Embed Size (px)
Citation preview

[CANCER RESEARCH56. 5484-5489. December I, 1996]
ABSTRACT
Fifty-four grade 1 tubular breast cancers and nine non-comedo ductal
carcinoma in situ samples have been analyzed for loss of heterozygosityusing a series of microsatellite markers Markers mapping to regions of
the genome for which loss of heterozygosity has been documented previously in higher-grade breast cancers were selected for this analysis. Even
within this group of good prognostic early breast cancers, genetic events
are very common. The highest levels of loss were observed for D3S1300,which maps within an Intron of the recently identified FHJT gene. Highlevels of loss were also observed within the ATM gene. These findings
indicate that allele loss at FHIT andATM may be an important early eventin the development of sporadic breast cancer.
INTRODUCTION
Despite the fact that breast cancer is a common disease afflicting 1in 10 women in the United Kingdom and 1 in 9 women in the UnitedStates, little is known about its etiology. Recently, significant progresshas been achieved in identifying some of the genes involved in theinherited forms of breast cancer with the cloning of BRCAJ onchromosome 17q (1) and BRCA2 on chromosome 13q (2). Mutationsin BRCAJ and BRCA2 may account for 90% of inherited breastcancers. Two other genes that may play a role in both inherited andsporadic forms of breast cancer are p53 (3) and ATM, the gene causingataxia telangiectasia (4). Germ-line mutations in the p53 gene producethe Li Fraumeni cancer susceptibility syndrome, which includes familial predisposition to breast cancer. Also, it has been reported thatfemales who are heterozygous for mutations in ATM are at increasedrisk of developing breast cancer (5) and that such heterozygousfemales may account for 20% of sporadic breast cancer patients.
Like most solid tumors, breast cancer is thought to arise through asequence of events involving the activation of oncogenes and theinactivation or loss of tumor suppressor genes by point mutation,deletion, or even whole chromosome loss. The loss of genetic materialin tumors is a common finding, and this can be exploited to identifygenomic intervals that may contain tumor suppressor genes by usinga LOH2 assay. LOH analysis has been used extensively in studies ofmost types of cancer, and it forms the basis of subsequent studies toidentify and clone the genes that are involved in tumor development.
DNA markers distributed throughout the entire genome have beenused in LOH studies on a range of breast cancer specimens to identifythe genes that are involved in the sporadic forms of the disease.Specific sites showing frequent LOH include lp, lq, 3p, 6q, 7q, 8p,11p. 1lq, I3q, l6q, 17p, l7q, and 18q (6—25).A range of tumor typesand grades has been analyzed in these studies, but for the most part,higher-grade invasive breast cancers have been examined. High-grade
Received 6/24/96; accepted 9/30/96.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
I To whom requests for reprints should be addressed, at Department of Genetics,Queen's Medical Centre, University of Nottingham. Nottingham NG7 2UH, UnitedKingdom. Phone: 44-0-I 15-924-9924, extension 42322; Fax: 44-0-I 15-970-9906; E-mail:[email protected].
2 The abbreviations used are: LOH, loss of heterozygosity; DCIS, ductal carcinoma(s)
in situ.
tumors represent more aggressive forms of the disease, which aremore likely to have sustained random genetic losses due to genomeinstability. It seems likely that in low-grade tumors, where cells arebetter differentiated, random losses will occur less frequently. However, several recent LOH studies on DCIS have reported that significant genetic change is already present in these noninvasive tumors(1 1, 22—25).For example Radford et al. (1 1) analyzed 61 samples ofDCIS for LOH on 39 autosomal arms and demonstrated the highestlevelsof losson8p,13q,l6q, andl7p. Similarly,Strattonetal. (22)examined DCIS specimens for LOH at selected loci that show loss ininvasive breast cancers. These authors report that losses are alreadypresent on l6q and l7p in in situ disease. In both reports, however, themajority ofthe tumors studied, although noninvasive, were high-gradecomedo type. With the introduction of mammographic screening,non-comedo DCIS and grade 1 invasive tumors are detected moreoften. This latter group, which represents around 2% of palpablelesions, accounts for 15% of tumors detected in some mammographicscreening programs (26).
To investigate early genetic change, we have performed LOHstudies in low-grade tubular cancers and non-comedo DCIS usinghighly informative microsatellite markers in a PCR-based assay. Wehave chosen markers on 10 chromosome arms, 9 of which have shownhigh levels of LOH in previous studies (1 1, 14, 15, 18, 19). Thisanalysis reveals that significant LOH is present even in these lowgrade tumors. Some of the highest levels of LOH have been found formarkers D11S1778, located within the ATM gene (4), and D3S1300,which is situated within an intron of the recently identified FHJT gene(27).
MATERIALS AND METHODS
Tumor Samples and Tumor Histology. Nine cases of non-comedoDCISand 54 cases of grade 1 invasive tubular cancer were identified from theNottingham Primary Breast Cancer Service. The tumor specimens and their
corresponding normal tissue were formalin-fixed and paraffin-embedded. Theparaffin blocks were selected to include a hemisphere of the bisected tumor,and a section would typically contain 70% tumor material. The histologicalclassification of tumor characteristics was performed according to established
criteria (26, 28—30). The invasive cases were all previously designated as
grade 1 carcinomas of pure tubular type or tubular/mixed type with a highproportion of tubules. On review, one case, after deeper sectioning, wasbiphasic with a focus of grade 2 invasive carcinoma. The remaining 53 caseswere of grade 1 histology: 37 cases were of pure tubular type; 12 weretubular/mixed type with over 70% tubular differentiation; 3 were tubular/
lobular type; and I was designated of no specific type with 100% tubular
differentiation but pleomorphic nuclear features. Of the nine DCIS cases, one
showed extensive florid hyperplasia on deeper sectioning; the other eight werepure non-comedo DCIS with cribriform and micropapillary subtypes. Ap
proval for this study was obtained from the Ethics Committee of NottinghamCity Hospital National Health Service Trust (Nottingham, United Kingdom),
and informed consent was given by participating patients.DNA Extractions. [email protected] were processedfor each specimen.
