Embed Size (px)
Citation preview

Guide to Master Formulae
WHO/FWC/IVB/QSS/VQR
2011

Guide to Master Formulae Guidance Document
1
This guidance document GUIDE TO MASTER FORMULAE is one of a series
developed by WHO/FWC/IVB Quality, Safety & Standards team upon request from the
manufacturers’ members of the Developing Countries Vaccine Manufacturers Network
(DCVMN), with funds of USAID.
A set of priority topics have been identified by the manufacturers for WHO to provide
guidance on expectations from the vaccine prequalification programme.
The guidance document GUIDE TO MASTER FORMULAE is targeted primarily at
manufacturers who are new to the prequalification of vaccines and who require detailed
guidance about the level of detail needed for the development of batch production
records. It may also be a useful guide to National Regulatory Authorities (NRAs) in
vaccine producing countries.
These are not official WHO documents but rather notes for guidance on expected
standards to be followed for the prequalification of vaccines. They are based on WHO
recommended requirements but providing further explanations with examples on how
these can be actually implemented.

Guide to Master Formulae Guidance Document
2
GUIDE TO MASTER FORMULAE
Table of Contents:
Page
1) Introduction 4
2) Terms for Master Formula (MF) 4
3) Definitions of Batch / Lot 5
4) Master Formulae needed 5
5) GMP guidelines on master documentation 5
6) Required Contents of a MF 6
7) MF and corresponding Batch Records 11
8) Formats for MF 11
9) Issuing of MF copy as a blank batch record 12
10) Electronic MF and Batch Records 13
11) Batch Records versus Master Formula 14
12) Batch record review checklist 14
Appendix 1: Extract from: World Health Organization, Technical Report
Series, No. 908, 2003; Annex 4: Good Manufacturing Practices for pharmaceutical
products: main principles.
Appendix 2: Extract from: EUDRALEX; Volume 4 - Medicinal Products for
Human and Veterinary Use: Good Manufacturing Practice: Chapter 4
Documentation.
Appendix 3: Extract from: Pharmaceutical Inspection Convention Co-operation
Scheme PE 009-3, 1 January 2006; Guide to Good Manufacturing Practice for
Medicinal Products; Documentation.
Appendix 4: Extract from Canadian GMP Guidelines, Health Canada, Health
Products and Food Branch Inspectorate. Good Manufacturing Practices Guidelines,
2002 Edition, Version 2.

Guide to Master Formulae Guidance Document
3
Appendix 5: Extracts from US Code of Federal Regulations (CFR) and US FDA
Guidelines.
App 5-1) US Regulations for Master Production Records for Finished
Pharmaceuticals. Extract from: CFR 21, Chapter I, Subchapter F: Biologics, Part 211 Current Good Manufacturing
Practice for Finished Pharmaceuticals; Subpart F--Production and Process Controls,
Sec. 211.100 Written procedures; deviations; and Subpart J--Records and Reports;
Sec. 211.186 Master production and control records.
App 5-2) US Regulations for Batch Records for Finished Pharmaceuticals:
Extract from: CFR 21, Chapter I, Subchapter F: Biologics; Subchapter C: Drugs General; Part 211
Current Good Manufacturing Practice for Finished Pharmaceuticals; Subpart J--
Records and Reports; Sec. 211.188 Batch production and control records.
App 5-3) US Regulations for Batch Records for Biological Products: Extract
from: CFR 21, Chapter I, Subchapter F: Biologics, Part 600 Biological Products: General;
subpart B Establishment Standards, Sec 600.12 Records
App 5-4) US FDA Guidelines for Batch Records for Sterile Products: Extract
from: Guidance for Industry. Sterile Drug Products Produced by Aseptic Processing —
Current Good Manufacturing Practice. U.S. Department of Health and Human
Services, Food and Drug Administration, Center for Drug Evaluation and Research
(CDER); Center for Biologics Evaluation and Research (CBER); Office of
Regulatory Affairs (ORA). September 2004 (Pharmaceutical cGMPs).
Appendix 6: Sample master formula for a hypothetical biological product
Appendix 7: Example one of a Master Formula
Appendix 8: Example two of a Master Formula
Appendix 9: Example three of a Master Formula

Guide to Master Formulae Guidance Document
4
1) Introduction
In the 1997 WHO guidance document: “WHO/VSQ/97.01: (A WHO guide to good
manufacturing practice (GMP) requirements. Part 1: Standard operating procedures and
master formulae)” some basic explanations and instructions were given for preparing
various documents required by Good Manufacturing Practice guidelines from WHO and
from several regulatory authorities.
GMP guidelines include the requirements for documents (individual), documentation (the
systems and formats for documents), and documenting (recording) of production and
control activities. Most GMP guidelines provide the same or very similar information as
the principles of Good Manufacturing Practice are now international in scope.
In this guidance document, the requirement for master manufacturing instructions and the
requirements as given in different GMP documents, different names for these documents
and various forms that they can take will be described. This is to guide vaccine
manufacturers who are applying for prequalification or re-qualification of their product(s)
in the preparation or improvement of current documents for manufacturing operations.
2) Terms for Master Formula (MF)
WHO identifies manufacturing instructions as “Master Formula. Other terms used in
GMP guidelines and regulations are “Manufacturing Formula”, “Master Production and
Control Record”, but all mean the same thing – an approved master document that
describes the full process of manufacturing for the batch of product with at least cross-
reference to the support operations for a batch of a specific product. Individual
companies may give internal names to these documents (manufacturing instructions,
monographs, etc). In this guidance document the WHO term Master Formula (or MF)
will be used.
The following are the extracted definitions from several guidelines:
• WHO GMP Guidelines: A formally authorized master formula should exist for
each product and batch size to be manufactured.
• EU and PIC GMP guidelines: “Formally authorised Manufacturing Formula
and Processing Instructions should exist for each product and batch size to be
manufactured. They are often combined in one document.”
• Health Canada GMP guidelines. MASTER FORMULA (formule-type) - A
document or set of documents specifying the raw materials with their quantities
and the packaging materials, together with a detailed description of the procedures
and precautions required to produce a specified quantity of a finished product as
well as the processing instructions, including the in-process controls.
• US CFR. To assure uniformity from batch to batch, master production and
control records for each drug product, including each batch size thereof, shall be
prepared, dated, and signed (full signature, handwritten) by one person and
independently checked, dated, and signed by a second person. The preparation of

Guide to Master Formulae Guidance Document
5
master production and control records shall be described in a written procedure
and such written procedure shall be followed.
3) Definitions of Batch / Lot:
A Master Formula is required for each batch and batch size. A “batch” or “lot” as defined
in the WHO GMP guideline (TRS 908 Annex 4) is”
“batch (or lot)
A defined quantity of starting material, packaging material, or product
processed in a single process or series of processes so that it is
expected to be homogeneous. It may sometimes be necessary to
divide a batch into a number of sub-batches, which are later brought
together to form a final homogeneous batch. In the case of terminal
sterilization, the batch size is determined by the capacity of the autoclave.
In continuous manufacture, the batch must correspond to a defined
fraction of the production, characterized by its intended homogeneity.
The batch size can be defined either as a fixed quantity or as the amount
produced in a fixed time interval”.
In general, the term “batch” more often refers to intermediates or final formulated bulks
which are in one or a few large containers, while “lot” usually refers to the final product
in the final container. They are, however, interchangeable as indicated in WHO’s GMP
guideline glossary.
4) Master Formulae needed:
As above, batch or lot will refer to all production intermediates, final formulated bulks
and final vialed product. Each master cell bank, viral seed lot, bulk concentrate or viral
harvest if stored and tested before release for further processing is a batch and a master
formula for its production is written and approved. Also, for different scales of
production of any batch or lot, a distinct master formula is prepared.
For final container product, as explained in the WHO definition above, a final “lot” will
be the product that is filled during the same continuous fill-run, and in the case of freeze-
dried products, the filled vials lyophilized in the same lyophilizer at the same time. These
should have unique numbers to identify them as having been processed exactly the same
way at the same time. On occasion, when only a part of a large final bulk is filled, the lot
numbers for these bulks may have a common identifier with a suffix (“-1” or “a”) to
show the separate fills. Similarly, a large fill lot with a unique lot number may be
lyophilized in different lyophilizers and the suffix would indicate the different freeze-
dryer. A master formula for a batch/lot of product with the possibility to select one of
several approved equipment items (e.g. a freeze dryer) should clearly indicate this choice
and provide space for the unique lot number designation.

Guide to Master Formulae Guidance Document
6
5) GMP guidelines on master documentation
To show the consistency of requirements for Master Formulae, the sections on master
documents and batch records have been extracted from various guidelines/regulations.
The extracted texts are provided in Appendices 1-5:
1) WHO:
World Health Organization Technical Report Series, No. 908, 2003, Annex 4.
Good Manufacturing Practices for Pharmaceutical Products: Main Principles
2) EU:
EUDRALEX; Volume 4 - Medicinal Products for Human and Veterinary Use:
Good Manufacturing Practice: Chapter 4: Documentation.
3) Pharmaceutical Inspection Convention (PIC):
Pharmaceutical Inspection Co-operation Scheme PE 009-3, 1 January 2006:
Guide to Good Manufacturing Practice for Medicinal Products. (© PIC/S January
2006)
4) Canada:
Health Canada, Health Products and Food Branch Inspectorate. Good
Manufacturing Practices Guidelines, 2002 Edition (Version 2).
5) USA:
US Code of Federal Regulations: Chapter 21, subparts 211 and 600.
And
US FDA: Guidance for Industry: Sterile Drug Products Produced by Aseptic
Processing — Current Good Manufacturing Practice. U.S. Department of Health
and Human Services, Food and Drug Administration, Center for Drug Evaluation
and Research (CDER), Center for Biologics Evaluation and Research (CBER),
Office of Regulatory Affairs (ORA), September 2004. Pharmaceutical cGMPs.
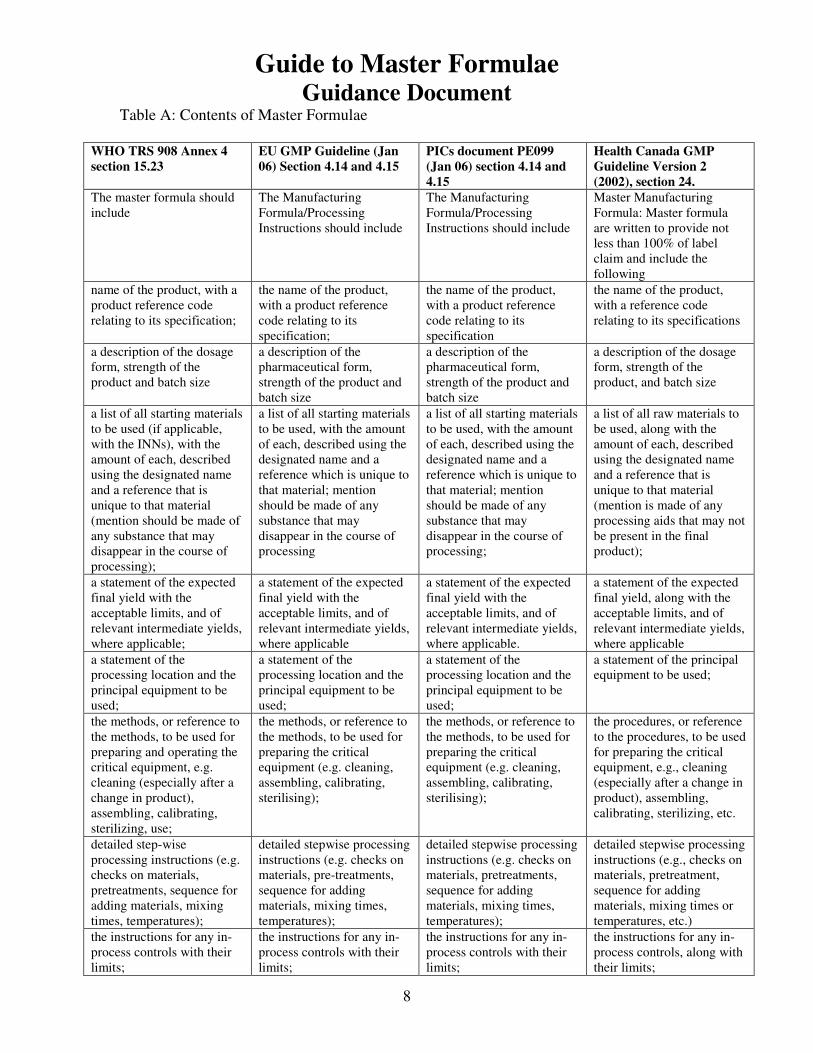
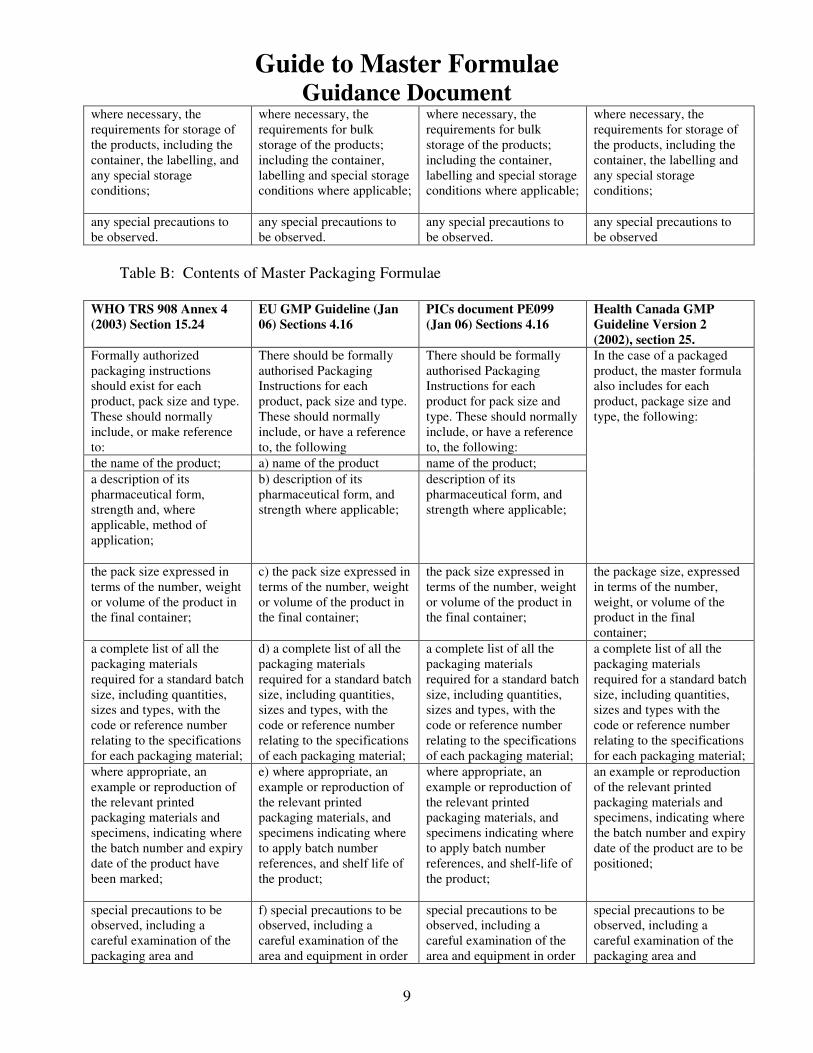
6) Required Contents of a MF
Each of the regulation or guidelines above gives a list of the requirements for the contents
of the MF. These are given in Table A for the Master (Production) Formula and Table B
for the Master Packaging Formula for WHO, EU, PICs and Health Canada. Table C gives
the contents Master Production and Control Records required by the USA.

Guide to Master Formulae Guidance Document
7
From Table A and B it is clear that the guidelines are harmonized and the requirements
are formatted the same way and with the same or very similar text. The USA regulations
cover the same information but in a different format and do not distinguish between
production and packaging master formulae.
Guide to Master Formulae Guidance Document
8
Table A: Contents of Master Formulae
WHO TRS 908 Annex 4
section 15.23
EU GMP Guideline (Jan
06) Section 4.14 and 4.15
PICs document PE099
(Jan 06) section 4.14 and
4.15
Health Canada GMP
Guideline Version 2
(2002), section 24.
The master formula should
include
The Manufacturing
Formula/Processing
Instructions should include
The Manufacturing
Formula/Processing
Instructions should include
Master Manufacturing
Formula: Master formula
are written to provide not
less than 100% of label
claim and include the
following
name of the product, with a
product reference code
relating to its specification;
the name of the product,
with a product reference
code relating to its
specification;
the name of the product,
with a product reference
code relating to its
specification
the name of the product,
with a reference code
relating to its specifications
a description of the dosage
form, strength of the
product and batch size
a description of the
pharmaceutical form,
strength of the product and
batch size
a description of the
pharmaceutical form,
strength of the product and
batch size
a description of the dosage
form, strength of the
product, and batch size
a list of all starting materials
to be used (if applicable,
with the INNs), with the
amount of each, described
using the designated name
and a reference that is
unique to that material
(mention should be made of
any substance that may
disappear in the course of
processing);
a list of all starting materials
to be used, with the amount
of each, described using the
designated name and a
reference which is unique to
that material; mention
should be made of any
substance that may
disappear in the course of
processing
a list of all starting materials
to be used, with the amount
of each, described using the
designated name and a
reference which is unique to
that material; mention
should be made of any
substance that may
disappear in the course of
processing;
a list of all raw materials to
be used, along with the
amount of each, described
using the designated name
and a reference that is
unique to that material
(mention is made of any
processing aids that may not
be present in the final
product);
a statement of the expected
final yield with the
acceptable limits, and of
relevant intermediate yields,
where applicable;
a statement of the expected
final yield with the
acceptable limits, and of
relevant intermediate yields,
where applicable
a statement of the expected
final yield with the
acceptable limits, and of
relevant intermediate yields,
where applicable.
a statement of the expected
final yield, along with the
acceptable limits, and of
relevant intermediate yields,
where applicable
a statement of the
processing location and the
principal equipment to be
used;
a statement of the
processing location and the
principal equipment to be
used;
a statement of the
processing location and the
principal equipment to be
used;
a statement of the principal
equipment to be used;
the methods, or reference to
the methods, to be used for
preparing and operating the
critical equipment, e.g.
cleaning (especially after a
change in product),
assembling, calibrating,
sterilizing, use;
the methods, or reference to
the methods, to be used for
preparing the critical
equipment (e.g. cleaning,
assembling, calibrating,
sterilising);
the methods, or reference to
the methods, to be used for
preparing the critical
equipment (e.g. cleaning,
assembling, calibrating,
sterilising);
the procedures, or reference
to the procedures, to be used
for preparing the critical
equipment, e.g., cleaning
(especially after a change in
product), assembling,
calibrating, sterilizing, etc.
detailed step-wise
processing instructions (e.g.
checks on materials,
pretreatments, sequence for
adding materials, mixing
times, temperatures);
detailed stepwise processing
instructions (e.g. checks on
materials, pre-treatments,
sequence for adding
materials, mixing times,
temperatures);
detailed stepwise processing
instructions (e.g. checks on
materials, pretreatments,
sequence for adding
materials, mixing times,
temperatures);
detailed stepwise processing
instructions (e.g., checks on
materials, pretreatment,
sequence for adding
materials, mixing times or
temperatures, etc.)
the instructions for any in-
process controls with their
limits;
the instructions for any in-
process controls with their
limits;
the instructions for any in-
process controls with their
limits;
the instructions for any in-
process controls, along with
their limits;
Guide to Master Formulae Guidance Document
9
where necessary, the
requirements for storage of
the products, including the
container, the labelling, and
any special storage
conditions;
where necessary, the
requirements for bulk
storage of the products;
including the container,
labelling and special storage
conditions where applicable;
where necessary, the
requirements for bulk
storage of the products;
including the container,
labelling and special storage
conditions where applicable;
where necessary, the
requirements for storage of
the products, including the
container, the labelling and
any special storage
conditions;
any special precautions to
be observed.
any special precautions to
be observed.
any special precautions to
be observed.
any special precautions to
be observed
Table B: Contents of Master Packaging Formulae
WHO TRS 908 Annex 4
(2003) Section 15.24
EU GMP Guideline (Jan
06) Sections 4.16
PICs document PE099
(Jan 06) Sections 4.16
Health Canada GMP
Guideline Version 2
(2002), section 25.
Formally authorized
packaging instructions
should exist for each
product, pack size and type.
These should normally
include, or make reference
to:
There should be formally
authorised Packaging
Instructions for each
product, pack size and type.
These should normally
include, or have a reference
to, the following
There should be formally
authorised Packaging
Instructions for each
product for pack size and
type. These should normally
include, or have a reference
to, the following:
In the case of a packaged
product, the master formula
also includes for each
product, package size and
type, the following:
the name of the product; a) name of the product name of the product;
a description of its
pharmaceutical form,
strength and, where
applicable, method of
application;
b) description of its
pharmaceutical form, and
strength where applicable;
description of its
pharmaceutical form, and
strength where applicable;
the pack size expressed in
terms of the number, weight
or volume of the product in
the final container;
c) the pack size expressed in
terms of the number, weight
or volume of the product in
the final container;
the pack size expressed in
terms of the number, weight
or volume of the product in
the final container;
the package size, expressed
in terms of the number,
weight, or volume of the
product in the final
container;
a complete list of all the
packaging materials
required for a standard batch
size, including quantities,
sizes and types, with the
code or reference number
relating to the specifications
for each packaging material;
d) a complete list of all the
packaging materials
required for a standard batch
size, including quantities,
sizes and types, with the
code or reference number
relating to the specifications
of each packaging material;
a complete list of all the
packaging materials
required for a standard batch
size, including quantities,
sizes and types, with the
code or reference number
relating to the specifications
of each packaging material;
a complete list of all the
packaging materials
required for a standard batch
size, including quantities,
sizes and types with the
code or reference number
relating to the specifications
for each packaging material;
where appropriate, an
example or reproduction of
the relevant printed
packaging materials and
specimens, indicating where
the batch number and expiry
date of the product have
been marked;
e) where appropriate, an
example or reproduction of
the relevant printed
packaging materials, and
specimens indicating where
to apply batch number
references, and shelf life of
the product;
where appropriate, an
example or reproduction of
the relevant printed
packaging materials, and
specimens indicating where
to apply batch number
references, and shelf-life of
the product;
an example or reproduction
of the relevant printed
packaging materials and
specimens, indicating where
the batch number and expiry
date of the product are to be
positioned;
special precautions to be
observed, including a
careful examination of the
packaging area and
f) special precautions to be
observed, including a
careful examination of the
area and equipment in order
special precautions to be
observed, including a
careful examination of the
area and equipment in order
special precautions to be
observed, including a
careful examination of the
packaging area and
Guide to Master Formulae Guidance Document
10
equipment in order to
ascertain the line clearance
before and after packaging
operations;
to ascertain the line
clearance before operations
begin;
to ascertain the line
clearance before operations
begin
equipment in order to
ascertain the line clearance
before operations begin;
a description of the
packaging operation,
including any significant
subsidiary operations, and
equipment to be used;
g) a description of the
packaging operation,
including any significant
subsidiary operations, and
equipment to be used;
a description of the
packaging operation,
including any significant
subsidiary operations, and
equipment to be used;
a description of the
packaging operations,
including any significant
subsidiary operations and
the equipment to be used
details of in-process
controls with instructions
for sampling and acceptance
limits.
h) details of in-process
controls with instructions
for sampling and acceptance
limits
details of in-process
controls with instructions
for sampling and acceptance
limits
details of in-process
controls, with instructions
for sampling and acceptance
limits.
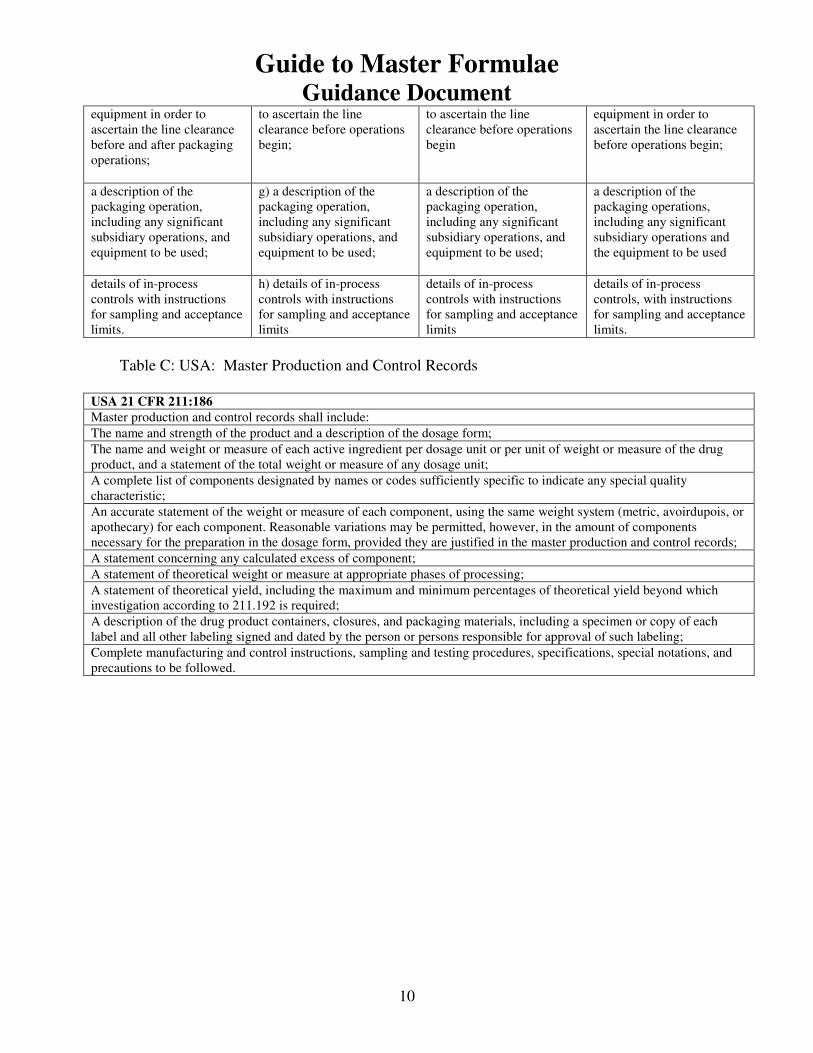
Table C: USA: Master Production and Control Records
USA 21 CFR 211:186
Master production and control records shall include:
The name and strength of the product and a description of the dosage form;
The name and weight or measure of each active ingredient per dosage unit or per unit of weight or measure of the drug
product, and a statement of the total weight or measure of any dosage unit;
A complete list of components designated by names or codes sufficiently specific to indicate any special quality
characteristic;
An accurate statement of the weight or measure of each component, using the same weight system (metric, avoirdupois, or
apothecary) for each component. Reasonable variations may be permitted, however, in the amount of components
necessary for the preparation in the dosage form, provided they are justified in the master production and control records;
A statement concerning any calculated excess of component;
A statement of theoretical weight or measure at appropriate phases of processing;
A statement of theoretical yield, including the maximum and minimum percentages of theoretical yield beyond which
investigation according to 211.192 is required;
A description of the drug product containers, closures, and packaging materials, including a specimen or copy of each
label and all other labeling signed and dated by the person or persons responsible for approval of such labeling;
Complete manufacturing and control instructions, sampling and testing procedures, specifications, special notations, and
precautions to be followed.

