Embed Size (px)
Citation preview

High-throughput measurement of mitochondrial membrane potentialin a neural cell line using a fluorescence plate reader
Alice Wong and Gino A. Cortopassi*
Department of Molecular Biosciences, University of California, 1311 Haring Hall, Davis, CA 95616, USA
Received 3 October 2002
Abstract
Mutations in mitochondrial genes cause mitochondrial genetic disease, which is often associated with deficiency of the mito-
chondrial membrane potential (MMP). We present a high-throughput method for measuring MMP in intact neural cells using
TMRM, a well-known potentiometric dye, in a 48-well plate format. Addition of known MMP depolarizing agents, FCCP or DNP,
resulted in a time- and concentration-dependent decrease in fluorescence, which was saturable, whereas the addition of drugs that
affect non-mitochondrial properties did not. A cell line deficient in mtDNA had decreased fluorescence, which was not further
depleted by a depolarizing agent. The high-throughput results are similar to those produced by more time-consuming and low-
throughput flow cytometry or microscopy methods. This plate-based system could facilitate the identification of cell-permeant small
molecules (i.e., drugs) that modify MMP, which could be used to enhance mitochondrial function, and also for screening small
populations of neural cells for mutations in nuclear or mtDNA genes that decrease MMP.
� 2002 Elsevier Science (USA). All rights reserved.
Keywords: High-throughput screen; TMRM; Mitochondria; mtDNA; Membrane potential; Apoptosis; Genetics; Rho-zero; Drug therapy
Mitochondria produce ATP through the process of
oxidative phosphorylation, which involves a series of
redox reactions that transfer electrons through multiple
protein complexes in the inner mitochondrial mem-
brane. As a result, protons are pumped out of the mi-
tochondria, generating the mitochondrial membrane
potential (MMP), which is not only harnessed to gen-
erate ATP, but is also responsible for mitochondrialCa2þ uptake [1,2], metabolite and protein transport
[3,4], production of reactive oxygen species [5,6], and has
also been related to the process of apoptosis [7–12].
Thus, the health and bioenergetic function of the mito-
chondria depend on its membrane potential.
Abnormal mitochondrial function has been attrib-
uted to cell death in several degenerative disorders in-
cluding Alzheimer�s and Parkinson�s diseases, as well astype 2 diabetes, stroke, and myocardial infarction
[13,14]. In addition, several debilitating diseases caused
by mutations in the mitochondrial genome have been
identified [15–17]. These mutations directly or indirectly
affect the electron transport chain, and thus, MMP.
Preservation of the MMP is essential during normal
conditions, and especially, during conditions of stress
and disease. Depolarization of the membrane results in a
reduction of ATP production and is also thought to
precipitate the release of pro-apoptotic factors in some
cell systems [7,10,11]. Furthermore, MMP is often defi-cient in mitochondria from patients with mitochondrial
genetic disease which is the result of mutations of the
mtDNA or of nuclear-encoded genes targeted to mito-
chondria. The phenotypes of such patients are pre-
dominantly neurological, i.e., preferentially affecting
neural and muscle cell types. Also, mitochondrial
oxidative stress has been invoked in several chronic,
progressive neurodegenerative diseases, including Alz-heimer�s, Parkinson�s, and ALS. Thus, there are goodreasons to search for therapeutic agents to increase
MMP in patients whose MMP is deficient, and for
therapeutic agents to decrease MMP as possible pro-
apoptotic drugs, for example, in the case of cancer.
Furthermore, there is no general assay for screening
Biochemical and Biophysical Research Communications 298 (2002) 750–754
www.academicpress.com
BBRC
* Corresponding author. Fax: 1-530-754-9342.
E-mail address: [email protected] (G.A. Cortopassi).
0006-291X/02/$ - see front matter � 2002 Elsevier Science (USA). All rights reserved.
