Embed Size (px)
Citation preview

Therapeutics, Targets, and Chemical Biology
Harnessing the Fcm Receptor for Potent and SelectiveCytotoxic Therapy of Chronic Lymphocytic Leukemia
B�ereng�ere Vire1, Martin Skarzynski1, Joshua D. Thomas2, Christopher G. Nelson2, Alexandre David3,Georg Aue1, Terrence R. Burke Jr2, Christoph Rader4,5,6, and Adrian Wiestner1
AbstractChronic lymphocytic leukemia (CLL) is a B-cell malignancy in need of new, effective, and safe therapies. The
recently identified IgM receptor FcmR is overexpressed on malignant B cells in CLL and mediates the rapidinternalization and lysosomal shuttling of IgM via its Fc fragment (Fcm). To exploit this internalization andtrafficking pathway for targeted drug delivery, we engineered an IgM-derived protein scaffold (Fcm) and linked itwith the cytotoxic agent monomethylauristatin F. This Fcm–drug conjugate was selectively toxic for FcmR-expressing cell lines in vitro and for CLL cells but not autologous normal T cells ex vivo. Notably, the cytotoxicactivity of the Fcm–drug conjugate wasmaintained in CLL cells carrying a 17p deletion, which predicts resistanceto standard chemotherapy. Next, we tested the possible therapeutic application of the Fcm–drug conjugate inimmunodeficient NOD/SCID/IL-2Rgnull (NSG) mice engrafted with peripheral blood cells from patients withleukemia. Three intravenous injections of the Fcm–drug conjugate over a 10-day period were well tolerated andselectively killed the human CLL cells but not the coengrafted autologous human T cells. In summary, wedeveloped a novel strategy for targeted cytotoxic therapy of CLL based on the unique properties of FcmR. FcmR-targeted drug delivery showed potent and specific therapeutic activity in CLL, thus providing proof of conceptfor FcmR as a valuable therapeutic target in CLL and for IgM-based antibody–drug conjugates as a new targetingplatform. Cancer Res; 74(24); 1–11. �2014 AACR.
IntroductionOn the basis of their ability to selectively deliver highly
cytotoxic drugs into tumor cells, antibody–drug conjugates(ADC) are among the most promising next-generation anti-body therapeutics for cancer therapy (1, 2). The promise ofADCs is the targeted delivery of a potent cytotoxic drugselectively into tumor cells, therebyminimizing toxicity toward
normal cells. The Food and Drug Administration (FDA)approval of brentuximab vedotin for the therapy of Hodgkinlymphoma and anaplastic large cell lymphoma in 2011 and oftrastuzumab emtansine for HER2þmetastatic breast cancer in2013 weremilestones that established the therapeutic utility ofADCs (3, 4). Brentuximab vedotin consists of a chimericmouse/human anti-human CD30 monoclonal antibody (mAb)in IgG1 format as the carrier protein conjugated to mono-methylauristatin E (MMAE) as the cytotoxic payload (5).MMAE is a synthetic antitubulin agent active at subnanomolarconcentrations. Each MMAE drug is linked to the antibodymolecule through a linker that harbors a valine–citrulline–para-aminobenzylcarbamate (Val–Cit–PABC) linker that iscleaved by lysosomal proteases such as cathepsin B. The linkeralso contains a maleimide group that reacts with thiol groupsin the IgG1 hinge region. This randomconjugation results in anADC mixture with an average drug-to-antibody ratio (DAR) of4:1 (range, 0:1 to 8:1). Trastuzumab emtansine is based on thehumanized anti-human HER2 mAb trastuzumab randomlyconjugated to an average of 3.5 maytansinoid drugs throughthe e-amino group of lysine (Lys) using a noncleavable linker(6). This new generation of ADCs has demonstrated twoimportant treatment advances; first, patients who relapse orare refractory to first-line therapy can be rescued using tar-geted cytotoxic drug delivery; and second, an ADC can replacesystemic cytotoxic therapy demonstrating higher efficacy withlower toxicity (7, 8).
Chronic lymphocytic leukemia (CLL), the most commonleukemia in Western countries, is characterized by the
1Hematology Branch, National Heart, Lung, and Blood Institute, NIH,Bethesda, Maryland. 2Chemical Biology Laboratory, Molecular DiscoveryProgram, Center for Cancer Research, National Cancer Institute, NIH,Frederick, Maryland. 3Laboratory of Viral Diseases, National Institute ofAllergy and Infectious Diseases, NIH, Bethesda, Maryland. 4ExperimentalTransplantation and Immunology Branch, Center for Cancer Research,National Cancer Institute, NIH, Bethesda, Maryland. 5Department of Can-cer Biology, The Scripps Research Institute, Scripps Florida, Jupiter,Florida. 6Department of Molecular Therapeutics, The Scripps ResearchInstitute, Scripps Florida, Jupiter, Florida.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
B. Vire and M. Skarzynski contributed equally to this article.
Corresponding Authors: Adrian Wiestner, Hematology Branch,National Heart, Lung, and Blood Institute, NIH, Building 10, CRC3-5140, 10 Center Drive, Bethesda, MD 20892-1202. Phone: 301-594-6855; Fax: 301-496-8396; E-mail: [email protected]; and ChristophRader, Department of Cancer Biology, Department of Molecular Thera-peutics, The Scripps Research Institute, Scripps Florida, 130 Scripps Way#2C1, Jupiter, FL 33458. Phone: 561-228-2053; Fax: 561-228-2169;E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-14-2030
�2014 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org OF1
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030

accumulation of mature monoclonal B cells in the blood, bonemarrow, spleen, and lymph nodes. Themedian age at diagnosisis 75 years, precluding use of allogeneic hematopoietic stemcell transplantation, the only curative treatment option, in themajority of patients. Current first-line treatment is chemoim-munotherapy with an anti-CD20 mAb and an alkylating agentwith or without the addition of a purine analog (9–11). One ofthe most commonly used regimens for younger patients is thecombination offludarabine, cyclophosphamide, and rituximab(FCR; refs. 11, 12). Although initial response rates are high,most patients relapse. FCR is less active in high-risk disease,less tolerable in elderly or frail patients, and increases the riskof infections. Fludarabine and cyclophosphamide are also toxicfor normal T cells and myeloid cells, leading to often long-lasting cytopenias. In contrast, the anti-CD20 mAbs rituximab,ofatumumab, and obinutuzumab selectively target B cells (13).These mAbs induce cell death mostly through immunologiceffector mechanisms such as complement-dependent cytotox-icity and antibody-dependent cellular cytotoxicity (13, 14). Assingle agents, the efficacy of anti-CD20 mAbs in CLL is limitedand they are used primarily in combination regimens. Notably,CD20 is expressed at higher levels on normal B cells than onCLL cells (15), and anti-CD20 mAbs effectively kill normal Bcells, potentially contributing to life-threatening viral, bacte-rial, and fungal infections (16). Recently, kinase inhibitors thattarget B-cell receptor (BCR) signal transduction have shownimpressive clinical activity in CLL (17–19). However, while welltolerated as single agents, these inhibitors achieve mostlypartial responses and have to be taken continuously. In fact,even after years of treatment with a kinase inhibitor, residualdisease is easily detected in virtually all patients and resistantclones emerge that lead to relapse. Thus, there is a great clinicalneed for targeted cytotoxic agents that could be combinedwithkinase inhibitors to eradicate the disease.
ADCs are also currently being tested as treatment optionsfor CLL. A phase II clinical trial (NCT01461538) is investigatingbrentuximab vedotin in CD30þ malignancies, includingCLL. In addition, a recently launched phase I clinical trial(NCT01290549) is based on an analogous ADC that targetsCD79B of the BCR complex in non-Hodgkin lymphoma andCLL. Notably, none of the cell-surface antigens targeted byADCs in clinical trials are overexpressed in CLL. On thecontrary, CD79B was not detected on the tumor cells of 43%of the patients with CLL tested, while it is expressed on normalB cells (20), and CD30 is expressed at higher levels on activatednormal B and T cells compared with CLL cells (21, 22). Thus,the current panel of clinically investigated ADCs for CLL doesnot bypass immunosuppression, underscoring the need forpursuing new ADCs that selectively target CLL.