Tissue sections were deparaffinized by three 1-h incubations in xylene, each
followed by washing in 100%ethanol. Cell pellets were dried and resuspendedin 500 p.1 of STE (pH 8.0; 10 mM Tris, 1 mt@iEDTA, and 100 mM NaC1) to
which 50 gd of 10% SDS and 16 pAproteinase K (20 mg/ml) were added.
5484
High Levels of Allele Loss at the FHIT and ATM Genes in Non-Comedo Ductal
Carcinoma in Situ and Grade I Tubular Invasive Breast Cancers
Somai Man, Ian 0. Ellis, Mark Sibbering, Roger W. Blarney, and J. David Brook'
Department of Surgery (S. M.. M. S., R. W. B.J. Centre for Medical Genetics [S. M., J. D. B.J, and Department of Histopathology (1. 0. El, City Hospital National Health ServiceTrust. Nottingham NG6 JPB, United Kingdom, and Department of Genetics, Queen ‘sMedical Centre, University of Nottingham, Nottingham NG7 2UH, United Kingdom (S. M.,J. D. B.)
on May 16, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

cancersAllelic
loss/Allelicloss/informativeinformativeChromosomeMicrosatellitecases
(%)cases(%)armmarkerDCIS―tubularcancers3pD3S12861/8
(12.5)2/28(7.1)3pD3S13004/9(44.4)16/36(44.4)6pD6S2711/4
(25.0)2/17(11.8)6pD6S295—1/14(7.1)7qD7S530—1/20(5.0)7qD7S486—2/25
(8.0)llqD11S9232/5(40.0)13/33(39.4)llqD11S17783/10(30.0)14/42(33.3)l2pDI2SIOO2/7
(28.6)1 1/41(26.8)l2pD12S772/5(40.0)9/34(26.5)l3qD1351552/8
(25.0)14/30(46.6)13qD13S1593/9(33.3)16/42(38.1)l5qDI551201/4
(25.0)5/28(17.9)l6pD1654030/7
(0.0)6/49(12.2)16qD165415—3/10(30)16qD16S4022/7
(28.6)12/27(44.4)l7qD17S7831/9(11.1)10/48(20.8)17qD17S7913/7(42.8)8/43(17.9)l7qD1757852/5(40.0)5/27(18.5)a
, reactions were not performed due to insufficient material.
ALLELE LOSS AT THE FlIT AND ATM GENES
Samples were incubated for 18 h, and two additional aliquots of proteinase Kwere added, each followed by a 6-h incubation. Two phenol-chloroform (1:1)extractions and one chloroform extraction were performed, and DNA was
precipitated using 2 volumes of 100% ethanol and 0.1 volume of 3 Msodiumacetate (pH 5.2) with tubes being placed at —20°Covernight. After a 30-mmspin, DNA pellets were dried and resuspended in 100 @lofdistilled water. Test
PCR assays were performed on each sample and, if necessary, further purification was performed using a Geneclean Kit II (BlO 101, Inc.). Purified pelletswere resuspended in 100 pJ of distilled water.
PCRConditions.PCRassayswerecarriedoutusing1—4ngof DNAinatotal volume of 13 @l.One primer of the pair was end-labeled using T4polynucleotide kinase (NBL Gene Sciences) and 3000 Ci/mmol y-ATP (Am
ersham). Reactions contained 10 pmol of each primer, 25 mr@iof each deoxynucleotide triphosphate (Pharmacia Biotech), 50 m@i KC1, 10 m@i Tris-CI
(pH 8.4), 1.5 mM MgCl2, 0.01% gelatin, and 1 unit of Taq polymerase(Boerhinger). Reactions were overlaid with mineral oil (Sigma) and placed in
an M. J. Research, Inc. thermal cycler once the heating block had reached80°C.PCR conditions were optimized for each primer pair but typicallyconsisted of 32 two-step cycles of 94°C for 40 s followed by annealing and
extension at 50°Cfor 30 s. The optimal number of PCR cycles was determinedempirically using DNA extracted from fixed paraffin-embedded tissue andDNA extracted from lymphocytes. Thus, it was possible to establish that theamplification of products lay within the linear phase of the PCR and that allelicratios were maintained. Amplified products were denatured at 80°Cfor 3 mmin loading dye (98% formamide, 10 mM EDTA, 0.025% bromphenol blue, and
0.025% xylene cyanol) and electrophoresed through an 8% denaturing polyacrylamide gel at 60 W for 3—6h. Gels were fixed, dried (at 80°Cfor 40 mm),and exposed to Phosphorlmager screens (Molecular Dynamics) for 12—48hbefore being scanned and quantified.
Microsatellites. Oligonucleotide primers corresponding to various mmcrosatellites were obtained from Research Genetics, Inc. The following loci were
analyzed: on 3p, D3S1286 (3p24.2), D3S1300 (3pl4.2), D3S1313 (3pl4.3),and D3S1287(3p14. 1-14.2);on 6p, D6S271 (6p2l .1-21.2) and D6S295 (6pl2);on 7q, D7S486 (7q3l) and D7S530 (7q3l-7q35);on Ilq,DJJS9OJ (11q14),
D11S1365 (llql3-llql4),D11S1354 (llql3-llql4),D11S1358 (llq2l-
1lq23.2),DJJSJ3IJ (11q21-l1q23.2),D11S920 (11q22-]1q23), D11S917
(11q21), D11S923 (1lq22-l1q23.3), DIJS900 (11q22-]1q23), D11S940
(11q22),D11S1818 (11q22-11q23),D11S1778 (11q22-]1q23),D11S1819
(11q22-11q23),D11S1817 (1lq22-11q23),and D11S927 (11q23);on l2p,
DJ2SJOO (l2pter—12p13.2)and D12S77 (l2pter—12pl3.2);on 13q,
(13q14.3-13q21.2)andD135159 (13q32);on 15q,D15S120 (l5qter);on 16p,
D16S403(l6pl2.3); on 16q,D16S402(16q24.2)andD16S415(16q12.1);andon llq,D17S783 (17q12-17q21),D17S791 (17q21.3),and D17S785 (17q24).
