Embed Size (px)
Citation preview

PART IV
REPRESENTATIVE GUIDELINES AND/OREXPERIMENTAL PROTOCOLS OFLC-MS BIOANALYSIS
Handbook of LC-MS Bioanalysis: Best Practices, Experimental Protocols, and Regulations, First Edition. Edited by Wenkui Li, Jie Zhang, and Francis L.S. Tse.C© 2013 John Wiley & Sons, Inc. Published 2013 by John Wiley & Sons, Inc.

34LC-MS BIOANALYSIS OF ESTER PRODRUGS AND OTHERESTERASE LABILE MOLECULES
Wenkui Li, Yunlin Fu, Jimmy Flarakos, and Duxi Zhang
34.1 INTRODUCTION
A prodrug is a “precursor” to an intended drug. Upon admin-istration, the prodrug is converted to the intended drug, theactive pharmacological agent, through normal in vivo bioac-tivation processes (metabolism). A prodrug can be usedto improve oral bioavailability of the intended drug thatis poorly absorbed in gastrointestinal tract. A prodrug canalso be used to improve the interaction selectivity of theintended drug with target cells (Stella et al., 1985; Rautioet al., 2008; Peterson and McKenna, 2009) in treatmentlike chemotherapy, which is often accompanied with someunwanted adverse effects (Rooseboom et al., 2004). Froma bioconversion perspective, a prodrug can be bioactivatedintracellularly (e.g., in therapeutic target tissues/cells ormetabolic tissues) or extracellularly (e.g., in the milieu of GIfluids, systemic circulation, and metabolic tissues) or both.The most common prodrugs are the ester-based ones thatare formed by derivatizing a phenol, hydroxyl, or carboxylgroup of the intended drug molecule (Stella et al., 1985;Rautio et al., 2008; Peterson and McKenna, 2009).
The cleavage process of an ester prodrug (Figure 34.1)is catalyzed by various esterases in vivo to form its activemolecule (Rooseboom et al., 2004; Liederer and Borchardt,2006). However, the same hydrolysis can occur ex vivo post-sample collection (including sample collection, processing,and storage and thawing prior to extraction). From a bio-analysis point of view, any ex vivo cleavage of prodrugmolecules in biological matrix is unwanted (Li et al., 2011).This ex vivo cleavage, if not controlled, can lead to overesti-mation of the active drug molecule and underestimation of the
prodrug. As a result, the safety and/or efficacy assessment ofthe prodrug and its active form in the intended studies can beinaccurate.
Over the past decades, bioanalytical scientists have madesignificant progress in preventing and/or blocking unwantedex vivo enzyme-catalyzed cleavage of ester prodrugs inLC-MS bioanalysis. This chapter highlights some importantprocedures for identifying and addressing the instability issueof an ester prodrug. The same approaches can be applied forthe analysis of other enzyme labile molecules that containamides, lactams, peptides, and lactones, and so on (Li et al.,2011). LC-MS of sulfhydryl (thiol) containing molecules isnot covered in this chapter.
34.2 COMMON ESTERASES THAT CATALYZEHYDROLYSIS OF ESTER PRODRUGS ANDOTHER MOLECULES
Esterases belong to a heterogeneous enzyme family con-sisting of three subgroups (esterases A, B, and C)(Aldrige, 1952; Bergmann et al., 1957). The most impor-tant esterases include carboxylesterase, acetylcholinesterase,butyrylcholinesterase, paraoxonase, cholinesterase, andarylesterase. These enzymes are widely distributed through-out the body, including systemic circulation (blood/plasma)and tissues (e.g., liver, brain, kidney, and lung) (Liederer andBorchardt, 2006).
Research has shown that different species have differ-ent amounts and types of esterases with different substratespecificities and different rates of hydrolysis. Within a given
Handbook of LC-MS Bioanalysis: Best Practices, Experimental Protocols, and Regulations, First Edition. Edited by Wenkui Li, Jie Zhang, and Francis L.S. Tse.C© 2013 John Wiley & Sons, Inc. Published 2013 by John Wiley & Sons, Inc.
431

432 LC-MS BIOANALYSIS OF ESTER PRODRUGS AND OTHER ESTERASE LABILE MOLECULES
Drug
O
O
R
Drug
OH
O
+ HOR
Esterase
H2O
Active form of drugProdrug
FIGURE 34.1 A representative scheme of esterase-catalyzedhydrolysis of an ester prodrug.
species, the enzyme activities can vary from one subject to theother. There may be more than a single esterase involved inthe hydrolysis of a given ester-containing compound. A givenester-containing compound may be subjected to hydrolysiscatalyzed by different esterase(s) in different species. In gen-eral, esterase activity in rodent (e.g., rat and mouse) bloodis much higher than that in nonrodent (e.g., dog and human)blood (Minagawa et al., 1995; Liederer and Borchardt, 2006;Koitka et al., 2010). For example, hydrolysis of cisatracuriumby rat plasma esterases is more rapid than that in humanplasma (Welch et al., 1995). While human plasma showedno significant issues associated with the decomposition of Ro64-0796 and Ro 64-0802, rodent plasma must be pretreatedwith dichlorvos, an esterase inhibitor, to prevent hydrolysis(Wiltshire et al., 2000). The high activity in rodent speciesbut low activity in nonrodent species of esterases often renderdifficulty for drug discovery and development in generatingaccurate preclinical model for the evaluation of prodrug can-didates. This difference can also be translated to that a sim-ple stabilization approach may be sufficient in dealing withsample analysis of human subjects, but a more sophisticatedapproach may be necessary for handling study samples fromrodent species.
34.3 METHODOLOGY AND APPROACHES
From an LC-MS bioanalysis perspective, the major chal-lenge for ester-containing prodrugs is the management oftheir instability in the intended biological matrices duringbiological sample collection, processing, and storage andsample preparation prior to LC-MS quantification.
34.3.1 Common Precautions
1. Temperature control: Temperature control plays a cru-cial role in assuring a reliable analysis of unstableanalytes in biological samples (Chen and Hsieh, 2005;Tokumura et al., 2005; Briscoe and Hage, 2009). Ingeneral, the enzyme-catalyzed degradation of an ana-lyte in biological matrix is significantly slower whenthe sample is processed on wet ice (∼0◦C to 4◦C)compared to at room temperature (∼22◦C) (Chen andHsieh, 2005). One general approach is to freeze the
blood sample right after collection if blood is thematrix of choice, or to process the blood sample at areduced temperature (i.e., centrifugation at ∼4◦C) forplasma, followed by immediate storage in a freezer.The duration between sample collection and storagein a freezer should be as short as possible to minimizepossible degradation of unstable molecules (Li et al.,2011).
2. Selection of anticoagulant: Anticoagulant and/or anti-coagulant counter ions can have an impact on com-pound stability (Evans et al., 2001; Bergeron et al.,2009). Heparin and ethylenediaminetetraacetic acid(EDTA) are common anticoagulants with differentmechanisms of action. Heparin, as a polysaccharide,accelerates the inactivation of thrombin (an enzymethat promotes clotting), whereas EDTA chelates cal-cium ions and interrupts the clotting cascade at multi-ple points. EDTA can prevent the activity of calcium-dependent phospholipases and ester hydrolases, whileheparin may not offer the same effect. In general,EDTA is preferred to heparin as anticoagulant inplasma samples for ester prodrugs (Li et al., 2011).
34.3.2 Stability Assessment of Prodrugs and/or OtherEsterase Labile Compounds
A quick stability assessment of an ester prodrug or otheresterase sensitive compound in an intended biological matrixis the first important step to determine whether one or morestabilization measures, in addition to the above (low tem-perature and proper anticoagulant), need to be evaluated andoptimized in the early stage of bioanalytical method devel-opment.
34.3.2.1 Stability Assessment in Plasma Plasma is themost common matrix for bioanalysis. In most cases, analytestability in plasma serves as a good indicator of its stabilityin whole blood (Freisleben et al., 2011). Compared to blood,plasma is more homogeneous, less viscous, and easier tohandle. Stability results using plasma is less subjected to thepossible impact due to blood/plasma partition of the ana-lyte of interest if liquid–liquid extraction other than proteinprecipitation is the method of choice for sample preparation.
