Embed Size (px)
Citation preview
LETTERS
Hax1-mediated processing of HtrA2 by Parl allowssurvival of lymphocytes and neuronsJyh-Rong Chao1, Evan Parganas1, Kelli Boyd2, Cheol Yi Hong1, Joseph T. Opferman1 & James N. Ihle1
Cytokines affect a variety of cellular functions, including regu-lation of cell numbers by suppression of programmed cell death1.Suppression of apoptosis requires receptor signalling through theactivation of Janus kinases and the subsequent regulation of mem-bers of the B-cell lymphoma 2 (Bcl-2) family. Here we demonstratethat a Bcl-2-family-related protein, Hax1, is required to suppressapoptosis in lymphocytes and neurons. Suppression requires theinteraction of Hax1 with the mitochondrial proteases Parl (pre-senilin-associated, rhomboid-like) and HtrA2 (high-temperature-regulated A2, also known as Omi). These interactions allow Hax1to present HtrA2 to Parl, and thereby facilitates the processingof HtrA2 to the active protease localized in the mitochondrialintermembrane space. In mouse lymphocytes, the presence ofprocessed HtrA2 prevents the accumulation of mitochondrial-outer-membrane-associated activated Bax, an event that initiatesapoptosis. Together, the results identify a previously unknownsequence of interactions involving a Bcl-2-family-related proteinand mitochondrial proteases in the ability to resist the inductionof apoptosis when cytokines are limiting.
In a screen for genes that suppress apoptosis of Janus kinase 2(Jak2)-null fetal liver cells, we identified Hax1 full-length clones.Expression of Hax1 rescued both erythroid-lineage-committed pro-genitors and, to a lesser extent, earlier multi-lineage progenitors(Supplementary Table 1). Hax1 was initially identified by its asso-ciation with HS1 (also known as Hcls1), a Src kinase substrate2. It hasanti-apoptotic activity and some structural similarities to Bcl-2family members, including the presence of BH1- and BH2-likedomains and a carboxyl transmembrane domain3. Like Bcl-2 familymembers, Hax1 expression is induced by cytokines (SupplementaryFig. 1a, b) and the protein is mitochondrial2. In mitochondria, it islocalized on the inner and outer mitochondrial membranes andexposed to the intermembrane space (unpublished data). Lastly,human mutations of Hax1 cause increased neutrophil apoptosis,resulting in autosomal recessive severe neutropenia4 and in somecases has been suggested to be the basis for neurological impair-ments5,6.
1Department of Biochemistry, 2Animal Resources Center, St Jude Children’s Research Hospital, Memphis, Tennessee 38105, USA.
Via
bili
ty (%
)
100
80
60
40
20
00 5 10
WT, n = 20Hax1 KO, n = 25Hax1/Bak DKO, n = 18Hax1/Bak DKO, n = 13Mnd2, n = 13
15 20 25
a
b
c
Time (weeks)
WT
WT
KO
KO
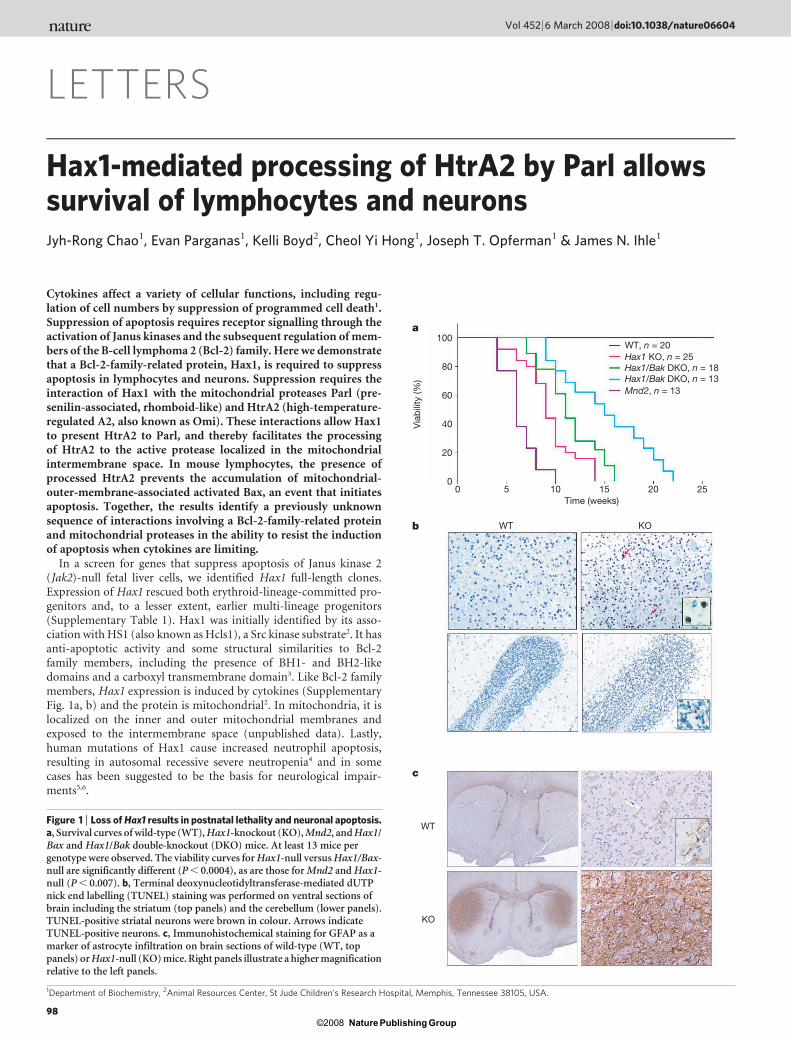
