Embed Size (px)
Citation preview

hnRNP U protein is required for normal pre-mRNAsplicing and postnatal heart development and functionJunqiang Yea, Nadine Beetzb,c, Sean O’Keeffea, Juan Carlos Tapiaa, Lindsey Macphersona, Weisheng V. Chena,Rhonda Bassel-Dubyb, Eric N. Olsonb, and Tom Maniatisa,1
aDepartment of Biochemistry and Molecular Biophysics, Columbia University College of Physicians and Surgeons, New York, NY 10032; bDepartment ofMolecular Biology, University of Texas Southwestern Medical Center, Dallas, TX 75390; and cInstitute of Experimental and Clinical Pharmacology andToxicology, University of Freiburg, 79104 Freiburg, Germany
Contributed by Tom Maniatis, April 30, 2015 (sent for review March 2, 2015; reviewed by Brenton R. Graveley)
We report that mice lacking the heterogeneous nuclear ribonucleo-protein U (hnRNP U) in the heart develop lethal dilated cardiomy-opathy and display numerous defects in cardiac pre-mRNA splicing.Mutant hearts have disorganized cardiomyocytes, impaired con-tractility, and abnormal excitation–contraction coupling activities.RNA-seq analyses of Hnrnpu mutant hearts revealed extensive de-fects in alternative splicing of pre-mRNAs encoding proteins knownto be critical for normal heart development and function, includingTitin and calcium/calmodulin-dependent protein kinase II delta(Camk2d). Loss of hnRNP U expression in cardiomyocytes also leadsto aberrant splicing of the pre-mRNA encoding the excitation–con-traction coupling component Junctin. We found that the proteinproduct of an alternatively spliced Junctin isoform is N-glycosylatedat a specific asparagine site that is required for interactions withspecific protein partners. Our findings provide conclusive evidencefor the essential role of hnRNP U in heart development and functionand in the regulation of alternative splicing.
heart development | alternative splicing | RNA-seq | dilatedcardiomyopathy | N-glycosylation
The expression of more than 95% of human genes is affected byalternative pre-mRNA splicing (AS) (1, 2). Differentially spliced
isoforms play distinct roles in a temporally and spatially specificmanner (3), and mutations that lead to aberrant splicing are the causeof many human genetic diseases (4). RNA-binding proteins (RBPs)play a central role in the regulation of alternative splicing duringdevelopment and disease. They function primarily by positively ornegatively regulating splice-site recognition by the spliceosome (1).Many RBPs are expressed in specific tissues, and AS is regulated bythe combinatorial activities of these factors on specific pre-mRNAsthrough their interactions with distinct regulatory sequences in pre-mRNA that function as splicing enhancers or silencers (5).The developing heart is one of the best studied systems where
splicing changes occur during normal development, and mutationsaffecting specific splicing outcomes contribute to cardiomyopathy(6, 7). Although these mutations can either disrupt splicing ele-ments or affect the expression of specific splicing factors, the lattermechanism is clearly responsible for the distinct splicing profilesat different developmental stages. For example, the dynamics ofalternative splicing during postnatal heart development correlatewith expression changes of many RBPs, including CUG-BP, Elav-like family member 1 (CELF1), Muscleblind-like 1 (MBNL1), andFOX proteins (8). Detailed biochemical studies have elucidatedthe mechanisms by which these splicing factors regulate splicing ina position- and context-dependent manner (9, 10). The function ofother RBPs during heart development has also been studied. Forexample, two of the muscle-specific splicing factors, RBM20 andRBM24, play distinct roles in splicing regulation. RBM20 mainlyacts as a splicing repressor, as its absence leads to multiple exoninclusion events in the heart. For example, the Titin gene is one ofthe major targets of RBM20 (11, 12). On the other hand, loss ofRBM24 expression gives rise to many exon skipping events (13),implicating RBM24 as a splicing activator. Strikingly, there is little
overlap between RBM20 and RBM24 splicing targets, suggestingthat RBM20 and RBM24 are involved in regulating splicing ofdistinct groups of pre-mRNAs and there is little cross-talk be-tween these two splicing factors.Distinct splicing activities have also been ascribed to general
splicing factors (1). There are two major types of ubiquitouslyexpressed RBPs: the heterogeneous nuclear ribonucleoproteins(hnRNPs) and serine/arginine (SR)-rich proteins. hnRNPs and SRproteins are generally believed to play opposite roles in splicing:SR proteins promote the inclusion of exons during splicing, whereashnRNP proteins repress inclusion (1). The function of certain SRproteins has been studied in the mouse heart through the con-ditional knockout approach. Srsf1 deletion in the heart leads tolethal dilated cardiomyopathy (DCM) (death occurs 6–8 wk afterbirth) (14). SRSF1 is required for the cardiac-specific splicing ofCypher (also called Ldb3) pre-mRNA, and the regulation of al-ternative splicing of calcium/calmodulin-dependent protein ki-nase II delta (Camk2d) and cardiac Troponin T (cTnT) duringheart development. In particular, the persistent splicing of aneuronal isoform of Camk2d and its ability to enhance excitationand contraction coupling (ECC) activity in Srsf1 mutant car-diomyocytes have been proposed as a possible cause of thephenotype in mutant mice (14). Ablation of another SR protein,SRSF10 (SRp38), from the mouse also leads to heart defects(15). SRSF10 has been shown to regulate the splicing of Triadin,an important component of ECC machinery (15). Interestingly,conditional deletion of Srsf2 from the heart leads to DCM withlittle splicing misregulation but instead affects the expression of
Significance
We studied the physiological function of the heterogeneous nu-clear ribonucleoprotein U (hnRNP U) by generating a conditionalknockout mouse in which the Hnrnpu gene is deleted in the heart.We found that hnRNP U is required for normal pre-mRNA splicingand postnatal heart development and function. Mutant mice de-velop severe dilated cardiomyopathy and die 2 wk after birth.Phenotypic characterization of mutant hearts coupled with RNA-seq data analyses revealed that mutant hearts display multiplecardiac defects as a result of misregulated gene expression andabnormal pre-mRNA splicing. We also identified the sarcoplasmicreticulummembrane protein Junctin as a splicing target of hnRNPU and provide an interesting example of alternative splicing incontrolling the modification and function of proteins.
Author contributions: J.Y., E.N.O., and T.M. designed research; J.Y., N.B., J.C.T., L.M., andW.V.C. performed research; J.Y., N.B., S.O., L.M., R.B.-D., E.N.O., and T.M. analyzed data;and J.Y. and T.M. wrote the paper.
Reviewers included: B.R.G., University of Connecticut Health Center.
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Gene Ex-pression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE68178).1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1508461112/-/DCSupplemental.
E3020–E3029 | PNAS | Published online May 26, 2015 www.pnas.org/cgi/doi/10.1073/pnas.1508461112

the calcium channel Ryr2 (16). It is striking that these SR proteinsaffect ECC activity in the heart by directly regulating the expres-sion/splicing of distinct players in this machinery. Because thesestudies were conducted before the advent of next-generation RNAsequencing, only a few splicing targets specifically regulated bythese SR proteins were identified. A more comprehensive studyof the effects of deleting the genes encoding these proteins fromthe heart on the splicing program has not been reported.In contrast to SR proteins, specific functions of hnRNP pro-
teins in cardiac pre-mRNA splicing have not been determined.In this report, we selectively inactivated the expression of one ofthe most abundant hnRNP proteins—hnRNP U—in the heart. Wefound that Hnrnpu mutant mice develop a lethal DCM phenotype,with death occurring around 2 wk after birth. There are multiplecardiac defects in mutant hearts accompanied by many splicingalterations. Some of these splicing targets are commonly regulatedby hnRNP U and other SR and RBM proteins. We also identifiedmany hnRNP U-specific splicing targets in the heart, including anECC component Junctin. The protein product of the alternativelyspliced Junctin isoform is N-glycosylated at a specific asparaginesite in Hnrnpu mutant cells and could contribute to abnormalcardiac function. Our study also enables comparisons of the rolesof different splicing factors in heart development and function.
ResultsGeneration of an Hnrnpu Conditional Knockout Mouse Line. hnRNPU is the highest molecular weight (MW) protein in the hnRNPcomplex and is one of the most abundant and ubiquitously expressedhnRNP proteins (17, 18). hnRNP U, which binds to both RNA andDNA, is thought to play a central role in transcription, RNA pro-cessing, and nuclear organization (17, 19, 20). Recently, hnRNPU has been shown to bind specifically to long noncoding RNAs(lncRNAs) and play a role in chromosome organization. For ex-ample, hnRNPU is required for normal X chromosome inactivation(21) and for the maintenance of specific nuclear interchromosomalconformation mediated by lncRNA Firre (22). hnRNP U has alsobeen shown to play an important role in splicing regulation in cul-tured cells (23, 24). However, different changes in splicing wereobserved in two studies in which hnRNP U was knocked down incells in culture. In one study, knocking down hnRNP U expressionwas reported to result in comparable levels of exon inclusion andskipping events (23), whereas in another study hnRNP U wasreported to preferentially promote exon inclusion (24).Here we report a study of hnRNP U function in mice. We
generated a conditional deletion allele of the mouse Hnrnpugene by homologous recombination in mouse ES cells. Two loxPsites were engineered to flank a region of Hnrnpu genomic DNAencoding exons 4–14 (Fig. 1A). The insertion of these sites doesnot affect the normal processing of the Hnrnpu gene, as micehomozygous for the conditional allele are phenotypically in-distinguishable from wild-type controls. Tissue-specific Cre-recom-binase (Cre)-mediated deletion removes more than 65% of thehnRNP U coding sequence, which includes the conserved SPRY(involved in protein–protein interactions) and the RNA-bindingRGG domains at the C terminus (25). The remaining N-terminaltranscript after the deletion can give rise to a truncated hnRNP Uprotein devoid of RNA-binding activity. Breeding the Hnrnpuconditional line with a number of Cre lines demonstrated thathnRNP U expression can be efficiently reduced in targeted tissues(Fig. 1 B and C). We also generated a constitutive Hnrnpu mutantallele by crossing the conditional knockout mice with a germ-line–expressing Sox2-Cre line. Consistent with a report that hnRNP Uis essential for mouse embryonic development (26), we failed torecover mice homozygous for the Hnrnpu mutant allele.
