Embed Size (px)
Citation preview

Hemolytic Anaemia
Dr Mere Kende MBBS, Mmed (Path), MAACB, MACTM,
MACRRM Division of Pathology, SMHS

Outline
• What is hemolysis/hemolytic Anaemia?
• Pathogenesis
• General Clinical Features of HA?
• What are the General Laboratory Features?
• Intrinsic vs extrinsic Causes
• Specific causes, diagnosis, investigation?
• References

Hemolysis
• Red Blood Cells disintegrate /break down releasing free hemoglobin+/- Anaemia.
• Hemolytic Anaemia –Anaemia caused by hemolysis
•

Pathogenesis
• Intrinsic Red cell Defects – Membrane Defect
– Hemoglobin Defects
– Enzyme defects
• Normal function depends on healthy membrane, normal hemoglobin, and normal enzyme system. Defective RBC , reduced survival due to rapid clearnance by RES
• Extrinsic Causes-Normal Red Cell – Immune mediated (IgG) – Transfusion – Infections – Splenomegaly

Clinical Features of Hemolytic Anaemia
• Signs of Anaemia & signs of hemolysis – Fatique/lethargy/Pale/dyspnoea/dark urine
– jaundice
• Features of initiating/primary cause – Fever/petechiae
– Medication hx
– Hx of prosthetic heart valve

General Laboratory Features
• Low Hemoglobin
• Reticulocytosis
• Reduced hemoglobin binding protein (haptoglobulin)
• Unconjugated/Indirect hyperbilirubinaemia
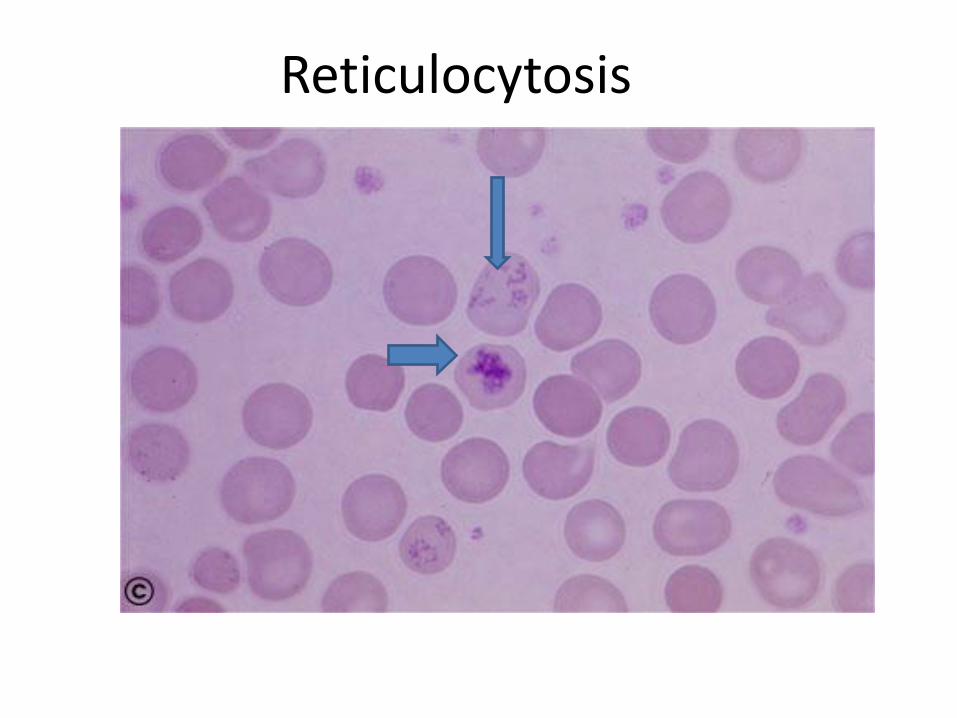
Reticulocytosis
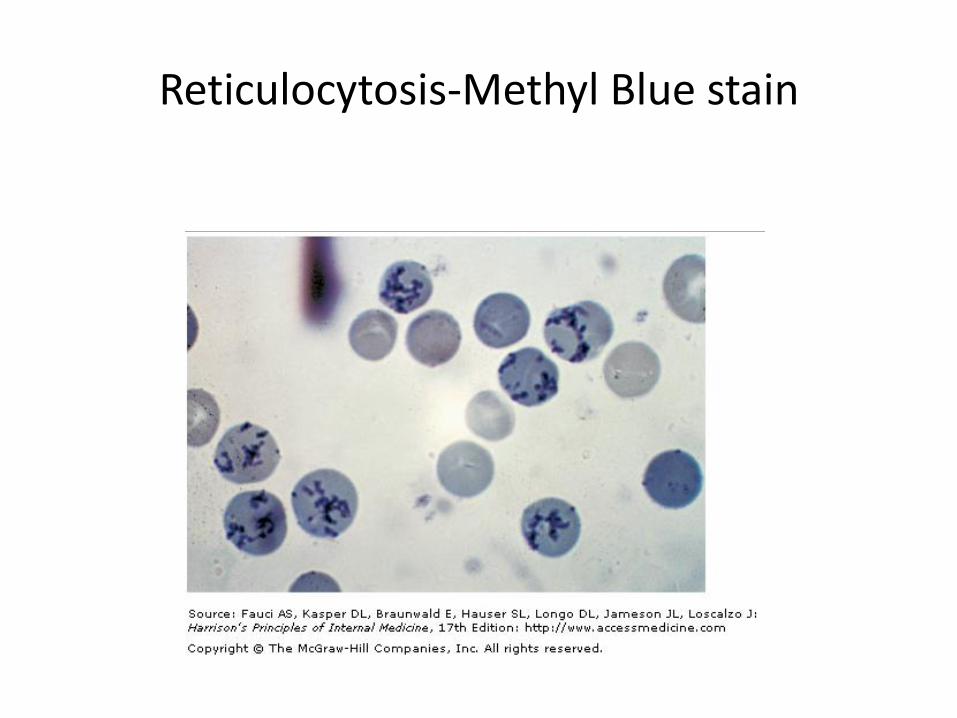
Reticulocytosis-Methyl Blue stain

Other Features
• Elevated LDH (x10)
• Elevated AST
• Hemoglobinuria but No biliruninuria
• Elevated MCV/MCH (reticulocytosis)
• Occasional nucleated RBC in blood film
• BM aspirate/biopsy- erythroid hyperplasia
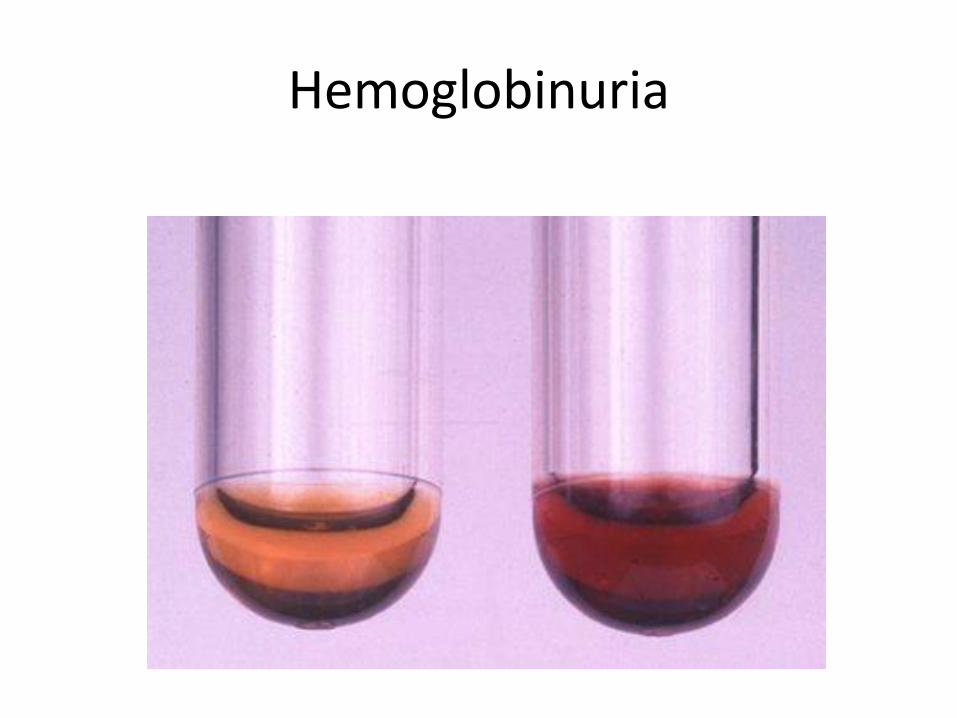
Hemoglobinuria
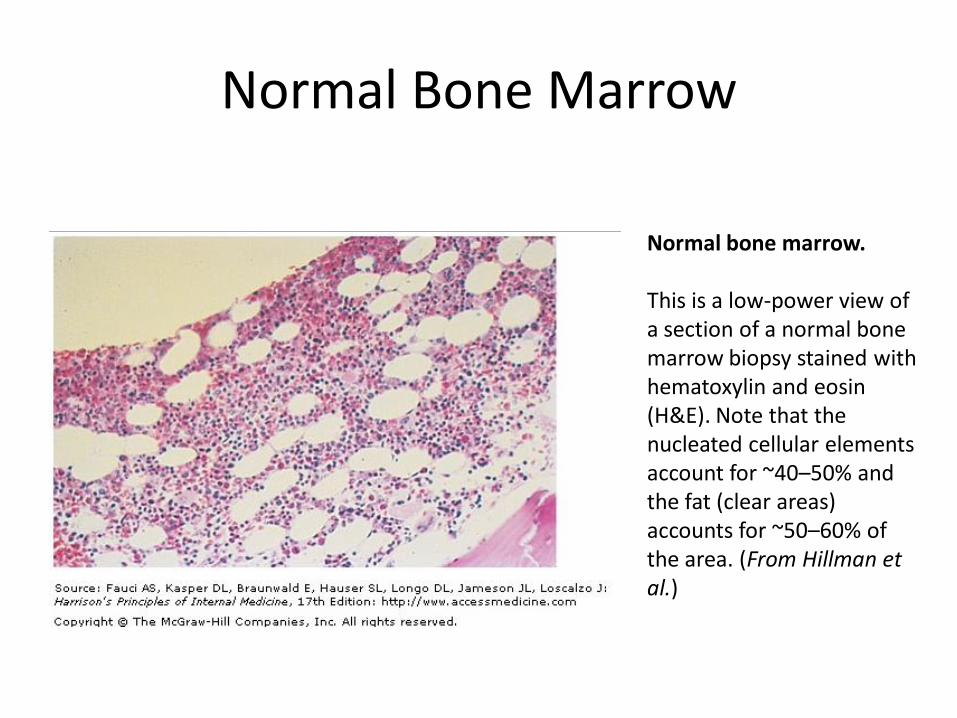
Normal Bone Marrow
Normal bone marrow. This is a low-power view of a section of a normal bone marrow biopsy stained with hematoxylin and eosin (H&E). Note that the nucleated cellular elements account for ~40–50% and the fat (clear areas) accounts for ~50–60% of the area. (From Hillman et al.)
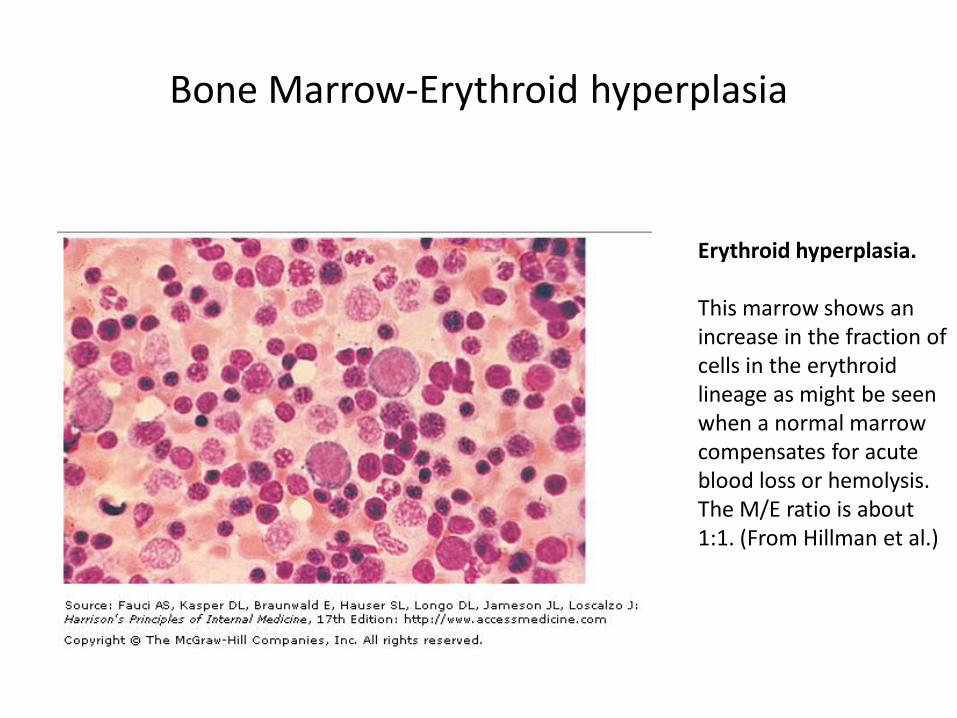
Erythroid hyperplasia. This marrow shows an increase in the fraction of cells in the erythroid lineage as might be seen when a normal marrow compensates for acute blood loss or hemolysis. The M/E ratio is about 1:1. (From Hillman et al.)
Bone Marrow-Erythroid hyperplasia

COMPENSATED HEMOLYSIS
• Features are the same except no anaemia
• Inherited Hemolysis may present with signs of hemolysis without anaemia
• Hemolytic Anaemia may be precipitated by other condition, Eg; Pregnancy/infection/folate deficiency/CRF

General Issues
• HD is amongst the least common causes of anaemia.
• Normal BM and recycling of iron compensates for loss (Reticulocytosis)
• Severity depends on primary cause
• Hemoglobinopathies (SS, thalassaemia) may or may not have reticulocytosis