Figure 1 | Loss of Hax1 results in postnatal lethality and neuronal apoptosis.a, Survival curves of wild-type (WT), Hax1-knockout (KO), Mnd2, and Hax1/Bax and Hax1/Bak double-knockout (DKO) mice. At least 13 mice pergenotype were observed. The viability curves for Hax1-null versus Hax1/Bax-null are significantly different (P , 0.0004), as are those for Mnd2 and Hax1-null (P , 0.007). b, Terminal deoxynucleotidyltransferase-mediated dUTPnick end labelling (TUNEL) staining was performed on ventral sections ofbrain including the striatum (top panels) and the cerebellum (lower panels).TUNEL-positive striatal neurons were brown in colour. Arrows indicateTUNEL-positive neurons. c, Immunohistochemical staining for GFAP as amarker of astrocyte infiltration on brain sections of wild-type (WT, toppanels) or Hax1-null (KO) mice. Right panels illustrate a higher magnificationrelative to the left panels.
Vol 452 | 6 March 2008 | doi:10.1038/nature06604
98Nature Publishing Group©2008
To assess the functions of Hax1, null mice were produced bygene targeting (Supplementary Fig. 2). Heterozygous mice wereviable and fertile whereas homozygous-null mice, although born atexpected frequencies, failed to survive longer than 14 weeks (Fig. 1a).Lethality was caused by loss of motor coordination and activity,ultimately leading to failure to eat or drink. Histological analysisidentified extensive apoptosis of neurons in the striatum and thecerebellum (Fig. 1b). Consistent with neuronal loss, staining for glialfibrillary acidic protein (GFAP) identified extensive infiltration ofastrocytes in the striatum (Fig. 1c). A comparable phenotype wasseen in Hax1/Rag2-null mice, ruling out a role for the immunesystem in neuronal apoptosis (data not shown).
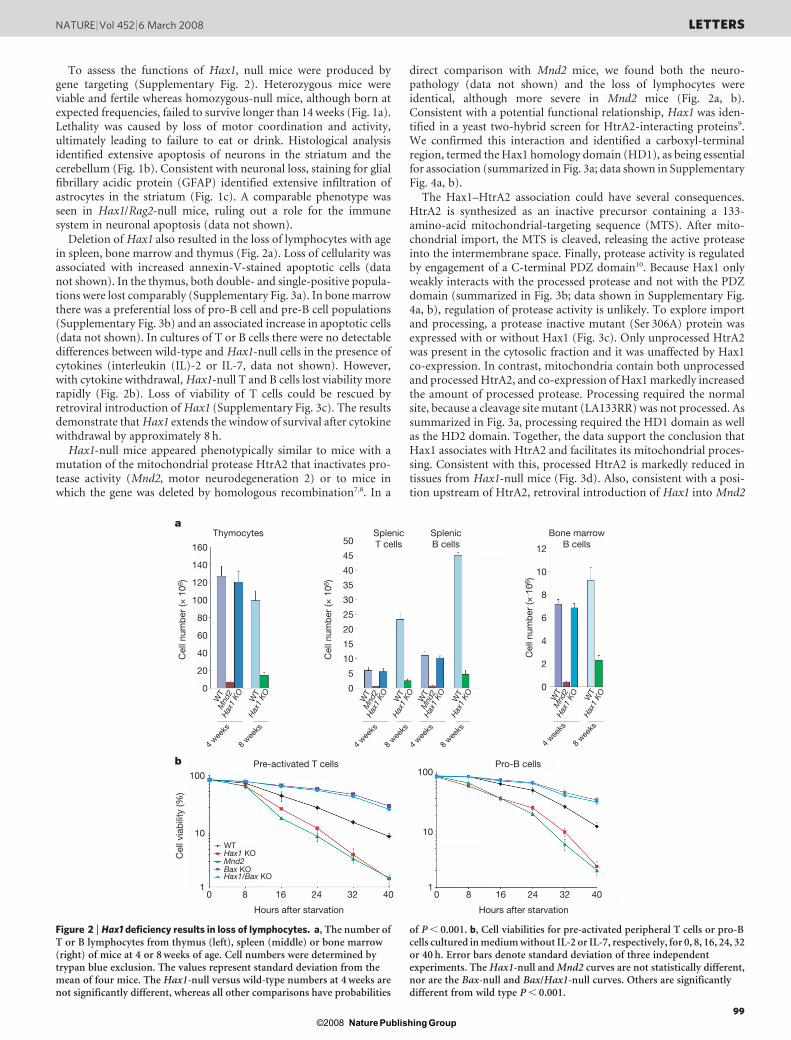
Deletion of Hax1 also resulted in the loss of lymphocytes with agein spleen, bone marrow and thymus (Fig. 2a). Loss of cellularity wasassociated with increased annexin-V-stained apoptotic cells (datanot shown). In the thymus, both double- and single-positive popula-tions were lost comparably (Supplementary Fig. 3a). In bone marrowthere was a preferential loss of pro-B cell and pre-B cell populations(Supplementary Fig. 3b) and an associated increase in apoptotic cells(data not shown). In cultures of T or B cells there were no detectabledifferences between wild-type and Hax1-null cells in the presence ofcytokines (interleukin (IL)-2 or IL-7, data not shown). However,with cytokine withdrawal, Hax1-null T and B cells lost viability morerapidly (Fig. 2b). Loss of viability of T cells could be rescued byretroviral introduction of Hax1 (Supplementary Fig. 3c). The resultsdemonstrate that Hax1 extends the window of survival after cytokinewithdrawal by approximately 8 h.