D16S403 was chosen as a negative control because previous studies have
demonstrated that there is little LOH in the corresponding region of I6p (11,
18, 19).Assessment of LOH. DNA quantitation was performed on a Molecular
Dynamics Phosphorlmager using ImageQuant software. LOH was measuredby comparing the relative intensity of alleles in the tumor DNA to the relativeintensity of alleles in the same individual's constitutional DNA. A differencein the allele ratios in excess of 20% was scored as LOH. Markers showing
LOH were repeated one or more times to confirm the result. Fisher's exact testwas used to test for concordant loss of alleles.
RESULTS
To assess the sensitivity and reliability of the PCR-based LOHassay, mixing experiments were performed using different proportionsof DNA extracted from the tissue of individuals known to be homozygous for different alleles at D6S295. These experiments, performedwith four replicates, indicate that the assay is sensitive and robust,consistently detecting losses of 10% or less (data not shown). Toensure that only genuine losses were scored, we chose our cutoff pointfor demonstration of LOH as a 20% difference in allele ratios in tumorDNA compared to normal tissue DNA. The median level of LOH wasaround 30%.
Table 1 shows the extent of LOH at each of the markers tested.More than 40% of DCIS demonstrate LOH at D3S]300, D11S923,
Table I Thefrequency ofWH at 19 loci in non-comedo DCIS and grade 1 tubular
D12S77, D17S791, and D17S785, and more than 40% of tubularcancers show LOH at D3S1300, D13S155, and D16S402. LOH atD11S1778 and D13S159 was observed in more than 30% of bothDCIS and tubular cancers. In addition, more than 30% of tubularcancers show LOH at D11S923 and D16S415. Statistical analysisrevealed that there was no significant difference in the extent of LOHfor DCIS and tubular cancers with each of the markers tested. MarkerD16S403 was used as a negative control in this study because therewas no previous evidence of LOH in breast cancer samples in theinterval to which it maps ( 11, 18, 19). To establish whether theobserved LOH at each of the other loci was significantly different tothe level of LOH at negative control D16S403, statistical analysis wasperformed on combined DCIS and tubular cancers using Fisher'sexact test, and the Bonferroni correction was applied. Significantdifferences were observed for D3S1300 (P = 0.00018), D11S923(P = 0.0019), D13S155 (P = 0.0009), D13S159 (P = 0.0014), andD16S402 (P = 0.0013). Fig. 1 shows representative examples of LOHatD6S271,D11S923,D]1S940,D13S155,D15S120,andDJ7S783.
To determine the total amount of genetic loss/tumor, the number ofinformative loci demonstrating LOH was summed for each sample.These data are shown in Table 2. The number of losses/tumor is quitevariable, with 3 tumors showing LOH at 7 or 8 of the 19 loci tested,and 6 tumors showing no losses at all. This was not simply a reflectionof the number of markers that were informative in each tumor becausetumors showing 0 or I loss were informative for 10 markers onaverage, compared to an average informativeness of 12.6 markers fortumors showing losses at 7 or 8 loci. The data set was tested forconcordant loss of markers in the tumor samples using Fisher' s exacttest. No statistically significant correlations were found after applyingthe Bonferroni correction. The tumors in this series were homogeneous with respect to their prognostic characteristics, and no relationships were found on statistical analysis between the level of LOH andtumor size, lymph node stage, presence of vascular invasion, age,menopausal status, and estrogen receptor status. There were very fewsubsequent clinical events with the follow-up range of 7—187months(the mean follow-up was 56 months, and median follow-up was 46months). There were three local recurrences, one local and distantrecurrence, and one death, with the latter events affecting the samepatient. This patient had a grade I carcinoma of tubular/lobular typethat showed a high level of LOH (7 of 13 informative markers). The
5485
on May 16, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
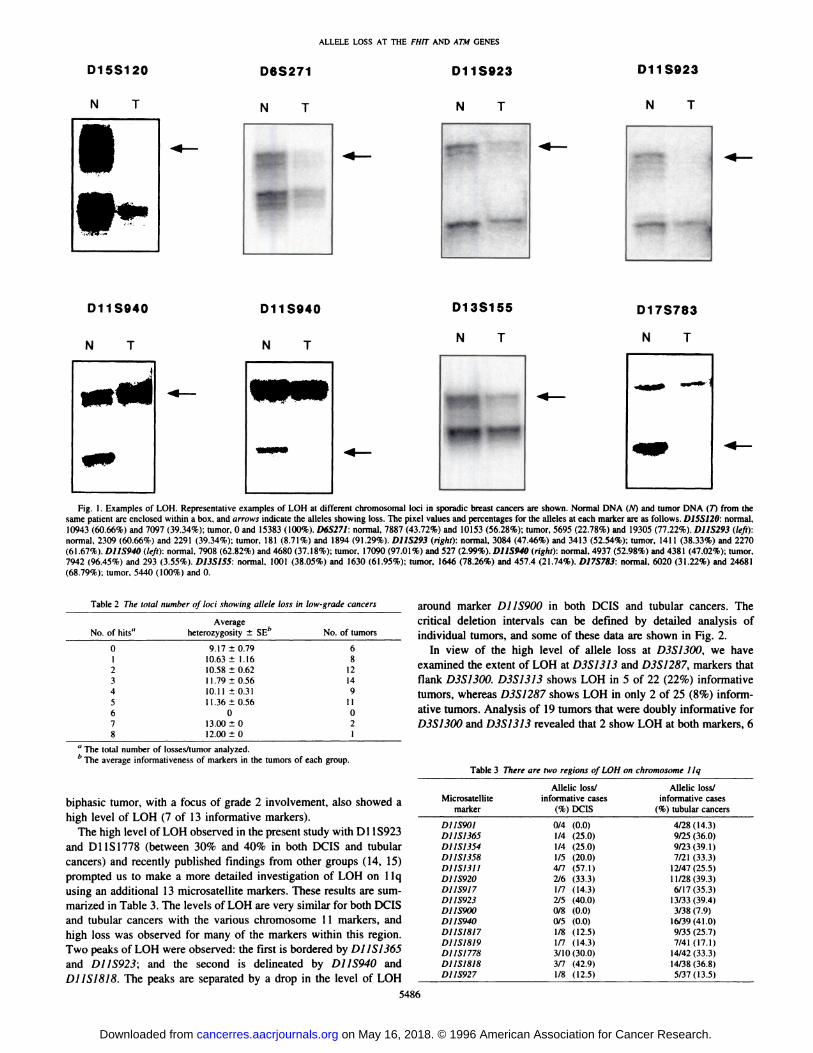
Table 2 The totalnumber of loci showing allele loss an low-gradecancersNo.