PII: S0006 -291X(02 )02546 -9
small populations of neural cells for nuclear or mtDNAmutations which decrease MMP. Thus there is a need
for high-throughput assays of living cells for drugs
which modify MMP and thus perforce mitochondrial
function. Several methods exist to measure MMP, many
of them using fluorescent dyes. All have their advanta-
ges and disadvantages. Besides measuring MMP, some
fluorescent dyes also measure the plasma membrane
potential, inhibit Complex I, or are photoreactive [18].We have developed an assay to measure MMP in
intact cells using tetramethylrhodamine methyl ester
(TMRM), a membrane-permeant, cationic fluorescent
dye that accumulates in the mitochondria according to
the Nernst equation [19–21]. When used at low con-
centrations, measurement of plasma membrane poten-
tial is minimal and the dye is the least toxic to
mitochondria [18,21–23]. The use of intact cells is ben-eficial, since permeabilization of the plasma membrane,
and potentially the mitochondrial membrane, can give
variable results. Using this assay, samples can be mea-
sured in a 48-well plate and respond to MMP-depolar-
izing agents.
Materials and methods
Cell culture. NT2 preneuronal and NT2 rho-zero cells were main-
tained in DMEM supplemented with 10% FBS, 50 lg/ml uridine, 1lMsodium pyruvate, penicillin, and streptomycin.
Measurement of mitochondrial membrane potential using the fluo-
rescence plate reader. Cells were isolated and washed in PBS before
resuspension in Hanks� buffered salt solution (HBSS) (1:0� 106 cells/ml) containing 50 nM TMRM. The cells were placed in a 48-well plate
and fluorescence was measured in a temperature controlled (37 �C)Cytofluor (PE Biosystems, Foster City, CA) plate reader for 3 cycles,
2min apart at 530–620nm. After the first 3 cycles, reagents (indicated
in the figure legends) were added to each well (indicated by an arrow)
and measurements were taken 2min apart for a total of 30min.
Background subtraction was conducted for each treatment and the
results shown are the means of three independent experiments per-
formed in triplicate. Error bars represent two standard error of the
mean.
Measurement of mitochondrial membrane potential by flow cytome-
try. Mitochondrial membrane potential was measured in digitonin
permeabilized cells using 50 nM TMRM [24]. Analysis was performed
on the FACSort flow cytometer (Becton–Dickinson, San Jose, CA),
with a 488 nm argon laser at the UC Davis Optical Biology facility.
Results and discussion
Known MMP depolarizers produce decreases in fluores-
cence in the high-throughput assay
A pharmacological proof that MMP was being
measured by the TMRM fluorescence was to use re-
agents known to abolish the membrane potential, FCCP
and DNP. After isolating cells and incubating them in
50 nM TMRM, fluorescence measurements were taken
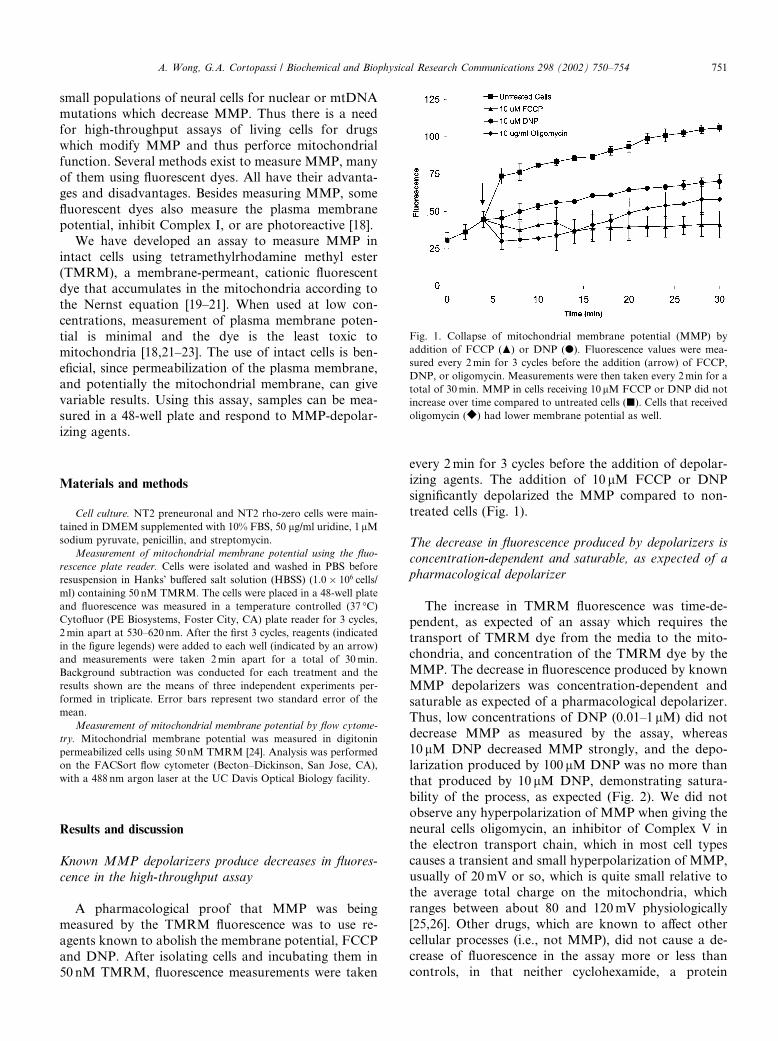
every 2min for 3 cycles before the addition of depolar-izing agents. The addition of 10 lM FCCP or DNP
significantly depolarized the MMP compared to non-
treated cells (Fig. 1).
