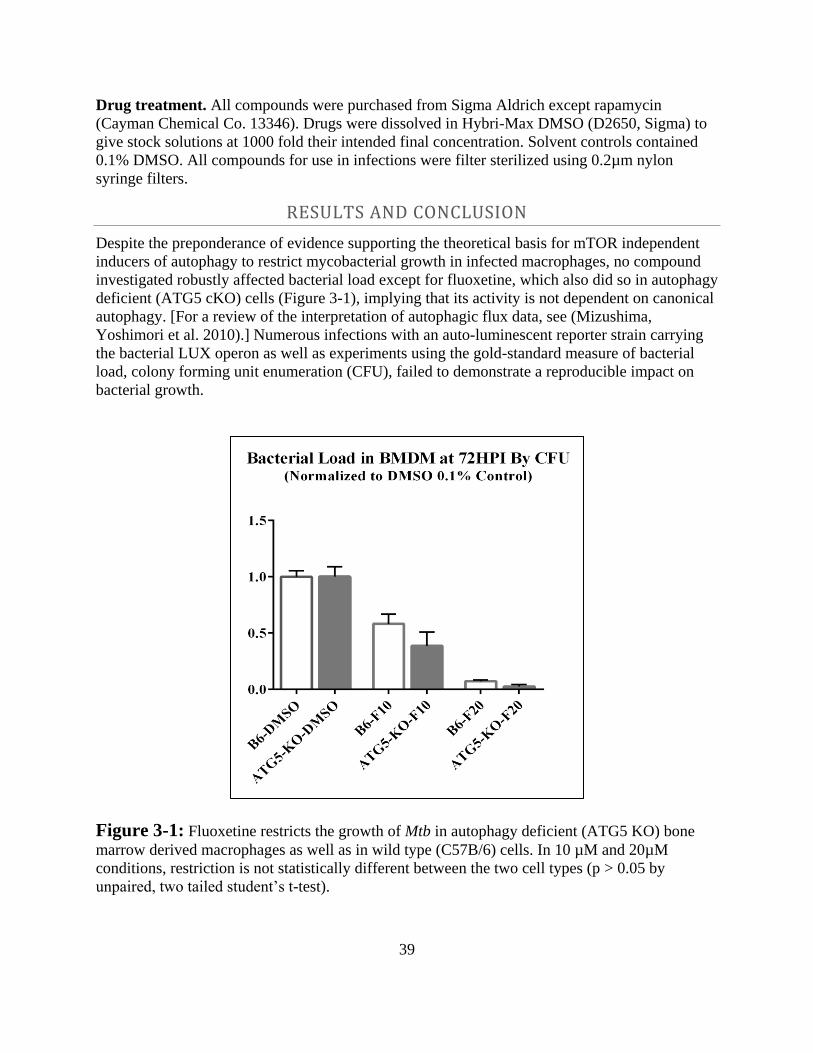
39
Drug treatment. All compounds were purchased from Sigma Aldrich except rapamycin
(Cayman Chemical Co. 13346). Drugs were dissolved in Hybri-Max DMSO (D2650, Sigma) to
give stock solutions at 1000 fold their intended final concentration. Solvent controls contained
0.1% DMSO. All compounds for use in infections were filter sterilized using 0.2µm nylon
syringe filters.
RESULTS AND CONCLUSION
Despite the preponderance of evidence supporting the theoretical basis for mTOR independent
inducers of autophagy to restrict mycobacterial growth in infected macrophages, no compound
investigated robustly affected bacterial load except for fluoxetine, which also did so in autophagy
deficient (ATG5 cKO) cells (Figure 3-1), implying that its activity is not dependent on canonical
autophagy. [For a review of the interpretation of autophagic flux data, see (Mizushima,
Yoshimori et al. 2010).] Numerous infections with an auto-luminescent reporter strain carrying
the bacterial LUX operon as well as experiments using the gold-standard measure of bacterial
load, colony forming unit enumeration (CFU), failed to demonstrate a reproducible impact on
bacterial growth.
Figure 3-1: Fluoxetine restricts the growth of Mtb in autophagy deficient (ATG5 KO) bone
marrow derived macrophages as well as in wild type (C57B/6) cells. In 10 µM and 20µM
conditions, restriction is not statistically different between the two cell types (p > 0.05 by
unpaired, two tailed student’s t-test).

40
The reason(s) for this failure are obscure. Some of the study compounds, notably carbamazepine,
were found to apparently upregulate autophagy by western blot for LC3 (Figure 3-2), a marker of
autophagy (Mizushima, Yoshimori et al. 2010), and by immuno-fluorescence microscopy for the
same target, in resting macrophages. Results did not appear to depend on the macrophage type,
solvent used (DMSO, ethanol), or time point post infection. The impact of handling, such as the
amount of time that macrophages were outside of the incubator for drug application or other
manipulation, was minimized but could have played a role in producing variability in results. For
all experiments, the multiplicity of infection (MOI) was set to one. Clumping of the bacteria,
however, could have affected the actual MOI achieved. Greater clumping could have reduced the
quantity of bacteria in successfully modified phagosomes, since it has been reported that clusters
of bacteria resist phagosomal maturation less effectively than single bacilli (de Chastellier,
Forquet et al. 2009).
Figure 3-2 : Western blot of LC3 protein from RAW264.7 murine macrophages treated as
labeled (A). Quantification of LC3B expression relative to LC3A or β actin shows apparent
increase in autophagic flux by addition of carbamazepine (CBZ) and chloroquine (Chlor.) versus
either treatment alone. For a review of the interpretation of autophagic flux data, see
(Mizushima, Yoshimori et al. 2010).
Drugs were selected based on their therapeutic index as gauged by the ratio of the concentrations
at which they have been reported to induce autophagy to the concentrations at which they are
used clinically, as well as their side effect profile and contraindications. The compounds were
tested at a target concentration which was chosen based on serum levels achieved in humans and
on the concentration at which they were reported to induce autophagy in the primary literature.
They were also tested at tenfold higher and lower concentrations (denoted 0.1x, 1x and 10x). For
the first set of experiments, the target concentrations were as follows: fluoxetine (FLX) 1µM,
verapamil (VER) 1µM, rilmenidine (RIL) 0.01µM, carbamazepine (CBZ) 25µM, minoxidil
(MNX) 1µM, and valproic acid (VPA) 500µM.
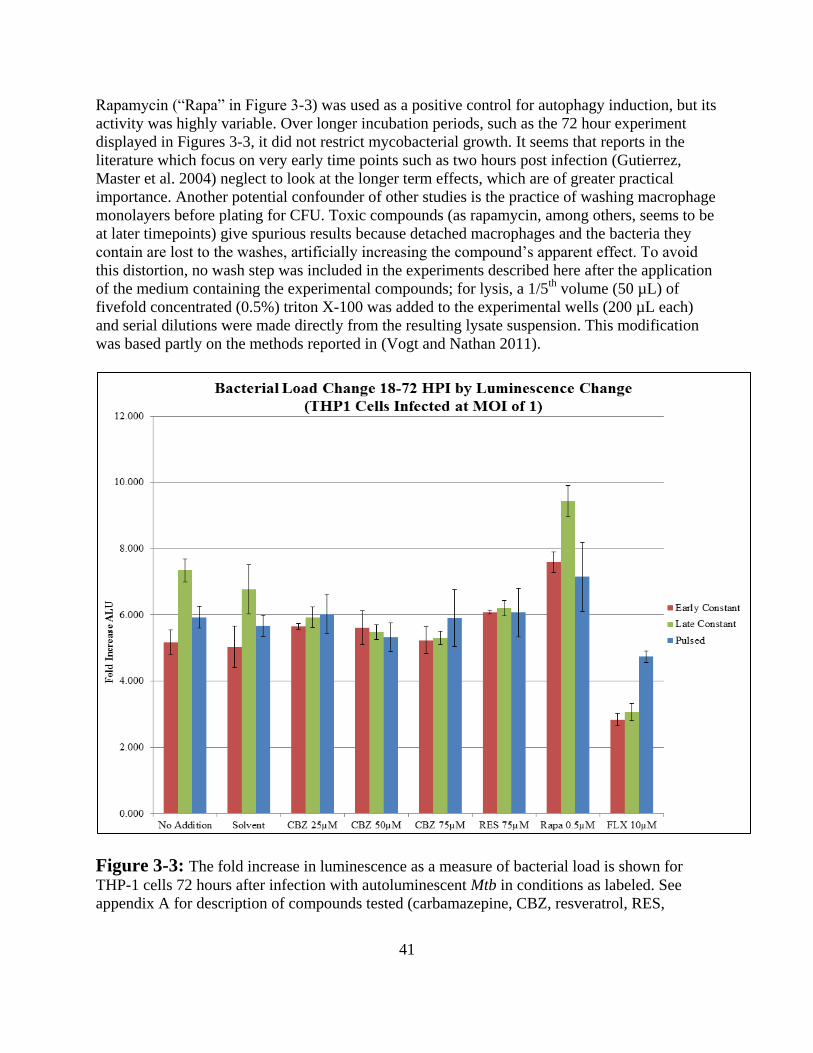
41
Rapamycin (“Rapa” in Figure 3-3) was used as a positive control for autophagy induction, but its
activity was highly variable. Over longer incubation periods, such as the 72 hour experiment
displayed in Figures 3-3, it did not restrict mycobacterial growth. It seems that reports in the
literature which focus on very early time points such as two hours post infection (Gutierrez,
Master et al. 2004) neglect to look at the longer term effects, which are of greater practical
importance. Another potential confounder of other studies is the practice of washing macrophage
monolayers before plating for CFU. Toxic compounds (as rapamycin, among others, seems to be
at later timepoints) give spurious results because detached macrophages and the bacteria they
contain are lost to the washes, artificially increasing the compound’s apparent effect. To avoid
this distortion, no wash step was included in the experiments described here after the application
of the medium containing the experimental compounds; for lysis, a 1/5th
volume (50 µL) of
fivefold concentrated (0.5%) triton X-100 was added to the experimental wells (200 µL each)
and serial dilutions were made directly from the resulting lysate suspension. This modification
was based partly on the methods reported in (Vogt and Nathan 2011).
Figure 3-3: The fold increase in luminescence as a measure of bacterial load is shown for
THP-1 cells 72 hours after infection with autoluminescent Mtb in conditions as labeled. See
appendix A for description of compounds tested (carbamazepine, CBZ, resveratrol, RES,
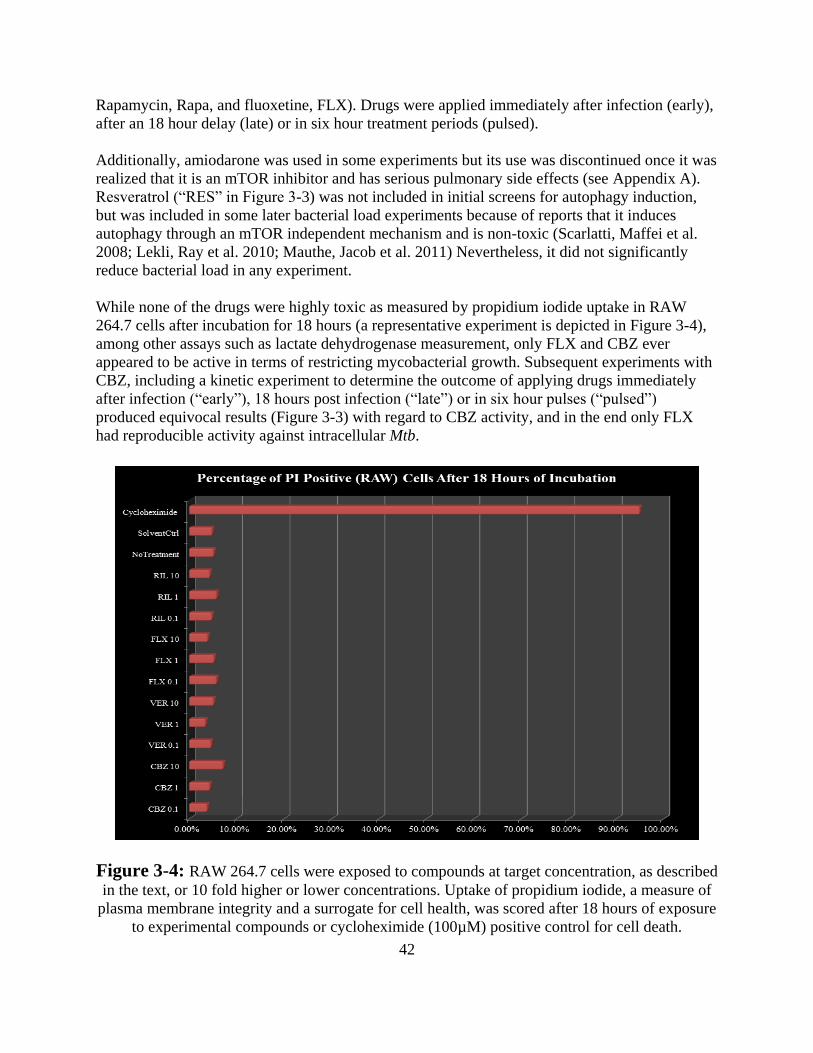
42
Rapamycin, Rapa, and fluoxetine, FLX). Drugs were applied immediately after infection (early),
after an 18 hour delay (late) or in six hour treatment periods (pulsed).
Additionally, amiodarone was used in some experiments but its use was discontinued once it was
realized that it is an mTOR inhibitor and has serious pulmonary side effects (see Appendix A).
Resveratrol (“RES” in Figure 3-3) was not included in initial screens for autophagy induction,
but was included in some later bacterial load experiments because of reports that it induces
autophagy through an mTOR independent mechanism and is non-toxic (Scarlatti, Maffei et al.
2008; Lekli, Ray et al. 2010; Mauthe, Jacob et al. 2011) Nevertheless, it did not significantly
reduce bacterial load in any experiment.
While none of the drugs were highly toxic as measured by propidium iodide uptake in RAW
264.7 cells after incubation for 18 hours (a representative experiment is depicted in Figure 3-4),
among other assays such as lactate dehydrogenase measurement, only FLX and CBZ ever
appeared to be active in terms of restricting mycobacterial growth. Subsequent experiments with
CBZ, including a kinetic experiment to determine the outcome of applying drugs immediately
after infection (“early”), 18 hours post infection (“late”) or in six hour pulses (“pulsed”)
produced equivocal results (Figure 3-3) with regard to CBZ activity, and in the end only FLX
had reproducible activity against intracellular Mtb.
Figure 3-4: RAW 264.7 cells were exposed to compounds at target concentration, as described
in the text, or 10 fold higher or lower concentrations. Uptake of propidium iodide, a measure of
plasma membrane integrity and a surrogate for cell health, was scored after 18 hours of exposure
to experimental compounds or cycloheximide (100µM) positive control for cell death.
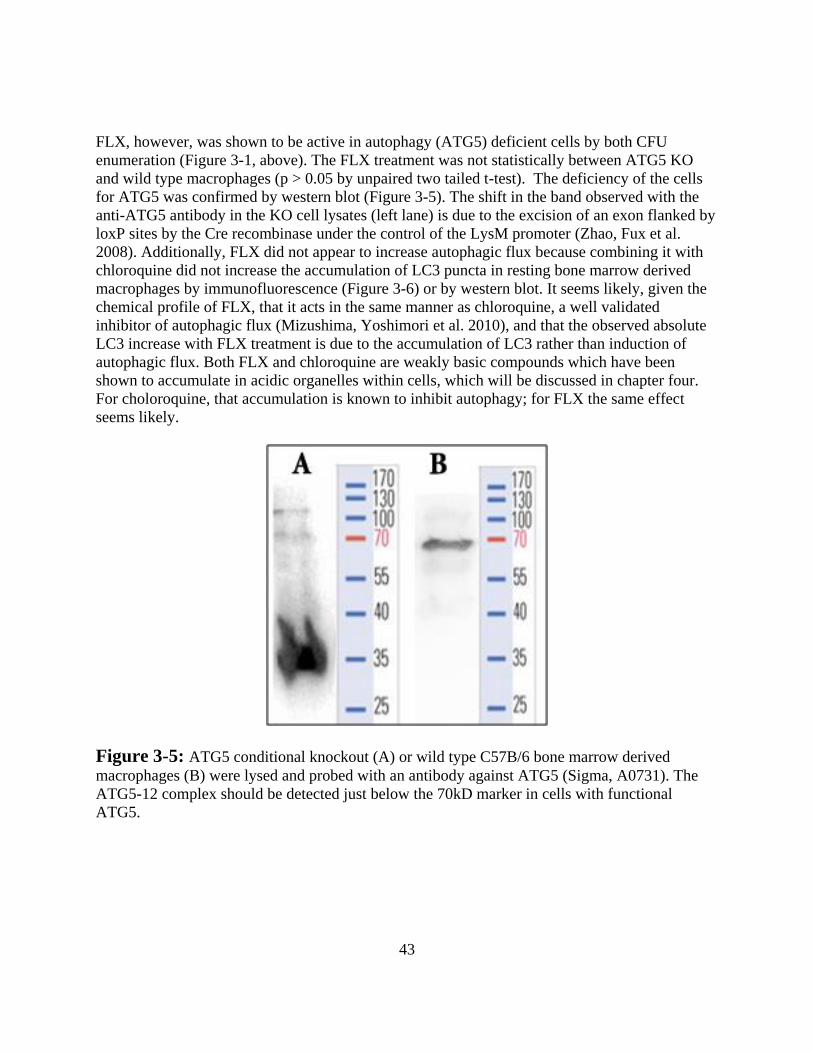
43
FLX, however, was shown to be active in autophagy (ATG5) deficient cells by both CFU
enumeration (Figure 3-1, above). The FLX treatment was not statistically between ATG5 KO
and wild type macrophages (p > 0.05 by unpaired two tailed t-test). The deficiency of the cells
for ATG5 was confirmed by western blot (Figure 3-5). The shift in the band observed with the
anti-ATG5 antibody in the KO cell lysates (left lane) is due to the excision of an exon flanked by
loxP sites by the Cre recombinase under the control of the LysM promoter (Zhao, Fux et al.
2008). Additionally, FLX did not appear to increase autophagic flux because combining it with
chloroquine did not increase the accumulation of LC3 puncta in resting bone marrow derived
macrophages by immunofluorescence (Figure 3-6) or by western blot. It seems likely, given the
chemical profile of FLX, that it acts in the same manner as chloroquine, a well validated
inhibitor of autophagic flux (Mizushima, Yoshimori et al. 2010), and that the observed absolute
LC3 increase with FLX treatment is due to the accumulation of LC3 rather than induction of
autophagic flux. Both FLX and chloroquine are weakly basic compounds which have been
shown to accumulate in acidic organelles within cells, which will be discussed in chapter four.
For choloroquine, that accumulation is known to inhibit autophagy; for FLX the same effect
seems likely.
Figure 3-5: ATG5 conditional knockout (A) or wild type C57B/6 bone marrow derived
macrophages (B) were lysed and probed with an antibody against ATG5 (Sigma, A0731). The
ATG5-12 complex should be detected just below the 70kD marker in cells with functional
ATG5.
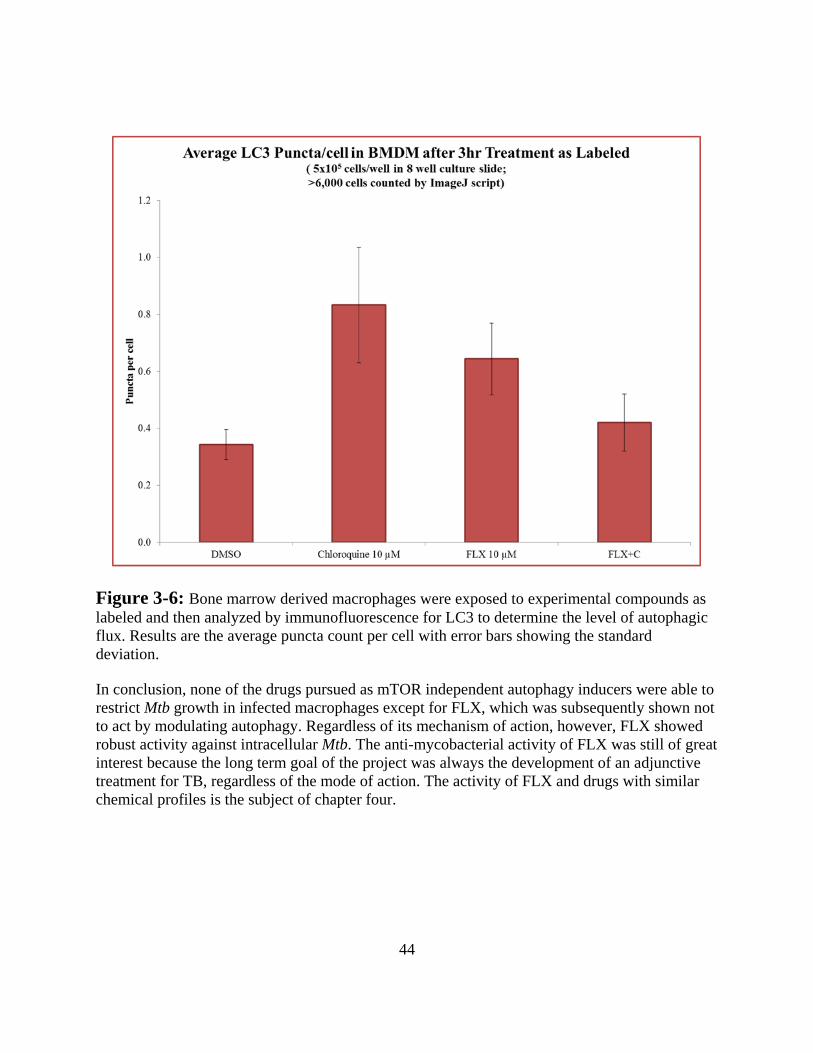
44
Figure 3-6: Bone marrow derived macrophages were exposed to experimental compounds as
labeled and then analyzed by immunofluorescence for LC3 to determine the level of autophagic
flux. Results are the average puncta count per cell with error bars showing the standard
deviation.
In conclusion, none of the drugs pursued as mTOR independent autophagy inducers were able to
restrict Mtb growth in infected macrophages except for FLX, which was subsequently shown not
to act by modulating autophagy. Regardless of its mechanism of action, however, FLX showed
robust activity against intracellular Mtb. The anti-mycobacterial activity of FLX was still of great
interest because the long term goal of the project was always the development of an adjunctive
treatment for TB, regardless of the mode of action. The activity of FLX and drugs with similar
chemical profiles is the subject of chapter four.

