Embed Size (px)
Citation preview

Chapter 30
ArrayOme- and tRNAcc-FacilitatedMobilome Discovery: ComparativeGenomics Approaches for IdentifyingRich Veins of Bacterial Novel DNASequences
Hong-Yu Ou and Kumar Rajakumar
30.1 INTRODUCTION
The bacterial genomics revolution kicked off in earnestwith the decoding of the Haemophilus influenzae genomein 1995 [Fleischmann et al., 1995]. In the relativelyshort period since 1995, it has assumed an explosivephase of growth driven by next-generation sequencingtechnologies [Ansorge, 2009; MacLean et al., 2009; seealso Chapter 18, Vol. I]. As of December 6, 2009, therewere 957 completely and 3570 ongoing sequenced bac-terial genome available that were representative of morethan 604 species spanning the full taxonomic breadth ofthe bacterial phylum [Liolios et al., 2009]. Remarkably,these numbers are yet to reflect the true impact ofhigh-throughput sequencing efforts because these data areonly just beginning to roll of sequencing production linesin staggering quantities at numerous centers worldwide.
An initial surprise that has since become a bacterialparadigm was the discovery of marked genome diversity(genodiversity) and plasticity within many individual bac-terial species, let alone genera or wider taxonomic group-ings [Canchaya et al., 2004; Dobrindt and Hacker, 2001;Dorrell et al., 2005]. The notion that two organisms mightbelong to the same species despite sharing less than 67%of DNA, as in the case of E. coli strains K-12 MG1655
Handbook of Molecular Microbial Ecology, Volume I: Metagenomics and Complementary Approaches, First Edition. Edited by Frans J. de Bruijn.© 2011 Wiley-Blackwell. Published 2011 by John Wiley & Sons, Inc.
against O157:H7 Sakai, continues to challenge our con-cept of speciation [Riley and Lizotte-Waniewski, 2009;Konstantinidis et al., 2006; see also Chapters 9–13, Vol.I]. Bioinformatics-facilitated comparative genomics stud-ies have also led at a population level to the concept ofthe metagenome or pan-genome specific to each bacterialspecies [Medini et al., 2005; Tettelin et al., 2008; see alsoChapter 10, Vol. I]. The pan-genome comprises the entireset of DNA sequences that are represented invariably,frequently, sporadically, or only rarely in members ofthe species. As a parallel theme, the genome of eachindividual bacterium can be divided into a core genome,common to the entire species, and an accessory genome.This latter optional genomic repertoire, which we termedthe “mobilome” (mobile genome) in 2005 [Ou et al.,2005], includes a myriad of short strain-specific sequencesand longer spans of episomal and integrative plasmids,transposons, integrons, gene cassettes, prophages, and agrowing list of genomic islands [Frost et al., 2005; Hackeret al., 1997; Canchaya et al., 2004]. The evolutionary andfunctional boundaries between integrative conjugativeelements (ICEs) [Churchward, 2008], integrative plas-mids, integrative nonconjugative islands, and prophages[Canchaya et al., 2004; Lima-Mendez et al., 2007] canat times be blurred, but evidence that these entities are
251

252 Chapter 30 ArrayOme- and tRNAcc-Facilitated Mobilome Discovery
horizontally acquired is irrefutable [Boyd et al., 2009].Intriguingly, a great diversity of alien DNA appears tomap to a limited number of species-specific insertion“hotspots” in many of the bacterial genomes that havebeen better characterized to date, with the commonestand most generic hotspots being tRNA and tmRNA sites[Ou et al., 2006; Mantri and Williams, 2004].
Despite the transformative nature of next-generationsequencing, the associated substantial equipment andanalytical costs and the daunting bioinformatics and com-puting challenge of dealing with the resultant tsunamiof digital DNA data restrict its ability to be appliedwidely to discover strain-specific DNA that is dispersedamongst hundreds or even thousands of members of eachbacterial species [Fux et al., 2005; Field et al., 2006]. Thescale of the challenge demands empowerment of a muchlarger community of researchers who would otherwise beconstrained by the above barriers. We have developed aparallel, high-throughput, comparative genomics strategythat readily facilitates experimental and in silico discov-ery of genomic islands and other foreign DNA. DubbedMobilomeFINDER [Ou et al., 2007a], our approachcombines ArrayOme-facilitated M icroarray-Assistedmobilome P rospecting (MAmP) to aid identification ofbacterial strains rich in novel DNA [Ou et al., 2005],tRNA gene contents and contexts analysis (tRNAcc)[Ou et al., 2006] to identify tRNA-associated hotspotsand to streamline design and validation of primers fortRIP (tRNA site I nterrogation for Pathogenicity islands,prophages, and other GIs) PCR-based identificationand characterization of new GIs in MAmP-selectedand/or otherwise chosen test strains [Ou et al., 2005].Novel islands can then be captured selectively by islandprobing [Rajakumar et al., 1997], long-range PCR,and/or the yeast-based homologous recombination systemdescribed by Wolfgang et al. [2003] and subjected tocost-effective barcode-facilitated, massively parallelfull-length sequencing. In this chapter we describethe MobilomeFINDER tools ArrayOme, tRNAcc, andmGenomeSubtractor [Ou et al., 2007a], provide examplesof their utility, and briefly outline selected wet-sciencediscoveries that have been achieved through use of thesimple and readily adopted tRIP PCR strategy.
30.2 MATERIALS AND METHODS
30.2.1 DatabasesExperimental microarray-derived comparative genomichybridization (CGH) data obtained using the Lactobacil-lus casei ATCC 334 microarray were recently reportedby Cai et al. [2009]. CGH data for 21 Lactobacilluscasei test strains isolated from various habitats (seven,
eight, and six from cheeses, plant materials, and humansources, respectively) were taken from Gene ExpressionOmnibus (GEO accession no. GSE15030). The followingcomplete and near-complete genome sequences andannotation information were downloaded from NCBI:Lactobacillus casei strains ATCC 334 (NC_008526) andBL23 (NC_010999); Lactobacillus paracasei 8700:02:00(NZ_ABQV00000000); Acinetobacter baumannii strainsATCC 17978 (NC_009085), SDF (NC_010400), ACICU(NC_010611), AB0057 (NC_011586), AB307-0294(NC_011595), and AYE (NC_010410); A. bauman-nii plasmids pAB1 (NC_009083), pAB2 (NC_0090-84), p1ABSDF (NC_010395), p2ABSDF (NC_010396),p3ABSDF (NC_010398), pACICU1 (NC_010605), pA-CICU2 (NC_010606), pAB0057 (NC_011585), p1ABA-YE (NC_010401), p2ABAYE (NC_010402), p4ABAYE(NC_010403), p3ABAYE (NC_010404), pABIR (NC_010481), pMMCU2 (NC_013506), pMMA2 (NC_0132-77), pABVA1 (NC_012813), and pMAC (NC_006877).
30.2.2 ArrayOme, tRNAccand mGenomeSubtractor ToolsThe MobilomeFINDER web-server (http://mml.sjtu.edu.cn/MobilomeFINDER/) comprises a flexible suiteof individual interactive tools that greatly facilitatehigh-throughput experimental and in silico discov-ery of bacterial mobilome in closely related bacteria(Table 30.1) Details on the use of these tools are availableon the MobilomeFINDER web server, and the preciserationale of the strategies are described in our originalpapers on ArrayOme [Ou et al., 2005], tRNAcc [Ou etal., 2006], mGenomeSubtractor [Shao et al., 2010], andMobilomeFINDER [Ou et al., 2007a].
