Embed Size (px)
Citation preview

Molecular and Cellular Pathobiology
Hedgehog-Producing Cancer Cells Respond to and RequireAutocrine Hedgehog Activity
Samer Singh1, Zhiqiang Wang1, Dennis Liang Fei1,5, Kendall E. Black1, John A. Goetz5, Robert Tokhunts1,5,Camilla Giambelli1, Jezabel Rodriguez-Blanco1, Jun Long1, Ethan Lee7, Karoline J. Briegel2,3,Pablo A. Bejarano4, Ethan Dmitrovsky5,6, Anthony J. Capobianco1,2, and David J. Robbins1,2
AbstractA number of Smoothened (SMO) pathway antagonists are currently undergoing clinical trials as anticancer
agents. These drugs are proposed to attenuate tumor growth solely through inhibition of Hedgehog (HH), whichis produced in tumor cells but acts on tumor stromal cells. The pivotal argument underlying this model is thatthe growth-inhibitory properties of SMO antagonists on HH-producing cancer cells are due to their off-targeteffects. Here, we show that the tumorigenic properties of such lung cancer cells depend on their intrinsic level ofHH activity. Notably, reducing HH signaling in these tumor cells decreases HH target gene expression. Takentogether, these results question the dogma that autocrine HH signaling plays no role in HH-dependent cancers,and does so without using SMO antagonists. Cancer Res; 71(13); 4454–63. �2011 AACR.
Introduction
Hedgehog (HH) signaling plays a critical role in the homeo-stasis of diverse adult tissues. Consistent with its importantrole in tissue maintenance, deregulated HH signaling has beenimplicated in common human cancers of different origins,such as melanoma, leukemia, glioblastoma, and those derivedfrom tumors of the breast, digestive tract, pancreas, lung, andkidney (1, 2). Mammals express 3 HH family members, Sonichedgehog (SHH), Desert hedgehog (DHH), and Indian hedge-hog (IHH), whose binding to their receptor Patched1 (PTCH1;refs. 3, 4) relieves PTCH1-mediated repression of Smoothened(SMO) activity (5–7). This active SMO modulates HH activitythrough regulation of the GLI family of transcription factorsGLI1, GLI2, and GLI3 (8–10) in a manner that is antagonizedby the GLI interacting protein suppressor of fused (SUFU; refs.11–13). Aberrant activation of the HH pathway in cancerresults from the overexpression of HH proteins, activating
mutations in downstream pathway components such as SMO,PTCH1, or SUFU, or gene amplification of GLI1—a knowntranscriptional effector and HH target gene (1, 2).
HH has a survival role for diverse cancers in which nomutations in HH signaling components were identified (14–20). Investigators used various SMO antagonists (21–24) toshow the HH signaling requirement of cancer cells. Recently, itwas suggested that these SMO antagonists exhibit off-targeteffects when used at the concentrations necessary to inhibitthe proliferation of HH-producing cancer cells (25). Thesearguments were based on the observation that a higherconcentration of SMO antagonist is required to inhibit tumorcell proliferation than that required to inhibit HH activity infibroblasts. Moreover, a lack of association between expressionof the HH target gene GLI1 and sensitivity of a large panel ofcancer cell lines to SMO antagonists was noted (25). Thisargument was developed in vivo, using human primary cancersor established cancer cell lines carried as xenografts in athymicmice, or mouse models of pancreatic cancer, to suggest thatSMO antagonists act exclusively on the tumor stroma (25–27).One of these studies also showed that, whereas SMO anta-gonists had marked effects in reducing the expression ofstromal derived mouse GLI1, it was unable to reduce humanGLI1 expression in the cancer cells themselves (25).
We previously reported that primary non–small cell lungcarcinomas (NSCLC) frequently elaborate a constitutivelyactive HH signaling pathway, which appeared to originate inthe cancer cells themselves (28, 29). Using 2 structurally dis-tinct SMO antagonists, we showed that a subset of NSCLC celllines require HH activity for their viability. Given the debatesurrounding the specificity of SMO antagonists, this studysought to elucidate the role HH signaling plays in regulatingthe tumorigenicity of NSCLC cells. This study reports thedependence of NSCLC cells on HH activity for their prolifera-tion and tumorigenesis, primarily using a loss-of-function
Authors' Affiliations: 1Molecular Oncology Program, Department of Sur-gery; 2Sylvester Cancer Center; 3Department of Biochemistry and Mole-cular Biology, Braman Family Breast Cancer Institute, Miller School ofMedicine, Miller School of Medicine, University of Miami, Miami, Florida;4Department of Pathology, Jackson Memorial Hospital, University ofMiami, Miami, Florida; 5Department of Pharmacology and Toxicology,Dartmouth Medical School, Hanover, New Hampshire; and 6Norris CottonCancer Center, Dartmouth Medical School and Dartmouth-HitchcockMedical Center, Lebanon, New Hampshire; 7Department of Cell andDevelopmental Biology, Vanderbilt University School of Medicine, Nash-ville, Tennessee
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author: David J. Robbins, Department of Surgery, MillerSchool of Medicine, University of Miami, 1042 RMSB, 1600 NW 10thAvenue, Miami, FL 33136. Phone: 305-243-5717; Fax: 305-243-2810;E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-10-2313
�2011 American Association for Cancer Research.
CancerResearch
Cancer Res; 71(13) July 1, 20114454
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313

approach that is independent of the use of SMO antagonists.Conversely, overexpression of HH proteins increased thetumorigenic properties of NSCLC cells. Furthermore, reducingGLI1 expression in NSCLC cells significantly reduced thetumorigenicity of NSCLC xenografts in vivo. These findingssuggest that NSCLC cells elaborate a highly active HH signalingpathway that is required for their in vitro growth and in vivotumorigenicity.
Materials and Methods
NSCLC cell lines HOP62, A549, H23, and H522, wereobtained from the Developmental Therapeutics Program(DTP), and U1752 and H157 were a gift from Dr. Neil Watkinsand Dr. Stephen Baylin (Johns Hopkins University, Baltimore,MD). These cell lines were maintained in RPMI-1640 mediumwith 10% FBS (Invitrogen) supplemented with penicillin andstreptomycin. SHH-Light2 cells (30) and SHH-I cells (23, 31)were cultured as described. The various chemical inhibitorswere obtained or purchased (DLF and derivatives: DTP; DMSOand Tomatidine: Sigma Aldrich; SANT1: Calbiochem), andtheir inhibition of HH (conditioned medium; SHH-I cells)signaling assayed in SHH-Light2 cells (30). All experimentswere repeated independently at least 3 times. Unless notedotherwise, data shown are representative, error bars representSD from the mean, and statistical significance was calculatedby Student 2-tailed t test. The values of P < 0.05 were consi-dered statistically significant and denoted by an asterisk (*).
AssaysExpression of the indicated genes was assessed by quanti-
tative real-time PCR (qRT-PCR) as described (ref. 32; andSupplementary Table S1). NSCLC cell lines were plated atdifferent cell densities (0.8–2� 103 cells) in 96-well plates. Thenext day, cells were transduced with short-hairpin RNA(shRNA) expressing lentiviruses, purchased from open biosys-tems (Supplementary Table S2), and packaged according tothe manufacturer's directions. Cell proliferation and apoptosiswere measured 5 days after transduction, using the CellTiter-Glo assay kit and Caspase-Glo 3/7 assay kit (Promega Inc.),respectively. The level of GLI1 protein in different shRNAtransduced NSCLC cell lines was determined by immunoblot-ting for GLI1 as described previously (32). The polyclonalHOP62 cells stably expressing human SHH or an insertlesscontrol pcDNA3.1 vector were engineered. In brief, HOP62cells were transfected with SHH or pcDNA3.1 and then drugselected [Geneticin (Gibco), 1,000 mg/mL] for 3 to 4 weeks, andthe early passage polyclonal cell lines expressing SHH (31)were grown in soft agar as described (28). The colonies formedafter 3 to 4 weeks of incubation were visualized by MTTstaining and quantitated (33).