Guide to Master Formulae Guidance Document
11
7) MF and corresponding Batch Records
Master Formula give the complete production instructions for a specific batch and batch
size of cell banks, virus seed lots, intermediates, final bulks, final formulated bulks or
final container product that are made in one production run with definite start to finish
steps. Blank spaces are provided for the entry of data as the production run progresses.
Identification or cross-reference to required supporting data is included in the step-by-
step instructions.
The batch production record (BPR) is the approved copy of the master document with
filled in data entries, signatures, dates, production locations, operators, and lot number,
records of all supporting data (autoclave records, cleaning records, equipment
identification and calibration dates, in-process test results, and QC results) appended. For
cell banks, intermediates and final bulks that are stored for significant periods, the BPR is
for that product. Once a final product has been produced, the batch record (BR) is
comprised of a single document of the sequential batch records for the starting cell banks
or virus seed lots, the intermediates, the final bulks, final formulated bulks and the final
container with all the supporting documents. If the product is a pool of several
intermediates or final bulks then the full batch record includes the individual batch
records of all the components. For combined vaccines, the complete batch record of the
final product is the composite of the batch records of the complete batch record of the
final bulks of each component, the final formulated bulk and the final product, again
including all the supporting data.
8) Formats for MF
The generally recommended MF format is to prepare a single continuous document that
provides step-by-step production instructions, raw materials, equipment used, locations of
production, dates, operators, etc for the product, with blank spaces to record the data and
sign and date all entries, and at least cross-references to all supporting SOPs and
operations. (See Tables A, B and C above).
An example of a Master Formula for a hypothetical biological product was provided in
the WHO Guidance document: WHO/VSQ/97.01: A WHO guide to good manufacturing
practice (GMP) requirements. Part 1: Standard operating procedures and master
formulae, Appendix 6: Sample master formula for a hypothetical biological product. A
copy of this is reproduced in Appendix 6 in this document. Many other formats are
possible for a MF and will depend on the production process and supporting activities, as
well as on the documentation system in place at the manufacturing company. Appendix
7, 8 and 9 provide examples of actual master formulae from several manufacturers with
details revised to protect confidential information.
For individual batches/lots of intermediates and bulks the MF is a complete document.
However, for the final container product, the full MF is the total of MF of each step.

Guide to Master Formulae Guidance Document
12
Therefore, for any final vaccine, a Master Formula Summary List is recommended. This
would be a listing of each MF document number and title for each batch size of final
product and would also include the options for any intermediates that are produced in
different batch sizes and for the individual bulk components of a combined antigen
vaccine.
Such a Master Formula Summary List would include as applicable:
MFs for:
• Master Cell Bank
• Working Cell Bank
• Fermentation or Culture harvests
• Harvest pool, or bulk concentrate
• Purified intermediate
• Final bulk
(The above would be duplicated for each antigen in a combined vaccine)
• Final formulated bulk (if stored)
• Filling or filling/lyophilization
• Labelling/Packaging
In some companies, manufacturing instructions have been prepared as a series of SOPs
which describe each step and each providing a record sheet for the data to be entered. In
this format, there is no continuous production instruction and recording document. If this
is the case, then the Master Formula will be a Master SOP List of the production SOPs in
order of their use i required to describe the overall master production process, with all the
SOPs and record sheets appended. A separate list would be required for each batch size.
Although many of the SOPs might be the same, some SOPs for various batch sizes may
be different. In this case, rather than the cross-referencing supporting SOPs in a
continuous MF, the SOPs for facility and equipment preparation, supporting sterilization
runs, in-process tests, etc would be included in the Master SOP List.
9) Issuing of MF copy as a blank batch record
While Master Formulae are almost invariably stored on the computer, the official signed
form is a paper copy. When a production order is made, QA is responsible to generate a
copy, usually adds the lot number and stamps each page of the reproduced MF which is
now the blank batch record for the production data for the assigned batch or lot. The MF
should make reference to in process tests, QC tests, production parameters that are
computer recorded (e.g. fermentation or lyophilization printouts), environmental
monitoring or water testing, autoclave run charts or depyrogenation oven charts, etc but
generally these supporting operations and records are not within a MF. The batch record,
however, includes the record sheets of all the production records and support records.

Guide to Master Formulae Guidance Document
13
Master formulae, once approved and signed, should remain under the control of QA.
Copies are not stored in the production areas for uncontrolled use. When revisions are
made (following the change control process, and document control process) a new
version is assigned a revision (or edition) number, the approval signatures and effective
dates are added, and the previous version is archived. Unlike routine testing SOPs which
have a fairly general distribution and are available in each laboratory or production area
using them, a copy of the currently approved MF is issued batch by batch on production
orders. When a series of SOPs are used for production operations, then the corresponding
SOPs record sheets should also be controlled by QA and issued on production orders.
Master copies of the MF (each numbered and recorded on a QA distribution list) can be
distributed to relevant departments if needed, but the MF issued for a production run
should be stamped by QA to ensure that the currently valid version is used. There will
obviously be company-by-company differences in the details of the procedures for QA
approval and issuing of MF.
10) Electronic MF and Batch Records
As mentioned above, the MF is invariably (in this electronic age) on the computer, and
should be under password control of QA. Because the MF master copy is a signed
document, the approved and signed original hard-copy of the MF becomes the official
copy and should remain with QA. Photocopies – stamped, numbered, and on a
distribution list - may be issued (see above) as reference copies to the relevant department
head. The electronic version may have the signature and date fields typed in, e.g.
“official copy signed by XXX”; “official copy dated ddmmyy”. If the electronic copy is
printed out as the blank batch record for each production run, the QA department must
stamp each page of the printout and sign that it is the approved current MF. The lot
number can be added electronically or by hand on each page by the responsible person in
QA. Alternately, the hard-copy can be photocopied for the production run, but will also
be stamped and the lot number added. Whatever method is decided by a company, the
MF must be issued by QA for each production run and controlled to ensure that only an
approved copy of the current MF (or series of SOP record sheets) is issued on a
production order.
Computerized batch records – e.g.: filling in the blank MF– are more complicated. The
computer program for permitting the entering of production data at the time of
performance of the production step will require computer access inside the cleanroom. In
this case, the MF would be issued electronically with safeguards to ensure that no
unofficial copies can be made, or pages replaced. Passwords for entering data, verifying
data or correcting data will need to be implemented. Specific procedures for recording
any changes made to data records or the recording of deviations to production procedures
must be validated and fully traceable retaining the original data and the corrected data.
The review of batch records should include the full review of all changes and corrections
made on the electronic forms. All of this process must be defined in SOPs for the

Guide to Master Formulae Guidance Document
14
procedures to be followed for electronic batch records. Specific guidelines on computer
data entry and validation have been published by PIC and the US which can be consulted
for detailed guidance.
For electronic batch records prepared by transcription from a hand-written record to a
computer batch record requires additional verification that the computerized entries have
been checked and are correct. This would require confirmation at the time of entry and
again verified by QA.
11) Batch Records versus Master Formula
In the regulations and guidelines (see appendices 1-4) there are also requirements for
completed batch records (for some reason never called “lot records”). The MF is
essentially the blank batch record for the production operations as discussed in 7) above.
The batch record (BR often called Batch Processing Record BPR) is the MF with all data
entered plus all the results of the supporting operations (in-process test results,
environmental monitoring, autoclave records, etc).
Details of the contents of the Batch Record are found in Appendix 1 (WHO); Appendix 2
(EU); Appendix 3 (PIC); and Appendix 4 (Health Canada) and Appendix 5 (US).
12) Batch record review checklist
For a continuous production instruction MF, all the supporting operations are included as
data fields or as cross-references within the document. For such a document, a list of the
records sheets that are expected to be present in the batch record are itemized in a
checklist which can also be a table of contents of the batch record.
For a production document using various SOPs to define the production process, the
Master SOP List may be essentially the same as the batch record checklist.

Guide to Master Formulae Guidance Document
15
Appendix 1: Extract from World Health Organization
WHO Technical Report Series, No. 908, 2003
Annex 4: Good Manufacturing Practices for pharmaceutical products: main
principles.
From the Glossary
batch records
All documents associated with the manufacture of a batch of bulk product or finished
product. They provide a history of each batch of product and of all circumstances
pertinent to the quality of the final product.
master formula
A document or set of documents specifying the starting materials with their quantities
and the packaging materials, together with a description of the procedures and
precautions required to produce a specified quantity of a finished product as well as the
processing instructions, including the in-process controls.
master record
A document or set of documents that serve as a basis for the batch documentation (blank
batch record).
standard operating procedure (SOP)
An authorized written procedure giving instructions for performing operations not
necessarily specific to a given product or material (e.g.: equipment operation,
maintenance and cleaning; validation; cleaning of premises and environmental control;
sampling and inspection). Certain SOPs may be used to supplement product-specific
master and batch production documentation.
15. Documentation
15.1 Principle. Good documentation is an essential part of the quality assurance system
and, as such, should exist for all aspects of GMP. Its aims are to define the specifications
and procedures for all materials and methods of manufacture and control; to ensure that
all personnel concerned with manufacture know what to do and when to do it; to ensure
that authorized persons have all the information necessary to decide whether or not to
release a batch of a drug for sale, to ensure the existence of documented evidence,
traceability, and to provide records and an audit trail that will permit investigation. It
ensures the availability of the data needed for validation, review and statistical analysis.
The design and use of documents depend upon the manufacturer. In some cases some or
all of the documents described below may be brought together, but they will usually be
separate.

Guide to Master Formulae Guidance Document
16
General
15.2 Documents should be designed, prepared, reviewed and distributed with care. They
should comply with the relevant parts of the manufacturing and marketing authorizations.
15.3 Documents should be approved, signed and dated by the appropriate responsible
persons. No document should be changed without authorization and approval.
15.4 Documents should have unambiguous contents: the title, nature and purpose should
be clearly stated. They should be laid out in an orderly fashion and be easy to check.
Reproduced documents should be clear and legible. The reproduction of working
documents from master documents must not allow any error to be introduced through the
reproduction process.
15.5 Documents should be regularly reviewed and kept up to date. When a document has
been revised, a system should exist to prevent inadvertent use of the superseded version.
Superseded documents should be retained for a specific period of time.
15.6 Where documents require the entry of data, these entries should be clear, legible and
indelible. Sufficient space should be provided for such entries.
15.7 Any alteration made to a document should be signed and dated; the alteration should
permit the reading of the original information. Where appropriate, the reason for the
alteration should be recorded.
15.8 Records should be made or completed when any action is taken and in such a way
that all significant activities concerning the manufacture of pharmaceutical products are
traceable. Records should be retained for at least one year after the expiry date of the
finished product.
15.9 Data (and records for storage) may be recorded by electronic data-processing
systems or by photographic or other reliable means. Master formulae and detailed
standard operating procedures relating to the system in use should be available and the
accuracy of the records should be checked. If documentation is handled by electronic
data-processing methods, only authorized persons should be able to enter or modify data
in the computer, and there should be a record of changes and deletions; access should be
restricted by passwords or other means and the entry of critical data should be
independently checked. Batch records stored electronically should be protected by back-
up transfer on magnetic tape, microfilm, paper print-outs or other means. It is particularly
important that, during the period of retention, the data are readily available.

Guide to Master Formulae Guidance Document
17
Documents required
Labels (sections 15.10-15.12 not extracted)
Specifications and testing procedures (sections 15.13-15.17 not extracted)
Specifications for starting and packaging materials (sections 15.18-15.21 not extracted)
Master formulae
15.22 A formally authorized master formula should exist for each product and batch size
to be manufactured.
15.23 The master formula should include:
(a) the name of the product, with a product reference code relating to its
specification;
(b) a description of the dosage form, strength of the product and batch size;
(c) a list of all starting materials to be used (if applicable, with the INNs), with the
amount of each, described using the designated name and a reference that is
unique to that material (mention should be made of any substance that may
disappear in the course of processing);
(d) a statement of the expected final yield with the acceptable limits, and of
relevant intermediate yields, where applicable;
(e) a statement of the processing location and the principal equipment to be used;
(f) the methods, or reference to the methods, to be used for preparing and
operating the critical equipment, e.g. cleaning (especially after a change in
product), assembling, calibrating, sterilizing, use;
(g) detailed step-wise processing instructions (e.g. checks on materials,
pretreatments, sequence for adding materials, mixing times, temperatures);
(h) the instructions for any in-process controls with their limits;
(i) where necessary, the requirements for storage of the products, including the
container, the labelling, and any special storage conditions;
(j) any special precautions to be observed.
Packaging instructions
15.24 Formally authorized packaging instructions should exist for each product, pack size
and type. These should normally include, or make reference to:
(a) the name of the product;
(b) a description of its pharmaceutical form, strength and, where applicable,
method of application;
(c) the pack size expressed in terms of the number, weight or volume of the
product in the final container;

Guide to Master Formulae Guidance Document
18
(d) a complete list of all the packaging materials required for a standard batch
size, including quantities, sizes and types, with the code or reference number
relating to the specifications for each packaging material;
(e) where appropriate, an example or reproduction of the relevant printed
packaging materials and specimens, indicating where the batch number and
expiry date of the product have been marked;
(f) special precautions to be observed, including a careful examination of the
packaging area and equipment in order to ascertain the line clearance before and
after packaging operations;
(g) a description of the packaging operation, including any significant subsidiary
operations, and equipment to be used;
(h) details of in-process controls with instructions for sampling and acceptance
limits.
Batch processing records
15.25 A batch processing record should be kept for each batch processed. It should be
based on the relevant parts of the currently approved specifications on the record. The
method of preparation of such records should be designed to avoid errors. (Copying or
validated computer programmes are recommended. Transcribing from approved
documents should be avoided.)
15.26 Before any processing begins, a check should be made that the equipment and
work station are clear of previous products, documents, or materials not required for the
planned process, and that the equipment is clean and suitable for use. This check should
be recorded.
15.27 During processing, the following information should be recorded at the time each
action is taken, and after completion the record should be dated and signed by the person
responsible for the processing operations:
(a) the name of the product;
(b) the number of the batch being manufactured;
(c) dates and times of commencement, of significant intermediate stages, and of
completion of production;
(d) the name of the person responsible for each stage of production;
(e) the initials of the operator(s) of different significant steps of production and,
where appropriate, of the person(s) who checked each of these operations (e.g.
weighing);
(f) the batch number and/or analytical control number and the quantity of each
starting material actually weighed (including the batch number and amount of any
recovered or reprocessed material added);
(g) any relevant processing operation or event and the major equipment used;

Guide to Master Formulae Guidance Document
19
(h) the in-process controls performed, the initials of the person(s) carrying them
out, and the results obtained;
(i) the amount of product obtained at different and pertinent stages of manufacture
(yield), together with comments or explanations for significant deviations from
the expected yield;
(j) notes on special problems including details, with signed authorization for any
deviation from the master formula.
Batch packaging records
15.28 A batch packaging record should be kept for each batch or part batch processed. It
should be based on the relevant parts of the approved packaging instructions, and the
method of preparing such records should be designed to avoid errors. (Copying or
validated computer programmes are recommended. Transcribing from approved
documents should be avoided.)
15.29 Before any packaging operation begins, checks should be made that the equipment
and work station are clear of previous products, documents or materials not required for
the planned packaging operations, and that equipment is clean and suitable for use.
These checks should be recorded.
15.30 The following information should be recorded at the time each action is taken, and
the date and the person responsible should be clearly identified by signature or electronic
password:
(a) the name of the product, the batch number and the quantity of bulk product to
be packed, as well as the batch number and the planned quantity of finished
product that will be obtained, the
quantity actually obtained and the reconciliation;
(b) the date(s) and time(s) of the packaging operations;
(c) the name of the responsible person carrying out the packaging operation;
(d) the initials of the operators of the different significant steps;
(e) the checks made for identity and conformity with the packaging instructions,
including the results of in-process controls;
(f) details of the packaging operations carried out, including references to
equipment and the packaging lines used, and, when necessary, the instructions for
keeping the product unpacked or a record of returning product that has not been
packaged to the storage area;
(g) whenever possible, samples of the printed packaging materials used, including
specimens bearing the approval for the printing of and regular check (where
appropriate) of the batch number, expiry date, and any additional overprinting;
(h) notes on any special problems, including details of any deviation from the
packaging instructions, with written authorization by an appropriate person;

Guide to Master Formulae Guidance Document
20
(i) the quantities and reference number or identification of all printed packaging
materials and bulk product issued, used, destroyed or returned to stock and the
quantities of product obtained to permit an adequate reconciliation.

Guide to Master Formulae Guidance Document
21
Appendix 2: Extract from: EUDRALEX; Volume 4 - Medicinal Products for
Human and Veterinary Use: Good Manufacturing Practice.
CHAPTER 4 DOCUMENTATION
Principle
Good documentation constitutes an essential part of the quality assurance system. Clearly
written documentation prevents errors from spoken communication and permits tracing
of batch history. Specifications, Manufacturing Formulae and instructions, procedures,
and records must be free from errors and available in writing. The legibility of documents
is of paramount importance.
General
4.1 Specifications describe in detail the requirements with which the products or
materials used or obtained during manufacture have to conform. They serve as a basis for
quality evaluation.
Manufacturing Formulae, Processing and Packaging Instructions state all the starting
materials used and lay down all processing and packaging operations.
Procedures give directions for performing certain operations e.g. cleaning, clothing,
environmental control, sampling, testing, equipment operation.
Records provide a history of each batch of product, including its distribution, and also of
all other relevant circumstances pertinent to the quality of the final product.
4.2 Documents should be designed, prepared, reviewed and distributed with care. They
should comply with the relevant parts of the manufacturing and marketing authorisation
dossiers.
4.3 Documents should be approved, signed and dated by appropriate and authorised
persons.
4.4 Documents should have unambiguous contents; title, nature and purpose should be
clearly stated. They should be laid out in an orderly fashion and be easy to check.
Reproduced documents should be clear and legible. The reproduction of working
documents from master documents must not allow any error to be introduced through the
reproduction process.
4.5 Documents should be regularly reviewed and kept up-to-date. When a document has
been revised, systems should be operated to prevent inadvertent use of superseded
documents.

