Embed Size (px)
Citation preview

Vol.19,n.2,pp.114-118 (Jun – Ago 2017) Brazilian Journal of Surgery and Clinical Research – BJSCR
BJSCR (ISSN online: 2317-4404) Openly accessible at http://www.mastereditora.com.br/bjscr
HIPOPARATIREOIDISMO PRIMÁRIO EM PACIENTE COMSÍNDROME DE RETT
HYPOPARATHYROIDISM IN PACIENT WITH RETT SYNDROME
JOÃO ROBERTO RESENDE FERNANDES1, RODRIGO DE OLIVEIRA MOREIRA2, IVY MENEZESMONTEIRO3, GIOVANNA LIMA VAZ¹, GABRIEL S. THIAGO CAVALLEIRO¹, ALINE GABRIELASANTOS COSTA¹, IBRAHIM YAHA EL SOMAILI¹, NÁDIA CHRISTINA MAIA MOREIRA4, ELAYNECHRISTINA MEIRELES MARTINS5, DANIEL ALMEIDA DA COSTA6*
1. Alunos do Curso de Graduação em Medicina. Faculdade de Medicina de Valença – FAA/CESVA; 2. Prof. da Faculdade de Medicina de Valença –FAA/CESVA. Graduado em Medicina pela Universidade Federal de Juiz de Fora, mestrado e doutorado em Medicina (Endocrinologia) pelaUniversidade Federal do Rio de Janeiro; 3. Médica Pneumologista. Docente do Curso de Graduação em Medicina da Faculdade de Medicina deValença.4.Aluna do Curso de Graduação em Medicina. Universidade Federal do Rio de Janeiro – UFRJ; 5. Médica graduada pela Faculdade deMedicina de Valença – FAA/CESVA; 6. Prof. da Faculdade de Medicina de Valença – FAA/CESVA. Graduado em Medicina pela Universidade GamaFilho, médico especialista em pediatria pela Pontifica Universidade Católica do Rio de Janeiro – PUC-RJ e Alergia e Imunologia pela UniversidadeFederal do Estado do Rio de Janeiro – UNI-RIO. Mestre em Ciências da Reabilitação pelo Centro Universitário de Caratinga.
* Hospital Escola Luiz Gioseffi Januzzi – Rua Dom José Costa Campos, 20, Centro, Valença, Rio de Janeiro, Brasil. CEP: [email protected]
Recebido em 03/06/2017. Aceito para publicação em 20/06/2017
RESUMOA síndrome de Rett (SR) é uma desordem dodesenvolvimento do sistema nervoso ligada ao cromossomoX, relacionada à mutações no gene MECP2, classicamente dosexo feminino. Instala-se por volta de um ano de vida e o lobofrontal é particularmente afetado. OPseudohipoparatireoidismo (PHP) é caracterizado porhipocalcemia e hiperfosfatemia, existindo pouca inibição nareabsorção tubular renal de fosfato e ausência de respostados órgãos-alvo ao paratormônio, sendo esta maior que aprópria deficiência hormonal. O quadro clínico clássico é debaixa estatura, fácies arredondada, obesidade, metacarposcurtos e calcificação ou ossificação do tecido subcutâneo. Asclássicas manifestações neurológicas descritas são deficiênciamental, alterações do comportamento e humor, e em casosmais raros, a epilepsia. No hipoparatireoidismo, há umaredução da secreção do paratormônio. O principal sintoma éo aumento da excitabilidade neuromuscular. É possívelespecular que a presença de hipoparatireoidismo podeagravar ou mascarar a apresentação clínica da SR. Relato decaso de paciente D.C.S.M, sexo feminino, 30 anos, natural deValença – RJ, portadora da SR, hipoparatireoidismo,epilepsia e lupus eritematoso sistêmico medicamentoso.Importante comprometimento cognitivo, semdesenvolvimento de fala. Atendida em 2010 na Faculdade deMedicina de Valença com possível Osteodistrofia de Albrighte suspeita de PHP, sem cariótipo. Encaminhada paraambulatório de paratireoide do Instituto Estadual deDiabetes e Endocrinologia e também para avaliação noambulatório de genética, no qual faz tratamento até hoje. Emagosto de 2012 foi diagnosticada osteonecrose de cabeça defêmur. A tomografia computadorizada de crânio demonstroucalcificação dos gânglios da base e a de bacia um tumor ósseoem cabeça de fêmur. Foi realizada dosagem de 25(OH)Vitamina D que evidenciou valores abaixo do limite dedetecção do método, assim como PTH baixo. Iniciadotratamento com cálcio e 25(OH) Vi tamina D3, com melhoraparcial do comprometimento cognitivo. Vem realizandoacompanhamento periódico a cada 6 meses. Pacientes comSR apresentam um quadro clínico que pode ser confundidocom o PHP. Em nossa paciente, a dosagem do PTH e da
Vitamina D permitiu o reconhecimento dehipoparatireoidismo, o que levou a suplementação de cálciocom vitamina D, contribuindo para uma possível melhoraglobal de uma paciente com uma rara e grave doençagenética.