The Fc receptor for IgM (FcmR), also known as TOSO or Fasapoptotic inhibitorymolecule 3 (FAIM3), is highly expressed onCLL cells (23–26). FcmR is an approximately 60-kDa type Isingle-pass transmembrane protein whose expression in nor-mal human cells and tissues is virtually restricted to thelymphoid lineage (23, 27). FcmR-deficient mice are viable andexhibit normal development (28–30). However, they havereduced numbers of marginal zone B cells (29), and FcmR-deficient B cells are less responsive to BCR stimulation and
more readily undergo apoptosis. FcmR is overexpressed on theCLL B cells compared with B cells from normal donors andcompared with the nonclonal T cells in the blood of patientswith CLL. FcmR may play a role in the pathogenesis of CLL,possibly by contributing to concomitant BCR and Toll-likereceptor (TLR) activation that could enhance leukemic cellproliferation and survival (26, 31, 32). However, exactly whatfunctional role, if any, FcmR has in the pathogenesis of CLLremains to be defined.
Fc receptors are highly effective in targeting specific mole-cules for binding to and internalization into select target cells.The existence of a variety of distinct Fc receptors with restrict-ed cellular expression provides the basis for selective targetingapproaches. For example, FcgRIII on dendritic cells has beenused for internalization of vaccine components (33). However,delivering cytotoxic agents through Fc receptor–mediatedinternalization as anticancer therapy has not been established.In previous work, we have shown that FcmR on CLL cells isfunctional, rapidly internalizes IgM, and delivers it to thelysosome where it is degraded (26). In fact, more than 50% ofIgM bound to the CLL cell surface is internalized via FcmRwithin 1 minute, and internalization is complete by 5 minutes.We hypothesized that the overexpression of FcmR on CLL cellsand its ability to internalize bound IgM could be used fortargeted delivery of antileukemic therapy.
Here, we describe the derivation of an FcmR-targeting strat-egy that can deliver a cytotoxic payload selectively into CLLcells and show that it has antitumor activity against primaryhuman tumor cells in vivo. On the basis of the work byKubagawa and colleagues (23), we knew that binding of IgMto FcmR critically depends on the integrity of sequences in theFc region of IgM (Fcm) in particular the Cm2-4 domains. Wetherefore engineered a protein scaffold consisting of Fcm andchose a site-directed conjugation technology we have previ-ously developed (34, 35). In this approach, the introduction of aC-terminal selenocysteine (Sec)makes site-specific as opposedto random conjugation of the payload possible, thereby pre-serving the structure of critical protein domains.
Materials and MethodsSupplementary Materials and Methods describe the (i)
cloning, expression, and purification of Fcµ-Sec; (ii) conju-gation of Fcµ-Sec; (iii) SDS-PAGE, Western blotting, andgel filtration chromatography; (iv) synthesis of MMAF com-pounds 1a and 1b; and (v) comparison of treatment naïve andpreviously treated patients.
Primary cells and cell linesWith written informed consent, peripheral blood was col-
lected from patients with treatment-na€�ve CLL at the ClinicalCenter, National Institutes of Health (Bethesda, MD; www.clinicaltrials.gov identifier NCT00923507). Peripheral bloodmononuclear cells (PBMC) were isolated by gradient centri-fugation (Lymphocyte Separation Medium; MP Biomedicals)and cultured in serum-free AIM-V medium (Life Technolo-gies). Mantle cell lymphoma (MCL) cell linesMino, JeKo-1, andHBL-2 were grown in RPMI-1640 medium (Life Technologies)andHeLa cells in DMEM (Lonza) supplemented with 10% (v/v)
Vire et al.
Cancer Res; 74(24) December 15, 2014 Cancer ResearchOF2
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030

fetal bovine serum (FBS; HyClone), 2 mmol/L L-glutamine,1,000 U/mL penicillin G, and 100 mg/mL streptomycin (allfrom Life Technologies).
Confocal immunofluorescence microscopyWe monitored Fcm–Sec internalization as previously
described (26). HeLa cells stably transfected with FcmR weregrown on coverslips, incubated for 15 minutes at 4�C withCm3–Cm4–Sec, washed with ice-cold PBS, and incubated at37�C with DMEM (Lonza) for 30 or 120 minutes. Cells werethen fixed with 3% (w/v) paraformaldehyde (Electron Micros-copy Sciences), washed with PBS, and incubated in StainingBuffer [0.05% (w/v) saponin, 10 mmol/L glycine, and 5% (v/v)FBS in PBS] for 15 minutes. Cells were costained with rabbitanti-human LAMP-1 polyclonal antibodies (pAb; Abcam) for 1hour, washed twice with PBS, and incubated with donkey anti-human Fcm pAbs conjugated to DyLight 488 and donkey anti-rabbit IgG pAbs conjugated to Cy5 (all from Jackson Immu-noResearch Laboratories). Cells were then washed twice withPBS and labeled for 5 minutes at room temperature with1 mg/mL Hoechst 33258 (Life Technologies) diluted in StainingBuffer. Subsequently, coverslips were washed twice with PBS,mounted with Fluoromount-G (SouthernBiotech), and visual-ized by confocal microscopy. Images were acquired using aLeica TCS SP5 laser scanning confocal microscope (LAS AFsoftware) using the HCX PLAPO 63X objective (numericalaperture, 1.4). Images were processed with Adobe Photoshopand analyzed using the same settings.
Flow cytometryMino, JeKo-1, and HBL-2 cells were stained with Cm3–Cm4–
Sec/DyLight 488 or Cm2–Cm3–Cm4–Sec/DyLight 488. FcmRexpression in cryopreserved PBMC samples from 30 patientswith CLL was measured with a mouse anti-human FcmR mAb(Abnova) and a FITC-conjugated goat anti-mouse secondaryantibody (Sigma-Aldrich) as previously described (26). B cellsand normal T cells were distinguishedwith anAPC-conjugatedmouse anti-human CD19 mAb and a PE-conjugated mouseanti-human CD3 mAb (BD Biosciences), respectively. All sam-ples were analyzed using a FACSCanto II flow cytometer(Becton-Dickinson) and FlowJo software (TreeStar).
In vitro cytotoxicity assaysCytotoxicity toward MCL cell lines in vitro was measured by
using the CellTiter 96 AQueous Cell Proliferation Assay (Pro-mega). Briefly, Mino or HBL2 cells were distributed in a 96-wellflat-bottomed plate (Corning) at a density of 5 � 104 cells perwell. Subsequently, compound 1a (0.001–1,000 nmol/L) orFcm–Sec protein selectively conjugated to compound 1b (1–100 nmol/L) were added to the cells. In parallel, unconjugatedFcm–Sec protein (1–100 nmol/L)was also tested. The cellswereexposed to the compound 1a and 1b reagents for 1 hour at37�C, washed three times with RPMI-1640 medium, and incu-bated for an additional 71 hours at 37�C. After adding 20-mLAssay Reagent to each well, the cells were further cultured for 3hours at 37�C and the absorbance at 490 nm was measuredusing a SpectraMax M5 microplate reader with SoftMax Prosoftware. Data were computed as mean � SD of triplicates.
Ex vivo cytotoxicity assaysCytotoxicity toward primary malignant B cells and primary
normal T cells ex vivo was measured by flow cytometry usingTO-PRO-3 and Annexin V staining. Briefly, cryopreservedPBMC samples from 30 different patients with CLL (5 � 105
cells in 100-mL serum-free AIM-V medium) were incubatedwith 100 nmol/L compound 1a, Cm2–Cm3–Cm4–Sec/com-pound 1b conjugate, Cm2–Cm3–Cm4–Sec alone, or were leftuntreated for 1 hour at 37�C, washed and then cytotoxicityagainst malignant B cells and normal T cells was evaluated byflow cytometry using PE-conjugated Annexin V, a PE–Cy5–conjugated mouse anti-human CD19 mAb, a FITC-conjugatedmouse anti-human CD3 mAb (all from BD Biosciences), andTO-PRO-3 (Invitrogen) according to the manufacturer'sinstructions.