30.3 RESULTS AND DISCUSSION
30.3.1 ArrayOme- andtRNAcc-Based Analyses of theClosely Related Lactobacillus caseiand Lactobacillus paracaseiGenomesMicroarray-derived CGH data identifies genes commonto both the microarray used and the genome of the strainunder investigation. This complement of genes representsan entity we previously defined as the microarray-v isualised genome (MVG) [Ou et al., 2005]. Briefly,ArrayOme produces an accurate estimate of MVG sizesby using a CGH dataset to generate “inferred contigs”(ICs) produced by merging adjacent genes classified as“present.” The sizes of novel mobilomes to which themicroarray would have been “blind” borne by individual

30.3 Results and Discussion 253
Table 30.1 Interactive Tools Available at the MobilomeFINDER Web Server(http://mml.sjtu.edu.cn/MobilomeFINDER/) that Facilitate the Process of BacterialGenomic Island or Mobile Genome (Mobilome) Discovery
Tool Descriptiona
Microarray-assistedmobilome prospecting
Identify strains that are particularly rich in novel mobilome DNA;underpinned by CGH, ArrayOme and PFGE
ArrayOme (Fig. 30.1 andTable 30.2)
Estimate the cumulative size of parts of the genome that harborgenes identified as “present” by microarray-based CGH; the size ofthe novel mobilome is thus predicted by comparing themicroarray-visualized genome size with the PFGE-measuredchromosome size
Island identificationb Identify GIs by tRNAcc analysis of closely related bacteria(Fig. 30.1B)
IslandScreen (Table 30.2) Identify putative islands by single-step crude analysis withcombination of IdentifyIsland, LocateHotspots, and DNAnalyzer.
IdentifyIsland Identify putative islands based on conserved flanking blocksrecognized by the multiple aligner Mauve 1.2.2 [Darling et al.,2004]
TabulateIsland Tabulate islands identified by IdentifyIsland following analysis ofdifferent subsets of genomes
LocateHotspots Locate proposed hotspots in non-annotated chromosomal sequencesusing BLASTN-based searches
Primer designb
ExtractFlank Generate ClustalW-derived MSA files that contain upstream ordownstream flanking regions of identified islands to serve as inputsfor Primaclade-facilitated [Gadberry et al., 2005] design ofconserved PCR primers
insilicotRIP Interrogate identified hotspots for the presence or absence of anintegrated element using a locally installed version of ElectronicPCR [Schuler, 1997]; generates 0- to 500-kb virtual fragments
Island analysisb
DNAnalyzer Calculate the GC content and dinucleotide bias of identified islands,and plot the negative cumulative GC profile of genomes
mGenomeSubtractor(Fig. 30.2 and Fig. 30.3)
High-throughput mpiBLASTN-based comparison of CDS sequencesagainst test genomes to identify strain-specific CDS based on thelevel of nucleotide similarity
a Abbreviations used: GI, genomic island; CGH, comparative genomic hybridization; PFGE, pulsed-fieldgel electrophoresis; MSA, multiple sequence alignment; CDS, protein coding sequence.bThese tools can also be used for the generic identification and preliminary characterization of putativegenomic islands located at other user-specified hotspots and for the analysis of cognate flanking sequences.
strains can subsequently be estimated by comparingArrayOme-estimated MVG sizes with PFGE-measuredphysical chromosome sizes to identify isolates rich innovel or highly divergent genetic material that are likelyto carry unique GIs or prophages. Recently, Cai et al.[2009] reported the CGH results for 21 Lactobacilluscasei strains isolated from various habitats using theL. casei strain ATCC 334 microarray that bore probesfor 2661 (97%) chromosomal and 17 (85%) plasmidannotated ATCC 334 CDS. We have estimated the ATCC
334-MVG sizes of the 21 test strains using these CGHdata (Table 30.2). The 2,895,264 bp chromosome ofL. casei ATCC 334, an Emmental cheese isolate, wasused as the reference template for this analysis. Basedon these CGH data, the Denmark cheese isolate 7A1was predicted to possess 2256 chromosomal and 16plasmid-borne CDS out of the total complement of 2678ATCC 334 CDS represented on the array. Entry of the7A1 CGH data into ArrayOme resulted in the generationof 139 inferred contigs (ICs) and an estimated MVG size
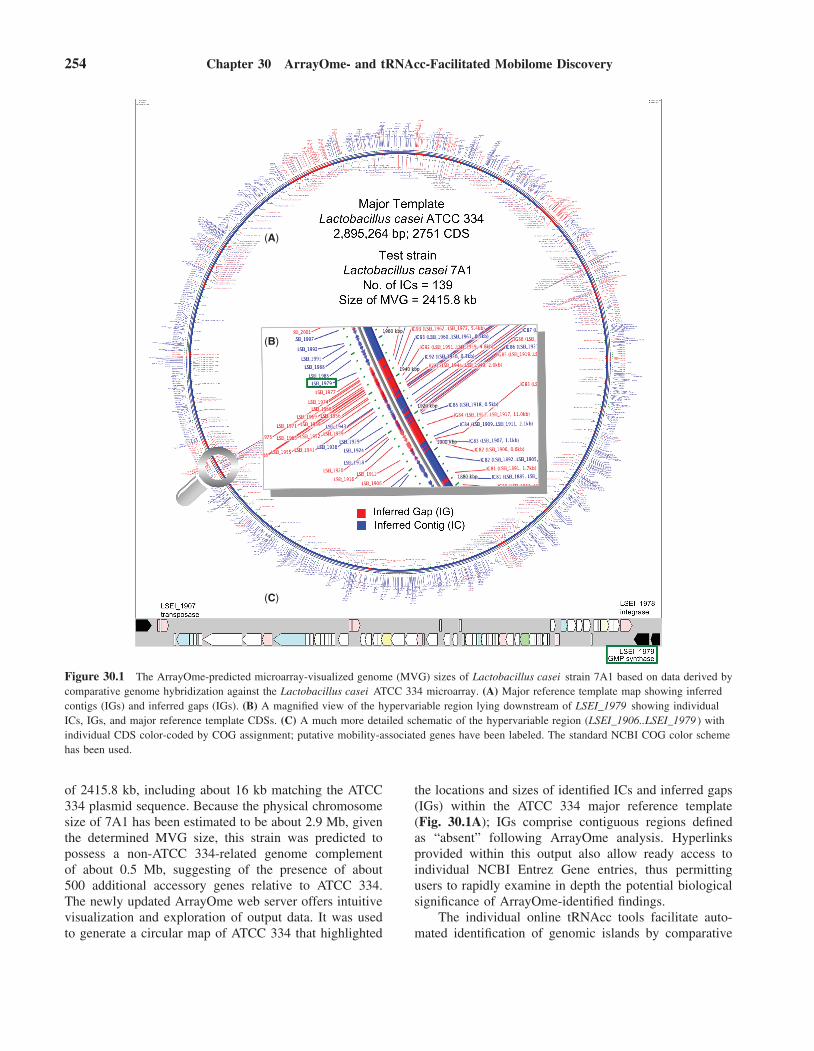
254 Chapter 30 ArrayOme- and tRNAcc-Facilitated Mobilome Discovery
(A)
(B)
(C)
Figure 30.1 The ArrayOme-predicted microarray-visualized genome (MVG) sizes of Lactobacillus casei strain 7A1 based on data derived bycomparative genome hybridization against the Lactobacillus casei ATCC 334 microarray. (A) Major reference template map showing inferredcontigs (IGs) and inferred gaps (IGs). (B) A magnified view of the hypervariable region lying downstream of LSEI_1979 showing individualICs, IGs, and major reference template CDSs. (C) A much more detailed schematic of the hypervariable region (LSEI_1906..LSEI_1979 ) withindividual CDS color-coded by COG assignment; putative mobility-associated genes have been labeled. The standard NCBI COG color schemehas been used.
of 2415.8 kb, including about 16 kb matching the ATCC334 plasmid sequence. Because the physical chromosomesize of 7A1 has been estimated to be about 2.9 Mb, giventhe determined MVG size, this strain was predicted topossess a non-ATCC 334-related genome complementof about 0.5 Mb, suggesting of the presence of about500 additional accessory genes relative to ATCC 334.The newly updated ArrayOme web server offers intuitivevisualization and exploration of output data. It was usedto generate a circular map of ATCC 334 that highlighted
the locations and sizes of identified ICs and inferred gaps(IGs) within the ATCC 334 major reference template(Fig. 30.1A); IGs comprise contiguous regions definedas “absent” following ArrayOme analysis. Hyperlinksprovided within this output also allow ready access toindividual NCBI Entrez Gene entries, thus permittingusers to rapidly examine in depth the potential biologicalsignificance of ArrayOme-identified findings.