General Issues
• Acute/Sudden HA (autoimmune hemolysis, G6PD defect)
• Lifelong clinical history typical of primary cause (Thalassaemia & SS)
• Complications from chronic hemolysis (gall stones, splenomegaly) eg HS
• Aplastic Crisis following infection
• Dx –good history, blood smear, electrphoresis, ant-globulin test (Coomb’s tests) and specific tests (G6PD)

Causes
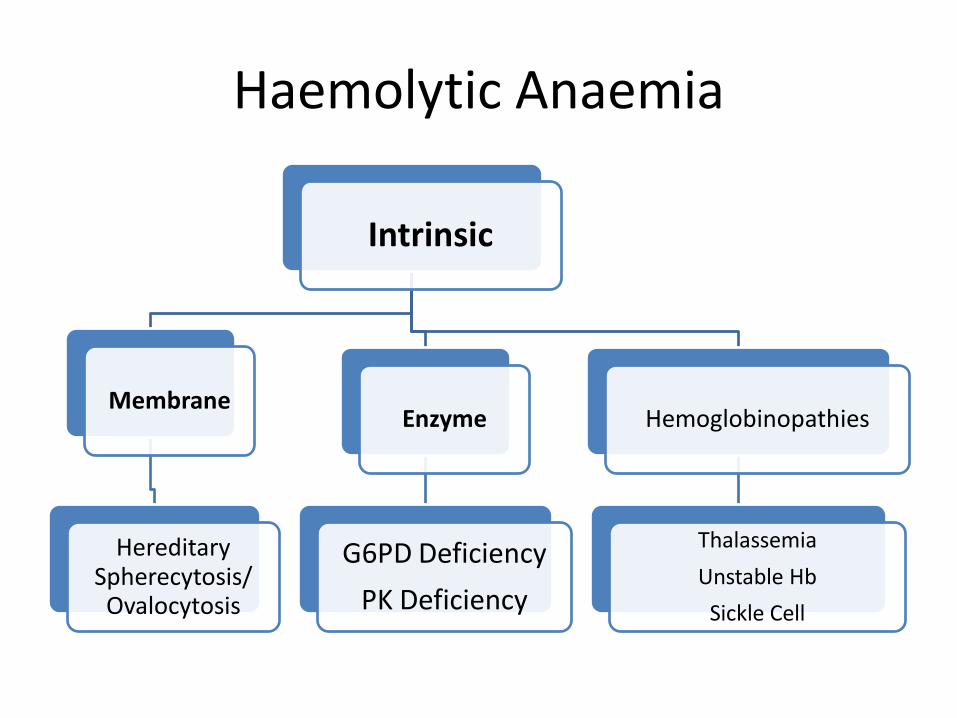
• Intrinsic RBC defects • Inherited
– Membrane defects: hereditary spherocytosis – Enzyme defects: G6PD deficiency – Hemoglobin Defects: sickle cells/Thalassaemia
• Acquired- – Paroxysmal nocturnal hemoglobinuria, – methhemoglobinuria
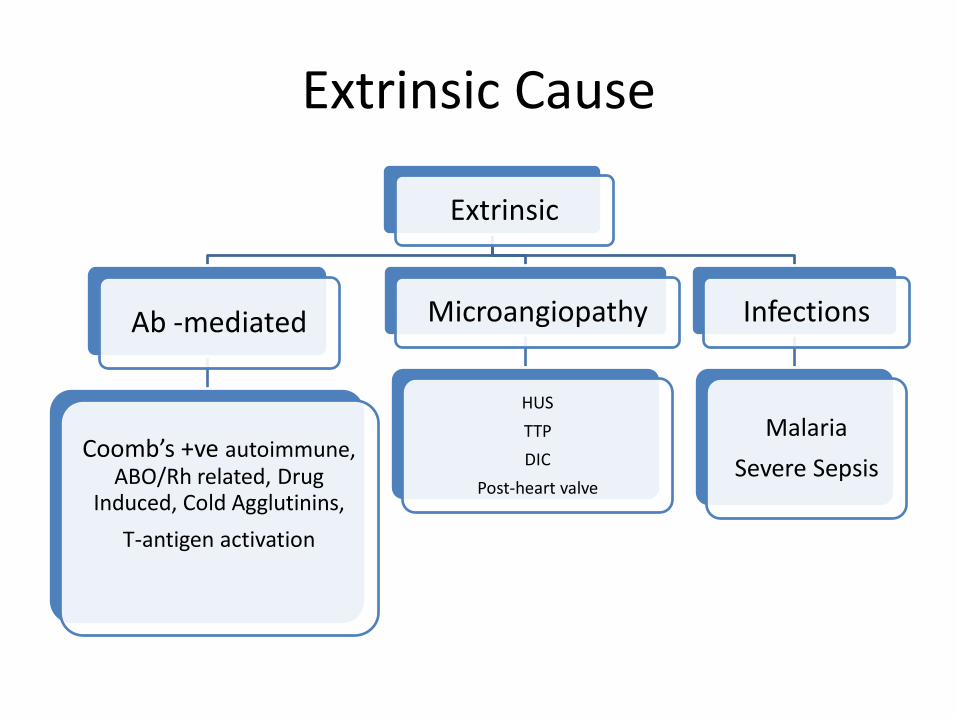
• Extrinsic factors
– Immune: autoimmune HA, drugs, lymphoproliferative ds – Microangiopathic: TTP, HUS, DIC, heart valves – Infections/Toxins: malaria, clostridium sepsis – Hypersplenisim – Burns (Extensive)
Haemolytic Anaemia
Intrinsic
Membrane
Hereditary
Spherecytosis/ Ovalocytosis
Enzyme
G6PD Deficiency
PK Deficiency
Hemoglobinopathies
Thalassemia
Unstable Hb
Sickle Cell
Extrinsic Cause
Extrinsic
Ab -mediated
Coomb’s +ve autoimmune, ABO/Rh related, Drug
Induced, Cold Agglutinins,
T-antigen activation
Microangiopathy
HUS
TTP
DIC
Post-heart valve
Infections
Malaria
Severe Sepsis

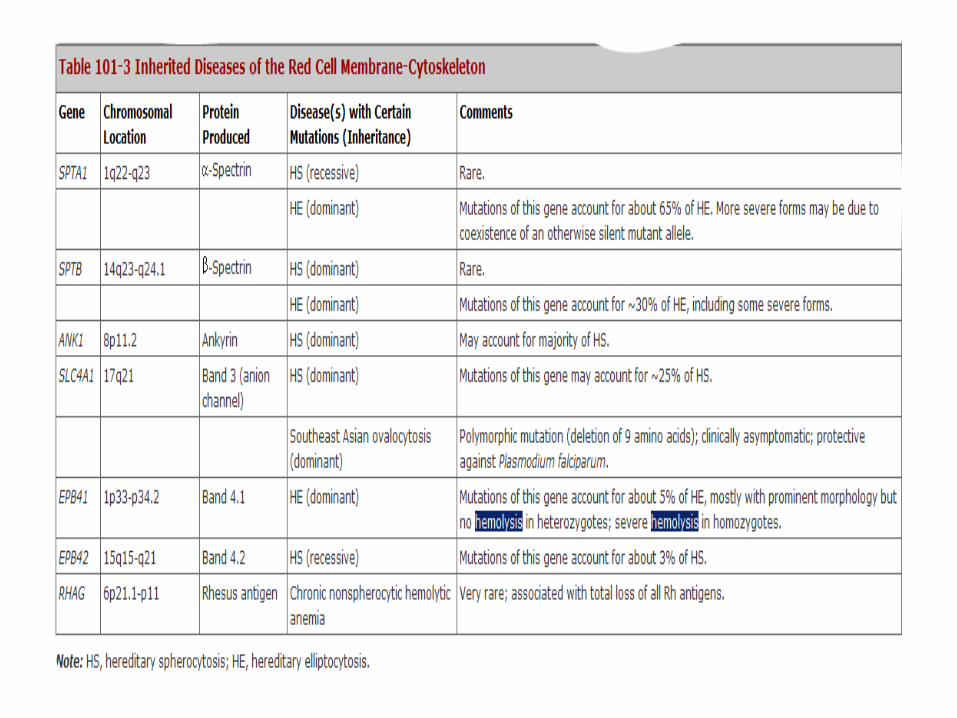
Inherited Hemolytic anaemias (RBC defects)
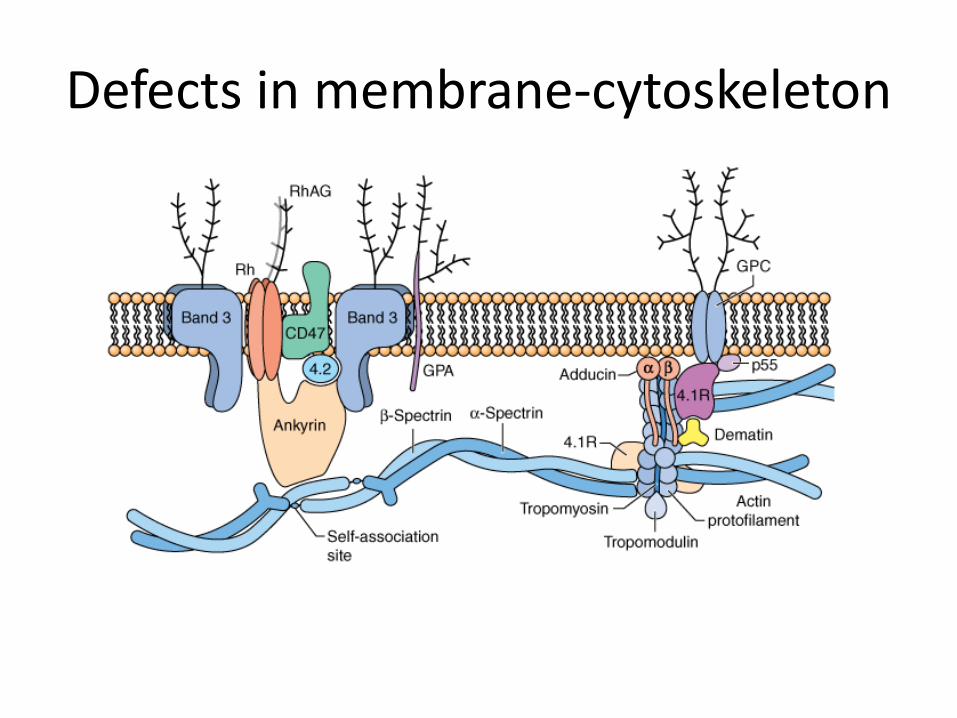
1. Defects in membrane-cytoskeleton
2. Defects in hemoglobin
3. Defects in enzymes/machinery required for maintaining 1 and 2 above
Defects in membrane-cytoskeleton

• highly integrated membrane structure
• Extracellular Glycoprotein antigen on the outside
• Band 3 most abundant protein
• Interruption of one protein results in structural failure-----alteration of RBC membrane ----prone to hemolysis e.g, Hereditary spherocytosis

General Pathophysiology • Abnormal RBC easily destroyed (hemolysed)
• RBC lacks mitochondria/ ribosomes for energy
(ATP) and maintenance of damaged structural proteins & Hb (NADPH)
• Easily damaged from stress of superoxides and oxygen radicals
• Protection of RBC depends of HMP pathway involving G6PD enzyme

Hereditary Spherocytosis
• Relatively common type of HA
• Frequency ~1 in 5000
• Discovered around 19 century
• autosomal dominant condition.
• Abundance of spherocytes in the peripheral blood
• RBCs abnormally susceptible to lysis in hypotonic media (osmotic fragility test)

• Genetically heterogeneous, i.e., it can arise from a variety of mutations in one of several genes
• Classically the inheritance of HS is autosomal dominant (heterozygous),
• some severe forms are instead autosomal recessive (homozygous).
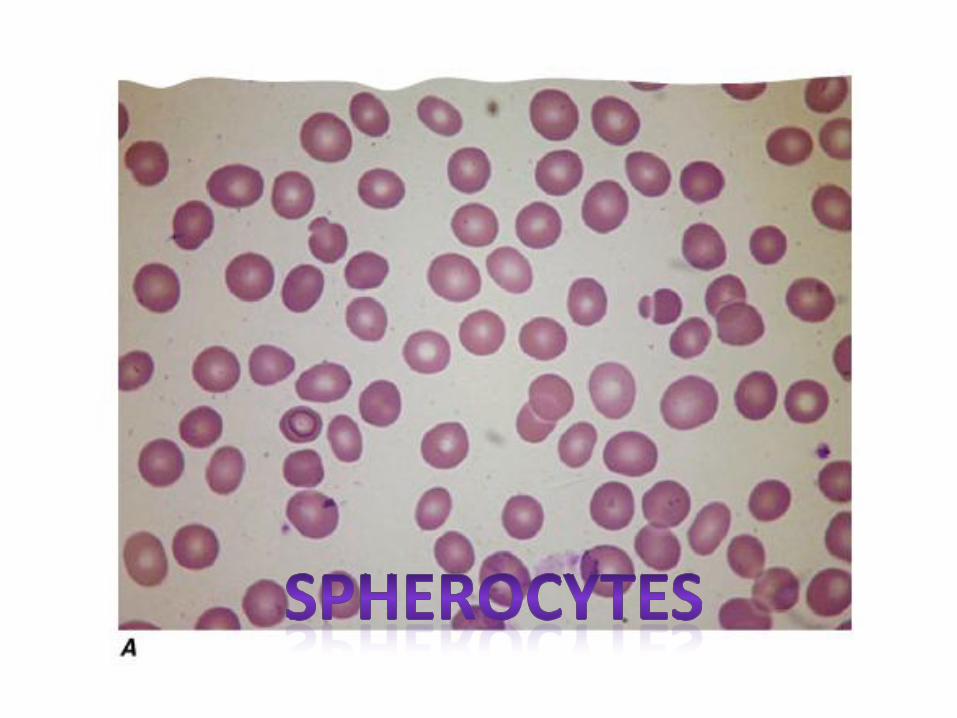
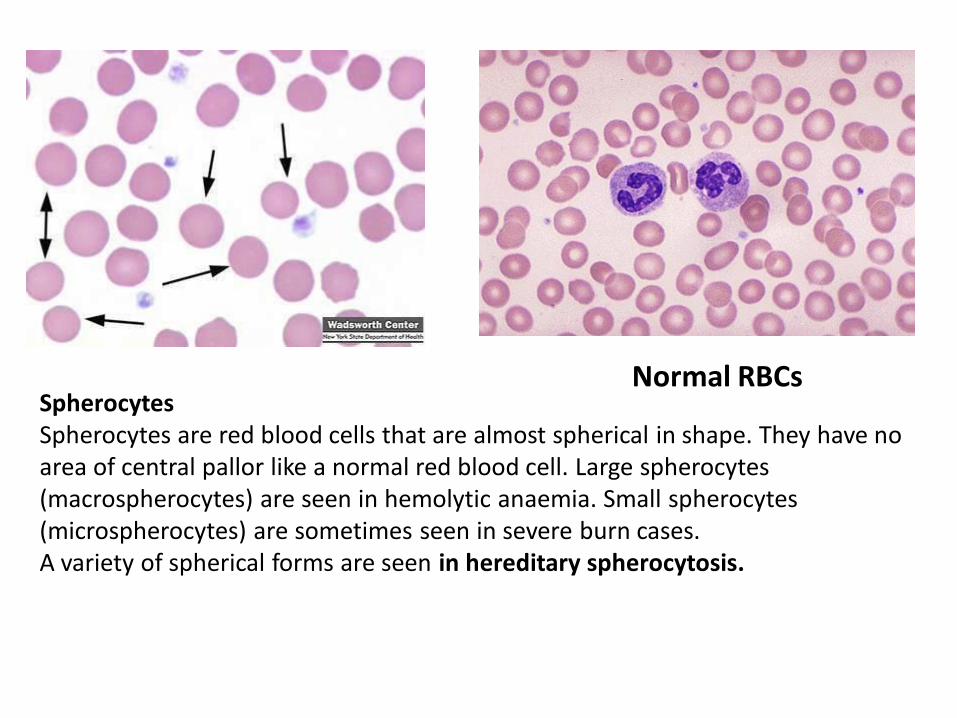
Spherocytes Spherocytes are red blood cells that are almost spherical in shape. They have no area of central pallor like a normal red blood cell. Large spherocytes (macrospherocytes) are seen in hemolytic anaemia. Small spherocytes (microspherocytes) are sometimes seen in severe burn cases. A variety of spherical forms are seen in hereditary spherocytosis.
Normal RBCs