Circulatory half-lifeTwo groups of two NOD/SCID/IL-2Rgnull (NSG) mice (JAX
strain 5557; The Jackson Laboratory) were injected i.v. (tailvein) or i.p. with 5mg/kg of the Fcm–carrier protein (Cm2–Cm3–Cm4–Sec). One control mouse was injected with PBS. Bloodwas collected via the tail vein at 0.5, 24, 48, and 72 hours afterinjection and serum was isolated. Sera were diluted 25-fold in1% (w/v) BSA in PBS and analyzed by sandwich ELISA. To dothis, 100 ng of rabbit anti-human Fcm pAbs (Jackson Immu-noResearch Laboratories) was coated on a 96-well Costar 3690plate (Corning). After blocking with 3% (w/v) BSA in PBS, thediluted sera or purified Cm2–Cm3–Cm4–Sec as standard wereadded to duplicate wells, washed 10 times with PBS, andincubated with a mouse anti-His6 mAb conjugated to horse-radish peroxidase (GenScript). All steps were carried out for 1hour at 37�C. The plate was washed 10 times with PBS andcolorimetric detection was performed using 2,20-azino-bis(3-ethylbenzthiazoline)-6-sulfonic acid (ABTS; Roche) as sub-strate according to the manufacturer's instructions. Absor-bance values were measured at 405 nm using a Victor3 platereader (PerkinElmer).
NSG/CLL xenograft modelThe NSG/CLL xenograft model was first introduced by
Bagnara and colleagues (36). We used a modified protocolestablished in our laboratory that replicates important aspectsof CLL biology and that we have successfully used in drugstudies (37).Micewere housed andhandled in accordancewiththe guidelines set by the Animal Care and Use Committee ofthe National Heart, Lung, and Blood Institute (Bethesda, MD).All animal experiments were carried out on an approvedanimal protocol. Thirty NSG mice were preconditioned withan i.p. injection of 25-mg/kg busulfan (Otsuka America Phar-maceutical) and on the following day (day 1) i.v. injected with5� 107 PBMC from 1 out of 4 patients with CLL. All mice werebled on day 4 and PBMC were isolated by gradient centrifu-gation using Lymphocyte Separation Medium. On day 4, 6, and8, different cohorts of mice were i.v. injected with 10 mg/kgCm2–Cm3–Cm4–Sec carrier protein or Cm2–Cm3–Cm4–Sec/compound 1b conjugate or an equal volume of PBS. On day11, retro-orbital puncture bleeds and spleens were harvestedfrom all mice. PBMC were isolated as above. Spleens were
Targeted Cytotoxic Drug Delivery through the Fcm Receptor
www.aacrjournals.org Cancer Res; 74(24) December 15, 2014 OF3
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030
homogenized using the gentleMACS Dissociator (MiltenyiBiotec). The homogenate was filtered through a 70-mm nyloncell strainer (BD Biosciences) and washed with ACK LysingBuffer (Quality Biological) to remove erythrocytes and plate-lets. PBMC and splenocytes were stained with a mouse anti-human CD45 mAb conjugated to V450, a mouse anti-humanCD19 mAb conjugated to PE-Cy5, a mouse anti-human CD3mAb conjugated to FITC, PE-conjugated Annexin V (all fromBD Biosciences), and TO-PRO-3 (Life Technologies), and thenanalyzed by flow cytometry as described above. Normalizedtiters of human lymphocyte populations in PBMC isolated ondays 4 and 11 were determined with 5.0- to 5.9-mmAccuCount-Blank Particles (Spherotech) according to the manufacturer'sinstructions.
ResultsGeneration and characterization of Fcm–Sec
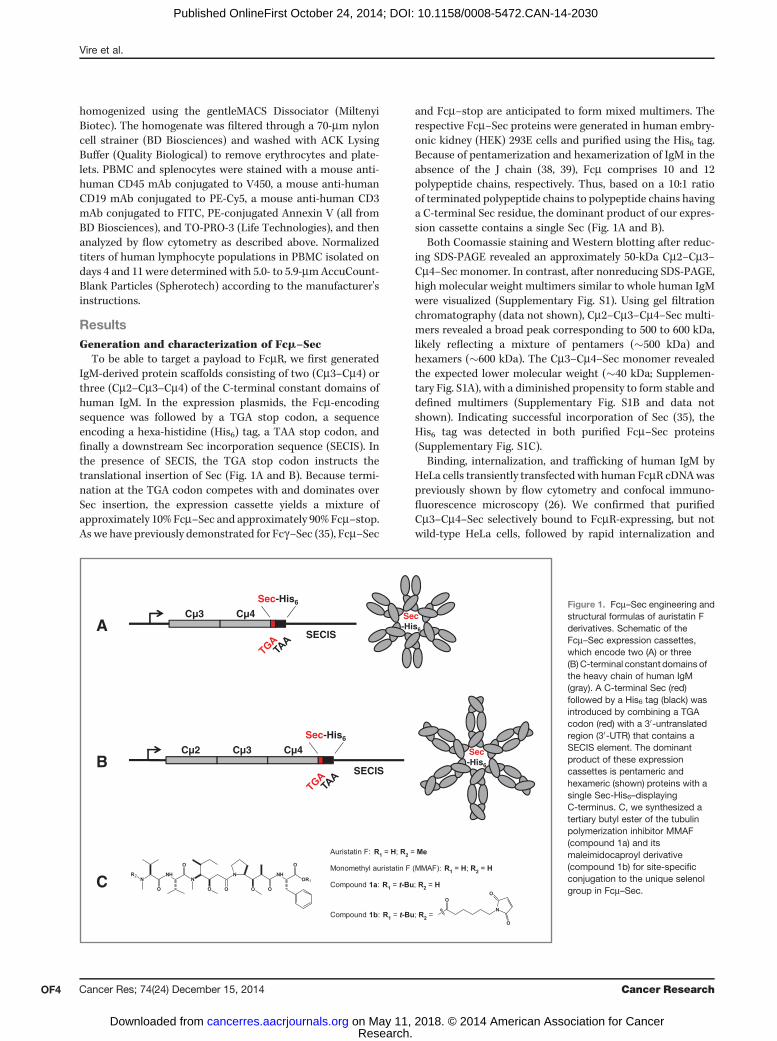
To be able to target a payload to FcmR, we first generatedIgM-derived protein scaffolds consisting of two (Cm3–Cm4) orthree (Cm2–Cm3–Cm4) of the C-terminal constant domains ofhuman IgM. In the expression plasmids, the Fcm-encodingsequence was followed by a TGA stop codon, a sequenceencoding a hexa-histidine (His6) tag, a TAA stop codon, andfinally a downstream Sec incorporation sequence (SECIS). Inthe presence of SECIS, the TGA stop codon instructs thetranslational insertion of Sec (Fig. 1A and B). Because termi-nation at the TGA codon competes with and dominates overSec insertion, the expression cassette yields a mixture ofapproximately 10% Fcm–Sec and approximately 90%Fcm–stop.As we have previously demonstrated for Fcg–Sec (35), Fcm–Sec
and Fcm–stop are anticipated to form mixed multimers. Therespective Fcm–Sec proteins were generated in human embry-onic kidney (HEK) 293E cells and purified using the His6 tag.Because of pentamerization and hexamerization of IgM in theabsence of the J chain (38, 39), Fcm comprises 10 and 12polypeptide chains, respectively. Thus, based on a 10:1 ratioof terminated polypeptide chains to polypeptide chains havinga C-terminal Sec residue, the dominant product of our expres-sion cassette contains a single Sec (Fig. 1A and B).
Both Coomassie staining and Western blotting after reduc-ing SDS-PAGE revealed an approximately 50-kDa Cm2–Cm3–Cm4–Sec monomer. In contrast, after nonreducing SDS-PAGE,high molecular weight multimers similar to whole human IgMwere visualized (Supplementary Fig. S1). Using gel filtrationchromatography (data not shown), Cm2–Cm3–Cm4–Sec multi-mers revealed a broad peak corresponding to 500 to 600 kDa,likely reflecting a mixture of pentamers (�500 kDa) andhexamers (�600 kDa). The Cm3–Cm4–Sec monomer revealedthe expected lower molecular weight (�40 kDa; Supplemen-tary Fig. S1A), with a diminished propensity to form stable anddefined multimers (Supplementary Fig. S1B and data notshown). Indicating successful incorporation of Sec (35), theHis6 tag was detected in both purified Fcm–Sec proteins(Supplementary Fig. S1C).