Hax1-null mice appeared phenotypically similar to mice with amutation of the mitochondrial protease HtrA2 that inactivates pro-tease activity (Mnd2, motor neurodegeneration 2) or to mice inwhich the gene was deleted by homologous recombination7,8. In a
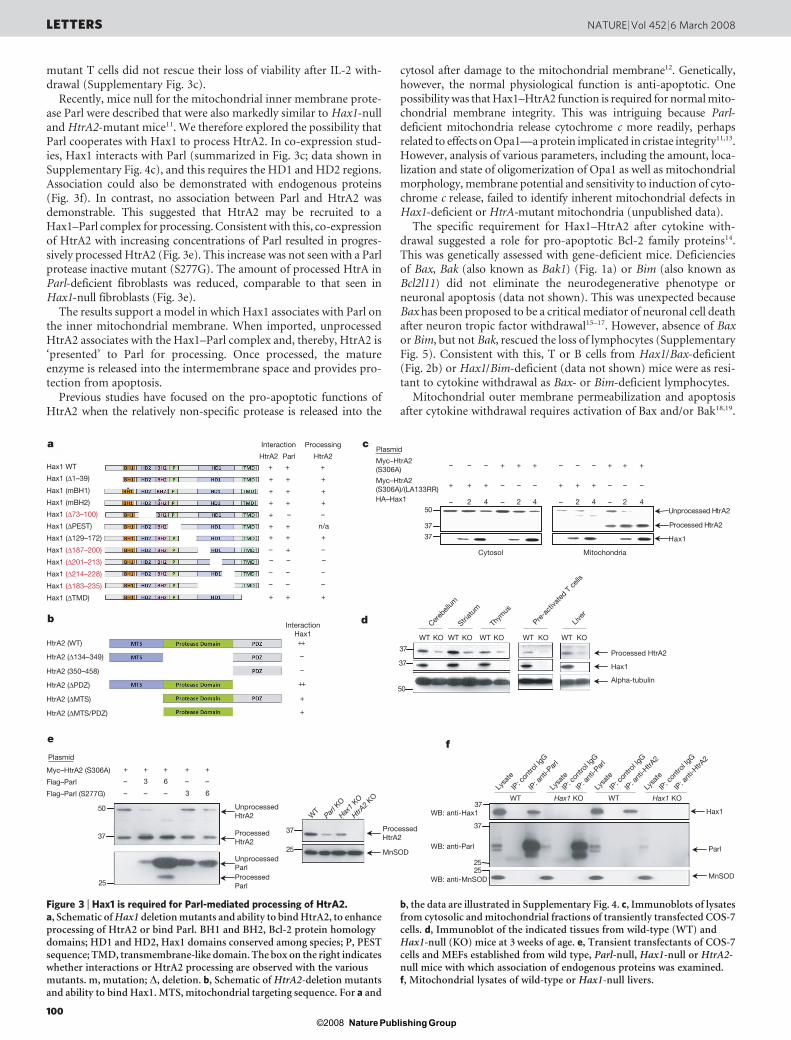
direct comparison with Mnd2 mice, we found both the neuro-pathology (data not shown) and the loss of lymphocytes wereidentical, although more severe in Mnd2 mice (Fig. 2a, b).Consistent with a potential functional relationship, Hax1 was iden-tified in a yeast two-hybrid screen for HtrA2-interacting proteins9.We confirmed this interaction and identified a carboxyl-terminalregion, termed the Hax1 homology domain (HD1), as being essentialfor association (summarized in Fig. 3a; data shown in SupplementaryFig. 4a, b).