45
REFERENCES
Aagaard, C., T. Hoang, et al. (2011). "A multistage tuberculosis vaccine that confers efficient
protection before and after exposure." Nature Medicine 17(2): 189-194.
Adams, K. N., K. Takaki, et al. (2011). "Drug tolerance in replicating mycobacteria mediated by
a macrophage-induced efflux mechanism." Cell 145(1): 39-53.
Alonso, S., K. Pethe, et al. (2007). "Lysosomal killing of Mycobacterium mediated by ubiquitin-
derived peptides is enhanced by autophagy." Proc Natl Acad Sci U S A 104(14): 6031-
6036.
Bertholet, S., G. C. Ireton, et al. (2010). "A Defined Tuberculosis Vaccine Candidate Boosts
BCG and Protects Against Multidrug-Resistant Mycobacterium tuberculosis." Science
Translational Medicine 2(53): 53ra74-53ra74.
Biswas, D., O. S. Qureshi, et al. (2008). "ATP-induced autophagy is associated with rapid killing
of intracellular mycobacteria within human monocytes/macrophages." BMC Immunol 9:
35.
Castillo, E. F., A. Dekonenko, et al. (2012). "Autophagy protects against active tuberculosis by
suppressing bacterial burden and inflammation." Proc Natl Acad Sci U S A.
Che, N., S. Li, et al. (2010). "Identification of a novel IRGM promoter single nucleotide
polymorphism associated with tuberculosis." Clin Chim Acta 411(21-22): 1645-1649.
Condos, R., W. N. Rom, et al. (1997). "Treatment of multidrug-resistant pulmonary tuberculosis
with interferon-gamma via aerosol." Lancet 349(9064): 1513-1515.
Criollo, A., M. C. Maiuri, et al. (2007). "Regulation of autophagy by the inositol trisphosphate
receptor." Cell Death and Differentiation.
Criollo, A., J. M. Vicencio, et al. (2007). "The inositol trisphosphate receptor in the control of
autophagy." Autophagy 3(4): 350-353.
de Chastellier, C., F. Forquet, et al. (2009). "Mycobacterium requires an all-around closely
apposing phagosome membrane to maintain the maturation block and this apposition is
re-established when it rescues itself from phagolysosomes." Cell Microbiol 11(8): 1190-
1207.
Decuypere, J.-P., G. Bultynck, et al. (2011). "A dual role for Ca2+ in autophagy regulation." Cell
Calcium 50(3): 242-250.
Gelband, H. (2000). "Regimens of less than six months for treating tuberculosis." Cochrane
Database Syst Rev(2): CD001362.
Gutierrez, M. G., S. S. Master, et al. (2004). "Autophagy is a defense mechanism inhibiting BCG
and Mycobacterium tuberculosis survival in infected macrophages." Cell 119(6): 753-
766.
Harris, J., S. A. De Haro, et al. (2007). "T helper 2 cytokines inhibit autophagic control of
intracellular Mycobacterium tuberculosis." Immunity 27(3): 505-517.
Hughes, T. and T. E. Rusten (2007). "Origin and evolution of self-consumption: autophagy."
Adv Exp Med Biol 607: 111-118.
Intemann, C. D., T. Thye, et al. (2009). "Autophagy gene variant IRGM -261T contributes to
protection from tuberculosis caused by Mycobacterium tuberculosis but not by M.
africanum strains." PLoS Pathog 5(9): e1000577.
Jagannath, C., D. R. Lindsey, et al. (2009). "Autophagy enhances the efficacy of BCG vaccine
by increasing peptide presentation in mouse dendritic cells." Nat Med 15(3): 267-276.