Binding, internalization, and trafficking of human IgM byHeLa cells transiently transfected with human FcmR cDNAwaspreviously shown by flow cytometry and confocal immuno-fluorescence microscopy (26). We confirmed that purifiedCm3–Cm4–Sec selectively bound to FcmR-expressing, but notwild-type HeLa cells, followed by rapid internalization and
Sec-His6A
Sec-His6
SECIS
Cµ3 Cµ4
B
Sec-His6
SECIS
Cµ2 Cµ3 Cµ4 Sec-His6
C
Figure 1. Fcm–Sec engineering andstructural formulas of auristatin Fderivatives. Schematic of theFcm–Sec expression cassettes,which encode two (A) or three(B) C-terminal constant domains ofthe heavy chain of human IgM(gray). A C-terminal Sec (red)followed by a His6 tag (black) wasintroduced by combining a TGAcodon (red) with a 30-untranslatedregion (30-UTR) that contains aSECIS element. The dominantproduct of these expressioncassettes is pentameric andhexameric (shown) proteins with asingle Sec-His6–displayingC-terminus. C, we synthesized atertiary butyl ester of the tubulinpolymerization inhibitor MMAF(compound 1a) and itsmaleimidocaproyl derivative(compound 1b) for site-specificconjugation to the unique selenolgroup in Fcm–Sec.
Vire et al.
Cancer Res; 74(24) December 15, 2014 Cancer ResearchOF4
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030

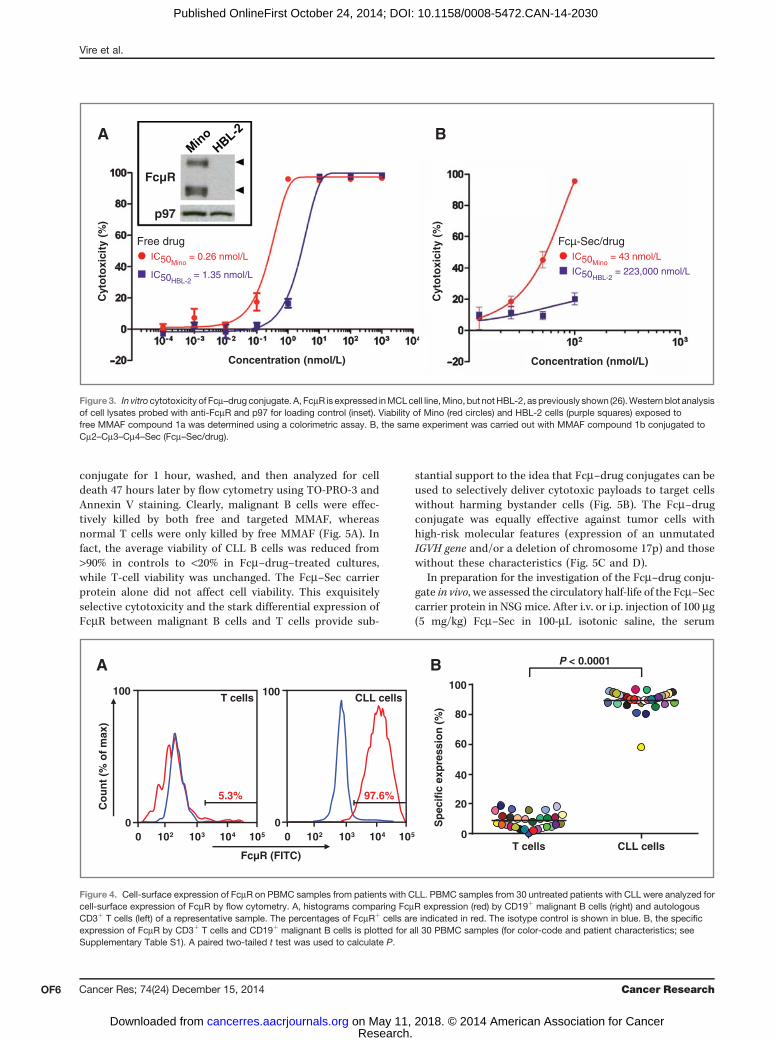
trafficking to lysosomes (Fig. 2). Thus, as described previously(23), the Fc fragment of IgM is sufficient for FcmR binding,internalization, and trafficking. We also confirmed the bindingof both purified Fcm–Sec proteins to the FcmR-expressingMCLcell line, Mino, using Fcm–Sec proteins that we labeled in aSec-dependent reactionwith amaleimide derivative of DyLight488. Two other MCL cell lines, JeKo-1 and HBL-2, which do notexpress FcmR, did not bind Fcm–Sec/DyLight 488 (Fig. 3A anddata not shown).
Generation and in vitro investigations of Fcm–drugconjugateAs the cytotoxic payload, we chose the tubulin polymeriza-
tion inhibitor monomethylauristatin F (MMAF). MMAF differsfromMMAE (the drug component of brentuximab vedotin) byhaving a C-terminal phenylalanine (40). As free drug control,we synthesized a tertiary butyl ester of MMAF (MMAF-tBu;compound 1a), which freely diffuses into cells through theplasma membrane (40), and for Fcm–Sec conjugation, wesynthesized a maleimide derivative with a stable alkyl linker(maleimidocaproyl–MMAF–tBu; compound 1b; Fig. 1C).To test the potency and specificity of free versus conjugated
drug, we used MCL cell lines Mino (FcmR-positive) and HBL-2(FcmR-negative; Fig. 3A). After exposure for 1 hour and chaseincubation for 71 hours, the free drug potently killed both MinoandHBL-2 cells with IC50 values of 0.26 and 1.35 nmol/L, respec-tively (Fig. 3A). As expected, the Fcm–drug conjugate was less
potent than the freely diffusing drug.Under the same conditions,the Fcm–Sec/MMAF conjugate killed Mino and HBL-2 cellswith IC50 values of 43 nmol/L and 223 mmol/L, respectively(Fig. 3B). Thus, FcmR-positive Mino cells were approximately5,200 times more sensitive to targeted MMAF than FcmR-neg-ative HBL-2 cells. Taking the different sensitivities to free druginto account, we observed a 1,000-fold increase in potencywhenFcmR, the target of the carrier protein, was present.
Ex vivo and in vivo investigations of Fcm–drug conjugateNext, we tested whether the Fcm–Sec/MMAF conjugate can
selectively kill primary CLL cells ex vivo. FcmR expression oncryopreserved PBMC from 30 patients with CLL (Supplemen-tary Table S1) was measured by flow cytometry. In patientswith CLL, there are very few or no normal B cells in circulation;therefore, >99% of CD19þ cells are CLL cells (31). As expected,virtually all CLL cells (CD19þ) and much fewer T cells (CD3þ)expressed FcmR (Fig. 4). These findings are consistent withpreviously published observations that FcmR is overexpressedin CLL and that some T cells, especially when activated, canalso express FcmR (23), albeit at low cell-surface levels (24).FcmR expression was equally high in treatment-na€�ve andpreviously treated patients (Supplementary Fig. S4), whichindicates for wide clinical applicability of the FcmR-targetingstrategy.
The same 30 CLL PBMC samples were then treated inparallel with 100-nmol/L free MMAF or the Fcm–Sec/MMAF
merge/HoechstFcµ-Sec LAMP1
0 min
30 min
120 min
Figure 2. Binding, internalization,and trafficking of Fcm–Sec. HeLacells stably transfected withhuman FcmR cDNA wereincubated with 25 mg/mL Cm3–Cm4–Sec (Fcm–Sec) for 30 minutesat 4�C. After washing, the cellswere kept at 4�Corwere incubatedat 37�C for 30 and 120 minutes.The cells were then fixed,permeabilized, and costained withrabbit anti-human LAMP1 pAbsfollowed by donkey anti-rabbit IgGpAbs conjugated to Cy5 (red) anddonkey anti-human Fcm pAbsconjugated to DyLight 488 (green).Nuclei were stained with Hoechst33258 (blue). Stained cellswere visualized by confocalimmunofluorescence microscopy.Note the colocalization (yellow) ofFcm–Sec and lysosomal markerLAMP1 following internalization.Scale bars, 10 mm.
