Embed Size (px)
Citation preview

Indian Journal of ChemistryVol. 33A, November 1994, pp. 969-977
Ground state vibrations of guanidinium and methylguanidinium ions-Anab initio study
D Chakraborty & S Manogaran*Department of Chemistry, Indian Institute of Technology, Kanpur 208 016, India
Received 27 December 1993; revised and accepted 15 June 1994
The results of ab initio SCFMO calculations using 6-31G* basis set for guanidinium - do,d6 andmethylguanidinium - do,ds .ions are presented. The optimized geometries from these calculationshave been compared with experimental bond distances and bond angles obtained from crystal struc-tures. The vibrational spectra (frequencies and intensities) for the compounds and their deuteratedisotopomers have been calculated using the same basis set. The predicted spectra have been com-pared with the experimental IR and Raman spectra of the molecules and their isotopomers in solidstate as well as in solution. The calculated potential energy distribution provides a basis for assigningthe experimental spectra. The isotopic frequency shifts for 2H substituted guanidinium ion and me-thylguanidinium ion are also found to be in close agreement with the experimental isotopic frequen-cy shift. The calculated IR intensity pattern also shows good agreement with the experimental spec-trum.
Guanidinium ion (GO+), [C(NH2)3l+, is a' simplelO-atom species that has been of much interest tobiochemists as a functional group present in theamino acid arginine and as a constituent of otherbiologically active molecules. Phosphate groupsare bound by GO +, including those constitutingthe side chains of arginyl residues in the enzymestaphylococcus nuclease, which has fundamentalimportance in structural biochemistry. GO +-car-boxylate interactions could prove to be the me-chanism through which tetrodotoxin and saxitoxinblock sodium channels in the nerve cell mem-brane. However, spectroscopists have shown littleinterest in this species and a very few referencesexist in the literature on the spectroscopy of thision. The reasons for this can be inferred from itsproperties. Because the ion exists only in polarsolvents or in the crystalline form, it is not verysuitable for IR studies, although there are a fewIR studies of GO" in crystalline form 1.2. Butseveral Raman studies in aqueous solution are re-ported in the literarure-:". Recently a resonanceRaman study of GO + and methylguanidinium(MGO ") ions has been reported by Sension andHudson".
The protonatcd form of guanidine (GO) (I), theguanidinium ion (GO +), belongs to a class of un-usually stable compounds often referred to as "y-aromatics". Indeed, the planar "V" shape allowsfor maximum dclocalization of positive chargethrough the p:-orhitals of nitrogen and carbon
atoms, thus givmg rise to a stable cation systemthrough substantial rr-overlap over the entiremolecule. This accounts for the fact that guani-dine is among the strongest of organic bases". The"Ydelocalization" of the GO and GO + is of im-portance in the stabilization and function of anumber of molecules of biological importance.These include the amino acid arginine, creatineand many purines including the nucleic acid baseguanine. Based on spectroscopic, crystallographicand theoretical studies, GO" has been assigned aplanar structure, with Dlh symmetry, consistentwith an electronic structure described by the fourresonance structures (II-IV) shown in Fig. l. Theground state electronic strucure" of GO"
indicates that the ion is in singlet spin state in itsground state with an overall charge of + 1. In thesubstituted GO + ,i.e., MGO" ion, the Dlh symme-try is destroyed due to the presence of methylgroup and C, symmetry results. Here, we addressourselves to the problem of possible differencesbetween the single GO + ion and arginine, usingMGD" as a model system for the latter.
GO + and MGO + ions have been the subject ofnumerous theoretical studies 1111 -; which have dealtprimarily with the torsional harrier for NH2 rota-tion II or with the hydrogen bonding interaction ofguanidinium ion with the carboxylate or phosph-ate I' I '. In their resonance Raman study, Scnsion

970 INDIAN J CHEM, SEe. A, NOVEMBER 1994
....HHII
H, .•••c..... ....HH HI IH H
H, ....H~ H•H, •...C ...•••... H
H HI IH H
II
v
H'H ....HI
H: ~c -, HH H ....I IH H
'"
H, ....HNI
H, .••.c... ~HH N....I IH H
IV
VI VII
Fig. I-Gaunidine(I), resonance structures of guanidiniumion(II-V); methylguanidinium ion(VI); and argininetVlf]
et a/.R calculated theoretically the excited electro-nic states of GD+ at the semiempirical level (IN-DO/S). But none of these theoretical studies in-clude a discussion of the vibrational frequenciesand their assignment in GD+ and MGD+, exceptthe normal coordinate analysis result reported byAngel et al), In recent years, reliable predictionof the vibrational spectra using MO theory hasproved to be very useful'v'? and it has helped tocorrect the possible misassignments of the ob-served vibrational frequencies. In this paper, wepresent an ab initio study of ground state vibra-tional frequencies and intensities of GD +, MGD +and their deuterated analogues. The agreementbetween the theoretical and experimental frequen-cies 1-8 is improved by the usual procedure of scal-ing the ab initio force constants. Further, the vi-brational frequencies of deuterated analogues ofGD+ and MGD+ have been calculated to checkwhether the force field is satisfactory to repro-duce the experimentally observed 2H shifts. An-other motivation for carrying out the presentwork was the realisation that study of GD+ andMGD+ was a key to the understanding of the vi-brational spectrum of arginine, for identifyingband characteristics of the guanidinium functionalgroup. All the results refer to the referencegeometry of the molecule obtained from the theo-retically fully optimized geometry.