Deletion of the Hnrnpu Gene from the Heart Leads to a Sudden DeathPhenotype.TheHnrnpu gene is expressed during all developmentalstages of the mouse heart (Fig. S1). However, the level of hnRNP
U expression gradually decreases from the embryonic and earlypostnatal stage to adulthood (Fig. S1). To study the role of hnRNPU in heart development and function, we crossed the conditionalHnrnpu knockout line with a cardiomyocyte-expressing Cre line,muscle creatine kinase-Cre (Ckmm-Cre), which is also active inskeletal muscle (Ckmm-Cre expression starts at E17.5 and con-tinues to adulthood) (27). The conditional Hnrnpu knockout micewere born at the expected Mendelian ratio with no apparent ab-normalities. Western blot analysis showed that full-length hnRNPU protein is expressed at a significantly reduced level in mutanthearts (Fig. 1C; greater than 80% reduction at P10 and P12). Weused Hnrnpu f/f, Ckmm-Cre+ mice as mutants (Ckmm-KO) andHnrnpu f/f, Ckmm-Cre–, or mice containing wild-type Hnrnpuallele as controls in this study. Residual levels of hnRNP U aremost likely due to expression in noncardiomyocytes in the heart.Deletion of Hnrnpu from the heart is confirmed by immunofluo-rescence staining of heart sections with antibodies that recognizeC-terminal hnRNP U (Fig. S2B). However, using antibodies thatspecifically recognize an N-terminal sequence of hnRNP U, wedetected a lower MW protein band exclusively in mutant samplesby Western blot analysis (Fig. S2C). Immunofluorescence stainingwith the same antibody also revealed the expression of the proteinin both wild-type and mutant cardiomyocytes (Fig. S2B). Thus, weconfirmed the existence of a truncated hnRNP U protein in ourHnrnpu mutant cells. The truncated protein of hnRNP U lackingSPRY and RGG domains can still interact with chromatin/DNA(through the N-terminal SAP domain) but not RNA (28). Thismutant mouse line can thus be used to study the specific effects ofeliminating hnRNP U–RNA interactions in vivo.Although mutant animals appear morphologically indistinguish-
able from control littermates, they all die abruptly from heartfailure around 14 d after birth. To examine the pathology in detail,hearts were dissected from surviving animals of different ages andsubjected to hematoxylin and eosin (H&E) staining. We observedincreased dilation of the ventricle chambers of the mutant heartsover time (dilation of the left ventricle chamber can be observed asearly as P7). One example of H&E-stained sections of P14 hearts ispresented in Fig. 2A. This staining clearly shows significantlyenlarged ventricle chambers accompanied with thinning of theventricle wall and septum. These are morphological features ob-served in mouse models with heart failure and characteristic ofhuman patients with DCM (29). Thus, deletion of hnRNP U inthe mouse heart leads to a lethal DCM phenotype, similar to thatobserved with deletion of the Srsf1 gene in heart cells. However,
Exon 1 2 3 4 5 6 7 8 910 111213 14
1 2 3 4 5 6 7 8 91011 13 14loxP loxPloxPNeo
Wild type allele
Targeting construct
Exon 1 2 3 4 5 6 7 8 910 111213 14loxP loxPConditional allele
f/f +/+ f/f f/f f/+
Conditional Wild type
A
B C
hnRNP U
PARP1
1kb
P10 P12
12SAP SPRY RGG
SAP SPRY RGG
Contro
lCkm
m-KO
Contro
lCkm
m-KO
Fig. 1. Generation of anHnrnpu conditional deletion allele. (A) Diagram of thegene targeting strategy. A triple loxP targeting construct was engineered forhomologous recombination in ES cells. Correctly targeted ES cells were trans-fected with a Cre-recombinase–expressing plasmid to selectively remove the Neocassette. Domains of hnRNP U encoded by different exons are also diagrammed.(B) An example of the characterization of the genotype of the Hnrnpu targetedallele. f, conditional allele; +, wild-type allele. (C) Conditional deletion of theHnrnpu gene from the heart. Shown are Western blots for hnRNP U and PARP1in heart protein lysates from control (Hnrnpu f/f; Ckmm-Cre–) and mutant(Ckmm-KO, Hnrnpu f/f; Ckmm-Cre+) animals.
Ye et al. PNAS | Published online May 26, 2015 | E3021
DEV
ELOPM
ENTA
LBIOLO
GY
PNASPL
US

hnRNP U-deficient mice die significantly earlier than that reportedfor Srsf1mutant mice (14). We did not observe a phenotype in miceheterozygous for the Hnrnpu allele and carrying the Ckmm-Cretransgene; thus, the loss of the protein–RNA interactions, insteadof the “gain-of-function” of the truncated hnRNP U protein, isresponsible for the DCM phenotype.As mentioned above, Ckmm-Cre is also active in skeletal muscle.
To exclude the possibility that Hnrnpu deletion in the skeletalmuscle by Ckmm-Cre also contributes to the observed suddendeath phenotype, we crossed the conditionalHnrnpu knockout micewith a cardiomyocyte-specific Cre line: cardiac myosin heavy chain-αCre (Myh6-Cre; its expression starts from E11.5 and continuesthroughout adulthood) (30). We observed a similar DCM phe-notype with mutant animals generated by either Myh6-Cre orCkmm-Cre lines. Strikingly, all of the mutant Hnrnpu micegenerated by Myh6-Cre die precisely 10 d after birth. Thus, thespecific loss of hnRNP U expression in cardiomyocytes is re-sponsible for the lethal DCM phenotype. The 4-d difference ofthe mutant animal survival between the two Cre lines is likely dueto different timing/efficiency/distribution of Cre-recombinase ex-pression in these lines (31). Because mutant Hnrnpu mice gener-ated by Ckmm-Cre can survive slightly longer, we exclusively usedthis deletion for subsequent studies.
Cardiomyocyte Disarray in hnRNP U-Deficient Hearts.Morphologicalalterations in Hnrnpu mutant hearts can be identified by H&Estaining of heart sections. In control hearts, cardiomyocytes aretightly packed with scattered interstitial fibroblasts in between(Fig. 2A, Top panels). This organization is required for efficientcommunication and synchronized contractility of cardiomyocytes.
By contrast, the distribution of cardiomyocytes in Hnrnpu mutantmice is markedly different. Mutant cardiomyocytes appear slightlyhypertrophic and are surrounded by more white space comparedwith control cells (Fig. 2A, Bottom). Trichrome staining revealedan increased blue signal between cardiomyocytes in mutant hearts,suggesting increased expression of the extracellular matrix inmutant hearts (Fig. 2A). The overall density of cardiomyocytes isreduced in mutant hearts compared with wild type, a likely resultof heart remodeling. Staining the heart sections with wheat germagglutinin (WGA), which highlights cell membranes, reveals apattern similar to that of the H&E staining (Fig. 2A, Far Rightpanels). This pattern is also observed in younger Hnrnpu mutantmice (P10) and becomes more obvious over time (Fig. S3).To determine whether deletion of hnRNPU from cardiomyocytes
causes cell death and contributes to heart remodeling, we stainedheart sections with antibodies against cleaved Caspase 3. Wedetected a slight increase in apoptosis in the mutant hearts (Fig.S4 A and B). This low level of apoptosis was also reflected inWestern blot analysis, as comparable levels of PARP1 proteinwere observed in mutant and control hearts (Fig. 1C). These ob-servations indicate that hnRNP U is not required for the survivalof cardiomyocytes in the early postnatal stage of development. Wenote that other splicing factors, including SRSF1 and SRSF2, arealso not essential for cardiomyocyte survival, although they arerequired for embryonic development of the whole animal (14, 16).
Impaired Sarcomere Relaxation in Hnrnpu Mutant Heart. We nextcharacterized the Hnrnpu mutant cardiomyocytes in more detail,by examining the structure of the sarcomere, the basic contractingunit in muscle cells. We carried out staining of heart sections withphalloidin and antibodies against Desmin, which reveal the actinfibers in the sarcomere and the sarcomere Z line, respectively. Incontrol cardiomyocytes, phalloidin staining showed regular-spacedsarcomeres, with adjacent actin fibers separated by clear gaps (Fig.2B). In contrast, although the pattern of sarcomere spacing wasdetected by phalloidin staining in mutant cells, the transition be-tween sarcomeres is not as clear as in control cells. There areconsiderably higher signals in the gap between neighboringsarcomeres. In addition, the length of individual sarcomeres inHnrnpu mutant cardiomyocytes appears slightly shorter; this isbetter revealed by Desmin staining for the sarcomere Z line.There are clearly more sarcomere units in a fixed length in mu-tants compared with control cells (Fig. 2B). These observationsindicate that deletion of Hnrnpu in the heart adversely affectsactin dynamics and sarcomere activities.We next examined sarcomere structure by electron micros-
copy. Heart samples were prepared under conditions of musclerelaxation, which expose the maximal length of the sarcomere.Structural differences in the sarcomere were not observed betweencontrol and mutant samples. However, the lengths of sarcomeresand I bands were considerably reduced in mutant cardiomyocytes.As shown in Fig. 2C of the P11 hearts, the average length of sar-comeres (Z line to Z line) is about 2.24 μm in control samples and1.68 μm in the Hnrnpu mutants. It thus appears that the ability ofsarcomeres to fully relax is impaired in mutant cardiomyocytes. Wealso observed slightly abnormal deposition of mitochondria alongsarcomeres in mutant cells, as some regions of mutant sarcomereslacked mitochondria coverage compared with control sarcomeres(Fig. 2C). A decrease in mitochondria will also compromise theability of cardiomyocytes to meet the increasing hemodynamicdemand of postnatal heart development. Taken together thesedata indicate that loss of hnRNP U expression in cardiomyocytesresults in impaired sarcomere contractility and cardiac function.These defects in conjunction with the abnormal cardiomyocyteorganization observed inHnrnpumutant hearts (Fig. 2A) are likelyto contribute significantly to heart failure.