Guide to Master Formulae Guidance Document
22
4.6 Documents should not be handwritten; although, where documents require the entry
of data, these entries may be made in clear, legible, indelible handwriting. Sufficient
space should be provided for such entries.
4.7 Any alteration made to the entry on a document should be signed and dated; the
alteration should permit the reading of the original information. Where appropriate, the
reason for the alteration should be recorded.
4.8 The records should be made or completed at the time each action is taken and in such
a way that all significant activities concerning the manufacture of medicinal products are
traceable. They should be retained for at least one year after the expiry date of the
finished product.
4.9 Data may be recorded by electronic data processing systems, photographic or other
reliable means, but detailed procedures relating to the system in use should be available
and the accuracy of the records should be checked. If documentation is handled by
electronic data processing methods, only authorised persons should be able to enter or
modify data in the computer and there should be a record of changes and deletions;
access should be restricted by passwords or other means and the result of entry of critical
data should be independently checked. Batch records electronically stored should be
protected by back-up transfer on magnetic tape, microfilm, paper or other means. It is
particularly important that the data are readily available throughout the period of
retention.
Documents required
Specifications (sections 4.10 to 4.13 not extracted)
Manufacturing Formula and Processing Instructions
Formally authorised Manufacturing Formula and Processing Instructions should exist for
each product and batch size to be manufactured. They are often combined in one
document.
4.14 The Manufacturing Formula should include:
a) the name of the product, with a product reference code relating to its
specification;
b) a description of the pharmaceutical form, strength of the product and batch
size;
c) a list of all starting materials to be used, with the amount of each, described
using the designated name and a reference which is unique to that material;
mention should be made of any substance that may disappear in the course of
processing;

Guide to Master Formulae Guidance Document
23
d) a statement of the expected final yield with the acceptable limits, and of
relevant intermediate yields, where applicable.
4.15 The Processing Instructions should include:
a) a statement of the processing location and the principal equipment to be used;
b) the methods, or reference to the methods, to be used for preparing the critical
equipment (e.g. cleaning, assembling, calibrating, sterilising);
c) detailed stepwise processing instructions (e.g. checks on materials, pre-
treatments,
sequence for adding materials, mixing times, temperatures);
d) the instructions for any in-process controls with their limits;
e) where necessary, the requirements for bulk storage of the products; including
the
container, labelling and special storage conditions where applicable;
f) any special precautions to be observed.
Packaging Instructions
4.16 There should be formally authorised Packaging Instructions for each product, pack
size and type. These should normally include, or have a reference to, the following:
a) name of the product;
b) description of its pharmaceutical form, and strength where applicable;
c) the pack size expressed in terms of the number, weight or volume of the
product in the final container;
d) a complete list of all the packaging materials required for a standard batch size,
including quantities, sizes and types, with the code or reference number relating to
the specifications of each packaging material;
e) where appropriate, an example or reproduction of the relevant printed
packaging materials, and specimens indicating where to apply batch number
references, and shelf life of the product;
f) special precautions to be observed, including a careful examination of the area
and equipment in order to ascertain the line clearance before operations begin;
g) a description of the packaging operation, including any significant subsidiary
operations, and equipment to be used;
h) details of in-process controls with instructions for sampling and acceptance
limits.
Batch Processing Records
4.17 A Batch Processing Record should be kept for each batch processed. It should be
based on the relevant parts of the currently approved Manufacturing Formula and
Processing Instructions. The method of preparation of such records should be designed

Guide to Master Formulae Guidance Document
24
to avoid transcription errors. The record should carry the number of the batch being
manufactured. Before any processing begins, there should be recorded checks that the
equipment and work station are clear of previous products, documents or materials not
required for the planned process, and that equipment is clean and suitable for use.
During processing, the following information should be recorded at the time each action
is taken and, after completion, the record should be dated and signed in agreement by the
person responsible for the processing operations:
a) the name of the product;
b) dates and times of commencement, of significant intermediate stages and of
completion of production;
c) name of the person responsible for each stage of production;
d) initials of the operator of different significant steps of production and, where
appropriate, of the person who checked each of these operations (e.g. weighing);
e) the batch number and/or analytical control number as well as the quantities of
each starting material actually weighed (including the batch number and amount
of any recovered or reprocessed material added);
f) any relevant processing operation or event and major equipment used;
g) a record of the in-process controls and the initials of the person(s) carrying
them out, and the results obtained;
h) the product yield obtained at different and pertinent stages of manufacture;
i) notes on special problems including details, with signed authorisation for any
deviation from the Manufacturing Formula and Processing Instructions.
Batch Packaging Records
4.18 A Batch Packaging Record should be kept for each batch or part batch processed. It
should be based on the relevant parts of the Packaging Instructions and the method of
preparation of such records should be designed to avoid transcription errors. The record
should carry the batch number and the quantity of bulk product to be packed, as well as
the batch number and the planned quantity of finished product that will be obtained.
Before any packaging operation begins, there should be recorded checks that the
equipment and work station are clear of previous products, documents or materials not
required for the planned packaging operations, and that equipment is clean and suitable
for use.
The following information should be entered at the time each action is taken and, after
completion, the record should be dated and signed in agreement by the person(s)
responsible for the packaging operations:
a) the name of the product;
b) the date(s) and times of the packaging operations;
c) the name of the responsible person carrying out the packaging operation;
d) the initials of the operators of the different significant steps;

Guide to Master Formulae Guidance Document
25
e) records of checks for identity and conformity with the packaging instructions
including the results of in-process controls;
f) details of the packaging operations carried out, including references to
equipment and the packaging lines used;
g) whenever possible, samples of printed packaging materials used, including
specimens of the batch coding, expiry dating and any additional overprinting;
h) notes on any special problems or unusual events including details, with signed
authorisation for any deviation from the Manufacturing Formula and Processing
Instructions;
i) the quantities and reference number or identification of all printed packaging
materials and bulk product issued, used, destroyed or returned to stock and the
quantities of obtained product, in order to provide for an adequate reconciliation.
Procedures and records (sections 4.19 to 4.29 not extracted)

Guide to Master Formulae Guidance Document
26
Appendix 3: Extracted from PHARMACEUTICAL INSPECTION CONVENTION
PHARMACEUTICAL INSPECTION CO-OPERATION SCHEME PE 009-3, 1
January 2006
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS.
© PIC/S January 2006 (Reproduction prohibited for commercial purposes. Reproduction
for internal use is authorised, provided that the source is acknowledged).
DOCUMENTATION
PRINCIPLE
Good documentation constitutes an essential part of the quality assurance system. Clearly
written documentation prevents errors from spoken communication and permits
tracing of batch history. Specifications, Manufacturing Formulae and instructions,
procedures, and records must be free from errors and available in writing. The
legibility of documents is of paramount importance.
GENERAL
4.1.
Specifications describe in detail the requirements with which the products or materials
used or obtained during manufacture have to conform. They serve as a basis for
quality evaluation.
Manufacturing Formulae, Processing and Packaging Instructions state all the starting
materials used and lay down all processing and packaging operations.
Procedures give directions for performing certain operations e.g. cleaning, clothing,
environmental control, sampling, testing, equipment operations.
Records provide a history of each batch of product, including its distribution, and also of
all other relevant circumstances pertinent for the quality of the final product.
DOCUMENTS REQUIRED
MANUFACTURING FORMULA AND PROCESSING NSTRUCTIONS
Formally authorised Manufacturing Formula and Processing Instructions should exist for
each product and batch size to be manufactured. They are often combined in one
document.
4.14. The Manufacturing Formula should include:
a) the name of the product, with a product reference code relating to its
specification;
b) a description of the pharmaceutical form, strength of the product and batch size;
c) a list of all starting materials to be used, with the amount of each, described using
the designated name and a reference which is unique to that material;

Guide to Master Formulae Guidance Document
27
mention should be made of any substance that may disappear in the course
of processing;
d) a statement of the expected final yield with the acceptable limits, and of relevant
intermediate yields, where applicable.
4.15. The Processing Instructions should include:
a) a statement of the processing location and the principal equipment to be used;
b) the methods, or reference to the methods, to be used for preparing the critical
equipment (e.g. cleaning, assembling, calibrating, sterilising);
c) detailed stepwise processing instructions (e.g. checks on materials, pretreatments,
sequence for adding materials, mixing times, temperatures);
d) the instructions for any in-process controls with their limits;
e) where necessary, the requirements for bulk storage of the products; including the
container, labelling and special storage conditions where applicable;
f) any special precautions to be observed.
PACKAGING INSTRUCTIONS
4.16. There should be formally authorised Packaging Instructions for each product for
pack size and type. These should normally include, or have a reference to, the
following:
a) name of the product;
b) description of its pharmaceutical form, and strength where applicable;
c) the pack size expressed in terms of the number, weight or volume of the product
in the final container;
d) a complete list of all the packaging materials required for a standard batch size,
including quantities, sizes and types, with the code or reference number
relating to the specifications of each packaging material;
e) where appropriate, an example or reproduction of the relevant printed packaging
materials, and specimens indicating where to apply batch number
references, and shelf-life of the product;
f) special precautions to be observed, including a careful examination of the area
and equipment in order to ascertain the line clearance before operations
begin;
g) a description of the packaging operation, including any significant subsidiary
operations, and equipment to be used;
h) details of in-process controls with instructions for sampling and acceptance
limits.
BATCH PROCESSING RECORDS
4.17. A Batch Processing Record should be kept for each batch processed. It should be
based on the relevant parts of the currently approved Manufacturing Formula and
Processing Instructions. The method of preparation of such records should be

Guide to Master Formulae Guidance Document
28
designed to avoid transcription errors. The record should carry the number of the
batch being manufactured.
Before any processing begins, there should be recorded checks that the equipment and
work station are clear of previous products, documents or materials not required
for the planned process, and that equipment is clean and suitable for use. During
processing, the following information should be recorded at the time each action
is taken and, after completion, the record should be dated and signed in agreement
by the person responsible for the processing operations:
a) the name of the product;
b) dates and times of commencement, of significant intermediate stages and of
completion of production;
c) name of the person responsible for each stage of production;
d) initials of the operator of different significant steps of production and, where
appropriate, of the person who checked each of these operations (e.g.
weighing);
e) the batch number and/or analytical control number as well as the quantities of
each starting material actually weighed (including the batch number and
amount of any recovered or reprocessed material added);
f) any relevant processing operation or event and major equipment used;
g) a record of the in-process controls and the initials of the person(s) carrying
them out, and the results obtained;
h) the amount of product yield obtained at different and pertinent stages of
manufacture;
i) notes on special problems including details, with signed authorization for any
deviation from the Manufacturing Formula and Processing Instructions.
BATCH PACKAGING RECORDS
4.18. A Batch Packaging Record should be kept for each batch or part batch processed. It
should be based on the relevant parts of the Packaging Instructions and the
method of preparation of such records should be designed to avoid transcription
errors. The record should carry the batch number and the quantity of bulk product
to be packed, as well as the batch number and the planned quantity of finished
product that will be obtained.
Before any packaging operation begins, there should be recorded checks that the
equipment and work station are clear of previous products, documents or
materials not required for the planned packaging operations, and that equipment is
clean and suitable for use.
The following information should be entered at the time each action is taken and, after
completion, the record should be dated and signed in agreement by the person(s)
responsible for the packaging operations:
a) the name of the product;
b) the date(s) and times of the packaging operations;
c) the name of the responsible person carrying out the packaging operation;
d) the initials of the operators of the different significant steps;

Guide to Master Formulae Guidance Document
29
e) records of checks for identity and conformity with the Packaging Instructions
including the results of in-process controls;
f) details of the packaging operations carried out, including references to
equipment and the packaging lines used;
g) whenever possible, samples of printed packaging materials used, including
specimens of the batch coding, expiry dating and any additional
overprinting;
h) notes on any special problems or unusual events including details with signed
authorization for any deviation from the Manufacturing Formula and
Processing Instructions;
i) the quantities and reference number or identification of all printed packaging
materials and bulk product issued, used, destroyed or returned to stock and
the quantities of obtained product, in order to provide for an adequate
reconciliation.

Guide to Master Formulae Guidance Document
30
Appendix 4: Extract from Canadian GMP Guidelines.
Health Canada, Health Products and Food Branch Inspectorate
GOOD MANUFACTURING PRACTICES GUIDELINES, 2002 EDITION, Version 2
From the GLOSSARY:
MANUFACTURING BATCH DOCUMENT (fiche de lot de fabrication) - Instructions
that outline in detail the materials and procedures required to fabricate, prepare, and
preserve a single lot or batch of a drug in dosage form.
MASTER FORMULA (formule-type) - A document or set of documents specifying the
raw materials with their quantities and the packaging materials, together with a detailed
description of the procedures and precautions required to produce a specified quantity of
a finished product as well as the processing instructions, including the in-process
controls.
MASTER PRODUCTION DOCUMENT (document-type de production) - a document
that includes specifications for raw material, for packaging material and for packaged
dosage form, master formula, sampling procedures, and critical processing related SOPs,
whether or not these SOPs are specifically referenced in the master formula.
MANUFACTURING CONTROL
REGULATION C.02.011
(1) Every fabricator, packager/labeller, distributor referred to in paragraph C.01A.003(b)
and importer of a drug shall have written procedures, prepared by qualified personnel, in
respect of the drug to ensure that the drug meets the specifications for use of that drug.
(2) Every person required to have written procedures referred to in subsection (1) shall
ensure that each lot or batch of the drug is fabricated, packaged/labelled and tested in
compliance with those procedures.
RATIONALE This Regulation requires that a number of measures be taken to maintain the integrity of a
drug product from the moment the various raw materials enter the plant to the time the
finished dosage form is released for sale. These measures seek to ensure that all
manufacturing processes are clearly defined, systematically reviewed in light of
experience, and shown to be capable of consistently manufacturing pharmaceutical
products of the required quality that comply with their established specifications.

Guide to Master Formulae Guidance Document
31
MANUFACTURING MASTER FORMULA
23. Processing operations are covered by master formulae, that are prepared by, and are
subject to independent checks by, persons who have the qualifications described under
Regulation C.02.006 Interpretation 1.
24. Master formulae are written to provide not less than 100% of label claim and
include the following:
24.1 the name of the product, with a reference code relating to its specifications;
24.2 a description of the dosage form, strength of the product, and batch size;
24.3 a list of all raw materials to be used, along with the amount of each,
described using the designated name and a reference that is unique to that
material (mention is made of any processing aids that may not be present in the
final product);
24.4 a statement of the expected final yield, along with the acceptable limits,
and of relevant intermediate yields, where applicable;
24.5 a statement of the principal equipment to be used;
24.6 the procedures, or reference to the procedures, to be used for preparing the
critical equipment, e.g., cleaning (especially after a change in product),
assembling, calibrating, sterilizing, etc.;
24.7 detailed stepwise processing instructions (e.g., checks on materials,
pretreatment, sequence for adding materials, mixing times or temperatures,
etc.);
24.8 the instructions for any in-process controls, along with their limits;
24.9 where necessary, the requirements for storage of the products, including
the container, the labelling and any special storage conditions; and
24.10 any special precautions to be observed.
PACKAGING MASTER FORMULA
25. In the case of a packaged product, the master formula also includes for each
product, package size and type, the following:
25.1 the package size, expressed in terms of the number, weight, or volume of
the product in the final container;
25.2 a complete list of all the packaging materials required for a standard batch
size, including quantities, sizes and types with the code or reference number
relating to the specifications for each packaging material;
25.3 an example or reproduction of the relevant printed packaging materials and
specimens, indicating where the batch number and expiry date of the product
are to be positioned;
25.4 special precautions to be observed, including a careful examination of the
packaging area and equipment in order to ascertain the line clearance before
operations begin;

Guide to Master Formulae Guidance Document
32
25.5 a description of the packaging operations, including any significant
subsidiary operations and the equipment to be used; and
25.6 details of in-process controls, with instructions for sampling and
acceptance limits.
MANUFACTURING BATCH DOCUMENT
26. Each batch processed is effectively governed by an individually numbered
manufacturing order prepared by qualified personnel from the master formula by such
means as to prevent errors in copying or calculation and verified by qualified personnel.
27. As it becomes available during the process, the following information is included
on or with the manufacturing order:
27.1 the name of the product;
27.2 the number of the batch being manufactured;
27.3 dates and times of commencement and completion of significant
intermediate stages, such as blending, heating, etc., and of production;
27.4 the batch number and/or analytical control number, as well as the quantity
of each raw material actually weighed and dispensed (for active raw material,
the quantity is to be adjusted if the assay value is less than 98% calculated on
“as is” basis and on which the master formula was based);
27.5 confirmation by qualified personnel of each ingredient added to a batch;
27.6 the identification of personnel performing each step of the process; and of
the person who checked each of these steps;
27.7 the actual results of the in-process quality checks performed at appropriate
stages of the process and the identification of the person carrying them out;
27.8 the actual yield of the batch at appropriate stages of processing and the
actual final yields, together with explanations for any deviations from the
expected yield;
27.9 detailed notes on special problems with written approval for any deviation
from the master formula; and
27.10 after completion, the signature of the person responsible for the
processing operations.
28. Batches are combined only with the approval of the quality control department and
according to pre-established written procedures.
28.1 The introduction of part of a previous batch, conforming to the required
quality, into the next batch of the same product at a defined stage of fabrication
is approved beforehand. This recovery is carried out in accordance with a
validated procedure and is recorded.
PACKAGING BATCH DOCUMENT
29. Packaging operations are performed according to comprehensive and detailed
written

Guide to Master Formulae Guidance Document
33
operating procedures or specifications, which include the identification of equipment
and
packaging lines used to package the drug, the adequate separation and if necessary, the
dedication of packaging lines that are packaging different drugs and disposal
procedures for unused printed packaging materials. Packaging orders are individually
numbered.
30. The method of preparing packaging orders is designed to avoid transcription errors.
31. Before any packaging operation begins, checks are made that the equipment and
work station are clear of previous products, documents, and materials that are not
required for the planned packaging operations and that equipment is clean and suitable
for use. These checks are recorded.
32. All products and packaging materials to be used are checked on receipt by the
packaging department for quantity, identity and conformity with the packaging
instructions.
33. Precautions are taken to ensure that containers to be filled are free from
contamination with extraneous material.
34. The name and batch number of the product being handled is displayed at each
packaging station or line.
35. Packaging orders include the following information (recorded at the time each
action is taken):
35.1 the date(s) and time(s) of the packaging operations;
35.2 the name of the product, the batch number, and the quantity of bulk
product to be packaged, as well as the batch number and the planned quantity of
finished product that will be obtained, the quantity actually obtained and the
reconciliation;
35.3 the identification of the personnel who are supervising packaging
operations and the withdrawal of bulks;
35.4 the identification of the operators of the different significant steps;
35.5 the checks made for identity and conformity with the packaging
instructions,
including the results of in-process controls;
35.6 the general appearance of the packages;
35.7 whether the packages are complete;
35.8 whether the correct products and packaging materials are used;
35.9 whether any on-line printing is correct;
35.10 the correct functioning of line monitors;
35.11 handling precautions applied to a partly packaged product;
35.12 notes on any special problems, including details of any deviation from the
packaging instructions with written approval by qualified personnel;
35.13 the quantity, lot number, and/or analytical control number of each
packaging material and bulk drug issued for use; and
35.14 a reconciliation of the quantity of printed packaging material and bulk
drug used, destroyed or returned to stock.
36. To prevent mix-ups, samples taken away from the packaging line are not returned.

Guide to Master Formulae Guidance Document
34
37. Whenever possible, samples of the printed packaging materials used, including
specimens bearing the batch number, expiry date, and any additional overprinting, are
attached to packaging orders.
38. Filling and sealing are followed as quickly as possible by labelling. If labelling is
delayed, procedures are applied to ensure that no mix-ups or mislabelling can occur.
39. Upon completion of the packaging operation, any unused batch-coded packaging
materials are destroyed, and their destruction is recorded. A procedure is followed if
non-coded printed materials are returned to stock.
40. Outdated or obsolete packaging materials are destroyed and their disposal is
recorded.
41. Products that have been involved in non-standard occurrences during packaging are
subject to inspection and investigation by qualified personnel. A detailed record is kept
of this operation.
42. Any significant or unusual discrepancy observed during reconciliation of the
amount of bulk product and printed packaging materials and the number of units
packaged is investigated and satisfactorily accounted for before release. Validated
electronic verification of all printed packaging materials on the packaging line may
obviate the need for their full reconciliation.
43. Printed packaging materials are
43.1 stored in an area to which access is restricted to designated personnel who
are
supervised by persons who have the qualifications outlined under Regulation
C.02.006 Interpretation 2;
43.2 withdrawn against a packaging order;
43.3 issued and checked by persons who have the qualifications outlined under
Regulation C.02.006 Interpretation 2; and
43.4 identified in such a way as to be distinguishable during the packaging
operations.
44. To prevent mix-ups, roll-fed labels are preferred to cut labels. Gang printing is
avoided.
45. Cut labels, cartons, and other loose printed materials are stored and transported in
separate closed containers.
46. Special care is taken when cut labels are used, when overprinting is carried out off-
line and in hand-packaging operations. On line verification of all labels by automated
electronic means can be helpful in preventing mix-ups. Checks are made to ensure that
any electronic code readers, label counters or similar devices are operating correctly.
47. The correct performance of any printing (e.g., of code numbers or expiry dates)
done
separately or in the course of the packaging is checked and recorded.
48. Raw materials, packaging materials, intermediates, bulk drugs and finished
products are (a) stored in locations that are separate and removed from immediate
manufacturing areas, and (b) transported under conditions designated by the quality
control department to preserve their quality and safety.
49. All intermediate and finished products are held in quarantine and are so identified in

Guide to Master Formulae Guidance Document
35
accordance with Interpretation 21, until released by the quality control department.
50. Every package of a drug is identified by a lot number.

Guide to Master Formulae Guidance Document
36
Appendix 5: Extracts from US Code of Federal Regulations (CFR) and US FDA
Guidelines.
App 5-1) US Regulations for Master Production Records for Finished
Pharmaceuticals. Extract from: CFR 21, Chapter I, Subchapter F: Biologics, Part 211 Current Good Manufacturing
Practice for Finished Pharmaceuticals;
Subpart F--Production and Process Controls:
Sec. 211.100 Written procedures; deviations.
(a) There shall be written procedures for production and process control designed to assure
that the drug products have the identity, strength, quality, and purity they purport or are
represented to possess. Such procedures shall include all requirements in this subpart. These
written procedures, including any changes, shall be drafted, reviewed, and approved by the
appropriate organizational units and reviewed and approved by the quality control unit.
(b) Written production and process control procedures shall be followed in the execution of
the various production and process control functions and shall be documented at the time of
performance. Any deviation from the written procedures shall be recorded and justified.
Subpart J--Records and Reports
Sec. 211.186 Master production and control records
(a) To assure uniformity from batch to batch, master production and control records
for each drug product, including each batch size thereof, shall be prepared, dated,
and signed (full signature, handwritten) by one person and independently checked,
dated, and signed by a second person. The preparation of master production and
control records shall be described in a written procedure and such written procedure
shall be followed.
(b) Master production and control records shall include:
(1) The name and strength of the product and a description of the dosage form;
(2) The name and weight or measure of each active ingredient per dosage unit or
per unit of weight or measure of the drug product and a statement of the total
weight or measure of any dosage unit;
(3) A complete list of components designated by names or codes sufficiently
specific to indicate any special quality characteristic;

Guide to Master Formulae Guidance Document
37
(4) An accurate statement of the weight or measure of each component, using the
same weight system (metric, avoirdupois, or apothecary) for each component.
Reasonable variations may be permitted, however, in the amount of components
necessary for the preparation in the dosage form, provided they are justified in the
master production and control records;
(5) A statement concerning any calculated excess of component;
(6) A statement of theoretical weight or measure at appropriate phases of
processing;
(7) A statement of theoretical yield, including the maximum and minimum
percentages of theoretical yield beyond which investigation according to 211.192 is
required;
(8) A description of the drug product containers, closures, and packaging materials,
including a specimen or copy of each label and all other labeling signed and dated
by the person or persons responsible for approval of such labeling;
(9) Complete manufacturing and control instructions, sampling and testing
procedures, specifications, special notations, and precautions to be followed.
Database Updated April 1, 2005
App 5-2) US Regulations for Batch Records for Finished Pharmaceuticals:
CFR 21, Chapter I, Subchapter F: Biologics; Subchapter C: Drugs General; Part 211
Current Good Manufacturing Practice for Finished Pharmaceuticals;
Subpart J--Records and Reports.
Sec. 211.188 Batch production and control records
Batch production and control records shall be prepared for each batch of drug
product produced and shall include complete information relating to the production
and control of each batch. These records shall include:
(a) An accurate reproduction of the appropriate master production or control record,
checked for accuracy, dated, and signed;
(b) Documentation that each significant step in the manufacture, processing,
packing, or holding of the batch was accomplished, including:

Guide to Master Formulae Guidance Document
38
(1) Dates;
(2) Identity of individual major equipment and lines used;
(3) Specific identification of each batch of component or in-process material used;
(4) Weights and measures of components used in the course of processing;
(5) In-process and laboratory control results;
(6) Inspection of the packaging and labeling area before and after use;
(7) A statement of the actual yield and a statement of the percentage of theoretical
yield at appropriate phases of processing;
(8) Complete labeling control records, including specimens or copies of all labeling
used;
(9) Description of drug product containers and closures;
(10) Any sampling performed;
(11) Identification of the persons performing and directly supervising or checking
each significant step in the operation;
(12) Any investigation made according to 211.192.
(13) Results of examinations made in accordance with 211.134.
Database Updated April 1, 2005
App 5-3) US Additional Regulations for Batch Records for Biological Products:
CFR 21, Chapter I, Subchapter F: Biologics, Part 600 Biological Products: General;
Subpart B Establishment Standards,
Sec 600.12 Records
(a) Maintenance of records. Records shall be made, concurrently with the
performance, of each step in the manufacture and distribution of products, in such a
manner that at any time successive steps in the manufacture and distribution of any

Guide to Master Formulae Guidance Document
39
lot may be traced by an inspector. Such records shall be legible and indelible, shall
identify the person immediately responsible, shall include dates of the various steps,
and be as detailed as necessary for clear understanding of each step by one
experienced in the manufacture of products.
(b) Records retention--(Not extracted)
(2) Records of recall. (Not extracted)
(3) Suspension of requirement for retention. (Not extracted)
(c) Records of sterilization of equipment and supplies. Records relating to the mode
of sterilization, date, duration, temperature and other conditions relating to each
sterilization of equipment and supplies used in the processing of products shall be
made by means of automatic recording devices or by means of a system of
recording which gives equivalent assurance of the accuracy and reliability of the
record. Such records shall be maintained in a manner that permits an identification
of the product with the particular manufacturing process to which the sterilization
relates.
(d) Animal necropsy records. (Not extracted)
(e) Records in case of divided manufacturing responsibility. If two or more
establishments participate in the manufacture of a product, the records of each such
establishment must show plainly the degree of its responsibility. In addition, each
participating manufacturer shall furnish to the manufacturer who prepares the
product in final form for sale, barter or exchange, a copy of all records relating to
the manufacturing operations performed by such participating manufacturer insofar
as they concern the safety, purity and potency of the lots of the product involved,
and the manufacturer who prepares the product in final form shall retain a complete
record of all the manufacturing operations relating to the product.
[38 FR 32048, Nov. 20, 1973, as amended at 49 FR 23833, June 8, 1984; 55 FR
11013, Mar. 26, 1990; 70 FR 14982, Mar. 24, 2005]
Database Updated April 1, 2005
App 5-4) Extract from US FDA Guidelines for Batch Records for Sterile Products:
Guidance for Industry. Sterile Drug Products Produced by Aseptic Processing —
Current Good Manufacturing Practice: U.S. Department of Health and Human Services,
Food and Drug Administration; Center for Drug Evaluation and Research (CDER);

Guide to Master Formulae Guidance Document
40
Center for Biologics Evaluation and Research (CBER); Office of Regulatory Affairs
(ORA). September 2004) Pharmaceutical cGMPs).
Manufacturers should build process and environmental control activities into their
aseptic processing operation. It is critical that these activities be maintained and
strictly implemented on a daily basis. The requirement for review of all batch
records and data for conformance with written procedures, operating parameters,
and product specifications prior to arriving at the final release decision for an
aseptically processed product calls for an overall review of process and system
performance for that given cycle of manufacture. All in-process and laboratory
control results must be included with the batch record documentation in accordance
with section 211.188. Review of environmental and personnel monitoring data, as
well as other data relating to acceptability of output from support systems (e.g.,
HEPA / HVAC, WFI, steam generator) and proper functioning of equipment (e.g.,
batch alarms report; integrity of various filters) are considered essential elements of
the batch release decision.
While interventions and/or stoppages are normally recorded in the batch record, the
manner of documenting these occurrences varies. In particular, line stoppages and
any unplanned interventions should be sufficiently documented in batch records
with the associated time and duration of the event. In addition to lengthened dwell
time of sterile product elements in the critical area, an extensive intervention can
increase contamination risk. Sterility failures have often been attributed to atypical
or extensive interventions that have occurred as a response to an undesirable event
during the aseptic process. Written procedures describing the need for line
clearances in the event of certain interventions, such as machine adjustments and
any repairs, should be established. Such interventions should be documented with
more detail than minor events. Interventions that result in substantial activity near
exposed product or container closures or that last beyond a reasonable exposure
time should, where appropriate, result in a local or full line clearance.
Any disruption in power supply, however momentary, that could affect product
quality is a manufacturing deviation and must be included in batch records
(211.100, 211.192).