Computational DetailsCalculations for GD+ and MGD+ ion were
performed at the Hartree-Fock level with Gaus-sian 90 program'S. The optimization of the mole-cules was performed by the use of geometry opti-mization facility with analytic gradient techniqueincluded in the program. Pople's!" 6-31G* split
valence basis set with six d-polarization functionson all the heavy atoms was used. The choice of6- 31G* basis set for optimization of the geometri-cal structure of the study system was made be-cause a basis set of this quality is required to pre-dict correctly the geometry of cationic molecule.The appropriate point group symmetry D3h forGD+ and C, for MGD+ was maintained through-out the calculation.
The HF cartesian force constants, vibrationalfrequencies and intensities were obtained analyti-cally at the optimized geometry; The cartesianforce constant matrix Fc was transformed to localcoordinate force constant matrix FL, through thetransformation 20:
A=M-I'Bl'G-1
where B' is the transpose of B matrix. Wilson's Band G matrices in internal coordinates for theoptimized structures were obtained through theuse of GMAT program of Schachtschneider ".The Band G matrices in internal coordinateswere transformed to B and G matrices in localcoordinates and the A matrix defined above wascomputed. The force field in local coordinates(FL) was obtained by the transformation,
FL= At'Fc'AThe scale factors used in the study to correctsystematic overestimation of the calculated forceconstants in the HF procedure were taken ac-cording to Durig et a/.22 Scale factors of 0.9 forstretching and 0.8 for bending and geometricmean of the scale factors for interaction forceconstants were applied.
The complete set of symmetry coordinates usedfor GD+ and MGD+ is listed in Tables 1 and 2.

CHAKRABORTY et al.:Ab inuio SCFMO CALCULATIONS FOR GD + & MGD + IONS 971
The force field in symmetry coordinates was ob-tained through appropriate transformation.' Thesecular equationIGF-lEI=Owas then solved using unsealed and scaled forceconstants to obtain the vibrational frequenciesalong with their potential energy distributionsamong the symmetry coordinates for the parentmolecule and its 2H isotopomer. The computa-tions were carried out at our Institute on a Con-vex C-220 computer system.
Results and Discussion(i) GeometryThe structures of GD+ and MGD+ were calcu-
lated using 6-31 G* basis set. The parameters ofthe optimized geometry are summarized in Table3: Since theoretical calculation dealt with the iso-lated molecule in the gas phase, the symmetry isconsidered to be D3h for GD +, which is due toY-aromaticity of the isolated system, while in thecrystalline phase symmetry is C3v due to the non-planarity (pyramidization) of NH2 groups. To res-
coordinateb
Table l-,Symmetry coordinates of GO· a
DescriptioncSpecies1.
Al / 2.J.1.
112/ 2.1.2.J.4.
E/ S.6.7.8.
9.10.11.12.
1.A //
I
A //1.2 2.
1.
2.E// J.