LV
RV
LVRV
P14
P14
A
EMBPhalloidin Desmin
H&E Trichrome WGA & DAPI
C
Con
trol
Ckm
m-K
OC
ontro
lC
kmm
-KO
Con
trol
Ckm
m-K
O
Fig. 2. Aberrant cardiomyocyte organization and contractility in Hnrnpumutant hearts. (A) Hnrnpu mutant hearts are dilated and display disorga-nized cardiomyocytes compared with controls. Shown is the H&E staining ofP14 heart sections of both control and mutant (Ckmm-KO) hearts. A low-mag-nification view of the heart and high-magnification views of cardiomyocyte ar-rangements are shown. Trichrome staining of P14 heart sections and WGA &DAPI staining for P11 heart sections are also shown. (Black scale bar, 40 μm;white scale bar in Far Right panels, 10 μm.) (B) Abnormal actin dynamics andshortened sarcomere in Hnrnpumutant hearts. Heart sections (P11) were stainedwith phalloidin and antibodies against Desmin to reveal actin fiber and sarco-mere Z line, respectively. (Scale bar, 10 μm.) (C) Mutant cardiomyocytes havecontractility defects. Control and mutant hearts (P11) were perfused underrelaxing conditions; heart sections were prepared and examined by electronmicroscopy. Two green lines spanning 8.772 μmwere drawn in both control andmutant samples to compare the unit length of sarcomeres.
E3022 | www.pnas.org/cgi/doi/10.1073/pnas.1508461112 Ye et al.
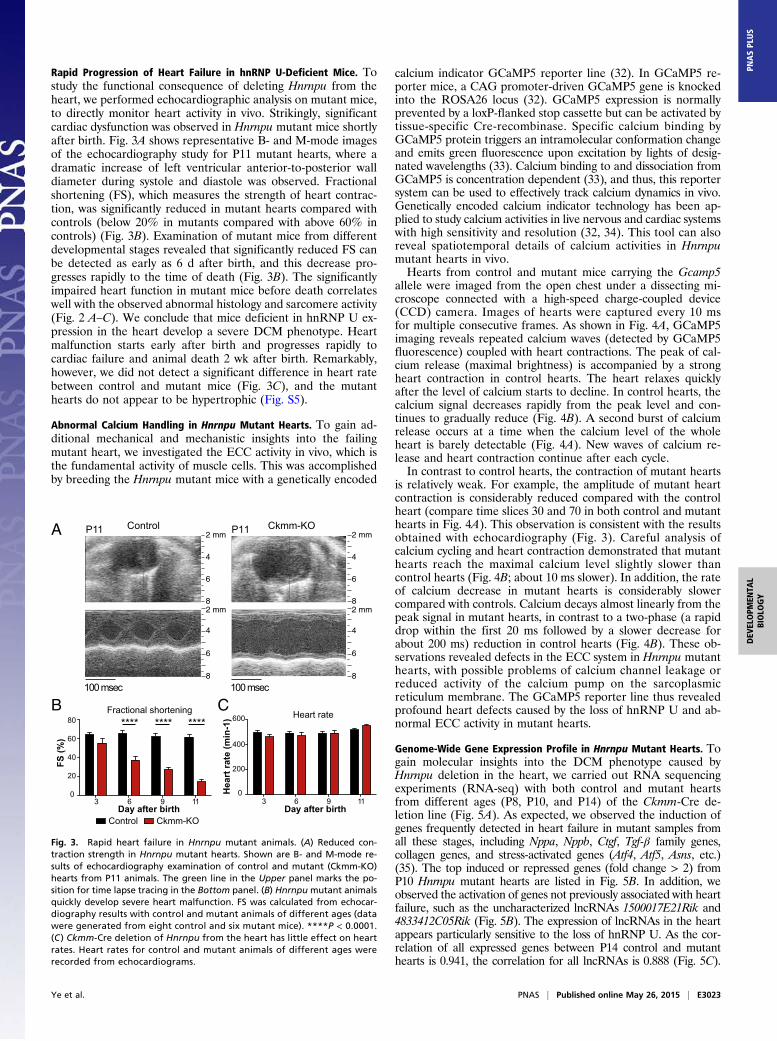
Rapid Progression of Heart Failure in hnRNP U-Deficient Mice. Tostudy the functional consequence of deleting Hnrnpu from theheart, we performed echocardiographic analysis on mutant mice,to directly monitor heart activity in vivo. Strikingly, significantcardiac dysfunction was observed in Hnrnpu mutant mice shortlyafter birth. Fig. 3A shows representative B- and M-mode imagesof the echocardiography study for P11 mutant hearts, where adramatic increase of left ventricular anterior-to-posterior walldiameter during systole and diastole was observed. Fractionalshortening (FS), which measures the strength of heart contrac-tion, was significantly reduced in mutant hearts compared withcontrols (below 20% in mutants compared with above 60% incontrols) (Fig. 3B). Examination of mutant mice from differentdevelopmental stages revealed that significantly reduced FS canbe detected as early as 6 d after birth, and this decrease pro-gresses rapidly to the time of death (Fig. 3B). The significantlyimpaired heart function in mutant mice before death correlateswell with the observed abnormal histology and sarcomere activity(Fig. 2 A–C). We conclude that mice deficient in hnRNP U ex-pression in the heart develop a severe DCM phenotype. Heartmalfunction starts early after birth and progresses rapidly tocardiac failure and animal death 2 wk after birth. Remarkably,however, we did not detect a significant difference in heart ratebetween control and mutant mice (Fig. 3C), and the mutanthearts do not appear to be hypertrophic (Fig. S5).
Abnormal Calcium Handling in Hnrnpu Mutant Hearts. To gain ad-ditional mechanical and mechanistic insights into the failingmutant heart, we investigated the ECC activity in vivo, which isthe fundamental activity of muscle cells. This was accomplishedby breeding the Hnrnpu mutant mice with a genetically encoded
calcium indicator GCaMP5 reporter line (32). In GCaMP5 re-porter mice, a CAG promoter-driven GCaMP5 gene is knockedinto the ROSA26 locus (32). GCaMP5 expression is normallyprevented by a loxP-flanked stop cassette but can be activated bytissue-specific Cre-recombinase. Specific calcium binding byGCaMP5 protein triggers an intramolecular conformation changeand emits green fluorescence upon excitation by lights of desig-nated wavelengths (33). Calcium binding to and dissociation fromGCaMP5 is concentration dependent (33), and thus, this reportersystem can be used to effectively track calcium dynamics in vivo.Genetically encoded calcium indicator technology has been ap-plied to study calcium activities in live nervous and cardiac systemswith high sensitivity and resolution (32, 34). This tool can alsoreveal spatiotemporal details of calcium activities in Hnrnpumutant hearts in vivo.Hearts from control and mutant mice carrying the Gcamp5
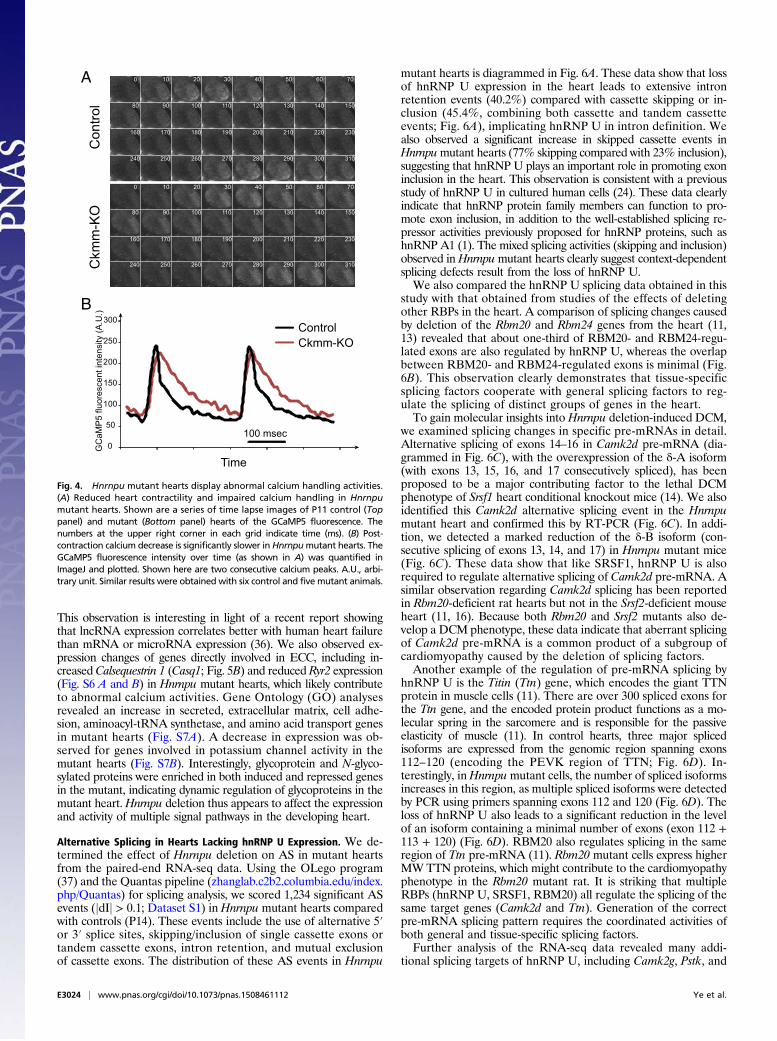
allele were imaged from the open chest under a dissecting mi-croscope connected with a high-speed charge-coupled device(CCD) camera. Images of hearts were captured every 10 msfor multiple consecutive frames. As shown in Fig. 4A, GCaMP5imaging reveals repeated calcium waves (detected by GCaMP5fluorescence) coupled with heart contractions. The peak of cal-cium release (maximal brightness) is accompanied by a strongheart contraction in control hearts. The heart relaxes quicklyafter the level of calcium starts to decline. In control hearts, thecalcium signal decreases rapidly from the peak level and con-tinues to gradually reduce (Fig. 4B). A second burst of calciumrelease occurs at a time when the calcium level of the wholeheart is barely detectable (Fig. 4A). New waves of calcium re-lease and heart contraction continue after each cycle.In contrast to control hearts, the contraction of mutant hearts
is relatively weak. For example, the amplitude of mutant heartcontraction is considerably reduced compared with the controlheart (compare time slices 30 and 70 in both control and mutanthearts in Fig. 4A). This observation is consistent with the resultsobtained with echocardiography (Fig. 3). Careful analysis ofcalcium cycling and heart contraction demonstrated that mutanthearts reach the maximal calcium level slightly slower thancontrol hearts (Fig. 4B; about 10 ms slower). In addition, the rateof calcium decrease in mutant hearts is considerably slowercompared with controls. Calcium decays almost linearly from thepeak signal in mutant hearts, in contrast to a two-phase (a rapiddrop within the first 20 ms followed by a slower decrease forabout 200 ms) reduction in control hearts (Fig. 4B). These ob-servations revealed defects in the ECC system in Hnrnpu mutanthearts, with possible problems of calcium channel leakage orreduced activity of the calcium pump on the sarcoplasmicreticulum membrane. The GCaMP5 reporter line thus revealedprofound heart defects caused by the loss of hnRNP U and ab-normal ECC activity in mutant hearts.