The individual online tRNAcc tools facilitate auto-mated identification of genomic islands by comparative
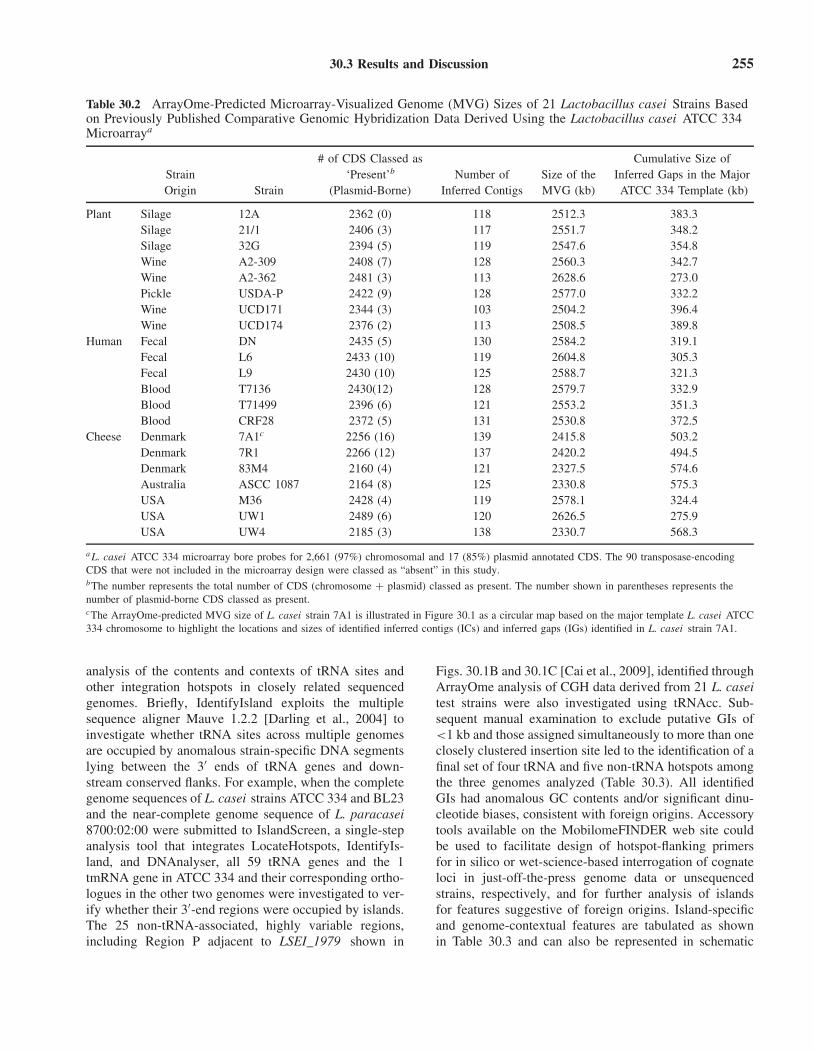
30.3 Results and Discussion 255
Table 30.2 ArrayOme-Predicted Microarray-Visualized Genome (MVG) Sizes of 21 Lactobacillus casei Strains Basedon Previously Published Comparative Genomic Hybridization Data Derived Using the Lactobacillus casei ATCC 334Microarraya
# of CDS Classed as Cumulative Size ofStrain ‘Present’b Number of Size of the Inferred Gaps in the MajorOrigin Strain (Plasmid-Borne) Inferred Contigs MVG (kb) ATCC 334 Template (kb)
Plant Silage 12A 2362 (0) 118 2512.3 383.3Silage 21/1 2406 (3) 117 2551.7 348.2Silage 32G 2394 (5) 119 2547.6 354.8Wine A2-309 2408 (7) 128 2560.3 342.7Wine A2-362 2481 (3) 113 2628.6 273.0Pickle USDA-P 2422 (9) 128 2577.0 332.2Wine UCD171 2344 (3) 103 2504.2 396.4Wine UCD174 2376 (2) 113 2508.5 389.8
Human Fecal DN 2435 (5) 130 2584.2 319.1Fecal L6 2433 (10) 119 2604.8 305.3Fecal L9 2430 (10) 125 2588.7 321.3Blood T7136 2430(12) 128 2579.7 332.9Blood T71499 2396 (6) 121 2553.2 351.3Blood CRF28 2372 (5) 131 2530.8 372.5
Cheese Denmark 7A1c 2256 (16) 139 2415.8 503.2Denmark 7R1 2266 (12) 137 2420.2 494.5Denmark 83M4 2160 (4) 121 2327.5 574.6Australia ASCC 1087 2164 (8) 125 2330.8 575.3USA M36 2428 (4) 119 2578.1 324.4USA UW1 2489 (6) 120 2626.5 275.9USA UW4 2185 (3) 138 2330.7 568.3
a L. casei ATCC 334 microarray bore probes for 2,661 (97%) chromosomal and 17 (85%) plasmid annotated CDS. The 90 transposase-encodingCDS that were not included in the microarray design were classed as “absent” in this study.bThe number represents the total number of CDS (chromosome + plasmid) classed as present. The number shown in parentheses represents thenumber of plasmid-borne CDS classed as present.cThe ArrayOme-predicted MVG size of L. casei strain 7A1 is illustrated in Figure 30.1 as a circular map based on the major template L. casei ATCC334 chromosome to highlight the locations and sizes of identified inferred contigs (ICs) and inferred gaps (IGs) identified in L. casei strain 7A1.
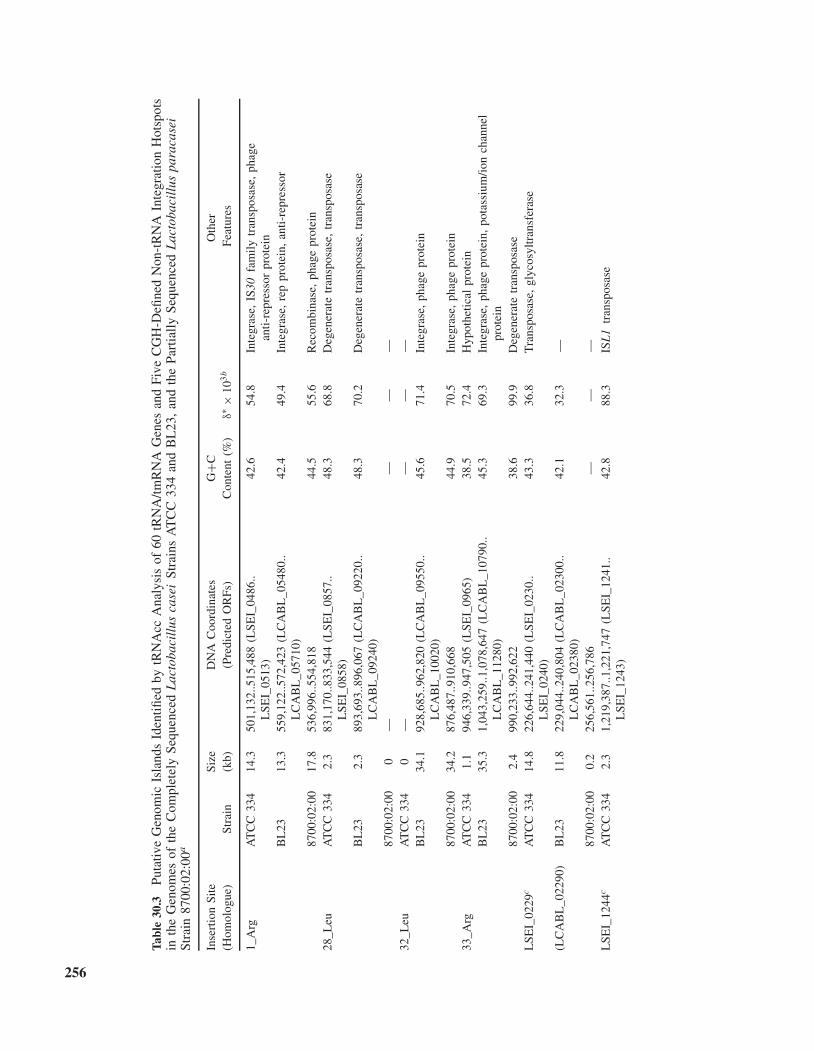
analysis of the contents and contexts of tRNA sites andother integration hotspots in closely related sequencedgenomes. Briefly, IdentifyIsland exploits the multiplesequence aligner Mauve 1.2.2 [Darling et al., 2004] toinvestigate whether tRNA sites across multiple genomesare occupied by anomalous strain-specific DNA segmentslying between the 3′ ends of tRNA genes and down-stream conserved flanks. For example, when the completegenome sequences of L. casei strains ATCC 334 and BL23and the near-complete genome sequence of L. paracasei8700:02:00 were submitted to IslandScreen, a single-stepanalysis tool that integrates LocateHotspots, IdentifyIs-land, and DNAnalyser, all 59 tRNA genes and the 1tmRNA gene in ATCC 334 and their corresponding ortho-logues in the other two genomes were investigated to ver-ify whether their 3′-end regions were occupied by islands.The 25 non-tRNA-associated, highly variable regions,including Region P adjacent to LSEI_1979 shown in
Figs. 30.1B and 30.1C [Cai et al., 2009], identified throughArrayOme analysis of CGH data derived from 21 L. caseitest strains were also investigated using tRNAcc. Sub-sequent manual examination to exclude putative GIs of<1 kb and those assigned simultaneously to more than oneclosely clustered insertion site led to the identification of afinal set of four tRNA and five non-tRNA hotspots amongthe three genomes analyzed (Table 30.3). All identifiedGIs had anomalous GC contents and/or significant dinu-cleotide biases, consistent with foreign origins. Accessorytools available on the MobilomeFINDER web site couldbe used to facilitate design of hotspot-flanking primersfor in silico or wet-science-based interrogation of cognateloci in just-off-the-press genome data or unsequencedstrains, respectively, and for further analysis of islandsfor features suggestive of foreign origins. Island-specificand genome-contextual features are tabulated as shownin Table 30.3 and can also be represented in schematic