The decrease in fluorescence produced by depolarizers is
concentration-dependent and saturable, as expected of a
pharmacological depolarizer
The increase in TMRM fluorescence was time-de-pendent, as expected of an assay which requires the
transport of TMRM dye from the media to the mito-
chondria, and concentration of the TMRM dye by the
MMP. The decrease in fluorescence produced by known
MMP depolarizers was concentration-dependent and
saturable as expected of a pharmacological depolarizer.
Thus, low concentrations of DNP (0.01–1 lM) did notdecrease MMP as measured by the assay, whereas10 lM DNP decreased MMP strongly, and the depo-
larization produced by 100 lM DNP was no more than
that produced by 10 lM DNP, demonstrating satura-
bility of the process, as expected (Fig. 2). We did not
observe any hyperpolarization of MMP when giving the
neural cells oligomycin, an inhibitor of Complex V in
the electron transport chain, which in most cell types
causes a transient and small hyperpolarization of MMP,usually of 20mV or so, which is quite small relative to
the average total charge on the mitochondria, which
ranges between about 80 and 120mV physiologically
[25,26]. Other drugs, which are known to affect other
cellular processes (i.e., not MMP), did not cause a de-
crease of fluorescence in the assay more or less than
controls, in that neither cyclohexamide, a protein
Fig. 1. Collapse of mitochondrial membrane potential (MMP) by
addition of FCCP (N) or DNP (d). Fluorescence values were mea-
sured every 2min for 3 cycles before the addition (arrow) of FCCP,
DNP, or oligomycin. Measurements were then taken every 2min for a
total of 30min. MMP in cells receiving 10 lM FCCP or DNP did not
increase over time compared to untreated cells (j). Cells that received
oligomycin (r) had lower membrane potential as well.
A. Wong, G.A. Cortopassi / Biochemical and Biophysical Research Communications 298 (2002) 750–754 751
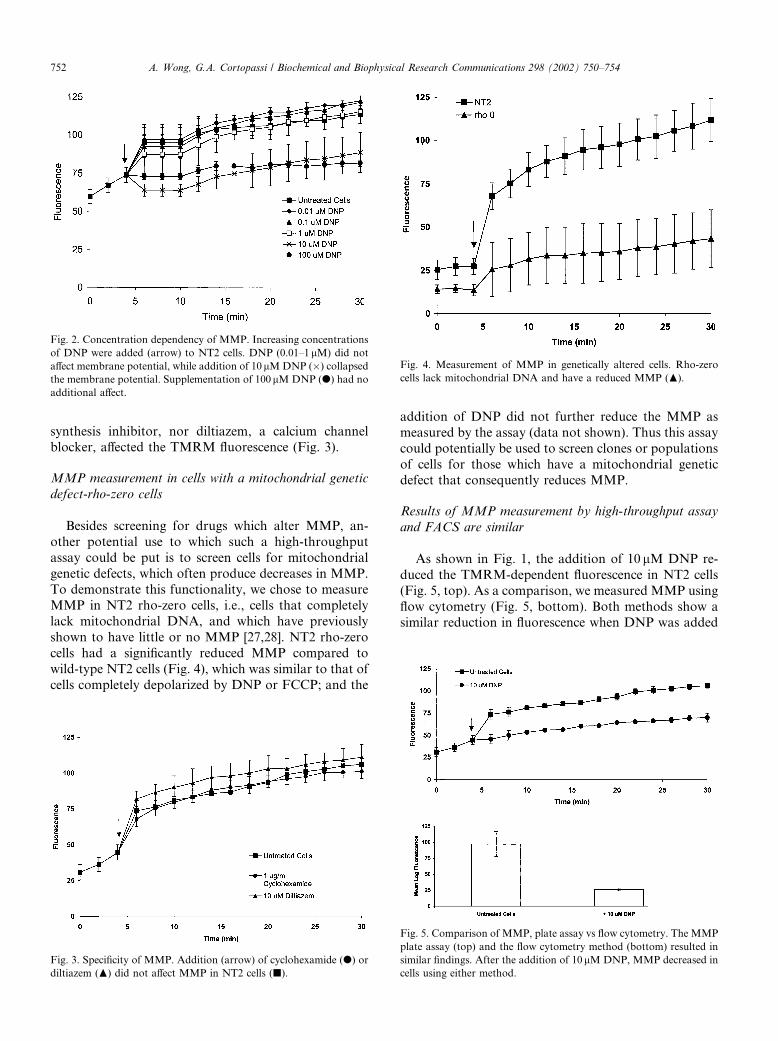
synthesis inhibitor, nor diltiazem, a calcium channelblocker, affected the TMRM fluorescence (Fig. 3).
MMP measurement in cells with a mitochondrial genetic
defect-rho-zero cells
Besides screening for drugs which alter MMP, an-
other potential use to which such a high-throughput
assay could be put is to screen cells for mitochondrial
genetic defects, which often produce decreases in MMP.To demonstrate this functionality, we chose to measure
MMP in NT2 rho-zero cells, i.e., cells that completely
lack mitochondrial DNA, and which have previously
shown to have little or no MMP [27,28]. NT2 rho-zero