11.+~+~r1+r2+r3+r4+r5+r6
2Pl+2~2+2~J-fl-f2-fJ-·4-f5-f6rl-r2+r3~r4+r5-r6f1-f2+~J-f4+f5-~62R1-R2-RJ
R2-RJ
2r1+2r2-rJ-r4-rS-r6rJ+r4-rS-r6
2r1-2r2-rJ+r4-rS+r6rJ-r4-rS+r6
2cx1-cx2-cxJcx2-cxJ
4~1-2/32-2PJ-2~1+~3+~S-2~2+·4+~62P2-fJ+fs-2/3J-f4+f62~1-2f2-'3+~4-f5+~6
'3-~4-'5+'6~5.2.1.J+~5.2.1.4+~6.2.1.3+~6.2.1.4
+r 7 .J•1.t~7 .3 .1.4+~ 8 •2 .1.3+~ 8 .2 .1.4+~ +1: +~ . +~9.4.1.2 9.4.1.J 10.4.1.2 10.4.1.J'
V. C~
V.NH26 NH (bend)V.NH2p NH (in-plane)v.CNJv.C~V.NH2-, NH:z
V.NH2-. NH:zo5.CNJ(skeletalo5.C~ deform.)o5.NH26.N~P NH2(in-p1ane)p NH II
~ CNJ
8 3 .1.4 .2+8 2 .1.4 •J+83.1. 2.485.2.6.1+87.3.6.1+89.4.10.1
28 -8 -85.2.6.1 7.3.8.1 9.4.10.18 -87.3.8.1 9.4.10.1
2~5.2.1.3+2~5.2.1.4+2~6.2.1.3+2~6.2.1.4-r -~ -~ -~7.3.1.2 7.3.1.4 8.3.1.2 8.3.1.4-~ -~ -~ -~9.4.1.2 9.4.1.3 10.4.1.2 10.4.1.3
~7.3.1.2+~7.3.1.4+~8.J.1.2+~8.3.1.4-~9.4.1.2-~9.4.1.J-~10.4.1.2-~10.4.1.3
a .bInternal coordinates as defined In Flg. 2.cUnnorl\lallzed
v-strtchlng. 6-bcn<11ng. p-rocklng. r-out-of-plane bend.T-torsion. s-syaaetric and a antlsyaaetric
4.
'7 C~
'7NH2'7NH2
'7NH2~ eN3
972 INDIAN J CHEM, SEe. A, NOVEMBER 1994
trict the symmetry, the eN bonds were allowed toexpand symmetrically, followed by systematic ex-pansion of the NH bonds and then a concertedangle optimization. The initial geometrical par-ameters have been taken from the 6-31G opti-mized geometry of Spase and Russel". The opti-mized geometrical parameters are in good agree-
ment with the experimental results'". The equilib-rium geometry of MGD+ has C, symmetry. Since,in C, symmetry the symmetry element present isonly the molecular plane, the geometry optimiza-tion of MGD + was performed by taking C( 1)-N(3,4) and C(1)-N(2) in separate group and alsoN(3,4)-H and N(2)-H separately. This results in
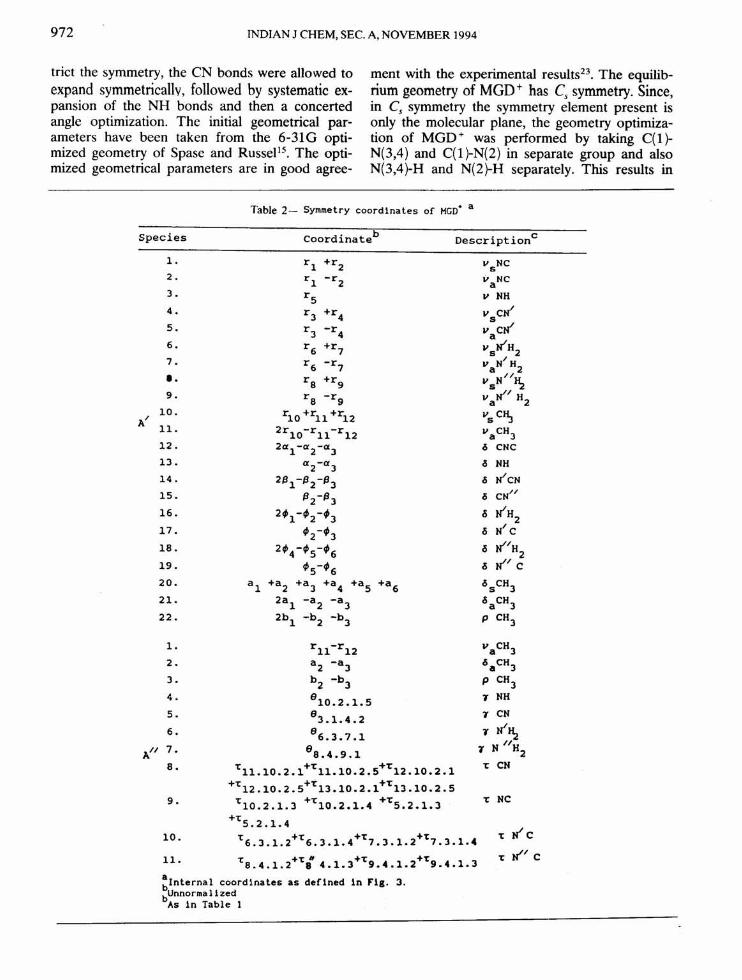
Table2- Symmetry coordinates of HGD+ a
Species coordinateb Description c
l. r1 +r2 VsNC2. r1 -r2 VaNC3. r5 v NH4. r3 +r4 vscJI5. r3 -r4 VacJI6. r6 +r7 VsJlH27. /r6 -r7 VaN H2I. ra +r9 VsN//~9. ra -rg vall/ H2
10. r10 +r11 +r12 VsC~A/ 1l. 2rl0-rll-r12 vaCH312. 2cx1-cx2-cx3 ~ CNC13. cx2-cx3 ~ NH14 . 2f31-f32-f33 s IIcN15. f32-f33 s CN//16. 2'1-'2-'3 <'iJlH217. '2-'3 <'iN/ Cla. 2'4-'5-'6 <'iJI/H