of hits―Average heterozygosity ±SE―No. oftumors09.17±0.796I10.63
±1.168210.58±0.6212311.79±0.5614410.11±0.319511.36±0.5611600713.00±02812.00±01
Table 3There are two regions of LOH onchromosomeIIqAllelic
loss/Allelicloss/Microsatelliteinformativecasesinformativecasesmarker(%)
DCIS(%) tubularcancersDJJS9OI0/4
(0.0)4/28(14.3)D11S13651/4(25.0)9/25(36.0)D11S13541/4(25.0)9/23(39.1)D11S13581/5(20.0)7/21(33.3)DJISI3II4(7(57.1)12/47(25.5)D11S9202/6(33.3)1 1/28(39.3)D11S9171/7(14.3)6/17(35.3)D11S9232/5(40.0)13/33(39.4)DJIS9tYi0/8(0.0)3/38(7.9)D11S9400/5
(0.0)16/39(41.0)D11S18171/8(12.5)9/35(25.7)D11S18191/7(14.3)7/41(17.1)D11S17783/10(30.0)14/42
(33.3)D11S18183/7(42.9)14/38(36.8)D11S9271/8(12.5)5/37 (13.5)
ALLELE LOSS AT THE FlIT AND ATM GENES
N T
‘a
D13S155 D17S783
N T N T
Fig. I . Examples of LOH. Representative examples of LOH at different chromosomal loci in sporadic breast cancers are shown. Normal DNA (N) and tumor DNA (7) from thesame patient are enclosed within a box, and arrows indicate the alleles showing loss. The pixel values and percentages for the alleles at each marker are as follows. D1SS12O: normal,10943 (60.66%) and 7097 (39.34%); tumor, 0 and 15383 (100%). D6S271: normal, 7887 (43.72%) and 10153 (56.28%); tumor, 5695 (22.78%) and 19305 (77.22%). D11S293 (left):normal, 2309 (60.66%) and 2291 (39.34%); tumor, 181 (8.71%) and 1894 (91.29%). D11S293 (right): normal, 3084 (47.46%) and 3413 (52.54%); tumor, 141 1 (38.33%) and 2270(61.67%). D11S940 (left): normal, 7908 (62.82%) and 4680 (37.18%); tumor, 17090 (97.01%) and 527 (2.99%). D11S940 (right): normal, 4937 (52.98%) and 4381 (47.02%); tumor.7942 (96.45%) and 293 (3.55%). D13S155: normal, 1001 (38.05%) and 1630 (61 .95%); tumor, 1646 (78.26%) and 457.4 (21.74%). D17S783: normal, 6020 (31.22%) and 24681(68.79%): tumor, 5440 (100%) and 0.
around marker DJJS900 in both DCIS and tubular cancers. Thecritical deletion intervals can be defined by detailed analysis ofindividual tumors, and some of these data are shown in Fig. 2.
In view of the high level of allele loss at D3S1300, we haveexamined the extent of LOH at D3S1313 and D3S1287, markers thatflank D3S1300. D3S1313 shows LOH in 5 of 22 (22%) informativetumors, whereas D3S1287 shows LOH in only 2 of 25 (8%) informative tumors. Analysis of 19 tumors that were doubly informative forD3S1300 and D3S1313 revealed that 2 show LOH at both markers, 6
a The total number of losses/tumor analyzed.
b The average informativeness of markers in the tumors of each group.
biphasic tumor, with a focus of grade 2 involvement, also showed ahigh level of LOH (7 of 13 informative markers).
The high level of LOH observed in the present study with Dl 15923and Dl 15 1778 (between 30% and 40% in both DCIS and tubularcancers) and recently published findings from other groups (14, 15)prompted us to make a more detailed investigation of LOH on 1lqusing an additional 13 microsatellite markers. These results are summarized in Table 3. The levels of LOH are very similar for both DCISand tubular cancers with the various chromosome 11 markers, andhigh loss was observed for many of the markers within this region.Two peaks of LOH were observed: the first is bordered by D11S1365and D11S923; and the second is delineated by D11S940 andDl 1S1818. The peaks are separated by a drop in the level of LOH
5486
Dl 5S1 20
N T
D11S923
N T
D11S923D6S271
N T
I,
D11S940
N T
D1IS94O
N T
@-
on May 16, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

ALLELE LOSS AT THE FlIT AND ATM GENES
I—
iI
12
13.113.213.313.413.5
14.1
14.214.32122.122.222.3
23.123.2
23.3
24
25
TUMOR
6 60 66 96 72 58 62 74 10 86 2 14 16 44 94
Fig. 2. LOH on chromosome 1lq. A schematicrepresentation of LOH on chromosome 1lq incases of non-comedo DCIS and grade 1 tubularbreast cancers is shown. Fifteen microsatellitemarkers were analyzed, and deletion intervals areapparent at the ATM gene at 11q23 and, moreproximally, at 1lq13.5-l4.l.