Nude mouse xenograftsAll mouse studies were done in accordance with the policies
of the University of Miami Institutional Animal Care and UseCommittee (IACUC). A549 cells transduced with GLI1 shRNA#1, GLI1 shRNA #5 or a control (Ctrl) scrambled shRNA weremixed with an equal volume of Matrigel and injected s.c. in the
flank of Nu/Nu nude mice (Crl:NU-Foxn1nu, Charles RiverLaboratories) at 1.5 � 106 cells per inoculation site (n ¼12). Tumor growth was measured for 6 weeks and the tumorvolume was calculated by using a modified ellipsoidal formula,Volume ¼ (S�S�L)/2, where S and L are the short and longdimensions, respectively.
Immunohistochemistry and in situ hybridizationTissue samples resected from NSCLC patients were fixed in
formalin, paraffin embedded, and cut into 5-mm thick sectionsto prepare slides according to an Institutional Review Board–approved protocol or purchased (US Biomax) and then probedfor the expression of SHH, PTCH, and GLI1 proteins by usinganti-SHH [H-160, Santa Cruz Biotechnology (SCBT)], anti-PTCH (C-20, SCBT), and anti-GLI1 antibody (32), respectively.RNA in situ hybridization on adjacent sections was done toexamine the expression of SHH, PTCH1, and GLI1 by usingdigoxigenin-labeled antisense RNA probes as previouslydescribed (20, 34). The plasmids used to make RNA probeswere a gift from Dr. Ariel Ruiz i Altaba (University of GenevaMedical School, Switzerland). All images were acquired byusing a Leica DMIRB microscope.
Results
DTP evaluates novel small molecule inhibitors of cancer cellproliferation (28). At the DTP, more than 70,000 differentcompounds have been screened for their ability to inhibitthe growth of a well-characterized panel of 60 cancer cell lines(NCI-60). The concentration of each compound that results ina 50% growth inhibition of each cell line (GI50) within the NCI-60 panel is determined. Taken together, individual GI50 acrossall 60 cell lines, for any specific compound, gives each com-pound a unique "signature" profile. When queried against theNCI-60 panel such a signature can be used to identify differentmolecules that inhibit similar biological processes. Previously,we had the growth-inhibitory signature of the SMO antagonistSANT1 (30) determined at the DTP. These results were sub-sequently confirmed in our laboratory, using the second SMOantagonist cyclopamine and a panel of NSCLC cell lines (28).This work provided a validated SMO inhibitor signature(Fig. 1A).
Contrary to what has been recently proposed (25), ourprevious results indicated that SMO antagonists inhibit theproliferation of NSCLC cells in a specific manner. If indeedthese SMO antagonists specifically attenuated NSCLC cellgrowth, we reasoned that compounds identified at the DTPbased on their similar GI50 signature to that of SANT1 wouldfunction to inhibit HH signaling. The DTP database was thenqueried for compounds that exhibit a similar growth-inhibi-tory signature to that of SANT1, and 4 compounds wereidentified that we designated DLF1–4. The ability of thesecompounds to inhibit HH-dependent activity was tested inSHH-Light2 cells (30), which stably express a HH-dependentluciferase reporter construct (Fig. 1B). Among the compoundstested, DLF3 reduced HH signaling with a similar potency tocyclopamine. Several structural derivatives of DLF3 wereavailable at the DTP, which we subsequently obtained and
Hedgehog Signaling Is Required in Lung Cancer
www.aacrjournals.org Cancer Res; 71(13) July 1, 2011 4455
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313
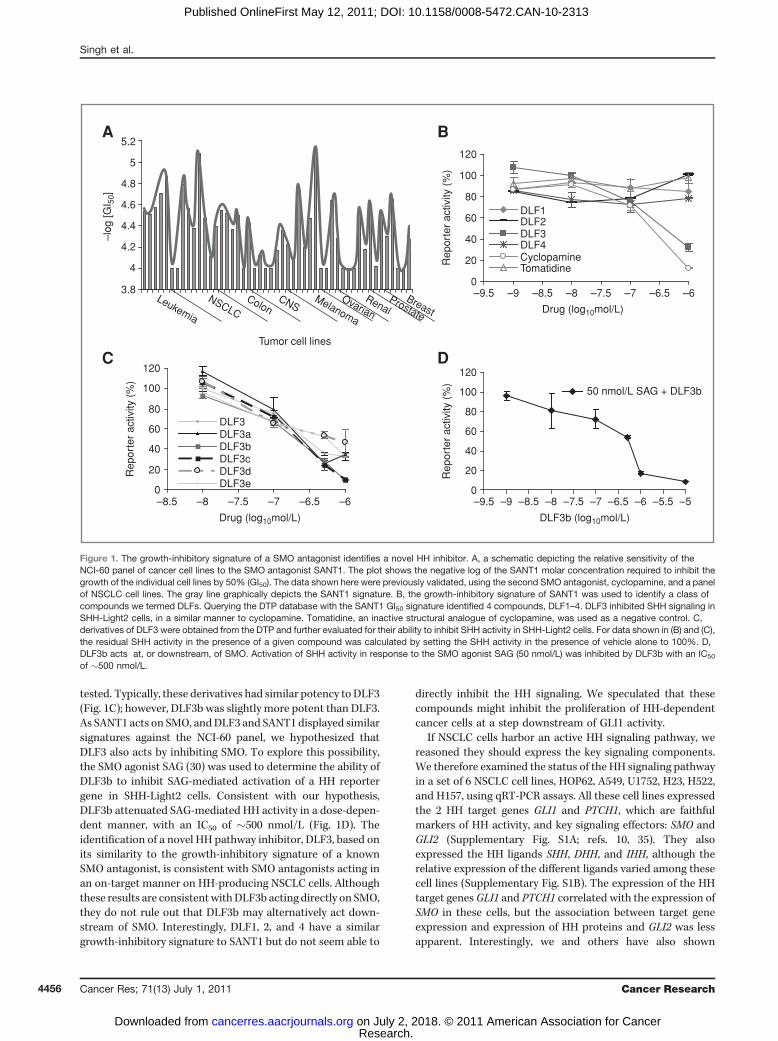
tested. Typically, these derivatives had similar potency toDLF3(Fig. 1C); however, DLF3b was slightly more potent than DLF3.As SANT1 acts on SMO, andDLF3 and SANT1 displayed similarsignatures against the NCI-60 panel, we hypothesized thatDLF3 also acts by inhibiting SMO. To explore this possibility,the SMO agonist SAG (30) was used to determine the ability ofDLF3b to inhibit SAG-mediated activation of a HH reportergene in SHH-Light2 cells. Consistent with our hypothesis,DLF3b attenuated SAG-mediated HH activity in a dose-depen-dent manner, with an IC50 of �500 nmol/L (Fig. 1D). Theidentification of a novel HH pathway inhibitor, DLF3, based onits similarity to the growth-inhibitory signature of a knownSMO antagonist, is consistent with SMO antagonists acting inan on-target manner on HH-producing NSCLC cells. Althoughthese results are consistentwithDLF3b acting directly on SMO,they do not rule out that DLF3b may alternatively act down-stream of SMO. Interestingly, DLF1, 2, and 4 have a similargrowth-inhibitory signature to SANT1 but do not seem able to
directly inhibit the HH signaling. We speculated that thesecompounds might inhibit the proliferation of HH-dependentcancer cells at a step downstream of GLI1 activity.