219. '5-'6 <'iJI/c20. a1 +a2 +a3 +a4 +a5 +a6 <'isCH32l. 2a1 -a2 -a3 <'iaCH322. 2b1 -b2 -b3 P CH3
l. r11-r12 VaCH32. a2 -a3 <'iaCH33. b2 -bJ P CH34. 810•2.1.5 T NH5. 83.1.4.2 T CN6. 86•3•7•1 T N/~
A// 7. 8a•4•9•1N //H
'1 28. Tll.l0.2.1+Tll.l0.2.S+T12.10.2.1 T CN
+T12.10.2.S+T13.10.2.1+T13.10.2.S9. T10•2.1.3 +T10• 2.1. 4 +TS•2•1•3 T NC
+TS. 2.1. 4T JI c10. T6.3.1.2+T6.3.1.4+T,.3.1.2+T,.3.1.4
11- Ta.4•1•2+Ti 4.1.3+T9.4.1.2+T9.4.1.3T JI/ C
a 1n Flg. 3.blnternal coordinates as def1nedbUnnormalized
As in Table 1

CHAKRABORTYet al.:Ab initio SCFMOCALCULATIONSFORGO+& MGO+IONS 973
a ' ,Table3- Geometricalparametersand energies for GD and HGD
Parameters GO' Expt. HGO' Expt.6-31Gto 6-31G••
Bond lengths(A)C-N 1 ••324 1.325 1.325 1.323
C-N(2) 1.319
N(2)-CH3 1.466 1.454
N-H 0.996 0.996N(2)-H 0.996C-H 1.081
Bond angles(deg)N-C-N 120.0 119.3 120.5 120.5N-C-N(2) 119.8C-N(2)-C 121.8 123.3H-N-C 120.0 121.6
H-N-H 120.0 116.8H-N(2)-C 121.8
N(2)-C-H 109.9Energy -204.521 -243.554a -18Energy in Hatree(lhR4.3589xlO per aolecule)
Fig.2-Intemal coordinatesof GO+;Rl-Cl- N2,R2-Cl-N3, R3-Cl-N4, rl-N2-H5, r2-N2-H6, r3-N3-H7,r4-N3-H8, r5-N4-H9, al-N3-Cl-N4, a2-N2-Cl-N4, a3-N2-Cl-N3, {J1-HS-N2-H6, {J2-H7-N3-H8, ~-H9-N4-H10, ,1-Cl-N2-HS, ,2-Cl-N2-H6, ;3-Cl-N3-H7, t64-Cl-N3-H8, ;5-Cl
-N4-H9, j66=Cl-N4- HIO
Fig.3-Intemal coordinatesof MGO+;rl-Cl- N2,r2""N2-ClO, r3-Cl-N3, r4-Cl-N4, r5-N2-H5, r6=N3-H6, r7-N3-H7, r8-N4-H8, r9-N4-H9, nn-rno-Hll, rll-ClO-H12, rl2-ClO-Hl3, al-CI-N2-C10, a2-Cl-N2-HS, a3-ClO-N2-HS, {J1-N2-Cl-N3, {J2-N3-Cl-N4, fJ3-N2-CI-N4, ;1-H6-N3-H7, ;2-CI-N3-H6, ;3-Cl-N3-H7, t64=H8-N4-H9, ;5-CI-N4-H8, fI6-CI-N4-H9, al =H12-CIO-H13, a2-Hll-ClO-H12, aJ-Hll-ClO-HI3, bl-N2-ClO-Hll, b2-N2-ClO-H12,
b3-N2-ClO-H13

974 INDIAN J CHEM, SEe. A, NOVEMBER 1994
C(1)-N(2) having a slightly higher double bondcharacter. Internal coordinates of Gd + andMGD+ are given in Figs 2 and 3 respectively. Abinitio calculations are expected to give a maxi-mum accuracy of ± 0.0l \A for bond lengths and± IO for angles in favourable cases 19.