MARKER
, D11S9O1/ D11S1365
D11S1354D11S1358D11S1311D11S920D11S917D11S923D11S900D11S940
D11S1817D11S1819D11S1778
%‘%,s.. D11S1818
.,,‘ D11S927
REGION OFLOH
show no LOH at either marker, and 10 show LOH at D3S1300 but notD3S1313, whereas only 1 tumor shows LOH at D3S1313 but notD3S1300. Seventeen tumors were doubly informative for D3S1300and D3S1287. Both of the tumors showing LOH at D3S1287 had lostD3S1300, whereas four tumors show LOH at D3S1300 but notD3S1287. Eleven tumors showed no LOH at either marker. Takentogether, these data indicate that in this region of chromosome 3p,LOH peaks at D3S1300.
DISCUSSION
To examine early genetic changes in breast cancer, we have performed LOH studies on a series of non-comedo DCIS and grade 1tubular cancers using markers on 10 chromosome arms, 9 of whichhave shown significant levels of loss in previous studies on highergrade tumors (6—25).We have found significant levels of LOH inthese low-grade cancers at frequencies comparable to previous reportson invasive and noninvasive breast cancers (6—25).The majority oftumors analyzed had accrued multiple losses, with 49 of 63 low-gradecancers showing losses at 2 or more loci. The high frequency of losseven in these low-grade tumors, which on histological grounds wouldbe predicted to have a good prognosis, is surprising. In some of thetumors, multiple losses were found, with three of the tumors showinglosses at seven or more of the loci tested.
Significantly high levels ofLOH were recorded at 3p, 1lq, l3q, andl6q, and the greatest loss was found with D3S1300, D11S923,D13S155, D13S159, and D16S402. The high frequency of allele lossat these loci in low-grade tumors is consistent with studies of moreadvanced tumor grades, indicating that these regions harbor genesinvolved in the development of sporadic breast cancer. D3S1300shows the highest level of LOH, with losses in 44.4% of both DCISand tubular cancers. LOH peaks at D3S1300 as flanking markersD3S1313 and D3S1287 show lower levels of LOH than D3S1300 intumors that are informative for both markers. Chen et a!. (30) suggested that two regions of 3p contained genes involved in the development of breast cancer. These are 3pl4.l (to which D3S1300 maps)
and 3p24-26 (to which D3S1287 maps). In view of the low level ofLOH at D3S1287, our data do not support the involvement of genesat 3p24.2 in early breast cancer. Recently, the fragile histidine triadgene (FHIT) has been identified at 3pl4 and shown to span a renalcarcinoma-associated translocation breakpoint (27). Abnormalities inFHIT have also been identified in digestive tract cancers (27) and lungcancer (32). In addition, the FHIT region is homozygously deleted insome breast carcinoma cell lines (33). It is of considerable interest tonote, therefore, that D3S1300 maps within intron 5 of the FHIT gene.Hence, the LOH observed at D3S1300 in our tumor set may reflect theimportance of FH!T in the development of sporadic breast cancer.Clearly, mutational analysis of FlIT in the tumor samples reportedhere and elsewhere will be of considerable value.
The initial analysis with markers at D11S923 and D11S1778 mdicated a high level of LOH on 1lq, in agreement with earlier reports inmore advanced tumors (13—15).Because this interval contains theATM gene, which has been implicated in the development of breastcancer (5), we performed a detailed analysis using an additional 13microsatellite markers that map between DJJS9OJ and D11S927.LOH is found in two peaks in the llql3.5-23.1 interval. The firstcommonly deleted region is bordered by D11S1365 and D11S923.The second interval, bordered by D11S940 and D11S1818, encompasses ATM. These two intervals are separated by DJJS900, whichshows loss in only 3 of 42 informative tumors. High levels of LOHhave been reported previously around llql3.5-14.1 (34, 35) and alsoin the ATM region in invasive breast cancers (14, 15). Ataxia telangiectasia heterozygotes show a predisposition to many types of cancerand particularly breast cancer, for which there is a 5-fold increasedrisk (5). With an estimated gene frequency of 1—3%,as many as 20%of female breast cancer patients may be ataxia telangiectasia heterozygotes (36). However, recent analysis of the ATM gene in breast cancersamples showing LOH on 1lq failed to identify mutations (37). Ourdata and that reported elsewhere (15) indicate that there are more thantwo regions of LOH on llq. The identification of other genes involved in breast cancer from this interval is therefore a priority.
5487
CHROMOSOME llq
:o: NOLOll UNINFORMATIVE
on May 16, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

ALLELE LOSS AT THE FHI'T AND ATM GENES
High LOH (16 of 38, 42.1%) was found in tumors informative forD13S155, which maps to l3q14.l. Other studies based on invasiveand noninvasive breast cancers report losses ranging from 18 —48%inthis region (1 1, 18), and possible candidate genes here include theretinoblastoma gene (Rb!) and BRCA2. D16S402, which maps to16q23-24, was lost in 41 .2% (13 of 34) of informative tumors, similarto the levels of LOH reported previously for invasive breast cancers(18, 19). The largest individual study reported to date found 44% (120of 269) LOH at l6q24 (38), whereas two studies on DCIS identifiedslightly lower frequencies of loss: 28% ( 11 of 39; Ref. 22) and 25.0%(5 of 20; Ref. 11) in this region. LOH at 17q is well documented inDCIS and invasive breast cancers with levels ranging from 15.9—63.0% (1 1, 39). Although there is no significant difference betweenDCIS and tubular cancers in the average level of LOH at each of thethree 17q markers tested, the levels do vary considerably. For example, the levels of loss for D17S791 and D17S785 in DCIS were morethan double the levels ofloss in tubular cancers. Conversely, DI7S 783was lost almost twice as often from tubular cancers than from DCIS.The significance of this observation is unclear.