Genome-Wide Gene Expression Profile in Hnrnpu Mutant Hearts. Togain molecular insights into the DCM phenotype caused byHnrnpu deletion in the heart, we carried out RNA sequencingexperiments (RNA-seq) with both control and mutant heartsfrom different ages (P8, P10, and P14) of the Ckmm-Cre de-letion line (Fig. 5A). As expected, we observed the induction ofgenes frequently detected in heart failure in mutant samples fromall these stages, including Nppa, Nppb, Ctgf, Tgf-β family genes,collagen genes, and stress-activated genes (Atf4, Atf5, Asns, etc.)(35). The top induced or repressed genes (fold change > 2) fromP10 Hnrnpu mutant hearts are listed in Fig. 5B. In addition, weobserved the activation of genes not previously associated with heartfailure, such as the uncharacterized lncRNAs 1500017E21Rik and4833412C05Rik (Fig. 5B). The expression of lncRNAs in the heartappears particularly sensitive to the loss of hnRNP U. As the cor-relation of all expressed genes between P14 control and mutanthearts is 0.941, the correlation for all lncRNAs is 0.888 (Fig. 5C).
A
B C
3 6 9 11 3 6 9 11
4
6
2 mm
8
4
6
2 mm
8
100 msec
2 mm
4
6
8
Control Ckmm-KO
2 mm
4
6
8
P11 P11
100 msec
****
Ckmm-KOControlDay after birthDay after birth
FS (%
)
Hea
rt ra
te (m
in-1
)**** ****Fractional shortening Heart rate
80
60
40
20
0
600
400
200
0
Fig. 3. Rapid heart failure in Hnrnpu mutant animals. (A) Reduced con-traction strength in Hnrnpu mutant hearts. Shown are B- and M-mode re-sults of echocardiography examination of control and mutant (Ckmm-KO)hearts from P11 animals. The green line in the Upper panel marks the po-sition for time lapse tracing in the Bottom panel. (B) Hnrnpu mutant animalsquickly develop severe heart malfunction. FS was calculated from echocar-diography results with control and mutant animals of different ages (datawere generated from eight control and six mutant mice). ****P < 0.0001.(C ) Ckmm-Cre deletion of Hnrnpu from the heart has little effect on heartrates. Heart rates for control and mutant animals of different ages wererecorded from echocardiograms.
Ye et al. PNAS | Published online May 26, 2015 | E3023
DEV
ELOPM
ENTA
LBIOLO
GY
PNASPL
US
This observation is interesting in light of a recent report showingthat lncRNA expression correlates better with human heart failurethan mRNA or microRNA expression (36). We also observed ex-pression changes of genes directly involved in ECC, including in-creased Calsequestrin 1 (Casq1; Fig. 5B) and reduced Ryr2 expression(Fig. S6 A and B) in Hnrnpu mutant hearts, which likely contributeto abnormal calcium activities. Gene Ontology (GO) analysesrevealed an increase in secreted, extracellular matrix, cell adhe-sion, aminoacyl-tRNA synthetase, and amino acid transport genesin mutant hearts (Fig. S7A). A decrease in expression was ob-served for genes involved in potassium channel activity in themutant hearts (Fig. S7B). Interestingly, glycoprotein and N-glyco-sylated proteins were enriched in both induced and repressed genesin the mutant, indicating dynamic regulation of glycoproteins in themutant heart. Hnrnpu deletion thus appears to affect the expressionand activity of multiple signal pathways in the developing heart.
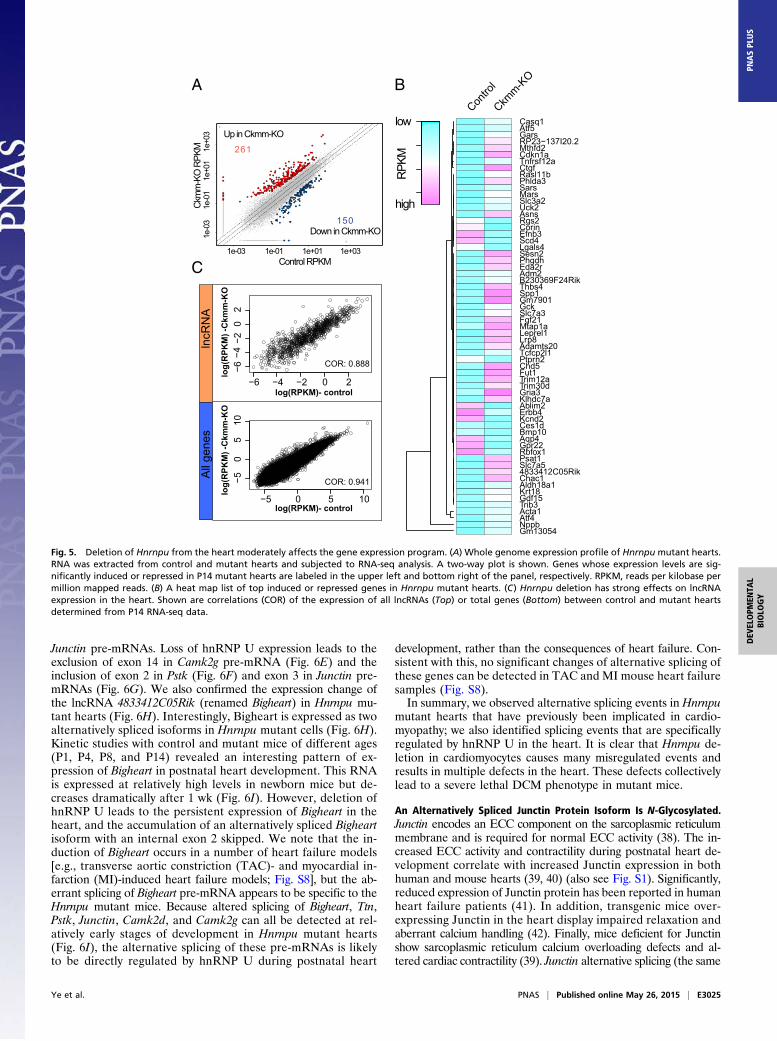
Alternative Splicing in Hearts Lacking hnRNP U Expression. We de-termined the effect of Hnrnpu deletion on AS in mutant heartsfrom the paired-end RNA-seq data. Using the OLego program(37) and the Quantas pipeline (zhanglab.c2b2.columbia.edu/index.php/Quantas) for splicing analysis, we scored 1,234 significant ASevents (jdIj > 0.1; Dataset S1) in Hnrnpu mutant hearts comparedwith controls (P14). These events include the use of alternative 5′or 3′ splice sites, skipping/inclusion of single cassette exons ortandem cassette exons, intron retention, and mutual exclusionof cassette exons. The distribution of these AS events in Hnrnpu
mutant hearts is diagrammed in Fig. 6A. These data show that lossof hnRNP U expression in the heart leads to extensive intronretention events (40.2%) compared with cassette skipping or in-clusion (45.4%, combining both cassette and tandem cassetteevents; Fig. 6A), implicating hnRNP U in intron definition. Wealso observed a significant increase in skipped cassette events inHnrnpumutant hearts (77% skipping compared with 23% inclusion),suggesting that hnRNP U plays an important role in promoting exoninclusion in the heart. This observation is consistent with a previousstudy of hnRNP U in cultured human cells (24). These data clearlyindicate that hnRNP protein family members can function to pro-mote exon inclusion, in addition to the well-established splicing re-pressor activities previously proposed for hnRNP proteins, such ashnRNP A1 (1). The mixed splicing activities (skipping and inclusion)observed inHnrnpumutant hearts clearly suggest context-dependentsplicing defects result from the loss of hnRNP U.We also compared the hnRNP U splicing data obtained in this
study with that obtained from studies of the effects of deletingother RBPs in the heart. A comparison of splicing changes causedby deletion of the Rbm20 and Rbm24 genes from the heart (11,13) revealed that about one-third of RBM20- and RBM24-regu-lated exons are also regulated by hnRNP U, whereas the overlapbetween RBM20- and RBM24-regulated exons is minimal (Fig.6B). This observation clearly demonstrates that tissue-specificsplicing factors cooperate with general splicing factors to reg-ulate the splicing of distinct groups of genes in the heart.To gain molecular insights into Hnrnpu deletion-induced DCM,
we examined splicing changes in specific pre-mRNAs in detail.Alternative splicing of exons 14–16 in Camk2d pre-mRNA (dia-grammed in Fig. 6C), with the overexpression of the δ-A isoform(with exons 13, 15, 16, and 17 consecutively spliced), has beenproposed to be a major contributing factor to the lethal DCMphenotype of Srsf1 heart conditional knockout mice (14). We alsoidentified this Camk2d alternative splicing event in the Hnrnpumutant heart and confirmed this by RT-PCR (Fig. 6C). In addi-tion, we detected a marked reduction of the δ-B isoform (con-secutive splicing of exons 13, 14, and 17) in Hnrnpu mutant mice(Fig. 6C). These data show that like SRSF1, hnRNP U is alsorequired to regulate alternative splicing of Camk2d pre-mRNA. Asimilar observation regarding Camk2d splicing has been reportedin Rbm20-deficient rat hearts but not in the Srsf2-deficient mouseheart (11, 16). Because both Rbm20 and Srsf2 mutants also de-velop a DCM phenotype, these data indicate that aberrant splicingof Camk2d pre-mRNA is a common product of a subgroup ofcardiomyopathy caused by the deletion of splicing factors.Another example of the regulation of pre-mRNA splicing by
hnRNP U is the Titin (Ttn) gene, which encodes the giant TTNprotein in muscle cells (11). There are over 300 spliced exons forthe Ttn gene, and the encoded protein product functions as a mo-lecular spring in the sarcomere and is responsible for the passiveelasticity of muscle (11). In control hearts, three major splicedisoforms are expressed from the genomic region spanning exons112–120 (encoding the PEVK region of TTN; Fig. 6D). In-terestingly, inHnrnpumutant cells, the number of spliced isoformsincreases in this region, as multiple spliced isoforms were detectedby PCR using primers spanning exons 112 and 120 (Fig. 6D). Theloss of hnRNP U also leads to a significant reduction in the levelof an isoform containing a minimal number of exons (exon 112 +113 + 120) (Fig. 6D). RBM20 also regulates splicing in the sameregion of Ttn pre-mRNA (11). Rbm20 mutant cells express higherMW TTN proteins, which might contribute to the cardiomyopathyphenotype in the Rbm20 mutant rat. It is striking that multipleRBPs (hnRNP U, SRSF1, RBM20) all regulate the splicing of thesame target genes (Camk2d and Ttn). Generation of the correctpre-mRNA splicing pattern requires the coordinated activities ofboth general and tissue-specific splicing factors.Further analysis of the RNA-seq data revealed many addi-
tional splicing targets of hnRNP U, including Camk2g, Pstk, and
0 10 20 30 40 50 60 70
80 90 100 110 120 130 140 150
160 170 180 190 200 210 220 230
240 250 260 270 280 290 300 310
0 10 20 30 40 50 60 70
80 90 100 110 120 130 140 150
160 170 180 190 200 210 220 230
240 250 260 270 280 290 300 310
A
B
0
100
200
GC
aMP
5 flu
ores
cent
inte
nsity
(A.U
.)