Tabl
e30
.3Pu
tativ
eG
enom
icIs
land
sId
entifi
edby
tRN
Acc
Ana
lysi
sof
60tR
NA
/tmR
NA
Gen
esan
dF
ive
CG
H-D
efine
dN
on-t
RN
AIn
tegr
atio
nH
otsp
ots
inth
eG
enom
esof
the
Com
plet
ely
Seq
uenc
edL
acto
baci
llus
case
iSt
rain
sA
TC
C33
4an
dB
L23
,an
dth
ePa
rtia
llySe
quen
ced
Lac
toba
cill
uspa
raca
sei
Stra
in87
00:0
2:00
a
Inse
rtio
nSi
teSi
zeD
NA
Coo
rdin
ates
G+C
Oth
er(H
omol
ogue
)St
rain
(kb)
(Pre
dict
edO
RFs
)C
onte
nt(%
)δ∗
×10
3b
Feat
ures
1_A
rgA
TC
C33
414
.350
1,13
2..5
15,4
88(L
SEI_
0486
..L
SEI_
0513
)42
.654
.8In
tegr
ase,
IS30
fam
ilytr
ansp
osas
e,ph
age
anti-
repr
esso
rpr
otei
nB
L23
13.3
559,
122.
.572
,423
(LC
AB
L_0
5480
..L
CA
BL
_057
10)
42.4
49.4
Inte
gras
e,re
ppr
otei
n,an
ti-re
pres
sor
8700
:02:
0017
.853
6,99
6..5
54,8
1844
.555
.6R
ecom
bina
se,
phag
epr
otei
n28
_Leu
AT
CC
334
2.3
831,
170.
.833
,544
(LSE
I_08
57..
LSE
I_08
58)
48.3
68.8
Deg
ener
ate
tran
spos
ase,
tran
spos
ase
BL
232.
389
3,69
3..8
96,0
67(L
CA
BL
_092
20..
LC
AB
L_0
9240
)48
.370
.2D
egen
erat
etr
ansp
osas
e,tr
ansp
osas
e
8700
:02:
000
——
——
32_L
euA
TC
C33
40
——
——
BL
2334
.192
8,68
5..9
62,8
20(L
CA
BL
_095
50..
LC
AB
L_1
0020
)45
.671
.4In
tegr
ase,
phag
epr
otei
n
8700
:02:
0034
.287
6,48
7..9
10,6
6844
.970
.5In
tegr
ase,
phag
epr
otei
n33
_Arg
AT
CC
334
1.1
946,
339.
.947
,505
(LSE
I_09
65)
38.5
72.4
Hyp
othe
tical
prot
ein
BL
2335
.31,
043,
259.
.1,0
78,6
47(L
CA
BL
_107
90..
LC
AB
L_1
1280
)45
.369
.3In
tegr
ase,
phag
epr
otei
n,po
tass
ium
/ion
chan
nel
prot
ein
8700
:02:
002.
499
0,23
3..9
92,6
2238
.699
.9D
egen
erat
etr
ansp
osas
eL
SEI_
0229
cA
TC
C33
414
.822
6,64
4..2
41,4
40(L
SEI_
0230
..L
SEI_
0240
)43
.336
.8T
rans
posa
se,
glyc
osyl
tran
sfer
ase
(LC
AB
L_0
2290
)B
L23
11.8
229,
044.
.240
,804
(LC
AB
L_0
2300
..L
CA
BL
_023
80)
42.1
32.3
—
8700
:02:
000.
225
6,56
1..2
56,7
86—
——
LSE
I_12
44c
AT
CC
334
2.3
1,21
9,38
7..1
,221
,747
(LSE
I_12
41..
LSE
I_12
43)
42.8
88.3
ISL
1tr
ansp
osas
e
256
(LC
AB
L_1
4750
)B
L23
11.9
1,38
5,42
8..1
,397
,364
(LC
AB
L_1
4580
..L
CA
BL
_147
40)
39.2
75.9
Tra
nspo
sase
,de
gene
rate
tran
spos
ase
8700
:02:
001.
21,
260,
051.
.1,2
61,2
8642
.484
.7—
LSE
I_19
79c
AT
CC
334
48.5
1,90
2,24
8..1
,950
,810
(LSE
I_19
06..
LSE
I_19
78)
44.5
54.5
Inte
gras
e,tr
ansp
osas
e,ph
age
Lca
1
(LC
AB
L_2
1540
)B
L23
20.7
2,05
8,00
7..2
,078
,741
(LC
AB
L_2
1270
..L
CA
BL
_215
30)
42.6
52.5
Inte
gras
eho
mol
og,
tran
spos
ase
8700
:02:
007.
81,
935,
918.
.1,9
43,7
6940
.076
.2T
rans
posa
seL
SEI_
2793
cA
TC
C33
40
——
——
(LC
AB
L_2
9990
)B
L23
2.4
2,94
6,18
7..2
,948
,618
(LC
AB
L_2
9970
..L
CA
BL
_299
80)
42.3
103.
5T
rans
posa
se
8700
:02:
000
——
——
LSE
I_10
93c
AT
CC
334
0—
——
—(L
CA
BL
_125
60)
BL
231.
81,
212,
697.
.1,2
14,4
67(L
CA
BL
_125
70..
LC
AB
L_1
2580
)50
.011
4.1
ISSt
h1tr
ansp
osas
e
8700
:02:
000
——
——
aT
hech
rom
osom
ele
ngth
sof
L.c
asei
AT
CC
334
and
BL
23ar
e28
95.2
6kb
and
3079
.2kb
,re
spec
tivel
y.E
ight
y-ni
neco
ntig
sof
L.p
arac
asei
8700
:02:
00av
aila
ble
atth
etim
eof
anal
ysis
wer
eco
mpa
red
with
the
AT
CC
334
geno
me
usin
gPi
pMak
er(S
chw
artz
etal
.,20
00)
and
orde
red
byco
ordi
nate
sof
best
alig
nmen
tto
prod
uce
a“v
irtu
al”
geno
me
with
the
size
of29
81.3
4kb
.bD
inuc
leot
ide
bias
anal
ysis
was
adap
ted
from
the
met
hod
prop
osed
byK
arlin
[200
1].