The Hax1–HtrA2 association could have several consequences.HtrA2 is synthesized as an inactive precursor containing a 133-amino-acid mitochondrial-targeting sequence (MTS). After mito-chondrial import, the MTS is cleaved, releasing the active proteaseinto the intermembrane space. Finally, protease activity is regulatedby engagement of a C-terminal PDZ domain10. Because Hax1 onlyweakly interacts with the processed protease and not with the PDZdomain (summarized in Fig. 3b; data shown in Supplementary Fig.4a, b), regulation of protease activity is unlikely. To explore importand processing, a protease inactive mutant (Ser 306A) protein wasexpressed with or without Hax1 (Fig. 3c). Only unprocessed HtrA2was present in the cytosolic fraction and it was unaffected by Hax1co-expression. In contrast, mitochondria contain both unprocessedand processed HtrA2, and co-expression of Hax1 markedly increasedthe amount of processed protease. Processing required the normalsite, because a cleavage site mutant (LA133RR) was not processed. Assummarized in Fig. 3a, processing required the HD1 domain as wellas the HD2 domain. Together, the data support the conclusion thatHax1 associates with HtrA2 and facilitates its mitochondrial proces-sing. Consistent with this, processed HtrA2 is markedly reduced intissues from Hax1-null mice (Fig. 3d). Also, consistent with a posi-tion upstream of HtrA2, retroviral introduction of Hax1 into Mnd2
a
b
Cel
l num
ber
(× 1
06)
Cel
l via
bili
ty (%
)
Hours after starvation
100
120
140
160
80
60
40
20
0
100
10
1
100
10
10 8 16 24 32 40
Hours after starvation
0 8 16 24 32 40
Cel
l num
ber
(× 1
06)
25
30
35
50
45
40
20
15
10
5
0
Cel
l num
ber
(× 1
06) 10
12
8
6
4
2
0
WT
4 wee
ks
8 wee
ks
4 wee
ks
8 wee
ks
4 wee
ks
8 wee
ks
Mnd
2H
ax1
KO WT
Hax
1 KO W
TM
nd2
Hax
1 KO W
TH
ax1
KO WT
Mnd
2H
ax1
KO WT
Hax
1 KO
4 wee
ks
8 wee
ks
WT
Mnd
2H
ax1
KO WT
Hax
1 KO
Thymocytes SplenicT cells
Pre-activated T cells Pro-B cells
SplenicB cells
Bone marrowB cells
WTHax1 KO
Bax KOHax1/Bax KO
Mnd2
Figure 2 | Hax1 deficiency results in loss of lymphocytes. a, The number ofT or B lymphocytes from thymus (left), spleen (middle) or bone marrow(right) of mice at 4 or 8 weeks of age. Cell numbers were determined bytrypan blue exclusion. The values represent standard deviation from themean of four mice. The Hax1-null versus wild-type numbers at 4 weeks arenot significantly different, whereas all other comparisons have probabilities
of P , 0.001. b, Cell viabilities for pre-activated peripheral T cells or pro-Bcells cultured in medium without IL-2 or IL-7, respectively, for 0, 8, 16, 24, 32or 40 h. Error bars denote standard deviation of three independentexperiments. The Hax1-null and Mnd2 curves are not statistically different,nor are the Bax-null and Bax/Hax1-null curves. Others are significantlydifferent from wild type P , 0.001.
NATURE | Vol 452 | 6 March 2008 LETTERS
99Nature Publishing Group©2008
mutant T cells did not rescue their loss of viability after IL-2 with-drawal (Supplementary Fig. 3c).
Recently, mice null for the mitochondrial inner membrane prote-ase Parl were described that were also markedly similar to Hax1-nulland HtrA2-mutant mice11. We therefore explored the possibility thatParl cooperates with Hax1 to process HtrA2. In co-expression stud-ies, Hax1 interacts with Parl (summarized in Fig. 3c; data shown inSupplementary Fig. 4c), and this requires the HD1 and HD2 regions.Association could also be demonstrated with endogenous proteins(Fig. 3f). In contrast, no association between Parl and HtrA2 wasdemonstrable. This suggested that HtrA2 may be recruited to aHax1–Parl complex for processing. Consistent with this, co-expressionof HtrA2 with increasing concentrations of Parl resulted in progres-sively processed HtrA2 (Fig. 3e). This increase was not seen with a Parlprotease inactive mutant (S277G). The amount of processed HtrA inParl-deficient fibroblasts was reduced, comparable to that seen inHax1-null fibroblasts (Fig. 3e).
The results support a model in which Hax1 associates with Parl onthe inner mitochondrial membrane. When imported, unprocessedHtrA2 associates with the Hax1–Parl complex and, thereby, HtrA2 is‘presented’ to Parl for processing. Once processed, the matureenzyme is released into the intermembrane space and provides pro-tection from apoptosis.
Previous studies have focused on the pro-apoptotic functions ofHtrA2 when the relatively non-specific protease is released into the
cytosol after damage to the mitochondrial membrane12. Genetically,however, the normal physiological function is anti-apoptotic. Onepossibility was that Hax1–HtrA2 function is required for normal mito-chondrial membrane integrity. This was intriguing because Parl-deficient mitochondria release cytochrome c more readily, perhapsrelated to effects on Opa1—a protein implicated in cristae integrity11,13.However, analysis of various parameters, including the amount, loca-lization and state of oligomerization of Opa1 as well as mitochondrialmorphology, membrane potential and sensitivity to induction of cyto-chrome c release, failed to identify inherent mitochondrial defects inHax1-deficient or HtrA-mutant mitochondria (unpublished data).