cells had a significantly reduced MMP compared to
wild-type NT2 cells (Fig. 4), which was similar to that of
cells completely depolarized by DNP or FCCP; and the
addition of DNP did not further reduce the MMP as
measured by the assay (data not shown). Thus this assay
could potentially be used to screen clones or populations
of cells for those which have a mitochondrial geneticdefect that consequently reduces MMP.
Results of MMP measurement by high-throughput assay
and FACS are similar
As shown in Fig. 1, the addition of 10 lM DNP re-
duced the TMRM-dependent fluorescence in NT2 cells
(Fig. 5, top). As a comparison, we measured MMP using
flow cytometry (Fig. 5, bottom). Both methods show asimilar reduction in fluorescence when DNP was added
Fig. 3. Specificity of MMP. Addition (arrow) of cyclohexamide (d) or
diltiazem (N) did not affect MMP in NT2 cells (j).
Fig. 4. Measurement of MMP in genetically altered cells. Rho-zero
cells lack mitochondrial DNA and have a reduced MMP (N).
Fig. 5. Comparison of MMP, plate assay vs flow cytometry. The MMP
plate assay (top) and the flow cytometry method (bottom) resulted in
similar findings. After the addition of 10lMDNP, MMP decreased in
cells using either method.
Fig. 2. Concentration dependency of MMP. Increasing concentrations
of DNP were added (arrow) to NT2 cells. DNP (0.01–1lM) did notaffect membrane potential, while addition of 10 lMDNP (�) collapsedthe membrane potential. Supplementation of 100lMDNP (d) had no
additional affect.
752 A. Wong, G.A. Cortopassi / Biochemical and Biophysical Research Communications 298 (2002) 750–754

to the cells. The high-throughput plate assay gave similarresults as the flow cytometry method, confirming by a
known method the utility of this assay to measure MMP.
Summary and conclusions
We have developed a high-throughput, plate-based
assay of MMP in neural cells using TMRM, a known
mitochondrial potentiometric dye. The increase in
TMRM fluorescence is time- and concentration-depen-dent, which presumably affects the transport of the dye
from media to mitochondria, and concentration of the
dye by the MMP. The assay reliably detects decreased
fluorescence produced by agents which are known to
depolarize mitochondria specifically and the effects of
the drugs are concentration-dependent and saturable
(Figs. 1 and 2). Similarly, the assay detected no changes
in fluorescence induced by drugs which have no knownmitochondrial effect (Fig. 3). Thus this assay is a rela-
tively sensitive, fast, and specific screening method for
drugs or other small molecules that modify MMP.