If NSCLC cells harbor an active HH signaling pathway, wereasoned they should express the key signaling components.We therefore examined the status of the HH signaling pathwayin a set of 6 NSCLC cell lines, HOP62, A549, U1752, H23, H522,and H157, using qRT-PCR assays. All these cell lines expressedthe 2 HH target genes GLI1 and PTCH1, which are faithfulmarkers of HH activity, and key signaling effectors: SMO andGLI2 (Supplementary Fig. S1A; refs. 10, 35). They alsoexpressed the HH ligands SHH, DHH, and IHH, although therelative expression of the different ligands varied among thesecell lines (Supplementary Fig. S1B). The expression of the HHtarget genes GLI1 and PTCH1 correlated with the expression ofSMO in these cells, but the association between target geneexpression and expression of HH proteins and GLI2 was lessapparent. Interestingly, we and others have also shown
A B
C D
0
20
40
60
80
100
120
–8.5 –8 –7.5 –7 –6.5 –6
Rep
orte
r ac
tivity
(%
)
DLF3DLF3aDLF3bDLF3cDLF3dDLF3e
0
20
40
60
80
100
120
–9.5 –9 –8.5 –8 –7.5 –7 –6.5 –6
Rep
orte
r ac
tivity
(%
)
DLF1DLF2DLF3DLF4CyclopamineTomatidine
Tumor cell lines
BreastLeukemia
NSCLCColon
CNSMelanoma
Ovarian
RenalProstate
5.2
5
4.8
4.6
4.4
4.2
4
3.8
–log
[GI 5
0]
0
20
40
60
80
100
120
–9.5 –9 –8.5 –8 –7.5 –7 –6.5 –6 –5.5 –5
Rep
orte
r ac
tivity
(%
)
50 nmol/L SAG + DLF3b
Drug (log10mol/L)
DLF3b (log10mol/L)Drug (log10mol/L)
Figure 1. The growth-inhibitory signature of a SMO antagonist identifies a novel HH inhibitor. A, a schematic depicting the relative sensitivity of theNCI-60 panel of cancer cell lines to the SMO antagonist SANT1. The plot shows the negative log of the SANT1 molar concentration required to inhibit thegrowth of the individual cell lines by 50% (GI50). The data shown here were previously validated, using the second SMO antagonist, cyclopamine, and a panelof NSCLC cell lines. The gray line graphically depicts the SANT1 signature. B, the growth-inhibitory signature of SANT1 was used to identify a class ofcompounds we termed DLFs. Querying the DTP database with the SANT1 GI50 signature identified 4 compounds, DLF1–4. DLF3 inhibited SHH signaling inSHH-Light2 cells, in a similar manner to cyclopamine. Tomatidine, an inactive structural analogue of cyclopamine, was used as a negative control. C,derivatives of DLF3 were obtained from the DTP and further evaluated for their ability to inhibit SHH activity in SHH-Light2 cells. For data shown in (B) and (C),the residual SHH activity in the presence of a given compound was calculated by setting the SHH activity in the presence of vehicle alone to 100%. D,DLF3b acts at, or downstream, of SMO. Activation of SHH activity in response to the SMO agonist SAG (50 nmol/L) was inhibited by DLF3b with an IC50
of �500 nmol/L.
Singh et al.
Cancer Res; 71(13) July 1, 2011 Cancer Research4456
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313
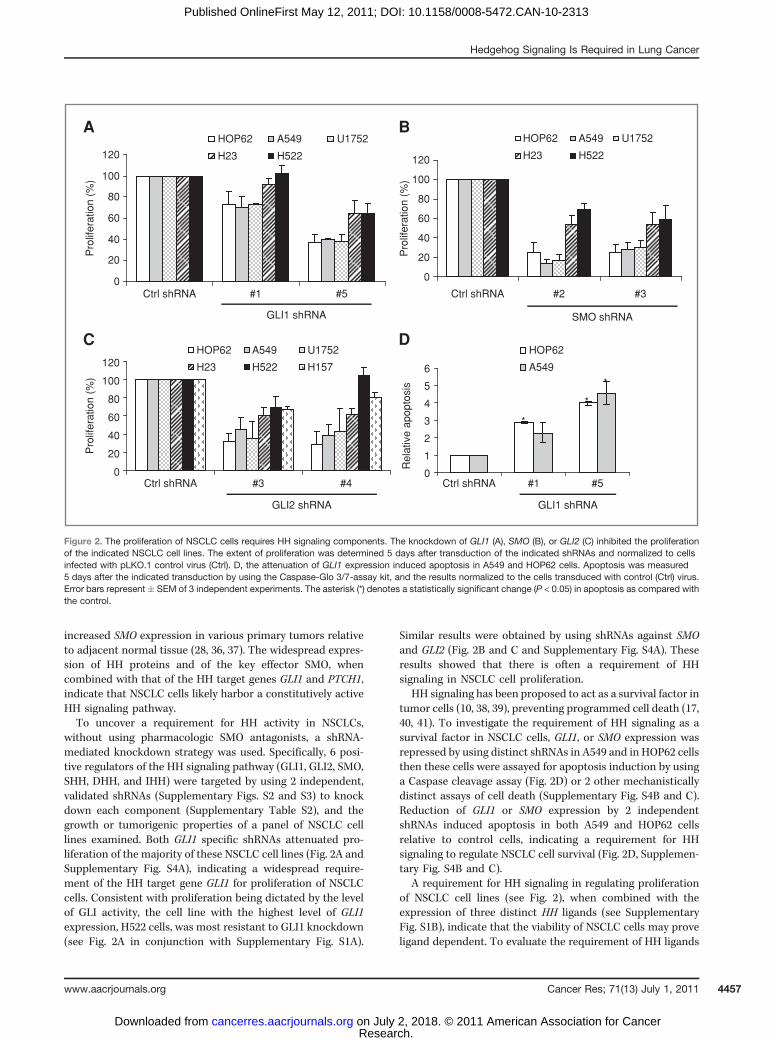
increased SMO expression in various primary tumors relativeto adjacent normal tissue (28, 36, 37). The widespread expres-sion of HH proteins and of the key effector SMO, whencombined with that of the HH target genes GLI1 and PTCH1,indicate that NSCLC cells likely harbor a constitutively activeHH signaling pathway.To uncover a requirement for HH activity in NSCLCs,
without using pharmacologic SMO antagonists, a shRNA-mediated knockdown strategy was used. Specifically, 6 posi-tive regulators of the HH signaling pathway (GLI1, GLI2, SMO,SHH, DHH, and IHH) were targeted by using 2 independent,validated shRNAs (Supplementary Figs. S2 and S3) to knockdown each component (Supplementary Table S2), and thegrowth or tumorigenic properties of a panel of NSCLC celllines examined. Both GLI1 specific shRNAs attenuated pro-liferation of the majority of these NSCLC cell lines (Fig. 2A andSupplementary Fig. S4A), indicating a widespread require-ment of the HH target gene GLI1 for proliferation of NSCLCcells. Consistent with proliferation being dictated by the levelof GLI activity, the cell line with the highest level of GLI1expression, H522 cells, was most resistant to GLI1 knockdown(see Fig. 2A in conjunction with Supplementary Fig. S1A).
Similar results were obtained by using shRNAs against SMOand GLI2 (Fig. 2B and C and Supplementary Fig. S4A). Theseresults showed that there is often a requirement of HHsignaling in NSCLC cell proliferation.
HH signaling has been proposed to act as a survival factor intumor cells (10, 38, 39), preventing programmed cell death (17,40, 41). To investigate the requirement of HH signaling as asurvival factor in NSCLC cells, GLI1, or SMO expression wasrepressed by using distinct shRNAs in A549 and in HOP62 cellsthen these cells were assayed for apoptosis induction by usinga Caspase cleavage assay (Fig. 2D) or 2 other mechanisticallydistinct assays of cell death (Supplementary Fig. S4B and C).Reduction of GLI1 or SMO expression by 2 independentshRNAs induced apoptosis in both A549 and HOP62 cellsrelative to control cells, indicating a requirement for HHsignaling to regulate NSCLC cell survival (Fig. 2D, Supplemen-tary Fig. S4B and C).