• The spectrum of clinical severity of HS is broad.
• Severe cases may present in infancy with severe anaemia
• Mild cases may present in young adults or even later in life.
• In women, HS is sometimes first diagnosed when anaemia is investigated during pregnancy.
• Main clinical findings: jaundice, an enlarged spleen, and often gallstones; frequently it is the finding of gallstones in a young person that triggers diagnostic investigations.
• The variability in clinical manifestations is related to different underlying molecular lesions

• Mutations –many different genes or various mutations of the same gene ----different clinical manifestations.
• Milder cases,--- compensated hemolysis ; de-compensation if precipitating event.
• Anaemia----is usually normocytic
• A characteristic feature is an increase in MCHC: this is almost the only condition in which high MCHC is seen.

• Family history may be present
• Diagnosis is confirmed on the basis of red cell morphology and a test for osmotic fragility, a modified version of which is called the "pink test.“
• Definitive diagnosis (some cases) -molecular studies demonstrating a mutation in one of the genes underlying HS.

Hereditary Spherocytosis: Treatment
• No definitive treatment
• Splenectomy after 4yrs old
• Avoid splenectomy in mild cases.
• Delay splenectomy until at least 4 years of age, after the risk of severe sepsis has peaked.
• Anti-pneumococcal vaccination before splenectomy is imperative, whereas penicillin prophylaxis postsplenectomy is controversial.
• Many will require cholecystectomy. It used to be considered mandatory to combine this procedure with splenectomy, but this may not be always necessary.

Hereditary Elliptocytosis
• HE is at least as heterogeneous as HS, both from the genetic
• Prevalence is same as HS
• Eliptical shaped RBCs
• No direct correlation between elliptocytic morphology and clinical severity exist
• Some mild or even asymptomatic cases may have nearly 100% elliptocytes,
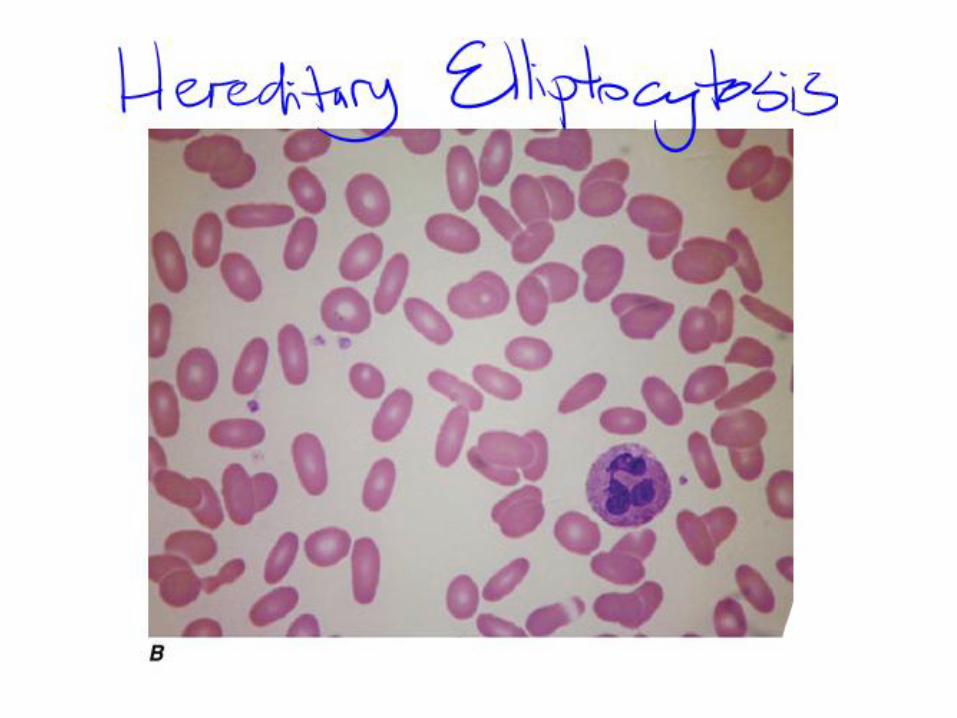

• Severe cases may have predominantly bizarre shaped poikilocytes
• Splenectomy may benefit severe cases
• An asymptomatic form, referred to as Southeast Asian ovalocytosis, has a frequency of up to 7% in certain populations, presumably as a result of malaria selection.

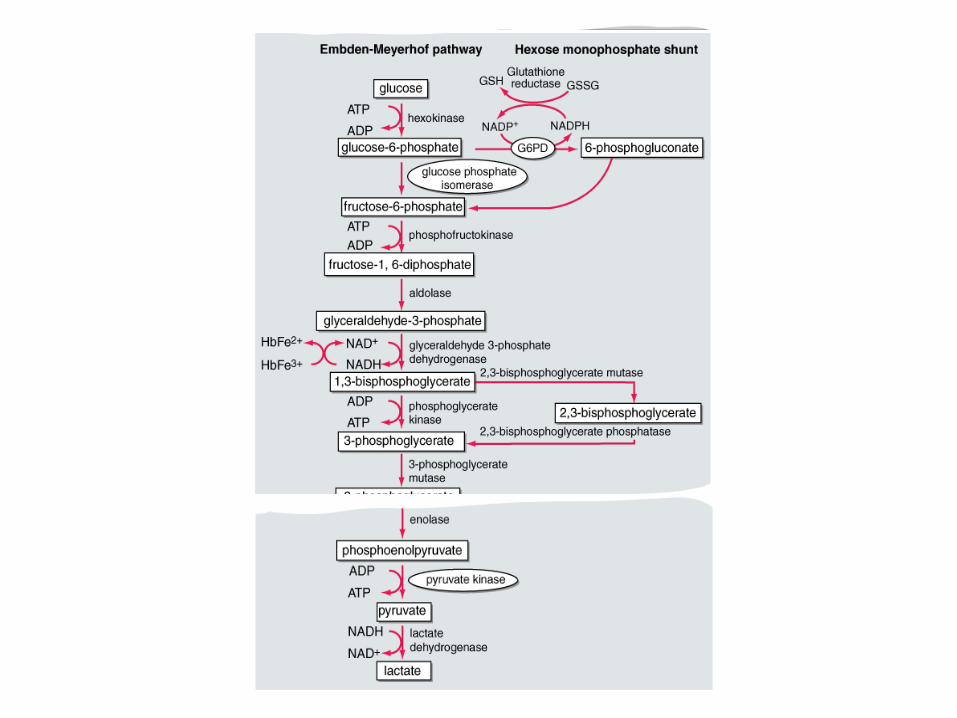
Enzyme Abnormalities
Importance of RBC enzymes:
Generate ATP (glycolytic pathway) – Anaerobic
– Provide most of the required ATP needed for cation transport against concentration gradient, maintain cell membranes of RBC
Generate NADPH (Hexose Monophophate Pathway) – Prevents oxidative damage to Hb

G6PD Deficient Hemolytic anaemia
• Hereditary enzyme defect
• Decreased ability of RBC to withstand oxidative stress-----episodic hemolytic anaemia
• Oxidized hemoglobin ----denatures -----Heinz bodies (precipitants).
• Heinz bodies ----------membrane damage --------- removal of these cells by the spleen.
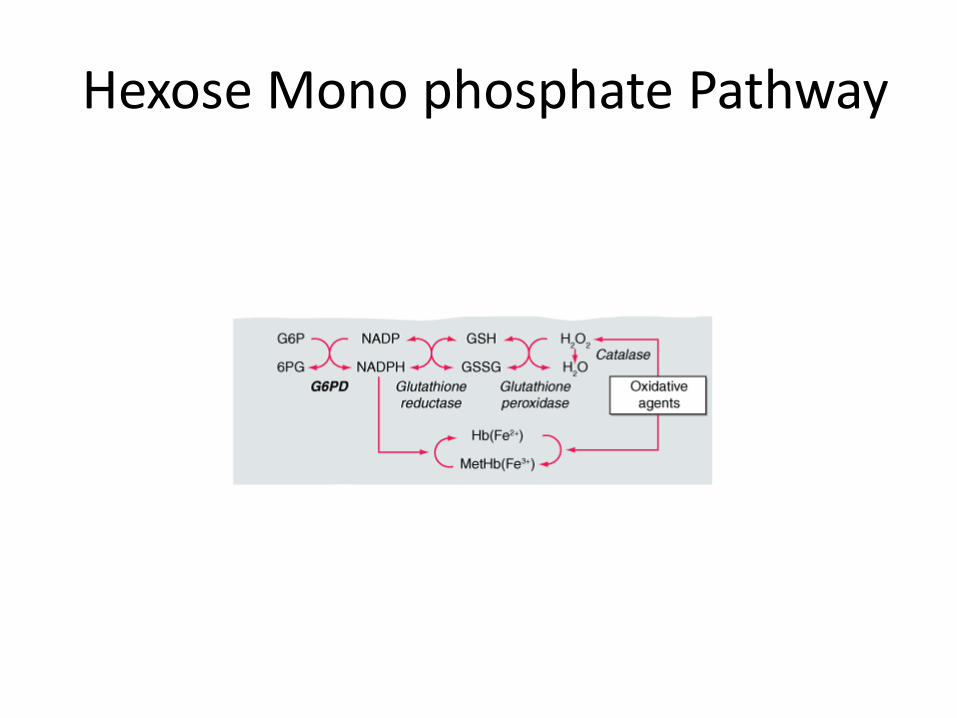
Hexose Mono phosphate Pathway

Genetic Considerations
• G6PD gene is X-linked,
• Males have only one G6PD gene (i.e., they are hemizygous for this gene),
• Females, having two G6PD genes, can be normal, deficient (homozygous), or intermediate (heterozygous).
• As a result of the phenomenon of X-chromosome inactivation, heterozygous females are genetic mosaics, with a highly variable ratio of G6PD-normal to G6PD-deficient cells and an equally variable degree of clinical expression;

• Some heterozygotes can be just as affected as hemizygous males.
• Gene defect: -mutations in the coding region of the G6PD gene.
• Almost all of the 140 different mutations known are single (missense )point mutations, entailing single amino acid replacements in the 514 amino acid G6PD protein.
• G6PD activity decreases with age of RBC so aging cells are more susceptible hemolysis

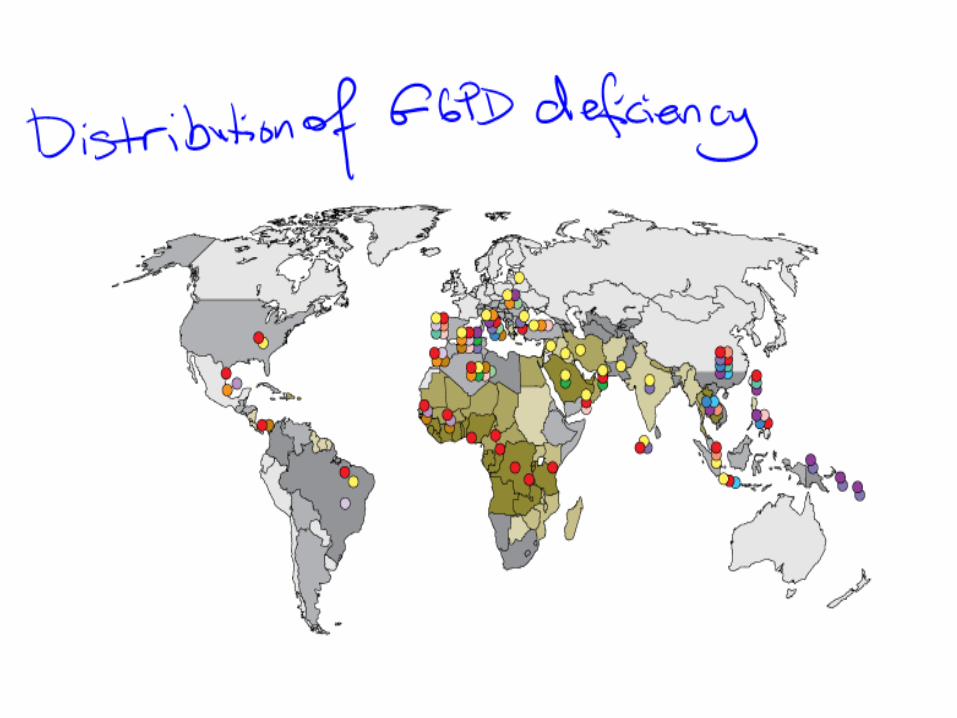
Epidemiology –G6PD • Worldwide distribution (tropical and subtropical)
• ~ 400 million people with defective gene.
• Frequency may be as high as 20% or more in some countries
• Confers relative resistance to plasmodium falciparum malaria (?natural selection)
• Different G6PD variants in different parts of the world.