In plasma stability assessment, one (middle), two (low andhigh), or three (low, middle, and high) concentration levelsof ester prodrug quality control (QC) samples are prepared inthe untreated plasma of the intended species. Although usingthree concentration levels may help generate a rich data seton analyte stability, assessment using the middle QC concen-tration level can be very efficient and the results should servewell for the follow-up step in LC-MS bioanalytical develop-ment process. A relatively large volume of sample may be

METHODOLOGY AND APPROACHES 433
needed. The prepared QC samples are placed on a labora-tory bench at room temperature (to mimic the most commonsituation of study sample analysis) and on wet ice (for com-parison). At various time points (e.g., 0, 0.5, 1, 2, 6, and 24h), two or more aliquots of each QC sample are taken andplaced in a deeper freezer (i.e., <−60◦C, to prevent or slowdown any further degradation) or immediately treated with afixed volume of organic (e.g., acetonirile or methanol, to stopenzyme activity) followed by vortex-mixing and storage in adeep freezer. Upon all the needed QC aliquots at the intendedtime points are taken and/or treated, stability assessment canbe conducted using one of the following approaches:
� Comparison of analyte/internal standard LC-MSresponse ratios over time: Upon removal of stabilitysamples (untreated or treated with organic) from thefreezer and thawing, a fixed volume of internal stan-dard (preferably stable-isotope labeled) working solu-tion in organic solvent is added to each sample. Addi-tional organic solvent may be needed to ensure thesame amount of organic or nonorganic in the sam-ple mixture prior to further processing. After propervortex-mixing and centrifugation, the supernatants aretransferred and evaporated. The resulting residues arereconstituted prior to injection onto LC-MS system. TheLC-MS response ratios of the analyte to the internalstandard for the QC samples at various time points areplotted against that of QC samples at time zero. Thetime zero QC samples may be freshly prepared priorto the above experiment to ensure accurate comparison.Other surrogate matrices (e.g., urine or organic solvent)that does not contain any esterase have been used (Zenget al., 2007). However, the possible difference in matrixeffect or signal suppression between the matrices needsto be taken into account for the evaluation of the sta-bility assessment outcomes when a surrogate matrix isused as control, otherwise surrogate matrix should notbe used.
� Quantification of analyte using freshly spiked cali-bration standards: The stability QC samples preparedabove can be analyzed against freshly spiked calibra-tion standards. In this process, an equal volume ofeach appropriate (lower limit of quantitation (LLOQ) toupper limit of quantification (ULOQ)) standard workingsolution (normally in 50% aqueous organic) is mixedwith blank plasma in the assigned well of the assayplate. As such, there is ∼25% of organic in the wellswhen the analyte is mixed with plasma and enzymeactivity, if any, is expected to be inhibited. Upon addi-tion of internal standard working solution and/or otherreagents for further processing prior to LC-MS analysis,the measured analyte concentrations in the stability QC
samples at various time points are compared to thenominal values or the values measured at time zero.To ensure the consistency of matrix effect and extrac-tion recovery between the QC samples and the freshlyspiked calibration standards, the final volume of matrix,organic and other nonmatrix components in all sam-ples should be the same and sample mixture should bevortex-mixed well prior to any supernatant transfer forfurther processing.
� Monitoring formation of hydrolysis product over time:This approach is similar to the above with exception ofthat, instead of employing the freshly spiked calibrationstandards of ester prodrug, the active form of the drug isused to prepare the calibration standards for the assess-ment (Fung et al., 2010). In this case, the active drugconcentration serves as a surrogate indicator of insta-bility of the prodrug. The stability or instability of theprodrug can be unveiled by plotting the measured activedrug concentration over time. The lower the active drugconcentration, the better the stability of the prodrug canbe. From an operational point of view, it is much eas-ier to monitor the formation of the active drug in theintended matrix than the prodrug itself as the activedrug molecule is, in general, much more stable than theprodrug.
34.3.2.2 Stability Assessment in Blood Although stabil-ity in plasma is considered as a good surrogate indicator ofanalyte stability in blood, there are exceptions (Freislebenet al., 2011) for certain chemical classes for which stabilitybehavior of the drug in blood versus plasma is expectedto be different based on the chemical structure. There-fore, performing blood stability assessment is necessary notonly to confirm the stability coverage but also to provideadequate sampling instructions for preclinical or clinicalfacilities.
All three approaches listed for plasma stability assessmentcan be implemented for assessing the stability of analyte inblood. The analyte of interest is spiked into fresh blood ofthe intended species at the intended middle QC concentra-tion level. It is worth noting that fresh blood should be usedas aged blood with reduced enzyme activities may result infalse outcomes. Upon a gentle mixing to ensure a properequilibrium (∼15 min) of the analyte of interest in the bloodat room temperature or 37◦C. The samples are incubated at4◦C (on wet ice), room temperature (∼22◦C) and/or 37◦Cas shown in Figure 34.2 for an investigational ester prodrugcompound A. At various time points, at least two aliquotsof blood sample are taken. The blood samples can be cen-trifuged for plasma or directly mixed with internal standard,followed by a conventional protein precipitation or liquid–liquid extraction process prior to LC-MS analysis.

434 LC-MS BIOANALYSIS OF ESTER PRODRUGS AND OTHER ESTERASE LABILE MOLECULES
(1) Starting at RT or 37°C
(2) Spike blood with analyte at middle concentration, followed by a ~15 min of equilibration
0–4°C RT and/or 37°C
Plasma Blood
Take aliquots at various time points for further processing
FIGURE 34.2 A representative scheme of stability assessment of prodrugs and/or other enzymelabile compounds using whole blood.
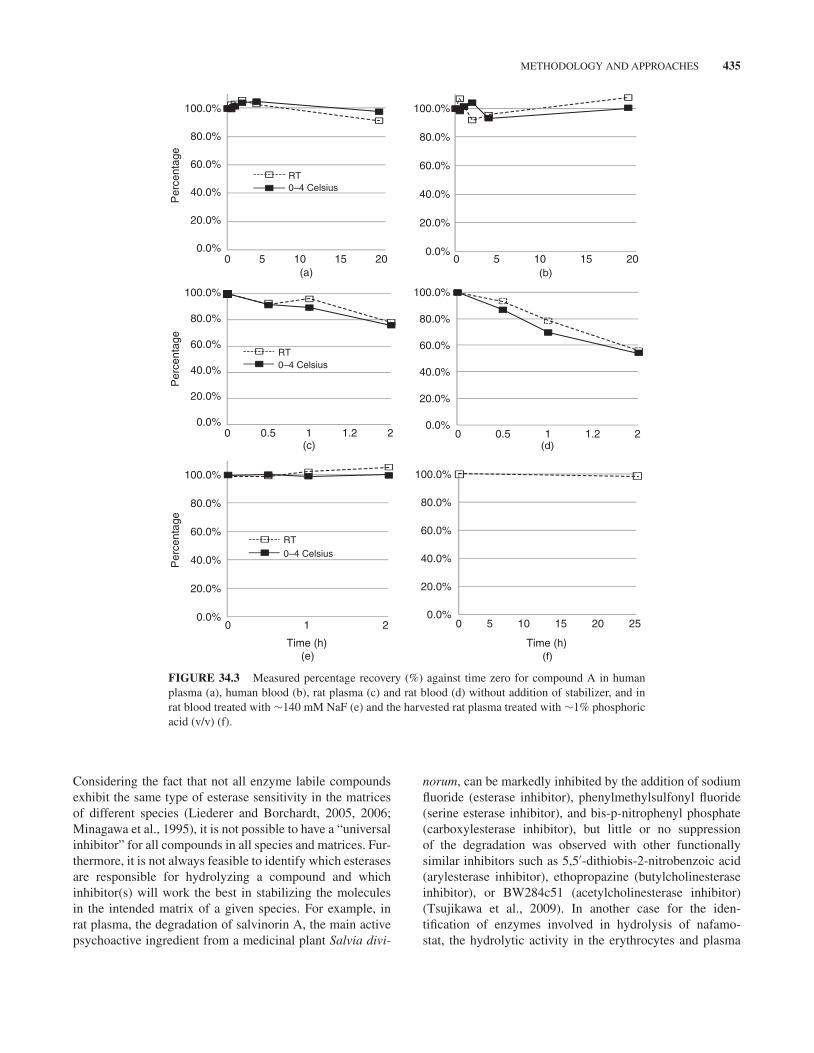
Considering the apparent difference in terms of samplehandling between plasma and blood samples, comparing LC-MS response ratios of the analyte to the internal standardacross the various time points might be more convenientthan other means in obtaining first-hand stability information.As shown in Figures 34.3a–d, compound A appears to bestable in human blood and plasma but not stable in rat bloodand plasma. Within 2 h of incubation, there are 20–40%losses of the compound in rat blood/plasma. On the otherhand, as shown in Figures 34.3c and d, the enzyme-catalyzedinstability of the compound in rat blood/plasma at a lowertemperature is generally better when the sample is placed onwet ice (∼0◦C to 4◦C) than at room temperature (∼22◦C)(Chen and Hsieh, 2005).