A requirement for HH signaling in regulating proliferationof NSCLC cell lines (see Fig. 2), when combined with theexpression of three distinct HH ligands (see SupplementaryFig. S1B), indicate that the viability of NSCLC cells may proveligand dependent. To evaluate the requirement of HH ligands
A B
C D
0
20
40
60
80
100
120
Ctrl shRNA #3 #4
Pro
lifer
atio
n (%
)
HOP62 A549 U1752
H23 H522 H157
GLI1 shRNA
0
1
2
3
4
5
6
Ctrl shRNA #1 #5
Rel
ativ
e ap
opto
sis
HOP62
A549
*
*
*
0
20
40
60
80
100
120
Ctrl shRNA #1 #5
Pro
lifer
atio
n (%
)
HOP62 A549 U1752
H23 H522
GLI1 shRNA
0
20
40
60
80
100
120
Ctrl shRNA #2 #3
Pro
lifer
atio
n (%
)
HOP62 A549 U1752
H23 H522
SMO shRNA
GLI2 shRNA
Figure 2. The proliferation of NSCLC cells requires HH signaling components. The knockdown of GLI1 (A), SMO (B), or GLI2 (C) inhibited the proliferationof the indicated NSCLC cell lines. The extent of proliferation was determined 5 days after transduction of the indicated shRNAs and normalized to cellsinfected with pLKO.1 control virus (Ctrl). D, the attenuation of GLI1 expression induced apoptosis in A549 and HOP62 cells. Apoptosis was measured5 days after the indicated transduction by using the Caspase-Glo 3/7-assay kit, and the results normalized to the cells transduced with control (Ctrl) virus.Error bars represent � SEM of 3 independent experiments. The asterisk (*) denotes a statistically significant change (P < 0.05) in apoptosis as compared withthe control.
Hedgehog Signaling Is Required in Lung Cancer
www.aacrjournals.org Cancer Res; 71(13) July 1, 2011 4457
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313
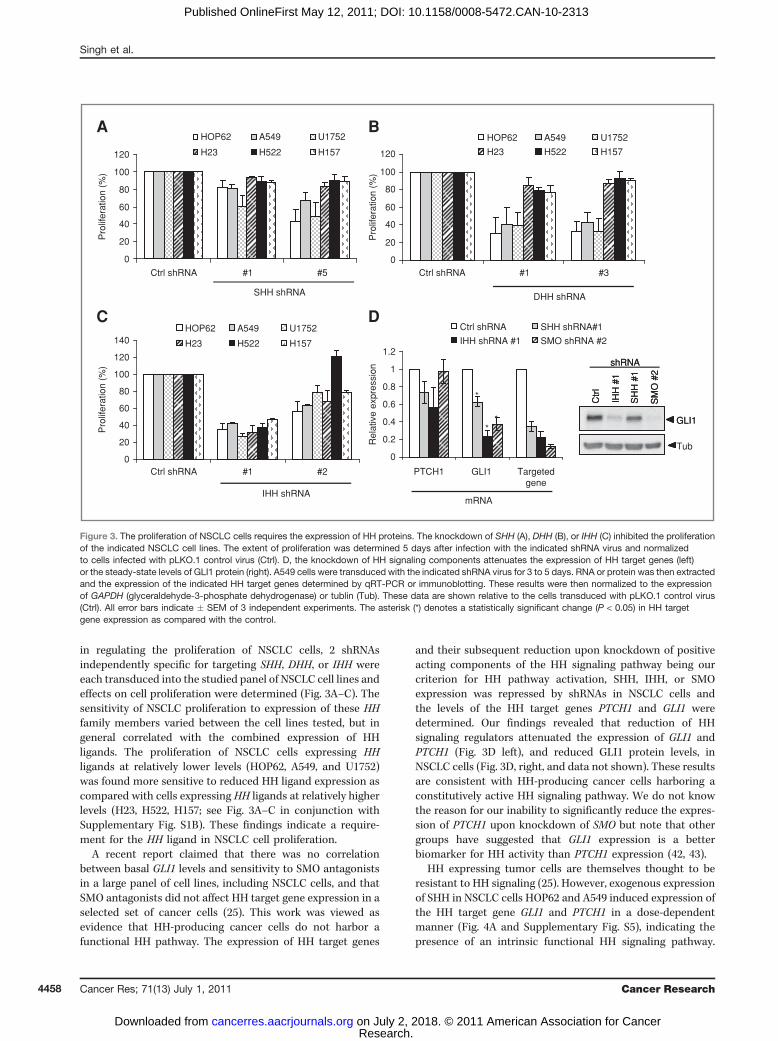
in regulating the proliferation of NSCLC cells, 2 shRNAsindependently specific for targeting SHH, DHH, or IHH wereeach transduced into the studied panel of NSCLC cell lines andeffects on cell proliferation were determined (Fig. 3A–C). Thesensitivity of NSCLC proliferation to expression of these HHfamily members varied between the cell lines tested, but ingeneral correlated with the combined expression of HHligands. The proliferation of NSCLC cells expressing HHligands at relatively lower levels (HOP62, A549, and U1752)was found more sensitive to reduced HH ligand expression ascompared with cells expressing HH ligands at relatively higherlevels (H23, H522, H157; see Fig. 3A–C in conjunction withSupplementary Fig. S1B). These findings indicate a require-ment for the HH ligand in NSCLC cell proliferation.
A recent report claimed that there was no correlationbetween basal GLI1 levels and sensitivity to SMO antagonistsin a large panel of cell lines, including NSCLC cells, and thatSMO antagonists did not affect HH target gene expression in aselected set of cancer cells (25). This work was viewed asevidence that HH-producing cancer cells do not harbor afunctional HH pathway. The expression of HH target genes
and their subsequent reduction upon knockdown of positiveacting components of the HH signaling pathway being ourcriterion for HH pathway activation, SHH, IHH, or SMOexpression was repressed by shRNAs in NSCLC cells andthe levels of the HH target genes PTCH1 and GLI1 weredetermined. Our findings revealed that reduction of HHsignaling regulators attenuated the expression of GLI1 andPTCH1 (Fig. 3D left), and reduced GLI1 protein levels, inNSCLC cells (Fig. 3D, right, and data not shown). These resultsare consistent with HH-producing cancer cells harboring aconstitutively active HH signaling pathway. We do not knowthe reason for our inability to significantly reduce the expres-sion of PTCH1 upon knockdown of SMO but note that othergroups have suggested that GLI1 expression is a betterbiomarker for HH activity than PTCH1 expression (42, 43).
HH expressing tumor cells are themselves thought to beresistant to HH signaling (25). However, exogenous expressionof SHH in NSCLC cells HOP62 and A549 induced expression ofthe HH target gene GLI1 and PTCH1 in a dose-dependentmanner (Fig. 4A and Supplementary Fig. S5), indicating thepresence of an intrinsic functional HH signaling pathway.
0
20
40
60
80
100
120
Pro
lifer
atio
n (%
)
HOP62 A549 U1752
H23 H522 H157
A B
C D
SHH shRNA
0
20
40
60
80
100
120
Pro
lifer
atio
n (%
)
HOP62 A549 U1752
H23 H522 H157
DHH shRNA
0
20
40
60
80
100
120
140
Ctrl shRNA #1 #2
Ctrl shRNA #1 #5 Ctrl shRNA #1 #3
Pro
lifer
atio
n (%
)
HOP62 A549 U1752
H23 H522 H157
IHH shRNA
0
0.2
0.4
0.6
0.8
1
1.2
PTCH1 GLI1 Targetedgene
Rel
ativ
e ex
pres
sion
Ctrl shRNA SHH shRNA#1
IHH shRNA #1 SMO shRNA #2
mRNA
**
* Ctr
l
IHH
#1
SH
H #
1
SM
O #
2
GLI1
shRNA
Ctr
l
IHH
#1
SH
H #
1
SM
O #
2
GLI1
Tub
shRNA
Figure 3. The proliferation of NSCLC cells requires the expression of HH proteins. The knockdown of SHH (A), DHH (B), or IHH (C) inhibited the proliferationof the indicated NSCLC cell lines. The extent of proliferation was determined 5 days after infection with the indicated shRNA virus and normalizedto cells infected with pLKO.1 control virus (Ctrl). D, the knockdown of HH signaling components attenuates the expression of HH target genes (left)or the steady-state levels of GLI1 protein (right). A549 cells were transduced with the indicated shRNA virus for 3 to 5 days. RNA or protein was then extractedand the expression of the indicated HH target genes determined by qRT-PCR or immunoblotting. These results were then normalized to the expressionof GAPDH (glyceraldehyde-3-phosphate dehydrogenase) or tublin (Tub). These data are shown relative to the cells transduced with pLKO.1 control virus(Ctrl). All error bars indicate � SEM of 3 independent experiments. The asterisk (*) denotes a statistically significant change (P < 0.05) in HH targetgene expression as compared with the control.