Time
100 msec
Ckmm-KOControl
300
Con
trol
Ckm
m-K
O
150
250
50
Fig. 4. Hnrnpu mutant hearts display abnormal calcium handling activities.(A) Reduced heart contractility and impaired calcium handling in Hnrnpumutant hearts. Shown are a series of time lapse images of P11 control (Toppanel) and mutant (Bottom panel) hearts of the GCaMP5 fluorescence. Thenumbers at the upper right corner in each grid indicate time (ms). (B) Post-contraction calcium decrease is significantly slower in Hnrnpumutant hearts. TheGCaMP5 fluorescence intensity over time (as shown in A) was quantified inImageJ and plotted. Shown here are two consecutive calcium peaks. A.U., arbi-trary unit. Similar results were obtained with six control and five mutant animals.
E3024 | www.pnas.org/cgi/doi/10.1073/pnas.1508461112 Ye et al.
Junctin pre-mRNAs. Loss of hnRNP U expression leads to theexclusion of exon 14 in Camk2g pre-mRNA (Fig. 6E) and theinclusion of exon 2 in Pstk (Fig. 6F) and exon 3 in Junctin pre-mRNAs (Fig. 6G). We also confirmed the expression change ofthe lncRNA 4833412C05Rik (renamed Bigheart) in Hnrnpu mu-tant hearts (Fig. 6H). Interestingly, Bigheart is expressed as twoalternatively spliced isoforms in Hnrnpu mutant cells (Fig. 6H).Kinetic studies with control and mutant mice of different ages(P1, P4, P8, and P14) revealed an interesting pattern of ex-pression of Bigheart in postnatal heart development. This RNAis expressed at relatively high levels in newborn mice but de-creases dramatically after 1 wk (Fig. 6I). However, deletion ofhnRNP U leads to the persistent expression of Bigheart in theheart, and the accumulation of an alternatively spliced Bigheartisoform with an internal exon 2 skipped. We note that the in-duction of Bigheart occurs in a number of heart failure models[e.g., transverse aortic constriction (TAC)- and myocardial in-farction (MI)-induced heart failure models; Fig. S8], but the ab-errant splicing of Bigheart pre-mRNA appears to be specific to theHnrnpu mutant mice. Because altered splicing of Bigheart, Ttn,Pstk, Junctin, Camk2d, and Camk2g can all be detected at rel-atively early stages of development in Hnrnpu mutant hearts(Fig. 6I), the alternative splicing of these pre-mRNAs is likelyto be directly regulated by hnRNP U during postnatal heart
development, rather than the consequences of heart failure. Con-sistent with this, no significant changes of alternative splicing ofthese genes can be detected in TAC and MI mouse heart failuresamples (Fig. S8).In summary, we observed alternative splicing events in Hnrnpu
mutant hearts that have previously been implicated in cardio-myopathy; we also identified splicing events that are specificallyregulated by hnRNP U in the heart. It is clear that Hnrnpu de-letion in cardiomyocytes causes many misregulated events andresults in multiple defects in the heart. These defects collectivelylead to a severe lethal DCM phenotype in mutant mice.
An Alternatively Spliced Junctin Protein Isoform Is N-Glycosylated.Junctin encodes an ECC component on the sarcoplasmic reticulummembrane and is required for normal ECC activity (38). The in-creased ECC activity and contractility during postnatal heart de-velopment correlate with increased Junctin expression in bothhuman and mouse hearts (39, 40) (also see Fig. S1). Significantly,reduced expression of Junctin protein has been reported in humanheart failure patients (41). In addition, transgenic mice over-expressing Junctin in the heart display impaired relaxation andaberrant calcium handling (42). Finally, mice deficient for Junctinshow sarcoplasmic reticulum calcium overloading defects and al-tered cardiac contractility (39). Junctin alternative splicing (the same
A B
Gm13054NppbAtf4Acta1Trib3Gdf15Krt18Aldh18a1Chac14833412C05RikSlc7a5Psat1Rbfox1Gpr22Aqp4Bmp10Ces1dKcnd2Erbb4Ablim2Klhdc7aGria3Trim30dTrim12aFut1Chd5Ptprn2Tcfcp2l1Adamts20Lrp8Leprel1Mtap1aFgf21Slc7a3GckGm7901Spp1Thbs4B230369F24RikAdm2Eda2rPhgdhSesn2Lgals4Scd4Efnb3CorinRgs2AsnsUck2Slc3a2MarsSarsPhlda3Rasl11bCtgfTnfrsf12aCdkn1aMthfd2RP23−137I20.2GarsAtf5Casq1
Contro
lCkm
m-KO
RPKM
low
high
Up in Ckmm-KO
Down in Ckmm-KO
Control RPKM1e-03 1e-01 1e+01 1e+03
Ckm
m-K
O R
PKM
1e-0
31e
-01
1e+0
11e
+03
−6 −4 −2 0 2
−6−2
02
log(
RPK
M) -
Ckm
m-K
O
log(RPKM)- control
COR: 0.888
−5 0 5 10
−50
510
COR: 0.941
lncR
NA
All
gene
s
261
150lo
g(R
PKM
) -C
kmm
-KO
log(RPKM)- control
−4
C
Fig. 5. Deletion of Hnrnpu from the heart moderately affects the gene expression program. (A) Whole genome expression profile of Hnrnpu mutant hearts.RNA was extracted from control and mutant hearts and subjected to RNA-seq analysis. A two-way plot is shown. Genes whose expression levels are sig-nificantly induced or repressed in P14 mutant hearts are labeled in the upper left and bottom right of the panel, respectively. RPKM, reads per kilobase permillion mapped reads. (B) A heat map list of top induced or repressed genes in Hnrnpu mutant hearts. (C) Hnrnpu deletion has strong effects on lncRNAexpression in the heart. Shown are correlations (COR) of the expression of all lncRNAs (Top) or total genes (Bottom) between control and mutant heartsdetermined from P14 RNA-seq data.