46
Johnson, J. L., R. M. Kamya, et al. (2000). "Randomized controlled trial of Mycobacterium
vaccae immunotherapy in non-human immunodeficiency virus-infected ugandan adults
with newly diagnosed pulmonary tuberculosis. The Uganda-Case Western Reserve
University Research Collaboration." J Infect Dis 181(4): 1304-1312.
Kaplan, G., F. A. Post, et al. (2003). "Mycobacterium tuberculosis growth at the cavity surface: a
microenvironment with failed immunity." Infect Immun 71(12): 7099-7108.
Koo, M. S., C. Manca, et al. (2011). "Phosphodiesterase 4 inhibition reduces innate immunity
and improves isoniazid clearance of Mycobacterium tuberculosis in the lungs of infected
mice." PLoS ONE 6(2): e17091.
Kroemer, G. and B. Levine (2008). "Autophagic cell death: the story of a misnomer." Nature
Reviews Molecular Cell Biology 9(12): 1004-1010.
Kumar, D., L. Nath, et al. (2010). "Genome-wide Analysis of the Host Intracellular Network that
Regulates Survival of Mycobacterium tuberculosis." Cell 140(5): 731-743.
Lekli, I., D. Ray, et al. (2010). "Co-ordinated autophagy with resveratrol and gamma-tocotrienol
confers synergetic cardioprotection." J Cell Mol Med 14(10): 2506-2518.
Lowrie, D. B., R. E. Tascon, et al. (1999). "Therapy of tuberculosis in mice by DNA
vaccination." Nature 400(6741): 269-271.
Maiga, M., N. Agarwal, et al. (2012). "Successful Shortening of Tuberculosis Treatment Using
Adjuvant Host-Directed Therapy with FDA-Approved Phosphodiesterase Inhibitors in
the Mouse Model." PLoS ONE 7(2): e30749.
Mauthe, M., A. Jacob, et al. (2011). "Resveratrol-mediated autophagy requires WIPI-1-regulated
LC3 lipidation in the absence of induced phagophore formation." Autophagy 7(12):
1448-1461.
Menzies, F. M., K. Moreau, et al. (2001). Measurement of Autophagic Activity in Mammalian
Cells. Current Protocols in Cell Biology, John Wiley & Sons, Inc.
Mihaylova, M. M. and R. J. Shaw (2011). "The AMPK signalling pathway coordinates cell
growth, autophagy and metabolism." Nat Cell Biol 13(9): 1016-1023.
Mitchison, D. A. (2005). "The diagnosis and therapy of tuberculosis during the past 100 years."
Am J Respir Crit Care Med 171(7): 699-706.
Mizushima, N. and M. Komatsu (2011). "Autophagy: Renovation of Cells and Tissues." Cell
147(4): 728-741.
Mizushima, N., T. Yoshimori, et al. (2010). "Methods in mammalian autophagy research." Cell
140(3): 313-326.
Munz, C. (2009). "Enhancing immunity through autophagy." Annu Rev Immunol 27: 423-449.
Napier, R. J., W. Rafi, et al. (2011). "Imatinib-sensitive tyrosine kinases regulate mycobacterial
pathogenesis and represent therapeutic targets against tuberculosis." Cell Host Microbe
10(5): 475-485.
Okada, M., Y. Kita, et al. (2009). "Novel prophylactic and therapeutic vaccine against
tuberculosis." Vaccine 27(25-26): 3267-3270.
Parihar, S. P., R. Guler, et al. (2013). "Statin Therapy Reduces the Mycobacterium tuberculosis
Burden in Human Macrophages and in Mice by Enhancing Autophagy and Phagosome
Maturation." J Infect Dis.
Petruccioli, E., A. Romagnoli, et al. (2012). "Specific T cells restore the autophagic flux
inhibited by Mycobacterium tuberculosis in human primary macrophages." J Infect Dis
205(9): 1425-1435.

47
Ponpuak, M., A. S. Davis, et al. (2010). "Delivery of Cytosolic Components by Autophagic
Adaptor Protein p62 Endows Autophagosomes with Unique Antimicrobial Properties."
Immunity 32(3): 329-341.
Renna, M., M. Jimenez-Sanchez, et al. (2010). "Chemical inducers of autophagy that enhance
the clearance of mutant proteins in neurodegenerative diseases." J Biol Chem 285(15):
11061-11067.
Rosas-Taraco, A. G., D. M. Higgins, et al. (2011). "Local pulmonary immunotherapy with
siRNA targeting TGFbeta1 enhances antimicrobial capacity in Mycobacterium
tuberculosis infected mice." Tuberculosis (Edinb) 91(1): 98-106.
Scarlatti, F., R. Maffei, et al. (2008). "Role of non-canonical Beclin 1-independent autophagy in
cell death induced by resveratrol in human breast cancer cells." Cell Death Differ 15(8):
1318-1329.
Singh, S. B. (2006). "Human IRGM Induces Autophagy to Eliminate Intracellular
Mycobacteria." Science 313(5792): 1438-1441.
Singh, S. B., A. S. Davis, et al. (2006). "Human IRGM Induces Autophagy to Eliminate
Intracellular Mycobacteria." Science 313(5792): 1438-1441.
Stanley, S. A., A. K. Barczak, et al. (2014). "Identification of Host-Targeted Small Molecules
That Restrict Intracellular <italic>Mycobacterium tuberculosis</italic> Growth." PLoS
Pathog 10(2): e1003946.
Sundaramurthy, V., R. Barsacchi, et al. (2013). "Integration of chemical and RNAi
multiparametric profiles identifies triggers of intracellular mycobacterial killing." Cell
Host Microbe 13(2): 129-142.
Vogt, G. and C. Nathan (2011). "In vitro differentiation of human macrophages with enhanced
antimycobacterial activity." The Journal of clinical investigation 121(10): 3889-3901.
Watson, R. O., P. S. Manzanillo, et al. (2012). "Extracellular M. tuberculosis DNA Targets
Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway." Cell 150(4):
803-815.
Williams, A., S. Sarkar, et al. (2008). "Novel targets for Huntington's disease in an mTOR-
independent autophagy pathway." Nat Chem Biol 4(5): 295-305.
Yan, Y., R. J. Flinn, et al. (2009). "hVps15, but not Ca2+/CaM, is required for the activity and
regulation of hVps34 in mammalian cells." Biochem J 417(3): 747-755.
Yang, Z. and D. J. Klionsky (2010). "Mammalian autophagy: core molecular machinery and
signaling regulation." Curr Opin Cell Biol 22(2): 124-131.
Youle, R. J. and D. P. Narendra (2011). "Mechanisms of mitophagy." Nat Rev Mol Cell Biol
12(1): 9-14.
Zhang, L., J. Yu, et al. (2007). "Small molecule regulators of autophagy identified by an image-
based high-throughput screen." Proc Natl Acad Sci U S A 104(48): 19023-19028.
Zhao, Z., B. Fux, et al. (2008). "Autophagosome-independent essential function for the
autophagy protein Atg5 in cellular immunity to intracellular pathogens." Cell Host
Microbe 4(5): 458-469.
Zullo, A. J. and S. Lee (2012). "Mycobacterial induction of autophagy varies by species and
occurs independently of mTOR inhibition." J Biol Chem.

48
CHAPTER 4
PARTITIONING OF ANTIMICROBIAL COMPOUNDS BY PH BASED ION TRAPPING CAN INCREASE INTRA-MACROPHAGE SELECTIVITY INDEX
AGAINST MYCOBACTERIUM TUBERCULOSIS

49
ABSTRACT
The efficacy of antimicrobial drugs against Mycobacterium tuberculosis (Mtb), an intracellular
bacterial pathogen, is generally first established by testing compounds against bacteria in culture
medium. However, inside of infected macrophages bacteria encounter an environment which
differs substantially from broth culture and are subject to important host-dependent
pharmacokinetic phenomena which can modulate drug activity. Here we describe how pH
dependent partitioning drives asymmetric antimicrobial compound distribution in Mtb infected
macrophages. Specifically, weak bases with moderate activity against Mtb were shown to
accumulate intracellularly due to differential permeability and relative abundance of their ionized
and un-ionized forms. Non-protonatable analogs of the test compounds did not show this effect.
Neutralization of acidic organelles directly with ammonium chloride or indirectly with
bafilomycin A1 partially abrogated the growth restriction of these drugs. We further
demonstrated that the efficacy of a clinically used compound, clofazimine, is augmented by pH
based partitioning. Because the parameters which govern this effect are well understood and are
amenable to chemical modification, this knowledge may enable the rational development of
more effective anti-infective drugs against tuberculosis.
INTRODUCTION
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), ranks with HIV as
the two pathogens which are the most prolific killers of humans (2015). As many as two billion
people are estimated to be latently infected with Mtb (Dye, Scheele et al. 1999), despite the
existence of effective antibioticss. Treatment of TB is challenging due to the long duration of
chemotherapy needed to sterilize infection (Blumberg, Burman et al. 2003) and the emergence
and spread of drug resistance (Gandhi, Nunn et al. 2010). The current standard of care for
uncomplicated, drug-susceptible cases is a minimum of 26 weeks of antibiotics given first as
eight weeks of an intensive, combined drug regimen and then a further 18 weeks of two drugs
(Blumberg, Burman et al. 2003). Shortening of this regimen is a major goal of current drug
development efforts while the development of drugs with novel mechanisms of action or
particularly favorable pharmacokinetic profiles is another priority to combat drug resistance and
improve compliance.
Standard TB treatment has been shortened before, from 9-12 months to the current 6 month
‘short-course’ period with the addition of pyrazinamide (PZA) (Blumberg, Burman et al. 2003;
Zhang and Mitchison 2003). Strikingly, PZA is inactive in-vitro at the neutral pH used in most
“whole-cell” screens and so would not have been identified by current approaches. It is only
because PZA was administered in a murine model of TB that its potent in-vivo activity was
discovered (Zhang and Mitchison 2003). This demonstrates the need for more robust systems for
screening potential novel antimicrobial compounds in relevant pharmacokinetic environments.
Screening compounds against Mtb in its natural niche of the macrophage phagosome is a step in
this direction, and this study derives from a hit from such an intra-macrophage screen (Stanley,
Grant et al. 2012).

50
Specifically, the antidepressant fluoxetine (FLX) was found to restrict Mtb growth inside
macrophages at concentrations much lower than those required for direct antibacterial effect
against bacteria in broth. While this effect was originally thought to indicate a host-mediated
effect, several experimental approaches suggest that direct action of the compound occurs due to
accumulation inside the acidic compartment of exposed cells.
Because weakly basic compounds (pKa ~ 8) of moderate lipophilicity have been shown to
accumulate in acidic organelles through a phenomenon called ion trapping (de Duve, de Barsy et
al. 1974; MacIntyre and Cutler 1988; Daniel and Wojcikowski 1997; Kaufmann and Krise 2007)
and because Mtb is known to reside in a relatively acidic compartment (Rohde, Yates et al. 2007;
Vandal, Nathan et al. 2009), we hypothesized that this mechanism explains the increased activity
of FLX and other chemically similar compounds when applied to infected macrophages relative
to their activity against bacteria in culture. We will refer to this class of compounds as “acidic
compartment accumulating” or ACA drugs. In addition to FLX, we examined two other drugs
based on their pKa and logP values, which are consistent with an ACA profile. They are the local
anesthetic dibucaine (DIB) and the antidepressant sertraline (SRT) (Mayer, Wong et al. 1988;
Hiemke and Hartter 2000).
MATERIALS AND METHODS
Macrophage cell culture. All experiments were performed on resting primary murine bone
marrow derived macrophages (BMDM) at a density of 5x104/well in 200µL of medium in 96
well cluster plates. BMDM medium was composed of DMEM supplemented with 10% FBS
(Gibco, ref. 11960 and 10082), glutamine as Glutamax (Gibco, ref. 35050) at 2mM, and 10%
3T3-MCSF cell conditioned medium. Macrophages were maintained at 37°C, 5% CO2 in a
humidified incubator and handling steps were executed as quickly as safely possible to minimize
exposure to ambient conditions. For cytotoxicity measurement, CellTox Green was used
according to manufacturer’s instructions (Promega, G8741).
Bacterial culture. All experiments were conducted with Mycobacterium tuberculosis grown to
mid-log phase. Bacteria were grown in 50mL of Middlebrook 7H9 broth (Difco, 271310)
supplemented with 0.5% (v/v) glycerol, 0.05% (w/v) Tween 80 (Sigma, P1754), and 10% (v/v)
oleate-albumin-dextrose-catalase enrichment (BBL Middlebrook OADC, catalog no. 212351).
Cultures were maintained at 37°C in 1L roller bottles. The Erdman strain of Mtb was used for all
experiments except for those involving the Rv2930c::Tn mutant, for which the strain matched
control was H37Rv.
MIC experiments. Minimum inhibitory concentration for study drugs were determined using a
standard microtiter dilution protocol (Palomino, Martin et al. 2002).
Infection. Bacteria were washed thrice in PBS, resuspended and centrifuged at 50g to pellet
clumps. Bacterial suspension was added to DMEM medium with 10% horse serum substituted
for FBS to produce infection medium at 5x105 CFU/mL in 100µL per well. Phagocytosis was
allowed to proceed for four hours, after which macrophages were washed twice with warm PBS
and returned to normal medium.