Six of the loci analyzed in this study (D3S1286 at 3p24, D6S295 at6pl2, D6S271 at 6p2l, D7S530 at 7q3l—q35,D7S486 at 7q3l, and
negative control D16S403 at l6pl2) show low levels of LOH. Highlevels of LOH have been described previously, however, in studies onregions around 3p24 and 7q3l. In the present study, 3 of 36 (8.3%)DCIS and tubular cancers informative for D3S1286 show LOH,compared with losses of 62% (12) and 39% (39) within 3p2 1-25 and45% within 3p24-26 (31) reported elsewhere. Furthermore, it has beenproposed that the 7q31 interval is the most commonly deleted regionin breast cancer, with 84% (22 of 26) of informative cases showingloss (10). This compares to only 8% (2 of 25) in the present study.This difference may reflect the fact that grade 2 and grade 3 tumorshave been analyzed in other studies (10), whereas only grade 1 tumorswere analyzed in the present study. Thus, this may indicate that genesat 3p24 and 7q31 are associated with tumor progression and metastasis rather than with the early initiating events of breast cancer.
Six of the tumors analyzed in our set show no losses at any of theloci tested. The significance of this is unclear, although it may indicatethat there are other loci, not tested in this study, that are important fortumor development. In addition to the loci selected for analysis in ourstudy, chromosome arms lp (6), lq (7), 8p (I 1), l7p (18—20,40), andI 8q (21 , 41) have been reported to harbor genes important in thedevelopment of breast cancer, and clearly, analysis of these and otherchromosomal regions will provide additional information on the extent of the genetic changes in our tumor set.
We have examined the correlation in our data set between patientsurvival and genetic loss. The types of tumors studied are recognizedto have an extremely good long-term prognosis, and it is not surprising that there were very few clinical events. The single case developing distant metastatic disease and dying of breast cancer did,however, have a high level of LOH, as did the case showing somegrade 2 histology, suggesting a possible relationship between prognosis and differentiation with level of LOH. In a large-scale study of457 invasive cancers (38), 3 loci (16q, lip, and llq) were examinedfor LOH, and 2 loci (erb2 and c-myc) were examined for geneamplification to correlate genetic change and clinicopathological features. The genetic changes identified were found more frequently inadvanced tumors, poorly differentiated tumors, and lymph nodepositive tumors (38). These authors suggest that a genetic diagnosismay provide a means of identifying the cohort of patients who are atan increased risk despite having a good prognosis on histopathologicalgrounds. The long-term follow-up of patients from our study shouldallow us to determine whether there is any correlation between geneticalteration and survival in our data set.
5488
REFERENCES
I. Mild, Y., Swensen, J., Shattuck-Eidens, D., Futreal, P. A., Harshman, K., Tavtigian,S., Liu, Q., Cochran, C., Bennett, L. M., Ding, W., Bell, R., Rosenthal, J., Hussey, C.,Tran. T., McClure, M., Frye, C.. Hattier. T., Phelps, R., Haugen-Strano, A., Katcher,H.,Yakumo,K.,Gholami,Z.,Shaffer,D.,Stone,S.,Bayer,S.,Wray,C.,Bodgen,R.,Dayananth,P.,Ward,J.,Tonin,P.,Narod,S.,Bnstow,P.K.,Norris,F.H.,Helvering,L., Morrison, P., Rosteck, P., Lai, M., Barren, J. C., Lewis, C., Neuhausen, S.,Cannon-Albright, L., Goldgar, D., Wiseman, R., Kamb, A., and Skolnick, M. H. Astrong candidate for the breast and ovarian cancer susceptibility gene BRCAI. Science(Washington DC), 266: 66-71, 1994.
2. Wooster, R., Bignell, G., Lancaster, J., Swift, S., Seal, S., Mangion, J., Collins, N.,Gregory, S., Gumbs, C., Micklem, G., Barfoot, R., Hamoudi, R., Patel, S., Rice, C.,Biggs, P., Hashim, Y., Smith, A., Connor, F., Arason, A., Gudmundsson, J., Ficenec,D.. Kelsell, D., Ford, D., Tonin, P., Bishop. D. T., Spurr, N. K., Ponder, B. A. J.,Eeles, R., Peto, J., Devilee, P., Cornelisse, C., Lynch, H., Narod, S., Lenoir, G.,Egilsson, V., Barkardottir, R. B., Easton, D. F., Bentley, D. R., Futreal, P. A.,Ashworth, A., and Stratton, M. R. ldentification of the breast cancer susceptibilitygene BRCA2. Nature (Lond.), 378: 789—792, 1995.
3. Lane, D. P., and Crawford, L. V. T antigen is bound to host protein in SV4O-transformed cells. Nature (Lond.), 278: 261—263,1979.
4. Savitsky, K., Bar-Shini, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L, Tagle. D. A.,Smith. S., Uziel, T., Sfez, S., Ashkenazi, M., Pecker, I., Frydman, M., Hansik, R.,Patanjali,S. R., Simmons,A., Clines, G. A., Sartiel, A., Gaul, R. A., Chessa, L., Sanal,0., Lavin, M. F., Jaspers, N. G. J., Taylor, A. M. R., Men, C. F., Mild, T., Weissman,S. M., Lovett. M., Collins, F. S., and Slsiloh, Y. A single ataxia telangiectasia gene witha product similar to P1-3 kinase. Science (Washington I)C), 268: 1749—1759.1995.
5. Swift, M., Reitnauer, P. J., Morrell, D., and Chase, C. L. Breast and other cancers infamilies with ataxia telangiectasia. N. EngI. J. Med., 316: 1289—1294,1987.
6. Genuardi, M., Tsihira, H., Anderson. D. E., and Saunders, G. F. Distal deletion ofchromosome lp in ductal carcinoma of the breast. Am. J. Hum. Genet., 45: 73—82,1989.