Clinical Findings
• X-linked recessive disorder –mainly affects males
• Female: carriers are rarely affected—only when an unusually high percentage of cells producing the normal enzyme is inactivated
• USA: 10–15% of American black males

Symptoms and Signs
• Patients are usually healthy, without chronic hemolytic anaemia or splenomegaly.
• Hemolysis occurs as a result of oxidative stress on the red blood cells, generated either by infection or exposure to certain drugs.
• Common drugs initiating hemolysis include dapsone, primaquine, quinidine, quinine, sulfonamides, and nitrofurantoin.
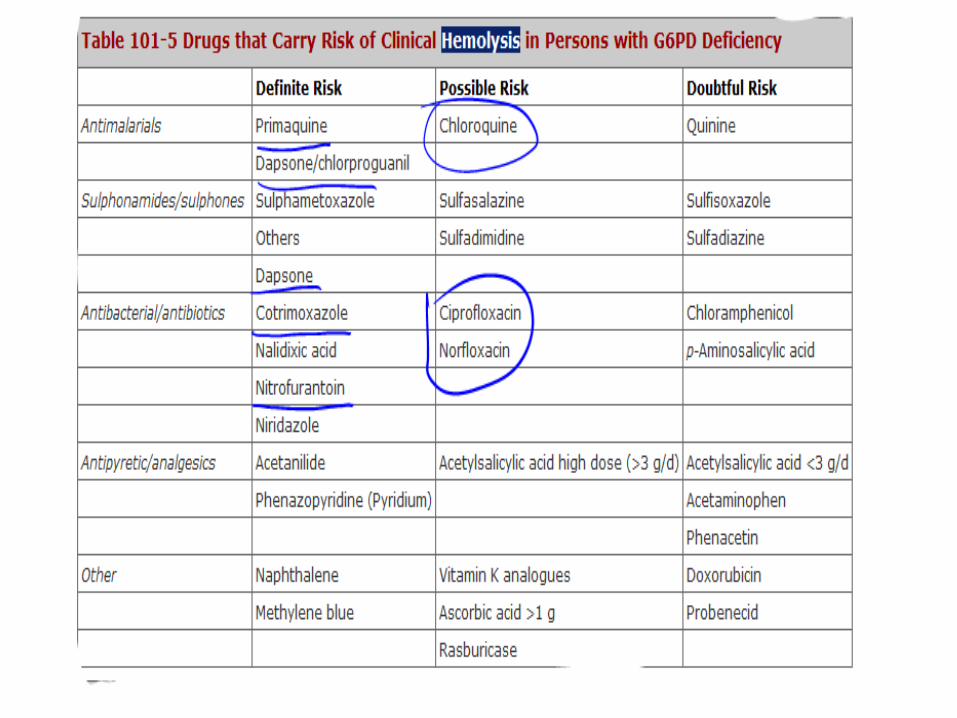

• Even with continuous use of the offending drug, the hemolytic episode is self-limited because older red blood cells (with low enzyme activity) are removed and replaced with a population of young red blood cells with adequate functional levels of G6PD.
• Mediterranean variants may produce a chronic hemolytic anaemia.

• Typically, a hemolytic attack starts with malaise, weakness, and abdominal or lumbar pain.
• After an interval of several hours to 2–3 days, – patient develops jaundice
– often dark urine (hemoglobinuria)
• Most serious threat is acute renal failure (exceedingly rare in children).

Laboratory Test
• Features of hemolysis (reticulocytosis, etc)
• The anaemia is moderate to extremely severe, usually normocytic and normochromic, and due partly to intravascular hemolysis;

Blood Film
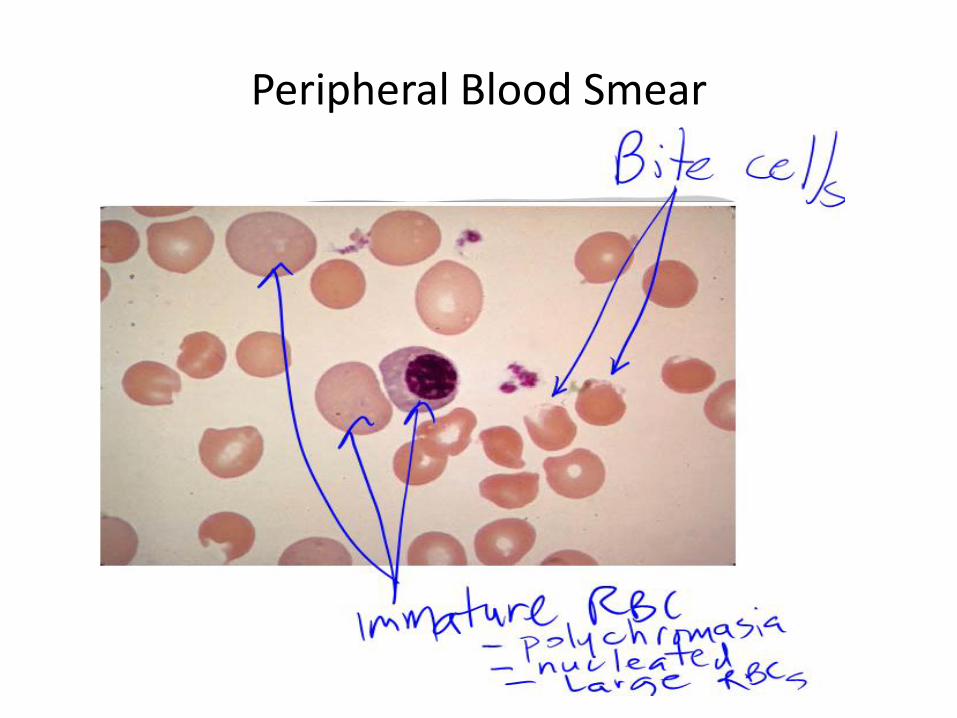
• Bite cells/immature red cells/henz bodies
• Blood films may show anisocytosis, and spherocytes
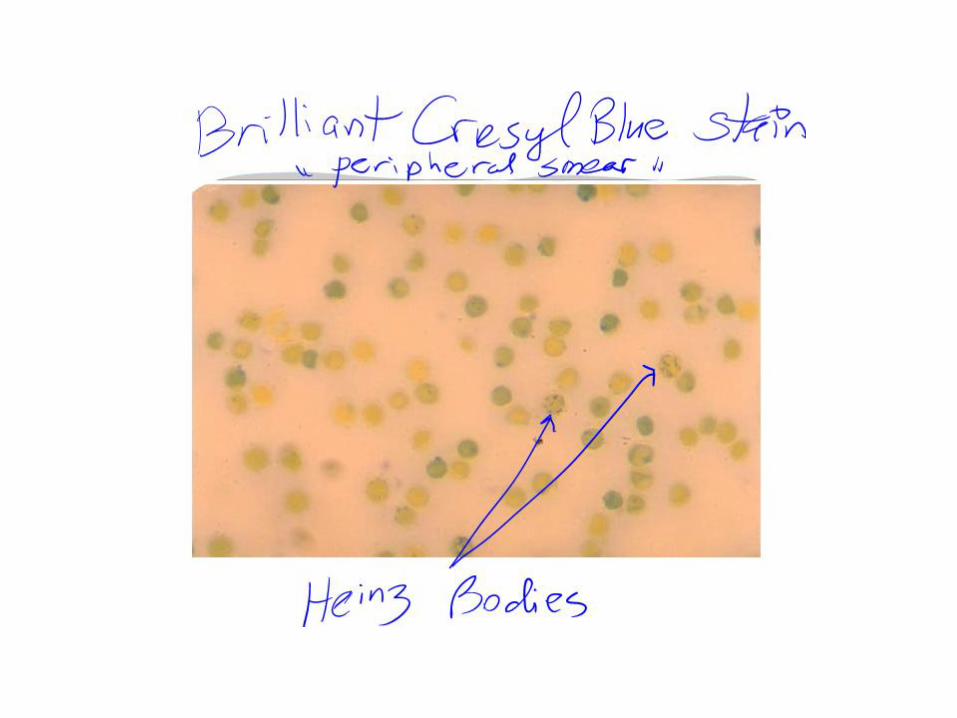
• Heinz Bodies: – A classic test, is supravital staining with methyl violet
(rarely done now) – Reveals the presence of Heinz bodies, – precipitates of denatured hemoglobin – Regarded as a signature of oxidative damage to red
cells
Peripheral Blood Smear

• Definitive Diagnosis
– G6PD assay: qualitative /quantitative test
– G6PD immediately after hemolysis may be normal due to increased G6PD activity in young red cells

Treatment
Treat anaemia & Infection
Avoid offending drug/oxidant

• Prognosis:
– Once the threat of acute anaemia is over, & in the absence of comorbidity,
– Full recovery from acute HA associated with G6PD deficiency is the rule.

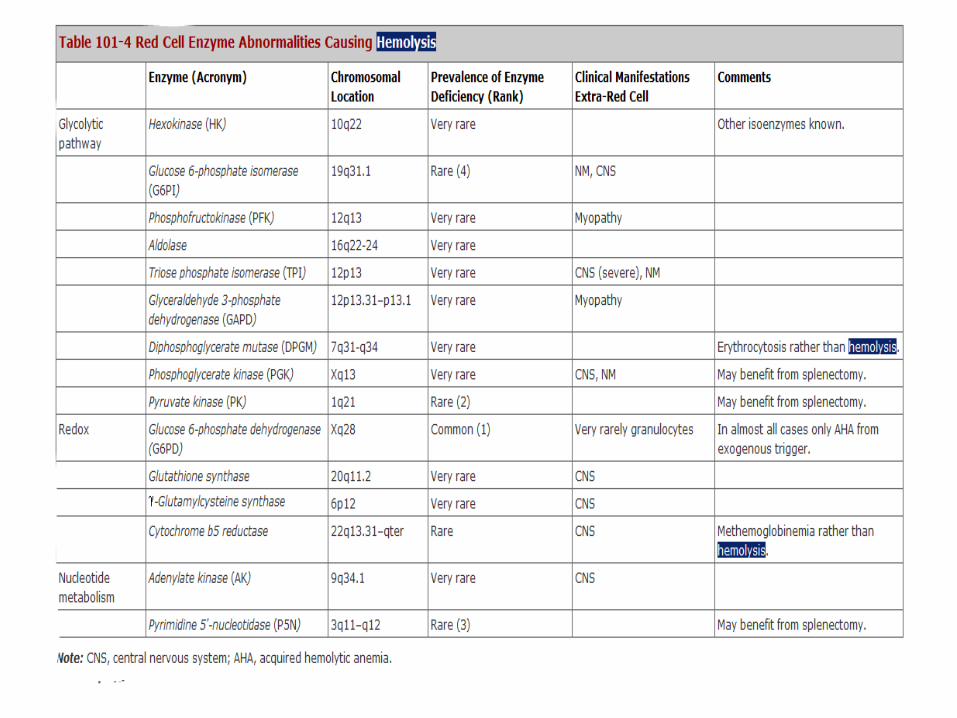
Pyruvate Kinase Deficiency • Abnormalities of the glycolytic pathway are all
inherited and all rare (Table 101-4).
• Among them, deficiency of pyruvate kinase (PK) is the least rare,
• An estimated prevalence of 1:10,000
• Newborn: neonatal jaundice + marked reticulocytosis.
• The anaemia is of variable severity

• Diagnosis may be delayed till young adults—for instance, in a woman during her first pregnancy, when the anaemia may get worse.
• Delayed diagnosis because – anaemia is remarkably well-tolerated or
– Block at the last step in glycolysis causes an increase in 2,3 DPG, a major effecter of the hemoglobin-oxygen dissociation curve i.e., O2 delivery to tissues increased.