51
Drug treatment. Immediately after infection, macrophages were returned to normal medium
with or without experimental compounds as appropriate. For experiments involving
neutralization of acidic organelles, macrophages were returned to 100µL of BMDM medium
with or without neutralization agent for one hour followed by a further 100µL of medium
containing 2X concentration of experimental compound or control dissolved in BMDM medium
of the same composition as the first 100µL.
Bacterial load quantification. For plating experiments, macrophages were lysed by the
addition of 50µL of 0.5% Triton X-100, yielding a final concentration of 0.1% detergent. Serial
dilutions were produced in complete 7H9 medium and plated on Middlebrook 7H10 agar plates
(Difco, 262710) containing 10% OADC and no antibiotics. CFU were counted after 17-21 days
of incubation at 37°C in a humidified incubator. For luminescence experiments, macrophages
were maintained in white 96 well cluster plates (Costar, 3610) with white vinyl sealing tape
attached to the bottom (Thermo Scientific, 236272) and read at 32°C with a Spectramax L
luminometer (Molecular Devices). Triplicate reads were averaged for each of three biological
replicates at each time point.
Reagents. The following compounds were purchased from Sigma Aldrich:, dibucaine (D0638),
resazurin (R7017), ammonium chloride (A-9434), bafilomycin A1 (B1793), isoniazid (I3377),
racemic fluoxetine (F132), serotonin (H9523), Triton X-100 (T8787) and clofazimine (C8895).
Fluoxetine enantiomers and the quaternary amine analog were generous gifts of Eli Lilly and Co.
Sertraline was from Enzo (BML-NS115) and the ketone analog was from Matrix Scientific
(072144). Hoechst 33342 was from Anaspec (AS-83218). Drugs for use in infection experiments
were dissolved in Hybri-Max DMSO (D2650, Sigma) to give stock solutions at 1000 fold their
intended final concentration and filter sterilized using 0.2µm nylon syringe filters. Solvent
controls contained 0.125% DMSO.
Imaging. BMDM were grown at a density of 1x105/well in eight chambered cover-glass culture
slides (Nunc, 155411) and treated as labeled overnight. For image acquisition, macrophages
were washed with warm PBS with calcium and magnesium and incubated at room temperature
with Hoechst 33342 at 10µM for five minutes. Nuclear counter stain solution was removed and
replaced with PBS at 4C and images were acquired promptly by wide field fluorescence
microscopy (Zeiss Axio Observer D1). Images were processed and analyzed using ImageJ
software (ImageJ, U. S. National Institutes of Health, Bethesda, MD).
Statistics. All statistical analysis was performed using Prism version 6.05 (GraphPad Software,
San Diego, CA).
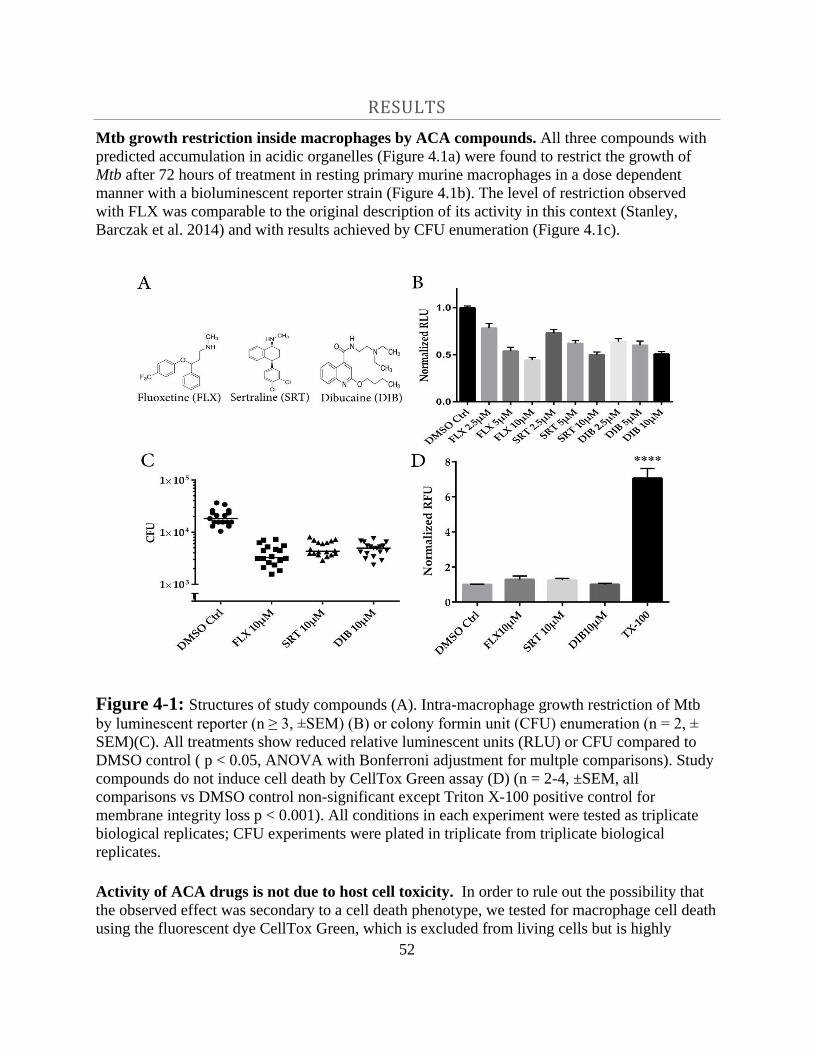
52
RESULTS
Mtb growth restriction inside macrophages by ACA compounds. All three compounds with
predicted accumulation in acidic organelles (Figure 4.1a) were found to restrict the growth of
Mtb after 72 hours of treatment in resting primary murine macrophages in a dose dependent
manner with a bioluminescent reporter strain (Figure 4.1b). The level of restriction observed
with FLX was comparable to the original description of its activity in this context (Stanley,
Barczak et al. 2014) and with results achieved by CFU enumeration (Figure 4.1c).
Figure 4-1: Structures of study compounds (A). Intra-macrophage growth restriction of Mtb
by luminescent reporter (n ≥ 3, ±SEM) (B) or colony formin unit (CFU) enumeration (n = 2, ±
SEM)(C). All treatments show reduced relative luminescent units (RLU) or CFU compared to
DMSO control ( p < 0.05, ANOVA with Bonferroni adjustment for multple comparisons). Study
compounds do not induce cell death by CellTox Green assay (D) (n = 2-4, ±SEM, all
comparisons vs DMSO control non-significant except Triton X-100 positive control for
membrane integrity loss p < 0.001). All conditions in each experiment were tested as triplicate
biological replicates; CFU experiments were plated in triplicate from triplicate biological
replicates.
Activity of ACA drugs is not due to host cell toxicity. In order to rule out the possibility that
the observed effect was secondary to a cell death phenotype, we tested for macrophage cell death
using the fluorescent dye CellTox Green, which is excluded from living cells but is highly
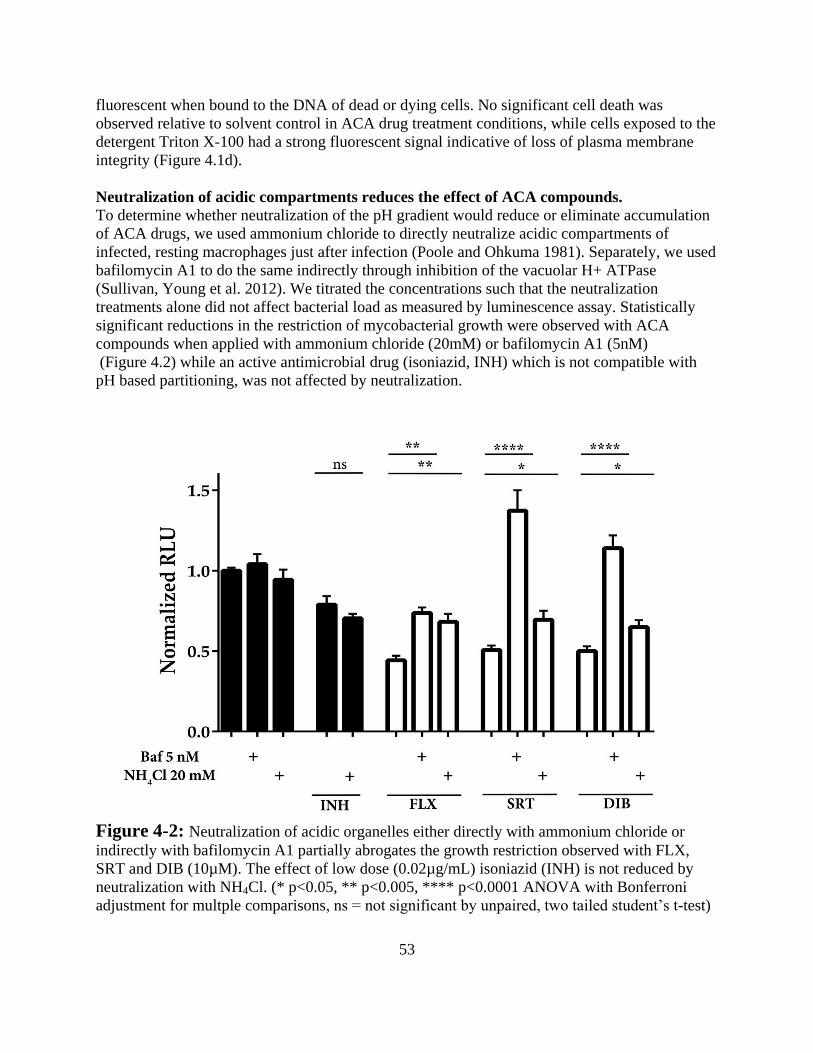
53
fluorescent when bound to the DNA of dead or dying cells. No significant cell death was
observed relative to solvent control in ACA drug treatment conditions, while cells exposed to the
detergent Triton X-100 had a strong fluorescent signal indicative of loss of plasma membrane
integrity (Figure 4.1d).
Neutralization of acidic compartments reduces the effect of ACA compounds.
To determine whether neutralization of the pH gradient would reduce or eliminate accumulation
of ACA drugs, we used ammonium chloride to directly neutralize acidic compartments of
infected, resting macrophages just after infection (Poole and Ohkuma 1981). Separately, we used
bafilomycin A1 to do the same indirectly through inhibition of the vacuolar H+ ATPase
(Sullivan, Young et al. 2012). We titrated the concentrations such that the neutralization
treatments alone did not affect bacterial load as measured by luminescence assay. Statistically
significant reductions in the restriction of mycobacterial growth were observed with ACA
compounds when applied with ammonium chloride (20mM) or bafilomycin A1 (5nM)
(Figure 4.2) while an active antimicrobial drug (isoniazid, INH) which is not compatible with
pH based partitioning, was not affected by neutralization.
Figure 4-2: Neutralization of acidic organelles either directly with ammonium chloride or
indirectly with bafilomycin A1 partially abrogates the growth restriction observed with FLX,
SRT and DIB (10µM). The effect of low dose (0.02µg/mL) isoniazid (INH) is not reduced by
neutralization with NH4Cl. (* p<0.05, ** p<0.005, **** p<0.0001 ANOVA with Bonferroni
adjustment for multple comparisons, ns = not significant by unpaired, two tailed student’s t-test)
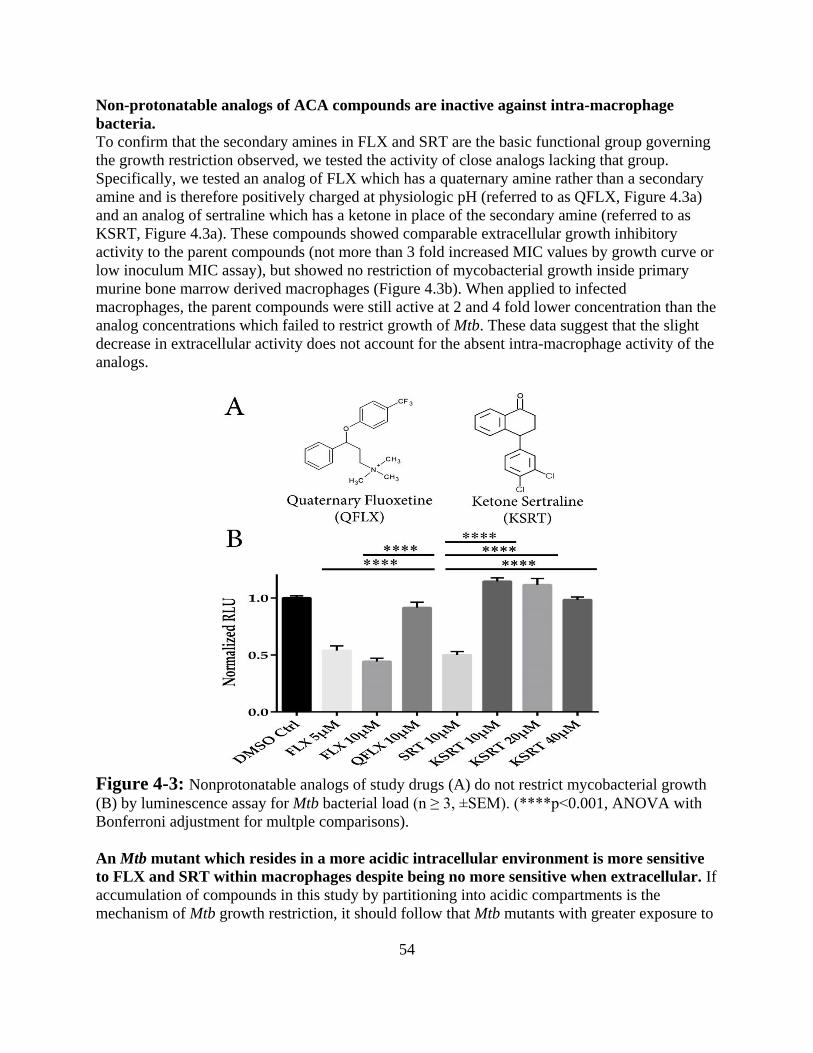
54
Non-protonatable analogs of ACA compounds are inactive against intra-macrophage
bacteria.
To confirm that the secondary amines in FLX and SRT are the basic functional group governing
the growth restriction observed, we tested the activity of close analogs lacking that group.
Specifically, we tested an analog of FLX which has a quaternary amine rather than a secondary
amine and is therefore positively charged at physiologic pH (referred to as QFLX, Figure 4.3a)
and an analog of sertraline which has a ketone in place of the secondary amine (referred to as
KSRT, Figure 4.3a). These compounds showed comparable extracellular growth inhibitory
activity to the parent compounds (not more than 3 fold increased MIC values by growth curve or
low inoculum MIC assay), but showed no restriction of mycobacterial growth inside primary
murine bone marrow derived macrophages (Figure 4.3b). When applied to infected
macrophages, the parent compounds were still active at 2 and 4 fold lower concentration than the
analog concentrations which failed to restrict growth of Mtb. These data suggest that the slight
decrease in extracellular activity does not account for the absent intra-macrophage activity of the
analogs.
Figure 4-3: Nonprotonatable analogs of study drugs (A) do not restrict mycobacterial growth
(B) by luminescence assay for Mtb bacterial load (n ≥ 3, ±SEM). (****p<0.001, ANOVA with
Bonferroni adjustment for multple comparisons).
An Mtb mutant which resides in a more acidic intracellular environment is more sensitive
to FLX and SRT within macrophages despite being no more sensitive when extracellular. If
accumulation of compounds in this study by partitioning into acidic compartments is the
mechanism of Mtb growth restriction, it should follow that Mtb mutants with greater exposure to
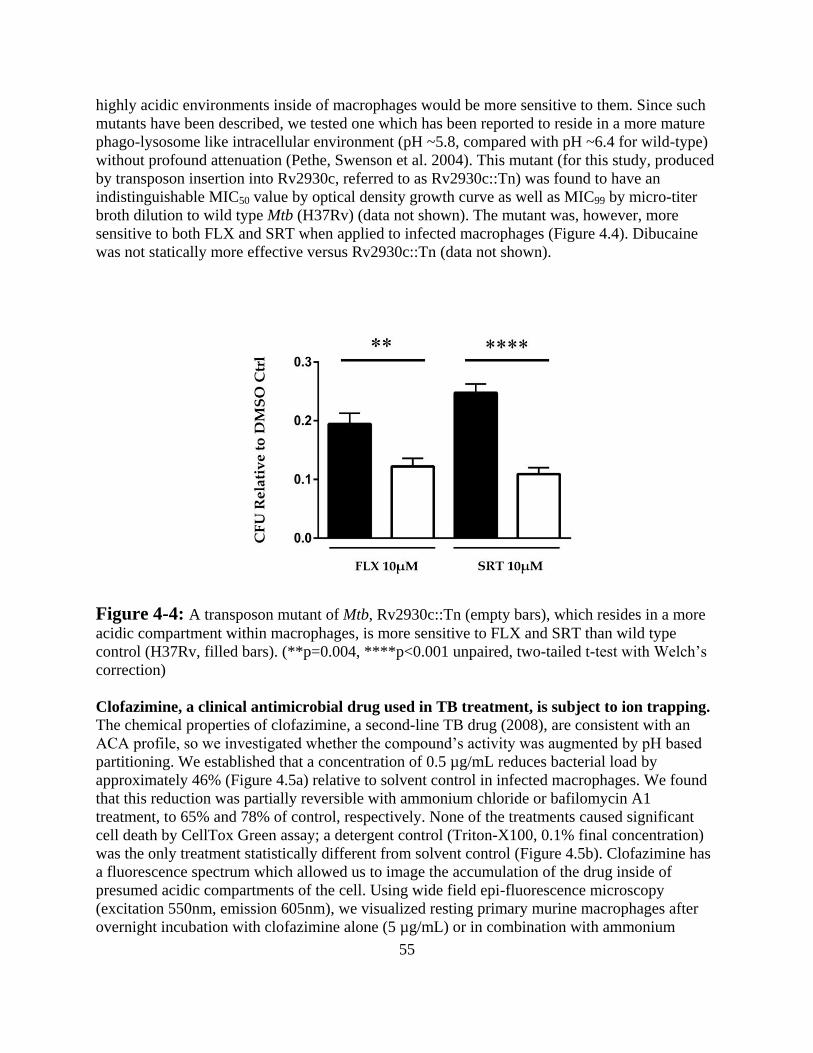
55
highly acidic environments inside of macrophages would be more sensitive to them. Since such
mutants have been described, we tested one which has been reported to reside in a more mature
phago-lysosome like intracellular environment (pH ~5.8, compared with pH ~6.4 for wild-type)
without profound attenuation (Pethe, Swenson et al. 2004). This mutant (for this study, produced
by transposon insertion into Rv2930c, referred to as Rv2930c::Tn) was found to have an
indistinguishable MIC50 value by optical density growth curve as well as MIC99 by micro-titer
broth dilution to wild type Mtb (H37Rv) (data not shown). The mutant was, however, more
sensitive to both FLX and SRT when applied to infected macrophages (Figure 4.4). Dibucaine
was not statically more effective versus Rv2930c::Tn (data not shown).
Figure 4-4: A transposon mutant of Mtb, Rv2930c::Tn (empty bars), which resides in a more
acidic compartment within macrophages, is more sensitive to FLX and SRT than wild type
control (H37Rv, filled bars). (**p=0.004, ****p<0.001 unpaired, two-tailed t-test with Welch’s
correction)
Clofazimine, a clinical antimicrobial drug used in TB treatment, is subject to ion trapping.
The chemical properties of clofazimine, a second-line TB drug (2008), are consistent with an
ACA profile, so we investigated whether the compound’s activity was augmented by pH based
partitioning. We established that a concentration of 0.5 µg/mL reduces bacterial load by
approximately 46% (Figure 4.5a) relative to solvent control in infected macrophages. We found
that this reduction was partially reversible with ammonium chloride or bafilomycin A1
treatment, to 65% and 78% of control, respectively. None of the treatments caused significant
cell death by CellTox Green assay; a detergent control (Triton-X100, 0.1% final concentration)
was the only treatment statistically different from solvent control (Figure 4.5b). Clofazimine has
a fluorescence spectrum which allowed us to image the accumulation of the drug inside of
presumed acidic compartments of the cell. Using wide field epi-fluorescence microscopy
(excitation 550nm, emission 605nm), we visualized resting primary murine macrophages after
overnight incubation with clofazimine alone (5 µg/mL) or in combination with ammonium
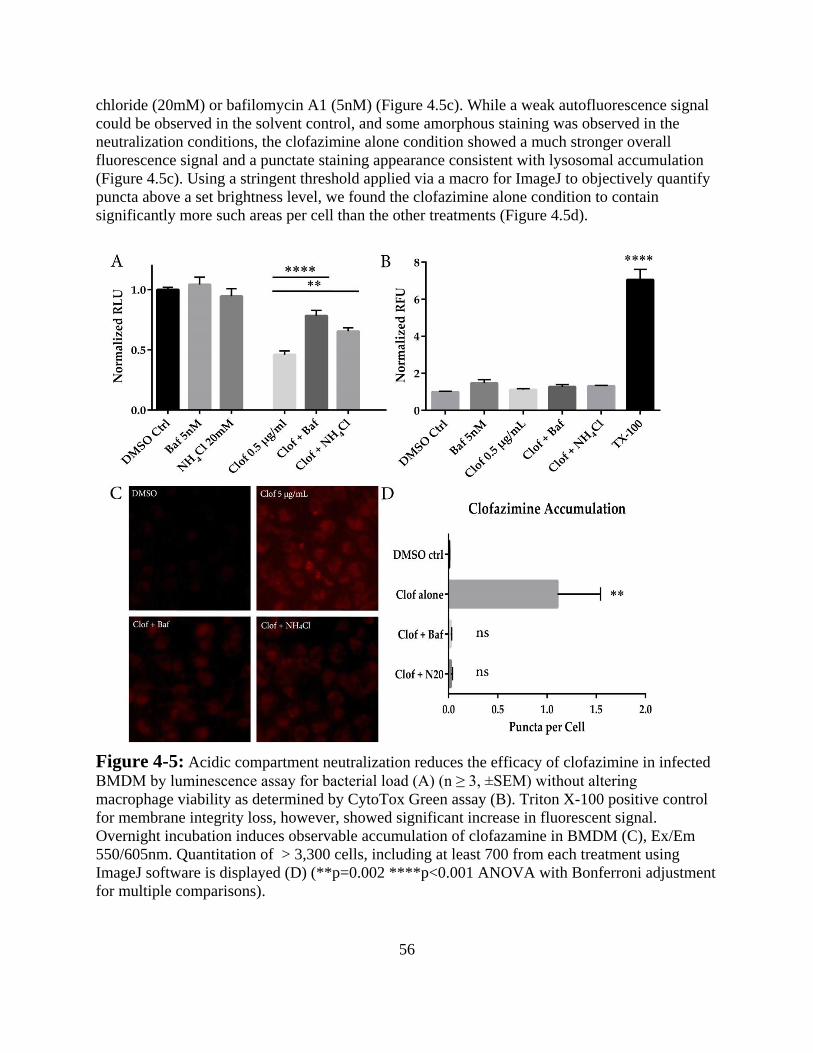
56
chloride (20mM) or bafilomycin A1 (5nM) (Figure 4.5c). While a weak autofluorescence signal
could be observed in the solvent control, and some amorphous staining was observed in the
neutralization conditions, the clofazimine alone condition showed a much stronger overall
fluorescence signal and a punctate staining appearance consistent with lysosomal accumulation
(Figure 4.5c). Using a stringent threshold applied via a macro for ImageJ to objectively quantify
puncta above a set brightness level, we found the clofazimine alone condition to contain
significantly more such areas per cell than the other treatments (Figure 4.5d).
Figure 4-5: Acidic compartment neutralization reduces the efficacy of clofazimine in infected
BMDM by luminescence assay for bacterial load (A) (n ≥ 3, ±SEM) without altering
macrophage viability as determined by CytoTox Green assay (B). Triton X-100 positive control
for membrane integrity loss, however, showed significant increase in fluorescent signal.
Overnight incubation induces observable accumulation of clofazamine in BMDM (C), Ex/Em
550/605nm. Quantitation of > 3,300 cells, including at least 700 from each treatment using
ImageJ software is displayed (D) (**p=0.002 ****p<0.001 ANOVA with Bonferroni adjustment
for multiple comparisons).