The
valu
eδ∗
deno
tes
the
dinu
cleo
tide
rela
tive
abun
danc
edi
ffer
ence
betw
een
the
isla
ndfr
agm
ent
and
the
refe
renc
ege
nom
e.T
hehi
ghδ∗
valu
esof
thes
efr
agm
ents
indi
cate
likel
yhe
tero
logo
usor
igin
s.To
avoi
dst
atis
tical
fluct
uatio
ns,δ∗
was
not
dete
rmin
ed(‘
—’)
for
GI
frag
men
tsle
ssth
an10
00bp
.T
hege
nom
e-av
erag
edδ∗
valu
ean
dst
anda
rdde
viat
ion
(SD
)w
asca
lcul
ated
usin
ga
20-k
bno
nove
rlap
ping
,sl
idin
gw
indo
wal
ong
the
entir
ege
nom
ese
quen
ce.
The
geno
me-
aver
aged
para
met
ers
for
the
thre
ege
nom
esar
eas
foll
ows:
L.c
asei
AT
CC
334,
G+
C=
46.6
%;δ
∗=
24.7
2(SD
=11
.84)
;L
.cas
eiB
L23
,G
+C
=46
.3%
,δ∗
=27
.4(S
D=
12.8
4);
L.p
arac
asei
8700
:02:
00,
G+
C=
46.2
%,δ
∗=
25.1
1(SD
=13
.41)
.cT
hefiv
eno
n-tR
NA
inse
rtio
nho
tspo
ts(L
SEI_
0229
,L
SEI_
1093
,L
SEI_
1244
,L
SEI_
1979
,an
dL
SEI_
2793
)su
bjec
ted
totR
NA
ccan
alys
isw
ere
iden
tified
base
don
prev
ious
lyre
port
edco
mpa
rativ
ege
nom
ichy
brid
izat
ion
data
deri
ved
follo
win
gan
alys
isof
21L
.cas
eist
rain
sag
ains
tth
eL
.cas
eiA
TC
C33
4ar
ray
[Cai
etal
.,20
09].
257

258 Chapter 30 ArrayOme- and tRNAcc-Facilitated Mobilome Discovery
and graphical forms using the newly incorporated visual-ization features found with IslandScreen.
30.3.2 The Use ofmGenomeSubtractor to HighlightGenomic Mosaicism, Examine StrainVariable Regions and Define theSpecies Gene Pool of AcinetobacterbaumanniiThe bacterial chromosomal backbone comprises a mosaicof core genomic sequences and horizontally acquiredDNA that has been termed the accessory genome ormobilome [Ou et al., 2005]. The latter flexible geno-mic repertoire is drawn from a gene pool dispersedamongst species-associated episomal plasmids, trans-posons, integrons, prophages, and a growing list of GIs.mGenomeSubtractor performs a simple and intuitivein silico “subtractive hybridization” by comparingselected closely related genomes to generate a list ofstrain-specific CDS which may provide clues as to thephenotype, specific environmental adaptation, and/ordisease-association linked with a particular bacterium.As previously, mGenomeSubtractor uses H -value criteriathat reflect the degree of similarity in terms of the lengthof match and degree of identity at a nucleotide levelbetween each CDS examined and the set of comparatorgenomes examined [Fukiya et al., 2004]. In the exampleshown in Figure 30.2, the genome map of A. baumanniistrain AYE was color-coded by mGenomeSubtractorbased on the number of comparator genomes that sharedeach AYE CDS. Gene sequences conserved across allsix fully sequenced A. baumannii genomes are shownin black with the legend label “5” to indicate that thecorresponding AYE CDS has homologues in the otherfive genomes examined. A. baumannii is an opportunistichuman pathogen which is an increasingly significantcause of hospital-associated infections, quite probablydue to its frequent multi-antibiotic resistant phenotype[Towner, 2009]. In addition, there are growing data tosupport the notion that the Acinetobacter genus exhibitshighly variable genomes and that interspecies, intergenus,interfamily, interorder, and even more remote DNAmigration events may be relatively frequent features ofthis genus [Fournier et al., 2006; Vallenet et al., 2008].These observations raise the realistic possibility of thefuture emergence of frankly pathogenic A. baumanniistrains that may, like many of their predecessors, evenassume endemic status [Peleg et al., 2008]. All 3607annotated AYE CDS were analyzed against the fivecompletely sequenced A. baumannii genomes. If allresulting H values for a given AYE CDS were ≤0.42,the CDS was classified as AYE strain-specific. Using thisapproach, 123 AYE CDS were found to be AYE-specific,
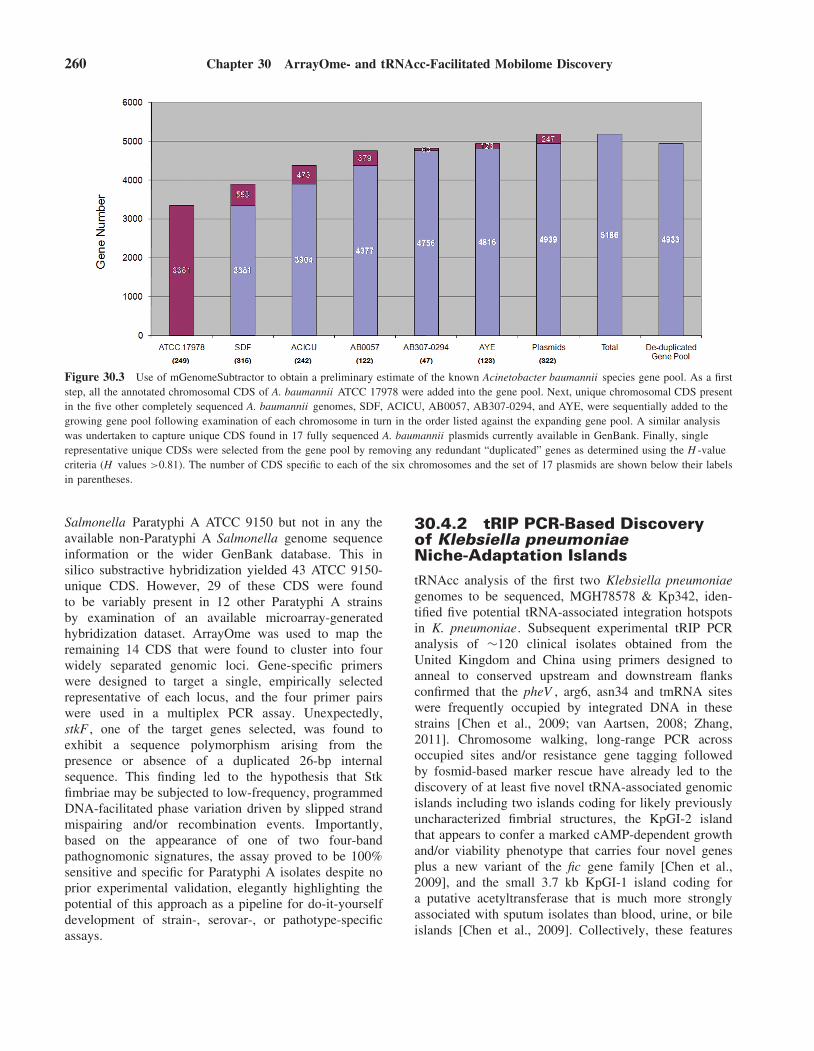
offering significant potential for the fine-tuning of traitsand/or for AYE-specific adaptations. The numbers ofstrain-specific CDS corresponding to each of the otherfive genomes and the set of 17 A. baumannii plasmidsexamined are shown in Fig. 30.3.