Targeted Cytotoxic Drug Delivery through the Fcm Receptor
www.aacrjournals.org Cancer Res; 74(24) December 15, 2014 OF5
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030
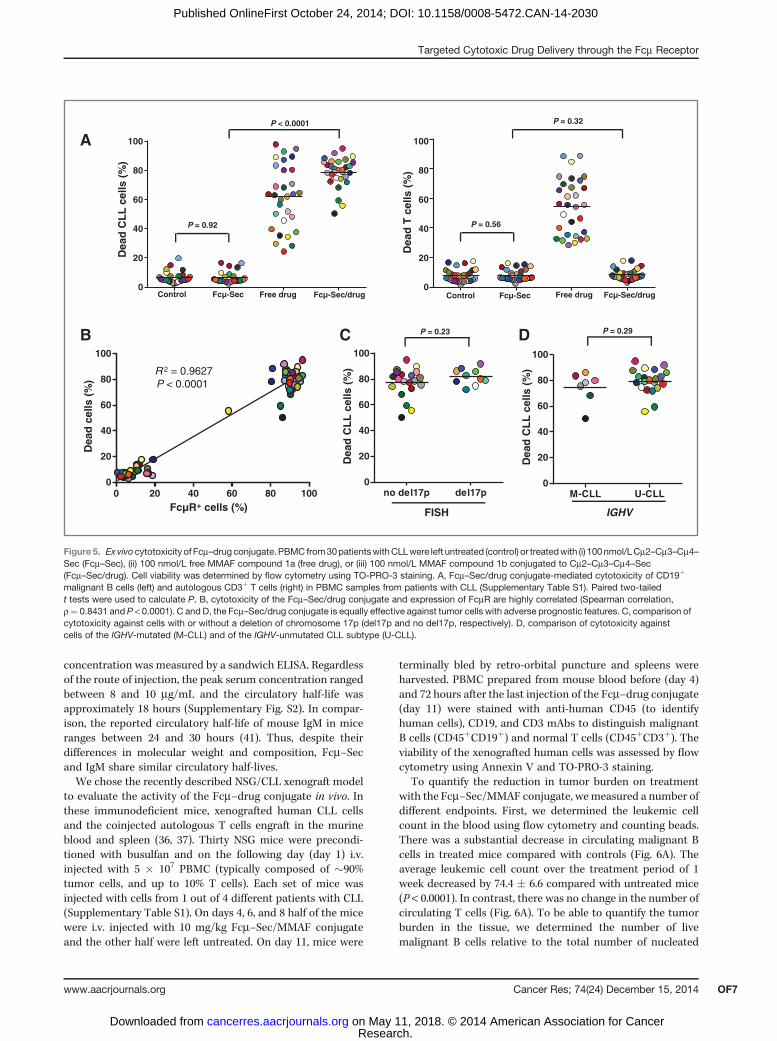
conjugate for 1 hour, washed, and then analyzed for celldeath 47 hours later by flow cytometry using TO-PRO-3 andAnnexin V staining. Clearly, malignant B cells were effec-tively killed by both free and targeted MMAF, whereasnormal T cells were only killed by free MMAF (Fig. 5A). Infact, the average viability of CLL B cells was reduced from>90% in controls to <20% in Fcm–drug–treated cultures,while T-cell viability was unchanged. The Fcm–Sec carrierprotein alone did not affect cell viability. This exquisitelyselective cytotoxicity and the stark differential expression ofFcmR between malignant B cells and T cells provide sub-
stantial support to the idea that Fcm–drug conjugates can beused to selectively deliver cytotoxic payloads to target cellswithout harming bystander cells (Fig. 5B). The Fcm–drugconjugate was equally effective against tumor cells withhigh-risk molecular features (expression of an unmutatedIGVH gene and/or a deletion of chromosome 17p) and thosewithout these characteristics (Fig. 5C and D).
In preparation for the investigation of the Fcm–drug conju-gate in vivo, we assessed the circulatory half-life of the Fcm–Seccarrier protein in NSG mice. After i.v. or i.p. injection of 100 mg(5 mg/kg) Fcm–Sec in 100-mL isotonic saline, the serum
Free drugIC50Mino
= 0.26 nmol/L
IC50HBL-2 = 1.35 nmol/L
FcµR
p97
BA
Concentration (nmol/L)
Cyt
oto
xici
ty (
%)
Fcµ-Sec/drugIC50Mino
= 43 nmol/L
IC50HBL-2 = 223,000 nmol/L
Cyt
oto
xici
ty (
%)
Concentration (nmol/L)
Figure3. In vitro cytotoxicity of Fcm–drug conjugate. A, FcmR is expressed inMCLcell line,Mino, but notHBL-2, as previously shown (26).Western blot analysisof cell lysates probed with anti-FcmR and p97 for loading control (inset). Viability of Mino (red circles) and HBL-2 cells (purple squares) exposed tofree MMAF compound 1a was determined using a colorimetric assay. B, the same experiment was carried out with MMAF compound 1b conjugated toCm2–Cm3–Cm4–Sec (Fcm–Sec/drug).
Co
un
t (%
of
max
)
100 2 103 104 105
0
100
100 2 103 104 105
0
100
FcµR (FITC)
P < 0.0001BA
CLL cellsT cells100
80
60
40
20
0T cells CLL cells
5.3% 97.6%
Sp
ecif
ic e
xpre
ssio
n (%
)
Figure 4. Cell-surface expression of FcmR on PBMC samples from patients with CLL. PBMC samples from 30 untreated patients with CLL were analyzed forcell-surface expression of FcmR by flow cytometry. A, histograms comparing FcmR expression (red) by CD19þ malignant B cells (right) and autologousCD3þ T cells (left) of a representative sample. The percentages of FcmRþ cells are indicated in red. The isotype control is shown in blue. B, the specificexpression of FcmR by CD3þ T cells and CD19þ malignant B cells is plotted for all 30 PBMC samples (for color-code and patient characteristics; seeSupplementary Table S1). A paired two-tailed t test was used to calculate P.
Vire et al.
Cancer Res; 74(24) December 15, 2014 Cancer ResearchOF6
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030
concentration was measured by a sandwich ELISA. Regardlessof the route of injection, the peak serum concentration rangedbetween 8 and 10 mg/mL and the circulatory half-life wasapproximately 18 hours (Supplementary Fig. S2). In compar-ison, the reported circulatory half-life of mouse IgM in miceranges between 24 and 30 hours (41). Thus, despite theirdifferences in molecular weight and composition, Fcm–Secand IgM share similar circulatory half-lives.We chose the recently described NSG/CLL xenograft model
to evaluate the activity of the Fcm–drug conjugate in vivo. Inthese immunodeficient mice, xenografted human CLL cellsand the coinjected autologous T cells engraft in the murineblood and spleen (36, 37). Thirty NSG mice were precondi-tioned with busulfan and on the following day (day 1) i.v.injected with 5 � 107 PBMC (typically composed of �90%tumor cells, and up to 10% T cells). Each set of mice wasinjected with cells from 1 out of 4 different patients with CLL(Supplementary Table S1). On days 4, 6, and 8 half of the micewere i.v. injected with 10 mg/kg Fcm–Sec/MMAF conjugateand the other half were left untreated. On day 11, mice were
terminally bled by retro-orbital puncture and spleens wereharvested. PBMC prepared from mouse blood before (day 4)and 72 hours after the last injection of the Fcm–drug conjugate(day 11) were stained with anti-human CD45 (to identifyhuman cells), CD19, and CD3 mAbs to distinguish malignantB cells (CD45þCD19þ) and normal T cells (CD45þCD3þ). Theviability of the xenografted human cells was assessed by flowcytometry using Annexin V and TO-PRO-3 staining.