The specific requirement for Hax1–HtrA2 after cytokine with-drawal suggested a role for pro-apoptotic Bcl-2 family proteins14.This was genetically assessed with gene-deficient mice. Deficienciesof Bax, Bak (also known as Bak1) (Fig. 1a) or Bim (also known asBcl2l11) did not eliminate the neurodegenerative phenotype orneuronal apoptosis (data not shown). This was unexpected becauseBax has been proposed to be a critical mediator of neuronal cell deathafter neuron tropic factor withdrawal15–17. However, absence of Baxor Bim, but not Bak, rescued the loss of lymphocytes (SupplementaryFig. 5). Consistent with this, T or B cells from Hax1/Bax-deficient(Fig. 2b) or Hax1/Bim-deficient (data not shown) mice were as resi-tant to cytokine withdrawal as Bax- or Bim-deficient lymphocytes.
Mitochondrial outer membrane permeabilization and apoptosisafter cytokine withdrawal requires activation of Bax and/or Bak18,19.
a
+++
+++
+++
+++
n/a++
+++
–+––––
–- ––
–––
+++
––+
++
–
–
++
+
+
b
+++––– +++–––
–––+++–––+++
42–42– 42–42–2ArtHdessecorpnU
2ArtHdessecorP
x1aH
c
05
73
73
73
73
05
d
73
73
5252
f
05
73
52
+++++
––63–
63–––
52
73
e
Hax1 WT
Interaction
InteractionHax1
HtrA2 HtrA2Parl
Processing
Hax1 (mBH1)
Hax1 (mBH2)
Hax1 (∆1–39)
HtrA2 (WT)
HtrA2 (∆134–349)
HtrA2 (350–458)
HtrA2 (∆PDZ)
HtrA2 (∆MTS)
HtrA2 (∆MTS/PDZ)
Myc–HtrA2 (S306A)
Flag–Parl
UnprocessedParlProcessedParl
UnprocessedHtrA2
ProcessedHtrA2
ProcessedHtrA2
WB: anti-Hax1
WB: anti-Parl
WB: anti-MnSOD
Hax1 KO
WT
Parl K
O
Hax1
KO
HtrA2
KO
MnSOD
Flag–Parl (S277G)
Hax1 (∆PEST)
Hax1 (∆73–100)
Hax1 (∆129–172)
Hax1 (∆187–200)
Hax1 (∆201–213)
Hax1 (∆214–228)
Hax1 (∆183–235)
Hax1 (∆TMD)
Plasmid
Myc–HtrA2(S306A)
Cytosol
WT KO WT KO WT KO WT KO WT KO
Cereb
ellum
Striat
um
Thym
us
Pre-a
ctiva
ted T
cells
Liver
Mitochondria
Processed HtrA2
Hax1
Alpha-tubulin
WTLy
sate
IP: c
ontro
l IgG
IP: a
nti-P
arl
Lysa
te
IP: c
ontro
l IgG
IP: a
nti-P
arl
Lysa
te
IP: c
ontro
l IgG
IP: a
nti-H
trA2
Lysa
te
IP: c
ontro
l IgG
IP: a
nti-H
trA2
Hax1 KOWT
Myc–HtrA2(S306A)/(LA133RR)HA–Hax1
Plasmid
Hax1
Parl
MnSOD
Figure 3 | Hax1 is required for Parl-mediated processing of HtrA2.a, Schematic of Hax1 deletion mutants and ability to bind HtrA2, to enhanceprocessing of HtrA2 or bind Parl. BH1 and BH2, Bcl-2 protein homologydomains; HD1 and HD2, Hax1 domains conserved among species; P, PESTsequence; TMD, transmembrane-like domain. The box on the right indicateswhether interactions or HtrA2 processing are observed with the variousmutants. m, mutation; D, deletion. b, Schematic of HtrA2-deletion mutantsand ability to bind Hax1. MTS, mitochondrial targeting sequence. For a and
b, the data are illustrated in Supplementary Fig. 4. c, Immunoblots of lysatesfrom cytosolic and mitochondrial fractions of transiently transfected COS-7cells. d, Immunoblot of the indicated tissues from wild-type (WT) andHax1-null (KO) mice at 3 weeks of age. e, Transient transfectants of COS-7cells and MEFs established from wild type, Parl-null, Hax1-null or HtrA2-null mice with which association of endogenous proteins was examined.f, Mitochondrial lysates of wild-type or Hax1-null livers.
LETTERS NATURE | Vol 452 | 6 March 2008
100Nature Publishing Group©2008

Activation of Bax is downstream of Bim activation, which occurs as aconsequence of loss of anti-apoptotic partners and/or receptor-induced phosphorylation promoting turnover20–23. We thereforeexamined the consequences of Hax1 deficiency or HtrA2 mutationon these events. Neither mutation affected Bim accumulation or totalBax. Nor did the mutations affect the expression patterns of anti-apoptotic proteins Bcl-2, Bcl-x or Mcl-1 (Supplementary Fig. 6).However, both mutations greatly affected the rate of appearance ofa conformation of Bax that is uniquely associated with the mitochon-drial outer membrane and is responsible for mitochondrial-outer-membrane permeabilization24,25. Increases in activated Bax occurredat least 8 h sooner than in wild-type cells and correlated directly withloss of viability (data provided in Supplementary Fig. 6).