Singh et al.
Cancer Res; 71(13) July 1, 2011 Cancer Research4458
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313
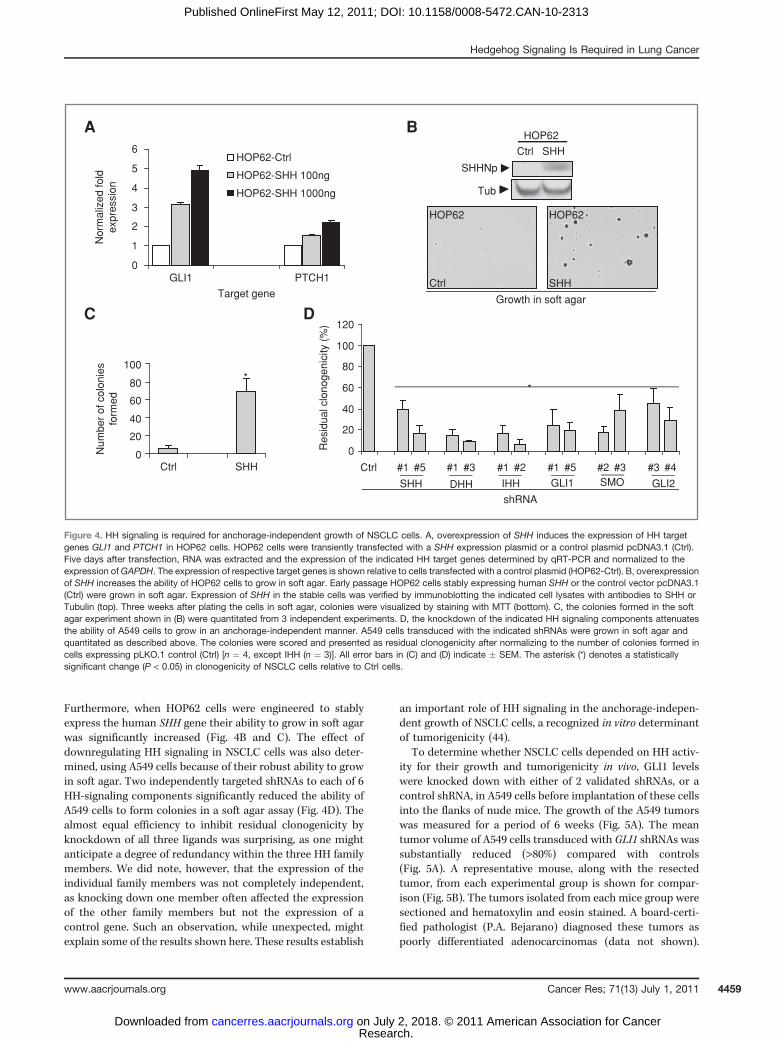
Furthermore, when HOP62 cells were engineered to stablyexpress the human SHH gene their ability to grow in soft agarwas significantly increased (Fig. 4B and C). The effect ofdownregulating HH signaling in NSCLC cells was also deter-mined, using A549 cells because of their robust ability to growin soft agar. Two independently targeted shRNAs to each of 6HH-signaling components significantly reduced the ability ofA549 cells to form colonies in a soft agar assay (Fig. 4D). Thealmost equal efficiency to inhibit residual clonogenicity byknockdown of all three ligands was surprising, as one mightanticipate a degree of redundancy within the three HH familymembers. We did note, however, that the expression of theindividual family members was not completely independent,as knocking down one member often affected the expressionof the other family members but not the expression of acontrol gene. Such an observation, while unexpected, mightexplain some of the results shown here. These results establish
an important role of HH signaling in the anchorage-indepen-dent growth of NSCLC cells, a recognized in vitro determinantof tumorigenicity (44).
To determine whether NSCLC cells depended on HH activ-ity for their growth and tumorigenicity in vivo, GLI1 levelswere knocked down with either of 2 validated shRNAs, or acontrol shRNA, in A549 cells before implantation of these cellsinto the flanks of nude mice. The growth of the A549 tumorswas measured for a period of 6 weeks (Fig. 5A). The meantumor volume of A549 cells transduced with GLI1 shRNAs wassubstantially reduced (>80%) compared with controls(Fig. 5A). A representative mouse, along with the resectedtumor, from each experimental group is shown for compar-ison (Fig. 5B). The tumors isolated from each mice group weresectioned and hematoxylin and eosin stained. A board-certi-fied pathologist (P.A. Bejarano) diagnosed these tumors aspoorly differentiated adenocarcinomas (data not shown).
A
C D
B
SHHNp
Tub
Ctrl SHH
HOP62
0
20
40
60
80
100
Ctrl SHH
Num
ber
of c
olon
ies
form
ed
*
Ctrl SHH
HOP62 HOP62
Growth in soft agar
0
20
40
60
80
100
120
Ctrl #1 #5 #1 #3 #1 #2 #1 #5 #2 #3 #3 #4
Res
idua
l clo
noge
nici
ty (
%)
SHH DHH IHH GLI1
*
GLI2SMO
shRNA
0
1
2
3
4
5
6
GLI1 PTCH1
Target gene
Nor
mal
ized
fold
expr
essi
on
HOP62-Ctrl
HOP62-SHH 100ng
HOP62-SHH 1000ng
Figure 4. HH signaling is required for anchorage-independent growth of NSCLC cells. A, overexpression of SHH induces the expression of HH targetgenes GLI1 and PTCH1 in HOP62 cells. HOP62 cells were transiently transfected with a SHH expression plasmid or a control plasmid pcDNA3.1 (Ctrl).Five days after transfection, RNA was extracted and the expression of the indicated HH target genes determined by qRT-PCR and normalized to theexpression ofGAPDH. The expression of respective target genes is shown relative to cells transfected with a control plasmid (HOP62-Ctrl). B, overexpressionof SHH increases the ability of HOP62 cells to grow in soft agar. Early passage HOP62 cells stably expressing human SHH or the control vector pcDNA3.1(Ctrl) were grown in soft agar. Expression of SHH in the stable cells was verified by immunoblotting the indicated cell lysates with antibodies to SHH orTubulin (top). Three weeks after plating the cells in soft agar, colonies were visualized by staining with MTT (bottom). C, the colonies formed in the softagar experiment shown in (B) were quantitated from 3 independent experiments. D, the knockdown of the indicated HH signaling components attenuatesthe ability of A549 cells to grow in an anchorage-independent manner. A549 cells transduced with the indicated shRNAs were grown in soft agar andquantitated as described above. The colonies were scored and presented as residual clonogenicity after normalizing to the number of colonies formed incells expressing pLKO.1 control (Ctrl) [n ¼ 4, except IHH (n ¼ 3)]. All error bars in (C) and (D) indicate � SEM. The asterisk (*) denotes a statisticallysignificant change (P < 0.05) in clonogenicity of NSCLC cells relative to Ctrl cells.