It is worth noting that if an analyte is stable in blood atroom temperature for more than 2 h, which is the typical dura-tion needed for harvesting plasma from blood, pretreatmentof blood collection tubes with inhibitors and/or pH modifiermight not be necessary. These agents can be added to theharvested plasma and then properly mixed prior to furtherprocessing. In general, treatment of plasma sample is mucheasier than that of blood sample.
34.3.3 Evaluation and Optimization ofStabilization Measures
As soon as instability is identified from any of the above pro-cesses, stabilization measures in addition to selection of rightanticoagulant and processing temperature have to be evalu-ated, optimized, and implemented for the best bioanlayticaloutcomes. The key stabilization measures include (1) pHcontrol, (2) use of esterase inhibitors, and (3) combination ofpH control and use of enzyme inhibitors.
34.3.3.1 pH Control It is well known that pH plays anessential role in acid-/base-catalyzed enzymatic reactionsand most enzymatic reactions have a narrow working pHwindow. A simple treatment of study samples with pH mod-ifiers can be very effective in stabilizing drug molecules (Liet al., 2011). In fact, a low pH can be much more effec-tive than a lower sample processing temperature in reducingesterase activities. From a bioanalytical operation perspec-tive, processing study samples at a low temperature is lessdesirable as it requires wet ice or chilled conditions, whichcreates challenges in using robotic liquid handler (Funget al., 2010). The most commonly used reagents for pHcontrol are formic acid, acetic acid, phosphoric acid, cit-ric acid or other buffers, including Tris buffer, at a variety ofconcentrations.
In practice, the strength and amount of the above pHmodifiers need to be adjusted to reduce the viscosity ofstudy samples. Therefore, it is desirable to use mild con-dition (e.g., pH 4–5). At a lower pH (e.g., pH 3 or lower),the treated plasma samples frequently become very viscous,making pipetting difficult and unreliable (Fung et al., 2010).A general recommendation is to treat the harvested plasmasamples with 0.5% formic acid (unpublished results), 0.5%phosphoric acid (unpublished results), 0.1 M HCl (Kameiet al., 2011), or treat blood samples with 0.5% citric acid(w/v) (Kamei et al., 2011) or 1 M citric acid at a final citricacid concentration of 20 mM (Fung et al., 2010). Otherwise,blood collection tubes with acid additives are commerciallyavailable for this purpose.
34.3.3.2 Use of Enzyme Inhibitors A variety of enzymeinhibitors (Table 34.1) can be used for stabilizing ester pro-drugs of interest and other esterase sensitive compounds.
METHODOLOGY AND APPROACHES 435
0.0%
20.0%
40.0%
60.0%
80.0%
100.0%
0.0%
20.0%
40.0%
60.0%
80.0%
100.0%
Per
cent
age
RT0–4 Celsius
(a) (b)
(c) (d)
(e) (f)
0.0%
20.0%
40.0%
60.0%
80.0%
100.0%
RT0–4 Celsius
0.0%
20.0%
40.0%
60.0%
80.0%
100.0%
0 0 5 10 15 20 25
0 0.5 1 1.2 2 0 0.5 1 1.2 2
0 5 10 15 20 0 5 10 15 20
1 2
Per
cent
age
0.0%
20.0%
40.0%
60.0%
80.0%
100.0%
Per
cent
age
RT0–4 Celsius
0.0%
20.0%
40.0%
60.0%
80.0%
100.0%
Time (h) Time (h)
FIGURE 34.3 Measured percentage recovery (%) against time zero for compound A in humanplasma (a), human blood (b), rat plasma (c) and rat blood (d) without addition of stabilizer, and inrat blood treated with ∼140 mM NaF (e) and the harvested rat plasma treated with ∼1% phosphoricacid (v/v) (f).
Considering the fact that not all enzyme labile compoundsexhibit the same type of esterase sensitivity in the matricesof different species (Liederer and Borchardt, 2005, 2006;Minagawa et al., 1995), it is not possible to have a “universalinhibitor” for all compounds in all species and matrices. Fur-thermore, it is not always feasible to identify which esterasesare responsible for hydrolyzing a compound and whichinhibitor(s) will work the best in stabilizing the moleculesin the intended matrix of a given species. For example, inrat plasma, the degradation of salvinorin A, the main activepsychoactive ingredient from a medicinal plant Salvia divi-
norum, can be markedly inhibited by the addition of sodiumfluoride (esterase inhibitor), phenylmethylsulfonyl fluoride(serine esterase inhibitor), and bis-p-nitrophenyl phosphate(carboxylesterase inhibitor), but little or no suppressionof the degradation was observed with other functionallysimilar inhibitors such as 5,5′-dithiobis-2-nitrobenzoic acid(arylesterase inhibitor), ethopropazine (butylcholinesteraseinhibitor), or BW284c51 (acetylcholinesterase inhibitor)(Tsujikawa et al., 2009). In another case for the iden-tification of enzymes involved in hydrolysis of nafamo-stat, the hydrolytic activity in the erythrocytes and plasma

436 LC-MS BIOANALYSIS OF ESTER PRODRUGS AND OTHER ESTERASE LABILE MOLECULES
TABLE 34.1 Commonly Used Esterase Inhibitors in Bioanalysis
Names Target enzymes References
Prazosin Carboxylesterase (CarbE) inhibitor Kudo et al. (2000)Thenoyltrifluoroacetone (TTFA) Carboxylesterase (also serving as a
chelating agent)Fung et al. (2010) and Zhang and Fariss
(2002)Acetylcholine (Ach) Competitive CarbE inhibitor Yoshigae et al. (1999)Bis(4-nitrophenyl)-phosphate (BNPP) Carboxylesterase inhibitor,
phosphodisterase inhibitorFung et al. (2010), Ishizuka et al. (2010),
Yoshigae et al. (1999), Kudo et al.(2000), Yamaori et al. (2006), andTsujikawa et al. (2009)
5, 5′-dithiobis-(2-nitrobenzoic acid)(DTNB, Ellman’s reagent)
Carboxylesterase inhibitor Yamaori et al. (2006), Tsujikawa et al.(2009), and Minagawa et al. (1995)
Ethopropazine (profenamine, parsidol,parsidan, parkin)
Butylcarboxylesterase inhibitor Yamaori et al. (2006) and Tsujikawaet al. (2009)
Phenylmethylsulfonyl fluoride (PMSF) Carboxylesterase inhibitor, Serineprotease inhibitor, but does notinhibitor all serine protease, rapidlydegraded in water
Fung et al. (2010), Ishizuka et al. (2010),Kim et al. (2011), Kudo et al. (2000),Yamaori et al. (2006), and Tsujikawaet al. (2009)
Diisopropyl fluorophosphate (DIFP, DFP) Irreversible serine hydrolase inhibitor Fung et al. (2010), Ishizuka et al. (2010),Yamaori et al. (2006), Minagawa et al.(1995), and Zeng et al. (2007)
2,2-Dichlorovinyl dimethyl phosphate(Dichlorvos, DDVP)
Carboxylesterase inhibitor,acetylcholinesterase inhibitor
Fung et al. (2010), Wiltshire et al. (2000),and Chang et al. (2009)
p-Nitrophenyl acetate (PNPA) Yoshigae et al. (1999)Paraoxon Acetylcholinesterase inhibitor Fung et al. (2010), Liederer and
Borchardt (2005), and Yang et al.(2002)
Eserine (physostigmine, erserine) Cholinesterase inhibitor,Acetylcholinesterase inhibitor
Fung et al. (2010), Ishizuka et al. (2010),Yoshigae et al. (1999), Minagawa et al.(1995), and Zhang et al. (2011)
BW284c51 Acetylcholinesterase inhibitor Olivera-Bravo et al. (2005), Tsujikawaet al. (2009), and Yamaori et al. (2006)
Sodium fluoride (NaF), Potassium fluoride(KF)
Phosphodiesterase inhibitor,Carboxylesterase inhibitor
Fung et al. (2010), Kim et al. (2011),Kromdijk et al. (2012), Salem et al.(2011), Skopp et al. (2001), and Wanget al. (2007)
Sodium arsenate Esterase inhibitor Salem et al. (2011)
Reproduced from Li et al. (2011) with permission of John Wiley & Sons Ltd.