(ii) FrequenciesSometimes the speculative assignment of the
observed bands of polyatomic molecules to spe-cific molecular vibrational modes can be con-firmed using theoretically calculated frequenciesand their assignments. These are given in Tables 4and 5 for both GD+ and MGD+ ions. In GD+,the symmetry considered is D3h and hence, all the24 vibrational modes are separated into6-symmetry blocks. These belong to 3A'1, 2A'2,12E' (all in-plane) and INI, 2Aw
2 and 4E- (allout-of-plane) irreducible representations. All thevibrational modes are not IR- and Raman-active,and so it is not possible to identify all of them ex-
perimentally. The A'2 (in-plane) and A-I (out-of-plane) modes are both IR- and Raman-inactive. Inconstructing the symmetry corodinate for GD +we changed the earlier assigned NH2 out-of-planetwist by the out-of-plane torsion around the CNbond. This shows an exact match with ab initiofrequencies.
In MGD + ion, the symmetry is C, and so allthe 33 vibrational modes belong to 22A' (in-plane) and UN (out-of-plane) irreducible repre-sentation. Since the only constraint in the C,symmetry is the molecular plane, in constructingthe symmetry coordinates, instead of taking allthe CN bonds as a group frequency (CN3), weseparated them as CN(3,4} and CN(2) and sim-ilarly the NH frequencies also. As a result, our as-signments differ from the earlier results':", whichwe think were rather oversimplified. In MGD + allthe modes are IR- and Raman-active. Calculationshave shown that values of harmonic vibrationalfrequencies computed with a split valence (6-
Table 4- V1bratlonal frequenc1es a of GD' and GD' (d6)
Asslgnment d' if c dSym. 6-31G. Expt.~ Raman C(NPJl Expt_, Shlftspecies scaled into act. 6-JIGI calc. (expt.
1. 3635 -, N~ 3328 0.0 164.5 2631 2410 1004 918(sh) (5)
A~ 2. 1662 c5 NH 16)0 0.0 0.) 1208 1257e )74 )7)· z
3. 1041 v CN 1007 0.0 15.1 929 916e 112 91• J (V5)
AI 1. 3744 V.N~ 0.0 0.0 2777 967z
2. 1028 P Nfl 0.0 0.0 776 2522
1- 3750 v NH 3410 235.4 44.7 2786 2564 964 846• z (5) (5h)
2. 3615 v NH 3275 220.1 0.3 2611 2410 1004 865• z (5) (5h)
EI 3. 1695 vCN +c5NH 1660 535.0 5.2 1642 1587 53 733 2 (5) (!?h)
4. 1570 c5NH +vCH 1553 31.3 0.5 1169 1157 401 3962 ) (w) (5)
5. 1115 pNH2 +vCtI 1120b 11.2 0.3 900 9,28 215 192)
6. 498 c5CN +PNHz 526 0.2 1-2 418 452 ):> 80 74) (w)
A" 1. 182 't CN3
0.0 0.0 129 53I
1- 717 7 CN) 724b 53.8 0.0 715 722 2 2A" 2. 390 ,. NH2 520 1059.8 0.0 290 1002
(w)1- 560 't CN 0.0 2.0 405 155 100
1/ )
E 2. 193 ,- NH 0.0 0.0 168 25)
a -1 b -1 c1n Ao4 llIIu-1dFrequencies 1n cm , In KIll molExperimental frequencies, IR Intensities and Raman activities
are taken from ref. 2. sh-shoulder, vs-very strong, s-strong,w-weak.
eAccording to ref. 1.