Guide to Master Formulae Guidance Document
41
Appendix 6: WHO/VSQ/97.01: A WHO guide to good manufacturing practice (GMP) requirements. Part 1: Standard operating procedures and master formulae,
Appendix 6: Sample master formula for a hypothetical biological product.
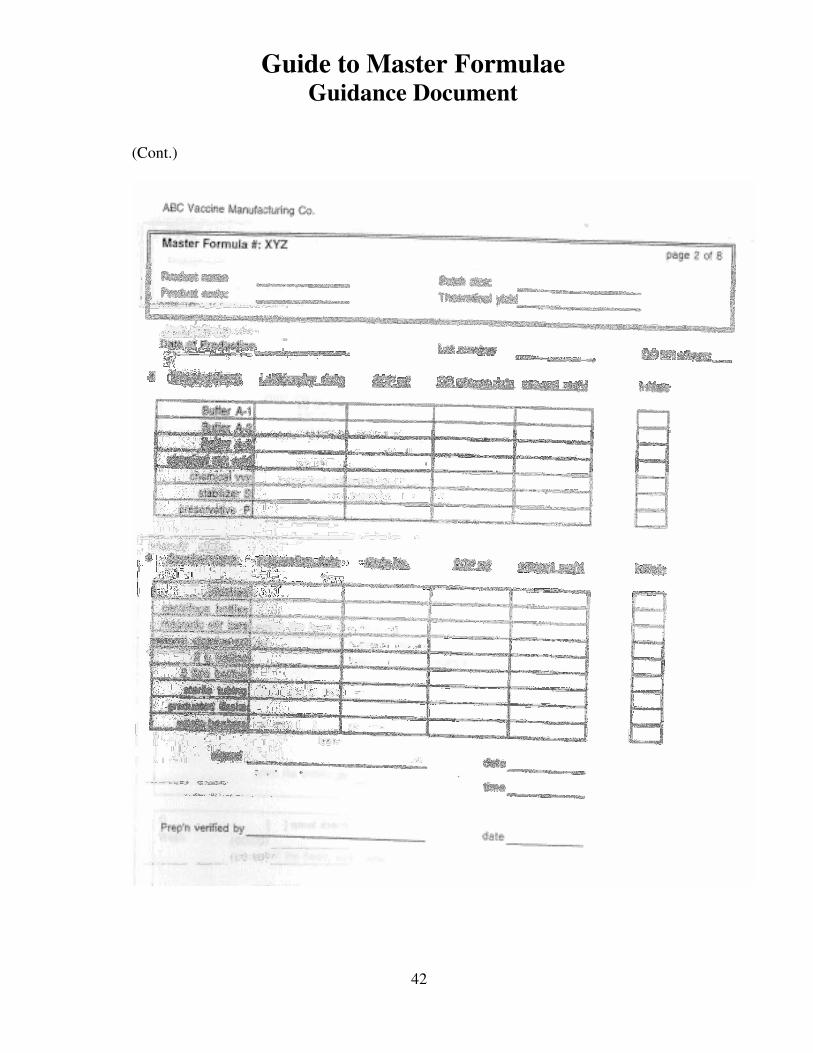
Guide to Master Formulae Guidance Document
42
(Cont.)

Guide to Master Formulae Guidance Document
43
(Cont.)

Guide to Master Formulae Guidance Document
44
(Cont.)

Guide to Master Formulae Guidance Document
45
(Cont.)

Guide to Master Formulae Guidance Document
46
(Cont.)

Guide to Master Formulae Guidance Document
47
(Cont.)

Guide to Master Formulae Guidance Document
48
(Cont.)

Guide to Master Formulae Guidance Document
49
Appendix 7: Example one of a Master Formula.
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 49 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
Issued by (Q.A.) : Date :
Production
Manager : Date :
Contents of Batch Manufacturing Record (BMR)
(Note: page numbers are from original document)
No. Description Page No.
1. Revival of lyophilized working cell bank 2
2. Transfer of revived culture to xx tube 3
3. Inoculation of Seed Bottle 4
4. Inoculation of Fermenter 5
5. Harvesting of Antigen X 7
6. Filtration details 9
7. Cleaning of system 10
8. BMR certification 12
Preparation of seed culture: Date: _____________
Culture seed preparation activities are to be performed as per the SOP ______________ “ XX
Seed Preparation”.
Master cell bank and working cell bank lot information
Strain used XXXXXXXXXXXX
Working cell bank lot no.
Effective Date:
(Approved by) Signature: Date:
Signature: Date:

Guide to Master Formulae Guidance Document
50
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 2 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
1) Revival of Lyophilized working cell bank: Draw one tube of Lyophilized working cell bank from Cold room No. 1 and disinfect it from
outside with absolute alcohol. Cut and open it under LAF. Transfer approximately x ml of xxx
medium to opened tube. Draw the suspension and transfer it to a vial containing xxx medium
(approx. x-x ml). Check purity by addition of a drop from vial on xxx agar and in xxx broth.
Incubate the vial and purity tubes at xx ± x °C for xx hrs.
Material Quantity Lot No. Checked by
Lyophilized culture tube
XX medium tube
XXXX agar tubes
XXXX broth tubes
Revival carried out on___________ by ____________.
Purity test details. SOP No.: ____________
XXX agar / broth tubes and vial incubated Purity checked Checked by
From To Date results
Deviations if any:
Date: _____________
2) Transfer of revived culture to XX tube.
Confirm the purity. Transfer x-x ml from the vial in step one to a tube containing XX medium.
Check purity by addition of a drop from vial on X agar and in X broth. Incubate the tube at xx ±
x °C for xx hrs. Incubate the purity tubes at xx ± x °C for xx hrs. Check purity also under
microscope.
Effective Date:
(Approved by) Signature: Date:
Signature: Date:

Guide to Master Formulae Guidance Document
51
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 3 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
Microscopic observations (Gram staining)._________________________________________
___________________________________________________________________________
Material Quantity Lot No. Checked by
XXXX agar tubes
XXXX broth tubes
XXXX medium tube
Passage carried out on___________ by __________. Incubation from________ to_______
Purity test details. SOP No.: ___________
XXXX agar / broth tubes
incubated
Purity checked Checked by
From To Date results
Deviations if any:
Date: _____________
3) Inoculation of Seed Bottle.
Confirm the purity on microscope. Transfer x-x ml culture from step 2 into Seed bottle
containing XXX medium. Use X L and X L medium for XX and XX L Fermenter batch
respectively. Check purity by addition of a drop from tube on xxx agar and in xxx broth.
Incubate the Seed bottle at xx ± x °C for xx hrs. Incubate the purity tubes at xx ± x °C for xx
hrs.
Seed bottle inoculated on __________ by __________.
Incubation of Seed bottle from __________ to ___________.
Effective Date:
(Approved by) Signature: Date:
Signature: Date:

Guide to Master Formulae Guidance Document
52
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 52 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
Purity test carried out as per. SOP No.: _________
Material Quantity Lot No. Checked by
XXXX agar tubes
XXXX broth tubes
XXXX agar / broth tubes
incubated
Purity checked Checked by
From To Date results
Deviations if any:
Date: _____________
4) Inoculation of Fermentor.
Transfer the contents of Seed bottle to Fermentor containing XX medium. Check purity by
addition of a drop from Seed bottle on XXX agar and in XXX broth. Incubate the Fermentor at
xx ± x °C for xx days. Incubate the purity tubes at xx ± x °C for xx hrs. Check purity also under
microscope. Adjust aeration and agitation as per SOP No. ________.
Fermentor No. ____ (Working vol. _____L. Sterilized on _____. containing XX Medium Lot
No.___________
SOP No.:____________ for media preparation SOP No.: ___________ for fermentor
sterilization
Effective Date:
(Approved by) Signature: Date:
Signature: Date:

Guide to Master Formulae Guidance Document
53
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 53 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
Air pressure in the fermentor checked _________ Pressure released _________
Microscopic observations of seed (gram staining):__________________________________
___________________________________________________________________________
Fermentor No.: ____ inoculated on ___________ by _________
Purity test details. SOP No.: _____________
Material Quantity Lot No. Checked by
XXXX agar tubes
XXXX broth tubes
XXXX agar / broth tubes
incubated
Purity checked Checked by
From To Date results
Incubation of Fermentor from _________ to __________.
Decontaminate the remnant culture, purity tubes and articles used for Culture transfer by
Autoclaving at xxx °C for xx min.
Decontamination charge No.: _____________ Date: _____________
Adjust the aeration and agitation as per SOP No.: ______________
Effective Date:
(Approved by) Signature: Date:
Signature: Date:
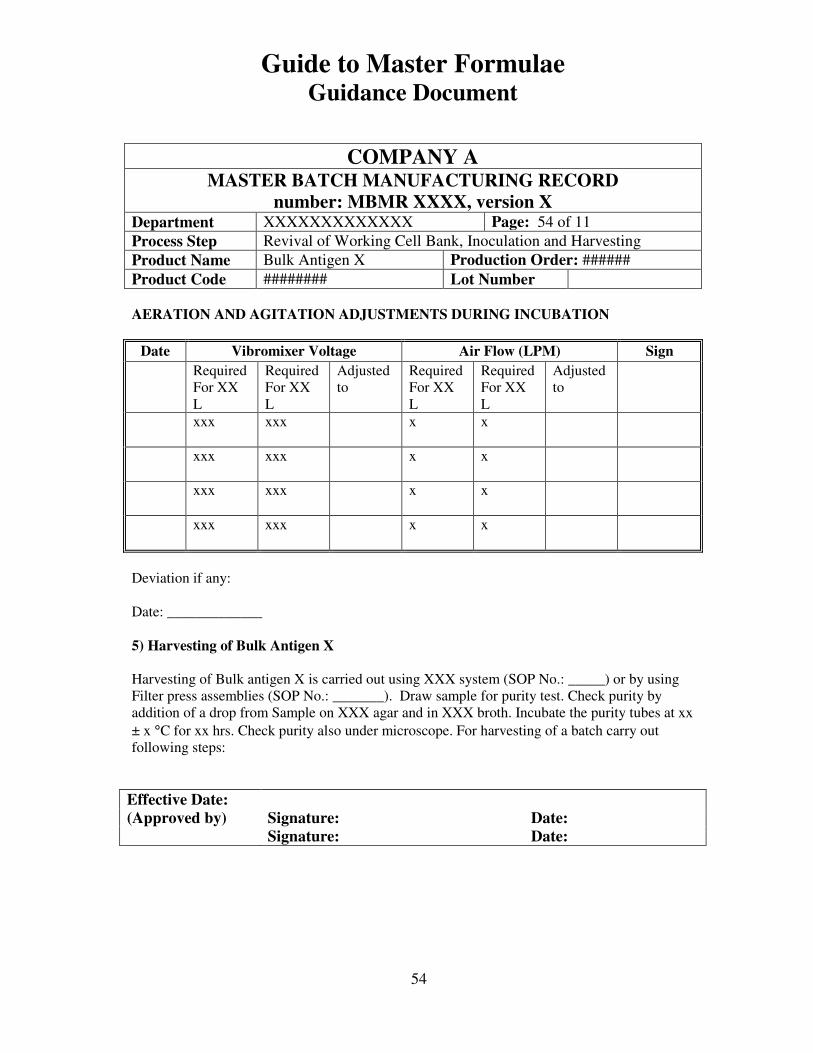
Guide to Master Formulae Guidance Document
54
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 54 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
AERATION AND AGITATION ADJUSTMENTS DURING INCUBATION
Date Vibromixer Voltage Air Flow (LPM) Sign
Required
For XX
L
Required
For XX
L
Adjusted
to
Required
For XX
L
Required
For XX
L
Adjusted
to
xxx xxx x x
xxx xxx x x
xxx xxx x x
xxx xxx x x
Deviation if any:
Date: _____________
5) Harvesting of Bulk Antigen X
Harvesting of Bulk antigen X is carried out using XXX system (SOP No.: _____) or by using
Filter press assemblies (SOP No.: _______). Draw sample for purity test. Check purity by
addition of a drop from Sample on XXX agar and in XXX broth. Incubate the purity tubes at xx
± x °C for xx hrs. Check purity also under microscope. For harvesting of a batch carry out
following steps:
Effective Date:
(Approved by) Signature: Date:
Signature: Date:

Guide to Master Formulae Guidance Document
55
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 55 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
Switch off temp. controller and recorder, Switch off Agitator controls, Stop Air flow
Temp. controller and recorder switched off
Agitator controls switched off
Air flow stopped
Sample drawn by
Purity test details. SOP No.: _______________
Material Quantity Lot No. Checked by
XXXX agar tubes
XXXX broth tubes
XXXX agar / broth tubes
incubated
Purity checked Checked by
From To Date results
Deviations, if any:
Date: _____________
Harvesting of Bulk antigen X Lot No.: ___________ on _____________.
Details of batch harvesting using XXXX system
Clean the XXXX system by flushing XXX L W.F.I. (Bulk)
Pre operation cleaning of system XXX L. W.F.I (bulk) flushed from _______to_______
Done by _____
Effective Date:
(Approved by) Signature: Date:
Signature: Date:

Guide to Master Formulae Guidance Document
56
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 8 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
Use of XXXX system for filtration of toxin.
Follow SOP No.: _______. Set inlet pressure in order to have initial filtrate rate of xxx to xxx LPH
Inlet Pressure set at _____bar. Initial filtrate flow-rate __________ LPH done by ________
FILTRATION DETAILS: Date: _____________
Time Filtration
Rate
(LPH)
Filtrate
Collected
(L)
Recirculation Details Observed/
Done by
Bulk Antigen is transferred to the Non- culture wing. Done by
____________________
Bulk Antigen Lot No.: __________ Filtration by XXXX System. Date: _____________
Effective Date:
(Approved by) Signature: Date:
Signature: Date:

Guide to Master Formulae Guidance Document
57
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 9 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
Cleaning of the System:
Wash the system with xxx L Potable water. Collect the washing in the fermentor vessel itself
by connecting the retentate and permeate pipes to the Fermentor. Thereafter disconnect the
pipes from the fermenter and keep these pipes in washing drum.
xxx L Potable Water wash: From : ________To ________ Checked by _____
I) Add xxx ml of x % XXX to xx L WFI (Bulk) and recirculate through XXX system for xx
minutes
xxx L WFI (Bulk) temp. ____°C + _______ml xxx % XXX (QC No. ) Checked by
:
Recirculation from : ________ to : __________ Checked by :
II) Add xx ml of xx % XXX to xx L WFI (Bulk) & recirculate through XXXX system for xx
minutes
xxx L WFI (Bulk) temp. ____°C + _______ml xxx % XXX (QC No. ) Checked by:
Recirculation from : ________ to : __________ Checked by :
III) Flush the system with W.F.I. (Bulk) xxx L
System flushed with ________W.F.I.(Bulk) From :________To : ______ Checked by ___
IV) XXX XXX wash :
Recirculate XXX XXX solution (Add xxx ml XXX XXX to xxx L W.F.I. (Bulk) temp. xx-
xx °C)
xxx L W.F.I. (Bulk) Temp. _____°C + ________ml XXX XXX (QC No. )
Checked by _______
Recirculation from : ________ to : __________ Checked by _______ Date :_____
V) Flush the system with W.F.I. (Bulk) xxx L
System flushed with _______W.F.I.(Bulk) From :_______To : ______ Checked by _______
Effective Date:
(Approved by) Signature: Date:
Signature: Date:

Guide to Master Formulae Guidance Document
58
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 10 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
Cleaning of the System: (continued)
VI) Check the Flow rate of W.F.I. (Bulk) at xxx bar inlet pressure. If the flow rate is less than
xxx LPH repeat washing procedure.
W.F.I.(Bulk) Flow-rate at xxx bar pressure : ______LPH (at ____°C) Checked by _______
All the washing from the above steps I to V connected to the drain.
VII) Storage of the system in xxx % XXX after recirculation for xxx minutes :
_______L WFI (Bulk) + ____ml XXX (Q. C. No. ________). Done by : _______
Recirculation from: ________ to: __________ Done by _________.
System disconnected from power supply at:
Operations supervised by:
Effective Date:
(Approved by) Signature: Date:
Signature: Date:

Guide to Master Formulae Guidance Document
59
COMPANY A
MASTER BATCH MANUFACTURING RECORD
number: MBMR XXXX, version X
Department XXXXXXXXXXXXX Page: 11 of 11
Process Step Revival of Working Cell Bank, Inoculation and Harvesting
Product Name Bulk Antigen X Production Order: ######
Product Code ######## Lot Number
BMR CERTIFICATION
01. MANUFACTURING DEPARTMENT:
The contents of this Document have been checked and verified by me.
The information contained herein is complete and true to the best of my
Knowledge. Deviations if any are reported.
Hence submitted to Quality Assurance Department.
Signature of Production Officer: ……………………………………..
Date of Completion: …………………………
Date of Submission: ………………………...
02. QUALITY ASSURANCE DEPARTMENT:
I hereby certify that, this batch record is reviewed by me, to ensure that the above
mentioned batch process has been carried out according to the Authorized Master
Formula and processing instructions.
All operational steps have been scrutinized & approved according to the
checklist (attached ) and have been found to be complete.
Signature of Quality Assurance Review Officer: …………………………
Date of Receipt: …………………………
Date of Approval: ……………………….
Effective Date:
(Approved by) Signature: Date:
Signature: Date:

Guide to Master Formulae Guidance Document
60
Appendix 8: Example two of a Master Formula.
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
PRODUCT X
FILL SIZE x mL
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
61
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
CERTIFICATION
CERTIFICATE OF QUALITY ASSURANCE
THIS IS TO CERTIFY THAT THE BATCH No. _______OF PRODUCT X (TRADE
NAME _______), SATISFIES THE REGULATORY AND PHARMACOPOEIAL
REQUIREMENTS FOR PRODUCT X VACCINE.
Signature of Quality Assurance:
Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
62
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
1. BATCH RECORD REVIEW AND APPROVAL REPORT
No Term Details
1 Name of the Product PRODUCT X
2 Batch No.
3 Date of Filling
4 Quantity Filled
5 Quantity Released
6 Mfg. Date
7 Exp. Date
Reviewed by QA (Analyst/Officer): Date: Approved by Head QA: Date:
2. CHECKLIST OF BATCH RECORD:
Signature of Supervisor: Date: Signature of QA: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:
No Description Date Document availability
checked by
Production QA
1 Product X Vaccine Blending
2 Primary packing materials procurement
3 Washing and sterilization of vials, stoppers and
vessels.
4 Sterilization of filling items
5 Filling
6 Filling particulars
7 Recording of deviations
8 Primary packing materials reconciliation record
9 Visual Inspection
11 Packing details for shipment to NCL
12 Finished goods transfer note to NCL