57
DISCUSSION
It has long been known that weak bases are more membrane permeable in their protonated, non-
ionized form (Milne, Scribner et al. 1958). This biophysical phenomenon, coupled with the
imbalance of ionized forms across membranes driven by actively maintained pH gradients, has
been shown to generate asymmetric distribution of weak bases. One such report used dibucaine
—a compound used in this study— as a model (Mayer, Wong et al. 1988). In his seminal 1974
work on the subject, Christian de Duve proposed that antibiotics with ‘lysosomotropic’
properties might be more effective against intracellular pathogens (de Duve, de Barsy et al.
1974). Despite the passage of 41 years, this is, to our knowledge, the first investigation of this
theory with regard to mycobacterial infection. Although a few groups have looked at the impact
of pH based partitioning on antibiotic pharmacokinetics (PK) at the cellular level (Lemaire, Van
Bambeke et al. 2009; Togami, Chono et al. 2010), in the field of mycobacteriology it has been
almost completely overlooked. Intracellular accumulation of drugs used in the treatment of TB
has been observed (Hand, Corwin et al. 1984; Pascual, Tsukayama et al. 1987) but the basis for
this accumulation was not determined.
Very few studies have investigated the activity of TB drugs in the intracellular environment.
When compared with extracellular activity, intra-macrophage activities can differ markedly
(Dhillon and Mitchison 1992; Rastogi, Labrousse et al. 1996) in mycobacterial infection models.
Isoniazid was selected as a negative control in this study because its chemical properties are not
consistent with an ACA profile and it has been shown to be present in similar concentrations
inside and outside of macrophages (Hand, Corwin et al. 1984). Clofazimine, on the other hand,
has been shown to be more active against intracellular bacteria in the context of Mycobacterium
avium infection (Rastogi, Potar et al. 1987), and in light of the findings presented here, pH based
partitioning is strongly supported as a causal mechanism. Physical accumulation, rather than
some impact on the infected cells is further supported by numerous observations in the clinical
literature (Hudson, Cox et al. 1988; Sandler, Ng et al. 1992; Silverman, Holter et al. 1993;
Harbeck, Worthen et al. 1999) wherein crystals of drug have repeatedly been observed in lung
tissue of treated patients, implying an accumulation phenotype. Interest in clofazimine has
recently intensified due to its successful use against multi-drug resistant Mtb (Caminero, Sotgiu
et al. 2010; Dey, Brigden et al. 2013), and the demonstration that it can shorten treatment in a
murine model of TB (Tyagi, Ammerman et al. 2015). Still, an effect on host cells cannot be
totally excluded and evidence exists to suggest that loading lysosomes with clofazimine impacts
the expression of lysosomal enzymes (Sarracent and Finlay 1982).
The impact of ACA drugs on the endogenous functions of the structures they accumulate within
is difficult to predict. For example, the partial neutralization of (phago)lysosomes by ACA drugs
due to their basicity may represent a countervailing effect to the direct antimicrobial effect of the
drugs themselves (Vandal, Nathan et al. 2009) and could account for the lack of additional effect
against the Rv2930c::Tn mutant demonstrated by dibucaine in this study (given its relatively low
antimicrobial activity but high accumulation). This effect, if present, would be expected to be
less of a concern with ACA drugs with greater antimicrobial activity, though, since lower doses
could be administered. Additionally, it has been shown that chloroquine, a known lysosomal