Hedgehog Signaling Is Required in Lung Cancer
www.aacrjournals.org Cancer Res; 71(13) July 1, 2011 4459
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313
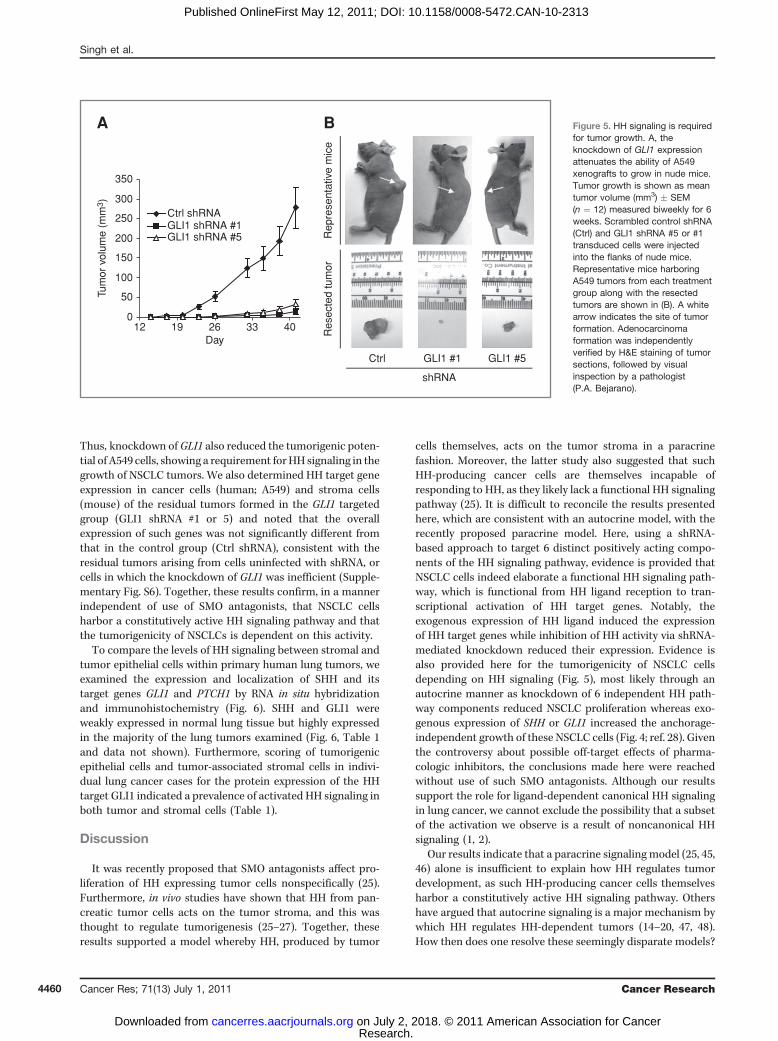
Thus, knockdown ofGLI1 also reduced the tumorigenic poten-tial of A549 cells, showing a requirement forHH signaling in thegrowth of NSCLC tumors. We also determined HH target geneexpression in cancer cells (human; A549) and stroma cells(mouse) of the residual tumors formed in the GLI1 targetedgroup (GLI1 shRNA #1 or 5) and noted that the overallexpression of such genes was not significantly different fromthat in the control group (Ctrl shRNA), consistent with theresidual tumors arising from cells uninfected with shRNA, orcells in which the knockdown of GLI1 was inefficient (Supple-mentary Fig. S6). Together, these results confirm, in a mannerindependent of use of SMO antagonists, that NSCLC cellsharbor a constitutively active HH signaling pathway and thatthe tumorigenicity of NSCLCs is dependent on this activity.
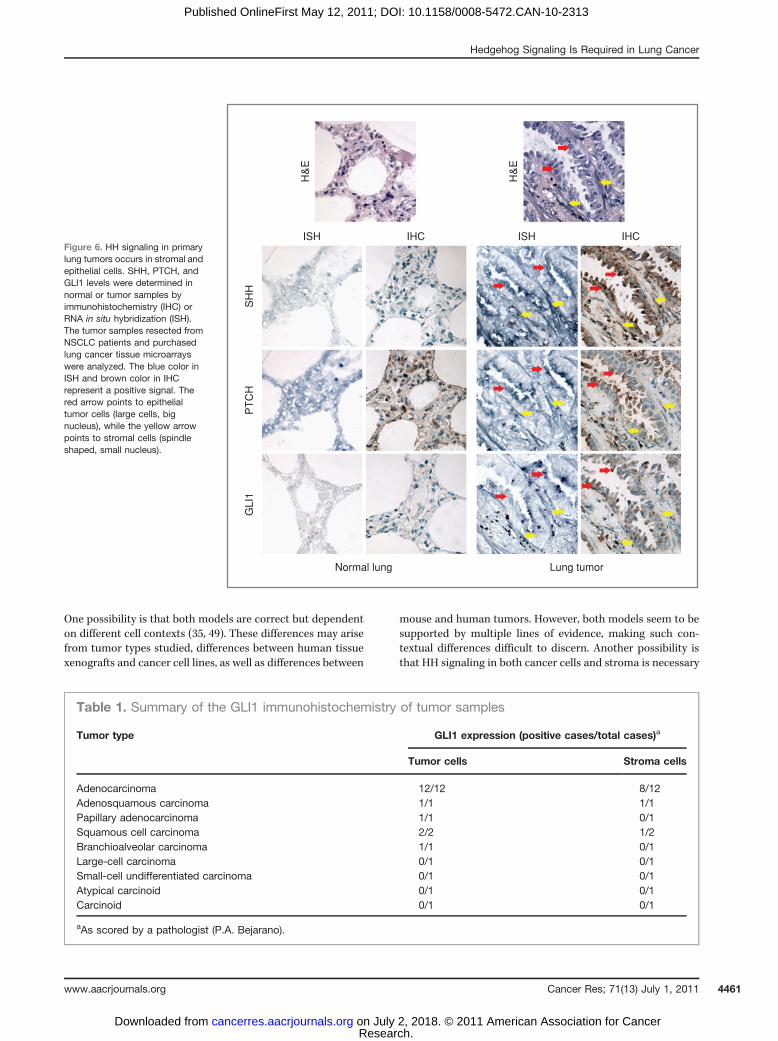
To compare the levels of HH signaling between stromal andtumor epithelial cells within primary human lung tumors, weexamined the expression and localization of SHH and itstarget genes GLI1 and PTCH1 by RNA in situ hybridizationand immunohistochemistry (Fig. 6). SHH and GLI1 wereweakly expressed in normal lung tissue but highly expressedin the majority of the lung tumors examined (Fig. 6, Table 1and data not shown). Furthermore, scoring of tumorigenicepithelial cells and tumor-associated stromal cells in indivi-dual lung cancer cases for the protein expression of the HHtarget GLI1 indicated a prevalence of activated HH signaling inboth tumor and stromal cells (Table 1).
Discussion
It was recently proposed that SMO antagonists affect pro-liferation of HH expressing tumor cells nonspecifically (25).Furthermore, in vivo studies have shown that HH from pan-creatic tumor cells acts on the tumor stroma, and this wasthought to regulate tumorigenesis (25–27). Together, theseresults supported a model whereby HH, produced by tumor
cells themselves, acts on the tumor stroma in a paracrinefashion. Moreover, the latter study also suggested that suchHH-producing cancer cells are themselves incapable ofresponding to HH, as they likely lack a functional HH signalingpathway (25). It is difficult to reconcile the results presentedhere, which are consistent with an autocrine model, with therecently proposed paracrine model. Here, using a shRNA-based approach to target 6 distinct positively acting compo-nents of the HH signaling pathway, evidence is provided thatNSCLC cells indeed elaborate a functional HH signaling path-way, which is functional from HH ligand reception to tran-scriptional activation of HH target genes. Notably, theexogenous expression of HH ligand induced the expressionof HH target genes while inhibition of HH activity via shRNA-mediated knockdown reduced their expression. Evidence isalso provided here for the tumorigenicity of NSCLC cellsdepending on HH signaling (Fig. 5), most likely through anautocrine manner as knockdown of 6 independent HH path-way components reduced NSCLC proliferation whereas exo-genous expression of SHH or GLI1 increased the anchorage-independent growth of these NSCLC cells (Fig. 4; ref. 28). Giventhe controversy about possible off-target effects of pharma-cologic inhibitors, the conclusions made here were reachedwithout use of such SMO antagonists. Although our resultssupport the role for ligand-dependent canonical HH signalingin lung cancer, we cannot exclude the possibility that a subsetof the activation we observe is a result of noncanonical HHsignaling (1, 2).