Pyruvate Kinase Deficiency: Treatment
Supportive
Folate
Iron chelation
Blood transufsion
Splenectomy (severe)
BM transplant

Hemoglobinopathies
• Thalassaemia
• Sickle Cell Disease
• Unstable hemoglobins
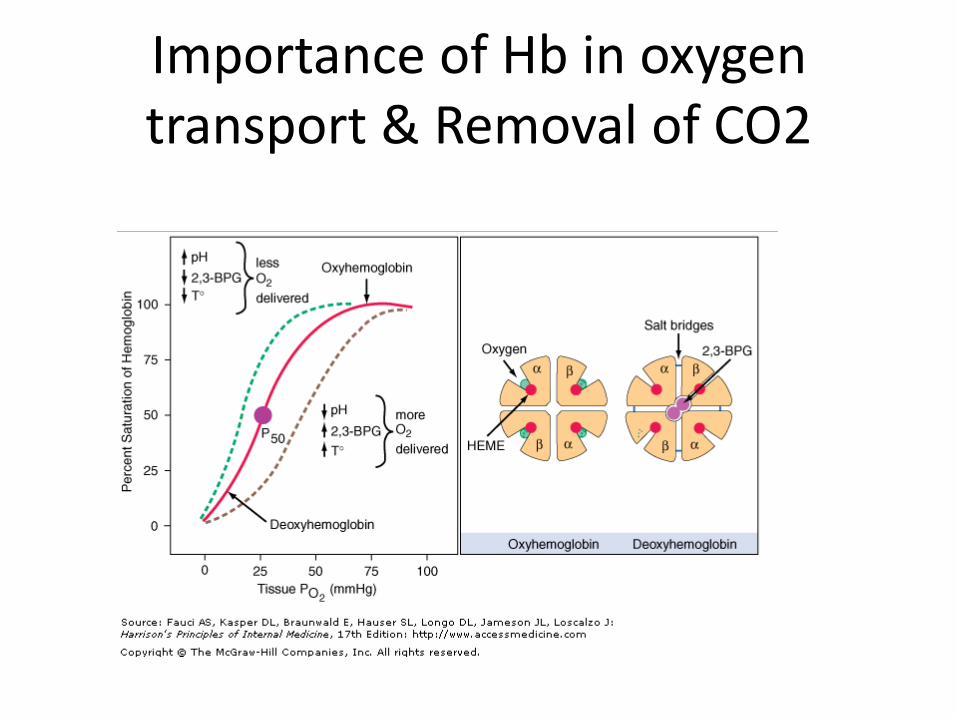
Importance of Hb in oxygen transport & Removal of CO2
Abnormal Hb = hemolysis
• Abnormal RBC membranes-hemolysis
• Abnormal Enzyme systems-hemolysis
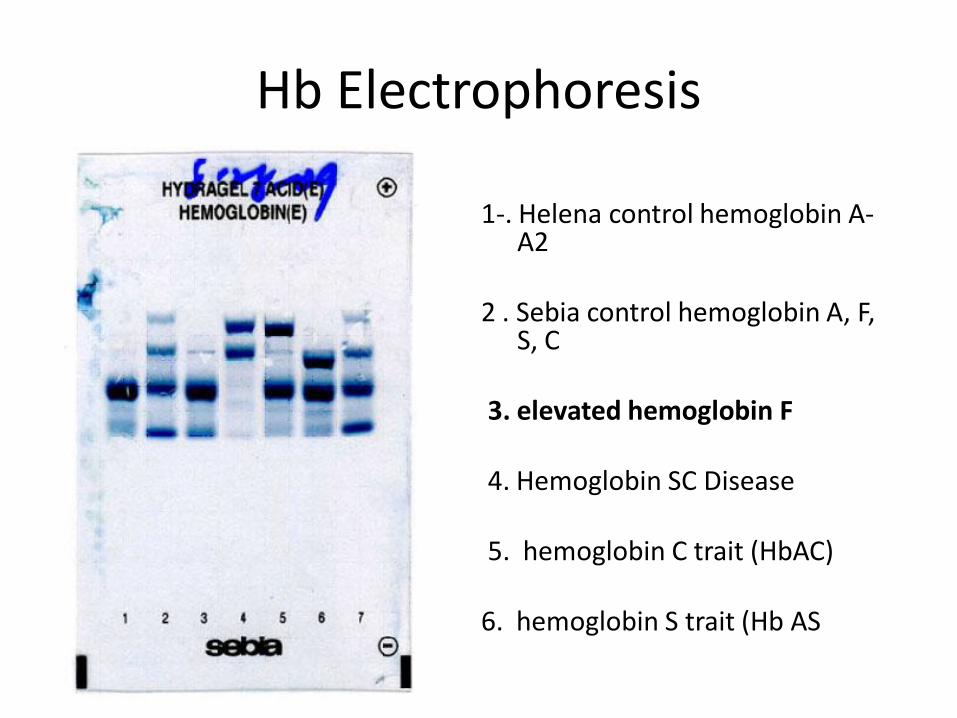
Hb Electrophoresis
1-. Helena control hemoglobin A-A2
2 . Sebia control hemoglobin A, F,
S, C 3. elevated hemoglobin F 4. Hemoglobin SC Disease 5. hemoglobin C trait (HbAC)
6. hemoglobin S trait (Hb AS
Quantization of Electrophorogram
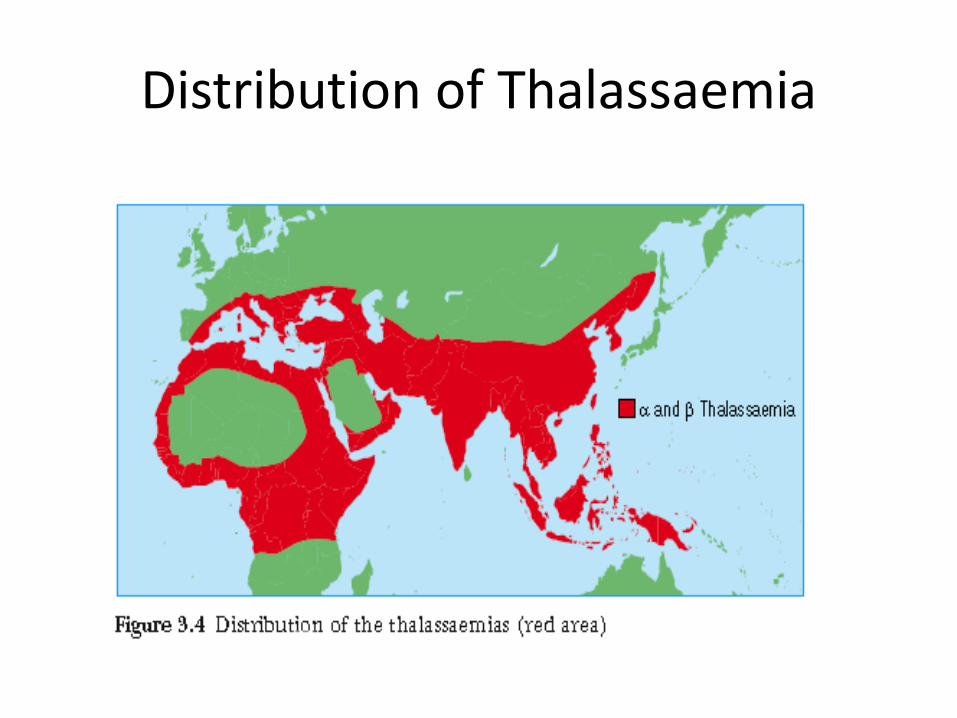
Distribution of Thalassaemia

Thalasaemia
• Normal HbA1 (2 2)
• <2% HbA2 (22)
• Individuals inherit one beta gene from each parents compared to 2 alpha genes
• Alpha thalassemia- alpha chain defect/gene deletion
• - Thalassemia-beta chain defect/reduced beta chain amount/qantitative defect

Thalassaemia Gene Mutation
• Can affect any step for Hb synthesis: – transcription,
– processing of the mRNA precursor
– translation,
– posttranslational metabolism of the -globin polypeptide chain, etc.
• The most common forms arise from mutations that derange splicing of the mRNA precursor or prematurely terminate translation of the mRNA

Pathogenesis
• Abnormally developed Hb chain,
• excess normal chains,
• results in accumulation of abnormal Hb pigments & disturbed normal BM red cell production,
• removal of abnormal RBC by spleen----- enhanced hemolysis

General Features
• Asymptomatic
• Hemolytic Anaemia
• Erythroid hyperplasia/Growth Failure
• Extramedullary hemopoiesis (hepatosplenomegaly)
• Microcytosis/hypochromia
• Iron Overload
• Need for repeated transfusion

Alpha Thalassaemia Syndromes
• -Globin Syndromes Haematocrit MCV
• genes
• 4 Normal Normal
• 3 Silent carrier Normal
• 2 Thalassemia minor 28–40% 60–75 fL
• 1 Hb H disease (4) 22–32% 60–70 fL
• 0 Hydrops fetalis/Hb Barts (γ4) stillbirths

Alpha Thalassaemia Trait/Minor
– 1 or 2 gene deletions
– relatively common in Asian populations
– Asymptomatic throughout life
– Microcytosis /target cells may be seen
– Hb electrophoresis –normal except decreased HbA2

HbH –3 gene deletions (β2β2)
– microcytic but asymptomatic when well
– Some normal Hb (~30%)
– Has chronic hemolytic Anaemia
– May develop anaemia when stressed
– Survive , do not depend on transfusion
– Heinz Body seen
– Gamma chains only, High Oxygen affinity—No tissue release of O2
– hydrops fetalis at birth or In utero, Incompatible with life

Beta Thalassemia Syndromes. •
-Globin Genes Hb A Hb A2 Hb F
• Normal Homozygous 97–99% 1–3% < 1%
• (transfusion required to survive)
• T. Major Homozygous 0 0% 4–10% 90–96%
• T. Major Homozygous + 0–10% 4–10% 90–96%
• (no transfusion required to survive)
• T. Intermedia Homozygous + 0–30% 0–10% 6–100%
• T. Minor Heterozygous 0 80–95% 4–8% 1–5%
Heterozygous + 80–95% 4–8% 1–5%

Thalassaemia Major (inadequate -chain)
– Alpha chain >beta chains
– Uncommon in 1st world due to increased antenatal screen & prenatal termination
– Present during second 6 months of life [switch of HbF (22) - HbA1 (2 2) at 1st 6months]
– Presentation: severe hemolytic anaemia, slow
growth, skeletal deformities, hepatosplenomegaly (always), heart failure

Laboratory Diagnosis
– Marked erythroblastosis & bizzare RBC forms on Blood Film
– hypochromic, microcytic anaemia, PCV <20%
– Increased HbF (22) and 2-fold increase in HbA2 (22)
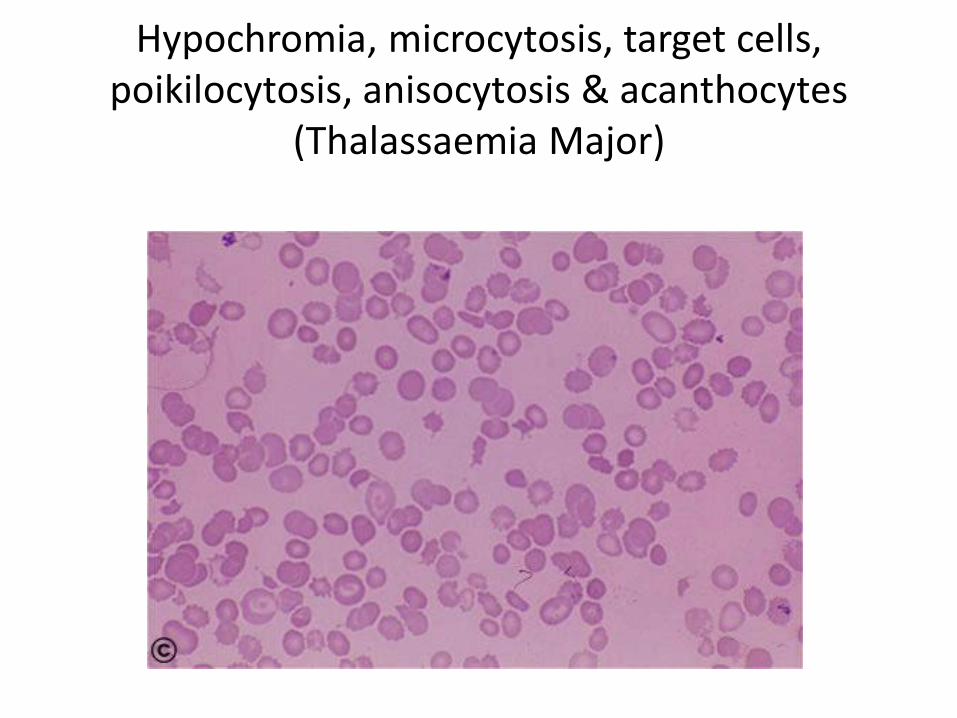
Hypochromia, microcytosis, target cells, poikilocytosis, anisocytosis & acanthocytes
(Thalassaemia Major)

Management
– Depend on transfusion/Require Folate supplements
– Reduced life expectancy (<30yr)
– Iron Overload & Endocrine disorders
– Infection/Gall bladder Disease
– Bone Marrow Transplant
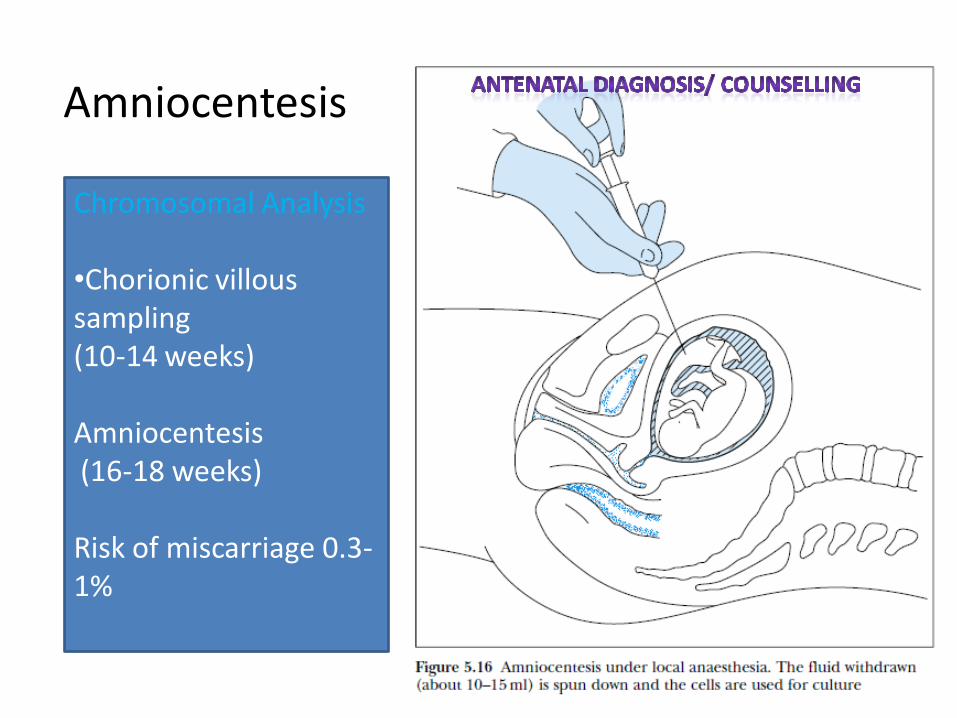
Amniocentesis
Chromosomal Analysis •Chorionic villous sampling (10-14 weeks) Amniocentesis (16-18 weeks) Risk of miscarriage 0.3-1%