was inhibited by 5, 5′-dithiobis (2-nitrobenzoic acid), anarylesterase inhibitor, in a concentration-dependent manner.By contrast, little or no suppression of the activity wasseen with phenylmethylsulfonyl fluoride, diisopropyl flu-orophosphate, bis(p-nitrophenyl)phosphate, BW284c51, orethopropazine (Yamaori et al., 2006).
As discussed, identification and use of the most suitableesterase inhibitor can be very critical. In this process, one ormore esterase inhibitor candidates at two or more concentra-tions (e.g., 0.1, 1, and 10 mM) are screened in the intendedmatrix against the ester prodrug. Considering the amount ofwork and numbers of inhibitor candidates to be screened,one single concentration level (i.e., middle QC level) for theanalyte should be sufficient as a starting point (Fung et al.,2010).
The screening process is similar to the plasma/blood sta-bility assessment described earlier. To the plasma or bloodtreated with inhibitor candidates, the ester prodrug is spikedand mixed. The samples are placed at room temperature(to mimic study sample analysis situation) and on wet ice(for comparison). At various time points up to 24 h post-preparation, two or more aliquots of the samples are takenand processed for LC-MS analysis. The same approach forplasma stability assessment (refer to Section 34.3.2.1) can beemployed for assessing the effectiveness of inhibitor candi-dates (Zeng et al., 2007; Fung et al., 2010). One needs to keepin mind that some organophosphates, for example, paraoxon,are potent neurotoxic reagents and require special handlingat toxicology assessment or clinical study sites. Wheneverpossible, less toxic inhibitors should be used.

METHODOLOGY AND APPROACHES 437
34.3.3.3 Combination of Addition of Enzyme Inhibitorsand Use of pH Modifiers In most cases, a simple adjust-ment of matrix pH (Kamei et al., 2011) or addition of anenzyme inhibitor (Chang et al., 2009; Kim et al., 2011;Lindegardh et al., 2006; Yang et al., 2002; Zeng et al., 2007)can be sufficient and effective in stopping esterase-catalyzedhydrolysis of an ester prodrug and/or other esterase labilecompound. However, there have been many cases wherecombined stabilization measures have to be utilized. Thiscombined measure includes pH control and use of inhibitor(Tang and Sojinu, 2012; Wang et al., 2007) or use of twoinhibitors (Salem et al., 2011), in addition to common pre-cautions, for example, low temperature and proper anticoag-ulant.
Blood collection tubes containing certain enzymeinhibitor (e.g., NaF) or pH modifier (e.g., citric acid) arecommercially available. This makes the stabilization processmuch easier as only one more necessary step is the treatmentof the harvested plasma samples with acid or inhibitor. Inpractice, regardless of whether the blood tubes containinginhibitor or acid is to be used in the first place, whole-bloodstability of the prodrug should be evaluated under optimizedplasma pH and processing temperature to establish the pro-cedures and conditions that are to be employed in the studysample collection and processing in preclinical or clinicalstudies (Fung et al., 2010).
Figures 34.3e and f depict the stability of compound A atmiddle QC concentration level in fresh rat blood treated withsodium fluoride (∼140 mM) and in the harvested rat plasmatreated with ∼1% phosphoric acid. Clearly, the compoundis stable for at least 2 h in rat blood and for at least 25 h inthe harvested plasma. Further evaluation of rat plasma QCsamples treated with NaF (∼140 mM) and phosphoric acid(∼1%) shows that compound A is stable after three freeze–thaw cycles and for at least 36 days following storage at ≤−60◦C (figure not shown). The established storage stabilitycovers the duration from the first sample collection to the lastsample analysis for a toxicokinetic study.
34.3.4 Confirmation of Effectiveness ofStabilization Measures in Method Validationand Sample Analysis
There is no exception that the health authority requirements(FDA, EMA, etc.) and the current industry practice shouldbe followed in validating an LC-MS method for quantita-tive analysis of both the prodrug and its active form in theintended matrix. It is also common that pooled QCs (prodrugand drug) are employed for assay validation and associatedstudy sample analysis. However, to confirm whether the pro-drug is stable in the QC samples, QC samples containingthe prodrug only need to be included in assay validation,where the prodrug alone QC samples go through the needed
freeze–thaw cycles, room temperature stay, and long-termstorage before analysis using freshly prepared calibrationstandards of both the prodrug and the active drug usingthe same matrix. By monitoring changes of both the par-ent and active drug molecule in those QC samples, a higherconfidence should be gained. Furthermore, using freshlyprepared calibration standards can for sure help gain thisconfidence.
One should always keep in mind that QC samples arenot incurred samples. The QC samples, prepared by indi-vidually spiking or serial dilution, might contain ≤5% ofnonmatrix component (Li et al., 2011). This small amount ofnon-matrix component, usually organic solvents, may pro-vide some unexpected stabilization effect to the prodrug. Onthe other hand, although a large portion (>95%) of the QCsample matrix is the same as the incurred samples, QC sam-ples do not contain the various drug metabolites, coadmin-istered drug(s) and their metabolites, and/or dosing vehiclesas found in incurred samples (Li et al., 2011). In particular,enzyme activity may be different between the incurred sam-ples and the blank matrix that is used for preparation of cali-bration and QC samples. The slow denaturation of esterasesin the aged matrix (whole blood, plasma, serum, etc.) oftenleads to a decreased hydrolysis activity. It is not unusual thata validated LC-MS method with an enzyme inhibitor con-centration optimized using the standards/QCs prepared fromaged matrix does not work well for the incurred samples.Heightened awareness among bioanalytical scientists of thedifferences between the QC samples and incurred sampleswould help reduce the risk of subsequent bioanalytical failure(Li et al., 2011; Meng et al., 2011). The use of representativeincurred samples is necessary in order to confirm that theenzyme inhibitor(s) and other stabilization measures workwell for both the calibration standards/QC samples and theincurred samples.