PALAVRAS-CHAVE: Síndrome de Rett,Hipoparatireoidismo, Valença.
ABSTRACTRett syndrome (SR) is a disorder of the development of thenervous system linked to the X chromosome, related tomutations in the MECP2 gene, classically female. It is installedaround one year of life and the frontal lobe is particularlyaffected. Pseudohypoparathyroidism (PHP) is characterized byhypocalcemia and hyperphosphatemia, with little inhibition inrenal tubular phosphate reabsorption and absence of response oftarget organs to parathyroid hormone, which is greater than thehormone deficiency itself. The classic clinical presentation is ofshort stature, rounded facies, obesity, short metacarpals andcalcification or ossification of the subcutaneous tissue. Theclassic neurological manifestations described are mentaldeficiency, changes in behavior and mood, and in rarer cases,epilepsy. In hypoparathyroidism, there is a reduction inparathyroid secretion. The main symptom is increased ofneuromuscular excitability. It is possible to speculate that thepresence of hypoparathyroidism may aggravate or mask theclinical presentation of SR. Case report of patient D.C.S.M,female, 30 years old, from Valença - RJ, with SR,hypoparathyroidism, epilepsy and systemic lupus erythematosus.Significant cognitive impairment without speech development.Attended in 2010 at the Faculty of Medicine of Valença withpossible Osteoarthritis of Albright and suspected of PHP, withoutkaryotype. She was referred to a parathyroid outpatient clinic ofthe State Institute of Diabetes and Endocrinology and also forevaluation in the genetics outpatient clinic, where she is currentlytreated. In August 2012, femoral head osteonecrosis wasdiagnosed. Computed tomography of the skull demonstratedcalcification of the basal ganglia and of the basal bone tumor inthe femoral head. A dose of 25 (OH) Vitamin D was performed,which showed values below the detection limit of the method, aswell as low PTH. Initiated treatment with calcium and 25 (OH)

Fernandes et al. / Braz. J. Surg. Clin. Res. V.19,n.2,pp.114-118 (Jun - Ago 2017)
BJSCR (ISSN online: 2317-4404) Openly accessible at http://www.mastereditora.com.br/bjscr
Vitamin D3, with partial improvement of cognitive impairment.It is performing periodic monitoring every 6 months. Patientswith SR have a clinical case that can be confused with PHP. Inour patient, the dosage of PTH and Vitamin D allowed therecognition of hypoparathyroidism, which led to calciumsupplementation with vitamin D, contributing to a possibleoverall improvement of a patient with a rare and serious geneticdisease.
KEYWORDS: Rett syndrome, Hypoparathyroidism, Valença.
1. INTRODUÇÃOA síndrome de Rett, descrita inicialmente por
Andréas Rett em 1966, é uma desordem dodesenvolvimento do sistema nervoso ligada aocromossomo X que acomete os portadores logo nosprimeiros anos de vida e que afeta, em sua formaclássica, o sexo feminino. A desordem se instala porvolta de um ano de vida e o lobo frontal éparticularmente afetado. Sendo assim, os portadoresapresentam desenvolvimento motor e comportamentalaté essa idade e após ocorre uma regressão, a criançavai se tornando cada vez mais introspectiva e perde ahabilidade de executar certos movimentos antes feitos.A doença tem por base genética em 80% dos casos,sendo relacionada às mutações no gene da proteínamethyl-CpG binding protein 2 (MeCP2)1.