Thalassemia minor /Thalassemia trait
– very common
– Rarely show significant anaemia and symptoms
– Causes microcytic, hypochromic Anaemia
– Clues on FBE include elevated RBC count/marked microcytosis
– Diagnosis: Hb electrophoresis- elevated HBA2
– Often treated unnecessarily with iron
– HbA2 levels may be corrected with iron therapy obscuring the dx

Essentials of Diagnosis of Thalassaemia
• Microcytosis out of proportion to the degree of anaemia.
• Positive family history or lifelong personal history of microcytic anaemia.
• Abnormal red blood cell morphology with microcytes, acanthocytes, and target cells.
• In -thalassemia, elevated levels of hemoglobin A2 or F.
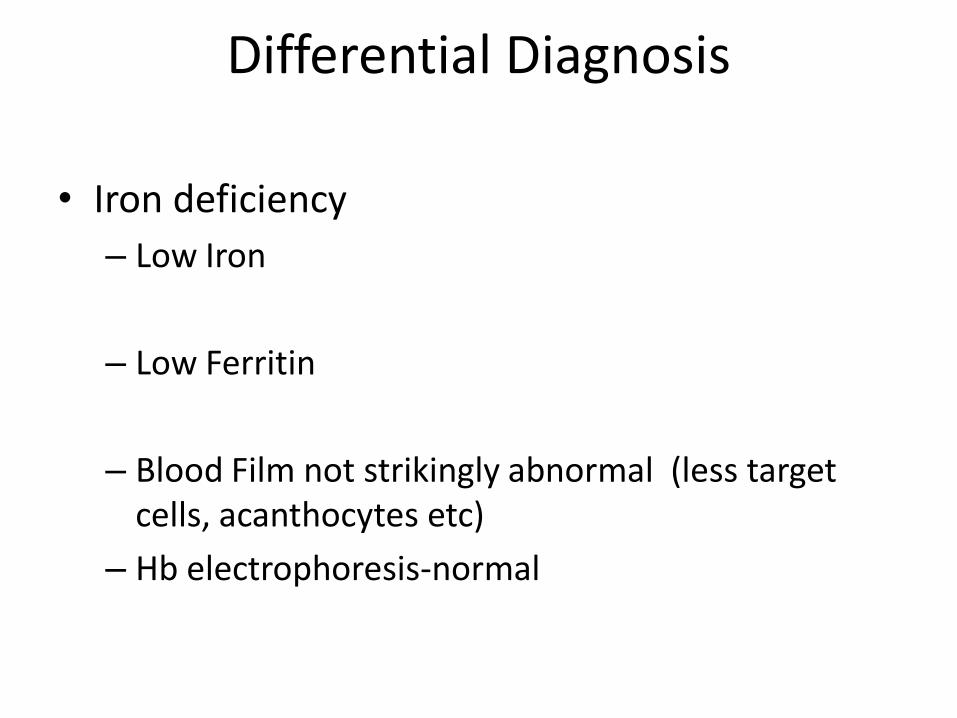
Differential Diagnosis
• Iron deficiency
– Low Iron
– Low Ferritin
– Blood Film not strikingly abnormal (less target cells, acanthocytes etc)
– Hb electrophoresis-normal

Sickle Cell Anaemia
• Homozygous (ss) or double heterozygous (HbS/-thalassemia)
• Defect in beta chain: substitution of valine for glutamic acid at 6th residue on beta chain
• Deoxygenated HbS cell sickles easily switch from biconcave disk to elongated crescent-shaped or sickled cell—
• Lack flexibility and rigid to transverse capillaries ---hemolysed
• 8% black Americans
• Up to 30% Central Africa (associated with malaria endemic areas)
• Gives slight protection from malaria
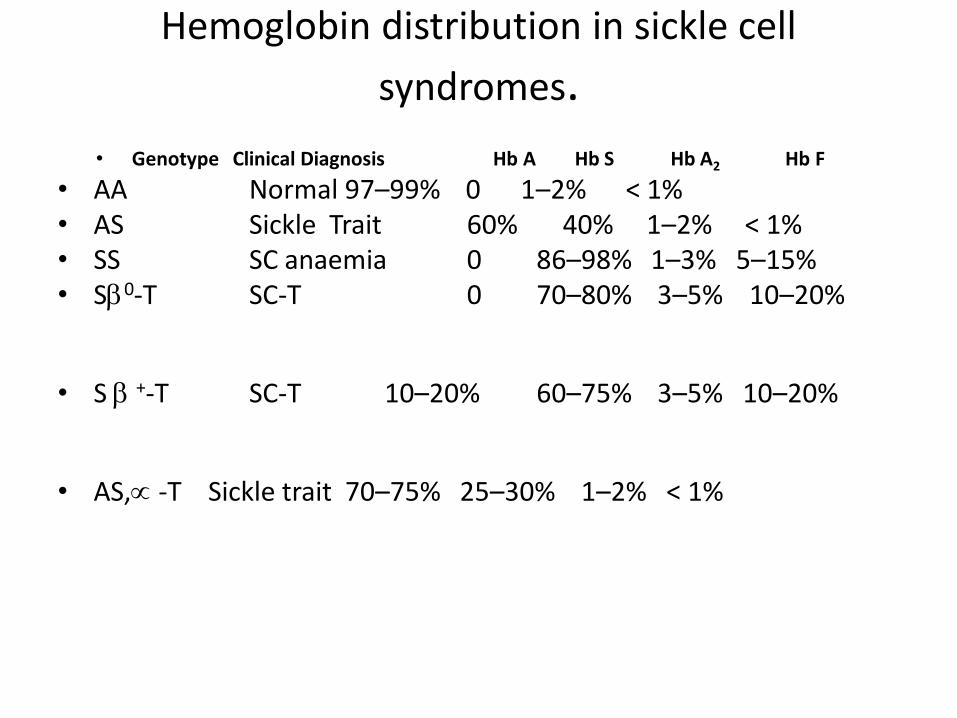
Hemoglobin distribution in sickle cell
syndromes.
• Genotype Clinical Diagnosis Hb A Hb S Hb A2 Hb F
• AA Normal 97–99% 0 1–2% < 1% • AS Sickle Trait 60% 40% 1–2% < 1% • SS SC anaemia 0 86–98% 1–3% 5–15% • S0-T SC-T 0 70–80% 3–5% 10–20%
• S +-T SC-T 10–20% 60–75% 3–5% 10–20%
• AS, -T Sickle trait 70–75% 25–30% 1–2% < 1%

• Clinically – Asymptomatic carrier
– Symptoms –onset at 6 months of age (HbFHbA switch)
– Multiple systems affected: CNS/Resp/skin/eyes/GUT/MSS etc
– Anaemia (hemolysis/aplasia)
– Half –life of red cells : 10-15 days
– Rapidly progressive anaemia with splenomegaly
– Delayed growth & Development
– Joint pains (small hand & Feet)

– Acute Chest syndrome (pneumonia-like)
– Arterial ischaemia stroke
– Painless haematuria
– Painful crisis (joint/abdomen)
– Sepsis/Sickling crisis in carriers– if severely hypoxic

Sickle Cell Trait
Heterozygous genotype (AS)
• Clinically and haematologically normal
• Experience acute painful episodes only under extreme conditions such as vigorous exertion at high altitudes (or in unpressurized aircraft).

• May have defect in renal tubular function
• Inability to concentrate the urine
• Experience episodes of gross hematuria.
• Sickling in the sluggish circulation of the renal medulla.
• Hb electrophoresis 40% HbS.
• No treatment is necessary.
• Genetic counselling is a reasonable strategy

Sickle Thalassemia
• Homozygous sickle cell anaemia and -thalassemia Associated with Milder form of hemolysis (slower rate of sickling) Reduced MCHC within the red blood cell.
• Double heterozygotes for sickle cell anaemia and –thalassemia
clinically affected with SC Syndromes.
• Sickle 0-thalassemia
Clinically very similar to homozygous SS disease. Vaso-occlusive crises may be somewhat less severe, Spleen is usually not infarcted. Hematologically, the MCV is usually low, in contrast to the normal MCV of sickle cell anaemia. No HbA but will show an increase in hemoglobin A2, which is not present in sickle cell
anaemia.
• Sickle +-thalassemia
milder disorder than homozygous SS disease, with fewer crises. Spleen is usually palpable. Hemolytic anaemia is less severe, Hematocrit is usually 30–38%, Reticulocytes of 5–10%. Electrophoresis show some Hb A.

Diagnosis of Sickle Cell
• Features of Hemolytic Anaemia
• Features of Sickle Cell +/-Thalassaemia • Blood Film-
– demonstrate sickling cells( 5-50% ) – Normocytic, normochromic anaemia,
Hct: usually 20–30%. Reticulocytosis (10–25%), Howell–Jolly bodies WBC elevated (12,000–15,000/mcL) Thombocytosis

Hb Electrophoresis-
– Hb S 76-96%
– HbF 2-20%
– HbA2 2-4%
– HbA1- 0%

Treatment
• Supportive
• Conservative
• Antenatal Counselling
• Prenatal Diagnosis (DNA analysis)

Unstable Hemoglobins
• Hb prone to oxidative denaturation
• Normal G6PD system.
• Autosomal dominant and of variable severity.
• Mostly mild chronic hemolytic anaemia
• Have splenomegaly, mild jaundice, and pigment (calcium bilirubinate) gallstones.
• Less severely affected patients are not anemic except under conditions of oxidative stress.

Diagnosis:
• Heinz bodies and a normal G6PD level.
• Normal Hb electrophoresis
• Usually no treatment is necessary.
• Treat folate supplementation and avoid known oxidative drugs.

Session 2
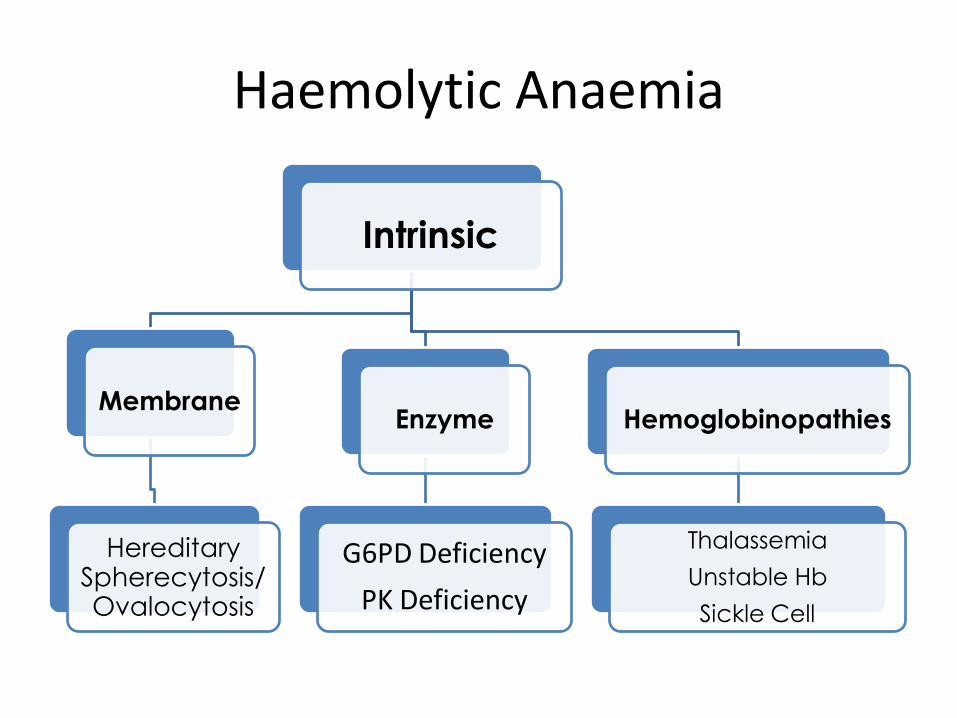
Haemolytic Anaemia
Intrinsic
Membrane
Hereditary
Spherecytosis/ Ovalocytosis
Enzyme
G6PD Deficiency
PK Deficiency
Hemoglobinopathies
Thalassemia
Unstable Hb
Sickle Cell

Extrinsic causes of hemolysis
Extrinsic
Ab -mediated
Coomb’s +ve autoimmune, ABO/Rh related, Drug Induced,
Cold Agglutinins,
T-antigen activation
Microangiopathy
HUS
TTP
DIC
Post-heart valve
Infections
Malaria
Severe Sepsis

Autoimmine Haemolytic anaemia
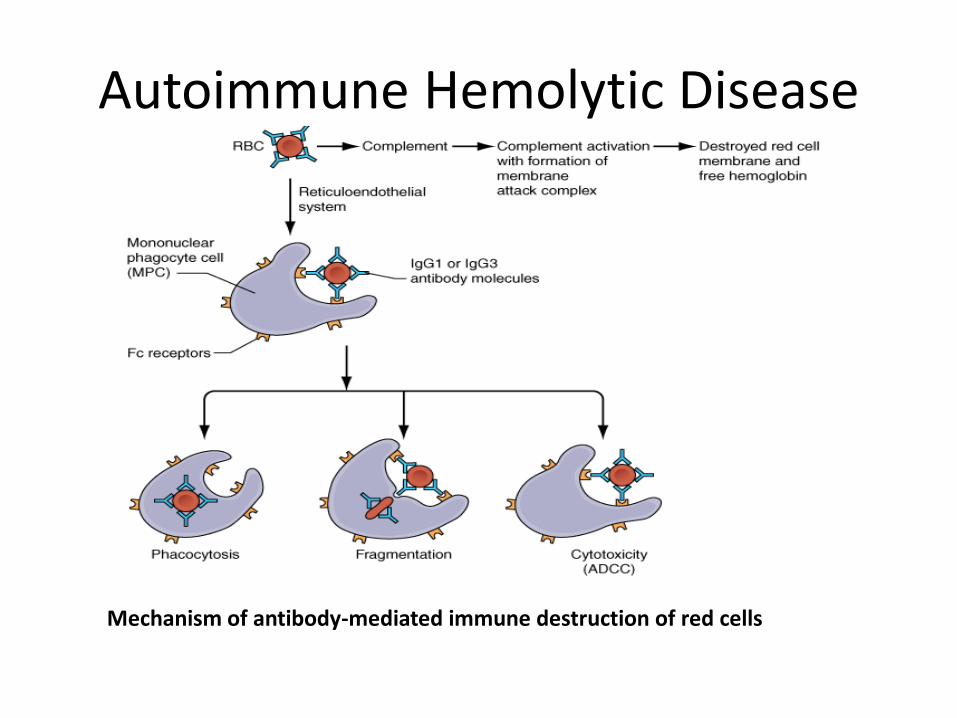
Autoimmune Hemolytic Disease
Mechanism of antibody-mediated immune destruction of red cells