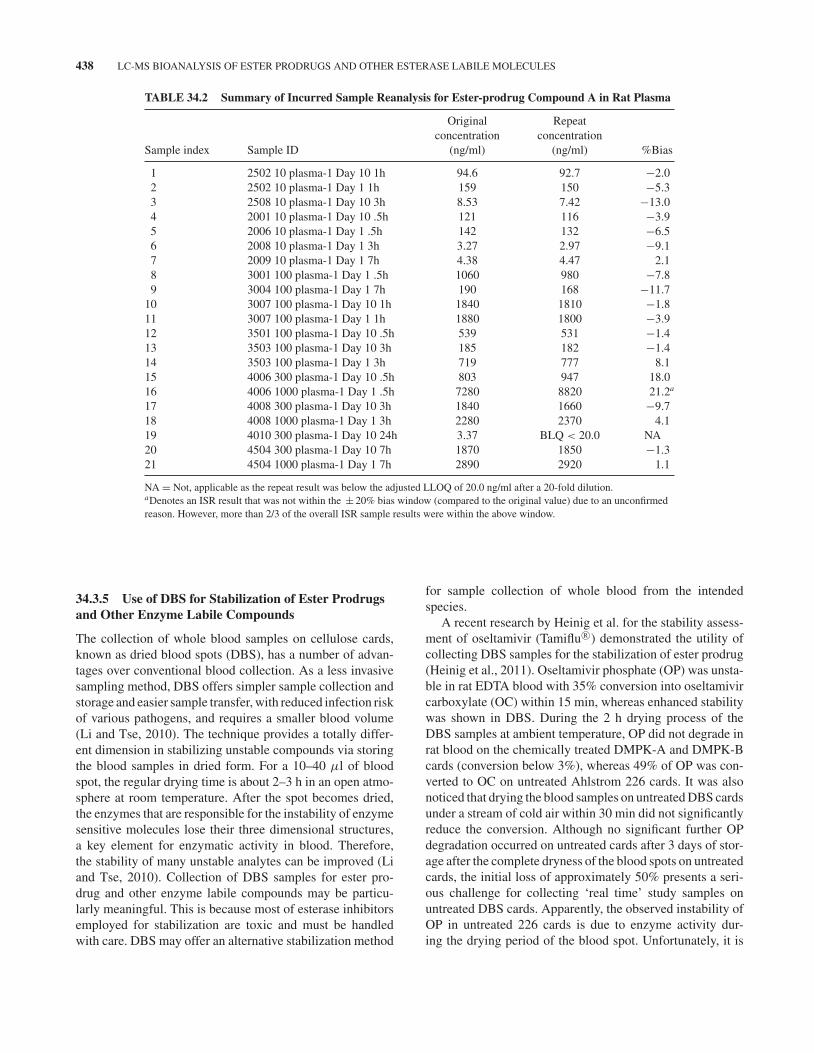
As soon as optimized stabilization measures are imple-mented in the first-in-animal or first-in-human studies,incurred sample reanalysis (ISR) and/or incurred sample sta-bility (ISS) assessment should be conducted. The differencein the measured prodrug concentrations between the repeatanalysis and initial analysis or the mean of two measurements(first and repeat) should be within ± 20% for two-thirds ofthe incurred samples selected. Any marked decrease in theprodrug concentrations would indicate prodrug instability inthe incurred samples. Under such circumstance, the stabiliza-tion measures will need to be modified prior to revalidationof the method and potential repeat of the preclinical study,which can be costly. Table 34.2 summarized the ISR resultsfor compound A in 21 randomly selected incurred samplesfrom a 2-week toxicokinetic study in rats. The results met theacceptance criteria, demonstrating collection of rat blood intoNaF/EDTA tubes and addition of phosphoric acid (∼1.0%,v/v) to the harvested plasma is effective in stabilizing theprodrug.
438 LC-MS BIOANALYSIS OF ESTER PRODRUGS AND OTHER ESTERASE LABILE MOLECULES
TABLE 34.2 Summary of Incurred Sample Reanalysis for Ester-prodrug Compound A in Rat Plasma
Original Repeatconcentration concentration
Sample index Sample ID (ng/ml) (ng/ml) %Bias
1 2502 10 plasma-1 Day 10 1h 94.6 92.7 −2.02 2502 10 plasma-1 Day 1 1h 159 150 −5.33 2508 10 plasma-1 Day 10 3h 8.53 7.42 −13.04 2001 10 plasma-1 Day 10 .5h 121 116 −3.95 2006 10 plasma-1 Day 1 .5h 142 132 −6.56 2008 10 plasma-1 Day 1 3h 3.27 2.97 −9.17 2009 10 plasma-1 Day 1 7h 4.38 4.47 2.18 3001 100 plasma-1 Day 1 .5h 1060 980 −7.89 3004 100 plasma-1 Day 1 7h 190 168 −11.7
10 3007 100 plasma-1 Day 10 1h 1840 1810 −1.811 3007 100 plasma-1 Day 1 1h 1880 1800 −3.912 3501 100 plasma-1 Day 10 .5h 539 531 −1.413 3503 100 plasma-1 Day 10 3h 185 182 −1.414 3503 100 plasma-1 Day 1 3h 719 777 8.115 4006 300 plasma-1 Day 10 .5h 803 947 18.016 4006 1000 plasma-1 Day 1 .5h 7280 8820 21.2a
17 4008 300 plasma-1 Day 10 3h 1840 1660 −9.718 4008 1000 plasma-1 Day 1 3h 2280 2370 4.119 4010 300 plasma-1 Day 10 24h 3.37 BLQ < 20.0 NA20 4504 300 plasma-1 Day 10 7h 1870 1850 −1.321 4504 1000 plasma-1 Day 1 7h 2890 2920 1.1
NA = Not, applicable as the repeat result was below the adjusted LLOQ of 20.0 ng/ml after a 20-fold dilution.aDenotes an ISR result that was not within the ± 20% bias window (compared to the original value) due to an unconfirmedreason. However, more than 2/3 of the overall ISR sample results were within the above window.
34.3.5 Use of DBS for Stabilization of Ester Prodrugsand Other Enzyme Labile Compounds
The collection of whole blood samples on cellulose cards,known as dried blood spots (DBS), has a number of advan-tages over conventional blood collection. As a less invasivesampling method, DBS offers simpler sample collection andstorage and easier sample transfer, with reduced infection riskof various pathogens, and requires a smaller blood volume(Li and Tse, 2010). The technique provides a totally differ-ent dimension in stabilizing unstable compounds via storingthe blood samples in dried form. For a 10–40 μl of bloodspot, the regular drying time is about 2–3 h in an open atmo-sphere at room temperature. After the spot becomes dried,the enzymes that are responsible for the instability of enzymesensitive molecules lose their three dimensional structures,a key element for enzymatic activity in blood. Therefore,the stability of many unstable analytes can be improved (Liand Tse, 2010). Collection of DBS samples for ester pro-drug and other enzyme labile compounds may be particu-larly meaningful. This is because most of esterase inhibitorsemployed for stabilization are toxic and must be handledwith care. DBS may offer an alternative stabilization method
for sample collection of whole blood from the intendedspecies.
A recent research by Heinig et al. for the stability assess-ment of oseltamivir (Tamiflu R©) demonstrated the utility ofcollecting DBS samples for the stabilization of ester prodrug(Heinig et al., 2011). Oseltamivir phosphate (OP) was unsta-ble in rat EDTA blood with 35% conversion into oseltamivircarboxylate (OC) within 15 min, whereas enhanced stabilitywas shown in DBS. During the 2 h drying process of theDBS samples at ambient temperature, OP did not degrade inrat blood on the chemically treated DMPK-A and DMPK-Bcards (conversion below 3%), whereas 49% of OP was con-verted to OC on untreated Ahlstrom 226 cards. It was alsonoticed that drying the blood samples on untreated DBS cardsunder a stream of cold air within 30 min did not significantlyreduce the conversion. Although no significant further OPdegradation occurred on untreated cards after 3 days of stor-age after the complete dryness of the blood spots on untreatedcards, the initial loss of approximately 50% presents a seri-ous challenge for collecting ‘real time’ study samples onuntreated DBS cards. Apparently, the observed instability ofOP in untreated 226 cards is due to enzyme activity dur-ing the drying period of the blood spot. Unfortunately, it is

A REPRESENTATIVE PROTOCOL-LC-MS BIOANALYSIS OF BMS-068645 AND ITS ACID METABOLITE IN HUMAN PLASMA 439
difficult to mix a small volume (10–40 μl) of fresh bloodsample with enzyme inhibitors and/or other stabilizers, suchas pH modifier, prior to blood spotting to prevent the enzy-matic, and/or chemical degradation.
Based on the aforementioned description, chemicallytreated cards appear to offer some stabilizing effect for theester prodrug. However, this effect is apparently compounddependent as the treated Whatman FTA Elute cards werereported not offering any better stability coverage for theinvestigated compounds than the untreated Whatman 903Protein Saver cards (D’Arienzo et al., 2010).
34.3.6 Representative Examples
Many research articles have been published on addressingthe stability issues of LC-MS bioanalysis of ester-based pro-drugs and other enzyme labile molecules. Some of the repre-sentative cases are captured in Table 34.3. These case studieshighlight both general and compound specific approaches.