To quantify the reduction in tumor burden on treatmentwith the Fcm–Sec/MMAF conjugate, wemeasured a number ofdifferent endpoints. First, we determined the leukemic cellcount in the blood using flow cytometry and counting beads.There was a substantial decrease in circulating malignant Bcells in treated mice compared with controls (Fig. 6A). Theaverage leukemic cell count over the treatment period of 1week decreased by 74.4 � 6.6 compared with untreated mice(P < 0.0001). In contrast, there was no change in the number ofcirculating T cells (Fig. 6A). To be able to quantify the tumorburden in the tissue, we determined the number of livemalignant B cells relative to the total number of nucleated
Control Free drug
Dea
d T
cel
ls (
%)
A
Dea
d C
LL
cel
ls (
%)
FISH
no del17p del17p0
20
40
60
80
100
Dea
d C
LL
cel
ls (
%)
IGHV
M-CLL U-CLL0
20
40
60
80
100
Dea
d c
ells
(%
)
FcµR+ cells (%)0 20 40 60 80 100
0
20
40
60
80
100
DCB
Fcµ-Sec Fcµ-Sec/drug
R 2 = 0.9627P < 0.0001
P = 0.23 P = 0.29
P = 0.32
P = 0.56
0
20
40
60
80
100
0
20
40
60
80
100
Control Free drug
Dea
d C
LL
cel
ls (
%)
P < 0.0001
Fcµ-Sec/drugFcµ-Sec
P = 0.92
Figure5. Exvivocytotoxicity of Fcm–drugconjugate.PBMC from30patientswithCLLwere left untreated (control) or treatedwith (i) 100nmol/LCm2–Cm3–Cm4–Sec (Fcm–Sec), (ii) 100 nmol/L free MMAF compound 1a (free drug), or (iii) 100 nmol/L MMAF compound 1b conjugated to Cm2–Cm3–Cm4–Sec(Fcm–Sec/drug). Cell viability was determined by flow cytometry using TO-PRO-3 staining. A, Fcm–Sec/drug conjugate-mediated cytotoxicity of CD19þ
malignant B cells (left) and autologous CD3þ T cells (right) in PBMC samples from patients with CLL (Supplementary Table S1). Paired two-tailedt tests were used to calculate P. B, cytotoxicity of the Fcm–Sec/drug conjugate and expression of FcmR are highly correlated (Spearman correlation,r¼ 0.8431 and P < 0.0001). C and D, the Fcm–Sec/drug conjugate is equally effective against tumor cells with adverse prognostic features. C, comparison ofcytotoxicity against cells with or without a deletion of chromosome 17p (del17p and no del17p, respectively). D, comparison of cytotoxicity againstcells of the IGHV-mutated (M-CLL) and of the IGHV-unmutated CLL subtype (U-CLL).
Targeted Cytotoxic Drug Delivery through the Fcm Receptor
www.aacrjournals.org Cancer Res; 74(24) December 15, 2014 OF7
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030

cells in single-cell suspensions prepared from spleens. Themajority of these nucleated cells are murine cells that havebeen used successfully as internal reference in other studies(36, 37). The tumor burden in spleens decreased on averageby 64.9� 9.7 on treatment with the Fcm–Sec/MMAF conjugate(P < 0.0001) but again there was no effect on T-cell numbers(Fig. 6B). The unconjugated Fcm–Sec carrier protein alone hadno effect on the viability of CLL cells nor T cells (SupplementaryFig. S3A and S3B). Taken together, these in vivo studies revealedsubstantial reductions of the total tumor burden in both blood(�74%) and spleens (�65%) of mice treated with Fcm–drugconjugate (Fig. 6C). Consistent with the desired selectivity ofthis targeted approach, there was no effect on T cells neither inblood nor spleen (Fig. 6C). Notably, none of the mice showedsigns of toxicity, and the viability of murine blood cells was notreduced in treated as compared with control mice (data notshown).
DiscussionFcmR is overexpressed in CLL and mediates the rapid
internalization of IgM by malignant B cells and its traffickingto lysosomes (24–26). In this study, we used an Fcm–drugconjugate to exploit FcmR for targeted drug delivery. In con-trast to conventional ADCs, our targeting platform is a recom-binant protein scaffold designed to mimic binding of the
natural ligand and not a mAb. In other respects, our Fcm–drugconjugate is built on important design principles shared withthe recently FDA-approved ADCs brentuximab vedotin andtrastuzumab emtansine (1–8). Specifically, the target antigenFcmR is overexpressed on tumor cells and rapidly internalizedupon ligand binding; the Fcm carrier protein was engineeredwith a C-terminal Sec residue for site-specific conjugation tothe potent antitubulin agent MMAF; finally, use of a nonclea-vable linker that canminimize systemic drug release is possiblebecause internalized FcmR travels to the lysosome where thecarrier protein is degraded releasing the cytotoxic payload.The resulting Fcm–Sec/MMAF conjugate selectively killedFcmR-expressing malignant B cells in vitro, ex vivo, and in vivo.Accordingly, our study provides proof-of-concept for FcmR asa therapeutic target in CLL and for Fcm carrier proteins asnew targeting devices.
As the carrier protein, we tested two formats of the Fcfragment of IgM carrying a C-terminal Sec residue. BothCm3–Cm4–Sec and Cm2–Cm3–Cm4–Sec were expressed, puri-fied, and conjugated in high yield, and both mediated selectivedrug delivery in vitro. However, for the subsequent ex vivo and invivo studies, we focused on the larger Cm2–Cm3–Cm4–Sec, as itmore closely resembled IgM with respect to the formation ofstable pentamers and hexamers, which are required for high-affinity binding to the target molecule (23). Despite theirdifferences in molecular weight and composition, Cm2–Cm3–
–100
–80
–60
–40
–20
0
20
40
–100
–80
–60
–40
–20
0
20
40
Cha
nge
intu
mor
bur
den
(%)
Blood Spleen Blood Spleen
CLL cells T cells
C
Cha
nge
inT
cel
l cou
nt (
%)
BA
Live
CLL
cel
ls/
1 µL
blo
od
Live
CLL
cel
ls/
tota
l spl
een
cells
(%
)
0
5
10
15
20 10 2.0
1.5
1.0
0.5
0.0
8
6
4
2
0
80
60
40
20
0Li
ve T
cel
ls/
1 µL
blo
od
Live
T c
ells
/to
tal s
plee
n ce
lls (
%)
Fcµ-Sec/drug
ControlFcµ-Sec/drug
Control Fcµ-Sec/drug
Control Fcµ-Sec/drug
Control
P < 0.0001 P = 0.23 P = 0.96P < 0.0001
Figure 6. In vivo cytotoxicity of the Fcm–Sec/drug conjugate. Thirty NSG mice were injected i.v. on day 1 with 5 � 107 PBMC from four different patientswith CLL and treated on days 4, 6, and 8 with 10 mg/kg Fcm–Sec/drug conjugate in PBS or with PBS alone (control) by i.v. injection. On day 11, the absolutenumbers of live (TO-PRO-3–negative) CD19þ malignant B cells and autologous CD3þ T cells in blood and spleen were quantified by flow cytometry.Two-way ANOVA was used to compare cell numbers in treated and control mice. A, the leukemic cell count in the blood of the xenografted NSG mice wassignificantly reduced by Fcm–Sec/drug, while there was no change in T-cell numbers. B, live CD19þ malignant B cells relative to the total numberof live nucleated cells in the spleen were also greatly reduced by Fcm–Sec/drug. Again, T cells were unchanged. Note that while the human cells engraft in thespleen, the majority of nucleated cells are of murine origin. C, the column graph summarizes the effect of the Fcm–Sec/drug conjugate on tumor burden (left)and T cells (right). Shown is the mean � SEM.
Vire et al.