General Considerations
• Acquired disorder in which an IgG autoantibody is formed that binds to the red blood cell membrane.
• Antibody is most commonly directed against a basic component of the Rh system present on most human red blood cells.
• Red Cell coded by IgG- • recognised by macrophages of RES/Spleen • –removal of RBC membrane • -formation of spherocytes • - removal of RBC by RES • --> hemolysis
•
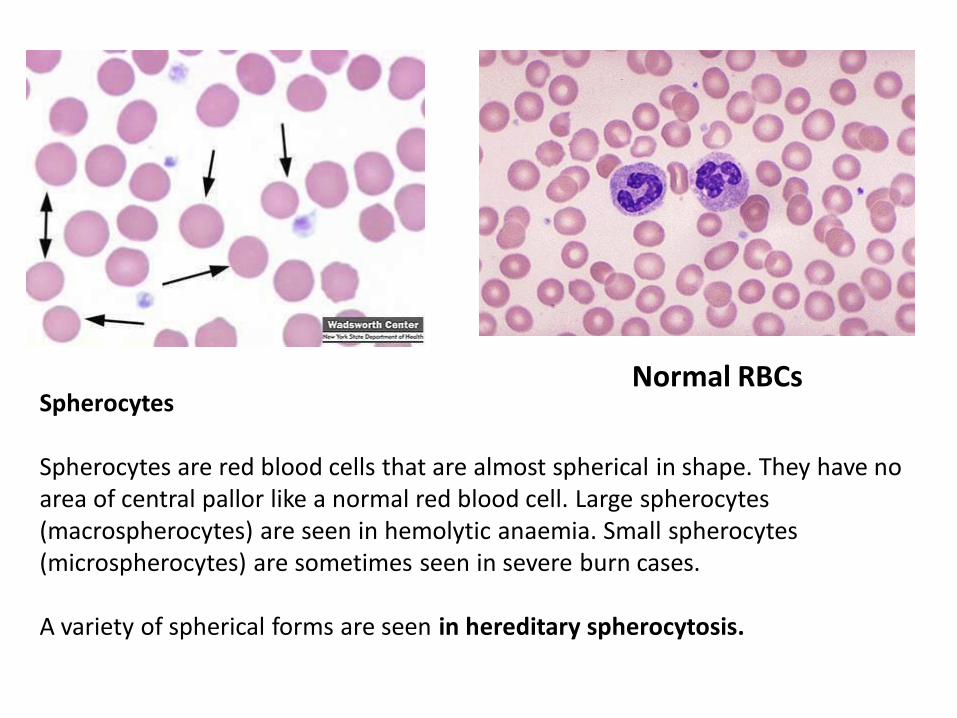
Spherocytes Spherocytes are red blood cells that are almost spherical in shape. They have no area of central pallor like a normal red blood cell. Large spherocytes (macrospherocytes) are seen in hemolytic anaemia. Small spherocytes (microspherocytes) are sometimes seen in severe burn cases. A variety of spherical forms are seen in hereditary spherocytosis.
Normal RBCs

Causes
• Idiopathic 50%
• Associated with SLE, CLL or lymphomas
• Transfusion incompatibility
• Note: drug-induced hemolytic anaemia. Eg penicillin (and other drugs) coats the red blood cell membrane, and the antibody is directed against the membrane–drug complex.

Coomb’s anti-globulin test
• Used to diagnose AIHA.
• Coomb's reagent – Is a rabbit IgM antibody raised against human IgG or human complement.
• Direct Coomb's test -
– mix the patient's red blood cells with the Coomb's reagent – Positive Agglutination – Presence of antibody on the red blood cell surface.
• Indirect Coomb's test
– Mixing the patient's serum with a panel of type O red blood cells. – Incubate test serum and panel O red blood cells, – Add Coomb's reagent – Positive Agglutination – Presence of free antibody in the patient's serum.

Symptoms and Signs
• Rapid Onset Anaemia
• May be life-threatening in severity.
• Complaints of fatigue
• May present with angina or congestive heart
failure.
• On examination, jaundice and splenomegaly are usually present.

Laboratory Findings
• Anaemia -variable severity
• Spherocytes are seen on the peripheral blood smear.
• Reticulocytosis is usually present,
• Nucleated RBCs (in cases of severe AIHA)
• Other lab signs of hemolysis
~ 10% have coincident immune thrombocytopenia (Evans syndrome).

Coomb's tests
• Direct Tests- Positive
• Indirect Test –may/may not be positive. If positive, indicates large amounts of antibodies saturated RBC binding sites
• Patient’s serum usually contains the autoantibody, it may be difficult to obtain a compatible cross-match with donor's cells.

Autoimmune Hemolytic anaemia
• Essentials of Diagnosis
– Acquired anaemia caused by IgG autoantibody.
– Spherocytes and reticulocytosis on peripheral blood smear.
– Positive Coombs test

Treatment • Prednisolone 1–2 mg/kg/d in divided doses.
• Most transfused blood will survive similarly to the patient's own red
blood cells.
• Because of difficulty in performing the cross-match, incompatible blood may be given.
• Splenectomy should be performed if no response
• Other agents: – Rituximab, a monoclonal antibody against the B cell antigen CD20, – Cyclophosphamide, – Azathioprine, or cyclosporine
• Prognosis: good if no underlying primary disorder
• Splenectomy is often successful in controlling the disorder.

Cold Agglutinin Disease

General Considerations
• Acquired disorder
• IgM autoantibody directed against the I antigen on all red blood cells.
• IgM autoantibodies does not react with cells at 37 °C but only at lower temperatures.
• Hemolysis results indirectly from attachment of IgM, which in the cooler parts of the circulation (fingers, nose, ears) binds and fixes complement ,Cb3.
• When the red blood cell returns to a warmer temperature, the IgM antibody dissociates, leaving complement on the cell.
• Lysis of cells rarely occurs. Rather, C3b present on the red cells is recognized by Kupffer cells (which have receptors for C3b), and red blood cell sequestration ensues.

Causes
• Idiopathic (majority)
• Associated with
– Waldenström's (IgM paraproteinaemia)
– Mycoplasma pneumonia
– EBV infection

Symptoms and Signs
• Related to RBC agglutination on exposure to
cold
• Mottled or numb fingers or toes.
• Hemolytic anaemia is rarely severe,
• Episodic hemoglobinuria may occur on exposure to cold.

Laboratory Findings
• Mild anaemia
• Reticulocytosis and spherocytes.
• Direct Coomb's test -positive for complement only.
• Bedside cold agglutinin test – place a glass slide in ice – Put a few drops of heparinized blood on it. – Observe for small clumps of agglutinated blood.

Cold Agglutinin Disease
• Essentials of Diagnosis
– Increased reticulocytes and spherocytes on peripheral blood smear.
– Coomb's test positive only for complement.
– Positive cold agglutinin test.

Treatment
• Largely symptomatic,
• Avoid exposure to cold.
• Splenectomy and prednisone are usually ineffective since hemolysis takes place in the liver.
• Rituximab, a monoclonal antibody directed against the CD20 antigen on B lymphocytes, is the treatment of choice.
• Other agents: cyclophosphamide+/-cyclosporine.
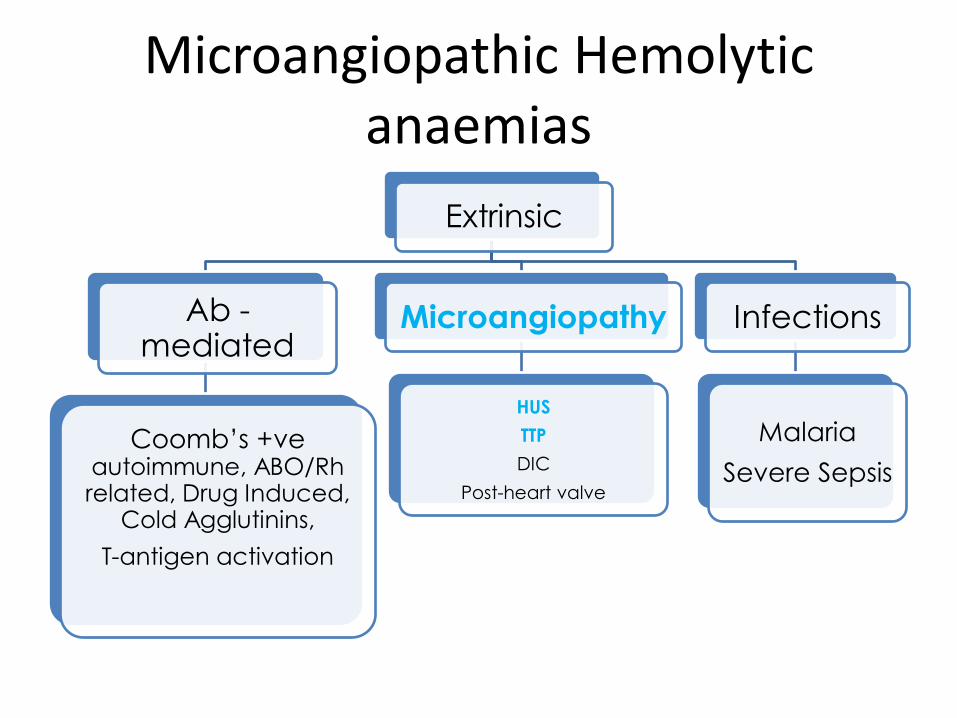
Microangiopathic Hemolytic anaemias
Extrinsic
Ab -mediated
Coomb’s +ve autoimmune, ABO/Rh related, Drug Induced,
Cold Agglutinins,
T-antigen activation
Microangiopathy
HUS
TTP
DIC
Post-heart valve
Infections
Malaria
Severe Sepsis

Microangiopathic Hemolytic anaemias
• Red Cell Fragmentation takes place intravascular space
• Hallmark of disorder is: finding of fragmented red blood cells (schistocytes, helmet cells) on the peripheral blood smear.
• Plus features of hemolysis
• In severe cases, methemalbuminemia.