7. Chen, L-C., Dollbaum, C., and Smith, H. S. Loss of heterozygosity on chromosomelq in human breast cancer. Proc. Nail. Acad. Sci. USA, 86: 7204—7207, 1989.
8. Ali, I. U., Lidereau, R., and Callahan, R. Presence oftwo members ofc-erbA receptorgene family (c-erbA@and c-erbA2) in smallest region of somatic homozygosity onchromosome 3p2l—p25in human breast carcinoma. J. Natl. Cancer Inst., 81: 1815—1820, 1989.
9. Devilee, P., van Vliet, M., van Sloun, P., Kuipers-Dijkshoorn, N., Hermans, J.,Pearson, P. L., and Cornelisse, C. J. Allelotype of human breast carcinoma: a secondmajor site for loss of heterozygosity is on chromosome 6q. Oncogene, 6: 1705—1711,1991.
10. Zenldusen, J. C., Bièche,I., Lidereau, R., and Conti, J. C. (C-A)n microsatellite repeatD7S522 is the most commonly deleted region in human primary breast cancer. Proc.Natl. Acad. Sci. USA, 91: 12155—12158, 1994.
11. Redford, D. M., Fair, K. L., Phillips, N. J., Riner, J. H.. Steinbrueck, T., Holt, M. S.,and Donis-Keller, H. Allelotyping of ductal carcinoma in situ of the breast: deletionof loci on 8p, 13q. 16q, lip, and 17q. Cancer Ret., 55: 3399—3405,1995.
12. Devilee, P., van den Broek, M., Kuipers-Dijkshoorn, N.. Kolluri, R., Khan, P. M.,Pearson, P. L., and Cornelisse, C. J. At least four different chromosomal regions areinvolved on loss of heterozygosity in human breast carcinoma. Genomics, 5: 554—560, 1989.
13. Ali, I. U.. Lidereau, R., Theillet, C., and Callahan, R. Reduction to homozygosity ofgenes on chromosome I I in human breast neoplasia. Science (Washington DC), 238:185—188,1987.
14. Caner, S. L., Negrini, M., Baffa, R., Gillum, D. R., Rosenberg, A. L., Schwartz, G. F.,and Croce, C. M. Loss of heterozygosity at 11q22—q23in breast cancer. Cancer Ret.,54: 6270—6274, 1994.
15. Negrini, M., Ratio, D., Hampton. G. M., Sabbioni, S., Rattan, S., Carter, S. L.,Rosenberg, A. L., Schwartz, G. F., Shiloh, Y., Cavenee, W. K., and Croce, C. M.Definition and refinement of chromosome I I regions of loss of heterozygosity inbreast cancer identification of a new region at 11q23.3.Cancer Ret., 55: 3093—3007,I995.
16. Lundberg, C., Skoog, L., Cavenee, W. K., and NordenskjOld, M. Loss of heterozygosity in human ductal breast tumors indicates a recessive mutation on chromosome13. Proc. Nail. Acad. Sci. USA, 84: 2372—2376, 1987.
17. Wooster, R., Neuhausen, S. L., Mangion, J., Quirk, Y., Ford, D., Collins, N., Nguyen,K.,Seal,S.,Tran,T., Averill,D.,Fields,P.,Marshall,G., Narod,S., Lenoir,G. M.,Lynch. H., Feunteun, J., Devilee, P., Cornelisse, C., Menko, F., Daly, P., Ormiston,W., Mcmanus, R., Pye, C., Lewis, C., Cannon-Albright, L., Peto, J., Ponder, B. A. J.,Skolnick, M. H., Easton, D., Goldgar, D., and Stratton, M. Localization of a breastcancer susceptibility gene, BRCA2, to chromosome l3ql2—l2. Science (WashingtonDC), 265: 2088—2090, 1994.
18. Sato, T., Tanigami, A., Yamakawa, K., Akiyama, F., Kasumi, F., Sakamoto, G., andNakamura, Y. Allelotype of breast cancer: cumulative allele losses promote tumorprogression in primary breast cancer. Cancer Res., 50: 7184—7189, 1990.
19. Sato, T., Akiyama, F., Sakamoto, G., Kasumi, F., and Nakamura, Y. Accumulation ofgenetic alterations and progression in primary breast cancer. Cancer Ret., 51: 5794—5799, 1991.
20. Mackay. J., Steel, C. M., Elder, P. A., Forrest, A. P. M., and Evans, H. J. Allele losson the shortarmof chromosome17in breastcancers.Lancet,2: 1384—1385,1988.
21. Cropp, C. S., Lidereau, R., Campbell, G., Champene, M. H.. and Callahan, R. Lossof heterozygosity on chromosomes I7 and 18 in breast carcinoma: two additionalregions identified. Proc. Natl. Acad. Sci. USA, 87: 7737—7741, 1990.
22. Stratton, M. R., Collins, N., Lakhani, S. R., and Sloane, J. P. Loss of heterozygosityin ducts] carcinoma in situ of the breast. J. Pathol., 175: 195—201,1995.
on May 16, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

ALLELE LOSS AT THE FHIT AND ATM GENES
23. Murphy, D. S., Hoare, S. F., Going, J. J., Mallon, E. E. A., George, W. D., Kaye,S. B., Brown, R., Black, D., and Keith, W. N. Characterization of extensive geneticalterations in ductal carcinoma in situ by fluorescence in situ hybridization andmolecular analysis. J. Nail. Cancer Inst., 87: 1694—1704, 1995.
24. Aldaz.C. M., Chen,T., Sahin,A., Cunningham,J., and Bondy,M. Comparativeallelotype of in situ and invasive human breast cancer high frequency of microsatellite instability in lobular breast carcinomas. Cancer Ret., 55: 3976—3981, 1995.
25. Radford,D. M.,Phillips,N.J., Fair,K. L, Ritter,J. H.,Holt,M.,andDonis-Keller,H.Alleliclost andprogressionof breastcancer.CancerRet..55: 5180—5183,1995.