CHAKRABORTY et al.: Ab initio SCFMO CALCULATIONS FOR GD + & MGD + IONS 975
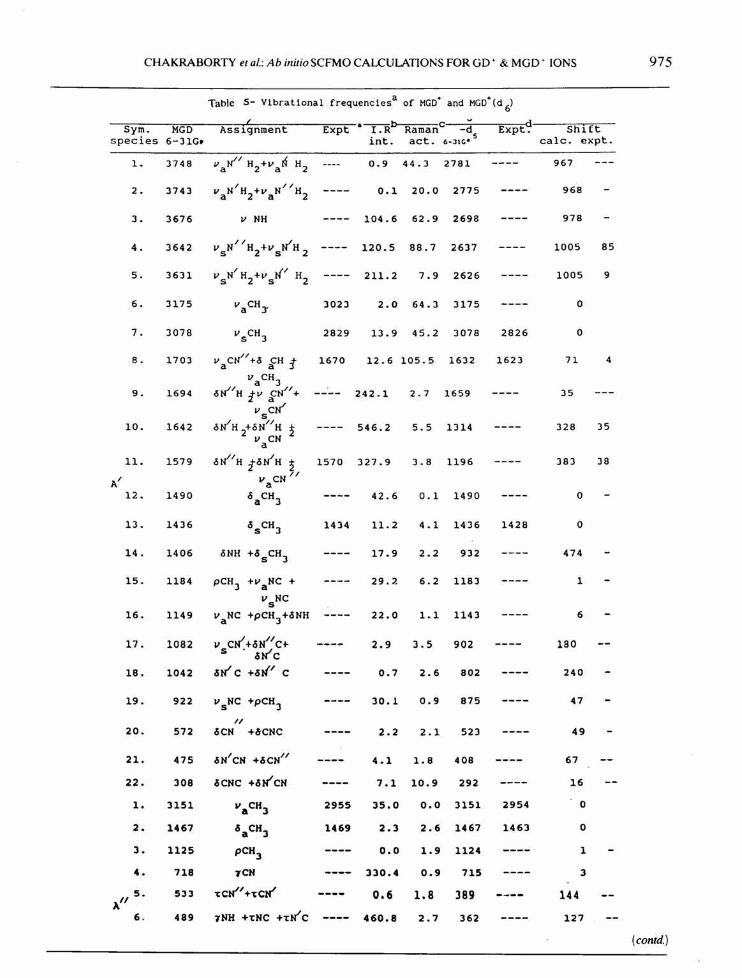
Table 5- Vlbratlonal frequencies a of MGO+ and MGO+(d 6)( a--b c dSym. MGO Assignment Expt l.R Raman -d Expt. Shirt
species 6-31G. into act. s calc. expt.6-:11Ce
1. 3748 Valil H2+v aN H2 0.9 44.3 2781 967
2. 3743 I II 0.1 20.0 2775 968VaN H2+VaN H2
3. 3676 V NH 104.6 62.9 2698 978
4. 3642 II Ii 120.5 88.7 2637 1005 85v sN H2+Vs H 2
5. 3631 I NI H2 211.2 7.9 2626 1005 9VsN H2+vs
6. 3175 vaCH)' 3023 2.0 64.3 3175 0
7. 3078 vSCH3 2829 13.9 45.2 3078 2826 0
8. 1703 V CN" +0 CH :t 1670 12.6 105.5 1632 1623 71 4a avaCH3
9. 1694 oli/H tv fN" + 242.1 2.7 1659 35V CWs
10. 1642 oWH +ON"H 2 546.2 5.5 1314 328 352 v CNa
1I. 1579 oli/H toN/H 2 1570 327.9 3.8 1196 383 38AI
v CN IIa12. 1490 °aCH3 42.6 0.1 1490 0
13. 1436 °sCH3 1434 11.2 4.1 1436 1428 0
14. 1406 oNH +.ssCH3 17.9 2.2 932 474
15. 1184 pCH3 +v NC + 29.7- 6.2 1183 1av NCs
16. 1149 v NC +pCH3+.sNH 22.0 1.1 1143 6a
17. 1082 v cN"+.sN"1 C+ 2.9 3.5 902 180s .sWC18. 1042 .sWC +.sN' C 0.7 2.6 802 240
19. 922 vsNC +pCH3 30.1 0.9 875 47I(
20. 572 .sCN +6CNC 2.2 2.1 523 49
21. 475 .sNICN +.sCNI( 4.1 1.8 408 67
22. 308 sese +.sN"CN 7.1 10.9 292 16
1. 3151 VaCH3 2955 35.0 0.0 3151 2954 0
2. 1467 c!5aCH3 1469 2.3 2.6 1467 1463 0
3. 1125 pCH3 0.0 1.9 1124 14. 718 TCN 330.4 0.9 715 35. 533 'tcN"+'tcN' 0.6 1.8 389 144A"6. 489 TNH +'tNC +'tN'C 460.8 2.7 362 127
(contd.)

976 INDIAN J CHEM, SEe. A, NOVEMBER 1994
Table 5- V1brat1onal frequenc1esa of MGD+ and MGD+ Cd 6) - Contd.
( a--b c d--~Ass Lqnment; Expt I.R Raman -.!:!s Expt. Sh~(t
into act. 6-JIC' calc. cxp t ,Sym. MGD
species 6-31G.
7. 396
8. 279
9. 246
10. 165
11. 95
rN//H trN/H 2 0.2 0.3 302 94
/ ~/rN H /1 H 2 28.5 0.1 201 76
rNH +1~/H 2+ 2.1 0.4 234 12"t~C
"tCN +rNH +"tN//C ---- 6.3 0.1 156 7
"tNC +"t~/ C+ 1.4 0.9 73 22"tN/C
~FreqUenC1es are 1n cm-1• b1n Km mol-1Exper1mental data comes from ref. 8.