Guide to Master Formulae Guidance Document
63
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
3. QUALITY CONTROL REPORTS AND MISCELLANEOUS DOCUMENTS CHECK
LIST:
No Name of the Report Q.C Ref.No. Availability checked by
Production QA
1 Formulation buffer
2 Final Blend report (1)
3 Final Blend report (2)
4 Filled vials report (1)
5 Filled vials report (2)
6 Thermo graphs of Autoclave NA
7 Vials Depyrogenation report NA
8 Membrane Integrity report NA
9 Environmental monitoring report NA
10 Particle count report NA
11 NCL report NA
12 WFI report of blending port NA
13 WFI report of washing port NA
Signature of Supervisor: Signature of Quality Assurance: Date: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
64
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
Product X VACCINE BLENDING:
4.1 Volume of blend: _____ Liters.
4.2 Details of blending materials:
Description of material Lot No / B. No Q.C. Ref No. Assay
(mg/mL)
A) Bulk antigen (xx
mg/mL)
(SOP # ___/ Spec # ___)
B) Formulation solution #1
(xx mg/mL) (SOP #_____)
C) Formulation solution #2
(xx mg/mL) (SOP # ____)
Signature of Supervisor: Date:
4.3 Bulk antigen requirement for blend:
4.3.1 Antigen requirement:
Calculate the antigen requirement for the blend as per SOP # _______
Blend volume in mL (A) Total antigen required in mg (A x XX) / 1000
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
65
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
4.3.2 Details of antigen:
Lot No.
of Bulk
Q.C. Ref
No. and
Date
Antigen
concentration in
mg/mL (A)
Volume in
mL (B)
Total antigen in mg A x B
As mentioned above
Total volume
Total rounded off to
Signature of Supervisor: Date:
4.4 Solution #1 (FS#1) requirement for the blend:
4.4.1 Solution #1 (FS#1) requirement: Calculate Formulation Solution #1 requirement for the blend as per SOP # ______
Blend volume in mL Total XXXX required in mg (Blend volume x XX
mg/mL)
4.4.2 Details of Formulation Solution #1 (FS#1)
No. Batch
No.
Q.C. Ref.
No.
and Date
FS#1
content
(mg/mL)
(A)
Volume in
mL
(B)
Total FS#1 in (mg)
(A x B)
as mentioned above
Total volume
Total round off to
Signature of Supervisor: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:
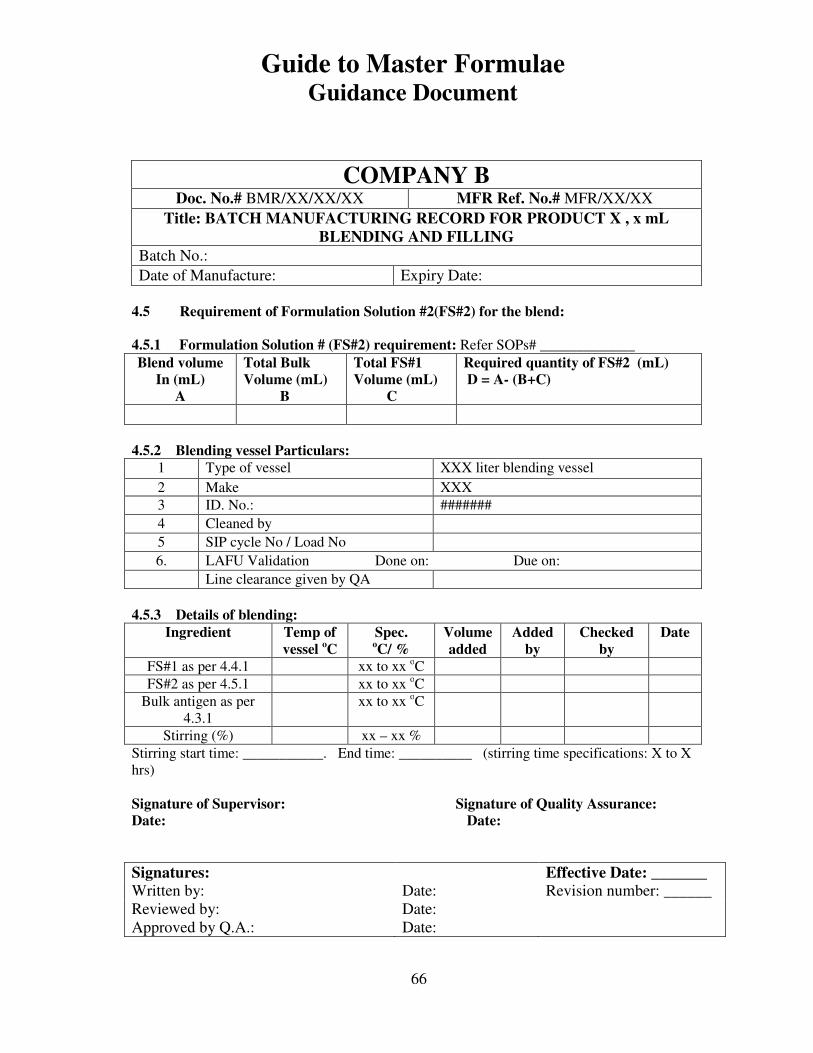
Guide to Master Formulae Guidance Document
66
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
4.5 Requirement of Formulation Solution #2(FS#2) for the blend:
4.5.1 Formulation Solution # (FS#2) requirement: Refer SOPs# _____________
Blend volume
In (mL)
A
Total Bulk
Volume (mL)
B
Total FS#1
Volume (mL)
C
Required quantity of FS#2 (mL)
D = A- (B+C)
4.5.2 Blending vessel Particulars:
1 Type of vessel XXX liter blending vessel
2 Make XXX
3 ID. No.: #######
4 Cleaned by
5 SIP cycle No / Load No
6. LAFU Validation Done on: Due on:
Line clearance given by QA
4.5.3 Details of blending:
Ingredient Temp of
vessel oC
Spec. oC/ %
Volume
added
Added
by
Checked
by
Date
FS#1 as per 4.4.1 xx to xx oC
FS#2 as per 4.5.1 xx to xx oC
Bulk antigen as per
4.3.1
xx to xx oC
Stirring (%) xx – xx %
Stirring start time: ___________. End time: __________ (stirring time specifications: X to X
hrs)
Signature of Supervisor: Signature of Quality Assurance:
Date: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
67
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
4.6 Final blend sampling details: For sample collection, labeling, storage details refer SOP# _________
Sampled
Quantity
Sampled
by
Date /
Time
Tests to be done SOP. No. /
Spec No.
QC Ref.
No. /
Date
Report
Date
xx mL
Description
pH
XXX content
XXX content
Bacterial Endotoxins
(LAL)
Sterility
XXX
Potency
Signature of Supervisor: Date: Signature of QA: Date:
5. PRIMARY PACKING MATERIALS PROCUREMENT:
No Name of
material
Spec.
No.
AR.
No.
Quantity
required
Quantity
issued
by store
Quantity
received
Checked by
Production
supervisor
Q.A.
in
charge
1 X mL vials
2 xx mm grey
butyl stoppers
3 xx mm
aluminium
seals
Remarks (if any):
Signature of Supervisor: Date: Signature of QA: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
68
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
6. WASHING AND STERILIZATION OF STOPPERS AND FILLING ITEMS: 6.1 Stoppers details: Size: xx mm Colour: grey butyl
6.1.1 Machine particulars:
Name of the machine: STOPPER WASHING MACHINE
I.D. No. xxxxxx
Validation Done on: Due on:
Cleaned by
Checked by
LAFU I.D. No: Validation Due on:
6.1.2 Treatment of stoppers: Treatment procedure of stoppers as per SOP # ________
6.1.2.1 Quantity of treatment solution (TS#1) required for stoppers:
Total volume of WFI (mL)
(A)
Total TS#1 required in mg at XX mg/mL
(A x XX)
6.1.2.2 Details of TS#1 treatment: (Final TS#1 concentration should be xx mg/mL)
No Ingredient STP.No./
Spec.No.
A.R. No. Qty
Required
Qty
weighed
Weighed
by
Checked by
1 TS#1 xxx mg
2 WFI NA xxxx mL
Volume of TS#1 solution
prepared
Volume of TS#1 solution
used
Volume of TS#1 solution discarded
Excess TS#1 solution discarded by: Checked by:
TS#1 treatment of stoppers done by: Date Checked by: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
69
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
6.1.3 20 mm Grey butyl rubber Stopper washing particulars: Stopper washing, WFI inspection procedure refer SOP # ______
6.1.3.1 Drain water of Stopper washing machine checked by___________
6.1.3.2 Lot No—I
Washing details:
No Date Operator
involved
in
washing
Washing Qty
Washed.
Final rinse drain
water inspected by Start
Time
End Time
1st rinse
2nd
rinse
3rd
rinse
Details of Stopper Collection into S.S cans for Stopper Lot No I:
Can
No
Date S.S can
cleaned
by
No. of
stoppers
collected
Collected
by
Thiomersal
solution
Checked by
Volume
added
Added
by
Repeat as necessary for as many lots of Stoppers that are required
6.1.4 Sterilization of stoppers: Sterilization procedure of stoppers and sterilization by autoclave refer SOP # ________.
6.1.4.1 Equipment particulars:
Name of the Machine and
Make
AUTOCLAVE XXXXXXX
I.D. No. #######
Validation Done on: Due on:
Cleaned by: Checked by:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
70
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
6.1.4.2 Details of stopper sterilization as per load pattern No.: 1
Date Load
No.
Can
No
Qty Loaded
by
Sterilization
temp
Sterilization
time
Specs. Un-
loaded by
Signature of Supervisor: Date:
6.2 Details of filling accessories sterilization as per load pattern No 2 Washing and sterilization of filling accessories refer SOP# _______
Date Load
No.
Items Qty Washed
&
Loaded
by
Sterilizat-
ion temp
Sterilizat-
ion time
Specs. Un-
loaded
by
Checke
d by
Filling
sets /
glass
syringes
#
xx
min at
XX oC
Stopper
Bowl
#
Chute #
Picker
wheel
#
Section
wheel
#
Glass
syringes
#
Forceps #
Silicon
tubes
#
20 L
vessel
#
SS tray #
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
71
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
Signature of Supervisor: Date:
6.3 Details of garments, gloves and wipes sterilization as per load pattern No 3
Dat
e
Loa
d
No.
Items Qt
y
Loade
d by
Sterilizati
on temp
Sterilizati
on time
Specifi
c-ation
Un-
loade
d by
Checke
d by
Garmen
ts
xx min
at
XX oC
Gloves
Wipes
Mask
Signature of Supervisor: Date:
6.4 Fumigation of blending and filling area: Fumigating procedure refer SOP# ___________
Area Date Formaldehyde
Quantity
WFI
Quantity
Fumigation
done by
Time Check-
ed by From To
Signature of Supervisor: Date:
Signature of QA: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
72
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
7. WASHING AND DEPYROGENATION OF VIALS. 7.1 Washing of vials: Vial washing, inspection and depyrogenation procedure as SOP #______.
7.1.1 Machine particulars:
Equipment Name: XXXXXXXX
Make XXXXXXX
ID.No. ###########
Validation Done on: Due on:
Cleaned by Checked by:
Parameter Results
Air pressure:
(Spec x to x Kg/cm2 )
Water pressure:
(Spec x to x Kg/cm2 )
Final rinsing WFI
Temperature:
(Spec xx to xx oC)
Flow of water in all
needles
Flow of air in all needles
Checked by
Date & Time
Signature of Supervisor: Date:
7.1.2 Vial washing particulars: LAFU I.D. No: ############# Validation Due on:
Date WFI sample
inspected by
Time of washing Vials
loaded
by
Washing
machine
operated by
Total No.
of vials
Washed
No. of
vials
broken Start
Time
End
Time
Total
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
73
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
7.1.3 Washed vials inspection (For particles): (Frequency every x hours)
Date Time Checked by Remarks
Signature of Supervisor: Date:
7.2 Depyrogenation of vials:
7.2.1 Machine particulars:
Equipment Name and Make XXXXXXX
I.D. No. ########
Validation Done on: Due on:
Cleaned by: Checked by:
7.2.2 Depyrogenation Details: Date Tunnel
start
time
Set
Temp
OC
Depyrog'n Tunne
l stop
time
Tunnel
Drive
mm/min
Temp
monitore
d by
Depyrog'n
data
enclosed
Yes/no
Checked
by
Quantity
depyrog'd Tem
p OC
(>
X)
Tim
e
Min
.
(≥
x)
Heater
1
Heater
2
Heater
3
Heater
4
< xxx
Signature of Supervisor: Date :
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
74
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
8.1 Filling line clearance : line clearance as per SOP# ____________
LAFU I.D. No: ######### Validation Due on:
8.1.1 Machine particulars and line clearance details:
Equipment Name Filling Machine Stoppering Machine Sealing Machine
Make XXXXX XXXXX XXXXX
I.D. No. ######## ######## ########
Room No.
Validation done on
Validation due on
Cleaned by
Checked by
Sterilization indicators on stoppers,
filling items, garments etc
Checked by Verified by QA
Filling area cleanliness
Fumigation details
Line clearance for filling given by QA:
8.2 Filling operation particulars:
8.2.1 Filling Operation: Filling operation refer SOP #____________
8.2.2 Filling Operation Details:
Date Time Filling
Machine
operator
Stoppering
Machine
operator
Sealing
Machine
operator
Filled vials
collected
by
Signature
of Prod.
Shift in
charge
Signature
of QA.
Shift in
charge
Signature of Supervisor: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
75
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
8.3 Final blend stirring: (Frequency of checking - Every x hour)
Stirring and temperature of vessel as per SOP# _______________
Date Time Stirring (%) Specification
(%)
Checked by
Production QA
xx - xx
Signature of Supervisor: Date:
8.4 Volume variation check during filling:
8.4.1 Filling operation: Filling operations, start up activities refer to SOP # ____ and to annex
for fill volume standards. Volume variations action limits refer SOP# ______
8.4.2 Volume check up Details: (Frequency every x to x hours)
Filling Date: Starting time: Closing time:
Pack Size X mL Syringe used for vol. Measurement: X mL
Fill Volume Spec xx.x – xx.x mL Calibration due on:
No Date Time
Volume drawn
Checked
by
Remarks
Nozzle
No-1
Nozzle
No-2
Nozzle
No-3
Nozzle
No-4
Nozzle
No-5
Nozzle
No-6
Start up volume checks in (mL)
1
2
3
In process Volume checks during filling in (mL)
1
2
3
Signature of Supervisor:
Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
76
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
8.5 Filling area monitoring: (Frequency every x hours)
Filling Date: Starting time:
Pack size: X mL Closing time:
Date Time Temp
(oC)
Spec
(oC)
Humidity
(%)
Spec
(%)
Checked
by
xx to xx oC
xx to xx%
Signature of Supervisor: Date:
8.6 Recording of Interferences/deviations during filling: Deviation action limits and procedures followed refer SOP# ____________
Note: This is the provision to record online interferences during filling if any.
Possible deviations: power failure, temperature out of specification, stirring of blend out of
specification, Fill volume out of specification, equipment problems etc.
Crate
No
Date Time Description of
Interference/Deviation
Recorded
By
Checked
by
Deviation report
No. if any
Note: This table helps to trace those vials that could have been filled during interferences (if
filled during interferences).
Signature of Supervisor: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
77
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
8.7 Filled vials sampling details for QC:
Collection of samples Refer SOP# _______and for number of samples Refer SOP ______
Date
Sampled
Quantity
Sampled
by QA
Tests to be done SOP/
Spec.
QC Ref. / Date
xx
Vials
Description
Identity
pH
XX content
XX content
Abnormal toxicity on guinea pigs
Abnormal toxicity on mice Report Date
Sterility
Bacterial endotoxins (LAL test)
Potency
Closure integrity test
Report checked by Prod. Date:
Report verified by QA Date:
Signature of Supervisor: Signature of Quality Assurance:
Date: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:
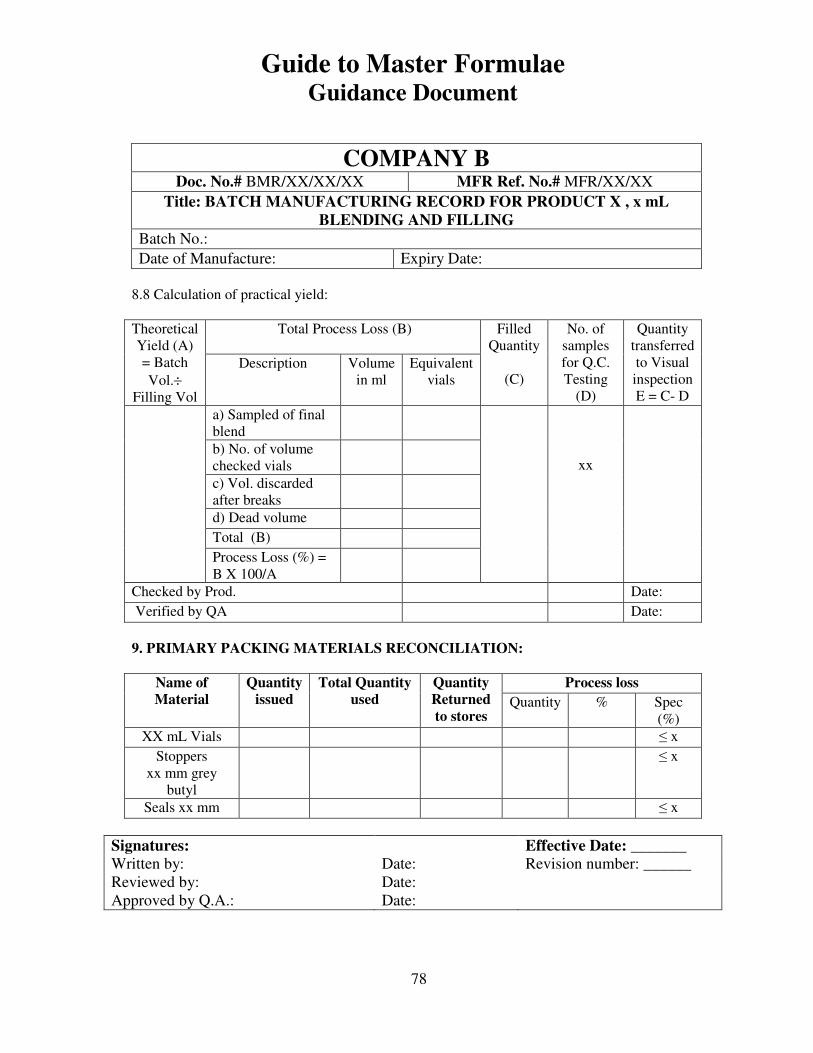
Guide to Master Formulae Guidance Document
78
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
8.8 Calculation of practical yield:
Theoretical
Yield (A)
= Batch
Vol.÷
Filling Vol
Total Process Loss (B)
Filled
Quantity
(C)
No. of
samples
for Q.C.
Testing
(D)
Quantity
transferred
to Visual
inspection
E = C- D
Description Volume
in ml
Equivalent
vials
a) Sampled of final
blend
xx
b) No. of volume
checked vials
c) Vol. discarded
after breaks
d) Dead volume
Total (B)
Process Loss (%) =
B X 100/A
Checked by Prod. Date:
Verified by QA Date:
9. PRIMARY PACKING MATERIALS RECONCILIATION:
Name of
Material
Quantity
issued
Total Quantity
used
Quantity
Returned
to stores
Process loss
Quantity % Spec
(%)
XX mL Vials ≤ x
Stoppers
xx mm grey
butyl
≤ x
Seals xx mm ≤ x
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
79
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
REMARKS (if any):
Signature of Supervisor: Signature of Quality Assurance:
Date: Date:
10. MATERIAL RETURN NOTE:
10.1 Material Return Note Number:
10.2 Material Return Details:
No Item code Description AR No. Indent
No.
Units Quantity Reason for
Returning
REMARKS (if any):
Returned By: Approved By: Received By:
Signature of Supervisor: Signature of Quality Assurance:
Date: Date:
11. VISUAL INSPECTION OF VIALS:
11.1 Line clearance for visual inspection: Refer Line clearance SOP# ________________
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
80
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
11.1.1 Cleaning of visual inspection area: Cleaning procedure refer SOP # ______________
Area cleaned by: Checked by:
Date: Date:
11.1.2 Line Clearance Details:
No Previous
product
Present
product
Previous
product
material
cleared
off from
the area
Yes /No
Checked.
By
Line
clearance
given by
QA
Date Time
Batch
No.
Pack
size
Batch
No.
Pack
size
Signature of Supervisor: Signature of Quality Assurance:
Date: Date:
11.2 Reconciliation of vials after Visual inspection: refer SOP# _____________
11.2.1 Quantity Received for Visual inspection: _____________________
11.2.2 Reconciliation Details: No Date
Time
Tested
by
Checked
by
Passed
Quantity
random
checked
by
Passed
Quantity Rejected vials as per possible defects
From To
Less
volume
(A)
Glass
pieces
(B)
Other
particles
(C)
Cap
Closures
defects
(D)
Total
(A+B+C+D)
1
2
3
Total Passed Quantity Total rejected Quantity
% rejection
Specification of optical rejection: ≤ x %
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
81
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
11.2.3 Good Quantity transferred to labeling after Visual inspection: ______________
Signature of Supervisor: Signature of Quality Assurance:
Date: Date:
12. DISCARD NOTE:
Date Item Quantity Reasons
for
destruction
Destruction
approved
by
Destruction
Destroyed
by
Supervised by
Production QA
Formulation
Solutions
Glass Vials
xx mm
stoppers
xx mm flip
top seals
Visual
inspection
rejections
Signature of Supervisor: Signature of Quality Assurance:
Date: Date:
13. BATCH ACCOUNT:
13.1 Details of batch account:
1 Theoretical yield
2 Filled quantity (b)
3 No. of vials given for Q.C + other samples
4 No. of vials given for visual inspection
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:

Guide to Master Formulae Guidance Document
82
COMPANY B Doc. No.# BMR/XX/XX/XX MFR Ref. No.# MFR/XX/XX
Title: BATCH MANUFACTURING RECORD FOR PRODUCT X , x mL
BLENDING AND FILLING
Batch No.:
Date of Manufacture: Expiry Date:
13.1 Details of batch account (cont.): 5 Total Optical rejections + breakages during
optical testing (e)
6 Passed quantity after Visual inspection.
7 No. of vials packed and sent to NCL
8 Net percentage Yield = (b - e X100)/b
9 Quantity remaining to be labeled and packed.
13.2 Batch packing details:
P.R.No.
Date Breakages Quantity
Packed
No. of
Samples
Quantity
Transferred
Quantity
remaining
1
2
3
4
5
Total
Signature of Supervisor: Signature of Quality Assurance:
Date: Date:
Signatures: Effective Date: _______ Written by: Date: Revision number: ______
Reviewed by: Date:
Approved by Q.A.: Date:
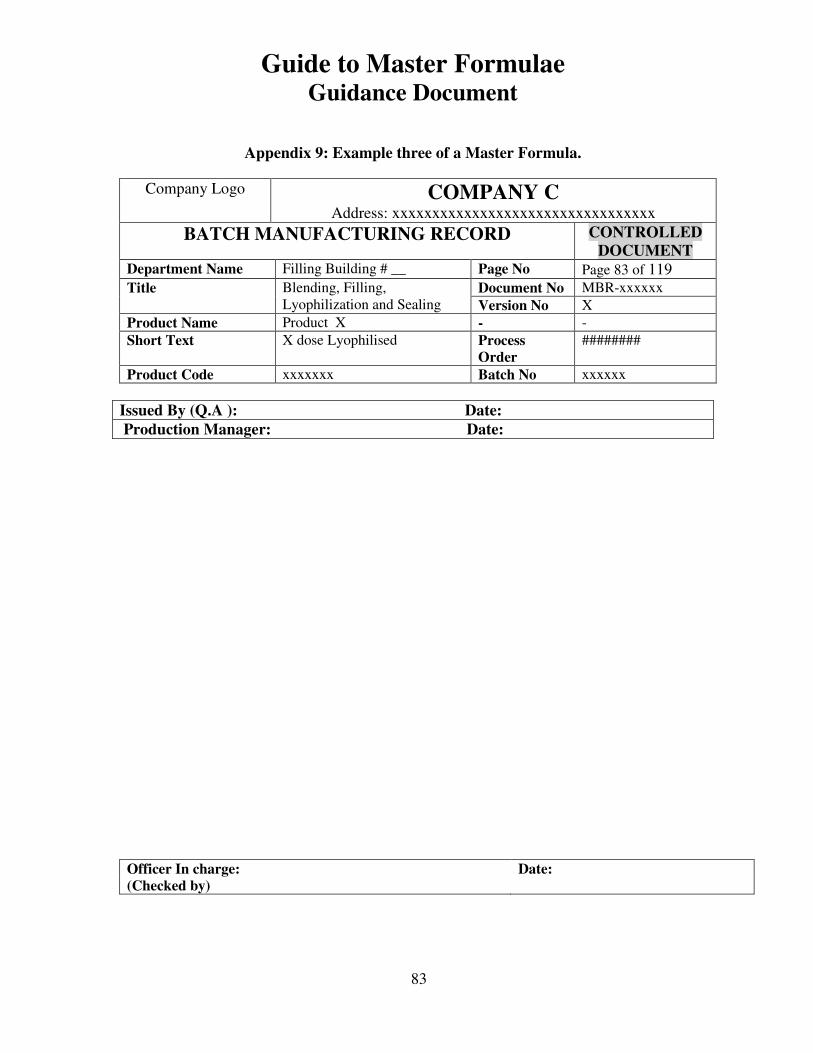
Guide to Master Formulae Guidance Document
83
Appendix 9: Example three of a Master Formula.
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department Name Filling Building # __ Page No Page 83 of 119
Title Blending, Filling,
Lyophilization and Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process
Order
########
Product Code xxxxxxx Batch No xxxxxx
Issued By (Q.A ): Date:
Production Manager: Date:
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
84
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department Name Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization and
Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
INDEX
No Description Page No
Summary of Process
Summary of Activities
Batch Formulation
Thawing of Bulk Antigen
Lyophilizer Trays Processing
Rubber Stopper Processing
Processing Product contact Material for Pooling & Filling
Vial Processing
Room Pressure Recording
Preparation of Lyophilizer
Verification of Bulk Antigen Container Weights.
Pooling & Clarification of Thawed Bulk and Preparation of Final Bulk
Integrity Testing of Product Filter
Filling
Integrity Test of Vent Filters
Loading of Lyophilizer
Lyophilization
Aluminum cap processing
Sealing of vials
Material Reconciliation record
Bulk reconciliation record
BMR Certification
Officer In charge: Date:
(Checked by)
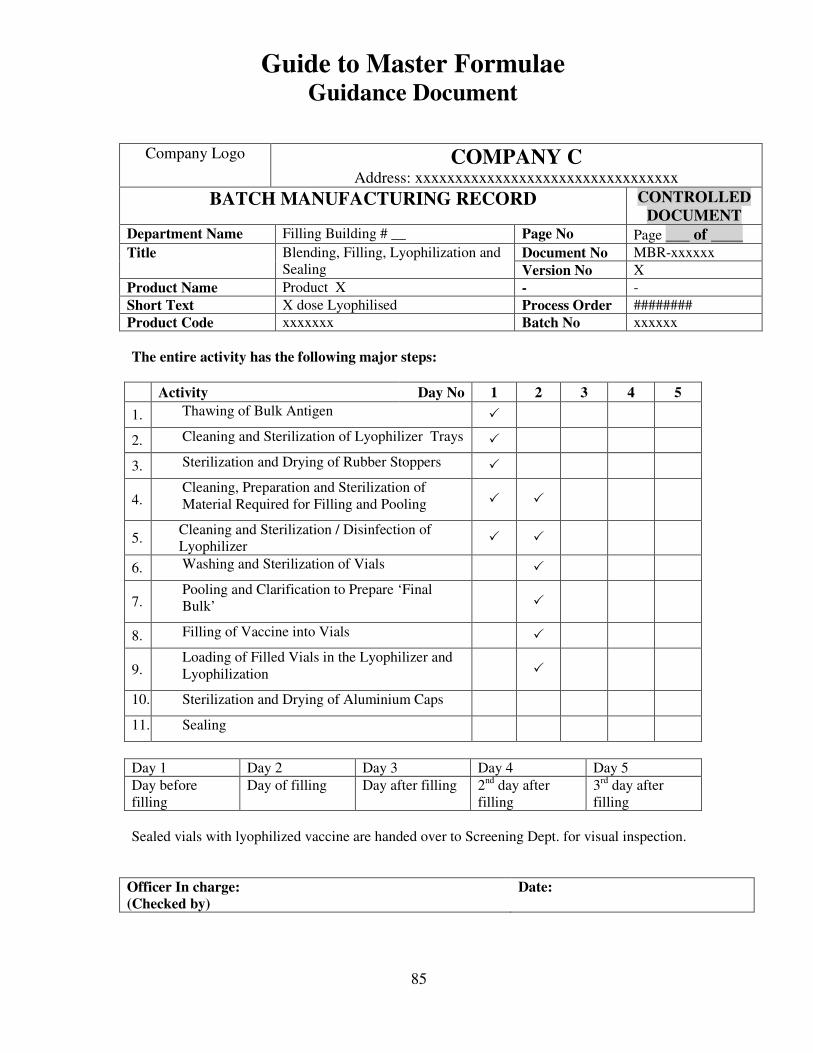
Guide to Master Formulae Guidance Document
85
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department Name Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization and
Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
The entire activity has the following major steps:
Activity Day No 1 2 3 4 5
1. Thawing of Bulk Antigen �
2. Cleaning and Sterilization of Lyophilizer Trays �
3. Sterilization and Drying of Rubber Stoppers �
4. Cleaning, Preparation and Sterilization of
Material Required for Filling and Pooling � �
5. Cleaning and Sterilization / Disinfection of
Lyophilizer � �
6. Washing and Sterilization of Vials �
7. Pooling and Clarification to Prepare ‘Final
Bulk’ �
8. Filling of Vaccine into Vials �
9. Loading of Filled Vials in the Lyophilizer and
Lyophilization �
10. Sterilization and Drying of Aluminium Caps
11. Sealing
Day 1 Day 2 Day 3 Day 4 Day 5
Day before
filling
Day of filling Day after filling 2nd
day after
filling
3rd
day after
filling
Sealed vials with lyophilized vaccine are handed over to Screening Dept. for visual inspection.
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
86
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department Name Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization and
Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Pages 5 and 6 to be issued to Bulk section who will fill the details and send it back.
Pages 25 To 27 to be issued to Lyophilization section who will fill the details and send it back.
SUMMARY OF ACTIVITIES
Target Lyophilizer Number XXXX
Lyophilizer Batch Capacity Vials
Volume per vial mL (23)
Batch Volume as Dispensed by Bulk Mfg Dept L (24)
Date of Filling dd/mm/yyyy
Fill volume per vial (xx x xx) (Overfill mid point at +X
%)
mL (25)
Batch Volume as Rechecked by Filling Dept L
Theoretical Batch Size Vials (28)
Approx Filling Yield. [Quantity loaded in
Lyophilizer]
Vials (22)
Approx Sealing Yield. [Quantity transferred to
screening]
Vials (19)
Note: Acceptable variation between 24 and 26 which is caused by removal of outer packaging /
double bagging should not be more than ± X %
Activity: BATCH FORMULATION Ref SOP No.: ______
Select the bulks for the batch on the basis of bulk Ag titre, volume, filling volume per vial, and
batch size. Filling personnel* to cross check in terms of Weights on receipt.
Officer In charge: Date:
(Checked by)
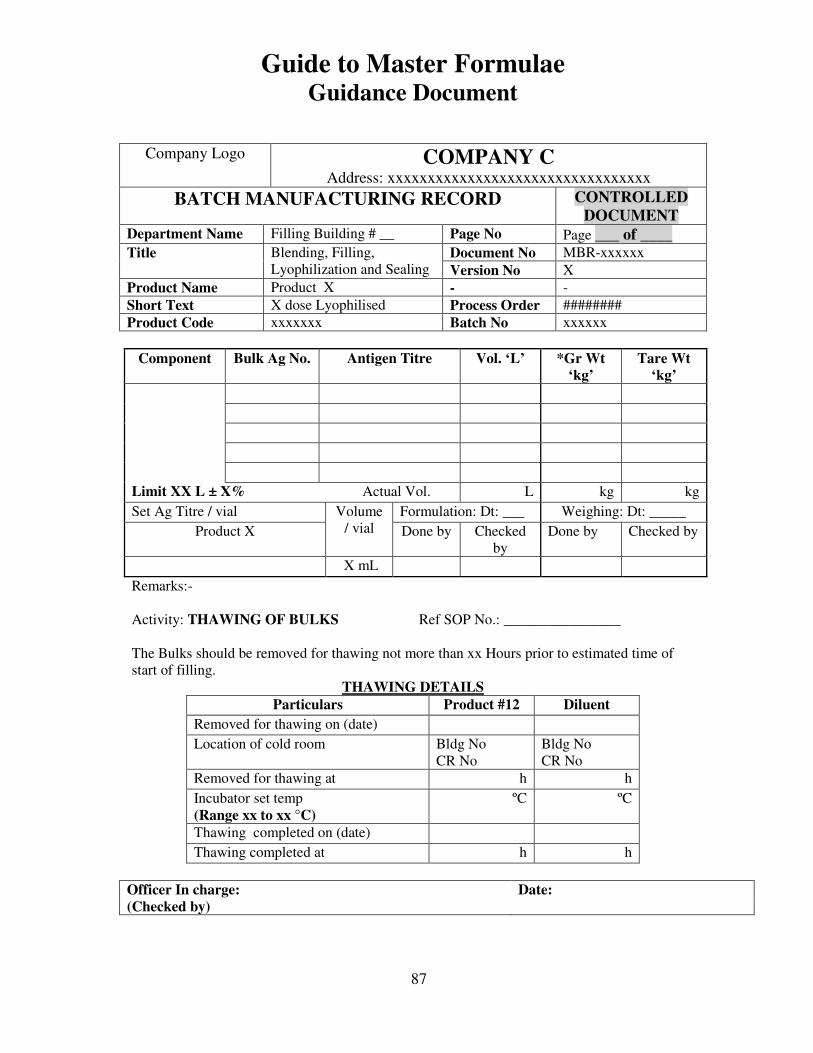
Guide to Master Formulae Guidance Document
87
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department Name Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization and Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Component Bulk Ag No. Antigen Titre Vol. ‘L’ *Gr Wt
‘kg’
Tare Wt
‘kg’
Limit XX L ± X% Actual Vol. L kg kg
Set Ag Titre / vial Volume
/ vial
Formulation: Dt: ___ Weighing: Dt: _____
Product X Done by Checked
by
Done by Checked by
X mL
Remarks:-
Activity: THAWING OF BULKS Ref SOP No.: ________________
The Bulks should be removed for thawing not more than xx Hours prior to estimated time of
start of filling.
THAWING DETAILS
Particulars Product #12 Diluent
Removed for thawing on (date)
Location of cold room Bldg No
CR No
Bldg No
CR No
Removed for thawing at h h
Incubator set temp
(Range xx to xx °C)
ºC ºC
Thawing completed on (date)
Thawing completed at h h
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
88
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
No Handling of Bulk Antigen Containers Done By Date
Name Initials
1
2
3
4
Bulk Antigen Containers Received By:
Remarks if any:
Activity: LYOPHILIZER TRAY PROCESSING Ref SOP No.: __________
1. Clean the trays to be used for collecting filled vials using a clean lint free mop wetted with
WFI/xx % Isopropyl alcohol.
2. The trays can be processed in either of two ways.
A) Load the cleaned trays on the dry heat sterilizer (DHS) trolley, as per validated max
loading pattern in DHS No xxxx and sterilize at xxx°C, xxx minutes.
B) Load the trays in Autoclave and sterilize at xxx °C ± x °C for XX minutes.
No of Trays
Pattern No
Pattern No
3. If option A above is selected, run the cycle to hold the trays at xxx°C for xxx minutes.
Normal variation –x ° to + x °C.
4. Write Product, B.No., Charge No & date on the sterilizer thermograph/printout (for Option
A only) sign it and attach it to this BMR.
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
89
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
No Description of Activity Done
By
Checked
By
Date
1 No of trays cleaned
_______
Quantity for batches: ____
2 Loaded ______ cleaned trays onto the trolley.
Loaded in Sterilizer Load pattern no Charge No
# xxxxxx xxxx xx
# xxxxxx xxxx xx
Checked Cycle parameters A) xxx °C, xxx minutes
B) xxx °C ± x°C for xx
minutes
3 Sterilization temperature xxx °C achieved
at (applicable for option A above)
h
4 Sterilization temperature xxx°C
maintained till (applicable for option A
above)
h
Remarks:-
Activity: RUBBER STOPPERS PROCESSING Ref SOP No: ___________
1. Verify that QC approved stoppers of correct type have been taken for de-cartoning.
2. For RFS stoppers, after de cartoning directly move to sterilization.
3. Load stoppers as per validated loading pattern given below.
RFS
Max No of stoppers / pouches xxxx
Pattern No for Autoclave X xxxx
Pattern No for Autoclave Y xxxx
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
90
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
4. The stoppers can be processed in either of the two ways:
A) Sterilization in Autoclave X (xxx to xxx °C) and drying in dry heat sterilizer (DHS)
No ___
B) Sterilization and drying in Autoclave Y.
In both the cases, carry out sterilization at 121°C for xxx minutes, Write Product, B. No.,
Charge No and date on the sterilizer printout / thermograph sign it and attach it to this BMR
5. If option A above is selected, then transfer the sterilized rubber stoppers from the autoclave
to the DHS from clean room side for drying. Start drying cycle: xxx°C for xxx mins.
Normal variation - x °C to + x °C/ ± x min. If option B above is selected then Sterilize
stoppers at xxx °C ± x °C for xxx minutes followed by a drying cycle for xxx mins in the
autoclave. In case of greater variation in temperature or time, the deciding factor will be the
moisture content. Write Product, B. No., Charge No and date on the sterilizer thermograph
/printout (for Option A only) sign it and attach it to this BMR.
6. Draw one sample for Moisture content analysis (Limit NMT xxx mg/stopper). In case
stoppers of 2 batches are sterilized in 1 load, draw only 1 sample, record both batch nos on
TRF, attach copies to both BMRs.
A) Stopper input: xx mm, Lyo Grey Butyl Rubber
1) Unprocessed b/f:
B. No________
2) Processed b/f: B.
No__________
3) Fresh quantity
issued
4) Fresh used from
issue
5) Option for
Process
� A =1+2+4
� B =1+4
Actually Loaded for Sterilization Unprocessed
Done by Checked by Date
9) No of stoppers Pouches
Remarks:
Activity: RUBBER STOPPERS PROCESSING, cont'd Ref SOP No.: ___________
7. Load pouches on the Autoclave trolley as per validated loading pattern.
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
91
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Autoclave
No. /
Equipment
No.
Charge
No.
Load
Pattern
No.
Program
No.
Quantity
for 1 / 2
batches
Done
by
Checked
by
Date
######## XX XX X
######### XX XX X
DHS No / Equip No #################### Charge
No.:
X
Drying temperature xxx °C for xxx minutes for drying in
DHS Done by Checked
by
Date
Achieved at : h
Maintained till: h
Sample for Moisture Content (11) Nos.
NOTE: Filling activity should not be started before the result of moisture content estimation is
received from Q.C.
Remarks:
Activity: PROCESSING PRODUCT CONTACT MATERIAL FOR POOLING AND
FILLING Ref SOP No.: _______________________
1. Use filters based on the following table for B. Size for Filtration> xx L
2. Carry out pre-filtration integrity test using the following data
Officer In charge: Date:
(Checked by)
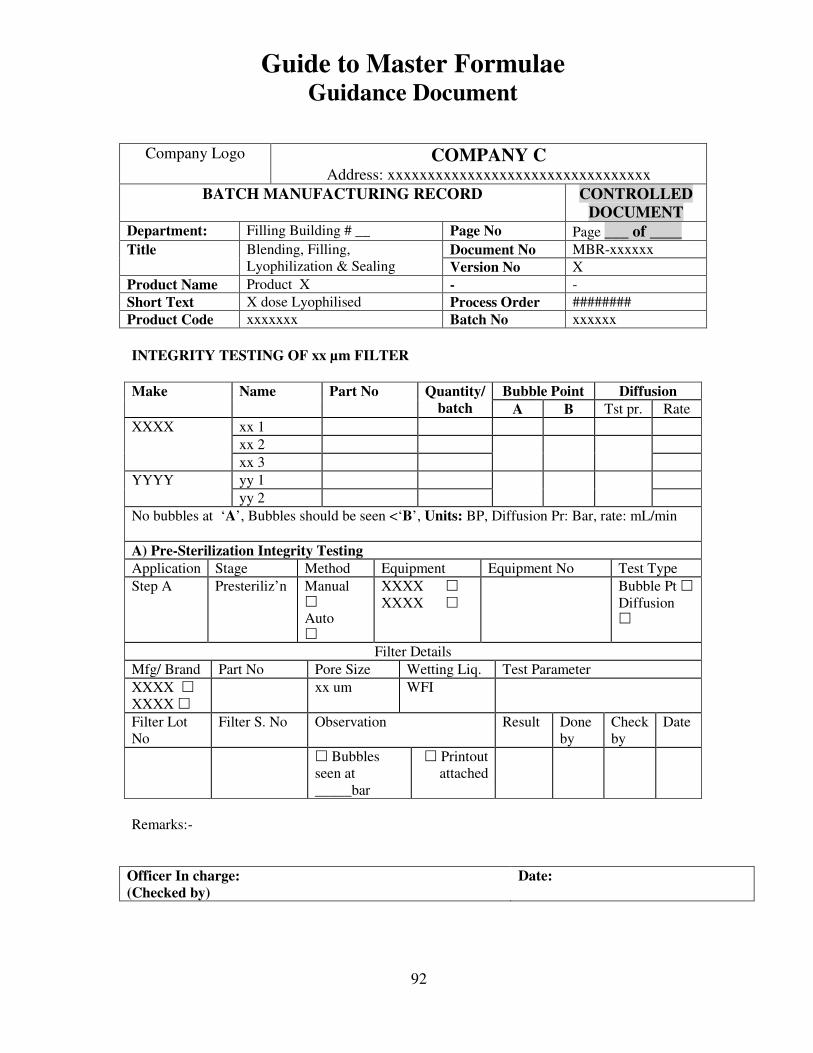
Guide to Master Formulae Guidance Document
92
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
INTEGRITY TESTING OF xx µm FILTER
Make Name Part No Quantity/
batch
Bubble Point Diffusion
A B Tst pr. Rate
XXXX xx 1
xx 2
xx 3
YYYY yy 1
yy 2
No bubbles at ‘A’, Bubbles should be seen <‘B’, Units: BP, Diffusion Pr: Bar, rate: mL/min
A) Pre-Sterilization Integrity Testing
Application Stage Method Equipment Equipment No Test Type
Step A Presteriliz’n Manual � Auto �
XXXX �
XXXX �
Bubble Pt �
Diffusion �
Filter Details
Mfg/ Brand Part No Pore Size Wetting Liq. Test Parameter
XXXX �
XXXX �
xx um WFI
Filter Lot
No
Filter S. No Observation Result Done
by
Check
by
Date
� Bubbles
seen at
_____bar
� Printout
attached
Remarks:-
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
93
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Activity: PROCESSING PRODUCT CONTACT MATERIAL FOR POOLING AND
FILLING Ref SOP No.: _______________________
1. Inactivation: Inactivate the Material used for Pooling and Filling in the Autoclave by
heating it to xxx°C for x minutes. Record in the autoclave Log Book. (Variation xx°C to
xx°C).
2. Preparation: Unload the inactivated material from the Autoclave on the Washing room side;
Wash inactivated material by passing Cooled WFI. Draw a sample from the tanks & check
conductivity. Draw samples from Tanks, Silicon tube to header, Header, Syringes, needles
& screen for particulates. In case the previous product is Media fill, draw additional
samples submit to QC with Product Changeover TRF. On clearance wrap in sterilization
pouches.
3. In case the vent filters on BV2 are new, carry out WIT and attach the results on opposite
page. In case they continue from the previous day, the Post use integrity test of the previous
batch will be treated as the pre-use integrity test of this batch.
4. Check the plan for the next day & select the loading pattern. Load the wrapped material on
the autoclave trolley as per validated loading pattern. Attach a list of material sterilized, to
this BMR. Write Product, B. No., Charge No and date on the sterilizer printout /
thermograph (for Autoclave XX only) sign it and attach it to this BMR.
Conductivity check on product tank rinse sample NMT 1 mS/cm
Check xx
mS/cm
BV1 BV2 Product C/O Done by Checked
by
Date
_____
mS/cm
____
mS/cm
____
mS/cm
� NA
� TRF attached
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
94
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Visual Inspection of Product Contact material rinse sample
Item BV 1 BV 2 Silicon
tube
Header Syringe Needle
Fibres
Particles
Sampled by: Screened by: Date:
STERILIZATION OF MATERIAL FOR POOLING AND FILLING
1 Autoclave No / Equip No ######### / ######## Done by Checked
by
Date
2 Load Pattern number XXX XXX