Our results indicate that a paracrine signaling model (25, 45,46) alone is insufficient to explain how HH regulates tumordevelopment, as such HH-producing cancer cells themselvesharbor a constitutively active HH signaling pathway. Othershave argued that autocrine signaling is a major mechanism bywhich HH regulates HH-dependent tumors (14–20, 47, 48).How then does one resolve these seemingly disparate models?
Ctrl GLI1 #5GLI1 #1
Rep
rese
ntat
ive
mic
eR
esec
ted
tum
or
shRNA
0
50
100
150
200
250
300
350
12 19 26 33 40Day
Tum
or v
olum
e (m
m3 )
Ctrl shRNA
A B
GLI1 shRNA #1GLI1 shRNA #5
Figure 5. HH signaling is requiredfor tumor growth. A, theknockdown of GLI1 expressionattenuates the ability of A549xenografts to grow in nude mice.Tumor growth is shown as meantumor volume (mm3) � SEM(n ¼ 12) measured biweekly for 6weeks. Scrambled control shRNA(Ctrl) and GLI1 shRNA #5 or #1transduced cells were injectedinto the flanks of nude mice.Representative mice harboringA549 tumors from each treatmentgroup along with the resectedtumors are shown in (B). A whitearrow indicates the site of tumorformation. Adenocarcinomaformation was independentlyverified by H&E staining of tumorsections, followed by visualinspection by a pathologist(P.A. Bejarano).
Singh et al.
Cancer Res; 71(13) July 1, 2011 Cancer Research4460
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313
One possibility is that both models are correct but dependenton different cell contexts (35, 49). These differences may arisefrom tumor types studied, differences between human tissuexenografts and cancer cell lines, as well as differences between
mouse and human tumors. However, both models seem to besupported by multiple lines of evidence, making such con-textual differences difficult to discern. Another possibility isthat HH signaling in both cancer cells and stroma is necessary
Table 1. Summary of the GLI1 immunohistochemistry of tumor samples
Tumor type GLI1 expression (positive cases/total cases)a
Tumor cells Stroma cells
Adenocarcinoma 12/12 8/12Adenosquamous carcinoma 1/1 1/1Papillary adenocarcinoma 1/1 0/1Squamous cell carcinoma 2/2 1/2Branchioalveolar carcinoma 1/1 0/1Large-cell carcinoma 0/1 0/1Small-cell undifferentiated carcinoma 0/1 0/1Atypical carcinoid 0/1 0/1Carcinoid 0/1 0/1
aAs scored by a pathologist (P.A. Bejarano).
Figure 6. HH signaling in primarylung tumors occurs in stromal andepithelial cells. SHH, PTCH, andGLI1 levels were determined innormal or tumor samples byimmunohistochemistry (IHC) orRNA in situ hybridization (ISH).The tumor samples resected fromNSCLC patients and purchasedlung cancer tissue microarrayswere analyzed. The blue color inISH and brown color in IHCrepresent a positive signal. Thered arrow points to epithelialtumor cells (large cells, bignucleus), while the yellow arrowpoints to stromal cells (spindleshaped, small nucleus).
IHCISHISH IHC
H&
E
H&
E
SH
HP
TC
HG
LI1
Normal lung Lung tumor
Hedgehog Signaling Is Required in Lung Cancer
www.aacrjournals.org Cancer Res; 71(13) July 1, 2011 4461
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313

for tumorigenesis but in mice the dominant effect is para-crine–attenuation of which debulks the tumor. Alternatively,autocrine HH-dependent cancer cells are a product of growingtumor cells in vitro and are therefore not relevant for under-standing tumor growth in vivo. We do not favor this possibilitybecause several groups, including ours, used immunohisto-chemistry or in situ hybridization analyses to find increasedHH target gene expression within the tumor cells themselves(Fig. 6; refs. 14–20, 28, 29, 34, 48, 50). Regardless of suchmodels, the findings presented here shows that the tumor-igenicity of human NSCLC cells often depend on HH signaling,and does so in a manner independent of the use of anypharmacologic SMO antagonist.
Disclosure of Potential Conflicts of Interest
E. Dmitrovsky is an American Cancer Society Professor supported by agenerous gift from the F. M. Kirby Foundation. The other authors disclose nopotential conflicts of interest.
Acknowledgments
We thank the members of the Robbins, Capobianco, and Dmitrovskylaboratories for thoughtful and lively discussions during the course of this work.
Grant Support
This work was supported by NIH grants GM64011 (D.J. Robbins), R03-CA132166 (E. Dmitrovsky), R01-CA087546 (E. Dmitrovsky), R01-CA111422 (E.Dmitrovsky), and R01-CA062275 (E. Dmitrovsky); and grants from SamuelWaxman Cancer Research Foundation (E. Dmitrovsky, A.J. Capobianco), Bank-head Coley Cancer Research Program, Florida State Department of Health 08-BB-03 (K.J. Briegel), Flight Attendant Medical Research Institute CIA_072080 (K.J. Briegel), and from the American Lung Association/LUNGevity Foundation (D.J. Robbins).
The costs of publication of this article were defrayed in part by the paymentof page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received June 28, 2010; revised April 13, 2011; accepted April 29, 2011;published OnlineFirst May 12, 2011.
References1. Teglund S, Toftgard R. Hedgehog beyondmedulloblastoma and basal
cell carcinoma. Biochim Biophys Acta 2010;1805:181–208.2. Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog-signaling
pathway in human cancer and the clinical implications. Oncogene2010;29:469–81.
3. Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL,et al. The tumour-suppressor gene patched encodes a candidatereceptor for Sonic hedgehog. Nature 1996;384:129–34.
4. Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemicalevidence that patched is the Hedgehog receptor. Nature 1996;384:176–9.
5. Hooper JE. Distinct pathways for autocrine and paracrine Winglesssignalling in Drosophila embryos. Nature 1994;372:461–4.
6. Chen Y, Struhl G. Dual roles for patched in sequestering and transdu-cing Hedgehog. Cell 1996;87:553–63.
7. Quirk J, van den Heuvel M, Henrique D, Marigo V, Jones TA, Tabin C,et al. The smoothened gene and hedgehog signal transduction inDrosophila and vertebrate development. Cold Spring Harb SympQuant Biol 1997;62:217–26.
8. Robbins DJ, Hebrok M. Hedgehogs: la dolce vita. Workshop onHedgehog-Gli Signaling in Cancer and Stem Cells. EMBO Rep2007;8:451–5.
9. Ruiz i Altaba A, Mas C, Stecca B. The Gli code: an information nexusregulating cell fate, stemness and cancer. Trends Cell Biol 2007;17:438–47.
10. Ingham PW, McMahon AP. Hedgehog signaling in animal develop-ment: paradigms and principles. Genes Dev 2001;15:3059–87.
11. Stone DM, Murone M, Luoh S, Ye W, Armanini MP, Gurney A, et al.Characterization of the human suppressor of fused, a negative reg-ulator of the zinc-finger transcription factor Gli. J Cell Sci 1999;112:4437–48.
12. Dunaeva M, Michelson P, Kogerman P, Toftgard R. Characterizationof the physical interaction of Gli proteins with SUFU proteins. J BiolChem 2003;278:5116–22.
13. MerchantM, Vajdos FF, UltschM,MaunHR,Wendt U, Cannon J, et al.Suppressor of fused regulates Gli activity through a dual bindingmechanism. Mol Cell Biol 2004;24:8627–41.
14. Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, BaylinSB. Hedgehog signalling within airway epithelial progenitors and insmall-cell lung cancer. Nature 2003;422:313–7.
15. Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, GerstenblithMR, Briggs K, et al. Widespread requirement for Hedgehog ligandstimulation in growth of digestive tract tumours. Nature 2003;425:846–51.
16. Kubo M, Nakamura M, Tasaki A, Yamanaka N, Nakashima H, NomuraM, et al. Hedgehog signaling pathway is a new therapeutic target forpatients with breast cancer. Cancer Res 2004;64:6071–4.
17. Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ,Lauwers GY, et al. Hedgehog is an early and late mediator ofpancreatic cancer tumorigenesis. Nature 2003;425:851–6.
18. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A.HEDGEHOG-GLI1 signaling regulates human glioma growth, can-cer stem cell self-renewal, and tumorigenicity. Curr Biol 2007;17:165–72.
19. Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A,et al. Hedgehog signalling in prostate regeneration, neoplasia andmetastasis. Nature 2004;431:707–12.
20. Sanchez P, Hernandez AM, Stecca B, Kahler AJ, DeGueme AM,Barrett A, et al. Inhibition of prostate cancer proliferation by inter-ference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad SciU S A 2004;101:12561–6.
21. Mas C, Ruiz i Altaba A. Small molecule modulation of HH-GLIsignaling: Current leads, trials and tribulations. Biochem Pharmacol2010;80:712–23
22. Stanton BZ, Peng LF. Small-molecule modulators of the SonicHedgehog signaling pathway. Mol Biosyst 2010;6:44–54.
23. Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediatedinhibition of target tissue response to Shh signaling. Science1998;280:1603–7.
24. Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenicVeratrum alkaloid cyclopamine inhibits sonic hedgehog signal trans-duction. Development 1998;125:3553–62.
25. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. Aparacrine requirement for hedgehog signalling in cancer. Nature2008;455:406–10.
26. Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, Scales SJ,et al. Hedgehog signaling is restricted to the stromal compartmentduring pancreatic carcinogenesis. Proc Natl Acad Sci U S A 2009;106:4254–9.
27. Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrinesignaling regulates metastasis and lymphangiogenesis in pancreaticcancer. Oncogene 2009;28:3513–25.
28. Yuan Z, Goetz JA, Singh S, Ogden SK, Petty WJ, Black CC, et al.Frequent requirement of hedgehog signaling in non-small cell lungcarcinoma. Oncogene 2007;26:1046–55.
29. Ma Y, Fiering S, Black C, Liu X, Yuan Z, Memoli VA, et al. Transgeniccyclin E triggers dysplasia and multiple pulmonary adenocarcinomas.Proc Natl Acad Sci U S A 2007;104:4089–94.
Singh et al.
Cancer Res; 71(13) July 1, 2011 Cancer Research4462
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313

30. Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small moleculemodulation of Smoothened activity. Proc Natl Acad Sci U S A2002;99:14071–6.
31. Goetz JA, Singh S, Suber LM, Kull FJ, Robbins DJ. A highly conservedamino-terminal region of sonic hedgehog is required for the formationof its freely diffusible multimeric form. J Biol Chem 2006;281:4087–93.
32. Fei DL, Li H, Kozul CD, Black KE, Singh S, Gosse JA, et al. Activationof Hedgehog signaling by the environmental toxicant arsenic maycontribute to the etiology of arsenic-induced tumors. Cancer Res2010;70:1981–8.
33. Lemieux P, Michaud M, Page M. A new formazan amplified clono-genic assay for cytotoxicity testing. Biotechnol. Techniques 1993;7:597–602.
34. Dahmane N, S�anchez P, Gitton Y, Palma V, Sun T, Beyna M, et al. TheSonic Hedgehog-Gli pathway regulates dorsal brain growth andtumorigenesis. Development 2001;128:5201–12.
35. Stecca B, Ruiz i Altaba A. Context-dependent regulation of the GLIcode in cancer by HEDGEHOG and non-HEDGEHOG signals. J MolCell Biol 2010;2:84–95.
36. Laurendeau I, Ferrer M, Garrido D, D'Haene N, Ciavarelli P, Basso A,et al. Gene expression profiling of the hedgehog signaling pathway inhuman meningiomas. Mol Med 2010;16:262–70.
37. Chen X, Horiuchi A, Kikuchi N, Osada R, Yoshida J, Shiozawa T, et al.Hedgehog signal pathway is activated in ovarian carcinomas, corre-lating with cell proliferation: it's inhibition leads to growth suppressionand apoptosis. Cancer Sci 2007;98:68–76.
38. Goetz JA, Suber LM, Zeng X, Robbins DJ. Sonic Hedgehog as amediator of long-range signaling. Bioessays 2002;24:157–65.
39. Singh S, Goetz JA, Robbins DJ. Sonic hedgehog. UCSD-NatureMolecule Pages 2006;doi:10.1038/mp.a002208.01.
40. Williams JA, Guicherit OM, Zaharian BI, Xu Y, Chai L,Wichterle H, et al.Identification of a small molecule inhibitor of the hedgehog signalingpathway: effects on basal cell carcinoma-like lesions. Proc Natl AcadSci U S A 2003;100:4616–21.
41. Athar M, Li C, Tang X, Chi S, Zhang X, Kim AL, et al. Inhibition ofsmoothened signaling prevents ultraviolet B-induced basal cell car-cinomas through regulation of Fas expression and apoptosis. CancerRes 2004;64:7545–52.
42. Dwyer JR, Sever N, Carlson M, Nelson SF, Beachy PA, Parhami F.Oxysterols are novel activators of the hedgehog signaling pathway inpluripotent mesenchymal cells. J Biol Chem 2007;282:8959–68.
43. Agren M, Kogerman P, Kleman MI, Wessling M, Toftgard R. Expres-sion of the PTCH1 tumor suppressor gene is regulated by alternativepromoters and a single functional Gli-binding site. Gene 2004;330:101–14.
44. Shin SI, Freedman VH, Risser R, Pollack R. Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with ancho-rage independent growth in vitro. Proc Natl Acad Sci U S A 1975;72:4435–9.
45. Theunissen JW, de Sauvage FJ. Paracrine Hedgehog signaling incancer. Cancer Res 2009;69:6007–10.
46. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D,Honess D, et al. Inhibition of Hedgehog signaling enhances delivery ofchemotherapy in a mouse model of pancreatic cancer. Science2009;324:1457–61.
47. Ruiz i Altaba A. Therapeutic inhibition of Hedgehog-GLI signaling incancer: epithelial, stromal, or stem cell targets? Cancer Cell 2008;14:281–3.
48. Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al.Melanomas require HEDGEHOG-GLI signaling regulated by interac-tions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl AcadSci U S A 2007;104:5895–900.
49. LauthM, Bergstr€omA, Shimokawa T, Tostar U, Jin Q, Fendrich V, et al.DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signal-ing by mutant RAS. Nat Struct Mol Biol 2010;17:718–25.
50. Huang S, He J, Zhang X, Bian Y, Yang L, Xie G, et al. Activation of thehedgehog pathway in human hepatocellular carcinomas. Carcinogen-esis 2006;27:1334–40.
Hedgehog Signaling Is Required in Lung Cancer
www.aacrjournals.org Cancer Res; 71(13) July 1, 2011 4463
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313

2011;71:4454-4463. Published OnlineFirst May 12, 2011.Cancer Res Samer Singh, Zhiqiang Wang, Dennis Liang Fei, et al. Autocrine Hedgehog ActivityHedgehog-Producing Cancer Cells Respond to and Require
Updated version
10.1158/0008-5472.CAN-10-2313doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2011/05/12/0008-5472.CAN-10-2313.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/71/13/4454.full#ref-list-1
This article cites 49 articles, 20 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/71/13/4454.full#related-urls
This article has been cited by 6 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/71/13/4454To request permission to re-use all or part of this article, use this link
Research. on July 2, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 12, 2011; DOI: 10.1158/0008-5472.CAN-10-2313





