Tipicamente feminina, o primeiro caso descrito domundo de um menino com 28 meses de idadeapresentando o fenótipo da SR clássica, com mutaçãono gene MECP2 e coexistência do cariótipo XXY, porpesquisadores brasileiros2.
A síndrome tem uma tríade característica:demência-ataxia-autismo, associada à movimentos dasmãos estereotipados característicos, há crianças queesfregam uma mão contra a outra com movimentosrotatórios como se as tivessem lavando; algumas outrasfrequentemente batem as palmas diante da face ou asleva à boca; levando a uma apraxia em que a criança éincapaz de executar simples gestos ou de usaradequadamente as mãos. Há desaceleração docrescimento cranial. Por volta do terceiro ano de vida, amaioria dos casos desenvolve crises convulsivas, dedifícil controle. Distúrbios ventilatórios também sãodescritos. A sobrevida pode ser considerada longa,existindo relatos de pacientes adolescentes e adultosjovens, nos quais o comportamento autista e as crisesconvulsivas são menos frequentes3.
Os critérios utilizados para o diagnóstico desíndrome de Rett incluem: (1) períodos pré e perinataisaparentemente normais; (2) desenvolvimentopsicomotor aparentemente normal durante os primeiros6 meses de idade; (3) perímetro cefálico normal aonascimento; (4) desaceleração do crescimento cranianoentre os 5 meses e os 4 anos de idade; (5) perda dosmovimentos voluntários manuais já adquiridos; (6)comprometimento severo da linguagem expressiva ereceptiva e presença de retardo psicomotor; (7)movimentos estereotipados das mãos; (8) ataxia damarcha e apraxia-ataxia do tronco entre 1 e 4 anos deidade; e (9) o diagnóstico é apenas de presunção até os
2 a 5 anos de idade4,5.Há também sinais e sintomas de exclusão, que
levam a pensar em outras desordens: (1) evidência deretardo de crescimento intrauterino; (2) visceromegaliaou outros indícios de doenças de depósito; (3)retinopatia ou atrofia óptica; (4) microcefalia aonascimento; (5) evidência de insulto cerebraladquirido; (6) doenças metabólicas ou neurológicasprogressivas; (7) condições neurológicas adquiridasdecorrentes de infecções severas ou traumatismoscrânio-encefálicos5.
A paratireoide é um órgão apresentado na forma dequatro nódulos, localizados, na maioria dos casos, doisno ápice dos lobos direito e esquerdo da tireoide e osoutros dois nos pólos inferiores. É a responsável pelasecreção do paratormônio, uma proteína constituída porcadeia simples de polipeptídio com 84 aminoácidos.Tem ação oposta à calcitonina advinda das células C datireoide6.
O paratormônio tem ação nas células tubularesrenais, onde inibem a reabsorção de fosfatos e regulama fosfatúria. Agem nos osteoclastos ósseos por ação deenzimas, reabsorvendo a matriz e solubilizando ocálcio. É de fundamental importância no equilíbrioentre a aposição e reabsorção óssea desse mineral, namanutenção sérica do mesmo em torno de 8,9 a 10 mg% e na absorção intestinal de cálcio6.
No hipoparatireoidismo, há uma redução desecreção do paratormônio, com consequentehipocalcemia e hiperfosfatemia. O principal sintoma éo aumento da excitabilidade neuro-muscular, que podeser observada pelos sinais semiológicos de Chvostek eTrousseau. Também podem existir alterações psíquicase neurológicas e calcificações anormais intracranianas.Pode ser causado devido à remoção inapropriada dasparatireoides durante tireoidectomias ou em casos delinfadectomias cervicais, ambas são as causas maiscomuns de hipoparatireoidismo, mas este tambémpode, mesmo sendo mais raro, ser adquirido pós-radiação ou pela supressão da produção hormonaldepois de uma prolongada hipercalcemia. Tambémexistem casos idiopáticos e formas autoimunesfamiliares, descritos em crianças6,7.