Our results provide mechanistic insights into one component ofthe events that determine how long lymphocytes can survive aftercytokine withdrawal. In particular, withdrawal of cytokines reducesthe levels of anti-apoptotic proteins including Hax1 (Fig. 4b). Withloss of Hax1, the production of the processed HtrA2 is reduced(Fig. 4b). Conversely, the loss of other anti-apoptotic proteins andthe accumulation of Bim contribute to the generation of activatedBax. Ultimately, it is the loss of HtrA2 activity that contributes to
the accumulation of sufficient amounts of activated Bax to inducecytochrome c release and cell death. Although we have focused onlymphocytes, Hax1 also contributes to suppression of apoptosis ofneutrophiles deprived of G-CSF (Supplementary Fig. 7) and mayfunction generally in protecting haematopoietic cells from cytokinewithdrawal.
Several challenging questions are raised by the results. Forexample, although activated Bax may be the target in lymphocytes,genetically, any role in the neuronal apoptosis is not evident.Therefore, it will be important to identify pathways and potentialmediators of apoptosis in neurons that are suppressed by Hax1–HtrA2 in both normal development and in conditions associatedwith Parkinson’s disease26. Also it will be important to determinehow HtrA2 suppresses the appearance of activated Bax. ActivatedBax may be a substrate of HtrA2, however it is equally possible thatBax-associated proteins are substrates. The results raise the possi-bility that other mitochondrial proteases may be important in regu-lating programmed cell death.
METHODS SUMMARY
Immunoprecipitations, immunoblots and northern blots used standard tech-
niques as described previously27. Expression constructs generally used pCDNA3
(Invitrogen), and deletion mutants were made by PCR-based, site-directed
mutation and confirmed by DNA sequencing. Transfections used FuGENE6
(Roche) or standard calcium-phosphate methods. Pre-activated T cells were
obtained by culture of splenocytes with anti-CD3e monoclonal antibody, fol-
lowed by culture in human IL-2 (200 U ml21) for 7 days. Pro-B cells were
obtained by culturing bone marrow cells in stem cell factor (50 ng ml21) and
IL-7 (10 ng ml21) for 14 days.
Full Methods and any associated references are available in the online version ofthe paper at www.nature.com/nature.
Received 24 October; accepted 21 December 2007.Published online 20 February 2008.
1. Ihle, J. N. Cytokine receptor signalling. Nature 377, 591–594 (1995).
2. Suzuki, Y. et al. HAX-1, a novel intracellular protein, localized on mitochondria,directly associates with HS1, a substrate of Src family tyrosine kinases. J. Immunol.158, 2736–2744 (1997).
3. Sharp, T. V. et al. K15 protein of Kaposi’s sarcoma-associated herpesvirus islatently expressed and binds to HAX-1, a protein with antiapoptotic function.J. Virol. 76, 802–816 (2002).
4. Klein, C. et al. HAX1 deficiency causes autosomal recessive severe congenitalneutropenia (Kostmann disease). Nature Genet. 39, 86–92 (2007).
5. Matsubara, K. et al. Severe developmental delay and epilepsy in a Japanesepatient with severe congenital neutropenia due to HAX1 deficiency.Haematologica 92, e123–e125 (2007).
6. Carlsson, G. & Fasth, A. Infantile genetic agranulocytosis, morbus Kostmann:Presentation of six cases from the original ‘Kostmann family’ and a review. ActaPaediatr. 90, 757–764 (2001).
7. Jones, J. M. et al. Loss of Omi mitochondrial protease activity causes theneuromuscular disorder of Mnd2 mutant mice. Nature 425, 721–727 (2003).
8. Martins, L. M. et al. Neuroprotective role of the Reaper-related serine proteaseHtrA2/Omi revealed by targeted deletion in mice. Mol. Cell. Biol. 24, 9848–9862(2004).
9. Cilenti, L. et al. Regulation of HAX-1 anti-apoptotic protein by Omi/HtrA2protease during cell death. J. Biol. Chem. 279, 50295–50301 (2004).
10. Li, W. et al. Structural insights into the pro-apoptotic function of mitochondrialserine protease HtrA2/Omi. Nature Struct. Biol. 9, 436–441 (2002).
11. Cipolat, S. et al. Mitochondrial rhomboid PARL regulates cytochrome c releaseduring apoptosis via OPA1-dependent cristae remodeling. Cell 126, 163–175(2006).
12. Vaux, D. L. & Silke, J. HtrA2/Omi, a sheep in wolf’s clothing. Cell 115, 251–253(2003).
13. Frezza, C. et al. OPA1 controls apoptotic cristae remodeling independently frommitochondrial fusion. Cell 126, 177–189 (2006).
14. Letai, A. Growth factor withdrawal and apoptosis: the middle game. Mol. Cell 21,728–730 (2006).
15. Deckwerth, T. L. et al. BAX is required for neuronal death after trophic factordeprivation and during development. Neuron 17, 401–411 (1996).