34.4 SUMMARY
Stability is a fundamental parameter that must be assessedand controlled in quantitative LC-MS bioanalysis of ester-based prodrugs and other enzyme labile molecules. In gen-eral, the instability of an ester-based prodrug is readily pre-dictable because of the presence of enzyme sensitive esterbonds in the molecule. However, the instability of many otherenzyme labile analytes may not be readily predictable. Fur-thermore, the instability of enzyme labile molecules withinthe same chemo-type can vary significantly in the sameintended matrix and the instability of a given enzyme labilemolecule can differ from one matrix to the other. Here aresome general recommendations:
� Conduct necessary search for stability information ofanalyte of interest or its analogs. Assume analyte isunstable when its stability in the intended biologicalmatrix is unknown.
� Conduct proper stability assessment of the analyte ofinterest in fresh plasma and/or blood of the intendedspecies.
� If instability is identified, a simple adjustment of plasmapH using 0.5% or 1% acid (formic acid, phosphoric acidor citric acid) should be tested.
� Screen and optimize the use of enzyme inhibitor(s).� If neither pH control nor inhibitor(s) alone is deemed
effective, a combination of pH modifier(s) andinhibitor(s) together with other means (e.g., tempera-ture control) should be evaluated.
� Minimize duration of study sample exposure to ambienttemperature during collection, processing, and extrac-tion for LC-MS analysis.
� Avoid using aged plasma or blood for stability assess-ment and/or the preparation of standards and QCs. Pre-pare QC samples with prodrug alone and monitor thepossible change in the prodrug concentration and/or for-mation of the active drug molecule after freeze–thaw, atroom temperature and after long-term storage.
� Avoid extreme conditions (strong base, strong acid, andhigh temperature) during sample extraction and/or chro-matography.
� Ensure baseline separation of the analyte of interestfrom its metabolites and other potential interfering com-ponents.
� Include incurred sample reanalysis and/or incurred sam-ple short-term stability assessment in method devel-opment and validation. This approach can help unveilany “hidden” instability issues that, if left unaddressed,could eventually lead to the invalidation of an otherwise“validated” method.
34.5 A REPRESENTATIVE PROTOCOL-LC-MSBIOANALYSIS OF BMS-068645 AND ITS ACIDMETABOLITE IN HUMAN PLASMA
BackgroundBMS-068645 is a selective adenosine 2A agonist that con-tains a methyl ester group that undergoes esterase hydrolysisto its acid metabolite, BMS-068645-acid (Zeng et al. 2007).A method is needed for simultaneous determination of boththe parent (BMS-068645) and its active acid (BMS-068645-acid), respectively in dynamic range of 0.02–10 ng/ml and0.05–10 ng/ml in human plasma.
Hydrolysis
BMS-068645 BMS-068645-acid

TA
BL
E34
.3R
epre
sent
ativ
eA
pplic
atio
nsof
Stab
iliza
tion
ofE
ster
-bas
edP
rodr
ugs
and
Oth
erE
ster
ase
Sens
itiv
eM
olec
ules
inB
iolo
gica
lMat
rice
s
Com
poun
dSt
ruct
ure
Type
ofin
stab
ility
App
roac
hof
stab
iliza
tion
Bri
efst
abili
zatio
npr
oced
ure
Spec
imen
Ref
eren
ce
Bet
amet
haso
neac
etat
ean
dbe
tam
etha
-so
neph
osph
ate
Hyd
roly
sis
ofes
ter
Add
ition
ofin
hibi
tors
Blo
odsa
mpl
esw
ere
colle
cted
into
prec
hille
dpl
astic
hepa
rini
zed
tube
sco
ntai
ning
10μ
l2M
sodi
umar
sena
teso
lutio
npe
rm
lblo
od.T
heha
rves
ted
plas
ma
was
siph
oned
into
prec
hille
dpl
astic
tube
sco
ntai
ning
10μ
lof
50%
(w/v
)po
tass
ium
fluor
ide
solu
tion
per
ml
plas
ma.
Hum
anpl
asm
aSa
lem
etal
.(2
011)
O
OH
H
OO
OH
HF
O
O
OH
H
OO
P3N
a 2O
H
HF
Caf
feic
acid
phen
ethy
les
ter
and
itsflu
orin
ated
deri
vativ
e
Est
eras
e-in
duce
dde
grad
atio
nA
dditi
onof
NaF
and
pHad
just
men
tA
dditi
onof
0.4%
sodi
umflu
orid
ean
dpH
was
adju
sted
to6.
Rat
plas
ma
Wan
get
al.
(200
7)
HO
OH
O
O
HO
OH
O
O
F
Col
lect
ing
stud
ybl
ood
sam
ples
into
stab
ilize
rtr
eate
dtu
bes
100
μla
liquo
tof
then
oyltr
ifluo
roac
eton
eso
lutio
n(5
mg/
mli
nM
eOH
/AC
N,1
:1,v
/v)
and
100
μla
liquo
tof
citr
icac
idso
lutio
n(5
0m
g/m
lin
MeO
H)
wer
epl
aced
ina
poly
prop
ylen
etu
bean
dev
apor
ated
todr
ynes
sun
der
age
ntle
nitr
ogen
stre
amat
40◦ C
.The
stab
ilize
r-tr
eate
dtu
bes
wer
est
ored
at−2
0◦ Cbe
fore
use
for
colle
ctio
nof
400
μld
ogbl
ood.
The
harv
este
dpl
asm
asa
mpl
e(1
00μ
l)w
astr
eate
dw
ith30
0μ
lmet
hano
land
stor
edat
−80◦ C
prio
rto
furt
her
proc
essi
ng.
Dog
plas
ma
Tang
and
Sojin
u(2
012)
KR
-629
80E
ster
ase-
indu
ced
inst
abili
tySt
ore
sam
ples
atve
rylo
wte
mpe
ratu
re(−
80◦ C
)or
add
este
rase
inhi
bito
rs
Add
ition
ofN
aF(1
0m
g/m
l)or
PMSF
(0.1
8m
g/m
l)si
gnifi
cant
lyde
crea
sed
the
degr
adat
ion
rate
.
Rat
plas
ma
Kim
etal
.(2
011)
OO
N+
O–
ON
O
TP3
00E
nzym
ein
duce
dde
grad
atio
nA
cidi
ficat
ion
ofhu
man
plas
ma
topr
even
tdeg
rada
tion
ofth
epr
odru
gan
dco
nver
sion
ofth
eac
tive
drug
from
lact
one
toca
rbox
ylat
efo
rm
Imm
edia
tely
afte
rco
llect
ion,
the
bloo
dw
astr
ansf
erre
din
totu
bes
cont
aini
nghe
pari
nan
dci
tric
acid
(∼0.
5%,w
/v)
and
stor
edin
are
frig
erat
oror
onw
etic
eun
tilce
ntri
fuga
tion.
The
harv
este
dpl
asm
aw
asth
enm
ixed
with
1M
HC
lata
ratio
of10
:1(v
:v)
and
stor
edat
−70◦ C
until
anal
ysis
.
Hum
anpl
asm
aK
amei
etal
.(2
011)
N
NN
N
O
O
O
ON
NH
2
O
Opi
oid
pept
ide
H-T
yr-D
-Ala
-Gly
-Phe
-D-L
eu-O
H(D
AD
LE
)E
ster
ase-
indu
ced
inst
abili
tyA
dditi
onof
este
rase
inhi
bito
rT
hebi
olog
ical
mat
rix
was
prei
ncub
ated
with
para
oxon
(fina
lcon
cent
ratio
nof
1m
M)
for
15m
inat
37◦ C
befo
reus
e.
Hum
anbl
ood,
ratb
lood
,rat
tissu
e
Yan
get
al.
(200
2)
Naf
amos
tat
Enz
ymat
icor
none
nzym
atic
degr
adat
ion
Qui
ckpl
asm
apr
epar
atio
nIm
med
iate
sepa
ratio
nof
plas
ma
at4◦ C
and
stor
eth
epl
asm
asa
mpl
esat
alo
wte
mpe
ratu
re.
Hum
anpl
asm
aC
aoet
al.