Causes
• Thrombotic thrombocytopenic purpura (TTP) • HUS • DIC • Heart Valve Prosthesis • Vasculitis
• Clinical features are variable and depend on the underlying
disorder. Coagulopathy and thrombocytopenia are variably present.
• Chronic microangiopathic hemolytic anaemia (such as is present with a malfunctioning cardiac valve prosthesis) may cause iron deficiency anaemia because of continuous low-grade hemoglobinuria.
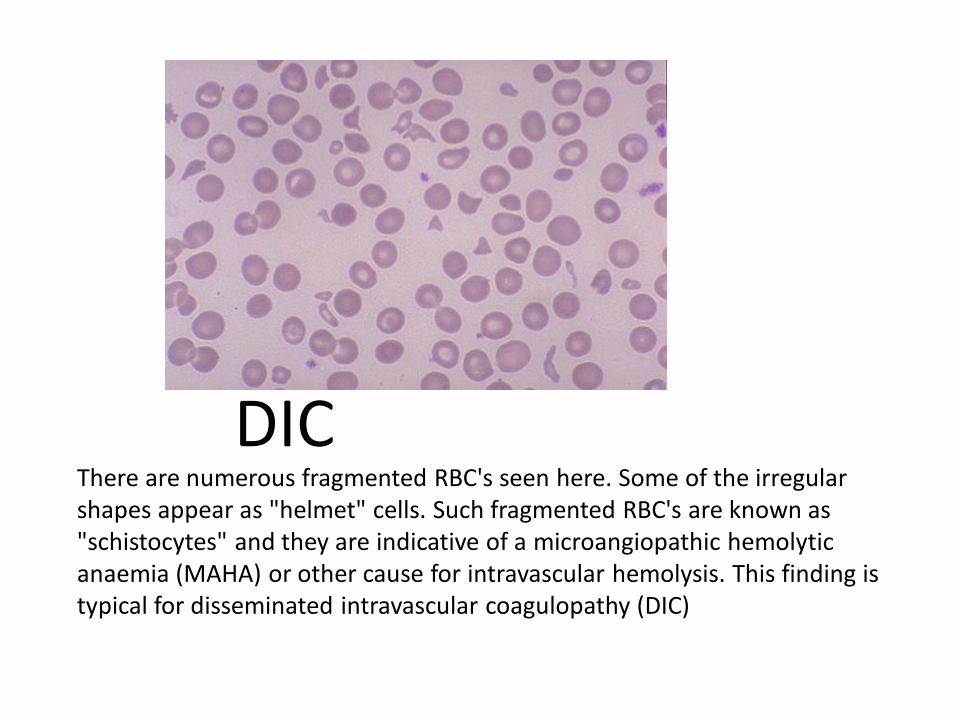
There are numerous fragmented RBC's seen here. Some of the irregular shapes appear as "helmet" cells. Such fragmented RBC's are known as "schistocytes" and they are indicative of a microangiopathic hemolytic anaemia (MAHA) or other cause for intravascular hemolysis. This finding is typical for disseminated intravascular coagulopathy (DIC)
DIC
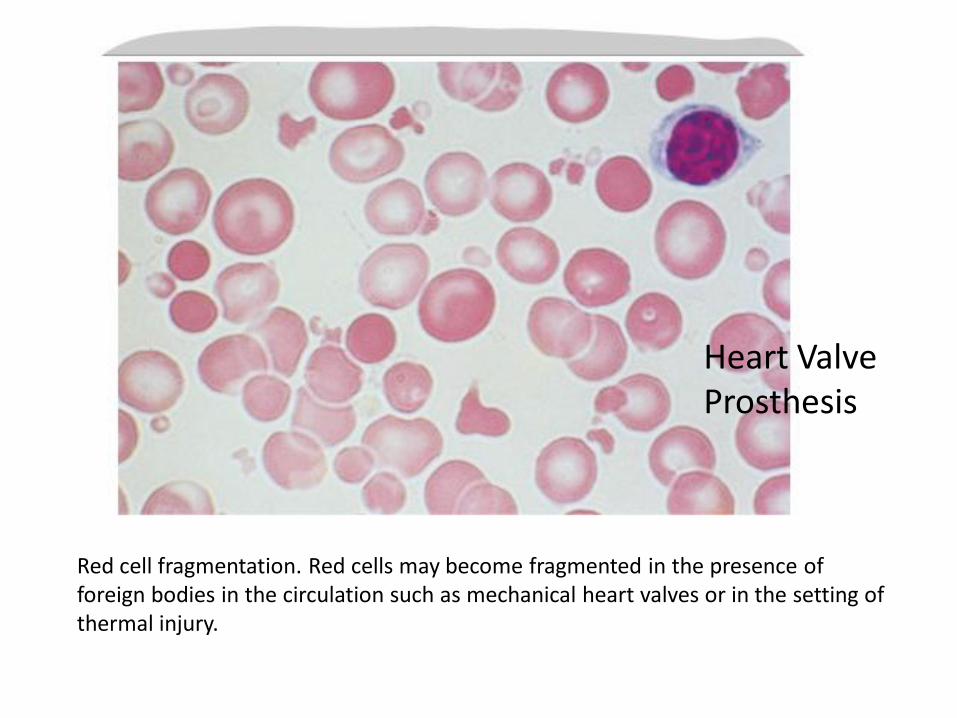
Red cell fragmentation. Red cells may become fragmented in the presence of foreign bodies in the circulation such as mechanical heart valves or in the setting of thermal injury.
Heart Valve Prosthesis
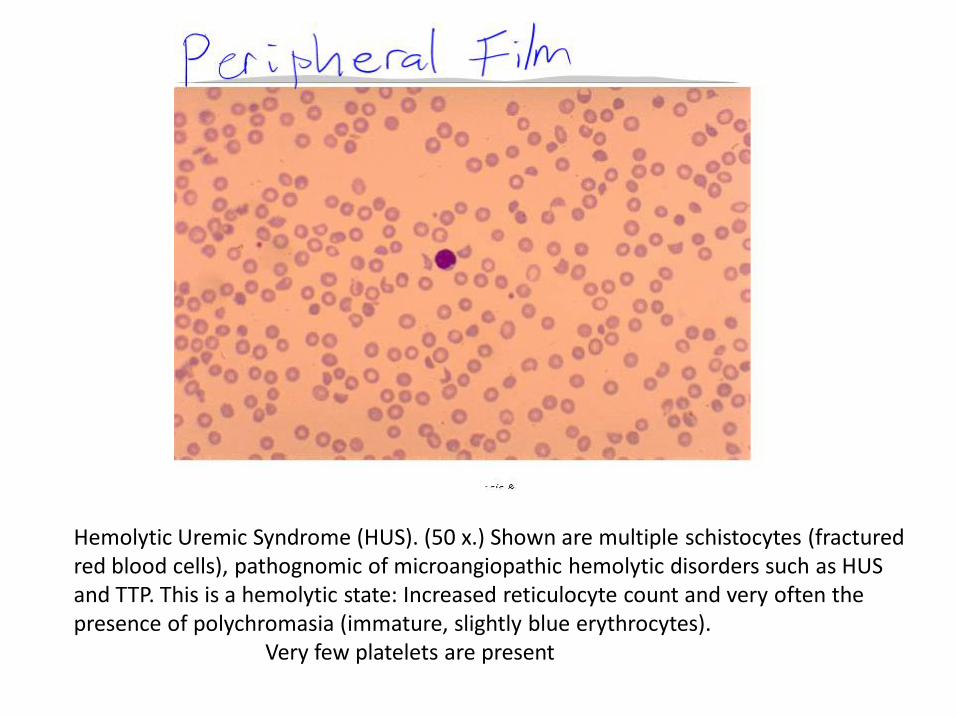
Hemolytic Uremic Syndrome (HUS). (50 x.) Shown are multiple schistocytes (fractured red blood cells), pathognomic of microangiopathic hemolytic disorders such as HUS and TTP. This is a hemolytic state: Increased reticulocyte count and very often the presence of polychromasia (immature, slightly blue erythrocytes). Very few platelets are present

Thrombotic Thrombocytopenic Purpura (TTP)

General Considerations • Is an uncommon syndrome
• No-ninfectious fever, neurologic disorders, and renal
abnormalities are less commonly seen.
• Pathogenesis: Adult TTP
– Deficiency of a von Willebrand factor-cleaving protease, ADAMTS13, in some cases due to an antibody directed against the protease.
– Results in accumulation of ultralarge multimers of vWF accumulate
– Lead to platelet agglutination and adhesion to endothelium.
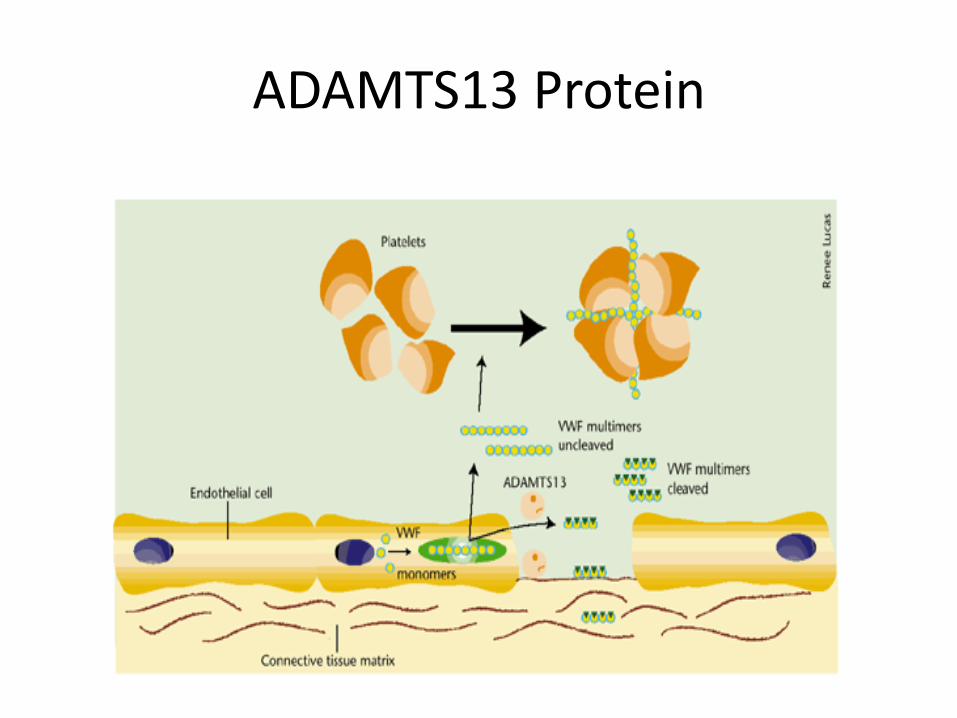
ADAMTS13 Protein
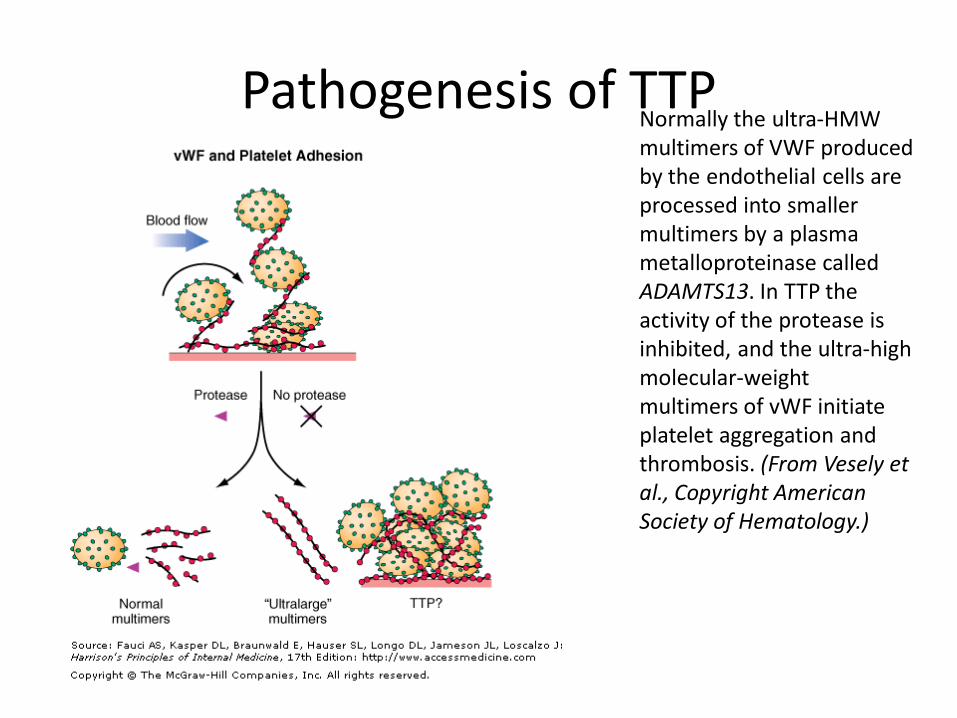
Pathogenesis of TTP Normally the ultra-HMW multimers of VWF produced by the endothelial cells are processed into smaller multimers by a plasma metalloproteinase called ADAMTS13. In TTP the activity of the protease is inhibited, and the ultra-high molecular-weight multimers of vWF initiate platelet aggregation and thrombosis. (From Vesely et al., Copyright American Society of Hematology.)

• TTP in young adult (20 and 50 years)
– F>M
– Precipitated by oestrogen use, pregnancy, drugs (quinine) and infections
• The syndrome may also occur as a complication of bone marrow transplantation or the use of cyclosporine or tacrolimus.
• Rarely familial cases

Symptoms and Signs
• Anaemia and Bleeding
• Neurological symptoms (headache, confusion, aphasia, and alterations in consciousness from lethargy to coma, or even hemiparesis and seizures)
• acutely ill and is usually febrile.
• Pallor, purpura, petechiae, and signs of neurologic dysfunction may be detected
• May have abdominal pain and tenderness due to pancreatitis

Laboratory Findings
• Signs of Hemolytic Anaemia
• Signs of MAHA (fragmented red blood cells-schistocytes, helmet cells, triangle forms)
• Coomb's test is negative.
• Coagulation tests (PT, APTT, Fibrinogen ) are normal
• Azotaemia (renal failure)
• ADAMTS13 is usually absent
• Thrombi in capillaries and small arteries (biopsy)

Thrombotic Thrombocytopenic Purpura
• Essentials of Diagnosis
– Thrombocytopenia.
– Microangiopathic hemolytic anaemia.
– Neurologic and renal abnormalities, fever.
– Reduced level of ADAMTS13.
– Normal coagulation tests.

Differential Diagnosis
• DIC (abnormal PT, APTT)
• HUS (more renal failure, no CNS symptoms)
• Vasculitis (skin biopsy)
• AIHA (Coomb's positive )
•

Treatment
• Plasmapheresis (1st choice)
• Antiplatelets (aspirin)
• Steroids (prednisolone)
• Immunosupression (cyclophopshamide)

Prognosis
• Complete recovery 80-90%.
•
• Neurologic abnormalities are almost always completely reversed.
• 20% of cases the disease will be chronic and relapsing.

Haemolytic Uraemic Syndrome (HUS)

General Considerations • HUS is an uncommon disorder
• Causes MAHA, thrombocytopenia and renal failure (haematuria & proteinuria)
• Cause is unclear.
• The disease is similar to TTP except that different vascular beds are involved.
• In fact, the two diseases are probably best considered as part of a spectrum of
HUS–TTP disorders.
• Pathogenesis of the two disorders is probably similar (platelet-agglutinating factor found in plasma may be involved)
• .

• In children,
– occurs after a diarrheal illness secondary to infections with Shigella, Salmonella, E coli strain O157:H7, or viral agents.
– The mortality rate of this form is low (< 5%).

• In adults
– Often precipitated by:
• estrogen use
• Postpartum state
• Drugs (high-dose corticosteroid therapy, cyclosporine or tacrolimus)
– Autologous bone marrow or stem cell transplantation
• Familial Type--members of a family have recurrent episodes over several years.

Symptoms and Signs
• Anaemia
• Bleeding
• Renal Failure
• In contrast to TTP, there are no neurologic manifestations other than those due to the uremic state.

Laboratory Findings • As in TTP
• Thrombocytopenia often less severe
• Coomb's test is negative.
• Coagulation tests are normal with the exception of elevated
FDP.
• As in TTP, levels of ADAMTS13 are usually low.
• Kidney biopsy -show endothelial hyaline thrombi in the afferent arterioles and glomeruli.
• Ischemic necrosis in the renal cortex may occur with obstruction from intravascular coagulation.

Haemolytic Uraemic Syndrome
• Essentials of Diagnosis
– Microangiopathic hemolytic anaemia.
– Thrombocytopenia and renal failure.
– Normal coagulation tests.
– Absence of neurologic abnormalities.

Differential Diagnosis
• DIC (abnormal PT, APTT)
• TTP (neurologic features)

Treatment • In children,
– is almost always self-limited ,
– Conservatively treat ARF
• In adults, if untreated,
– there is a high rate of permanent renal insufficiency
and death.
– Daily plasmapheresis + FFP treatment of choice

Prognosis
• Children –good
• Adults -remains unclear.
– No treatment- up to 40% died, and 80% have had chronic renal insufficiency.
– Early aggressive treatment - improved survival.

Infections- hemolysis
• Sepsis
• Malaria
• Viral infections

References
• Harrisons Textbook of Medicine 17th edition
• Current Medical Diagnosis & Treatment 2008
• Emedicine.medscape.com





