Ye et al. PNAS | Published online May 26, 2015 | E3025
DEV
ELOPM
ENTA
LBIOLO
GY
PNASPL
US

cassette event as we identified here; Figs. 6G and 7A) was reportedin human samples (43), but its function and potential link to heartfailure is not known. We therefore decided to study the alternativesplicing of Junctin in more detail.First, we confirmed that the alternatively spliced Junctin (JAS)
protein isoform is expressed in Hnrnpu mutant hearts. Usingantibodies specific for Junctin protein, we detected a band inHnrnpu mutant hearts in addition to the normal Junctin proteinby Western blotting (Fig. 7B). Surprisingly, the hnRNP U-spe-cific band migrates slower than expected for a protein containingjust 15 additional amino acids (encoded by the 45 bp alterna-tively spliced exon) compared with the normal Junctin (JCT)protein. It therefore seemed likely that the JCT protein encodedby JAS pre-mRNA is posttranslationally modified. The alterna-tively spliced exon 3 is highly conserved across evolution (Fig.7A), and motif analysis by NetNGlyc algorithm (www.cbs.dtu.dk/services/NetNGlyc/) predicted that the asparagine residue atposition 64 (N64, within the full-length JAS protein) is likely to beN-glycosylated, as it is contained within the consensus sequence(N-X-S/T) for this posttranslational modification. To test this pos-sibility, we cloned cDNAs encoding JCT and JAS from Hnrnpumutant hearts and expressed them in 293T cells. We foundthat JAS migrates as two bands in SDS/PAGE (Fig. 7C), with
one band slightly slower than JCT, corresponding to un-modified JAS (contains 15 more amino acids than JCT). Theother slower band migrates to a similar position as the additionalJunctin band detected in Hnrnpu mutant hearts. To determinewhether this JAS protein isoform is indeed N-glycosylatedat N64, we carried out two lines of experiments. First, we in-troduced a number of point mutations into the JAS expressionconstruct. Strikingly, the slower migrating band was abolished bymutating the N64 residue to encode isoleucine (N64I) or glutamine(N64Q), whereas mutating an adjacent leucine 65 to isoleucine(L65I) had no effect (Fig. 7C). These experiments demonstrate thatN64 is required for the modification. In addition, we showed thatPNGase F, an enzyme that specifically removes N-glycosylatedpolysaccharides from modified proteins, specifically removed themodification from JAS (Fig. 7D). Taken together, these data showthat Junctin protein generated by AS is, indeed, N-glycosylated atamino acid N64.To investigate whether JAS interacts with different proteins
and potentially contributes to different physiological propertiesin cardiomyocytes, we made use of the mouse cardiomyocyte cellline HL-1. We generated stable HL-1 cells that overexpressedJunctin, JAS, or JAS N64I by infecting parental cells with lentivi-ruses encoding these proteins. JCT, JAS, and JAS N64I proteins
C 13 14 15 16 17
δA δB δC13 15 16 17 13 14 17 13 17AS:
Exon:
Camk2d
1 1 12
+ - + -
δAδB
δC
3 4
Exon: 112 113 116 118 120
Titin
D E
I
2 3 4
2 34 2 4AS:
Exon:
Junctin
F
AS:
1 2 3 4 5 6
Pstk
Exon:
AS: 1 2 3 1 3
1 2 3
1 3
2 342 4
Camk2g
12 13 14 15 16 17
13 16 13 1614 13 1614 1513 1615AS:
13 16
13 1614
13 1614 15
13 1615
Exon:
G
Camk2d
Camk2g
Junctin
Pstk
Ttn
Bigheart
P1 P4 P8 P14+ - + -+ - + -hnRNP U
hnRNP U
+ - + -hnRNP U+ - + -hnRNP U
1 2 3 4 41 1 1
+ - + -hnRNP U
+ - + -hnRNP U
H 1 2 3
1 2 3 1 3AS:
Exon:
+ - + -hnRNP U
Bigheart 1 2 31 3
A B
388
28
49
31
4401
RBM24 RBM20
hnRNP U
Mutual exclusion
2.4%
Cassette Inc/Skip36.3%
Tandem cassette
9.1%
Intron retention
40.2%
Alternative 5'4.1% Alternative 3'
7.9%
δA
δB
δC
Fig. 6. Hnrnpu deletion affects AS in the heart. (A) Distribution of various alternative splicing events in Hnrnpu mutant hearts. Calculations were conductedwith the total 1,234 scored events from the P14 data. (B) hnRNP U coregulates the splicing of a distinct subgroup of pre-mRNAs with RBM20 and RBM24 in theheart. hnRNP U-regulated cassette RNA splicing events were analyzed together with published RBM20 and RBM24 exon splicing events. The overlaps betweeneach of the datasets are diagramed. (C) hnRNP U regulates alternative splicing of exons 14–16 of the Camk2d pre-mRNA. The exon position, various al-ternative splicing products, and RT-PCR primer positions are diagramed. Plus and minus signs above the gel picture indicate control and mutant samples,respectively. (D) Ttn pre-mRNA splicing is regulated by hnRNP U. (E–H) Examples of additional splicing targets of hnRNP U: Camk2g (E), Pstk (F), Junctin (G),and Bigheart (H). (I) Time course of splicing changes of selected genes in Hnrnpumutant hearts. RT-PCR was conducted with P1, P4, P8, and P14 heart samplesusing primers designed for the detection of specific splicing products. Some lanes in C, F, and G were spliced and rearranged from the same gels to keep aconsistent loading order.
E3026 | www.pnas.org/cgi/doi/10.1073/pnas.1508461112 Ye et al.
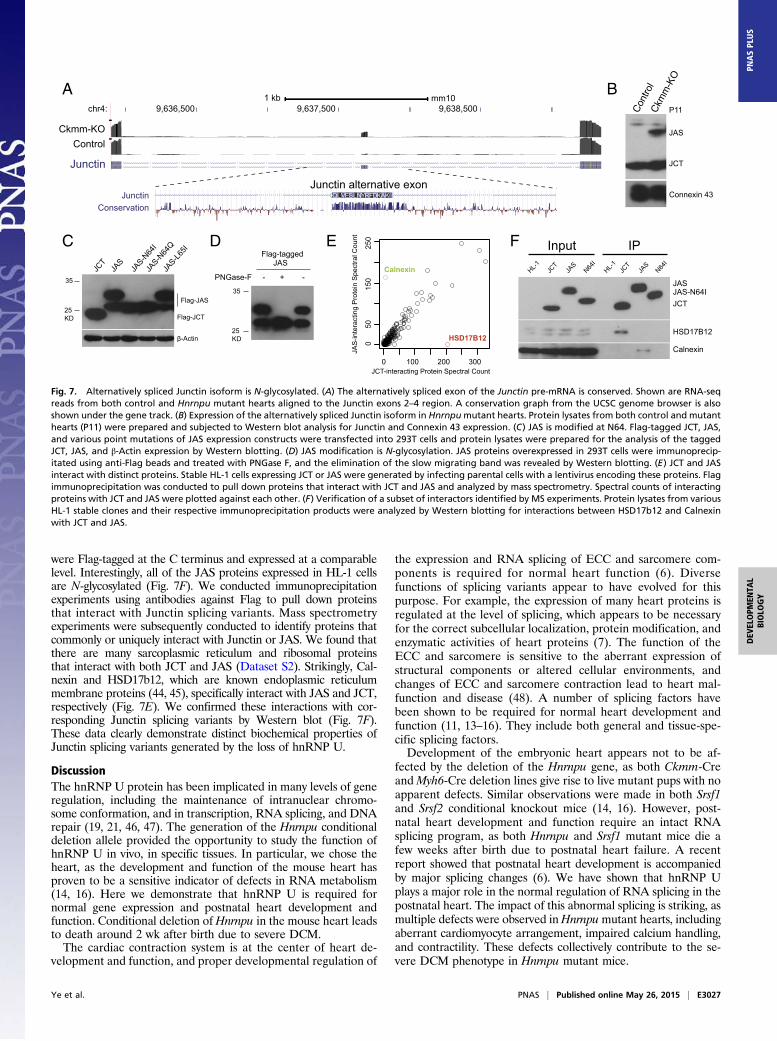
were Flag-tagged at the C terminus and expressed at a comparablelevel. Interestingly, all of the JAS proteins expressed in HL-1 cellsare N-glycosylated (Fig. 7F). We conducted immunoprecipitationexperiments using antibodies against Flag to pull down proteinsthat interact with Junctin splicing variants. Mass spectrometryexperiments were subsequently conducted to identify proteins thatcommonly or uniquely interact with Junctin or JAS. We found thatthere are many sarcoplasmic reticulum and ribosomal proteinsthat interact with both JCT and JAS (Dataset S2). Strikingly, Cal-nexin and HSD17b12, which are known endoplasmic reticulummembrane proteins (44, 45), specifically interact with JAS and JCT,respectively (Fig. 7E). We confirmed these interactions with cor-responding Junctin splicing variants by Western blot (Fig. 7F).These data clearly demonstrate distinct biochemical properties ofJunctin splicing variants generated by the loss of hnRNP U.
DiscussionThe hnRNP U protein has been implicated in many levels of generegulation, including the maintenance of intranuclear chromo-some conformation, and in transcription, RNA splicing, and DNArepair (19, 21, 46, 47). The generation of the Hnrnpu conditionaldeletion allele provided the opportunity to study the function ofhnRNP U in vivo, in specific tissues. In particular, we chose theheart, as the development and function of the mouse heart hasproven to be a sensitive indicator of defects in RNA metabolism(14, 16). Here we demonstrate that hnRNP U is required fornormal gene expression and postnatal heart development andfunction. Conditional deletion ofHnrnpu in the mouse heart leadsto death around 2 wk after birth due to severe DCM.The cardiac contraction system is at the center of heart de-
velopment and function, and proper developmental regulation of
the expression and RNA splicing of ECC and sarcomere com-ponents is required for normal heart function (6). Diversefunctions of splicing variants appear to have evolved for thispurpose. For example, the expression of many heart proteins isregulated at the level of splicing, which appears to be necessaryfor the correct subcellular localization, protein modification, andenzymatic activities of heart proteins (7). The function of theECC and sarcomere is sensitive to the aberrant expression ofstructural components or altered cellular environments, andchanges of ECC and sarcomere contraction lead to heart mal-function and disease (48). A number of splicing factors havebeen shown to be required for normal heart development andfunction (11, 13–16). They include both general and tissue-spe-cific splicing factors.Development of the embryonic heart appears not to be af-
fected by the deletion of the Hnrnpu gene, as both Ckmm-CreandMyh6-Cre deletion lines give rise to live mutant pups with noapparent defects. Similar observations were made in both Srsf1and Srsf2 conditional knockout mice (14, 16). However, post-natal heart development and function require an intact RNAsplicing program, as both Hnrnpu and Srsf1 mutant mice die afew weeks after birth due to postnatal heart failure. A recentreport showed that postnatal heart development is accompaniedby major splicing changes (6). We have shown that hnRNP Uplays a major role in the normal regulation of RNA splicing in thepostnatal heart. The impact of this abnormal splicing is striking, asmultiple defects were observed inHnrnpumutant hearts, includingaberrant cardiomyocyte arrangement, impaired calcium handling,and contractility. These defects collectively contribute to the se-vere DCM phenotype in Hnrnpu mutant mice.