16. Deckwerth, T. L., Easton, R. M., Knudson, C. M., Korsmeyer, S. J. & Johnson, E. M.Jr. Placement of the BCL2 family member BAX in the death pathway ofsympathetic neurons activated by trophic factor deprivation. Exp. Neurol. 152,150–162 (1998).
WT
Activated Bax
Activated Bax
Activated Bax
Activated Bax
Activated Bax
Hax1 KO
Bax KO
Bim KO
Mnd2
WT
Hax1 KO
Bax KO
Bim KO
Mnd2
42618025
25
25
25
25
37HtrA2
Hax1
HtrA2
Hax1
HtrA2
Hax1
HtrA2
Hax1
HtrA2
Hax1
37
37
37
37
37
37
37
37
37
Time (h)
a
b
–IL-2
426180Time (h) –IL-2
Figure 4 | HtrA2 has a role in controlling accumulation of activated Bax inperipheral lymphocytes. Pre-activated T cells from wild-type, Hax1-null,Bax-null, Mnd2 or Bim-null mice were cultured in media without IL-2 for 0,8, 16 or 24 h. Lysates were immunoblotted with the indicated antibodies.a, Activated Bax was detected by lysing cells in 1% CHAPS lysis buffer (1%CHAPS, 20 mM Tris-Cl, pH 7.4, 137 mM NaCl, 10% glycerol and 1 mMEDTA), immunoprecipitation with mouse monoclonal anti-Bax antibody(6A7) and immunoblotting with rabbit polyclonal anti-Bax antibody.b, Immunoblots for detection of HtrA2 or Hax-1. Loading controls (b-actin)and the results examining Bim, total Bax, Mcl-1, Bcl-x, Bcl-2 and Parl, as wellas the percentage of viable T cells are presented in Supplementary Fig. 6.
NATURE | Vol 452 | 6 March 2008 LETTERS
101Nature Publishing Group©2008

17. White, F. A., Keller-Peck, C. R., Knudson, C. M., Korsmeyer, S. J. & Snider, W. D.Widespread elimination of naturally occurring neuronal death in Bax-deficientmice. J. Neurosci. 18, 1428–1439 (1998).
18. Lindsten, T. et al. The combined functions of proapoptotic Bcl-2 family membersbak and bax are essential for normal development of multiple tissues. Mol. Cell 6,1389–1399 (2000).
19. Wei, M. C. et al. tBID, a membrane-targeted death ligand, oligomerizes BAK torelease cytochrome c. Genes Dev. 14, 2060–2071 (2000).
20. Harada, H., Quearry, B., Ruiz-Vela, A. & Korsmeyer, S. J. Survival factor-inducedextracellular signal-regulated kinase phosphorylates BIM, inhibiting itsassociation with BAX and proapoptotic activity. Proc. Natl Acad. Sci. USA 101,15313–15317 (2004).
21. Ley, R. et al. Extracellular signal-regulated kinases 1/2 are serum-stimulated‘‘Bim(EL) kinases’’ that bind to the BH3-only protein Bim(EL) causing itsphosphorylation and turnover. J. Biol. Chem. 279, 8837–8847 (2004).
22. Luciano, F. et al. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes itsdegradation via the proteasome pathway and regulates its proapoptotic function.Oncogene 22, 6785–6793 (2003).
23. Seward, R. J., von Haller, P. D., Aebersold, R. & Huber, B. T. Phosphorylation of thepro-apoptotic protein Bim in lymphocytes is associated with protection fromapoptosis. Mol. Immunol. 39, 983–993 (2003).
24. Nechushtan, A., Smith, C. L., Hsu, Y. T. & Youle, R. J. Conformation of the BaxC-terminus regulates subcellular location and cell death. EMBO J. 18, 2330–2341(1999).
25. Wolter, K. G. et al. Movement of Bax from the cytosol to mitochondria duringapoptosis. J. Cell Biol. 139, 1281–1292 (1997).
26. Plun-Favreau, H. et al. The mitochondrial protease HtrA2 is regulated byParkinson’s disease-associated kinase PINK1. Nature Cell Biol. 9, 1243–1252(2007).
27. Parganas, E. et al. Jak2 is essential for signaling through a variety of cytokinereceptors. Cell 93, 385–395 (1998).
Supplementary Information is linked to the online version of the paper atwww.nature.com/nature.
Acknowledgements This work was supported by grants from the NationalInstitutes of Health to J.N.I. and the American Lebanese and Syrian AssociatedCharities (ALSAC) of SJCRH (St Jude Children’s Research Hospital). We thankB. De Strooper for providing reagents and information regarding the properties ofthe Parl-null mice and L. Pellegrini for antisera against Parl. J.T.O. is a Pew Scholarin the Biomedical Sciences.
Author Contributions J.-R.C. planned the project, performed experimental workand helped to prepare the manuscript. E.P. and C.Y.H. performed experimentalwork. K.B. performed the animal histology. J.T.O. and J.N.I. planned and directedthe project, analysed data and wrote the manuscript.