58
accumulator, does not decrease the activity of pyrazinamide, a drug only active at acidic pH,
when applied to infected macrophages (Crowle and May 1990). This implies that strong
alkalization of Mtb containing structures does not occur with that compound and may be
generalizable. That finding, and our results with ammonium chloride and bafilomycin treatment
alone, which were similar to published results reported by another group (Sullivan, Young et al.
2012), suggests that acidic compartment neutralization alone does not profoundly impact the
course of infection of macrophages with wild type Mtb. So while mutants which traffic to more
mature phagolysosomes may be saved by neutralization [our results and figure 7c in (Sullivan,
Young et al. 2012)], wild type bacteria are less affected . An impact on autophagy, in particular,
could be a concern given that bafilomycin is an inhibitor of that process and was shown to
partially reverse the action of ACA drugs. A role for canonical autophagy is essentially ruled out,
however, by our observations of unaffected activity of FLX and SRT in autophagy deficient
(ATG5 conditional knockout) cells (see chapter 3, figure 3.1).
Conversely, host-beneficial effects may result from ACA drug deposition and subsequent effects
on the host cell such as phopholipidosis (Mesens, Steemans et al. 2010), for example by
interference with the close apposition of the bacterial cell wall to the host membrane which has
been reported to be necessary for phagosomal maturation arrest (de Chastellier, Forquet et al.
2009), a key survival strategy of Mtb. A multifactorial mechanism of restriction of mycobacterial
growth by high-accumulating drugs seems likely.
The antimicrobial activities of fluoxetine and sertraline have been described before (Munoz-
Bellido, Munoz-Criado et al. 2000; Coban, Tanriverdi Cayci et al. 2009; Bohnert, Szymaniak-
Vits et al. 2011; Zhai, Wu et al. 2012) but only for cryptococcal infection has the therapeutic
index of one of the drugs (i.e., SRT) been suggested to be compatible with clinical treatment
(Zhai, Wu et al. 2012). Our contention here is not that FLX, SRT or DIB are viable drugs for the
treatment of TB, but rather that they are exemplars of a phenomenon (i.e., ion trapping) which
could increase the local concentration of antimicrobial compounds to which Mtb is exposed
intracellularly. Rational modification of antibiotics to endow them with an ACA profile has been
demonstrated; Renard and colleagues modified penicillin G (an otherwise non-accumulating,
predominantly extracellular drug) by removing a carboxylic acid moiety and replacing it with a
basic, amine containing group (Renard, Vanderhaeghe et al. 1987). That alteration was sufficient
to cause a shift toward accumulation of the drug in the acidic compartment – primarily
lysosomes as identified by co-fractionation.
As an antidepressant, fluoxetine is thought to work by blocking serotonin uptake from synapses
(Wong, Perry et al. 2005). Though murine macrophages may express the serotonin transporter
(Jackson, Walker et al. 1988), the level of serotonin in culture medium did not impact the
restriction of mycobacterial growth we observed in our infections at any physiologically
plausible concentration, and medium made with dialyzed, serotonin-free fetal bovine serum
(FBS) performed no differently from standard FBS (data not shown). Also, the concentration
range used in this study was around three orders of magnitude greater than the Ki of the drug for
the serotonin transporter (Koch, Perry et al. 2002), so essentially all binding sites should be
saturated and a dose-wise response should not be observed. It remained possible that FLX was
directly binding serotonin receptors, rather than the transporter, because such affinity has been
reported (Koch, Perry et al. 2002). However, the enantiomers of FLX, which have differing
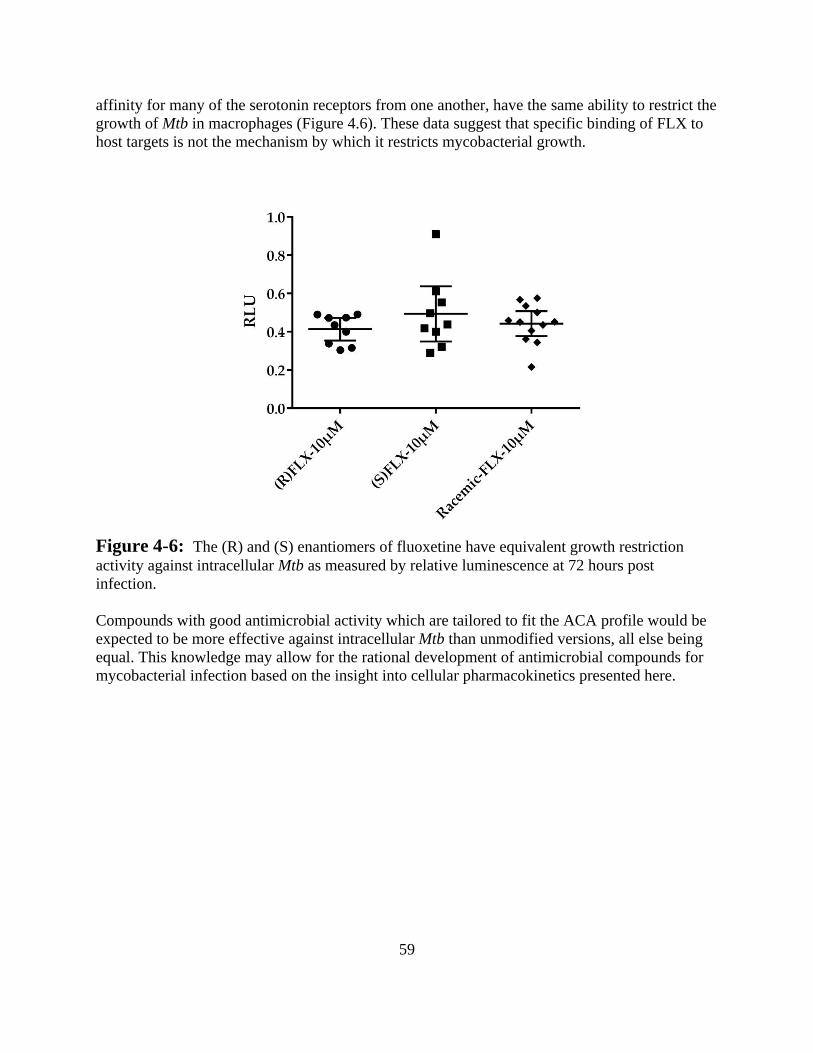
59
affinity for many of the serotonin receptors from one another, have the same ability to restrict the
growth of Mtb in macrophages (Figure 4.6). These data suggest that specific binding of FLX to
host targets is not the mechanism by which it restricts mycobacterial growth.
Figure 4-6: The (R) and (S) enantiomers of fluoxetine have equivalent growth restriction
activity against intracellular Mtb as measured by relative luminescence at 72 hours post
infection.
Compounds with good antimicrobial activity which are tailored to fit the ACA profile would be
expected to be more effective against intracellular Mtb than unmodified versions, all else being
equal. This knowledge may allow for the rational development of antimicrobial compounds for
mycobacterial infection based on the insight into cellular pharmacokinetics presented here.

60
REFERENCES
(2008). "Clofazimine." Tuberculosis 88(2): 96-99.
(2015). "Global, regional, and national age-sex specific all-cause and cause-specific mortality for
240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease
Study 2013." Lancet 385(9963): 117-171.
Blumberg, H. M., W. J. Burman, et al. (2003). "American Thoracic Society/Centers for Disease
Control and Prevention/Infectious Diseases Society of America: treatment of
tuberculosis." Am J Respir Crit Care Med 167(4): 603-662.
Bohnert, J. A., M. Szymaniak-Vits, et al. (2011). "Efflux inhibition by selective serotonin
reuptake inhibitors in Escherichia coli." J Antimicrob Chemother 66(9): 2057-2060.
Caminero, J. A., G. Sotgiu, et al. (2010). "Best drug treatment for multidrug-resistant and
extensively drug-resistant tuberculosis." The Lancet Infectious Diseases 10(9): 621-629.
Coban, A. Y., Y. Tanriverdi Cayci, et al. (2009). "[Investigation of antibacterial activity of
sertralin]." Mikrobiyol Bul 43(4): 651-656.
Crowle, A. J. and M. H. May (1990). "Inhibition of tubercle bacilli in cultured human
macrophages by chloroquine used alone and in combination with streptomycin, isoniazid,
pyrazinamide, and two metabolites of vitamin D3." Antimicrob Agents Chemother
34(11): 2217-2222.
Daniel, W. A. and J. Wojcikowski (1997). "Contribution of lysosomal trapping to the total tissue
uptake of psychotropic drugs." Pharmacol Toxicol 80(2): 62-68.
de Chastellier, C., F. Forquet, et al. (2009). "Mycobacterium requires an all-around closely
apposing phagosome membrane to maintain the maturation block and this apposition is
re-established when it rescues itself from phagolysosomes." Cell Microbiol 11(8): 1190-
1207.
de Duve, C., T. de Barsy, et al. (1974). "Lysosomotropic agents." Biochem Pharmacol 23(18):
2495-2531.
Dey, T., G. Brigden, et al. (2013). "Outcomes of clofazimine for the treatment of drug-resistant
tuberculosis: a systematic review and meta-analysis." J Antimicrob Chemother 68(2):
284-293.
Dhillon, J. and D. A. Mitchison (1992). "Activity in vitro of rifabutin, FCE 22807, rifapentine,
and rifampin against Mycobacterium microti and M. tuberculosis and their penetration
into mouse peritoneal macrophages." Am Rev Respir Dis 145(1): 212-214.
Dye, C., S. Scheele, et al. (1999). "Consensus statement. Global burden of tuberculosis:
estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and
Monitoring Project." JAMA 282(7): 677-686.
Gandhi, N. R., P. Nunn, et al. (2010). "Multidrug-resistant and extensively drug-resistant
tuberculosis: a threat to global control of tuberculosis." Lancet 375(9728): 1830-1843.
Hand, W. L., R. W. Corwin, et al. (1984). "Uptake of antibiotics by human alveolar
macrophages." Am Rev Respir Dis 129(6): 933-937.
Harbeck, R. J., G. S. Worthen, et al. (1999). "Clofazimine crystals in the cytoplasm of
pulmonary macrophages." Ann Pharmacother 33(2): 250.
Hiemke, C. and S. Hartter (2000). "Pharmacokinetics of selective serotonin reuptake inhibitors."
Pharmacol Ther 85(1): 11-28.

61
Hudson, V., F. Cox, et al. (1988). "Pulmonary clofazimine crystals in a child with acquired
immunodeficiency syndrome and disseminated Mycobacterium avium-intracellulare
infection." Pediatr Infect Dis J 7(12): 880-882.
Jackson, J. C., R. F. Walker, et al. (1988). "Specific uptake of serotonin by murine
macrophages." Life Sciences 42(17): 1641-1650.
Kaufmann, A. M. and J. P. Krise (2007). "Lysosomal sequestration of amine-containing drugs:
analysis and therapeutic implications." J Pharm Sci 96(4): 729-746.
Koch, S., K. W. Perry, et al. (2002). "R-fluoxetine increases extracellular DA, NE, as well as 5-
HT in rat prefrontal cortex and hypothalamus: an in vivo microdialysis and receptor
binding study." Neuropsychopharmacology 27(6): 949-959.
Lemaire, S., F. Van Bambeke, et al. (2009). "Cellular accumulation and pharmacodynamic
evaluation of the intracellular activity of CEM-101, a novel fluoroketolide, against
Staphylococcus aureus, Listeria monocytogenes, and Legionella pneumophila in human
THP-1 macrophages." Antimicrob Agents Chemother 53(9): 3734-3743.
MacIntyre, A. C. and D. J. Cutler (1988). "The potential role of lysosomes in tissue distribution
of weak bases." Biopharm Drug Dispos 9(6): 513-526.
Mayer, L. D., K. F. Wong, et al. (1988). "Influence of ion gradients on the transbilayer
distribution of dibucaine in large unilamellar vesicles." Biochemistry 27(6): 2053-2060.
Mesens, N., M. Steemans, et al. (2010). "Screening for phospholipidosis induced by central
nervous drugs: Comparing the predictivity of an in vitro assay to high throughput in silico
assays." Toxicology in Vitro 24(5): 1417-1425.
Milne, M. D., B. H. Scribner, et al. (1958). "Non-ionic diffusion and the excretion of weak acids
and bases." Am J Med 24(5): 709-729.
Munoz-Bellido, J. L., S. Munoz-Criado, et al. (2000). "Antimicrobial activity of psychotropic
drugs: selective serotonin reuptake inhibitors." Int J Antimicrob Agents 14(3): 177-180.
Palomino, J. C., A. Martin, et al. (2002). "Resazurin microtiter assay plate: simple and
inexpensive method for detection of drug resistance in Mycobacterium tuberculosis."
Antimicrob Agents Chemother 46(8): 2720-2722.
Pascual, A., D. Tsukayama, et al. (1987). "Uptake and activity of rifapentine in human peritoneal
macrophages and polymorphonuclear leukocytes." Eur J Clin Microbiol 6(2): 152-157.
Pethe, K., D. L. Swenson, et al. (2004). "Isolation of Mycobacterium tuberculosis mutants
defective in the arrest of phagosome maturation." Proc Natl Acad Sci U S A 101(37):
13642-13647.
Poole, B. and S. Ohkuma (1981). "Effect of weak bases on the intralysosomal pH in mouse
peritoneal macrophages." J Cell Biol 90(3): 665-669.
Rastogi, N., V. Labrousse, et al. (1996). "In vitro activities of fourteen antimicrobial agents
against drug susceptible and resistant clinical isolates of Mycobacterium tuberculosis and
comparative intracellular activities against the virulent H37Rv strain in human
macrophages." Curr Microbiol 33(3): 167-175.
Rastogi, N., M.-C. Potar, et al. (1987). "Intracellular growth of pathogenic mycobacteria in the
continuous murine macrophage cell line J-774: Ultrastructure and drug-susceptibility
studies." Current Microbiology 16(2): 79-92.
Renard, C., H. J. Vanderhaeghe, et al. (1987). "Influence of conversion of penicillin G into a
basic derivative on its accumulation and subcellular localization in cultured
macrophages." Antimicrob Agents Chemother 31(3): 410-416.

62
Rohde, K., R. M. Yates, et al. (2007). "Mycobacterium tuberculosis and the environment within
the phagosome." Immunol Rev 219: 37-54.
Sandler, E. D., V. L. Ng, et al. (1992). "Clofazimine crystals in alveolar macrophages from a
patient with the acquired immunodeficiency syndrome." Arch Pathol Lab Med 116(5):
541-543.
Sarracent, J. and C. M. Finlay (1982). "The action of Clofazimine on the level of lysosomal
enzymes of cultured macrophages." Clin Exp Immunol 48(1): 261-267.
Silverman, J. F., J. F. Holter, et al. (1993). "Negative images due to clofazimine crystals
simulating MAI infection in a bronchoalveolar lavage specimen." Diagn Cytopathol 9(5):
534-539; discussion 539-540.
Stanley, S. A., A. K. Barczak, et al. (2014). "Identification of Host-Targeted Small Molecules
That Restrict Intracellular <italic>Mycobacterium tuberculosis</italic> Growth." PLoS
Pathog 10(2): e1003946.
Stanley, S. A., S. S. Grant, et al. (2012). "Identification of novel inhibitors of M. tuberculosis
growth using whole cell based high-throughput screening." ACS Chem Biol 7(8): 1377-
1384.
Sullivan, J. T., E. F. Young, et al. (2012). "The Mycobacterium tuberculosis SecA2 system
subverts phagosome maturation to promote growth in macrophages." Infect Immun
80(3): 996-1006.
Togami, K., S. Chono, et al. (2010). "Intracellular pharmacokinetics of telithromycin, a ketolide
antibiotic, in alveolar macrophages." J Pharm Pharmacol 62(1): 71-75.
Tyagi, S., N. C. Ammerman, et al. (2015). "Clofazimine shortens the duration of the first-line
treatment regimen for experimental chemotherapy of tuberculosis." Proc Natl Acad Sci U
S A.
Vandal, O. H., C. F. Nathan, et al. (2009). "Acid resistance in Mycobacterium tuberculosis." J
Bacteriol 191(15): 4714-4721.
Wong, D. T., K. W. Perry, et al. (2005). "The Discovery of Fluoxetine Hydrochloride (Prozac)."
Nat Rev Drug Discov 4(9): 764-774.
Zhai, B., C. Wu, et al. (2012). "The antidepressant sertraline provides a promising therapeutic
option for neurotropic cryptococcal infections." Antimicrob Agents Chemother 56(7):
3758-3766.
Zhang, Y. and D. Mitchison (2003). "The curious characteristics of pyrazinamide: a review." Int
J Tuberc Lung Dis 7(1): 6-21.