mGenomeSubtractor analysis also allowed for theready definition of the A. baumannii species known genepool based on the six completely sequenced A. baumanniichromosomes and 17 completely sequenced A. baumanniiplasmids as the input. Firstly, all the 3351 annotated CDSpresent on the A. baumannii ATCC 17978 chromosomewere included in the gene pool. Then each annotatedCDS on the A. baumannii SDF chromosome was usedas a query in a BLASTN similarity search against theATCC 17978 chromosomal sequence that had been storedin the gene pool. The 553 SDF-specific CDS identifiedby mGenomeSubtractor (H values ≤0.42) based on thistwo chromosome comparison were added to the genepool. A similar reiterative process was used to analyzeeach of the remaining four fully sequenced A. baumanniichromosomes, leading to the sequential inclusion of afurther 1035 genes into the growing gene pool. Next, the247 unique CDS present on 17 completely sequencedA. baumannii plasmids currently available in Gen-Bank, pAB1, pAB2, p1ABSDF, p2ABSDF, p3ABSDF,pACICU1, pACICU2, pAB0057, p1ABAYE, p2ABAYE,p4ABAYE, p3ABAYE, pABIR, pMMCU2, pMMA2,pABVA1, and pMAC were also identified and placedinto the gene pool. Finally, single representative uniqueCDSs were selected from the gene pool by removingany redundant “duplicated” genes as determined usingthe H -value criteria (H values >0.81). Hence, basedon mGenomeSubtractor analysis of the six complete A.baumannii chromosomes and 17 completely sequencedA. baumannii plasmids, 4933 unique CDS, including4695 chromosomal and 238 plasmid-borne CDS, weredefined in the presently known A. baumannii gene pool, anumber that was 33–89% greater than that of the uniquegene complement of each of the six fully sequenced A.baumannii chromosomes under study.
30.4 OTHER APPLICATIONS
30.4.1 Rapid Developmentof a Salmonella enterica SerovarParatyphi A Multiplex PCR Assay:A MobilomeFINDER-EnabledPipeline for Future Strain-, Serovar-,or Pathotype-Specific AssaysOu et al. [2007b] described a simple MobilomeFINDER-enabled strategy that utilized mGenomeSubtractor toidentify protein coding sequences that were present in

30.4 Other Applications 259
(A)
(B)
(C)
Figure 30.2 Comparative genomics based in silico “subtractive hybridization” using the mpiBLAST-based mGenomeSubtractor tool. (A)Genome map of Acinetobacter baumannii strain AYE with CDS color-coded based on the number of comparator A. baumannii genomesidentified as harboring a nucleotide sequence-conserved homologue; CDS shown in black (5) are conserved across the five other full-sequencedA. baumannii comparator genomes, while at the other extreme those shown in white (0) are unique to strain AYE. AYE strain-specific CDSwere identified based on an H -value cutoff of less than 0.42. The H value reflects the degree of similarity in terms of the length of match andthe degree of identity at a nucleotide level between the matching reference genome sequence and the CDS examined. (B) An expanded view ofthe hypervariable region highlighted by a red rectangle in part A corresponding to the 86-kb TnAbaR1 resistance mega-transposons of AYE andits 2 kb upstream/downstream flanking regions. A. baumannii strain AYE CDS are color-coded based on their COG assignment. The fivecomparator genomes are shown below. Gray bars indicate the extent of AYE sequences within this region that are also present in the individualcomparator genomes. The black bars at the end represent the ends of the region selected for the expanded view. A G+C profile of the selectedAYE region is shown topmost. (C) A zoom-in view of the region of the AYE genome highlighted by the red rectangle in part B showingselected TnAbaR1 CDS coding for an integrase, transposases, and antibiotic resistance genes.
260 Chapter 30 ArrayOme- and tRNAcc-Facilitated Mobilome Discovery
Figure 30.3 Use of mGenomeSubtractor to obtain a preliminary estimate of the known Acinetobacter baumannii species gene pool. As a firststep, all the annotated chromosomal CDS of A. baumannii ATCC 17978 were added into the gene pool. Next, unique chromosomal CDS presentin the five other completely sequenced A. baumannii genomes, SDF, ACICU, AB0057, AB307-0294, and AYE, were sequentially added to thegrowing gene pool following examination of each chromosome in turn in the order listed against the expanding gene pool. A similar analysiswas undertaken to capture unique CDS found in 17 fully sequenced A. baumannii plasmids currently available in GenBank. Finally, singlerepresentative unique CDSs were selected from the gene pool by removing any redundant “duplicated” genes as determined using the H -valuecriteria (H values >0.81). The number of CDS specific to each of the six chromosomes and the set of 17 plasmids are shown below their labelsin parentheses.
Salmonella Paratyphi A ATCC 9150 but not in any theavailable non-Paratyphi A Salmonella genome sequenceinformation or the wider GenBank database. This insilico substractive hybridization yielded 43 ATCC 9150-unique CDS. However, 29 of these CDS were foundto be variably present in 12 other Paratyphi A strainsby examination of an available microarray-generatedhybridization dataset. ArrayOme was used to map theremaining 14 CDS that were found to cluster into fourwidely separated genomic loci. Gene-specific primerswere designed to target a single, empirically selectedrepresentative of each locus, and the four primer pairswere used in a multiplex PCR assay. Unexpectedly,stkF , one of the target genes selected, was found toexhibit a sequence polymorphism arising from thepresence or absence of a duplicated 26-bp internalsequence. This finding led to the hypothesis that Stkfimbriae may be subjected to low-frequency, programmedDNA-facilitated phase variation driven by slipped strandmispairing and/or recombination events. Importantly,based on the appearance of one of two four-bandpathognomonic signatures, the assay proved to be 100%sensitive and specific for Paratyphi A isolates despite noprior experimental validation, elegantly highlighting thepotential of this approach as a pipeline for do-it-yourselfdevelopment of strain-, serovar-, or pathotype-specificassays.
30.4.2 tRIP PCR-Based Discoveryof Klebsiella pneumoniaeNiche-Adaptation IslandstRNAcc analysis of the first two Klebsiella pneumoniaegenomes to be sequenced, MGH78578 & Kp342, iden-tified five potential tRNA-associated integration hotspotsin K. pneumoniae. Subsequent experimental tRIP PCRanalysis of ∼120 clinical isolates obtained from theUnited Kingdom and China using primers designed toanneal to conserved upstream and downstream flanksconfirmed that the pheV , arg6, asn34 and tmRNA siteswere frequently occupied by integrated DNA in thesestrains [Chen et al., 2009; van Aartsen, 2008; Zhang,2011]. Chromosome walking, long-range PCR acrossoccupied sites and/or resistance gene tagging followedby fosmid-based marker rescue have already led to thediscovery of at least five novel tRNA-associated genomicislands including two islands coding for likely previouslyuncharacterized fimbrial structures, the KpGI-2 islandthat appears to confer a marked cAMP-dependent growthand/or viability phenotype that carries four novel genesplus a new variant of the fic gene family [Chen et al.,2009], and the small 3.7 kb KpGI-1 island coding fora putative acetyltransferase that is much more stronglyassociated with sputum isolates than blood, urine, or bileislands [Chen et al., 2009]. Collectively, these features

30.5 Summary 261
suggest possible niche-adaptation roles for these islandsand provide ample bioinformatics-derived substrate fortargeted, hypothesis-driven experimental investigations.