Author Information Reprints and permissions information is available atwww.nature.com/reprints. Correspondence and requests for materials should beaddressed to J.N.I. ([email protected]).
LETTERS NATURE | Vol 452 | 6 March 2008
102Nature Publishing Group©2008

METHODSIsolation of mitochondrial and cytosolic fractions. Mitochondria were isolated
by suspending cells in hypotonic butter (250 mM sucrose, 0.1 mM EGTA,
10 mM HEPES-KOH, pH 7.4) and homogenization or passing through a 27G
syringe. The homogenate was centrifuged for 10 min at 700g to remove cell
debris. The supernatant was centrifuged for 10 min at 7,000g to pellet the mito-
chondria. The resulting supernatant was further centrifuged for 10 min at
15,000g to obtain a cytosolic fraction.
Immunoprecipitations and immunoblots. Immunoprecipitations and
immunoblots used standard techniques that have been described previously27.Antibodies included mouse monoclonal anti-Hax1 (BD Biosciences and Santa
Cruz Biotechnology), mouse monoclonal anti-Bcl-X (BD Biosciences), mouse
monoclonal anti-Bcl-2 (BD Biosciences), rabbit polyclonal anti-Bax (Upstate)
and rabbit polyclonal anti-Mcl-1 (Rockland Immunochemical). Other antibod-
ies used are anti-HA (3F10 from Roche), anti-Flag (M2 from Sigma), anti-Myc
(Sigma), anti-HtrA2 (R&D and Santa Cruz Biotechnology), anti-Parl (obtained
from B. D. Strooper and L. Pellegrini), anti-Bim (BD Sciences). Activated Bax
was detected by lysing cells in 1% CHAPS lysis buffer (1% CHAPS, 20 mM Tris-
Cl, pH 7.4, 137 mM NaCl, 10% glycerol and 1 mM EDTA). Lysates were
immunoprecipitated with mouse monoclonal anti-Bax antibody (6A7 from
BD Biosciences) followed by immunoblotting with rabbit polyclonal anti-Bax
antibody (Upstate).
Derivation of pre-activated T and pro-B cells. Pre-activated T cells were
obtained by culturing splenocytes with anti-CD3e monoclonal antibody, fol-
lowed by 7 days of culture in medium containing 200 units ml–1 human IL-2.
Pro-B cells were obtained by culturing bone marrow cells in the medium con-
taining stem cell factor and IL-7 for 14 days.
Analysis of neuronal apoptosis and astrocyte infiltration. For brain sections,mice were perfused with PBS followed by 10% buffered formalin. Brains were
dissected and embedded in paraffin, and were sectioned by a microtome. The
sections were stained with hematoxylin and eosin or TUNEL, or were immuno-
stained for GFAP using a rabbit anti-human GFAP antibody (Dako).
HtrA2 mutant mice and Parl-deficient fibroblasts. Mnd2 mice were provided
by G. Cox and are a consequence of a spontaneous recessive mutation in C57Bl/6
mice. Parl-deficient fibroblasts were provided by B. De Strooper and were
obtained from mice created from E14 embryonic stem cells injected into
C57Bl/6 blastocysts11.
doi:10.1038/nature06604
Nature Publishing Group©2008

CORRIGENDUMdoi:10.1038/nature06871
A photosynthetic alveolate closely related toapicomplexan parasitesRobert B. Moore, Miroslav Obornık, Jan Janouskovec,Tomas Chrudimsky, Marie Vancova, David H. Green,Simon W. Wright, Noel W. Davies, Christopher J. S. Bolch,Kirsten Heimann, Jan Slapeta, Ove Hoegh-Guldberg,John M. Logsdon Jr & Dee A. Carter
Nature 451, 959–963 (2008)
In Fig. 2c of this Article, the GenBank accession number DQ174732was incorrectly cited. The correct accession number is DQ174731(Chromera velia).
ERRATUMdoi:10.1038/nature06872
Hax1-mediated processing of HtrA2 by Parlallows survival of lymphocytes and neuronsJyh-Rong Chao, Evan Parganas, Kelli Boyd, Cheol Yi Hong,Joseph T. Opferman & James N. Ihle
Nature 452, 98–102 (2008)
In Fig. 1a of this Letter, the light blue line was incorrectly labelledas Hax1/Bak DKO, n 5 13. The light blue line should be labelledHax1/Bax DKO, n 5 13.
ERRATUMdoi:10.1038/nature06898
Strong dispersive coupling of a high-finessecavity to a micromechanical membraneJ. D. Thompson, B. M. Zwickl, A. M. Jayich, Florian Marquardt,S. M. Girvin & J. G. E. Harris
Nature 452, 72–72 (2008)
In the print and HTML versions of this Letter, Fig. 2b was printedincorrectly. The PDF version published online is correct. The cor-rected Fig. 2b is shown below.
Membrane displacement (nm)1,2008004000
100
102
101
1.0
2.0
0.0
3.0
Lase
r d
etun
ing
(GH
z)
b
CORRECTIONS & AMENDMENTS NATUREjVol 452j17 April 2008
900Nature Publishing Group©2008





