Similarly, the assay detected decreased fluorescence in
cells with a deficiency of mitochondrial DNA, and
which are known to have a deficiency of MMP. Cur-
rently there are no general methods for rapid screening
(or selection) of colonies or small groups of mammaliancells for mitochondrial mutations. This genetic proof-
of-principle demonstrates that the assay could be used
to identify colonies or groups of cells with genetic
modifications of the mtDNA, and thus, could be used
for genetic screens of cells bearing deleterious mtDNA
mutations. A number of samples can be measured
concurrently using this rapid assay, and the dye used,
TMRM, is the least toxic to cells of the mitochondrialpotentiometric dyes [21] and can be incorporated into
mitochondria rapidly [19,29].
In summary, the simple and rapid method presented
specifically measures MMP in intact cells and could be
used to identify either drugs that affect mitochondrial
function, or as a screening method for colonies of cells
with mutations that alter mitochondrial properties, that
consequently decrease MMP.
Acknowledgments
We thank Dr. Heidi Gross for her helpful discussions and Carol
Oxford for her assistance with flow cytometry. This work was sup-
ported by USPHS Grant Ey12245.
References
[1] D.G. Nicholls, S.L. Budd, Mitochondria and neuronal glutamate
excitotoxicity, Biochim. Biophys. Acta 1366 (1998) 97–112.
[2] J.A. Dykens, Isolated cerebral and cerebellar mitochondria
produce free radicals when exposed to elevated Ca2þ and Naþ:
implications for neurodegeneration, J. Neurochem. 63 (1994)
584–591.
[3] C.K. Suzuki, M. Rep, J.M. van Dijl, K. Suda, L.A. Grivell, G.
Schatz, ATP-dependent proteases that also chaperone protein
biogenesis, Trends Biochem. Sci. 22 (1997) 118–123.
[4] W. Neupert, Protein import into mitochondria, Annu. Rev.
Biochem. 66 (1997) 863–917.
[5] A. Boveris, N. Oshino, B. Chance, The cellular production of
hydrogen peroxide, Biochem. J. 128 (1972) 617–630.
[6] R. van Belzen, A.B. Kotlyar, N. Moon, W.R. Dunham, S.P.
Albracht, The iron–sulfur clusters 2 and ubisemiquinone radicals
of NADH: ubiquinone oxidoreductase are involved in energy
coupling in submitochondrial particles, Biochemistry 36 (1997)
886–893.
[7] N. Zamzami, P. Marchetti, M. Castedo, D. Decaudin, A. Macho,
T. Hirsch, S.A. Susin, P.X. Petit, B. Mignotte, G. Kroemer,
Sequential reduction of mitochondrial transmembrane potential
and generation of reactive oxygen species in early programmed
cell death, J. Exp. Med. 182 (1995) 367–377.
[8] N. Zamzami, P. Marchetti, M. Castedo, C. Zanin, J.L. Vayssiere,
P.X. Petit, G. Kroemer, Reduction in mitochondrial potential
constitutes an early irreversible step of programmed lymphocyte
death in vivo, J. Exp. Med. 181 (1995) 1661–1672.
[9] J. Yang, X. Liu, K. Bhalla, C.N. Kim, A.M. Ibrado, J. Cai, T.I.
Peng, D.P. Jones, X. Wang, Prevention of apoptosis by Bcl-2:
release of cytochrome c from mitochondria blocked, Science
275 (1997) 1129–1132.
[10] S.A. Susin,H.K.Lorenzo,N.Zamzami, I.Marzo,B.E. Snow,G.M.
Brothers, J. Mangion, E. Jacotot, P. Costantini, M. Loeffler, N.
Larochette, D.R. Goodlett, R. Aebersold, D.P. Siderovski, J.M.
Penninger, G. Kroemer, Molecular characterization of mitochon-
drial apoptosis-inducing factor, Nature 397 (1999) 441–446.
[11] J.C. Goldstein, N.J. Waterhouse, P. Juin, G.I. Evan, D.R. Green,
The coordinate release of cytochrome c during apoptosis is rapid,
complete and kinetically invariant, Nat. Cell Biol. 2 (2000)
156–162.