(200
8)
NH
NH
2
O
NHN
H
NH
2
O
Ace
tylc
holin
eA
cety
lch
olin
este
rase
indu
ced
degr
adat
ion
Add
ition
ofin
hibi
tor
Tre
atsa
mpl
ew
ith25
mM
ofes
erin
eR
atC
SFZ
hang
etal
.(2
011)
O
O
N+
(con
tinu
ed)
TA
BL
E34
.3(C
ontin
ued)
Com
poun
dSt
ruct
ure
Type
ofin
stab
ility
App
roac
hof
stab
iliza
tion
Bri
efst
abili
zatio
npr
oced
ure
Spec
imen
Ref
eren
ce
Ose
ltam
ivir
Est
eras
eca
taly
zed
degr
adat
ion
Add
ition
ofin
hibi
tor
tosa
mpl
etu
bepr
ior
tosa
mpl
eco
llect
ion
Prio
rto
bloo
dsa
mpl
e(2
00μ
l)co
llect
ion,
toth
etu
bes
was
adde
d2.
5μ
lof
dich
lorv
osin
norm
alsa
line
solu
tion
(8m
g/m
l)to
afin
alco
ncen
trat
ion
of20
0μ
g/m
l
Rat
bloo
dC
hang
etal
.(2
009)
NH
O
O
NH
2
O
O
Col
lect
hum
anbl
ood
into
4m
lNaF
/Na 2
ED
TA(6
.0m
g)tu
bes
for
furt
her
proc
essi
ng
Hum
anpl
asm
aK
rom
dijk
etal
.(2
012)
Plas
ma
(5m
l)w
astr
ansf
erre
din
toa
tube
cont
aini
ngdi
chlo
rvos
at20
0μ
g/m
l(N
ote:
sign
ifica
ntin
ter-
indi
vidu
alva
riat
ion
was
obse
rved
for
the
sam
ples
colle
cted
with
outa
dditi
onof
the
inhi
bito
r)
Hum
anbl
ood/
plas
ma
Lin
dega
rdh
etal
.(2
006)
Rep
rodu
ced
from
Lie
tal.
(201
1)w
ithpe
rmis
sion
ofJo
hnW
iley
&So
nsL
td.

REFERENCES 443
Inhibitor ScreeningThe instability of BMS-068645 in human blood with or with-out inhibitors, urine, and methanol (control) was evaluated:
� Esterase inhibitors, diisopropyl fluorophosphate (DFP)and paraoxon in water, phenylmethylsulfonyl fluoride(PMSF) and eserine in DMSO, were added to freshhuman blood (heparin treated) to a final concentrationof 10 mM of each inhibitor.
� BMS-068645 was added to (1) blood only, (2) bloodwith 10 mM inhibitors, (3) human urine, and (4)methanol to a final concentration of 25.0 ng/ml. Theblood and methanol samples were maintained for 4 hat room temperature and the urine samples were main-tained at room temperature for 96 h.
� Two milliliters of acetonitrile containing internal stan-dard were added to completely quench any esteraseactivity ending the incubation.
� The samples were vortex mixed, centrifuged, and thesupernatant was removed and dried under nitrogen at37◦C. The samples were reconstituted with 0.1% formicacid in water and analyzed by LC-MS.
Clinical Study Sample Collection ProcedureBased on the aforementioned preliminary assessment, DFPwas chosen as the stabilizer for the compound in humanplasma. A detailed clinical sample collection procedure wasgenerated as below. This was followed by (1) stability assess-ment of the ester prodrug and the active drug in both plasmaand whole blood treated as below and (2) stability assessmentof the inhibitor in the blood collection tubes:
� The plastic vacutainer tubes (6 ml, K2EDTA) weretreated by the injection of 21 μl of DFP with asyringe with a sharp, thin needle (needle gauge around22–26).
� After the addition of the inhibitor, the tubes were storedat −30◦C and only warmed to room temperature lessthan half an hour prior to use.
� Blood samples (approximately 6 ml per sample) werecollected in the tubes resulting in a final DFP concen-tration in blood of 20 mM.
� Immediately after collection, each blood sample wasgently inverted several times to ensure complete mixingwith the anticoagulant (K2EDTA) and DFP and thenplaced in chipped ice.
� The blood samples were centrifuged for 15 min at 1000× g at 4◦C to obtain plasma.
� The separated plasma samples were stored at −30◦Cuntil analyzed.
Preparation of Cs/QCs in the Human Plasma
� A 40 μl portion of the 1000 ng/ml stock solution wasdiluted to 4.0 ml with control human K2EDTA plasmacontaining 20 mM DFP to yield a combined stock solu-tion of 10.0 ng/ml for each analyte.
� The plasma pool was diluted with plasma containing 20mM DFP to obtain the appropriate final concentrationsfor both the standards and QC samples.
� Calibration standards were freshly prepared daily.� Additional QC samples with the ester prodrug alone
were prepared for monitoring the extent of the possibleconversion to the active drug.
Sample Processing Procedure for Human Plasma
� Isotope-labeled internal standards, D5-BMS-068645and D5-BMS-068645-acid, respectively at 1.0 and 5.0ng/ml in methanol/water (50/50, v/v) were employed.
� After the addition of 100 μl of the IS working solutionand 0.5 ml of 0.1N HCl solution, 4 ml of methyl-tert-butyl ether were added to 0.5 ml of each calibrationstandard, QC sample, and clinical sample.
� The samples were shaken for 20 min, and then cen-trifuged to separate the liquid phases.
� The organic layer from each sample was transferred toa clean tube and evaporated to dryness.
� The dried extracts were redissolved in 100 μl of thereconstitution solution containing 0.1% formic acid inacetonitrile/water (40/60, v/v) and transferred to injec-tion vials.
� A 10 μl aliquot of the reconstituted samples was injectedinto the LC-MS system.
REFERENCES
Aldrige WN. Serum esterases. Biochem J 1952;53:110–117.
Bergeron M, Bergeron A, Furtado M, Garofolo F. Impact ofplasma and whole-blood anticoagulant counter ion choice ondrug stability and matrix effects during bioanalysis. Bioanalysis2009;1(3):537–548.
Bergmann F, Segal R, Rimon S. A new type of esterase in hog-kidney extract. Biochem J 1957;67(3):481–486.
Briscoe CJ, Hage DS. Factors affecting the stability of drugsand drug metabolites in biological matrices. Bioanalysis2009;1(1):205–220.
Cao YG, Zhang M, Yu D, Shao JP, Chen YC, Liu XQ. A method forquantifying the unstable and highly polar drug nafamostat mesi-late in human plasma with optimized solid-phase extraction andESI-MS detection: more accurate evaluation for pharmacoki-netic study. Anal Bioanal Chem 2008;391(3):1063–1071.

444 LC-MS BIOANALYSIS OF ESTER PRODRUGS AND OTHER ESTERASE LABILE MOLECULES
Chang Q, Chow MS, Zuo Z. Studies on the influence of esteraseinhibitor to the pharmacokinetic profiles of oseltamivir andoseltamivir carboxylate in rats using an improved LC/MS/MSmethod. Biomed Chromatogr 2009;23(8):852–857.
Chen J, Hsieh Y. Stabilizing drug molecules in biological samples.Ther Drug Monit 2005;27(5):617–624.
D’Arienzo CJ, Ji QC, Discenza L, et al. DBS sampling can be usedto stabilize prodrugs in drug discovery rodent studies withoutthe addition of esterase inhibitors. Bioanalysis 2010;2(8):1415–1422.
Evans MJ, Livesey JH, Ellis MJ, Yandle TG. Effect of anticoagu-lants and storage temperatures on stability of plasma and serumhormones. Clin Biochem 2001;34(2):107–112.
Freisleben A, Brudny-Kloppel M, Mulder H, de Vries R, de ZwartM, Timmerman P. Blood stability testing: European bioanaly-sis forum view on current challenges for regulated bioanalysis.Bioanalysis 2011;3(12):1333–1336.
Fung EN, Zheng N, Arnold ME, Zeng J. Effective screen-ing approach to select esterase inhibitors used for stabilizingester-containing prodrugs analyzed by LC-MS/MS. Bioanalysis2010;2(4):733–743.
Heinig K, Wirz T, Bucheli F, Gajate-Perez A. Determination ofoseltamivir (Tamiflu R©) and oseltamivir carboxylate in driedblood spots using offline or online extraction. Bioanalysis2011;3(4):421–437.
Ishizuka T, Fujimori I, Kato M, et al. Human car-boxymethylenebutenolidase as a bioactivating hydrolase ofolmesartan medoxomil in liver and intestine. J Biol Chem2010;285(16):11892–11902.