31G*) basis set are generally 10-15% higher whencompared with the experimental frequencies dueto the effect of electron correlation and anhar-monicity". The predicted wave numbers and rela-tive intensities of the absorption bands generallyagree with the experimental values depending onthe region of absorption.
(a) Guanidinium ion (CD + )
The calculated and experimental spectral resultsof GD+ and GD+(d6) are given in Table 4. InGD + we consider CN) as a group vibration. Cal-culated values of CN) symmetric stretching [1041cm-I (A'I)' 1695 cm-I (E')], CN) angle deforma-tion (in-plane skeletal deformation) [498 em - I
(E')] and CN) out-of-plane angle deformation [717em - 1 (A" 2)] are in good agreement with the exper-imental numbers, i.e., 1007, 1660, 526, 724 cm - I
respectively. For NH2 group vibration, the calcu-lated values of NH 2 symmetric stretching are[3615 cm-I (E') and 3635 cm-I (A'I)] while theexperimentally observed values are 3275, and3328 cm -I. The calculated values of NH2 anti-symmetric stretching are [3744 cm -I (A'2) and3750 cm-I (E')]; the latter is experimentally ob-served at 3410 cm - I and the former is not ob-served experimentally, since, this band is IR- aswell as Raman-inactive according to selection ruleof D)h symmetry. NH2 rocking and deformationmodes are calculated at [1028 cm-I (A'2)' 1115em -I (E'), 1570 ern -I (E') and 1662 em -I (A'I)];the last three are experimentally observed at1120, 1553, and 1630 cm-I, and the first one isan inactive mode. Though ab initio frequenciesand assignments are best possible theoretical re-sults obtained through exhaustive calculation, stillthere are some limitations in predicting the lower
frequency torsional and out-of-plane bendingmodes. Thus the broad band observed at 520em - I in the IR spectra of Angell et al.I was as-signed earlier as NHz out-of-plane bending mode;it is calculated to be at 390 em -I (A"2)' but theo-retically it appears as one of the strongest peaks.Since theoretical intensities are reliable only forcomparative study, the earlier assignment may notbe correct. Angell et ai: have also assigned twovery weak bands as NH2 twisting modes at 830em - I (E") and 500 em - I (E"'), but we did not getany theoretical relevance of such bands; rather,two E'" symmetry frequencies are assigned here asteN) and rNH2 at 560 and 193 cm-I respect-ively.
For a definite evidence in support of the assign-ment for the vibrational spectra of a compound,isotopic frequency calculation is necessary. In thedeuterated spectra of GD + ion, the agreement inthe shifts for the calculated and observed frequen-cies is very good. All the NH2 group vibrationsshifted to lower value in - d6 compound, as ex-pected. Lesser or even no shifts in the in and out-of-plane vibrations of CN 3 group indicate the cor-rect assignment for the compound.
The calculated 1R intensities and Raman activit-ies with 6-31G* basis set for the compound arealso included in Table 4. Though the calculatedIR intensities and Raman activities are qualitative,still these can be used to compare the activities ofbands, i.e., whether these are IRiRaman-active ornot and in this respect the calculated intensitiesagree well with the experimental spectra. Othertheoretically most intense IR bands are observedat 3750, 3615 and 1695 cm-I. Experimentalspectra of guanidinium chloride 1,2 also show veryintense peaks at 3410, 3275 and 1660 em-I. The

CHAKRABORTY et al.: Ab initio SCFMO CALCULATIONS FOR GO + & MGD + IONS 977
experimental Raman activity- also matches wellwith the calculated one except for the bands at1007 (vs) and 1553 cm-I (s) which indicate thatthe theoretical Raman activities are still not goodenough to produce a one-to-one correspondencewith the experimental spectra, but can be used fora qualitative comparison.
(b) Methylguanidinium ion (MGD+)Theoretically calculated results of MGD+ and
its - d5 analogue are given in Table 5. The loss ofsymmetry in methylated GD+ makes a detailedvibrational analysis much more complicated. TheCH3 symmetric stretching is calculated at 3078em - 1 (X), and is observed at 2829 ern - I, whileCH3 antisymmetric stretchings are calculated at[3175 cm-I (X) and 3151 cm' (X)] and are ob-served at 3023 and 2955 cm-I respectively. TheCH3 symmetric and antisymmetric deformationand rocking modes are calculated at [1436 cm-I(X), 1467 cm-I (X) and 1184 cm "! (X), 1125em -I (X)] while the observed values are 1434(bsCH3, X) and 1469 cm-I (baCH3, AW). Asym-metric CN stretching is observed at 1670 cm-Iwhile calculated value is 1703 cm-I (X), showinggood agreement with the experimental spectrum.Since for MGD + very few experimental data areavailable from literatures, mainly the CH3 groupvibrations, and remaining important NH2 groupand CN 3 group vibrational frequencies (experi-mental) are compared with the results for GD+ion in Table 4. A good match is observed. On se-lective deuteration [C(ND2hNDCH3], all the NH2group vibrations shifted to lower values, while theCN 3 and CH3 group vibrational shifts were com-paratively less and sometimes almost zero, indi-cating correct assignment of the normal modes.IR intensity and Raman activity for the com-pounds are also included in Table 4. Since wehave used MGD + as a model for the guanidiniumpart of the arginine molecule, due to their structu-ral similarity, this complete theoretical study ofMGD+ gives us a flavour of the arginine spectralassignment and our calculated values are quiteclose to those observed in arginine spectrum".