[12] H. Stridh, M. Kimland, D.P. Jones, S. Orrenius, M.B. Hampton,
Cytochrome c release and caspase activation in hydrogen
peroxide- and tributyltin-induced apoptosis, FEBS Lett. 429
(1998) 351–355.
[13] T. Ozawa, Mitochondrial genome mutation in cell death and
aging, J. Bioenerg. Biomembr. 31 (1999) 377–390.
[14] C.B. Wollheim, b-Cell mitochondria in the regulation of insulinsecretion: a new culprit in type II diabetes, Diabetologia 43 (2000)
265–277.
[15] D.C. Wallace, M.T. Lott, J.M. Shoffner, M.D. Brown, Diseases
resulting from mitochondrial DNA point mutations, J. Inherit.
Metab. Dis. 15 (1992) 472–479.
[16] S. DiMauro, C.T. Moraes, Mitochondrial encephalomyopathies,
Arch. Neurol. 50 (1993) 1197–1208.
[17] E.A. Schon, M. Hirano, S. DiMauro, Mitochondrial encephal-
omyopathies: clinical and molecular analysis, J. Bioenerg. Bio-
membr. 26 (1994) 291–299.
[18] D.G. Nicholls, M.W. Ward, Mitochondrial membrane potential
and neuronal glutamate excitotoxicity: mortality and millivolts,
Trends Neurosci. 23 (2000) 166–174.
[19] B. Ehrenberg, V. Montana, M.D. Wei, J.P. Wuskell, L.M. Loew,
Membrane potential can be determined in individual cells from
the nernstian distribution of cationic dyes, Biophys. J. 53 (1988)
785–794.
[20] J.C. Smith, Potential-sensitive molecular probes in membranes of
bioenergetic relevance, Biochim. Biophys. Acta 1016 (1990) 1–28.
[21] R.C. Scaduto Jr., L.W. Grotyohann, Measurement of mitochon-
drial membrane potential using fluorescent rhodamine derivatives,
Biophys. J. 76 (1999) 469–477.
A. Wong, G.A. Cortopassi / Biochemical and Biophysical Research Communications 298 (2002) 750–754 753

[22] M.W. Ward, A.C. Rego, B.G. Frenguelli, D.G. Nicholls,
Mitochondrial membrane potential and glutamate excitotoxicity
in cultured cerebellar granule cells, J. Neurosci. 20 (2000) 7208–
7219.
[23] J.A. Dykens, A.K. Stout, Assessment of mitochondrial membrane
potential in situ using single potentiometric dyes and a novel
fluorescence resonance energy transfer technique, Methods Cell
Biol. 65 (2001) 285–309.
[24] A. Wong, L. Cavelier, H.E. Collins-Schramm, M.F. Seldin, M.
McGrogan, M.L. Savontaus, G.A. Cortopassi, Differentiation-
specific effects of LHON mutations introduced into neuronal NT2
cells, Hum. Mol. Genet. 11 (2002) 431–438.
[25] I.D. Scott, D.G. Nicholls, Energy transduction in intact
synaptosomes. Influence of plasma-membrane depolarization
on the respiration and membrane potential of internal
mitochondria determined in situ, Biochem. J. 186 (1980) 21–
33.
[26] J.B. Hoek, D.G. Nicholls, J.R. Williamson, Determination of the
mitochondrial proton motive force in isolated hepatocytes, J. Biol.
Chem. 255 (1980) 1458–1464.
[27] T.B. Sherer, P.A. Trimmer, J.K. Parks, J.B. Tuttle, Mitochondrial
DNA-depleted neuroblastoma (Rho degrees) cells exhibit altered
calcium signaling, Biochim. Biophys. Acta 1496 (2000) 341–355.
[28] K. Buchet, C. Godinot, Functional F1-ATPase essential in
maintaining growth and membrane potential of human mito-
chondrial DNA-depleted rho degrees cells, J. Biol. Chem. 273
(1998) 22983–22989.
[29] D.L. Farkas, M.D. Wei, P. Febbroriello, J.H. Carson, L.M.
Loew, Simultaneous imaging of cell and mitochondrial membrane
potentials, Biophys. J. 56 (1989) 1053–1069.
754 A. Wong, G.A. Cortopassi / Biochemical and Biophysical Research Communications 298 (2002) 750–754





