Kamei T, Uchimura T, Nishimiya K, Kawanishi T. Method develop-ment and validation of the simultaneous determination of a noveltopoisomerase 1 inhibitor, the prodrug, and the active metabolitein human plasma using column-switching LC-MS/MS, and itsapplication in a clinical trial. J Chromatogr B Analyt TechnolBiomed Life Sci 2011;879(30):3415–3422.
Kim MS, Song JS, Roh H, et al. Determination of a perox-isome proliferator-activated receptor γ agonist, 1-(trans-methylimino-N-oxy)-6-(2-morpholinoethoxy-3-phenyl-1H-indene-2-carboxylic acid ethyl ester (KR-62980) in rat plasmaby liquid chromatography-tandem mass spectrometry. J PharmBiomed Anal 2011;54(1):121–126.
Koitka M, Hochel J, Gieschen H, Borchert HH. Improving the exvivo stability of drug ester compounds in rat and dog serum:inhibition of the specific esterases and implications on theiridentity. J Pharm Biomed Anal 2010;51(3):664–678.
Kromdijk W, Rosing H, van den Broek MP, Beijnen JH, HuitemaAD. Quantitative determination of oseltamivir and oseltamivircarboxylate in human fluoride EDTA plasma including the exvivo stability using high-performance liquid chromatographycoupled with electrospray ionization tandem mass spectrometry.J Chromatogr B Analyt Technol Biomed Life Sci 2012;891-892:57–63.
Kudo S, Umehara K, Hosokawa M, Miyamoto G, Chiba K, SatouhT. Phenacetin deacetylase activity in human liver microsomes:distribution, kinetics, and chemical inhibition and stimulation. JPharmacol Exp Ther 2000;294(1):80–87.
Li W, Tse FL. Dried blood spot sampling in combination with LC-MS/MS for quantitative analysis of small molecules. BiomedChromatogr 2010;24(1):49–65.
Li W, Zhang J, Tse FL. Strategies in quantitative LC-MS/MS anal-ysis of unstable small molecules in biological matrices. BiomedChromatogr 2011;25(1-2):258–277.
Liederer BM, Borchardt RT. Stability of oxymethyl-modifiedcoumarinic acid cyclic prodrugs of diasteromeric opioid pep-tides in biological media from various animal species includinghuman. J Pharm Sci 2005;94(10):2198–2206.
Liederer BM, Borchardt RT. Enzymes involved in the bioconver-sion of ester-based prodrugs. J Pharm Sci 2006;95(6):1177–1195.
Lindegardh N, Davies GR, Tran TH, et al. Rapid degradation ofoseltamivir phosphate in clinical samples by plasma esterases.Antimicrob Agents Chemother 2006;50(9):3197–3199.
Meng M, Reuschel S, Bennett P. Identifying trends and developingsolutions for incurred sample reanalysis failure investigations ina bioanalytical CRO. Bioanalysis 2011;3(4):449–465.
Minagawa T, Kohno Y, Suwa T, Tsuji A. Species differences inhydrolysis of isocarbacyclin methyl ester (TEI-9090) by bloodesterases. Biochem Pharmacol 1995;49(10):1361–1365.
Olivera-Bravo S, Ivorra I, Morales A. The acetylcholinesteraseinhibitor BW284c51 is a potent blocker of Torpedo nicotinicAchRs incorporated into the Xenopus oocyte membrane. Br JPharmacol 2005;144(1):88–97.
Peterson LW, McKenna CE. Prodrug approaches to improving theoral absorption of antiviral nucleotide analogues. Expert OpinDrug Deliv 2009;6(4):405–420.
Rautio J, Kumpulainen H, Heimbach T, et al. Prodrugs: designand clinical applications. Nat Rev Drug Discov 2008;7(3):255–270.
Rooseboom M, Commandeur JN, Vermeulen NP. Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol Rev2004;56(1):53–102.
Salem II, Alkhatib M, Najib N. LC–MS/MS determination ofbetamethasone and its phosphate and acetate esters in humanplasma after sample stabilization. J Pharm Biomed Anal2011;56(5):983–991.
Skopp G, Klingmann A, Potsch L, Mattern R. In vitro stabilityof cocaine in whole blood and plasma including ecgonine as atarget analyte. Ther Drug Monit 2001;23(2):174–181.
Stella VJ, Charman WN, Naringrekar VH. Prodrugs: Do they haveadvantages in clinical practice? Drugs 1985;29(5):455–473.
Tang C, Sojinu OS. Simultaneous determination of caffeic acidphenethyl ester and its metabolite caffeic acid in dog plasmausing liquid chromatography tandem mass spectrometry. Talanta2012;94:232–239.
Tokumura T, Muraoka A, Masutomi T, Machida Y. Stability ofspironolactone in rat plasma: strict temperature control of bloodand plasma samples is required in rat pharmacokinetic studies.Biol Pharm Bull 2005;28(6):1126–1128.
Tsujikawa K, Kuwayama K, Miyaguchi H, Kanamori T, Iwata YT,Inoue H. In vitro stability and metabolism of salvinorin A in ratplasma. Xenobiotica 2009;39(5):391–398.

REFERENCES 445
Wang X, Bowman PD, Kerwin SM, Stavchansky S. Stability ofcaffeic acid phenethyl ester and its fluorinated derivative in ratplasma. Biomed Chromatogr 2007;21(4):343–350.
Welch RM, Brown A, Ravitch J, Dahl R. The in vitro degradationof cisatracurium, the R, cis-R′-isomer of atracurium, in humanand rat plasma. Clin Pharmacol Ther 1995;58(2):132–142.
Wiltshire H, Wiltshire B, Citron A, et al. Development of a high-performance liquid chromatographic-mass spectrometric assayfor the specific and sensitive quantification of Ro 64-0802, ananti-influenza drug, and its pro-drug, oseltamivir, in human andanimal plasma and urine. J Chromatogr B Biomed Sci Appl2000;745(2):373–388.
Yamaori S, Fujiyama N, Kushihara M, et al. Involvementof human blood arylesterase and liver microsomal car-boxylesterase in nafamostat hydrolysis. Drug Metab Pharma-cokinet 2006;21(2):147–155.
Yang JZ, Chen W, Borchardt RT. In vitro stability and in vivopharmacokinetic studies of a model opioid peptide, H-Tyr-D-Ala-Gly-Phe-D-Leu-OH (DADLE), and its cyclic prodrugs. JPharmacol Exp Ther 2002;303(2):840–848.
Yoshigae Y, Imai T, Taketani M, Otagiri M. Characterizationof esterases involved in the steroselective hydrolysis of ester-type prodrugs of propranolol in rat liver and plasma. Chirality1999;11:10–13.
Zeng J, Onthank D, Crane P, et al. Simultaneous determinationof a selective adenosine 2A agonist, BMS-068645, and its acidmetabolite in human plasma by liquid chromatography-tandemmass spectrometry—evaluation of the esterase inhibitor, diiso-propyl fluorophosphate, in the stabilization of a labile ester-containing drug. J Chromatogr B Analyt Technol Biomed LifeSci 2007;852(1–2):77–84.
Zhang JG, Fariss MW. Thenoyltrifluoroacetone, a potent inhibitorof carboxylesterase activity. Biochem Pharmacol 2002;63:751–754.
Zhang Y, Tingley FD 3rd, Tseng E, et al. Development andvalidation of a sample stabilization strategy and a UPLC-MS/MS method for the simultaneous quantitation of acetyl-choline (ACh), histamine (HA), and its metabolites in rat cere-brospinal fluid (CSF). J Chromatogr B Analyt Technol BiomedLife Sci 2011;879(22):2023–2033.





![Part I Introduction to MS in bioanalysis - Wiley-VCH · before the direct interfacing of liquid chromatography with mass spectrometry (LC-MS) was described by Arpino et al. [5]. With](https://img.pdfslide.us/doc/110x75/5ecf35a64ccdb1799a4b6a04/part-i-introduction-to-ms-in-bioanalysis-wiley-vch-before-the-direct-interfacing.jpg)