O Pseudohipoparatireoidismo (PHP) foi descritoprimeiramente em 1942 por Albright, e é caracterizadopor hipocalcemia e hiperfosfatemia, existindo poucainibição na reabsorção tubular renal de fosfato eausência de resposta dos órgãos-alvo ao paratormônio,sendo esta maior que a própria deficiência hormonal. Oquadro clínico clássico é de baixa estatura, fáciesarredondada, obesidade, metacarpos curtos ecalcificação ou ossificação do tecido subcutâneo. Asclássicas manifestações neurológicas descritas sãodeficiência mental, alterações do comportamento ehumor, e em casos mais raros, a epilepsia8.
É possível especular que a presença deHipoparatireoidismo pode agravar ou mascarar aapresentação clínica da Síndrome de Rett. Além disso,pacientes com Síndrome de Rett podem apresentarcaracterísticas fenotípicas que se assemelham ao PHP,

Fernandes et al. / Braz. J. Surg. Clin. Res. V.19,n.2,pp.114-118 (Jun - Ago 2017)
BJSCR (ISSN online: 2317-4404) Openly accessible at http://www.mastereditora.com.br/bjscr
dificultando ainda mais o diagnóstico, principalmenteem pacientes com cálcio abaixo do limite danormalidade. Este trabalho teve como objetivo relatar ocaso de paciente Valenciana portadora da Síndrome deRett com Hipoparatireoidismo, observando a evoluçãoda doença e da paciente após diagnóstico eacompanhamento médico, com tratamento adequado.
Procurou-se também discutir com a literaturapertinente o caso da paciente, assim como, descrever asalterações ósseas da mesma e seu diagnósticodiferencial clínico com Pseudohipoparatireoidismo.
2. CASO CLÍNICO
Este foi um trabalho exploratório. A análise do casofoi feita através de uma interpretação dasintomatologia, evolução, exames realizados e oporquê da exclusão de diagnósticos diferenciais;obtidos por uma revisão do prontuário da paciente,correlacionando-o com os achados da literatura.
Enquanto procedimento, o trabalho foi realizadoatravés de uma observação direta, analisando o perfilda paciente e todas as suas variáveis de influênciasobre a patologia. O material coletado juntamente coma sua análise foi organizado neste presente artigo.
D.C.S.M, sexo feminino, 30 anos, natural eresidente de Valença – RJ, portadora da Síndrome deRett, hipoparatireoidismo, epilepsia e lupus eritematososistêmico (LES) medicamentoso, por uso decarbamazepina.
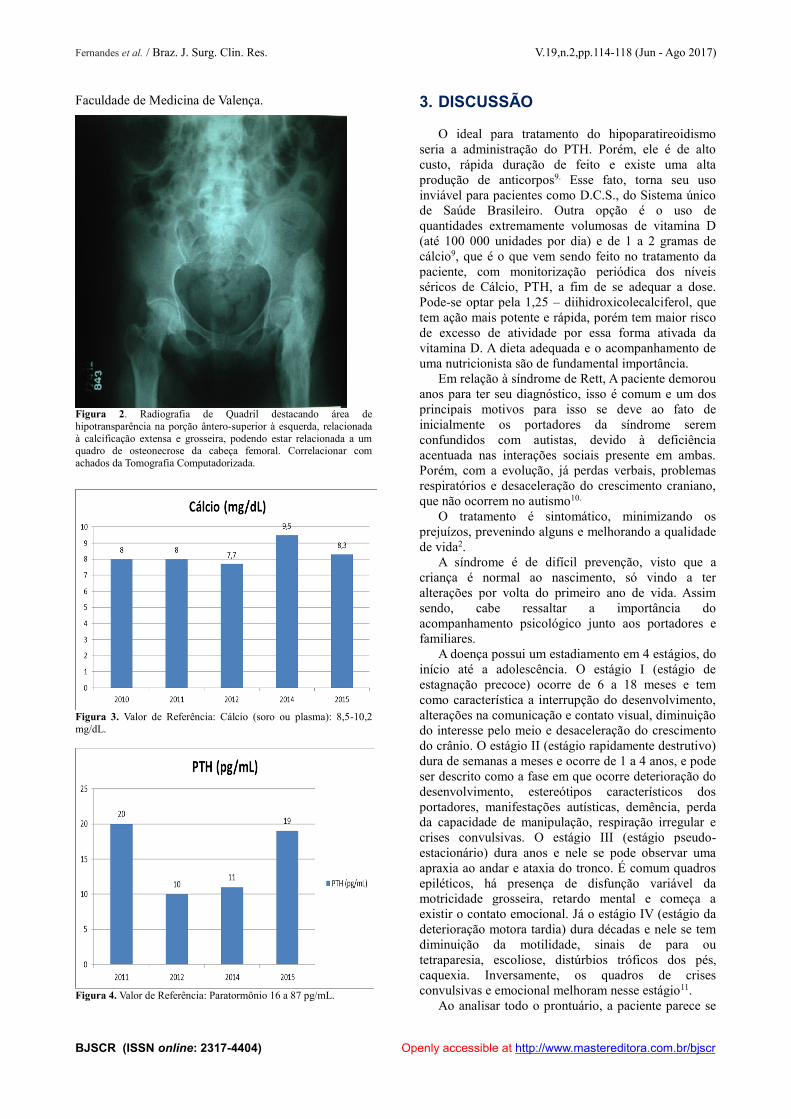
Figura 1. Tomografia computadorizada de quadril mostrandoheterogeneidade com acentuação do trabeculado ósseo medular emambas as cabeças femorais, destacando-se traço hipodenso subcondralna porção ântero-superior à esquerda, com aparente retificação dacortical. O aspecto descrito pode estar relacionado a um quadro deosteonecrose das cabeças femorais. Exuberante e extensa calcificaçãogrosseira, lobulada, que mede cerca de 20,0 x 4,4 x 4,6 cm, e se estendeinferiormente a partir da cortical anterior da asa do ilíaco esquerdo até aregião da diáfise proximal do fêmur, através das interfaces mioadiposasanteriores do quadril e com envolvimento semicircunferencial da