63
DISSERTATION SUMMARY AND CONCLUSIONS
The studies described in this dissertation were embarked upon with the goal of improving the
therapy of tuberculosis by changing the dynamics of the host-pathogen interface. Whether by
shaping the adaptive immune response using a vaccine as in chapter one, by augmenting innate
immune function by pharmacologic intervention, as in chapter two, or by leveraging
characteristics of the intracellular niche occupied by Mtb to improve chemotherapy, as in chapter
four, the goal was always to tip the outcome of infection in favor of containment and eradication
of the pathogen.
The vaccine approach pursued in chapter two failed to protect against reactivation or relapse
disease in a murine model of TB despite promising data from an earlier study which showed
strong protection. After a thorough assessment of the outcomes and differences in execution of
the two studies, it seems most likely that the cause of the disparate results was the change in
challenge strain of Mtb. Because the subunit vaccine used in both studies was composed of a
protein which is overexpressed by the strain of Mtb used in the original report, the protection
demonstrated in that study was probably an artifact of antigen overexpression in the mutant
strain. The failure to recapitulate the earlier result with wild type Mtb was disappointing, but
further study of the resolution of infection seen in that system may still be worth pursuing in
pursuit of correlates of protection against reactivation or relapse TB.
In chapter three, the application of compounds reported to upregulate autophagy, a host-cell
process with reported mycobactericidal activity, was described. Despite a strong theoretical basis
for expecting these compounds to restrict mycobacterial growth, none robustly did so. One
compound, however, was confirmed to have reproducible activity against intracellular Mtb. That
compound, fluoxetine, was found not to operate through canonical autophagy, but to act at least
partially through an accumulation phenomenon which was described in depth in chapter four.
Finally, in chapter four, the accumulation of weakly basic antimicrobial drugs was demonstrated
to be due to pH based ion trapping. The accumulation of several model compounds was shown to
depend on intracellular accumulation in acidic organelles. This accumulation was reversible by
neutralization of the acidic compartment and did not occur with analogs of the study drugs which
were modified to have non-basic pKa values. It was further demonstrated that a mutant of Mtb
which resides in more acidic phagosomes was more sensitive to two of the study drugs. A
clinically used compound compatible with the pH based partitioning effect, clofazimine, was
shown to benefit from ion trapping, as well, and due to its fluorescence spectrum, its
accumulation was shown by fluorescence microscopy.
The findings described in chapter four may enable medicinal chemists to rationally develop new
anti-infective drugs for TB to have more favorable pharmacokinetic profiles based on their
cellular level distribution. This aspect of host-factor enhancement, namely asymmetric drug
distribution due to pH gradients, was discovered serendipitously and represents the major
contribution of this work to future development of therapies for tuberculosis.
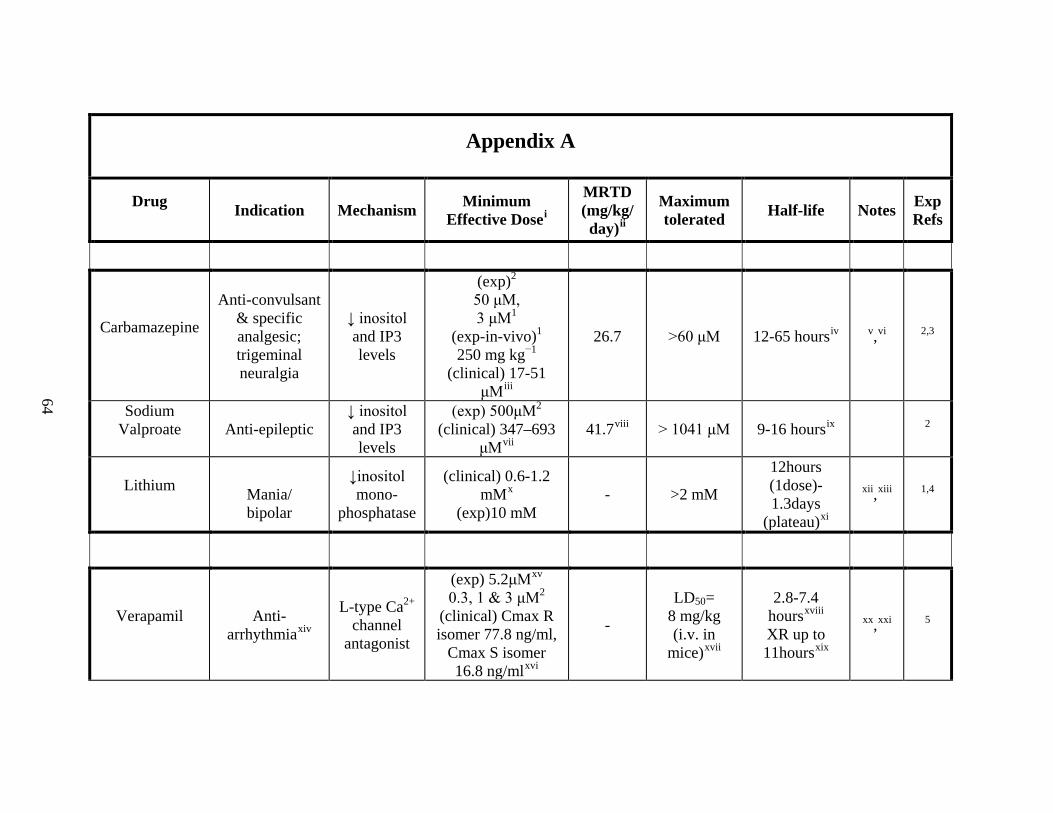
64
Appendix A
Drug Indication Mechanism Minimum
Effective Dosei
MRTD (mg/kg/ day)ii
Maximum tolerated Half-life Notes Exp
Refs
Carbamazepine
Anti-convulsant & specific analgesic; trigeminal neuralgia
↓ inositol and IP3 levels
(exp)2 50 μM, 3 μM1
(exp-in-vivo)1 250 mg kg−1
(clinical) 17-51 μMiii
26.7 >60 μM 12-65 hoursiv v,vi 2,3
Sodium Valproate
Anti-epileptic
↓ inositol and IP3 levels
(exp) 500μM2 (clinical) 347–693
μMvii 41.7viii > 1041 μM 9-16 hoursix 2
Lithium
Mania/ bipolar
↓inositol mono-
phosphatase
(clinical) 0.6-1.2 mMx
(exp)10 mM - >2 mM
12hours (1dose)- 1.3days
(plateau)xi
xii,xiii 1,4
Verapamil
Anti-arrhythmiaxiv
L-type Ca2+ channel
antagonist
(exp) 5.2μMxv 0.3, 1 & 3 μM2
(clinical) Cmax R isomer 77.8 ng/ml,
Cmax S isomer 16.8 ng/mlxvi
-
LD50= 8 mg/kg (i.v. in
mice)xvii
2.8-7.4 hoursxviii XR up to 11hoursxix
xx,xxi 5
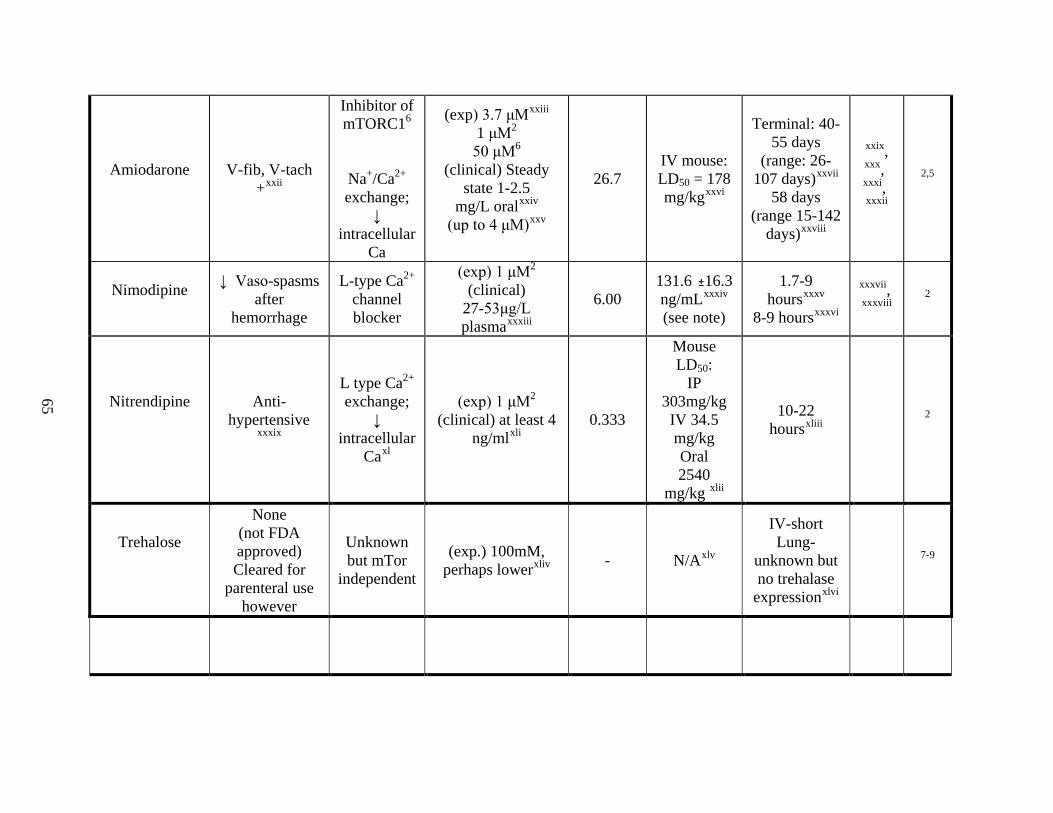
65
Amiodarone
V-fib, V-tach +xxii
Inhibitor of mTORC16
Na+/Ca2+ exchange;
↓ intracellular
Ca
(exp) 3.7 μMxxiii 1 μM2 50 μM6
(clinical) Steady state 1-2.5
mg/L oralxxiv (up to 4 μM)xxv
26.7 IV mouse: LD50 = 178 mg/kgxxvi
Terminal: 40-55 days
(range: 26-107 days)xxvii
58 days (range 15-142
days)xxviii
xxxii
xxix,xxx, xxxi,
2,5
Nimodipine
↓ Vaso-spasms after
hemorrhage
L-type Ca2+ channel blocker
(exp) 1 μM2 (clinical)
27-53μg/L plasmaxxxiii
6.00 131.6 16.3 ng/mLxxxiv (see note)
1.7-9 hoursxxxv
8-9 hoursxxxvi
xxxvii
xxxviii,
2
Nitrendipine
Anti-hypertensive
xxxix
L type Ca2+ exchange;
↓ intracellular
Caxl
(exp) 1 μM2 (clinical) at least 4
ng/mlxli 0.333
Mouse LD50:
IP 303mg/kg IV 34.5 mg/kg Oral 2540
mg/kg xlii
10-22 hoursxliii 2
Trehalose
None (not FDA approved) Cleared for
parenteral use however
Unknown but mTor
independent
(exp.) 100mM, perhaps lowerxliv - N/Axlv
IV-short Lung-
unknown but no trehalase
expressionxlvi
7-9
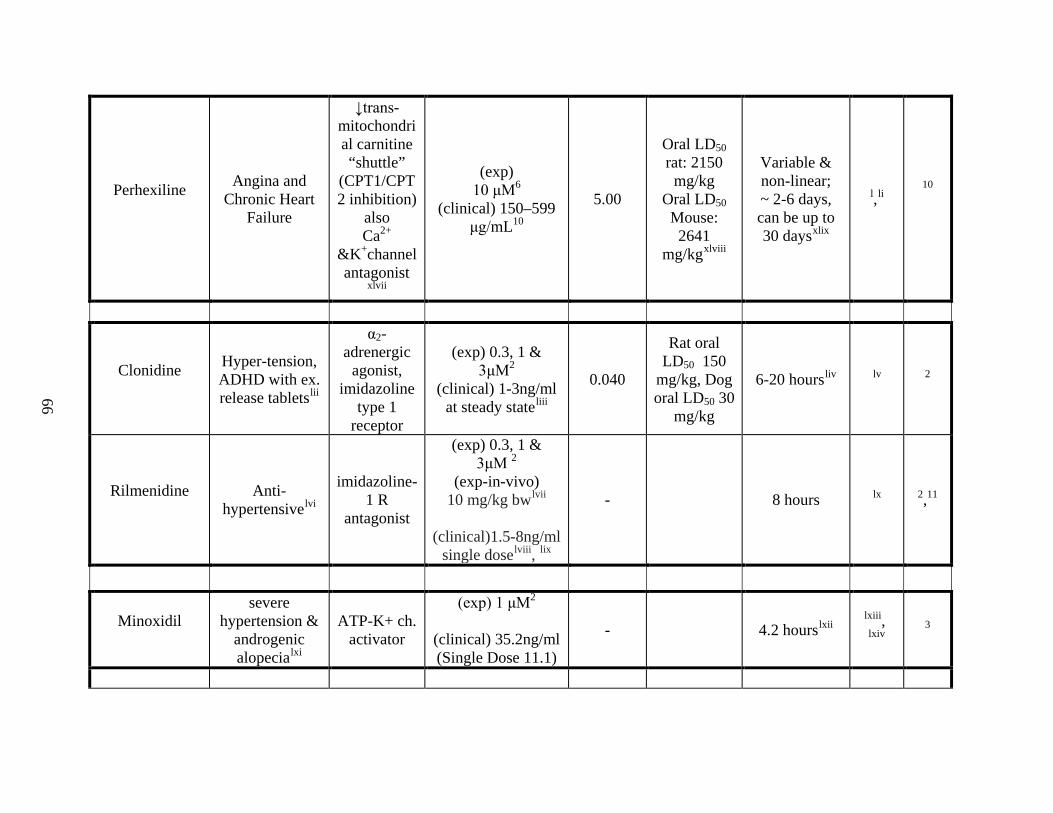
66
Perhexiline
Angina and Chronic Heart
Failure
↓trans-mitochondrial carnitine “shuttle”
(CPT1/CPT2 inhibition)
also Ca2+
&K+channel antagonist
xlvii
(exp) 10 μM6
(clinical) 150–599 μg/mL10
5.00
Oral LD50 rat: 2150
mg/kg Oral LD50
Mouse: 2641
mg/kgxlviii
Variable & non-linear; ~ 2-6 days, can be up to 30 daysxlix
l,li 10
Clonidine
Hyper-tension, ADHD with ex. release tabletslii
α2-adrenergic
agonist, imidazoline
type 1 receptor
(exp) 0.3, 1 & 3μM2
(clinical) 1-3ng/ml at steady stateliii
0.040
Rat oral LD50 150
mg/kg, Dog oral LD50 30
mg/kg
6-20 hoursliv lv 2
Rilmenidine
Anti- hypertensivelvi
imidazoline-1 R
antagonist
(exp) 0.3, 1 & 3μM 2
(exp-in-vivo) 10 mg/kg bwlvii
(clinical)1.5-8ng/ml
single doselviii, lix
- 8 hours lx 2,11
Minoxidil
severe hypertension &
androgenic alopecialxi
ATP-K+ ch. activator
(exp) 1 μM2
(clinical) 35.2ng/ml (Single Dose 11.1)
- 4.2 hourslxii lxiii, lxiv
3