30.4.3 TnAbaR1-like ResistanceMega-transposons Are WidelyDisseminated Among ClinicalMulti-Drug-Resistant Acinetobacterbaumannii StrainsIn 2006, through full-length sequencing of two Acine-tobacter baumannii genomes, Fournier et al. describedAbaR1, a large 86-kb chromosomal island harboringmore than 40 recognized resistance genes present in strainAYE [Fournier et al., 2006]. These authors also reported amuch smaller, entirely unrelated island, integrated into anidentical site within the comM gene in the second strainsequenced. Indeed, according to Fournier et al. [2006]this was an unexpected discovery. Using a targeted tRIPPCR strategy, we recently interrogated the comM loci of50 multidrug-resistant A. baumannii isolates; 40 isolatesfailed to yield comM tRIP PCR amplicons suggestiveof the presence of integrated islands within the gene.Subsequent chromosome walking into the predictedisland from the 5′ and/or 3′ ends of the split comMgene confirmed that eight of ten genotypically distinct A.baumannii isolates predicted to carry comM -integratedelements did indeed carry such entities. However, toour surprise based on the available data, six of theeight newly discovered islands bore termini that werehighly similar to those of AbaR1, a seventh possessed adivergent but closely related AbaR1-like terminus at the5′-comM end, and only one appeared to be entirely novel[Shaikh et al., 2009]. These data suggested that >60%of clinical multidrug-resistant A. baumannii isolatesharbored comM -borne AbaR1-like elements. Recentdetailed bioinformatics analyses of the terminal segmentsof these elements drawn from multiple strains [Fournieret al., 2006; Post and Hall, 2009; Iacono et al., 2008;Adams et al., 2008], coupled with preliminary experi-mental investigations, have led us to hypothesize thatthese elements comprise a novel transposon family thatis distantly related to Tn7 [Rose, 2009]. Designated asTnAbaR1 -like transposons, typical members have accu-mulated multiple transposons, integrons, and resistancegene cassettes, often in a nested fashion, and, like thehighly promiscuous Tn7 , potentially exploit two distinctmobilization pathways: stable integration into a uniquechromosomal site or homing onto conjugative plasmidsfor onward transfer to a new host [Peters and Craig,2001; Parks and Peters, 2009]. We have hypothesizedthat this complex, well-orchestrated process is mediatedby five novel TnAbaR1 transposition proteins and arecurrently investigating this idea.
30.4.4 Comparative Mappingof Members of the ShigellaResistance Locus Island Family:Clues to Improved Future StrategiesLuck et al. [2001] described the full-length charac-terization of the Shigella resistance locus island inShigella flexneri 2a strain YSH6000. The SRL islandwas found to code for both multidrug resistance and awell-recognized virulence-associated attribute—the latterthrough the eight-gene fec locus that mediates ferricdicitrate uptake, thus offering an advantage under invivo conditions of iron limitation. Based on a study ofa collection of strains representative of the four Shigellaspecies, SRL-like islands were subsequently shown tohave largely, if not completely, displaced plasmids as thestandard vehicle for dissemination of genes conferringresistance to the quartet of ampicillin, streptomycin,tetracycline, and chloramphenicol [Turner et al., 2003].These findings prompted a comparative analysis of SRL-like islands in representative strains facilitated largelyby an overlapping, tiling PCR approach [Turner et al.,2003; Al-Hasani et al., 2001]. We have applied a similarapproach to study TnAbaR1 -like transposons; but facedwith the much more substantial challenge of mappinglarge, highly divergent cargo-gene-bearing central spansof these islands, we have used mGenomeSubtractorto identify genes unique to each element, common tovarious subsets and those universally present amongstthe full complement of TnAbaR1 -like elements that havebeen fully characterized to date. These data have beenused to synthesize ∼50 oligonucleotide primer pairs tar-geting known TnAbaR1 -like genes and selected other A.baumannii resistance genes, and they are currently beingused in various semi-random permutations to facilitatea shot-gun, combinatorial, long-range PCR approachaimed at generating overlapping amplicons that span thefull lengths of uncharacterized TnAbaR1 -like elements.Long-range PCR amplicons obtained will be subjectedto additional PCR mapping and/or barcode-facilitatedhigh-throughput sequencing to complete the comparativeanalysis dataset.
30.5 SUMMARY
Even with current high-throughput genomic sequencingfacilities, it is not feasible to sequence hundreds ofisolates to identify and decode the global gene poolaccessible to a single bacterial species. Instead, wepropose that the application of a strategy such asMobolimeFINDER, underpinned by ArrayOme andtRNAcc, followed by either random or targeted gene-discovery studies focused on selected strains bearing

262 Chapter 30 ArrayOme- and tRNAcc-Facilitated Mobilome Discovery
mobilome-rich regions, would make a major contributionto this effort. The highly intuitive mGenomeSubtractortool, which offers visualization of the genomic mosaic,zoom-in options, and linkages to other databases, is idealfor the identification and exploration of the most dynamicregions of bacterial genomes. The tRIP PCR strategy thatarises directly from tRNAcc analysis offers a powerful,readily accessible approach to allow for widespreadengagement in the bacterial metagenome discovery game.Selection of strains representative of a greater diversityof ecological, geographic, temporal, animal–host, and/ordisease-association categories could well result in theidentification of dramatically more novel islands. Fur-thermore, initial analysis of a greater number of referencegenomes by tRNAcc and mGenomeSubtractor and/oradditional input from ArrayOme-analysed comparativehybridization datasets should allow for the identificationof many more tRNA and non-tRNA potential hotspots,which in turn could be targeted by a broader tRIP PCRscan. Collectively, these approaches should not justhelp reveal the wider DNA blueprints of individualbacterial species but play a significant role in definingthe functional contributions of bacterial mobilomes.
INTERNET RESOURCES
NCBI Microbial Genomes (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi)
The Genomes On Line Database (GOLD) (http://www.genomesonline.org/)
MobilomeFINDER (http://mml.sjtu.edu.cn/MobilomeFINDER/)
mGenomeSubtractor (http://bioinfo-mml.sjtu.edu.cn/mGS/)
Islander (http://kementari.bioinformatics.vt.edu/cgi-bin/islander.cgi)
xBASE (http://xbase.bham.ac.uk/)
webACT (http://www.webact.org/)
Mauve (http://gel.ahabs.wisc.edu/mauve/)
IS Finder (http://www-is.biotoul.fr/)
δρ–web (http://deltarho.amc.nl/)
PipMaker (http://pipmaker.bx.psu.edu/pipmaker/)
Primaclade (http://www.umsl.edu/services/kellogg/primaclade.html)
AcknowledgmentsThis work was supported by grants from NationalNatural Science Foundation of China to HYO (30700013,30871345, and 30821005), as well as by the Royal
Society—National Natural Science Foundation of ChinaInternational Joint Program (2007/R3), British Society forAntimicrobial Chemotherapy (GA803), Action MedicalResearch (SP4255), and Medisearch to KR.
REFERENCES
Adams MD, Goglin K, Molyneaux N, Hujer KM, Lavender H,et al. 2008 Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii . J. Bacteriol . 190:8053–8064.
Al-Hasani K, Adler B, Rajakumar K, Sakellaris H. 2001. Dis-tribution and structural variation of the she pathogenicity island inenteric bacterial pathogens. J. Med. Microbiol . 50:780–786.
Ansorge WJ. 2009. Next-generation DNA sequencing techniques. N.Biotechnol . 25:195–203.
Boyd EF, Almagro-Moreno S, Parent MA. 2009. Genomic islandsare dynamic, ancient integrative elements in bacterial evolution.Trends Microbiol . 17:47–53.
Cai H, Thompson R, Budinich MF, Broadbent JR, Steele JL. 2009.Genome sequence and comparative genome analysis of Lactobacilluscasei : Insights into their niche-associated evolution. Genome Biol.Evol . 1:239–257.
Canchaya C, Fournous G, Brussow H. 2004. The impact ofprophages on bacterial chromosomes. Mol. Microbiol . 53:9–18.
Chen N, Ou HY, van Aartsen JJ, Jiang X, Li M, et al. 2009. ThepheV phenylalanine tRNA gene in Klebsiella pneumoniae clinical iso-lates is an integration hotspot for possible niche-adaptation GenomicIslands. Curr. Microbiol., 60:210–216.
Churchward G. 2008. Back to the future: The new ICE age. Mol.Microbiol . 70:554–556.
Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: Mul-tiple alignment of conserved genomic sequence with rearrangements.Genome Res . 14:1394–1403.
Dobrindt U, Hacker J. 2001. Whole genome plasticity in pathogenicbacteria. Curr. Opin. Microbiol . 4:550–557.
Dorrell N, Hinchliffe SJ, Wren BW. 2005. Comparative phyloge-nomics of pathogenic bacteria by microarray analysis. Curr. Opin.Microbiol . 8:620–626.
Field D, Wilson G, van der Gast C. 2006. How do we comparehundreds of bacterial genomes? Curr. Opin. Microbiol . 9:499–504.