cortical óssea femoral, destacando-se junto à inserção tendínea doiliopsoas. Há área hipodensa central na porção superior, nãocalcificada. Junto a asa do ilíaco nota-se foco gasoso na interface entrea calcificação e a cortical óssea do ilíaco, esta com importanteespessamento, esboçando neoarticulação. O aspecto pode estarrelacionado à sequela traumática, como calcificação heterotópica oumiosite ossificante, admitindo diferencial com hematoma crônicocalcificado. Finais calcificações nos ventres musculares do glúteomínimo e médio, que apresentam redução da densidade das suas fibrasmusculares. Demais estruturas ósseas, espações articulares e partesmoles preservados.
Paciente acompanhada no ambulatório deNeurologia com epilepsia desde 1989, com 1ª criseconvulsiva com 2 anos e 9 meses, parcial e deausência. Diagnosticada com LES medicamentoso poruso de carbamazepina desde 2009. Importantecomprometimento cognitivo, sem desenvolvimento defala, cobre o rosto o tempo todo. Histórico depneumonias de repetição, otite média recorrente, rinitealérgica, alergia medicamentosa a váriosmedicamentos. Queixas recorrentes de tosse produtiva,ouvido seco e fezes ressecadas com presença dedistensão abdominal. Apresenta 3º,4º,5º metacarposcurtos.
Atendida em 2010 no Ambulatório de ClínicaMédica da Faculdade de Medicina de Valença (FMV)com possível Osteodistrofia de Albright e suspeita dePseudohipoparatireoidismo, sem cariótipo.
Apresentava sinais clínicos de Cushing (devido aocorticoide utilizado para o tratamento do LES induzidopela carbamazepina), amenorreia há 6 meses, retardomental e dificuldade para interação.
Em fevereiro de 2011, foi encaminhada paraambulatório do Hospital Universitário ClementinoFraga Filho (HUCFF), onde faz acompanhamento atéhoje. Em agosto de 2012 foi diagnosticadaosteonecrose de cabeça de fêmur (Figuras 1 e 2). Foirealizada dosagem de 25(OH) Vitamina D queevidenciou valores abaixo do limite de detecção dométodo, assim como PTH baixo. Iniciado tratamentocom cálcio e 25(OH) Vi tamina D3, com melhoraparcial do comprometimento cognitivo.A última consulta no ambulatório da paciente foi em09/03/15, data na qual estava em uso de fenobarbital 44gotas pela manhã, hidroxicloroquina 400 mg pelamanhã, carbonato de Cálcio uma vez ao dia, ômega 3 -1000mg uma vez ao dia, colecalciferol 4000 UI gota –4 gotas/semana, topiramato 25 mg à noite efenobarbital 100 mg à noite. A mesma realiza dosagensseriadas de Cálcio Plasmático e PTH, desde o início dotratamento (Figuras 3, 4).