GQLVESLNYRFDKAKAJunctinConservation
Junctin alternative exon
JCT
JAS
JAS-N
64I
JAS-N
64Q
JAS-L6
5I
PNGase-F - + -
Flag-tagged
35
25KD
35
25KD
JAS
A B
C
Flag-JCT
chr4:1 kb mm10
9,636,500 9,637,500 9,638,500
Junctin
Ckmm-KOControl
JCT
JAS
Connexin 43
Cont
rol
Ckm
m-K
O
D
P11
JCT-interacting Protein Spectral Count
JAS
-inte
ract
ing
Pro
tein
Spe
ctra
l Cou
nt
Calnexin
HSD17B12
E
Flag-JAS
0 100 200 3000
5015
025
0
HSD17B12
Calnexin
Input IP
HL-1 JCT
JAS
N64I
HL-1 JCT
JAS
N64I
JASJAS-N64IJCT
F
β-Actin
Fig. 7. Alternatively spliced Junctin isoform is N-glycosylated. (A) The alternatively spliced exon of the Junctin pre-mRNA is conserved. Shown are RNA-seqreads from both control and Hnrnpu mutant hearts aligned to the Junctin exons 2–4 region. A conservation graph from the UCSC genome browser is alsoshown under the gene track. (B) Expression of the alternatively spliced Junctin isoform in Hnrnpumutant hearts. Protein lysates from both control and mutanthearts (P11) were prepared and subjected to Western blot analysis for Junctin and Connexin 43 expression. (C) JAS is modified at N64. Flag-tagged JCT, JAS,and various point mutations of JAS expression constructs were transfected into 293T cells and protein lysates were prepared for the analysis of the taggedJCT, JAS, and β-Actin expression by Western blotting. (D) JAS modification is N-glycosylation. JAS proteins overexpressed in 293T cells were immunoprecip-itated using anti-Flag beads and treated with PNGase F, and the elimination of the slow migrating band was revealed by Western blotting. (E) JCT and JASinteract with distinct proteins. Stable HL-1 cells expressing JCT or JAS were generated by infecting parental cells with a lentivirus encoding these proteins. Flagimmunoprecipitation was conducted to pull down proteins that interact with JCT and JAS and analyzed by mass spectrometry. Spectral counts of interactingproteins with JCT and JAS were plotted against each other. (F) Verification of a subset of interactors identified by MS experiments. Protein lysates from variousHL-1 stable clones and their respective immunoprecipitation products were analyzed by Western blotting for interactions between HSD17b12 and Calnexinwith JCT and JAS.
Ye et al. PNAS | Published online May 26, 2015 | E3027
DEV
ELOPM
ENTA
LBIOLO
GY
PNASPL
US

Coordination of RNA Splicing by hnRNP and Other RBPs. Analyses ofsplicing changes in Hnrnpu mutant hearts showed that splicingtargets are either hnRNP U-specific or shared among selectedsplicing factors. Camk2d is one example of pre-mRNAs that areregulated by multiple factors, including SRSF1, RBM20, andhnRNP U. Ttn alternative splicing is also regulated by bothhnRNP U and RBM20. For many hnRNP U-specific splicingtargets—for example, Pstk and Junctin—it is likely that theirsplicing also requires the concerted activities of other RBPs.Proper splicing requires coordination of the activities of bothgeneral and tissue-specific splicing factors. Remarkably, RBM20and RBM24, both muscle-specific splicing factors, regulate mostlynonoverlapping splicing targets. However, ∼30% of RBM20 andRBM24 splicing targets are also regulated by hnRNP U. In gen-eral, it appears that the activities of tissue-specific and generalsplicing factors function together to regulate the alternative splicingof specific sets of target pre-mRNAs. The splicing identity of aspecific tissue is determined by the combined activities of dis-tinct splicing complexes formed on different groups of pre-mRNAs.It is important to point out that splicing factors can regulate ASdirectly through protein–RNA interactions or indirectly throughaffecting the expression of other RBPs. Examination of the re-lationship between specific RBP–RNA interactions in the pres-ence or absence of other splicing factors coupled with subsequentsplicing outcomes should provide interesting insights into themechanisms by which distinct splicing factors function togetherto regulate splicing.
Alternative Splicing of ECC Components and Protein Glycosylation.Many components of the ECC machinery are subjected to splicingregulation during development or under disease conditions (7).Alternative splicing of Ryr2, Triadin, and Atp2a2 has been reported(7), and protein products of splicing variants affect calcium han-dling in myocytes in general. We show here that hnRNP Uspecifically regulates alternative splicing of Junctin pre-mRNA,as the deletion of Hnrnpu leads to an aberrant splicing productof Junctin. The JAS protein is N-glycosylated at a specific as-paragine site (N64). JAS glycosylation occurs at the luminal side ofthe sarcoplasmic reticulum; thus, glycosylation could potentiallyaffect the interactions between Junctin and other sarcoplasmicreticulum proteins. We showed that N-glycosylated JAS is spe-cifically recognized by Calnexin, and it disrupts the normal in-teractions between JCT and HSD17B12. The interaction betweenglycosylated JAS and Calnexin is not surprising, as Calnexin playsan important role in glycoprotein folding and maturation (49). Itcould also potentially lead to different turnover rates of Junctinsplicing variants and thus affect calcium dynamics in cardiomyocytes.We also identified an interesting interaction partner, HSD17B12,for Junctin in this study. HSD17B12 is highly expressed in neuraltissues as well as in the heart, and it is required for mouse em-bryonic organogenesis (50). HSD17B12 is an ER membraneprotein known to play an important role in steroid and fat metab-olism (44, 51). The interaction between Junctin and HSD17B12
(Fig. 7 E and F) suggests a possible coupling between lipid me-tabolism and calcium handling in cardiomyocytes.It is interesting to note that about half of the Triadin proteins
are also N-glycosylated at a specific asparagine site (N75) in theheart (52). Glycosylation of Triadin appears to increase its sta-bility (53) and possibly other functions. Glycosylation is also knownto occur during the maturation of the sarcoplasmic reticulum cal-cium-binding protein Calsequestrin, and altered Calsequestrin gly-cosylation has been linked to heart failure in canine models of heartdiseases (54). Thus, it appears that glycosylation is an importantregulatory mechanism in cardiomyocyte physiology. Further studiesare required to determine the physiological function of JAS glyco-sylation and its potential contribution to heart physiology. However,the hnRNP U-dependent alternative RNA splicing of Junctin pre-mRNA provides an interesting example of the role of splicingregulation in controlling the modification and function of proteins.
Materials and MethodsMouse Strains. The generation of Hnrnpu conditional deletion allele is de-scribed in detail in SI Materials and Methods. All animal experimental pro-cedures were in accordance with protocols approved by the InstitutionalAnimal Care and Use Committees of Columbia University Medical Center.
Histology, Immunofluorescence Staining, and Electron Microscopy. Mousehearts from animals of different ageswere dissected and fixed with 4% (wt/vol)formaldehyde. H&E and trichrome stainings were conducted according tostandard procedures with paraffin-embedded heart sections. Immunofluo-rescence staining was performed with heart sections prepared by Leicavibratome with agarose embedding. Electron microscopy analysis of heartswas conducted by the conventional osmium–uranium–lead method, withheart samples perfused first with a heparin-containing Krebs–HenseleitBuffer (10 U/mL heparin in 118 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4,1.2 mM MgSO4, 25 mM NaHCO3, 11 mM Glucose) and followed by standardprocedures. Images were acquired by Zeiss SIGMA Field Emission ScanningElectron Microscope. The average length of the sarcomere (Z line to Z line)was determined from at least 20 units.
RNA-Seq for Expression and Splicing Analysis. Total RNA from dissected con-trol and mutant hearts was isolated by TRIzol reagent (Invitrogen). Thesequencing library was prepared with the Nextera sample preparation kit(Illumina) and subjected to HiSEQ paired-end 100 bp plus sequencing.ExpressionPlot (55) pipeline was used for expression analysis; OLego (37) andQuantas pipelines were used for alternative splicing analysis.
Cell Culture, Molecular Cloning, Transfection, Western Blot, and LentivirusInfection. These assays were performed according to routine procedures.Please refer to SI Materials and Methods for details.
ACKNOWLEDGMENTS. We thank Wei Tan for performing TAC and MIsurgeries. We also thank members of the T.M. laboratory for discussions andcomments on the manuscript. This work was supported by US NationalInstitutes of Health (NIH) Pioneer Grant 8DP1NS082099 (to T.M.) and startupfunds from the Columbia University Medical Center. E.N.O. was supportedby NIH Grants HL-077439, HL-111665, HL-093039, DK-099653, and U01-HL-100401; the Foundation Leducq Networks of Excellence; the Cancer Pre-vention and Research Institute of Texas; and Robert A. Welch FoundationGrant 1-0025. N.B. was supported by a Marie Curie International OutgoingFellowship within the 7th European Community Framework Program.
1. Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu RevBiochem 72:291–336.
2. Maniatis T, Tasic B (2002) Alternative pre-mRNA splicing and proteome expansion inmetazoans. Nature 418(6894):236–243.
3. Grabowski PJ, Black DL (2001) Alternative RNA splicing in the nervous system. ProgNeurobiol 65(3):289–308.
4. Wang GS, Cooper TA (2007) Splicing in disease: Disruption of the splicing code andthe decoding machinery. Nat Rev Genet 8(10):749–761.
5. Fu XD, Ares M, Jr (2014) Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15(10):689–701.
6. Giudice J, et al. (2014) Alternative splicing regulates vesicular trafficking genes incardiomyocytes during postnatal heart development. Nat Commun 5:3603.
7. Lara-Pezzi E, Gómez-Salinero J, Gatto A, García-Pavía P (2013) The alternative heart:Impact of alternative splicing in heart disease. J Cardiovasc Transl Res 6(6):945–955.
8. Giudice J, Cooper TA (2014) RNA-binding proteins in heart development. Adv ExpMed Biol 825:389–429.
9. Lambert N, et al. (2014) RNA Bind-n-Seq: Quantitative assessment of the sequenceand structural binding specificity of RNA binding proteins. Mol Cell 54(5):887–900.
10. Yeo GW, et al. (2009) An RNA code for the FOX2 splicing regulator revealed bymapping RNA-protein interactions in stem cells. Nat Struct Mol Biol 16(2):130–137.