26. Ellis,I. 0., Galea,M.H.,Locker,A.,Roebuck,E.J., Elston,C. W.,Blamey.R.W.,and Wilson, A. R. M. Early experience in breast cancer screening: emphasis ondevelopment of protocols for triple assessment. The Breast. 2: 148—153,1993.
27. Ohta, M., Inoue, H., Cotticelli, M. G., Kastury, K., Baffa, R., Palazzo, J., Siprashvili,z.,Mori,M.,McCue,P.,Druck,T.,Croce,C.M.,andHuebner,K.TheFHJTgene,spanning the chromosome 3pl4.2 fragile site and renal carcinoma associated t(3;8)breakpoint, is abnormal in digestive tract cancers. Cell, 84: 587-597, 1996.
28. Elston, C. W., and Ellis, I. 0. Pathological prognostic factors in breast cancer. I. Thevalue of histological grade in breast cancer: experience from a large study withlong-term follow-up. Histopathol. (Oxford), 19: 403—410, 1991.
29. Ellis,I. 0., Galea,M.,Broughton,N.,Locker,A.,Blamey,R.W.,andElston,C.W.Pathological prognostic factors in breast cancer. II. Histological type. Relationshipwith survival in a large study with long-term follow-up. Histopathol. (Oxford), 20:479—489,1992.
30. Royal College of Pathologists Working Group. Pathology reporting in breast cancerscreening. J. Clin. Pathol. (Lond.), 44: 710—725, 1991.
31. Chen,L-C.,Matsumura,K., Deng,0.. Kurisu,W., Ljung,B-M.,Lerman,M. I.,Waldman,F. M.,andSmith,H. S. Deletionof twoseparateregionsonchromosome3p in breast cancers. Cancer Ret., 54: 3021—3024,1994.
32. Sozzi, 0., Veronese, M. L., Negrini, M., Baffa, R., Cotticelli, M. G., Inoue, H.,Tornielli, S.. Pilotti, S., Dc Gregorio, L., Pastorino, U., Pierotti, M. A., Ohta, M.,Huebner,K.,andCroce,C. M.TheFlIT geneat 3pl4.2is abnormalin lungcancer.Cell,85: 17—26,1996.
33. Kastury, K., Baffa, R.. Druck, T., Ohta, M., Cotticelli, M. C.. Inouc, H.. Negrini. M.,Rugge. M., Huang, D., Croce, C. M., Palazzo, J., and Huebner, K. Potential gastrointestinal tumor suppressor locus at the 3pl4.2 FRAB3 site identified by homozygousdeletiont in tumor cell lines. Cancer Res., 56: 978—983,1996.
34. Gudmundsson, J., Barkardottir. R. B., Eiriksdottir, G., Baldursson, T., Arason, A.,Egilsson, V.. and Ingvarsson, S. Loss of heterozygosity at chromosome I I in breastcancer: association of prognostic factors with genetic alterations. Br. J. Cancer, 72:696—701, 1995.
35. Zhuang, Z., Merino, M. J., Chuaqui, R., Liofta, L. A., and Emmett-Buck, M. R.Identical allelic loss on chromosome I 1q13 in microdissected in situ and invasivehuman breast cancer. Cancer Res., 55: 467—471, 1995.
36. Swift, M., Morrell, D., Massey, R. B., and Chase, C. L. Incidence ofcancer in 161families affected by ataxia telangiectasia. N. EngI. 3. Med., 325: 1831—1836,I991.
37. Vorechovsky, I., Ratio, D., Luo, L, Monaco, C., Hammarstrom, L., Webster, D. B.,Zaloudik, J., Barbanti-Brodano, G., James, J., Russo, G., Croce, C. M., and Negrini,M. The ATM gene susceptibility to breast cancer analysis of 38 breast tumors revealsno evidence for mutation. Cancer Res., 56: 2726—2732, 1996.
38. Harada, Y., Katagiri. T., Ito, I., Akiyama, F., Sakamoto, 0., Kasumi, F., Nakamura.Y.,andEmi,M.Geneticstudiesof 457breastcancers.Clinicopathologicparameterscompared with genetic alterations. Cancer (Phila.), 74: 2281—2286, 1994.
39. Futreal, P. A., Soderkvist, P., Marks, J. R., Iglehart, J. D., Cochran, C., Barrett, J. C.,and Wiseman, R. W. Detection of frequent allelic loss on proximal chromosome 17qin sporadic breast carcinoma using microsatellite length polymorphisms. Cancer Ret..52: 2624—2627,1992.
40. Deng, G., Chen, L-C., Schott, D. R., Thor, A., Bhargava, V., Ljung, B-M.. Chew, K.,and Smith, H. S. Loss of heterozygosity and p53 gene mutations in breast cancer.Cancer Res., 54: 499—505, 1994.
41 . Devilee, P., van den Broek, M., Mannens, M., Slater, R., Cornelisse, C. J.,Westerveld, A.. and Khan, P. M. Differences in patterns of allelic lots between 2common types of adult cancer, breast and colon carcinoma. and Wilma' tumour ofchildhood. Int. J. Cancer, 47: 817—821, 1991.
5489
on May 16, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

1996;56:5484-5489. Cancer Res Somai Man, Ian O. Ellis, Mark Sibbering, et al. Invasive Breast Cancers
and Grade I Tubularin SituNon-Comedo Ductal Carcinoma Genes inATM and FHITHigh Levels of Allele Loss at the
Updated version
http://cancerres.aacrjournals.org/content/56/23/5484
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/56/23/5484To request permission to re-use all or part of this article, use this link
on May 16, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
























![Working With Asbestos Guide 5484[1] Copia 2](https://img.pdfslide.us/doc/110x75/55cf9482550346f57ba27df8/working-with-asbestos-guide-54841-copia-2.jpg)