A última consulta no ambulatório da paciente foiem 09/03/15, data na qual estava em uso defenobarbital 44 gotas pela manhã, hidroxicloroquina400 mg pela manhã, carbonato de Cálcio uma vez aodia, ômega 3 - 1000mg uma vez ao dia, colecalciferol4000 UI gota – 4 gotas/semana, topiramato 25 mg ànoite e fenobarbital 100 mg à noite. A mesma realizadosagens seriadas de Cálcio Plasmático e PTH, desde oinício do tratamento (Figuras 3, 4).
Paciente queixa-se apenas de distensão abdominal egazes. Vem realizando acompanhamento periódico acada 6 meses no Ambulatório de Endocrinologia da
Fernandes et al. / Braz. J. Surg. Clin. Res. V.19,n.2,pp.114-118 (Jun - Ago 2017)
BJSCR (ISSN online: 2317-4404) Openly accessible at http://www.mastereditora.com.br/bjscr
Faculdade de Medicina de Valença.
Figura 2. Radiografia de Quadril destacando área dehipotransparência na porção ântero-superior à esquerda, relacionadaà calcificação extensa e grosseira, podendo estar relacionada a umquadro de osteonecrose da cabeça femoral. Correlacionar comachados da Tomografia Computadorizada.
Figura 3. Valor de Referência: Cálcio (soro ou plasma): 8,5-10,2mg/dL.
Figura 4. Valor de Referência: Paratormônio 16 a 87 pg/mL.
3. DISCUSSÃO
O ideal para tratamento do hipoparatireoidismoseria a administração do PTH. Porém, ele é de altocusto, rápida duração de feito e existe uma altaprodução de anticorpos9. Esse fato, torna seu usoinviável para pacientes como D.C.S., do Sistema únicode Saúde Brasileiro. Outra opção é o uso dequantidades extremamente volumosas de vitamina D(até 100 000 unidades por dia) e de 1 a 2 gramas decálcio9, que é o que vem sendo feito no tratamento dapaciente, com monitorização periódica dos níveisséricos de Cálcio, PTH, a fim de se adequar a dose.Pode-se optar pela 1,25 – diihidroxicolecalciferol, quetem ação mais potente e rápida, porém tem maior riscode excesso de atividade por essa forma ativada davitamina D. A dieta adequada e o acompanhamento deuma nutricionista são de fundamental importância.
Em relação à síndrome de Rett, A paciente demorouanos para ter seu diagnóstico, isso é comum e um dosprincipais motivos para isso se deve ao fato deinicialmente os portadores da síndrome seremconfundidos com autistas, devido à deficiênciaacentuada nas interações sociais presente em ambas.Porém, com a evolução, já perdas verbais, problemasrespiratórios e desaceleração do crescimento craniano,que não ocorrem no autismo10.
O tratamento é sintomático, minimizando osprejuízos, prevenindo alguns e melhorando a qualidadede vida2.
A síndrome é de difícil prevenção, visto que acriança é normal ao nascimento, só vindo a teralterações por volta do primeiro ano de vida. Assimsendo, cabe ressaltar a importância doacompanhamento psicológico junto aos portadores efamiliares.
A doença possui um estadiamento em 4 estágios, doinício até a adolescência. O estágio I (estágio deestagnação precoce) ocorre de 6 a 18 meses e temcomo característica a interrupção do desenvolvimento,alterações na comunicação e contato visual, diminuiçãodo interesse pelo meio e desaceleração do crescimentodo crânio. O estágio II (estágio rapidamente destrutivo)dura de semanas a meses e ocorre de 1 a 4 anos, e podeser descrito como a fase em que ocorre deterioração dodesenvolvimento, estereótipos característicos dosportadores, manifestações autísticas, demência, perdada capacidade de manipulação, respiração irregular ecrises convulsivas. O estágio III (estágio pseudo-estacionário) dura anos e nele se pode observar umaapraxia ao andar e ataxia do tronco. É comum quadrosepiléticos, há presença de disfunção variável damotricidade grosseira, retardo mental e começa aexistir o contato emocional. Já o estágio IV (estágio dadeterioração motora tardia) dura décadas e nele se temdiminuição da motilidade, sinais de para outetraparesia, escoliose, distúrbios tróficos dos pés,caquexia. Inversamente, os quadros de crisesconvulsivas e emocional melhoram nesse estágio11.