67
Experimental References
1. Hidvegi, T., et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 329, 229-232 (2010).
2. Sarkar, S., et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 170, 1101-1111 (2005). 3. Williams, A., et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 4,
295-305 (2008). 4. Sarkar, S., et al. A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum
Mol Genet 17, 170-178 (2008). 5. Zhang, L., et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad
Sci U S A 104, 19023-19028 (2007). 6. Balgi, A.D., et al. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling.
PLoS ONE 4, e7124 (2009). 7. Sarkar, S., Davies, J.E., Huang, Z., Tunnacliffe, A. & Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy
enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 282, 5641-5652 (2007). 8. Ishihara, R., et al. Molecular cloning, sequencing and expression of cDNA encoding human trehalase. Gene 202, 69-74 (1997). 9. Oesterreicher, T.J., Markesich, D.C. & Henning, S.J. Cloning, characterization and mapping of the mouse trehalase (Treh)
gene. Gene 270, 211-220 (2001). 10. Ashrafian, H., Horowitz, J.D. & Frenneaux, M.P. Perhexiline. Cardiovasc Drug Rev 25, 76-97 (2007). 11. Rose, C., et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington's disease. Hum
Mol Genet 19, 2144-2153 (2010).

68
Appendix A Notes
[Note: all hyperlinks current as of April 2012]
i Abbreviations: “exp” denotes a value from the basic science literature while “clinical” denotes a value from clinical practice. iiMaximum Recommended Therapeutic Dose – according to: http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm092199.html iii Timing of serum samples: Absorption is slow, peak levels occur 6-8 hours after ingestion of the first dose; the half-life ranges from 8-60 hours, therefore, steady-state is achieved in 2-5 days Therapeutic levels: 4-12 mcg/mL (SI: 17-51 μmol/L) Toxic concentration: >15 mcg/mL; patients who require higher levels of 8-12 mcg/mL (SI: 34-51 μmol/L) should be watched closely. Side effects including CNS effects occur commonly at higher dosage levels. If other anticonvulsants are given therapeutic range is 4-8 mcg/mL. [http://www.merckmanuals.com/professional/lexicomp/carbamazepine.html] iv Highly dependent on previous dosing - extended release available (keep in mind it autoinduces its own metabolism) : “Following a single extended release dose of carbamazepine, the average half-life range from 35-40 hours and 12-17 hours on repeated dosing.” http://www.fda.gov/downloads/Drugs/DrugSafety/DrugSafetyNewsletter/UCM148014.pdf v Isoniazid: May decrease the metabolism of Carbamazepine [http://www.merckmanuals.com/professional/lexicomp/carbamazepine.html] vi Carbamazepine may enhance the adverse/toxic effect of Lithium [http://www.merckmanuals.com/professional/lexicomp/carbamazepine.html] vii http://www.merckmanuals.com/media/professional/pdf/Table_214-5.pdf?qt=valproate&alt=sh viiiValproic acid

69
ix http://www.drugbank.ca/drugs/DB00313 xi Goodnick PJ, Fieve RR, Meltzer HL, Dunner DL. Lithium elimination half-life and duration of therapy. Clin Pharmacol Ther. 1981 Jan;29(1):47-50. xii Carbamazepine may enhance the adverse/toxic effect of lithium [http://www.merckmanuals.com/professional/lexicomp/carbamazepine.html] xivOral: Treatment of hypertension; angina pectoris (vasospastic, chronic stable, unstable); supraventricular tachyarrhythmia (PSVT, atrial fibrillation/flutter [rate control]) I.V.: Supraventricular tachyarrhythmia (PSVT, atrial fibrillation/flutter [rate control]) http://www.merckmanuals.com/professional/lexicomp/Verapamil.html xv Zhang, L., et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A 104, 19023-19028 (2007). xvi Mean Cmax of the R isomer was 77.8 ng/ml and 16.8 ng/ml for the S isomer; AUC (0-24h) of the R isomer was 1037 ng•h/ml and 195 ng•h/ml for the S isomer. http://www.rxlist.com/verelan_pm-drug.htm xviihttp://www.drugbank.ca/drugs/DB00661 xviiihttp://www.drugbank.ca/drugs/DB00661 xixHalf life: Single dose: 3-7 hours, Multiple doses: 4.5-12 hours Extended release ~11 hours, drug release delayed ~ 4-5 hours Sustained release: 5.21 hours 7-9 hours http://www.merckmanuals.com/professional/lexicomp/Verapamil.html xx(Williams et al - 2008)

70
xxiRifamycin Derivatives: May increase the metabolism of Calcium Channel Blockers. This primarily affects oral forms of calcium channel blockers. Management: The labeling for some U.S. and Canadian calcium channel blockers contraindicate use with rifampin however recommendations vary. Consult appropriate labeling. Risk D: Consider therapy modification http://www.merckmanuals.com/professional/lexicomp/Verapamil.html xxiiAmiodarone Labeled Indications: Management of life-threatening recurrent ventricular fibrillation (VF) or hemodynamically-unstable ventricular tachycardia (VT) refractory to other antiarrhythmic agents or in patients intolerant of other agents used for these conditions… xxiv Steady-state amiodarone concentrations of 1 to 2.5 mg/L have been associated with antiarrhythmic effects and acceptable toxicity following chronic oral amiodarone therapy. http://www.rxlist.com/amiodarone-hcl-injection-drug.htm xxv Therapeutic levels: 0.5-2.5 mg/L (SI: 1-4 μmol/L) http://www.merckmanuals.com/professional/lexicomp/Amiodarone.html#N2B1DC xxvihttp://www.drugbank.ca/drugs/DB01118 xxviihttp://www.merckmanuals.com/professional/lexicomp/Amiodarone.html#N2B085 xxviiihttp://www.drugbank.ca/drugs/DB01118 xxixRifamycin Derivatives: May increase the metabolism of Amiodarone. Risk C: Monitor therapy http://www.merckmanuals.com/professional/lexicomp/Amiodarone.html xxx “Following chronic oral administration of the drug in humans, amiodarone and N-desethylamiodarone are distributed extensively into many body tissues and fluids, including adipose tissue, liver, lung, spleen, skeletal muscle, bone marrow, adrenal glands, kidneys, pancreas, testes, semen, saliva, lymph nodes, myocardium, thyroid gland, skin, and brain.” http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?sid=24277818#x50 xxxi Potentially fatal pulmonary toxicity is the most severe adverse effect associated with oral amiodarone therapy. More information is available: http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?sid=24277818#x94

71
xxxii Essentially irreversibly inhibits mTORC1 activity according to: Baek, K.H., Park, J. & Shin, I. Autophagy-regulating small molecules and their therapeutic applications. Chem Soc Rev (2012). xxxiii Langley MS, Sorkin EM. Nimodipine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in cerebrovascular disease. Drugs. 1989 May;37(5):669-99. Review. xxxiv Blardi P, Urso R, De Lalla A, Volpi L, Perri TD, Auteri A. Nimodipine: drug pharmacokinetics and plasma adenosine levels in patients affected by cerebral ischemia. ClinPharmacolTher. 2002 Nov;72(5):556-61. PubMed PMID: 12426519. xxxvhttp://www.drugbank.ca/drugs/DB00393 xxxvihttp://www.rxlist.com/nimotop-drug.htm xxxviiRifampin: From experience with the calcium antagonist nifedipine it is to be expected that rifampin accelerates the metabolism of nimodipine capsules due to enzyme induction. Thus, efficacy of nimodipine capsules could be reduced when concomitantly administered with rifampin. http://www.rxmed.com/b.main/b2.pharmaceutical/b2.1.monographs/CPS-%20Monographs/CPS-%20%28General%20Monographs-%20N%29/NIMOTOP.html xxxviii Antiepileptic Drugs: A pharmacokinetic study in epileptic patients receiving long-term treatment has shown that concurrent administration of oral nimodipine and antiepileptic drugs (phenobarbital, phenytoin and/or carbamazepine) reduces the bioavailability of nimodipine by about 80%. In those patients receiving sodiumvalproate and oral nimodipine, the bioavailability of the nimodipine increased by about 50%. Therefore, the concomitant use of oral nimodipine and these antiepileptic drugs requires close monitoring and appropriate adjustment of the dosage of nimodipine. http://www.rxmed.com/b.main/b2.pharmaceutical/b2.1.monographs/CPS-%20Monographs/CPS-%20%28General%20Monographs-%20N%29/NIMOTOP.html xxxix A calcium channel blocker with marked vasodilator action. It is an effective antihypertensive agent and differs from other calcium channel blockers in that it does not reduce glomerular filtration rate and is mildly natriuretic, rather than sodium retentive. http://www.drugbank.ca/drugs/DB01054

72
xlNitrendipine, a dihydropyridine calcium-channel blocker, is used alone or with an angiotensin-converting enzyme inhibitor, to treat hypertension, chronic stable angina pectoris, and Prinzmetal's variant angina. Nitrendipine is similar to other peripheral vasodilators. Nitrendipine inhibits the influx of extra cellular calcium across the myocardial and vascular smooth muscle cell membranes possibly by deforming the channel, inhibiting ion-control gating mechanisms, and/or interfering with the release of calcium from the sarcoplasmic reticulum. The decrease in intracellular calcium inhibits the contractile processes of the myocardial smooth muscle cells, causing dilation of the coronary and systemic arteries, increased oxygen delivery to the myocardial tissue, decreased total peripheral resistance, decreased systemic blood pressure, and decreased afterload. http://www.drugbank.ca/drugs/DB01054 xli Chen, X., Zhong, D., Yang, H., Luan, Y. and Xu, H. (2001), Quantitative determination of nitrendipine and its metabolite dehydronitrendipine in human plasma using liquid chromatography–tandem mass spectrometry. Biomedical Chromatography, 15: 518–524. doi: 10.1002/bmc.104 xliihttp://chem.sis.nlm.nih.gov/chemidplus/jsp/common/Toxicity.jsp xliiihttp://www.mims.com/Philippines/drug/info/nitrendipine/nitrendipine?type=full&mtype=generic xliv 100mM trehalose was about equal to 0.2µM rapamycin for autophagy induction (Sakar et al 2007) xlv In mice, oral LD50 was > 5,000 (mg/kg bw) Intravenous: > 1,000 (mg/kg bw) Atkinson & Thomas (1994) xlvi “In general, the fate of ingested or parenterally administered trehalose corresponds to that of glucose since trehalose is rapidly hydrolysed to glucose by the enzyme trehalase.” http://www.inchem.org/documents/jecfa/jecmono/v46je05.htm However, trehalase is only found in the kidney and small intestine according to T.J.Oesterreicher et al. I Gene 270(2001)211-220 xlvii(see Ashrafian et al 2007) xlviiihttp://www.drugbank.ca/drugs/DB01074 xlixhttp://www.drugbank.ca/drugs/DB01074 l Seldom used in US – common in Australia and New Zealand. (Ashrafian et al 2007) li Serious adverse effects are generally only seen after 3 months or more of treatment. (Ashrafian et al 2007)

73
liihttp://www.rxlist.com/jenloga-drug.htm liiiHogan MJ, Wallin JD, Chu LC. Plasma clonidine concentration and pharmacologic effect. ClinPharmacolTher. 1981 Dec;30(6):729-34. PubMed PMID: 7307423. livhttp://www.drugbank.ca/drugs/DB00575 lv (Rose et al - 2010) lviReid JL. Rilmenidine: a clinical overview. Am J Hypertens. 2000 Jun;13(6 Pt2):106S-111S. Review. PubMed PMID: 10921529. Also describes good long term safety/acceptability and fewer side effects relative to clonidine. lviii The American Journal of Cardiology Volume 61, Issue 7, 24 February 1988, Pages D47-D53 A Symposium: Rilmenidine-A Novel Alpha2 Agonist Antihypertensive Agent lixvan der Post JP, de Visser SJ, Schoemaker RC, Cohen AF, van Gerven JM. Pharmacokinetic/pharmacodynamic assessment of tolerance to central nervous system effects of a 3 mg sustained release tablet of rilmenidine in hypertensive patients. J Psychopharmacol. 2004 Jun;18(2):221-7. lx (Rose et al - 2010) lxihttp://www.drugbank.ca/drugs/DB00350 lxiiPettinger WA. Minoxidil and the treatment of severe hypertension. N Engl J Med. 1980 Oct 16;303(16):922-6. Review. PubMed PMID: 6997744. lxiii Shown to help clear protein aggregates (as in Huntington’s disease) See (Williams et al 2008) lxiv Clonidine in small doses (0.2 to 0.8 mg daily) also helps to prevent the compensatory reflex effects produced by minoxidil, since clonidine suppresses sympathetic outflow from the brain. Pettinger WA. Minoxidil and the treatment of severe hypertension. N Engl J Med. 1980 Oct 16;303(16):922-6. Review.





