Fleischmann RD, Adams MD, White O, Clayton RA, KirknessEF, et al. 1995. Whole-genome random sequencing and assembly ofHaemophilus influenzae Rd. Science 269:496–512.
Fournier PE, Vallenet D, Barbe V, Audic S, Ogata H, et al.2006. Comparative genomics of multidrug resistance in Acinetobacterbaumannii . PLoS Genet . 2:e7.
Frost LS, Leplae R, Summers AO, Toussaint A. 2005. Mobilegenetic elements: The agents of open source evolution. Nat. Rev.Microbiol . 3:722–732.
Fukiya S, Mizoguchi H, Tobe T, Mori H. 2004. Extensive genomicdiversity in pathogenic Escherichia coli and Shigella Strains revealedby comparative genomic hybridization microarray. J. Bacteriol .186:3911–3921.
Fux CA, Shirtliff M, Stoodley P, Costerton JW. 2005. Can lab-oratory reference strains mirror “real-world” pathogenesis? TrendsMicrobiol . 13:58–63.
Gadberry MD, Malcomber ST, Doust AN, Kellogg EA. 2005.Primaclade—A flexible tool to find conserved PCR primers acrossmultiple species. Bioinformatics 21:1263–1264.
Hacker J, Blum-Oehler G, Muhldorfer I, Tschape H. 1997.Pathogenicity islands of virulent bacteria: Structure, function andimpact on microbial evolution. Mol. Microbiol . 23:1089–1097.

References 263
Iacono M, Villa L, Fortini D, Bordoni R, Imperi F, et al. 2008.Whole-genome pyrosequencing of an epidemic multidrug-resistantAcinetobacter baumannii strain belonging to the European clone IIgroup. Antimicrob. Agents Chemother . 52:2616–2625.
Karlin S. 2001. Detecting anomalous gene clusters and pathogenicityislands in diverse bacterial genomes. Trends Microbiol . 9:335–343.
Konstantinidis KT, Ramette A, Tiedje JM. 2006. The bacterialspecies definition in the genomic era. Philos. Trans. R. Soc. Lond.B. Biol. Sci . 361:1929–1940.
Lima-Mendez G, Toussaint A, Leplae R. 2007. Analysis of thephage sequence space: The benefit of structured information. Virology365:241–249.
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, HugenholtzP, et al. 2009. The Genomes On Line Database (GOLD) in 2009:status of genomic and metagenomic projects and their associatedmetadata. Nucleic. Acids Res.. 38:D346–D354.
Luck SN, Turner SA, Rajakumar K, Sakellaris H, Adler B.2001. Ferric dicitrate transport system (Fec) of Shigella flexneri 2aYSH6000 is encoded on a novel pathogenicity island carrying multi-ple antibiotic resistance genes. Infect. Immun . 69:6012–6021.
MacLean D, Jones JD, Studholme DJ. 2009. Application of ‘next-generation’ sequencing technologies to microbial genetics. Nat. Rev.Microbiol . 7:287–296.
Mantri Y, Williams KP. 2004. Islander: A database of integrativeislands in prokaryotic genomes, the associated integrases and theirDNA site specificities. Nucleic Acids Res . 32:D55–D58.
Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. 2005.The microbial pan-genome. Curr. Opin. Genet. Dev . 15:589–594.
Ou HY, Smith R, Lucchini S, Hinton J, Chaudhuri RR, et al.2005. ArrayOme: a program for estimating the sizes of microarray-visualized bacterial genomes. Nucleic Acids Res . 33:e3.
Ou HY, Chen LL, Lonnen J, Chaudhuri RR, Thani AB, et al.2006. A novel strategy for the identification of genomic islands bycomparative analysis of the contents and contexts of tRNA sites inclosely related bacteria. Nucleic Acids Res . 34:e3.
Ou HY, He X, Harrison EM, Kulasekara BR, Thani AB, et al.2007a. MobilomeFINDER: web-based tools for in silico and exper-imental discovery of bacterial genomic islands. Nucleic Acids Res .35:W97–W104.
Ou HY, Ju CT, Thong KL, Ahmad N, Deng Z, et al. 2007b. Transla-tional genomics to develop a Salmonella enterica serovar Paraty-phi A multiplex polymerase chain reaction assay. J. Mol. Diagn .9:624–630.
Parks AR, Peters JE. 2009. Tn7 elements: engendering diversity fromchromosomes to episomes. Plasmid 61:1–14.
Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter bauman-nii: Emergence of a successful pathogen. Clin. Microbiol. Rev .21:538–582.
Peters JE, Craig NL. 2001. Tn7 : Smarter than we thought. Nat. Rev.Mol. Cell. Biol . 2:806–814.
Post V, Hall RM. 2009. AbaR5, a large multiple-antibiotic resis-tance region found in Acinetobacter baumannii . Antimicrob AgentsChemother . 53:2667–2671.
Rajakumar K, Sasakawa C, Adler B. 1997. Use of a novel approach,termed island probing, identifies the Shigella flexneri she pathogenic-ity island which encodes a homolog of the immunoglobulinA protease-like family of proteins. Infect. Immun . 65:4606–4614.
Riley MA, Lizotte-Waniewski M. 2009. Population genomicsand the bacterial species concept. Methods Mol. Biol . 532:367–377.
Rose A. 2009. TnAbaR1: A novel Tn7 -related transposon in Acine-tobacter baumannii that contributes to the accumulation and dis-semination of large repertoires of resistance genes. Biosci. Horizons3:40–48.
Schuler GD. 1997. Sequence mapping by electronic PCR. GenomeRes . 7:541–550.
Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, et al. 2000.PipMaker—a web server for aligning two genomic DNA sequences.Genome Res . 10:577–586.
Shaikh F, Spence RP, Levi K, Ou HY, Deng Z, et al. 2009. ATPasegenes of diverse multidrug-resistant Acinetobacter baumannii iso-lates frequently harbour integrated DNA. J. Antimicrob. Chemother .63:260–264.
Shao Y, He X, Harrison EM, Tai C, Ou HY, Rajakumar K,Deng Z, 2010. mGenomeSubtractor: a web-based tool for parallelin silico subtractive hybridization analysis of multiple bacterialgenomes. Nucleic. Acids. Res . 38:W194–200.
Tettelin H, Riley D, Cattuto C, Medini D. 2008. Compara-tive genomics: The bacterial pan-genome. Curr. Opin. Microbiol .11:472–477.
Towner KJ. 2009. Acinetobacter : An old friend, but a new enemy.J. Hosp. Infect . 73:355–363.
Turner SA, Luck SN, Sakellaris H, Rajakumar K, Adler B. 2003.Molecular epidemiology of the SRL pathogenicity island. Antimicrob.Agents Chemother . 47:727–734.
Vallenet D, Nordmann P, Barbe V, Poirel L, Mangenot S, etal. 2008. Comparative analysis of Acinetobacters: Three genomes forthree lifestyles. PLoS One 3:e1805.
van Aartsen JJ. 2008. The Klebsiella pheV tRNA locus: a hotspotfor integration of alien genomic islands. Biosci. Horizons 1:51–60.
Wolfgang MC, Kulasekara BR, Liang X, Boyd D, Wu K, et al.2003. Conservation of genome content and virulence determinantsamong clinical and environmental isolates of Pseudomonas aerugi-nosa . Proc. Natl. Acad. Sci. USA 100:8484–8489.
Zhang J, van Aartsen JJ, Jiang X, Shao Y, Tai C, He X, TanZ, Deng Z, Jia S, Rajakumar K, Ou HY, 2011. Expansion of theknown Klebsiella pneumoniae species gene pool by characterizationof novel alien DNA islands integrated into tmRNA gene sites. J,Microbiol Methods . 84:283–289.
















![[13.07.07] albertsen mewe13 metagenomics](https://img.pdfslide.us/doc/110x75/554f4770b4c905524c8b46ef/130707-albertsen-mewe13-metagenomics.jpg)
![[2013.10.29] albertsen genomics metagenomics](https://img.pdfslide.us/doc/110x75/554a2539b4c90520578b4861/20131029-albertsen-genomics-metagenomics.jpg)