3 Charge No XX XX
4 Program No XXXXX XXXXX
Remarks:-
Activity: VIAL PROCESSING – WASHING Ref SOP No.: _________
1. Verify that QC approved vials of correct type have been taken for de-cartoning.
2. De-carton the Vials, transfer them into clean SS trays and pass them to the washing area.
3. Ensure WFI cooling assembly is sanitized before commencing Washing of xst batch. Ensure
pressure of Fresh and Recycled WFI and compressed air, conductivity of Fresh WFI are
within limits. Ensure area is free of previous product vials.
4. Perform Test for Adequacy using X washed vials.
5. After operation is completed, drain the WFI assembly and clean the machine.
Officer In charge: Date:
(Checked by)
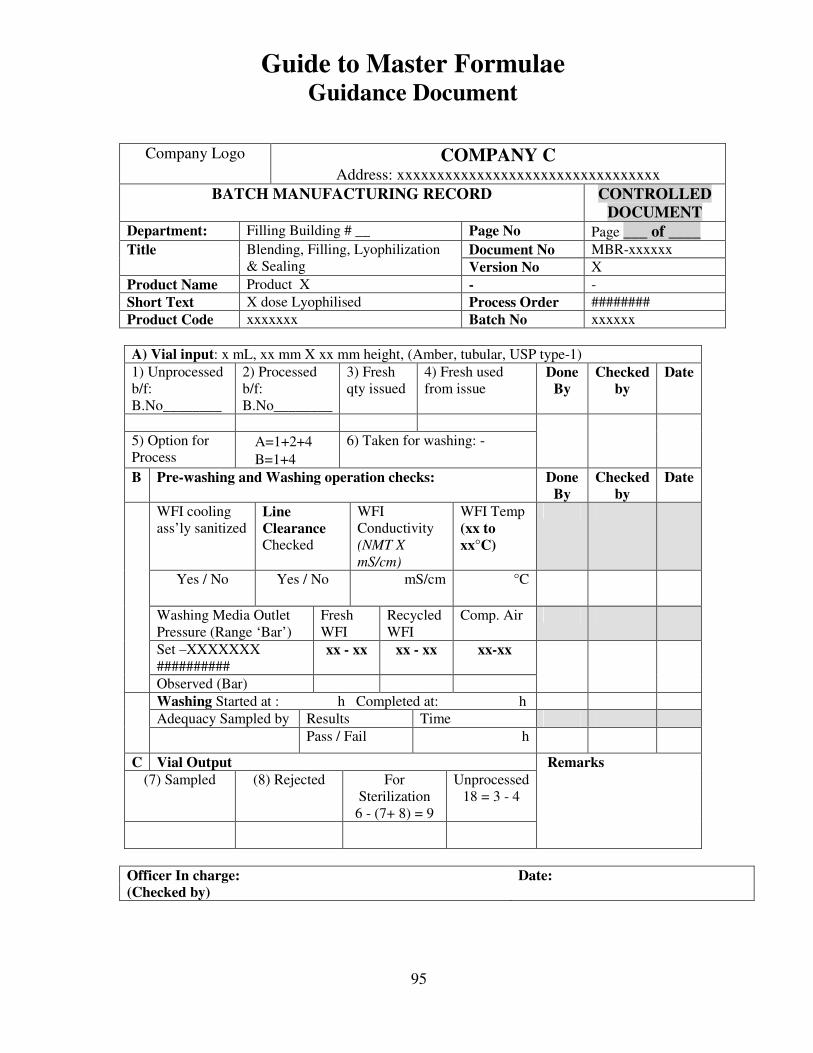
Guide to Master Formulae Guidance Document
95
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
A) Vial input: x mL, xx mm X xx mm height, (Amber, tubular, USP type-1)
1) Unprocessed
b/f:
B.No________
2) Processed
b/f:
B.No________
3) Fresh
qty issued
4) Fresh used
from issue Done
By
Checked
by
Date
5) Option for
Process � A=1+2+4
� B=1+4
6) Taken for washing: -
B Pre-washing and Washing operation checks: Done
By
Checked
by
Date
WFI cooling
ass’ly sanitized Line
Clearance Checked
WFI
Conductivity
(NMT X
mS/cm)
WFI Temp
(xx to
xx°C)
Yes / No Yes / No mS/cm °C
Washing Media Outlet
Pressure (Range ‘Bar’)
Fresh
WFI
Recycled
WFI
Comp. Air
Set –XXXXXXX
########## xx - xx xx - xx xx-xx
Observed (Bar)
Washing Started at : h Completed at: h
Adequacy Sampled by Results Time
Pass / Fail h
C Vial Output Remarks
(7) Sampled (8) Rejected For
Sterilization
6 - (7+ 8) = 9
Unprocessed
18 = 3 - 4
Officer In charge: Date:
(Checked by)
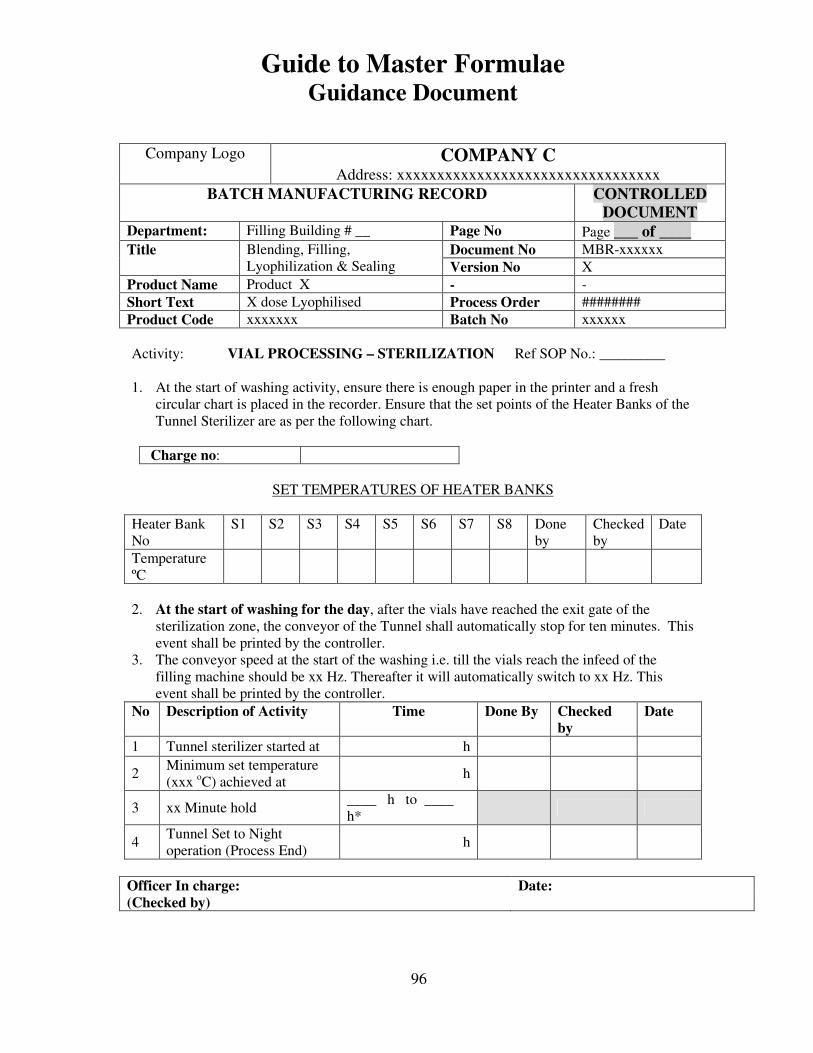
Guide to Master Formulae Guidance Document
96
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Activity: VIAL PROCESSING – STERILIZATION Ref SOP No.: _________
1. At the start of washing activity, ensure there is enough paper in the printer and a fresh
circular chart is placed in the recorder. Ensure that the set points of the Heater Banks of the
Tunnel Sterilizer are as per the following chart.
Charge no:
SET TEMPERATURES OF HEATER BANKS
Heater Bank
No
S1 S2 S3 S4 S5 S6 S7 S8 Done
by
Checked
by
Date
Temperature
ºC
2. At the start of washing for the day, after the vials have reached the exit gate of the
sterilization zone, the conveyor of the Tunnel shall automatically stop for ten minutes. This
event shall be printed by the controller.
3. The conveyor speed at the start of the washing i.e. till the vials reach the infeed of the
filling machine should be xx Hz. Thereafter it will automatically switch to xx Hz. This
event shall be printed by the controller.
No Description of Activity Time Done By Checked
by
Date
1 Tunnel sterilizer started at h
2 Minimum set temperature
(xxx oC) achieved at
h
3 xx Minute hold ____ h to ____
h*
4 Tunnel Set to Night
operation (Process End) h
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
97
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
4. Attach the printout and circular chart to the BMR.
Operators – Washing and Sterilization:-
Remarks: - * xst / x
nd batch of the day.
Activity: VIAL PROCESSING – STERILIZATION, continued Ref SOP No.:
_________
1. Check the pressures of the three zones of the tunnel, at least once, preferably at start of
washing. If the pressures are not within the range given below, immediately stop washing
activity and inform the Officer in charge.
2. At the end of washing, perform sanitization of the bulk WFI cooling assembly.
3. Write the Product, B. No., Charge no and date on the sterilizer printout / graph, sign it and
attach it with this page of BMR.
CHART SHOWING THE PRESSURES OF THE VARIOUS ZONES OF THE TUNNEL
#XXX
Pressure
Range
Drying Zone
(P1) mm wg
Sterilization Zone
(P2)
mm wg
Cooling
Zone (P3)
mm wg
Done by Checked
by
Time
xx --xx xx --xx xx --xx
Observed
Observed
Remarks:-
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
98
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Activity: ROOM PRESSURE RECORDING Ref SOP No.: ____________
1. Record ‘Room differential pressure’ before entering the clean area for pooling and filling.
No Room & range
Observed value
(Pa)
Done
by
Checked
by
Date Time
1. PAL 1 (xx-xx)
2. PAL 2 (xx-xx)
3. PAL 3 (xx-xx)
4. Filling (xx-xx)
5. Tunnel Room (xx-xx)
6. Sealing (xx-xx)
7. Pooling (xx-xx)
Remarks:
Activity: PREPARATION OF LYOPHILIZER Ref SOP No.: ________
1. After Unloading, Carry out cleaning of Lyophilizer and record in the BMR.
2. Carry out Sterilization / Disinfection as below:
A week1 denotes 6 working days with the day the Lyophilizer gets Steam
Sterilized/Steamed being declared the 1st day of the week and the batch filled after steam
sterilization/ steaming the 1st batch. If the third batch is to be loaded later than the 6
th day of
the week, it is treated as the first batch of the next week. The gap between
Sterilization/disinfection step and loading should not be more than xx hours.
Officer In charge: Date:
(Checked by)
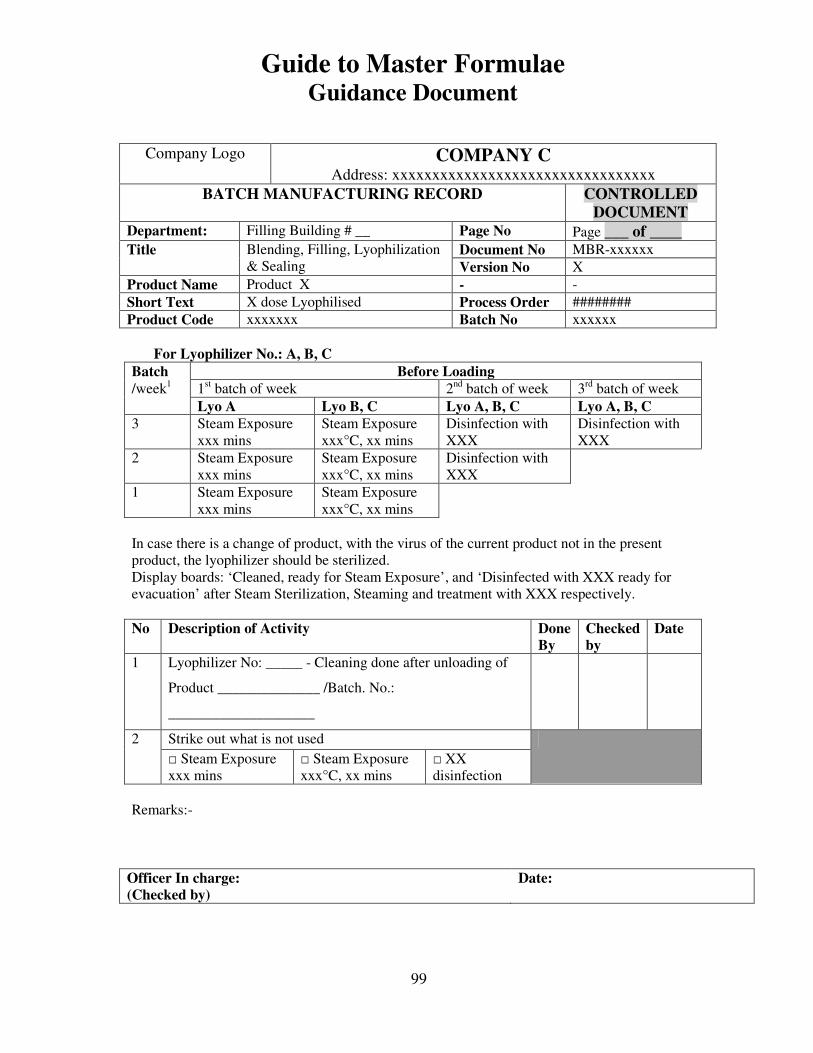
Guide to Master Formulae Guidance Document
99
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
For Lyophilizer No.: A, B, C
Batch Before Loading
/week1 1
st batch of week 2
nd batch of week 3
rd batch of week
Lyo A Lyo B, C Lyo A, B, C Lyo A, B, C
3 Steam Exposure
xxx mins
Steam Exposure
xxx°C, xx mins
Disinfection with
XXX
Disinfection with
XXX
2 Steam Exposure
xxx mins
Steam Exposure
xxx°C, xx mins
Disinfection with
XXX
1 Steam Exposure
xxx mins
Steam Exposure
xxx°C, xx mins
In case there is a change of product, with the virus of the current product not in the present
product, the lyophilizer should be sterilized.
Display boards: ‘Cleaned, ready for Steam Exposure’, and ‘Disinfected with XXX ready for
evacuation’ after Steam Sterilization, Steaming and treatment with XXX respectively.
No Description of Activity Done
By
Checked
by
Date
1 Lyophilizer No: _____ - Cleaning done after unloading of
Product ______________ /Batch. No.:
____________________
2 Strike out what is not used
□ Steam Exposure
xxx mins
□ Steam Exposure
xxx°C, xx mins
□ XX
disinfection
Remarks:-
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
100
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Activity: VERIFICATION OF BULK ANTIGEN CONTAINER WEIGHTS Ref SOP
No.: _____
1. Unwrap the outer packing of the thawed bulk antigen containers.
2. Check the Bulk antigen container numbers and record their gross Weights in the Batch
Formulation Sheet.
3. Carry out surface disinfection and transfer them to clean room, through hatch.
Net Weight of
Formulation
(Gross – Tare on
Page 5)
Weight in g /mL
(B) (From Page
20) (Same as kg
/L)
Net Volume of
formulation
(Net Wt / kg/L)
Done
by
Checked
by
Date
___________ kg ___________ kg/L (26) _______
L
Remarks:-
Activity: POOLING & CLARIFICATION OF THAWED BULK AND PREPARATION
OF FINAL BULK Ref SOP No.: ________
1. The contents of the Bulk antigen containers are pooled in a sterilized, SS blending vessel, with
the jacket at a temperature NMT XX°C. Start the magnetic stirrer (xxx to xxx rpm) after the
level of bulk antigen inside the tank has gone above the impeller. If the temperature of the
pool exceeds xxoC, immediately shut off the heating immersion circulator and the magnetic
stirrer.
2. The pool of bulk antigens is clarified using a sterilized xx µm filter capsule/s. The outlet of the
xx µm filter/s is connected to a 2nd
sterilized SS blending vessel with the jacket at a
temperature of XX – XX°C. Clarification should be started only after the temperature of the
pool is in the range of XX –XX°C for xx minutes. The contents of the second SS blending
vessel will be the final bulk for the batch.
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
101
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
No Particulars Done by Checked by Date
1 Bath temp before Pooling
Pooling: xx-xx°C Start End Blending
vessel
°C ______
h
______ h A
B
2 Temperature of Stirrer Clarification: A, B
Pool Final
Bulk
RPM Start time End time
xx -
xx°C
xx - xx°C xxx-xxx
°C °C ______ h ______ h
3 Filtration Residue
(29)
Sampled Bulk (30)
mL XX mL x X bottles = XX mL
Remarks:-
Activity: POOLING & CLARIFICATION OF THAWED BULK AND PREPARATION
OF FINAL BULK Ref SOP No.: _________________________
1. Check the integrity of xx µm filter capsule by bubble point method. Connect the SS
blending vessel with the final bulk to the glass header only after the integrity test is done
and the filter capsule has passed the test.
2. Inactivation: Inactivate the Material used for Pooling in the Autoclave by heating it to
XXX °C for X minutes. (Variation permitted XX °C to XX °C).
3. Perform the environmental monitoring activities as per SOP No.: _______. This includes
exposing settle plates, contact plates and Air sampling as per location map, Swabs of
critical surfaces, Finger dabs of operators involved in pooling & filling operation.
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
102
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
FILTER INTEGRITY TESTING
Application Stage Method Equipment Equipment No Test Type
Clarification Post-Clarific’n XXXXX XXXXX ######## Bubble Pt
Filter Details
Mfg/ Brand Part No Pore Size Wetting
Liq.
Test Parameter
Filter Lot
No
Filter S. No. Observation:
bubbles @
Result Done by Checked
by
Date
_______________
bar
Pooling material
Inactivation
Autoclave
No:
Charge no: Done by Checked
by
Date
Remarks:-
Activity: FILLING Ref SOP No.: _________________
1. Volume per vial (23) is indicated on page 5 of this BMR. The Volume per vial is the lower
limit of the permitted volume range. The upper range is calculated by adding X % of the
Volume per vial. The weight range is derived from this volume range by multiplying it
with weight/mL of the bulk. Calibrate the balance to be used for verifying the Fill volume.
2. Check for absence of previous product components. Carry out the priming of the header
and the pumps, collect the priming liquid and send as sample of final bulk to Q.C.
Officer In charge: Date:
(Checked by)
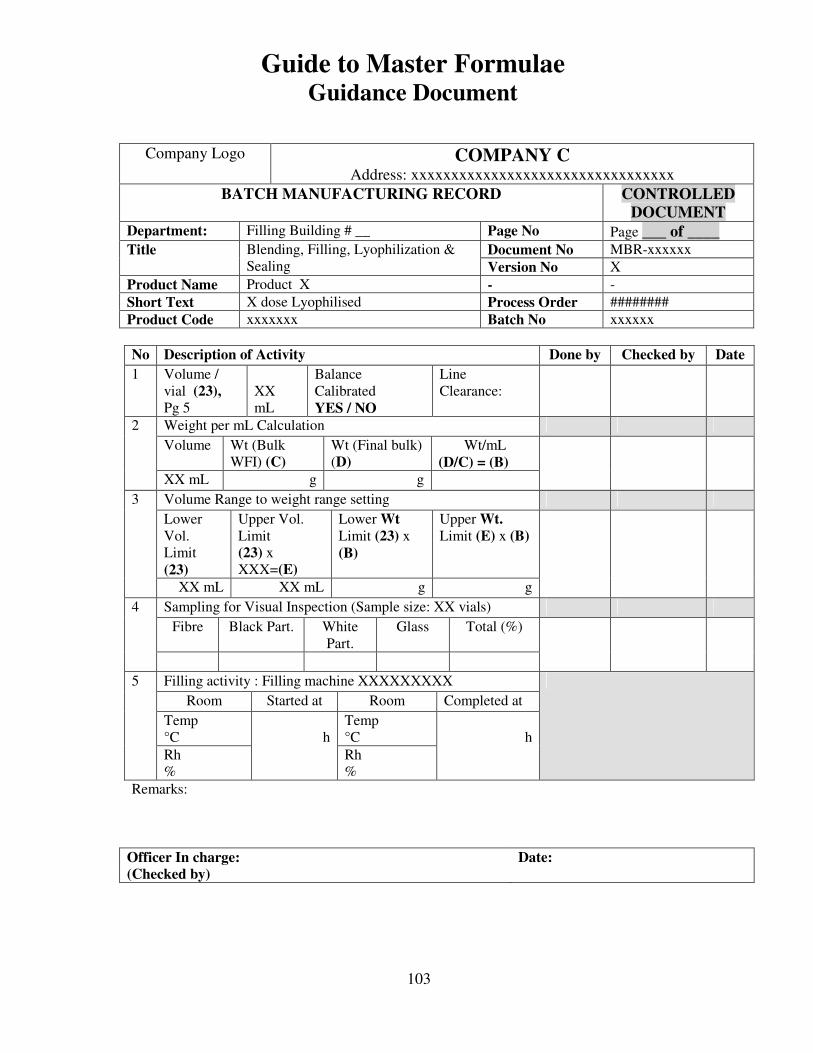
Guide to Master Formulae Guidance Document
103
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization &
Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
No Description of Activity Done by Checked by Date
1 Volume /
vial (23),
Pg 5
XX
mL
Balance
Calibrated
YES / NO
Line
Clearance:
2 Weight per mL Calculation
Volume Wt (Bulk
WFI) (C)
Wt (Final bulk)
(D)
Wt/mL
(D/C) = (B)
XX mL g g
3 Volume Range to weight range setting
Lower
Vol.
Limit
(23)
Upper Vol.
Limit
(23) x
XXX=(E)
Lower Wt
Limit (23) x
(B)
Upper Wt.
Limit (E) x (B)
XX mL XX mL g g
4 Sampling for Visual Inspection (Sample size: XX vials)
Fibre Black Part. White
Part.
Glass Total (%)
5 Filling activity : Filling machine XXXXXXXXX
Room Started at Room Completed at
Temp
°C
h
Temp
°C
h
Rh
%
Rh
%
Remarks:
Officer In charge: Date:
(Checked by)
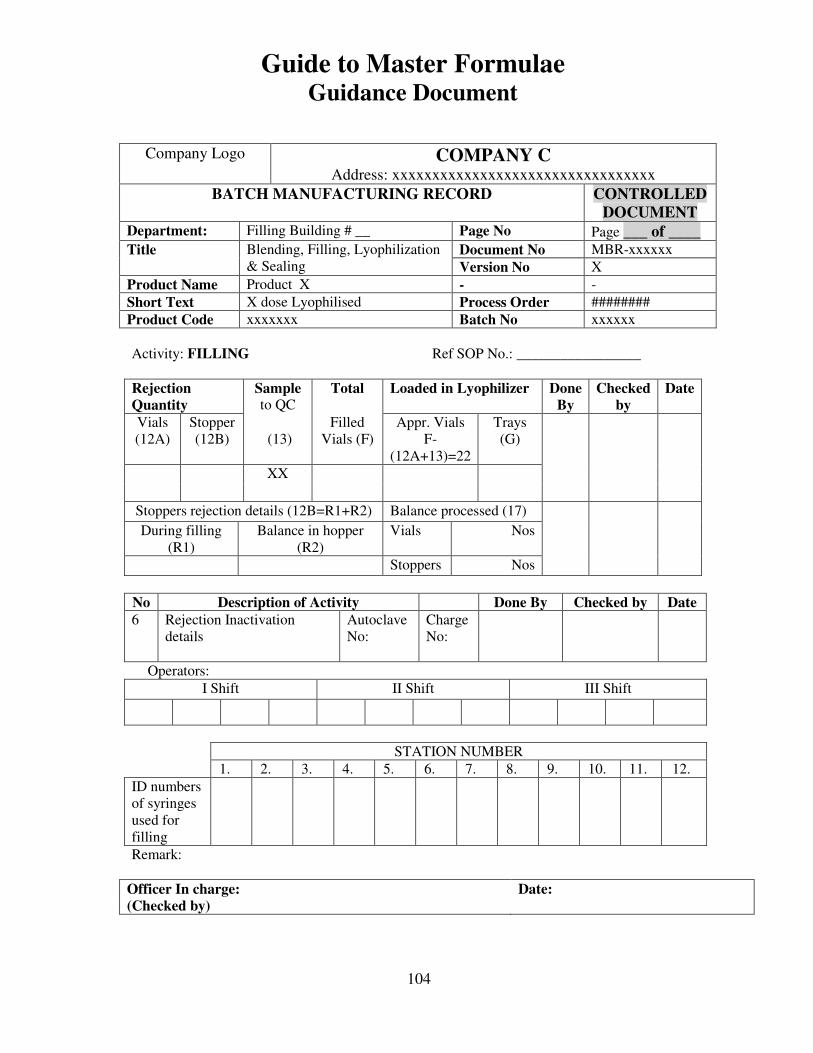
Guide to Master Formulae Guidance Document
104
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Activity: FILLING Ref SOP No.: _________________
Rejection
Quantity
Sample to QC
Total Loaded in Lyophilizer Done
By
Checked
by
Date
Vials
(12A)
Stopper
(12B)
(13)
Filled
Vials (F)
Appr. Vials
F-
(12A+13)=22
Trays
(G)
XX
Stoppers rejection details (12B=R1+R2) Balance processed (17)
During filling
(R1)
Balance in hopper
(R2)
Vials Nos
Stoppers Nos
No Description of Activity Done By Checked by Date
6 Rejection Inactivation
details
Autoclave
No:
Charge
No:
Operators:
I Shift II Shift III Shift
STATION NUMBER
1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12.
ID numbers
of syringes
used for
filling
Remark:
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
105
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Activity: FILLING Ref SOP No.: _____________________
1. At the start of filling, reject the vials used for checking the centering of filling needles,
volume setting etc. (Reject Not less than first xx vials).
Syringe
Number
Attach Fill Volume Print or record manually, on weight basis, every xx
Minutes
Time (h)
1
2
3
4
5
6
7
8
9
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
106
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Water
bath
temp
xx-xx 0C
Done by
*Checked
by
* Fill in case no printout is available, otherwise sign on printout
Remarks:-
Activity: INTEGRITY TESTING VENTS FILTERS Ref SOP No.:
________________
1. On completion of the Filling activity, carry out integrity testing of the two vent filters
(XXXX) of the Blending vessel tank attached to the filling machine by WIT method.
2. Fill the upstream side with WFI at xx-xx oC. A hold at xxxx mbar should not produce an
intrusion rate greater than xxx mL/min.
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
107
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
3. Attach printouts below:-
Remarks:-
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
108