Cancer Res; 74(24) December 15, 2014 Cancer ResearchOF8
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030

Cm4–Sec and IgM shared similar circulatory half-lives (�1 day),which are relatively short comparedwith IgG (>7 days), but longcompared with scFv and other antibody fragments (<1 hour).Although providing sufficient time for on-target toxicity, acirculatory half-life of approximately 1 day may diminish off-target toxicity. We conclude that the pharmacokinetic proper-ties of Cm2–Cm3–Cm4–Sec are suitable for targeted drugdelivery.The rationale for using our Sec technology (34, 35) was to
enable site-specific as opposed to random attachment of thedrug payload. Site-specific conjugationminimizes interferencewith functional domains of the Fcm scaffold and has beenshown to increase the therapeutic index compared with con-ventional drug conjugation strategies (42, 43). Given a Secinsertion rate of approximately 10% (35), the dominant fractionof the purified Fcm–Sec pentamers and hexamers contains oneC-terminal Sec residue, affording a drug-to-carrier ratio of 1:1.Although this proved to be sufficient for killing malignant Bcells in vitro, ex vivo, and in vivo, it is lower than the ratio of 4:1considered ideal for ADCs generated using non–site-specifictechniques (2). Although the lower drug-to-carrier ratio maydecrease the potency of the current Fcm–drug conjugate,improvements in site-specific conjugation technologies thatincrease the number of drug molecules per carrier couldfurther increase the potency of the Fcm–drug conjugate. Forexample, we have recently shown that proteins carrying twoC-terminal Sec residues can be conjugated with two drugmolecules (44).The linker between carrier protein and payload has to be
stable to minimize systemic toxicity while at the same timeensuring effective intracellular drug release. Drug releasefrom noncleavable linkers, such as the alkyl linker, used hereand in trastuzumab emtansine requires antibody degrada-tion within lysosomes (45). The use of this technology in thetargeting of FcmR is made possible by the effective delivery ofthe internalized complex to the lysosome where it is degrad-ed (26, 46).FcmR is the only Ig receptor that exclusively binds IgM
(23). In addition, the polymeric Ig receptor (PIGR) and theFca/m receptor (FCAMR) bind and internalize both IgM andIgA. Whereas binding of IgA and IgM to PIGR requires theJ chain, which is not present in our Fcm–drug conjugates,FCAMR can mediate IgA and IgM binding in the absenceof the J chain (47, 48). As is the case for FcmR, the expressionof FCAMR is virtually restricted to specialized immunecells, including follicular B cells and follicular dendriticcells of germinal centers (49). Nonetheless, an assessmentof on-target and off-target toxicities of Fcm–drug conjugatesin preclinical and clinical investigations will have to takeboth FcmR and FCAMR expression by normal cells intoconsideration.For the in vivo studies, we chose a recently established
adoptive transfer model of human primary PBMC frompatients with CLL injected into NSG mice, which recapitulateskey aspects of tumor biology as seen in patients (36, 37). Bothmalignant B cells and the corresponding autologous T cells ofthe patient engraft in the blood and spleen of the mouse, thelatter demonstrating tumor cell aggregates reminiscent of
human lymph nodes. Thus, themodel allows testing of potencyand selectivity of the Fcm–drug conjugate in a partially human-ized environment. Furthermore, variability in tumor biologyamong individual patients is, at least partially, reproducedby injecting cells from different patients into separate cohortsof mice. A caveat of this model is that the survival of the miceis not determined by tumor progression and that over timeT-cell expansion dominates and can lead to the demise ofthe animals. We therefore chose the impact of the Fcm–drugconjugate on tumor burden as the clinical endpoint.
Here, we demonstrated effectiveness and selectivity ofFcm–drug conjugates by comparing the effect on malignantB cells and normal T cells from patients with CLL side-by-side ex vivo and in vivo. With only three injections of theFcm–Sec/MMAF over 1 week, we consistently obtainedobjective responses quantified as >60% reduction in tumorburden. Notably, the tumor samples studied here wereprimarily from patients with high-risk disease characterizedby advanced Rai stage and the expression of unmutatedIGHV genes, an indicator of more rapid disease progressionand reduced benefit from standard treatment. The potentactivity of Fcm–Sec/MMAF against tumor cells from thesehigh-risk patients is promising. Using this partially human-ized xenograft model, we could also verify a degree ofselectivity in vivo; while CLL B cells were killed, we did notobserve a decrease in the viability of the autologous T cells.This is consistent with the fact that the malignant B cellsoverexpress the FcmR while the normal T cells of the patientswith CLL as well as the normal B and normal T cells ofhealthy donors have considerably lower expression levels(24, 26). This suggests that FcmR targeting with ADCs will beless damaging to normal cells and tissues than currenttreatment options in CLL.
Collectively, our study provides proof-of-concept for thetherapeutic targeting of the recently identified FcmR with anovel IgM-derived protein–drug conjugate. We established theutility of lead components and compositions of Fcm–drugconjugates that provide opportunities for further optimizationof FcmR-targeted drug delivery and translation of this approachinto the clinic.
Disclosure of Potential Conflicts of InterestC. Rader, T.R. Burke Jr, and J.D. Thomas have ownership interest in the U.S.
patent application 20100104510. No potential conflicts of interest were disclosedby the other authors.
Authors' ContributionsConception and design: B. Vire,M. Skarzynski, A. David, T.R. Burke Jr, C. Rader,A. WiestnerDevelopment of methodology: B. Vire, M. Skarzynski, G. Aue, T.R. Burke Jr,C. Rader, A. WiestnerAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): M. Skarzynski, C.G. Nelson, G. Aue, C. Rader,A. WiestnerAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): B. Vire, M. Skarzynski, C.G. Nelson, A. David, G. Aue,C. Rader, A. WiestnerWriting, review, and/or revision of the manuscript: B. Vire, M. Skarzynski,C.G. Nelson, T.R. Burke Jr, C. Rader, A. WiestnerAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): M. SkarzynskiStudy supervision: C. Rader, A. WiestnerOther (synthesized reagents used in bioconjugation): J.D. Thomas
Targeted Cytotoxic Drug Delivery through the Fcm Receptor
www.aacrjournals.org Cancer Res; 74(24) December 15, 2014 OF9
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030

AcknowledgmentsThe authors thank Dr. William K. Gillette and his team at the Protein
Expression Laboratory, SAIC-Frederick, for custom expression and purificationof Fcm–Sec, and Dr. David J. Fitzgerald for helpful comments on the article.
Grant SupportThis work was funded by the Intramural Research Programs of the National
Heart, Lung, and Blood Institute and the National Cancer Institute, NIH, and bythe NIH U01 grant CA174844.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicate thisfact.
Received July 9, 2014; revised September 24, 2014; accepted October 13, 2014;published OnlineFirst October 24, 2014.
References1. Adair JR, Howard PW, Hartley JA,WilliamsDG, Chester KA. Antibody–
drug conjugates—a perfect synergy. Expert Opin Biol Ther 2012;12:1191–206.
2. Sievers EL, Senter PD. Antibody–drug conjugates in cancer therapy.Annu Rev Med 2013;64:15–29.
3. de Claro RA, McGinn K, Kwitkowski V, Bullock J, Khandelwal A,Habtemariam B, et al. U.S. Food and Drug Administration approvalsummary: brentuximab vedotin for the treatment of relapsed Hodgkinlymphoma or relapsed systemic anaplastic large-cell lymphoma. ClinCancer Res 2012;18:5845–9.
4. Baron JM, Boster BL, Barnett CM. Ado-trastuzumab emtansine (T-DM1): a novel antibody–drug conjugate for the treatment of HER2-positive metastatic breast cancer. J Oncol Pharm Pract 2014 Mar 27.[Epub ahead of print].
5. Senter PD,SieversEL. Thediscovery anddevelopment of brentuximabvedotin for use in relapsed Hodgkin lymphoma and systemic anaplas-tic large cell lymphoma. Nat Biotechnol 2012;30:631–7.
6. LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastu-zumab emtansine: a unique antibody–drug conjugate in developmentfor human epidermal growth factor receptor 2-positive cancer. ClinCancer Res 2011;17:6437–47.
7. Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ,et al. Results of a pivotal phase II study of brentuximab vedotin forpatients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol2012;30:2183–9.
8. Hurvitz SA, Dirix L, Kocsis J, Bianchi GV, Lu J, Vinholes J, et al. Phase IIrandomized study of trastuzumab emtansine versus trastuzumab plusdocetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2013;31:1157–63.
9. Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM,et al. Obinutuzumab plus chlorambucil in patients with CLL andcoexisting conditions. N Engl J Med 2014;370:1101–10.
10. Gribben JG, O'Brien S. Update on therapy of chronic lymphocyticleukemia. J Clin Oncol 2011;29:544–50.
11. Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J,et al. Addition of rituximab to fludarabine and cyclophosphamide inpatients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164–74.
12. Tam CS, Keating MJ. Chemoimmunotherapy of chronic lymphocyticleukemia. Nat Rev Clin Oncol 2010;7:521–32.
13. Jaglowski SM, Alinari L, Lapalombella R, Muthusamy N, Byrd JC. Theclinical application of monoclonal antibodies in chronic lymphocyticleukemia. Blood 2010;116:3705–14.
14. Weiner GJ. Rituximab: mechanism of action. Semin Hematol 2010;47:115–23.
15. Tembhare PR,Marti G, Wiestner A, Degheidy H, Farooqui M, KreitmanRJ, et al.Quantification of expressionof antigens targetedbyantibody-based therapy in chronic lymphocytic leukemia. Am J Clin Pathol2013;140:813–8.
16. Gea-Banacloche JC. Rituximab-associated infections. Semin Hema-tol 2010;47:187–98.
17. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al.Targeting BTKwith ibrutinib in relapsed chronic lymphocytic leukemia.N Engl J Med 2013;369:32–42.
18. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM,HillmenP,et al. Idelalisib and rituximab in relapsedchronic lymphocytic leukemia.N Engl J Med 2014;370:997–1007.
19. Wiestner A. Emerging role of kinase-targeted strategies in chroniclymphocytic leukemia. Blood 2012;120:4684–91.
20. Garcia Vela J, Delgado I, Benito L, Monteserin M, Garcia Alonso L,Somolinos N, et al. CD79b expression in B cell chronic lymphocyticleukemia: its implication for minimal residual disease detection.Leukemia 1999;13:1501–5.
21. Trentin L, Zambello R, Sancetta R, Facco M, Cerutti A, Perin A, et al. Blymphocytes from patients with chronic lymphoproliferative disordersare equipped with different costimulatory molecules. Cancer Res1997;57:4940–7.
22. DeutschYE, Tadmor T, PodackER,Rosenblatt JD.CD30: an importantnew target in hematologic malignancies. Leuk Lymphoma 2011;52:1641–54.
23. KubagawaH, OkaS, Kubagawa Y, Torii I, TakayamaE, Kang DW, et al.Identity of the elusive IgM Fc receptor (FcmuR) in humans. J Exp Med2009;206:2779–93.
24. Pallasch CP, Schulz A, Kutsch N, Schwamb J, Hagist S, Kashkar H,et al. Overexpression of TOSO in CLL is triggered by B-cell receptorsignaling and associated with progressive disease. Blood 2008;112:4213–9.
25. Proto-Siqueira R, Panepucci RA, Careta FP, Lee A, Clear A, Morris K,et al. SAGE analysis demonstrates increased expression of TOSOcontributing to Fas-mediated resistance in CLL. Blood 2008;112:394–7.
26. Vire B, David A, Wiestner A. TOSO, the Fcmicro receptor, is highlyexpressed on chronic lymphocytic leukemia B cells, internalizes uponIgM binding, shuttles to the lysosome, and is downregulated inresponse to TLR activation. J Immunol 2011;187:4040–50.
27. Shima H, Takatsu H, Fukuda S, OhmaeM, Hase K, Kubagawa H, et al.Identification of TOSO/FAIM3 as an Fc receptor for IgM. Int Immunol2010;22:149–56.
28. Choi SC, Wang H, Tian L, Murakami Y, Shin DM, Borrego F, et al.Mouse IgM Fc receptor, FCMR, promotes B cell development andmodulates antigen-driven immune responses. J Immunol 2013;190:987–96.
29. HonjoK,KubagawaY, JonesDM,DizonB,ZhuZ,OhnoH, et al. AlteredIg levels and antibody responses in mice deficient for the Fc receptorfor IgM (FcmuR). Proc Natl Acad Sci U S A 2012;109:15882–7.
30. Ouchida R, Mori H, Hase K, Takatsu H, Kurosaki T, Tokuhisa T, et al.Critical role of the IgM Fc receptor in IgM homeostasis, B-cell survival,and humoral immune responses. Proc Natl Acad Sci U S A 2012;109:E2699–706.
31. Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B,et al. The lymph node microenvironment promotes B-cell receptorsignaling, NF-kappaB activation, and tumor proliferation in chroniclymphocytic leukemia. Blood 2011;117:563–74.
32. Leadbetter EA, Rifkin IR,HohlbaumAM,BeaudetteBC, ShlomchikMJ,Marshak-Rothstein A. Chromatin-IgG complexes activate B cells bydual engagement of IgM and Toll-like receptors. Nature 2002;416:603–7.
33. Mende I, Hoffmann P,Wolf A, LutterbuseR, Kopp E, Baeuerle PA, et al.Highly efficient antigen targeting toM-DC8þ dendritic cells via Fcgam-maRIII/CD16-specific antibody conjugates. Int Immunol 2005;17:539–47.
34. Hofer T, Skeffington LR, Chapman CM, Rader C. Molecularly definedantibody conjugation through a selenocysteine interface. Biochemis-try 2009;48:12047–57.
Vire et al.
Cancer Res; 74(24) December 15, 2014 Cancer ResearchOF10
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030

35. Hofer T, Thomas JD, Burke TR Jr, Rader C. An engineered seleno-cysteine defines a unique class of antibody derivatives. ProcNatl AcadSci U S A 2008;105:12451–6.
36. Bagnara D, Kaufman MS, Calissano C, Marsilio S, Patten PE, SimoneR, et al. A novel adoptive transfer model of chronic lymphocyticleukemia suggests a key role for T lymphocytes in the disease. Blood2011;117:5463–72.
37. Herman SE, Sun X, McAuley EM, Hsieh MM, Pittaluga S, Raffeld M,et al. Modeling tumor-host interactions of chronic lymphocytic leuke-mia in xenografted mice to study tumor biology and evaluate targetedtherapy. Leukemia 2013;27:1769–73.
38. Azuma Y, Ishikawa Y, Kawai S, Tsunenari T, Tsunoda H, Igawa T, et al.Recombinant human hexamer-dominant IgM monoclonal antibody toganglioside GM3 for treatment of melanoma. Clin Cancer Res2007;13:2745–50.
39. Tchoudakova A, Hensel F, Murillo A, Eng B, Foley M, Smith L, et al.High level expression of functional human IgMs in human PER.C6cells. MAbs 2009;1:163–71.
40. Doronina SO, Mendelsohn BA, Bovee TD, Cerveny CG, Alley SC,Meyer DL, et al. Enhanced activity of monomethylauristatin F throughmonoclonal antibody delivery: effects of linker technology on efficacyand toxicity. Bioconjug Chem 2006;17:114–24.
41. Hughey CT, Brewer JW, Colosia AD, Rosse WF, Corley RB. Pro-duction of IgM hexamers by normal and autoimmune B cells:implications for the physiologic role of hexameric IgM. J Immunol1998;161:4091–7.
42. Junutula JR, Flagella KM, GrahamRA, Parsons KL, Ha E, RaabH, et al.Engineered thio-trastuzumab-DM1 conjugate with an improvedtherapeutic index to target human epidermal growth factor receptor2–positive breast cancer. Clin Cancer Res 2010;16:4769–78.
43. Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD,Weir S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves thetherapeutic index. Nat Biotechnol 2008;26:925–32.
44. Li X, Yang J, RaderC. Antibody conjugation via one and twoC-terminalselenocysteines. Methods 2014;65:133–8.
45. Senter PD. Potent antibody drug conjugates for cancer therapy. CurrOpin Chem Biol 2009;13:235–44.
46. Ivanov A, Beers SA, Walshe CA, Honeychurch J, Alduaij W, Cox KL,et al. Monoclonal antibodies directed to CD20 and HLA-DR canelicit homotypic adhesion followed by lysosome-mediated celldeath in human lymphoma and leukemia cells. J Clin Invest 2009;119:2143–59.
47. Yoo EM, Trinh KR, Lim H, Wims LA, Morrison SL. Characterization ofIgA and IgM binding and internalization by surface-expressed humanFcalpha/mu receptor. Mol Immunol 2011;48:1818–26.
48. Kaetzel CS. The polymeric immunoglobulin receptor: bridging innateand adaptive immune responses at mucosal surfaces. Immunol Rev2005;206:83–99.
49. Honda S, Kurita N, Miyamoto A, Cho Y, Usui K, Takeshita K, et al.Enhanced humoral immune responses against T-independent anti-gens in Fc alpha/muR-deficient mice. Proc Natl Acad Sci U S A2009;106:11230–5.
www.aacrjournals.org Cancer Res; 74(24) December 15, 2014 OF11
Targeted Cytotoxic Drug Delivery through the Fcm Receptor
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030

Published OnlineFirst October 24, 2014.Cancer Res Bérengère Vire, Martin Skarzynski, Joshua D. Thomas, et al. Cytotoxic Therapy of Chronic Lymphocytic Leukemia
Receptor for Potent and SelectiveµHarnessing the Fc
Updated version
10.1158/0008-5472.CAN-14-2030doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2014/10/24/0008-5472.CAN-14-2030.DC1
Access the most recent supplemental material at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/early/2014/12/05/0008-5472.CAN-14-2030To request permission to re-use all or part of this article, use this link
Research. on May 11, 2018. © 2014 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 24, 2014; DOI: 10.1158/0008-5472.CAN-14-2030





