Ao analisar todo o prontuário, a paciente parece se

Fernandes et al. / Braz. J. Surg. Clin. Res. V.19,n.2,pp.114-118 (Jun - Ago 2017)
BJSCR (ISSN online: 2317-4404) Openly accessible at http://www.mastereditora.com.br/bjscr
encaixar entre os estágios III e IV.
4. CONCLUSÃO
Pacientes com Síndrome de Rett apresentam umquadro clínico que pode ser confundido com oPseudohipoparatireoidismo. Em nossa paciente, adosagem do PTH e da Vitamina D permitiu oreconhecimento de Hipoparatireoidismo, o que levou asuplementação de cálcio com vitamina D, contribuindopara uma possível melhora global de uma paciente comuma rara e grave doença genética.
Pode-se concluir a relevância do caso, visto que asíndrome de Rett é uma das causas mais frequentes dedeficiência múltipla severa no sexo feminino e ohipoparatireoidismo deve ser rapidamentediagnosticado e tratado, devido às suas funçõesessenciais para o adequado nível sérico principalmentedo cálcio, indispensável a diversos mecanismos,principalmente a estabilidade neuromuscular. Tambémé de suma importância nesses pacientes uma dietaadequada. São doenças que devem interessar todos osprofissionais relacionados à saúde, principalmentepediatras, devido à importância de um diagnóstico etratamento precoces.
5. REFERÊNCIAS[1] Farage L. Síndrome de Rett: achados clínicos,
genéticos e por ressonância magnética. Radiol Bras,São Paulo , v. 42, n. 4, p. 254, Aug. 2009 .
[2] Schwartzan JS. et al. Fenótipo Rett em paciente comcariótipo XXY: relato de caso. Arq. Neuro-Psiquiatr.,São Paulo , v. 56, n. 4, p. 824-828, Dec. 1998.
[3] Rosemberg S. et al. Síndrome de Rett: análise dosprimeiros cinco casos diagnosticados no Brasil. Arq.Neuro-Psiquiatr., São Paulo , v. 45, n. 2, p. 143-152, June 1987.
[4] Hagberg B. et al. — Rett syndrome: criteria forinclusion and exclusion. Brain D e v . 7:372, 1985.
[5] Trevathan E, Naidu S. The clinical recognition anddifferential doagnosis of Rett syndrome. J Child Neurol1988;3(Suppl.):S6-S16.
[6] Prospero JD. et al. Paratireóides: estrutura, funções epatologia. Acta ortop. bras., São Paulo , v. 17, n. 2, p.53-57, 2009.
[7] Robbins SL, Cotran RS, Kumar V. Patologia estruturale funcional. Rio de Janeiro: Interamerica; 1986.
[8] Rocha C, Gonfinetti NV, Pelluci LA. Aspectos clínicose Neurorradiológicos do pseudo-hipoparatireoidismo:relato de caso.Arq. Neuro-Psiquiatr., São Paulo , v. 55,n. 1, p. 139-143, 1997.
[9] Guyton & Hall. Tratado de Fisiologia Médica. 11ªEdição. São Paulo: Elsevier, 2006. 1115 páginas.
[10] Associação Americana de Psiquiatria. Manual deEstatística e Diagnóstica de Transtornos Mentais (DSMIV TM) 4. Ed. Porto Alegre: Artes Médicas, 1995.
[11] Hagberg B & Witt, Engerström I. Rett syndrome: asuggested staging system for describing impairmentprofile with increasing age towards adolescence. Amer .J. med. Genet. 24:47, 1986.





















![Hipocalcemia exp[1][1]](https://img.pdfslide.us/doc/110x75/55ba1494bb61ebad3d8b4753/hipocalcemia-exp11.jpg)