11. Guo W, et al. (2012) RBM20, a gene for hereditary cardiomyopathy, regulates titinsplicing. Nat Med 18(5):766–773.
12. Maatz H, et al. (2014) RNA-binding protein RBM20 represses splicing to orchestratecardiac pre-mRNA processing. J Clin Invest 124(8):3419–3430.
13. Yang J, et al. (2014) RBM24 is a major regulator of muscle-specific alternative splicing.Dev Cell 31(1):87–99.
14. Xu X, et al. (2005) ASF/SF2-regulated CaMKIIdelta alternative splicing temporally re-programs excitation-contraction coupling in cardiac muscle. Cell 120(1):59–72.
15. Feng Y, et al. (2009) SRp38 regulates alternative splicing and is required for Ca(2+)handling in the embryonic heart. Dev Cell 16(4):528–538.
16. Ding JH, et al. (2004) Dilated cardiomyopathy caused by tissue-specific ablation ofSC35 in the heart. EMBO J 23(4):885–896.
E3028 | www.pnas.org/cgi/doi/10.1073/pnas.1508461112 Ye et al.

17. Dreyfuss G, Matunis MJ, Piñol-Roma S, Burd CG (1993) hnRNP proteins and the bio-genesis of mRNA. Annu Rev Biochem 62:289–321.
18. Kamma H, Portman DS, Dreyfuss G (1995) Cell type-specific expression of hnRNPproteins. Exp Cell Res 221(1):187–196.
19. Kukalev A, Nord Y, Palmberg C, Bergman T, Percipalle P (2005) Actin and hnRNP Ucooperate for productive transcription by RNA polymerase II. Nat Struct Mol Biol12(3):238–244.
20. Romig H, Fackelmayer FO, Renz A, Ramsperger U, Richter A (1992) Characterizationof SAF-A, a novel nuclear DNA binding protein from HeLa cells with high affinity fornuclear matrix/scaffold attachment DNA elements. EMBO J 11(9):3431–3440.
21. Hasegawa Y, et al. (2010) The matrix protein hnRNP U is required for chromosomallocalization of Xist RNA. Dev Cell 19(3):469–476.
22. Hacisuleyman E, et al. (2014) Topological organization of multichromosomal regionsby the long intergenic noncoding RNA Firre. Nat Struct Mol Biol 21(2):198–206.
23. Xiao R, et al. (2012) Nuclear matrix factor hnRNP U/SAF-A exerts a global control ofalternative splicing by regulating U2 snRNP maturation. Mol Cell 45(5):656–668.
24. Huelga SC, et al. (2012) Integrative genome-wide analysis reveals cooperative regu-lation of alternative splicing by hnRNP proteins. Cell Reports 1(2):167–178.
25. Kiledjian M, Dreyfuss G (1992) Primary structure and binding activity of the hnRNP Uprotein: Binding RNA through RGG box. EMBO J 11(7):2655–2664.
26. Roshon MJ, Ruley HE (2005) Hypomorphic mutation in hnRNP U results in post-implantation lethality. Transgenic Res 14(2):179–192.
27. Brüning JC, et al. (1998) A muscle-specific insulin receptor knockout exhibits featuresof the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell2(5):559–569.
28. Göhring F, Schwab BL, Nicotera P, Leist M, Fackelmayer FO (1997) The novel SAR-binding domain of scaffold attachment factor A (SAF-A) is a target in apoptotic nu-clear breakdown. EMBO J 16(24):7361–7371.
29. Seidman JG, Seidman C (2001) The genetic basis for cardiomyopathy: From mutationidentification to mechanistic paradigms. Cell 104(4):557–567.
30. Gaussin V, et al. (2002) Endocardial cushion and myocardial defects after cardiacmyocyte-specific conditional deletion of the bone morphogenetic protein receptorALK3. Proc Natl Acad Sci USA 99(5):2878–2883.
31. Davis J, Maillet M, Miano JM, Molkentin JD (2012) Lost in transgenesis: A user’s guidefor genetically manipulating the mouse in cardiac research. Circ Res 111(6):761–777.
32. Zariwala HA, et al. (2012) A Cre-dependent GCaMP3 reporter mouse for neuronalimaging in vivo. J Neurosci 32(9):3131–3141.
33. Nakai J, Ohkura M, Imoto K (2001) A high signal-to-noise Ca(2+) probe composed of asingle green fluorescent protein. Nat Biotechnol 19(2):137–141.
34. Tallini YN, et al. (2006) Imaging cellular signals in the heart in vivo: Cardiac expressionof the high-signal Ca2+ indicator GCaMP2. Proc Natl Acad Sci USA 103(12):4753–4758.
35. Frey N, Olson EN (2003) Cardiac hypertrophy: The good, the bad, and the ugly. AnnuRev Physiol 65:45–79.
36. Yang KC, et al. (2014) Deep RNA sequencing reveals dynamic regulation of myocar-dial noncoding RNAs in failing human heart and remodeling with mechanical circu-latory support. Circulation 129(9):1009–1021.
37. Wu J, Anczuków O, Krainer AR, Zhang MQ, Zhang C (2013) OLego: Fast and sensitivemapping of spliced mRNA-Seq reads using small seeds. Nucleic Acids Res 41(10):5149–5163.
38. Fan GC, Yuan Q, Kranias EG (2008) Regulatory roles of junctin in sarcoplasmic re-ticulum calcium cycling and myocardial function. Trends Cardiovasc Med 18(1):1–5.
39. Yuan Q, et al. (2007) Sarcoplasmic reticulum calcium overloading in junctin deficiencyenhances cardiac contractility but increases ventricular automaticity. Circulation115(3):300–309.
40. Jung DH, Lee CJ, Suh CK, You HJ, Kim DH (2005) Molecular properties of excitation-contraction coupling proteins in infant and adult human heart tissues.Mol Cells 20(1):51–56.
41. Gergs U, et al. (2007) On the role of junctin in cardiac Ca2+ handling, contractility,and heart failure. Am J Physiol Heart Circ Physiol 293(1):H728–H734.
42. Kirchhefer U, et al. (2003) Impaired relaxation in transgenic mice overexpressingjunctin. Cardiovasc Res 59(2):369–379.
43. Lim KY, Hong CS, Kim DH (2000) cDNA cloning and characterization of human cardiacjunctin. Gene 255(1):35–42.
44. Moon YA, Horton JD (2003) Identification of two mammalian reductases involved inthe two-carbon fatty acyl elongation cascade. J Biol Chem 278(9):7335–7343.
45. Wada I, et al. (1991) SSR alpha and associated calnexin are major calcium bindingproteins of the endoplasmic reticulum membrane. J Biol Chem 266(29):19599–19610.
46. Göhring F, Fackelmayer FO (1997) The scaffold/matrix attachment region bindingprotein hnRNP-U (SAF-A) is directly bound to chromosomal DNA in vivo: A chemicalcross-linking study. Biochemistry 36(27):8276–8283.
47. Hegde ML, et al. (2012) Enhancement of NEIL1 protein-initiated oxidized DNA baseexcision repair by heterogeneous nuclear ribonucleoprotein U (hnRNP-U) via directinteraction. J Biol Chem 287(41):34202–34211.
48. Harvey PA, Leinwand LA (2011) The cell biology of disease: Cellular mechanisms ofcardiomyopathy. J Cell Biol 194(3):355–365.
49. Helenius A, Aebi M (2004) Roles of N-linked glycans in the endoplasmic reticulum.Annu Rev Biochem 73:1019–1049.
50. Rantakari P, et al. (2010) Hydroxysteroid (17beta) dehydrogenase 12 is essential formouse organogenesis and embryonic survival. Endocrinology 151(4):1893–1901.
51. Luu-The V, Tremblay P, Labrie F (2006) Characterization of type 12 17beta-hydroxy-steroid dehydrogenase, an isoform of type 3 17beta-hydroxysteroid dehydrogenaseresponsible for estradiol formation in women. Mol Endocrinol 20(2):437–443.
52. Kobayashi YM, Jones LR (1999) Identification of triadin 1 as the predominant triadinisoform expressed in mammalian myocardium. J Biol Chem 274(40):28660–28668.
53. Milstein ML, McFarland TP, Marsh JD, Cala SE (2008) Inefficient glycosylation leads tohigh steady-state levels of actively degrading cardiac triadin-1. J Biol Chem 283(4):1929–1935.
54. Jacob S, et al. (2013) Altered calsequestrin glycan processing is common to diversemodels of canine heart failure. Mol Cell Biochem 377(1-2):11–21.
55. Friedman BA, Maniatis T (2011) ExpressionPlot: A web-based framework for analysisof RNA-Seq and microarray gene expression data. Genome Biol 12(7):R69.
56. Garg A, et al. (2014) KLHL40 deficiency destabilizes thin filament proteins and pro-motes nemaline myopathy. J Clin Invest 124(8):3529–3539.
Ye et al. PNAS | Published online May 26, 2015 | E3029
DEV
ELOPM
ENTA
LBIOLO
GY
PNASPL
US








![Supplementary Table 1 - hnRNP K co-immunoprecipitated ... · Elmo3 Rattus norvegicus engulfment and cell motility 3 (Elmo3), mRNA [NM_001030028] 97.64 Grlf1 PREDICTED: Rattus norvegicus](https://img.pdfslide.us/doc/110x75/5f165050ace2765afb16af7c/supplementary-table-1-hnrnp-k-co-immunoprecipitated-elmo3-rattus-norvegicus.jpg)









![Neuronal Activity Induces Synaptic Delivery of hnRNP A2/B1 ... · noncoding BC1 RNA and PKMf mRNA to distal dendritic domains [8,9]. Given the nature of some of the hnRNP A2/B1-associated](https://img.pdfslide.us/doc/110x75/6115de56ebc5246c75038bff/neuronal-activity-induces-synaptic-delivery-of-hnrnp-a2b1-noncoding-bc1-rna.jpg)