ConclusionThe ab initio calculations of the vibrational
spectra of GD + and MGD + and their 2H isotop-omers at the Hartree-Fock level with 6-31G*basis set indicate that the ab initio force field is agood theoretical basis for calculating the vibra-tional frequencies and intensities of the cationiccompounds. The calculated isotopic frequencyshifts are in very good agreement with the experi
mental isotopic shifts. The results further revealthe usefulness of the theoretical intensity data inthe assignment of the molecular vibrations.
AcknowledgementWe thank prof. DN Sathyanarayana, I I Sc.,
Bangalore for a copy ofVECEIG program.
References1 Angell C L, Shepard N, Yamaguchi A, Shimanouchi T,
Miyazawa T & Mizushima S, Trans Faraday Soc. 53(1957) 589.
2 Bonner 0 0 & Jordon C F, Spectrochim Acta, 32 (1976)1243.
3 Edshall J T, J phys Chern, 41 (1937) 133.4 Otvos J W & Edshall J T, J chern Phys, 7 (1939) 632.5 Gupta J,1 Indian chern Soc. 13 (1936) 575.6 Bonner 0 0, J phys Chern, 81 (1977) 2247.7 Ananthakrishnan R, Proc indian Acad Sci A, 5 (1937)
200.8 Sension R J, Hudson B & Callis P R, J phys Chern, 94
(1990)4015.9 Pauling L, The nature of the chemical bond, 3rd Edn
(Cornell University, Ithaca, NY) 1960,286.10 Kollman P, Mackelvey J 8. Gund P J, J Am chem Soc, 97
(1975) 1640.11 Capitani J J & Pedersen L, Chern Phys Leu, 54 (1978)
547.12 Spase A M & Massa L J,1 org Chern, 45 (1980) 719.13 Cotton F A, Hazen (Jr) E E, Day V W, Larsen S, Nor-
man J G, Wong S T K & Johnson K H, J Am chem Soc,95 (1973) 2367.
14 Herzig L, Massa L J, Santoro A & Spase A M, J orgChern, 46 (1981 i2330.
15 Spase A M & Russel C S, Int J quantum Chern, 26(1984) 91.
16 Hess (Jr) B A, Schaad L J, Carsky P & Zahradnik R,Chern Rev, 86 ( 1986) 709.
17 Fogarasi G & Pulay P, Vibrational spectra and structure,Vol 14, edited by J R Durig (Elsevier, Amsterdam) 1985and references therein.
18 Gaussian 90, Frisch M J, Head-Gordon M, Trucks G W,Forseman J B, Schlegal H B, Raghavachari K, Robb M,Binkley J S, Gonzalez C, Defrees 0 J, Fox 0 J, WhitesideR A, Seeger R, Melius C F, Backer J, Martin R L, KahnL R, Stewart J J P, Topiol S & Pople J A (Gaussian Inc,Pittsburgh PAl 1990.
19 Hehre W J, Radom L, Schleyer P V R & Pople J A, Abinitio molecular orbital theory, (Wiley, NY) 1986.
20 Crawford B L & Fletcher W H, J chem Phys, 19 (1954)141.
21 Schachtschneider J H, Vibrational analysis of polyatomicmolecules, Part-5, Technical Report No. 23164 (ShellDevelopment Company, Emeryville, CA) 1964.
22 Durig J R, Wang A Y, Little J S & Brletic P A, J chemPhys, 93 (1990) 905.
23 Sundaralingam M & Chwang A K, Carbonium ions, Vol5, edited by G A Olah & P V R Schleyer (Wiley, NY)1976,2427.
24 Pople J A, Schlegal H B, Krishnan R, Defrees 0 J, Bin-kley J S, Frisch M J, Whiteside R A, Hout R A & HehreW J,Int] quantum Chern Symp, 15 (1981) 269.
25 Krishnan R S, Sankaranarayanan V N & Krishnan K, JIndian Inst Sci, SS (973)66.