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Activity: LOADING OF LYOPHILIZER Ref SOP No.: _________________
1. Ensure that the Lyophilizer to be loaded has been cleaned and Sterilized/disinfected, the
date of loading should not be later than the date on the board (Page. no 16).
2. The trays containing filled and half stoppered vials are to be transported to the Lyophilizer
in a mobile class 100 trolley.
3. Ensure that the temperature of the Lyophilizer shelves is –xx ºC before loading of trays
containing filled and half-stoppered vials.
4. After all the trays have been loaded in the lyophilizer, inform lyophilizer operator on duty.
He will check the loading of the lyophilizer, insert product temperature probes and close
the door of the lyophilizer.
No Description of Activity Done by Checked by Date
1. Checked, that Lyophilizer is □ Sterilized / □
Disinfected
2. Checked that Class 100 trolley is Switched “ON”
3. Vials Loaded in Lyophilizer No: _______
Remarks:-
Vol. Per
Vial (23) xx mL
Lyo.
No:-
Loading
date
Recipe
No.
Sterilization
Details (see
page 15)
□ Lyophilizer B, C:
1st Batch Steam
Steriliz’n.
□ Lyophilizer A: 1st
Batch Steaming.
□ 2nd
Batch / 3rd
Batch
All Lyophililzers XX
disinfection
For Filling Dept:
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
109
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
LYOPHILIZER CHAMBER CLEANING, DISINFECTION AND STERILIZATION: 1. For Lyophilizers B and C, If Status board ‘Cleaned, Ready for Steam exposure’ is
displayed on clean room side, evacuate chamber to xxx mbar (Water ring vac pump: WP),
followed by steam exposure at xxx - xxx °C for xx minutes.
2. For Lyophilizer A, If Status board ‘Cleaned, Ready for Steam exposure’ is displayed on
clean room side, evacuate chamber to xxx mbar (WP), followed by steaming for xx mins.
3. Above steps are followed by drying of chamber with WP for minimum xx mins.
4. For all Lyophilizers, If status board ‘Disinfected with XXX, Start evacuation’ is displayed
on clean room side, carry out evacuation as per SOP _____ (XXX cycle: xx min
evacuation by vacuum pump with shelf at xx °C).
No Description of Activity Done By Date
1. Status board: Lyo B and C:'Cleaned, Ready for Steam Exposure’
Process xxx - xxx °C maintained Process
Start Start End End
h h h h
2 Status board: Lyo A:Cleaned, Ready for Steam Exposure’
Vacuum Pull down Started xxx min Exposure Drying
Start End Steam Start End End
h h h h h h
Drain temperature at the end of Steaming: _______°C
3 Status board: ‘Disinfected with XXX, Start evacuation’
Shelf temp.
+xx°C
Evacuation with vac pump
attained at Start End
h h h
4. Display Board: ‘Ready for Loading up to: _________’
Officer In charge: Date:
(Checked by)
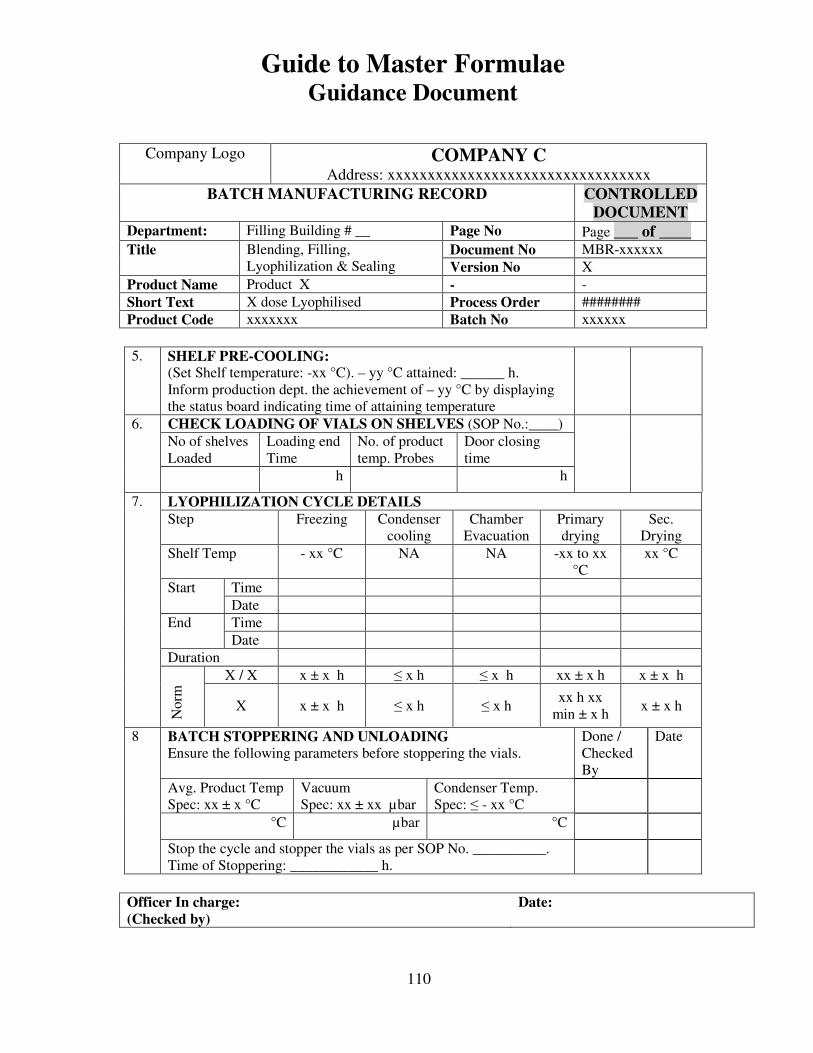
Guide to Master Formulae Guidance Document
110
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
5. SHELF PRE-COOLING:
(Set Shelf temperature: -xx °C). – yy °C attained: ______ h.
Inform production dept. the achievement of – yy °C by displaying
the status board indicating time of attaining temperature
6. CHECK LOADING OF VIALS ON SHELVES (SOP No.:____)
No of shelves
Loaded
Loading end
Time
No. of product
temp. Probes
Door closing
time
h h
7. LYOPHILIZATION CYCLE DETAILS
Step Freezing Condenser
cooling
Chamber
Evacuation
Primary
drying
Sec.
Drying
Shelf Temp - xx °C NA NA -xx to xx
°C
xx °C
Start Time
Date
End Time
Date
Duration
No
rm X / X x ± x h ≤ x h ≤ x h xx ± x h x ± x h
X x ± x h ≤ x h ≤ x h xx h xx
min ± x h x ± x h
8 BATCH STOPPERING AND UNLOADING Ensure the following parameters before stoppering the vials.
Done /
Checked
By
Date
Avg. Product Temp
Spec: xx ± x °C
Vacuum
Spec: xx ± xx µbar
Condenser Temp.
Spec: ≤ - xx °C
°C µbar °C
Stop the cycle and stopper the vials as per SOP No. __________.
Time of Stoppering: ____________ h.
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
111
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
9 Chamber vacuum released by sterile air at _______ h.
Instruct production department to unload the vials after breaking
the vacuum by air.
DEVIATIONS DURING LYOPHILIZATION (if any):____________
Activity: ALUMINUM CAPS PROCESSING Ref SOP No.: __________________
1. Verify that QC approved Aluminum Caps of correct colour have been taken for de-
cartoning.
2. Remove the outer package and transfer the inner plastic bag to the sterilizer loading area.
3. The caps can be processed in either of the two ways: A) Sterilization in Autoclave X and
drying in dry heat sterilizer (DHS) XX; B) Sterilization and drying in Autoclave Y. If
option A is selected, then transfer the caps into clean SS trays, load into the autoclave as
per validated loading pattern and sterilize it at XXX °C to XXX °C for xx minutes.
Pattern No 1 II
No of Caps/trays
4. After sterilization, transfer the trays containing the Aluminium caps from the autoclave to
the DHS via clean room side for drying at xxx °C for xx mins Normal variation – x °C to
+ x °C. 5. If option B is selected then process the caps at xxx °C ± x °C for xx minutes in the
autoclave.
5. Write /verify Product, B. No., Charge No and date on the sterilizer thermograph / printout
(for Option A only) sign it and attach it to this BMR.
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
112
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
A) Aluminum Cap input: 13 mm, Colour ______ Done
by
Checked
by
Date
1) Unprocessed b/f:
B.No_____________
2) Processed b/f:
B.No___________
3) Fresh
quantity
issued
4) Fresh
used
from issued
Nos Nos Nos Nos
5) Option of Process
� A =1+ 2 + 4
� B =1+ 4
Unprocessed
quantity
from this batch
(18) (3 - 4)
6) Taken for Sterility =
9
(For 1 / 2 batch/es)
Nos
No Description of Activity
B) Loaded in Autoclave No. ################
Charge No. Load Pattern No Prog No.
C) Aluminum Cap Drying at xxx °C in DHS No: ###########
Charge No. xxx °C Achieved at xxx °C Held till
h h
Remarks: - .
Activity: SEALING OF VIALS Ref SOP No.: ____________
Lyophilizer Unloading:
1. After the lyophilization operator on duty has informed you about completion of
lyophilization cycle, open the door of the Lyophilizer &unload the trays containing the
lyophilized vaccine.
Officer In charge: Date:
(Checked by)
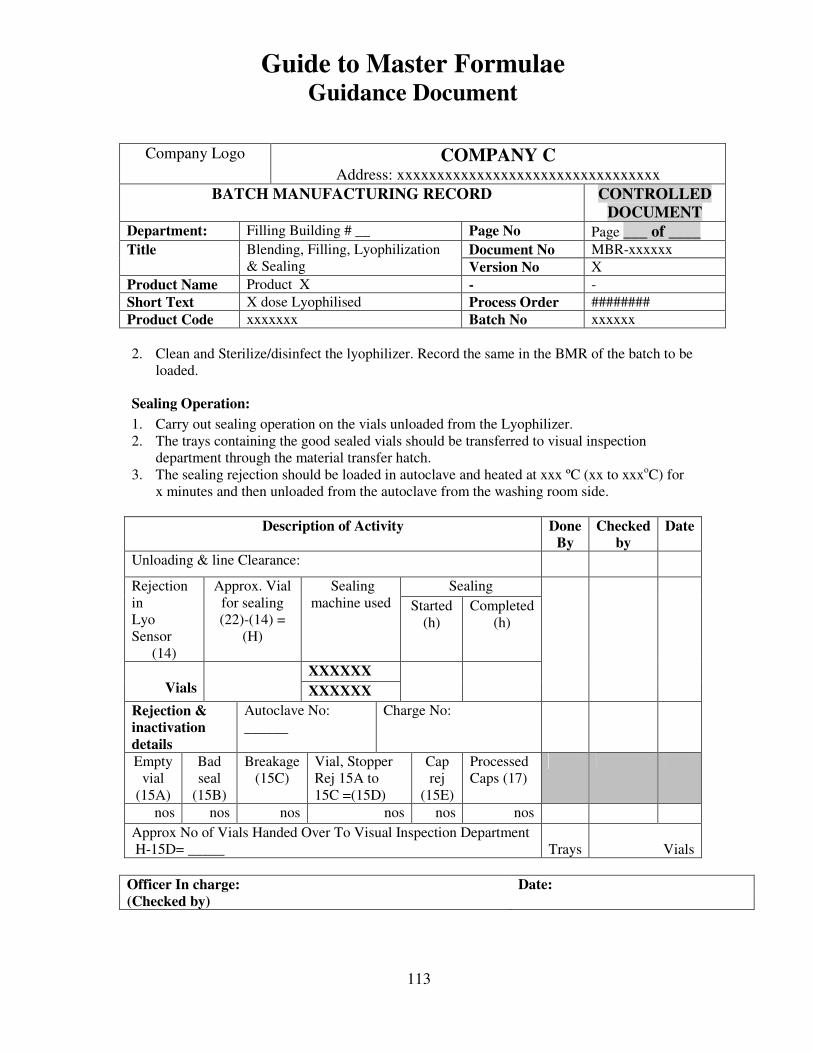
Guide to Master Formulae Guidance Document
113
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
2. Clean and Sterilize/disinfect the lyophilizer. Record the same in the BMR of the batch to be
loaded.
Sealing Operation:
1. Carry out sealing operation on the vials unloaded from the Lyophilizer.
2. The trays containing the good sealed vials should be transferred to visual inspection
department through the material transfer hatch.
3. The sealing rejection should be loaded in autoclave and heated at xxx ºC (xx to xxxoC) for
x minutes and then unloaded from the autoclave from the washing room side.
Description of Activity Done
By
Checked
by
Date
Unloading & line Clearance:
Rejection
in
Lyo
Sensor
(14)
Approx. Vial
for sealing
(22)-(14) =
(H)
Sealing
machine used
Sealing
Started
(h)
Completed
(h)
Vials
XXXXXX
XXXXXX
Rejection &
inactivation
details
Autoclave No:
______
Charge No:
Empty
vial
(15A)
Bad
seal
(15B)
Breakage
(15C)
Vial, Stopper
Rej 15A to
15C =(15D)
Cap
rej
(15E)
Processed
Caps (17)
nos nos nos nos nos nos
Approx No of Vials Handed Over To Visual Inspection Department
H-15D= _____
Trays
Vials
Officer In charge: Date:
(Checked by)
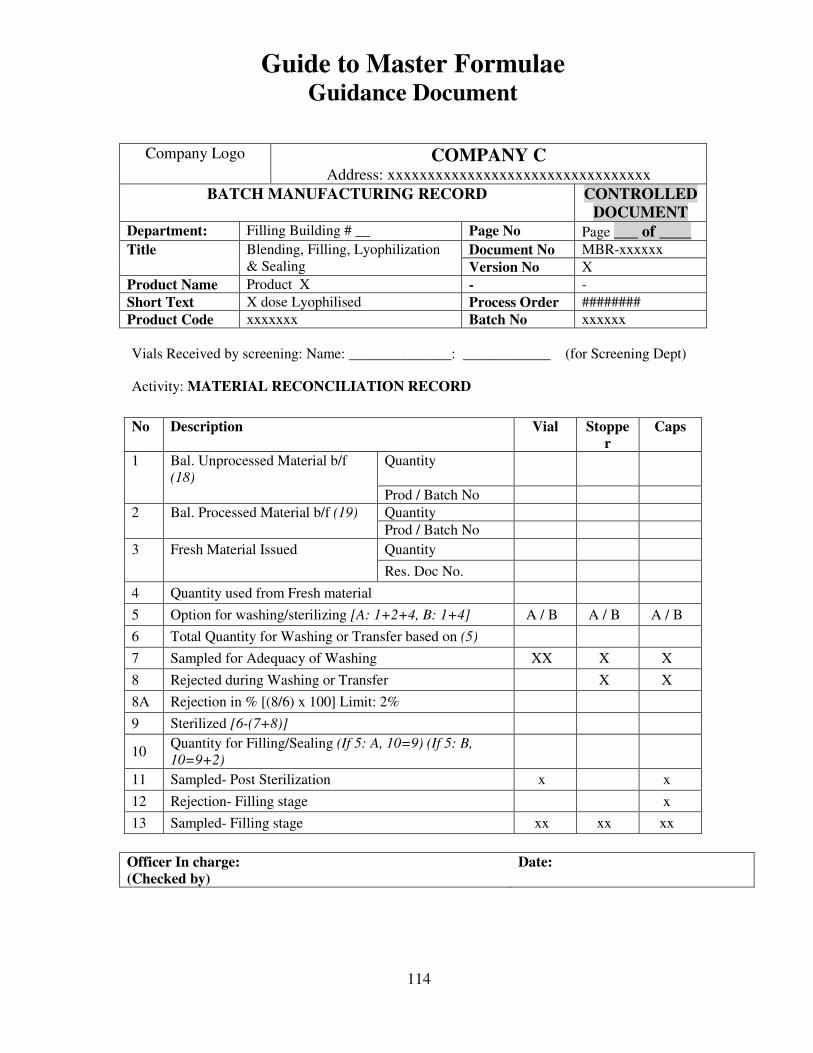
Guide to Master Formulae Guidance Document
114
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
Vials Received by screening: Name: ______________: ____________ (for Screening Dept)
Activity: MATERIAL RECONCILIATION RECORD
No Description Vial Stoppe
r
Caps
1 Bal. Unprocessed Material b/f
(18)
Quantity
Prod / Batch No
2 Bal. Processed Material b/f (19) Quantity
Prod / Batch No
3 Fresh Material Issued Quantity
Res. Doc No.
4 Quantity used from Fresh material
5 Option for washing/sterilizing [A: 1+2+4, B: 1+4] A / B A / B A / B
6 Total Quantity for Washing or Transfer based on (5)
7 Sampled for Adequacy of Washing XX X X
8 Rejected during Washing or Transfer X X
8A Rejection in % [(8/6) x 100] Limit: 2%
9 Sterilized [6-(7+8)]
10 Quantity for Filling/Sealing (If 5: A, 10=9) (If 5: B,
10=9+2)
11 Sampled- Post Sterilization x x
12 Rejection- Filling stage x
13 Sampled- Filling stage xx xx xx
Officer In charge: Date:
(Checked by)
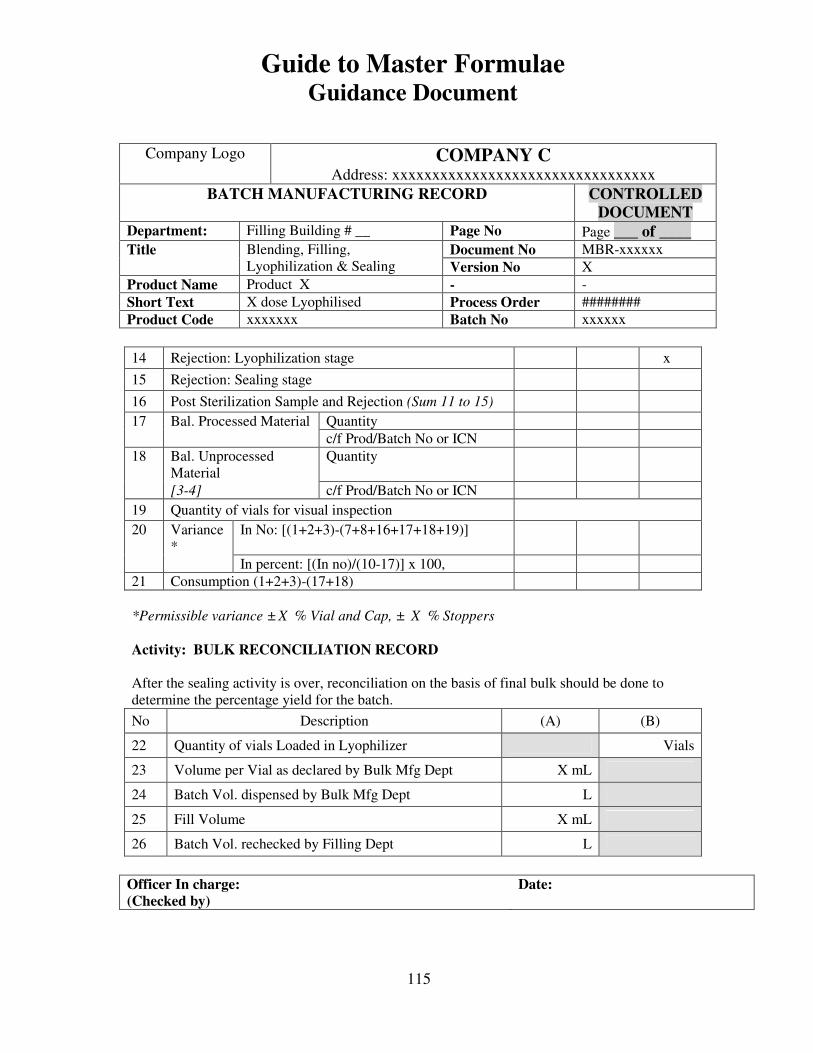
Guide to Master Formulae Guidance Document
115
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
14 Rejection: Lyophilization stage x
15 Rejection: Sealing stage
16 Post Sterilization Sample and Rejection (Sum 11 to 15)
17 Bal. Processed Material Quantity
c/f Prod/Batch No or ICN
18 Bal. Unprocessed
Material
Quantity
[3-4] c/f Prod/Batch No or ICN
19 Quantity of vials for visual inspection
20 Variance
*
In No: [(1+2+3)-(7+8+16+17+18+19)]
In percent: [(In no)/(10-17)] x 100,
21 Consumption (1+2+3)-(17+18)
*Permissible variance ± X % Vial and Cap, ± X % Stoppers
Activity: BULK RECONCILIATION RECORD
After the sealing activity is over, reconciliation on the basis of final bulk should be done to
determine the percentage yield for the batch.
No Description (A) (B)
22 Quantity of vials Loaded in Lyophilizer Vials
23 Volume per Vial as declared by Bulk Mfg Dept X mL
24 Batch Vol. dispensed by Bulk Mfg Dept L
25 Fill Volume X mL
26 Batch Vol. rechecked by Filling Dept L
Officer In charge: Date:
(Checked by)
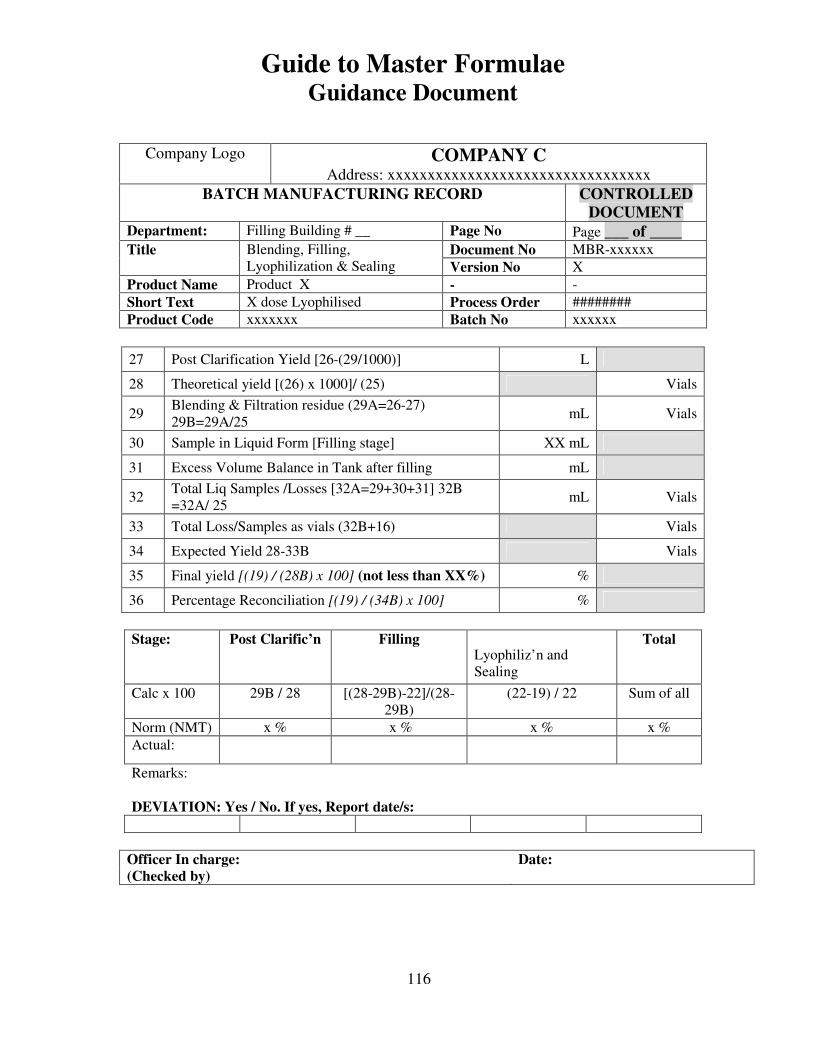
Guide to Master Formulae Guidance Document
116
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling,
Lyophilization & Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
27 Post Clarification Yield [26-(29/1000)] L
28 Theoretical yield [(26) x 1000]/ (25) Vials
29 Blending & Filtration residue (29A=26-27)
29B=29A/25 mL Vials
30 Sample in Liquid Form [Filling stage] XX mL
31 Excess Volume Balance in Tank after filling mL
32 Total Liq Samples /Losses [32A=29+30+31] 32B
=32A/ 25 mL Vials
33 Total Loss/Samples as vials (32B+16) Vials
34 Expected Yield 28-33B Vials
35 Final yield [(19) / (28B) x 100] (not less than XX%) %
36 Percentage Reconciliation [(19) / (34B) x 100] %
Stage: Post Clarific’n Filling Lyophiliz’n and
Sealing
Total
Calc x 100 29B / 28 [(28-29B)-22]/(28-
29B)
(22-19) / 22 Sum of all
Norm (NMT) x % x % x % x %
Actual:
Remarks:
DEVIATION: Yes / No. If yes, Report date/s:
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
117
Company Logo COMPANY C Address: xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
BATCH MANUFACTURING RECORD CONTROLLED
DOCUMENT Department: Filling Building # __ Page No Page ___ of ____
Title Blending, Filling, Lyophilization
& Sealing Document No MBR-xxxxxx
Version No X
Product Name Product X - -
Short Text X dose Lyophilised Process Order ########
Product Code xxxxxxx Batch No xxxxxx
BMR CERTIFICATION
01. MANUFACTURING DEPARTMENT:
The contents of this Document have been checked and verified by me.
The information contained herein is complete and true to the best of my
acknowledge. Deviations if any are reported.
Hence submitted to Quality Assurance Department.
Signature of Production Officer: ____________________________
Date of Completion : ___________________________
Date of Submission : ___________________________
02. QUALITY ASSURANCE DEPARTMENT:
I hereby certify that, this batch record is reviewed by me, to ensure that the above
mentioned batch process has been carried out according to the Authorized Master
formula and processing instructions.
All operational steps have been scrutinized and approved according to the checklist
(Attached) and have been found to be complete.
Signature of Quality Assurance Review Officer: __________________________
Date of Receipt : __________________________
Date of Approval : __________________________
Officer In charge: Date:
(Checked by)

Guide to Master Formulae Guidance Document
118
WHO Quality Safety & Standards of Vaccines and Biologicals
WHO has played a key role in developing WHO guidelines and recommendations on
the production and control of biological products and technologies. These
recommendations are aimed to assist WHO Member States in ensuring the quality and
safety of biological medicines worldwide, and are based on scientific consensus
achieved through international consultations as well as involving close collaboration
with the international scientific and professional communities, regional and national
regulatory authorities, manufacturers and expert laboratories worldwide.
Written guidelines and recommendations describe procedures for the manufacture and
quality control testing of biological medicinal products to ensure safe and effective
products. Guidance documents, like this one, provide more general information on a
range of topics of interest to National Regulatory Authorities (NRAs) and
manufacturers. It also advises NRAs and manufacturers on the control of biological
products, with the aim of establishing a harmonized regulatory framework for products
moving in international markets.





























